 Chi-E Qiu
Chi-E Qiu Lei Lei1†
Lei Lei1† Guosong Jiang
Guosong Jiang- 1Hematology Department, Northeast Yunnan Central Hospital, Zhaotong, Yunnan, China
- 2Department of Pulmonary and Critical Care Medicine, The First People’s Hospital of Zhaotong & The Zhaotong Affiliated Hospital of Kunming Medical University, Zhaotong, Yunnan, China
- 3Clinical Laboratory, The First People’s Hospital of Zhaotong & The Zhaotong Affiliated Hospital of Kunming Medical University, Zhaotong, Yunnan, China
Hereditary spherocytosis (HS) is a genetic hemolytic disorder primarily characterized by hemolytic anemia, jaundice, splenomegaly, and frequent complications, including cholelithiasis, accompanied by the presence of spherocytes in the peripheral blood. This disorder predominantly follows an autosomal dominant inheritance pattern; however, certain cases exhibit an autosomal recessive mode of inheritance. HS is the most prevalent disorder associated with defects in the red blood cell membrane. Primary myelofibrosis (PMF), a chronic myeloproliferative neoplasm (MPN) characterized by splenomegaly resulting from extramedullary hematopoiesis, is associated with the JAK2 V617F mutation. Currently, there are no documented instances of co-occurrence of HS and PMF in the literature. We report the case of a 37-year-old male who experienced recurrent abdominal distension and splenomegaly over the past decade, along with elevated platelet counts over the past nine years. The patient tested positive for the JAK2V617F mutation, and bone marrow smears revealed the presence of teardrop-shaped erythrocytes. Peripheral blood smears indicated the presence of approximately 20% of spherocytes. The morphology of the bone marrow biopsy specimen was consistent with an MPN, classified as MF-2 grade. The highly specific eosin-5’-maleimide binding assay demonstrated a reduced mean fluorescence intensity of 25.73%. The patient was managed with aspirin and ruxolitinib and continued to be monitored through follow-up evaluations.
1 Introduction
Hereditary spherocytosis (HS) is an inherited hemolytic disorder that can present at any age and is characterized by hemolytic anemia, jaundice, and splenomegaly, frequently complicated by gallstone formation, with the presence of spherocytes in peripheral blood smears. The incidence of HS is estimated to be approximately 1 in 2,000–3,000 individuals (1). Although specific incidence data for China are not available, HS is recognized as the most prevalent disorder of red blood cell membrane defects in the region (2). The clinical severity of HS varies, with the condition being predominantly inherited in an autosomal dominant manner, although certain cases demonstrate autosomal recessive inheritance (3). Diagnosis is primarily based on clinical presentation, family history, and laboratory evaluations, including peripheral blood spherocyte (PBS) count and the osmotic fragility test (OFT). Nevertheless, due to atypical clinical presentations in certain patients, the absence of family history, the lack of definitive diagnostic thresholds for PBS count, and suboptimal sensitivity and specificity of the OFT, there are significant rates of missed and misdiagnosed cases associated with HS (2).
Primary myelofibrosis (PMF) is a type of chronic myeloproliferative neoplasm (MPN) characterized by an insidious onset, often marked by significant splenomegaly, prominent hyperplasia of megakaryocytes and granulocytes in the bone marrow, reactive fibroconnective tissue deposition, and extramedullary hematopoiesis. Peripheral blood exhibits nucleated red blood cells and immature granulocytes, while bone marrow aspirate often yields a dry tap and displays hypoplasia. Its pathogenesis involves driver mutations in JAK2, CALR, and MPL, leading to the hyperactivation of the JAK-STAT signaling pathway, immune system dysregulation, and chronic inflammation. Immune system dysregulation is a major pathogenic mechanism in MF and serves as a key mediator of disease progression (4). The JAK2 V617F mutation is one of the critical molecular markers for PMF diagnosis (5). Although not all patients with PMF carry this mutation, its high incidence makes it an important reference for diagnosis (6).
Splenomegaly is intricately linked to a range of hematologic disorders and frequently manifests as a clinical symptom of these conditions. As the largest lymphoid organ, the spleen is integral to blood filtration, the removal of senescent or abnormal blood cells, and the facilitation of immune responses (1). In the context of hematologic diseases, splenic enlargement can occur due to various pathological mechanisms. In cases of hemolytic anemia, increased destruction of red blood cells is the primary contributor to splenomegaly (7). This enhanced erythrocyte destruction, stemming from various etiologies, stimulates hematopoietic activity in the spleen, resulting in its enlargement (8). In myeloproliferative disorders, such as PMF, the normal hematopoietic function of the bone marrow is compromised, prompting the spleen to serve as a site for extramedullary hematopoiesis. This activation of extramedullary hematopoiesis leads to a significant increase in splenic size (9).
In this patient, the presence of splenomegaly was attributed to both HS and PMF. HS does not result in elevated white blood cell or platelet counts, while PMF seldom produces a substantial number of spherocytes. Given that a singular diagnosis could not account for the entirety of the patient’s clinical manifestations, additional diagnostic evaluation was performed using the highly specific eosin-5’-maleimide (EMA) binding assay. This assay confirmed the coexistence of HS and PMF.
2 Case description
A 37-year-old male patient with a history of hypertension and no reported family history of anemia presented with a decade-long history of recurrent abdominal distension and splenomegaly, accompanied by elevated platelet counts persisting for nine years. Abdominal ultrasonography identified severe splenomegaly, characterized by a thickness of approximately 10 cm and a longitudinal diameter of approximately 20 cm, with the lower edge extending below the umbilical level and the medial border reaching the anterior midline. A patchy hypoechoic region was noted at the lower pole of the spleen, the etiology of which remains indeterminate but is most likely suggestive of an infarction (Figure 1).
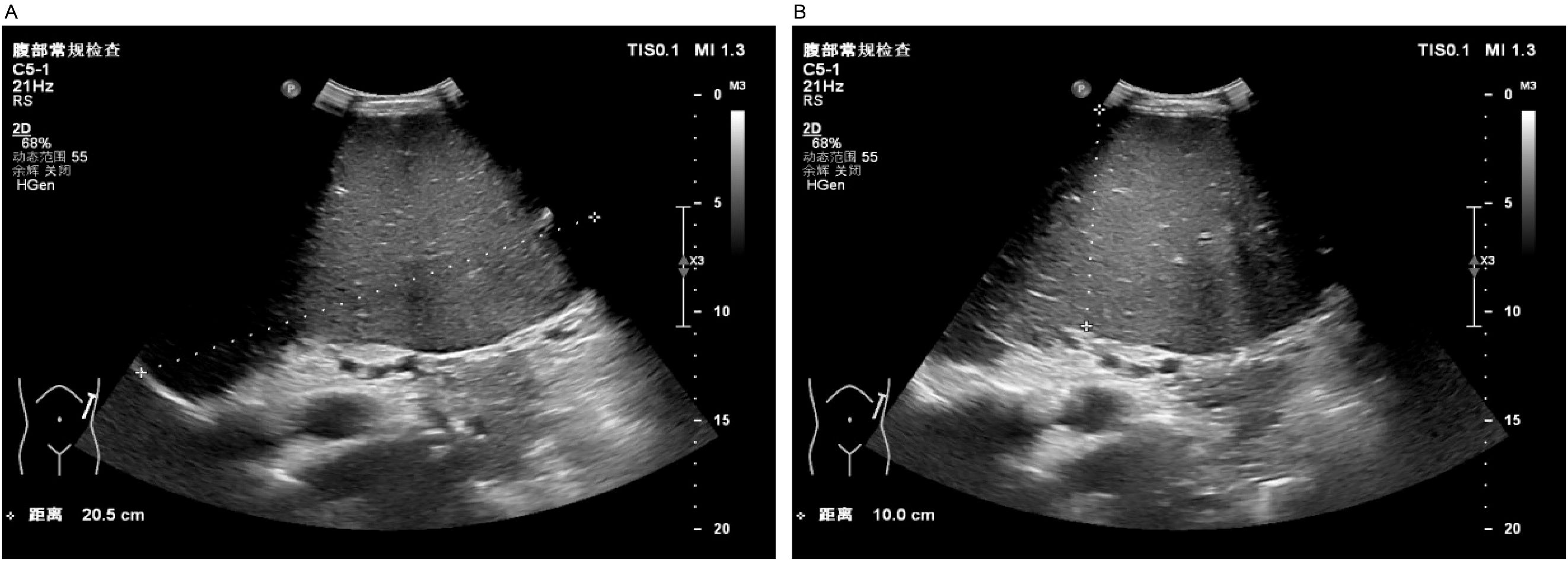
Figure 1. Abdominal ultrasound. Severe splenomegaly (approximately 10 cm in thickness, approximately 20 cm in the upper pole diameter, with the lower margin extending below the umbilical level and the medial border reaching the anterior midline). Panel (A) shows the craniocaudal diameter of the spleen, while Panel (B) illustrates the thickness of the spleen.
Initial laboratory investigations revealed a platelet count of 772 × 109/L, a white blood cell count of 15.40 × 109/L, and a lactate dehydrogenase level of 315.00 U/L. The initial hemoglobin concentration was 101 g/L; however, during the clinical workup for his progressive symptoms, his hemoglobin level was documented to have declined to 74 g/L, indicating disease-related anemia. Peripheral blood smear analysis demonstrated hyperplasia across granulocytic, erythroid, and megakaryocytic lineages, with increased platelets in both the bone marrow and peripheral circulation, along with teardrop-shaped erythrocytes (Figure 2). Bone marrow smear analysis revealed the presence of approximately 20% spherocytes (Figure 3). The EMA mean fluorescence intensity exhibited a reduction of 25.73% (Figure 4). According to the pertinent literature, a reduction exceeding 16% compared to healthy individuals is indicative of HS, whereas a reduction of 16% or less is not (10). The osmotic fragility test yielded a negative result, as did the G-6-PD fluorescent spot test. The direct antiglobulin test results were also negative. Comprehensive screening for thalassemia gene mutations did not reveal any variants within the detectable range for both α- and β-thalassemia. Morphological analysis of the bone marrow biopsy was consistent with a myeloproliferative neoplasm of MF-2 grade (Figure 5). Allele-specific quantitative PCR identified the JAK2 V617F mutation with an allele burden of 53.167%. Subsequent cytogenetic analysis of myeloproliferative disorders did not detect any additional genetic alterations, including mutations in CALR, MPL, BCR-ABL1, or other JAK2 mutations, aside from JAK2 V617F. Although myelofibrosis could account for the patient’s elevated white blood cell count, thrombocytosis, and splenomegaly, it did not explain the presence of 20% spherocytes in the peripheral blood. Therefore, a highly specific EMA binding test was conducted. The diagnoses were as follows: (1) HS; (2) PMF; (3) Hypertension. Regarding treatment and outcome, the patient was administered aspirin and ruxolitinib. However, due to a demanding work schedule, the patient did not consistently adhere to the prescribed medication regimen or attend regular follow-up appointments, leaving treatment efficacy under ongoing evaluation during subsequent follow-ups.
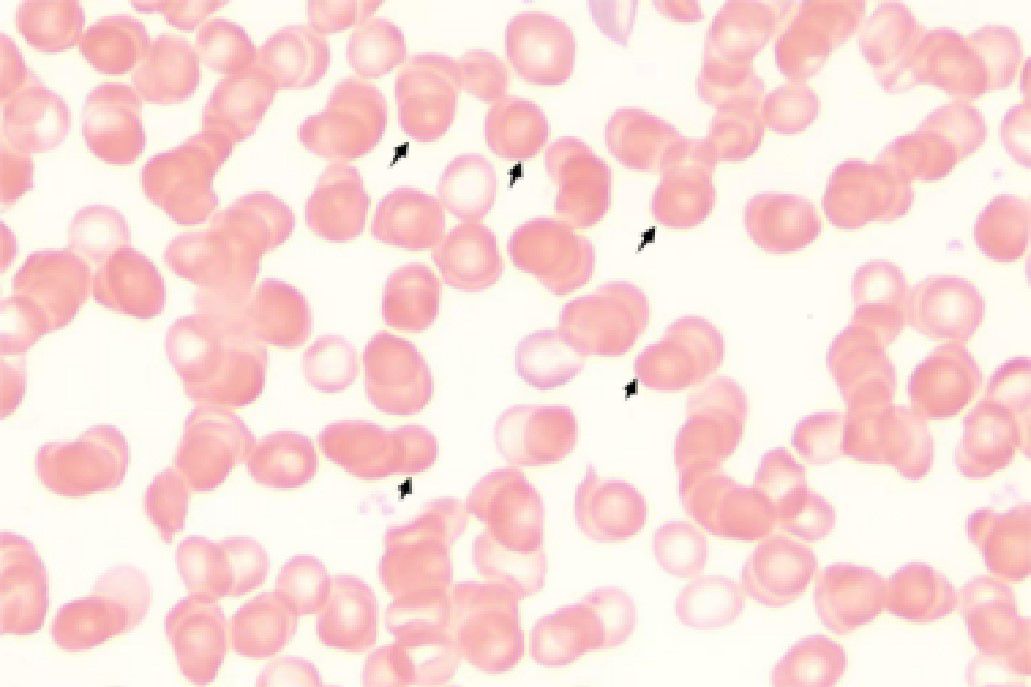
Figure 2. Peripheral blood smear. Approximately 20% spherocytes.
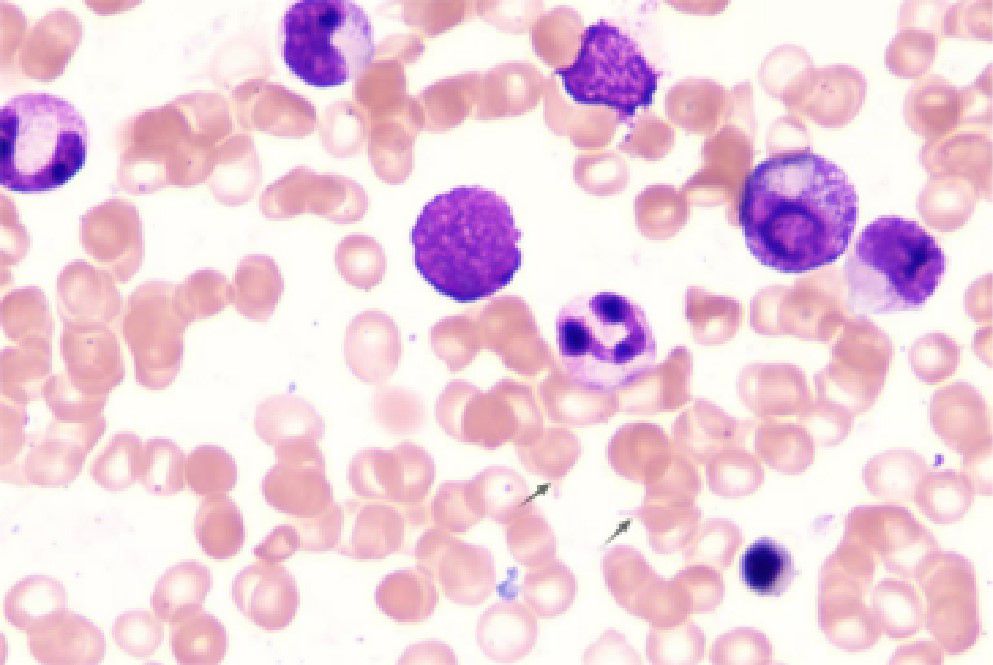
Figure 3. Bone marrow smear. Increased proliferation of granulocytic, erythroid, and megakaryocytic lineages in the bone marrow, with elevated platelets in both bone marrow and peripheral blood and the presence of teardrop-shaped red blood cells. Bone marrow biopsy morphology is consistent with myeloproliferative neoplasm (MF-2 grade).

Figure 4. EMA binding test result. The excerpt from the laboratory report shows a mean fluorescence intensity reduction of 25.73%.
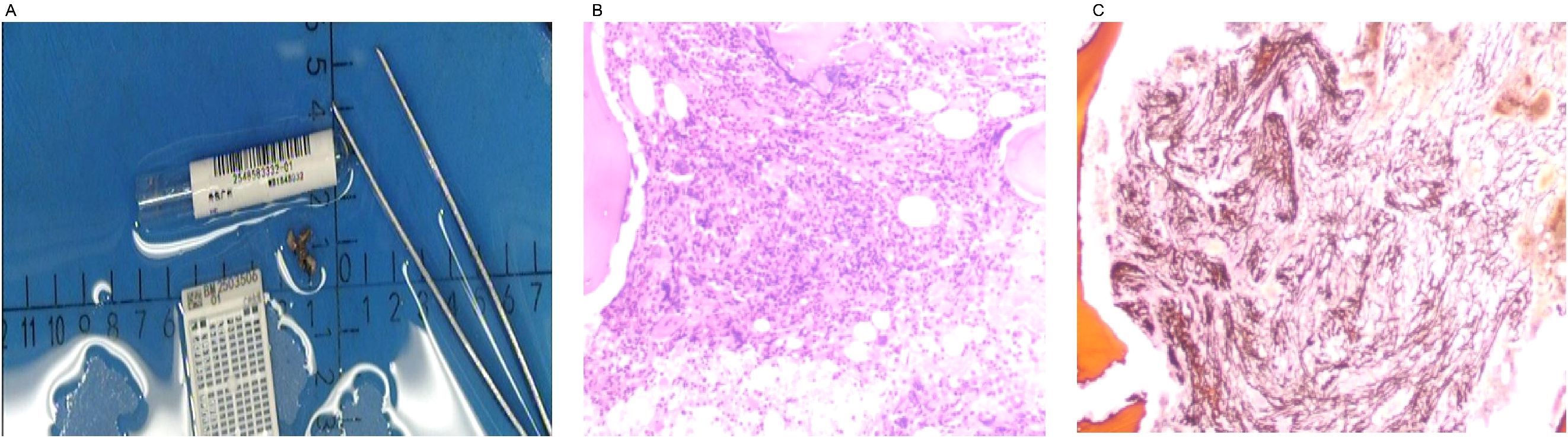
Figure 5. Bone marrow biopsy. The bone marrow biopsy morphology is consistent with myeloproliferative neoplasm (MF-2 grade). Panel (A) shows the bone marrow biopsy tissue specimen. Panel (B) displays the bone marrow biopsy tissue under the microscope. Panel (C) illustrates the reticulin fiber staining of the bone marrow biopsy tissue under the microscope.
3 Discussion
This report describes the first documented case of HS with JAK2 V617F-positive PMF. This case underscores the importance of peripheral blood smear examination in identifying the etiology of splenomegaly. Peripheral blood smears are instrumental in the diagnosis of hematologic disorders, as they provide detailed insights into cellular morphology (11). This case emphasizes the need for rigorous clinical reasoning when evaluating diseases with overlapping clinical manifestations to prevent potential diagnostic oversights.
This case illustrates the coexistence of HS PMF, characterized by marked splenomegaly, elevated leukocyte and platelet counts, mild anemia, and increased reticulocyte percentage. Peripheral blood analysis revealed numerous spherocytes that were not singularly attributed to either HS or PMF. A few patients with HS encounter diagnostic difficulties due to atypical clinical presentations, inconclusive laboratory results, or absence of familial history (3, 12). Both mean corpuscular volume (MCV) and mean corpuscular hemoglobin concentration (MCHC) were below the normal range in this case. Research indicates that only 8% of patients with HS exhibit subnormal MCV, while merely 32% have MCHC values above normal, suggesting that these parameters should not be used as definitive diagnostic criteria for HS (2). Blood smear analysis identified a significant presence of spherocytes and abnormal erythrocytes characterized by reduced size, spherical shape, and the absence of central pallor (13). These findings represent significant hematological abnormalities that may be indicative of multiple disorders. A thorough assessment of peripheral blood smears, along with pertinent laboratory tests, facilitates the precise diagnosis and implementation of suitable therapeutic interventions (14). HS continues to be one of the predominant causes of spherocyte formation.
In this case, the highly sensitive and specific EMA binding test (15) was used, significantly enhancing the reliability of the clinical diagnosis and confirming HS in the patient. This highlights the importance of blood smear examination in conjunction with bone marrow biopsy and genetic testing for the diagnosis of myeloproliferative disease to distinguish HS from PMF. Peripheral blood smears are crucial diagnostic tools for the assessment of myelofibrosis. Typically, peripheral blood smears in cases of myelofibrosis reveal characteristic abnormalities, including teardrop-shaped red blood cells, nucleated red blood cells, and immature granulocytes (16). In the current case, the peripheral blood smear demonstrated a marked increase in spherocytes and teardrop-shaped red blood cells, with an absence of immature granulocytes and nucleated red blood cells. The lack of immature granulocytes and nucleated red blood cells in myelofibrosis may be associated with the extent of fibrosis, disease progression, specific genetic mutations, and potential HS, warranting further investigation. Although peripheral blood smear examination is a vital tool for evaluating PMF, the absence of immature granulocytes and nucleated red blood cells should not be the sole criterion to exclude PMF (17). A definitive diagnosis of PMF necessitates comprehensive evaluation, including bone marrow biopsy, cytogenetic analysis, molecular biology testing, and clinical manifestations (18).
In this case, the patient had exhibited splenomegaly for more than a decade. Two prior bone marrow aspiration examinations were inconclusive in determining the etiology, as the patient reported a loss of medical records. A subsequent bone marrow examination conducted after a 9-year interval revealed the presence of teardrop-shaped red blood cells in the marrow, while a peripheral blood smear indicated approximately 20% spherocytes. Bone marrow biopsy demonstrated markedly active hyperplasia of nucleated cells, with an estimated hematopoietic capacity of 60%, accompanied by a diffuse and dense increase in reticulin fibers and extensive cross-linking, classified as MF-2 grade. Further genetic testing for MPN was performed, revealing a positive JAK2 V617F mutation, and EMA flow cytometry indicated a 25.73% reduction in average fluorescence intensity. The concurrent diagnosis of overt PMF and HS in this patient was established through a comprehensive evaluation. The diagnosis of overt PMF was definitively confirmed as the patient’s findings fulfilled all three major and all four minor criteria of the 2022 World Health Organization (WHO) classification (19). Following this diagnosis, a formal risk stratification was conducted to guide therapeutic management, in line with the NCCN Clinical Practice Guidelines in Oncology for Myeloproliferative Neoplasms (Version 3.2022) (20). The patient’s clinical status met the criteria for two adverse factors within the Dynamic International Prognostic Scoring System (DIPSS): hemoglobin <100 g/L (2 points) and the presence of constitutional symptoms, manifested as severe abdominal distension (1 point). This yielded a total DIPSS score of 3, placing the patient in the Intermediate-2 (Int-2) risk category. For symptomatic patients with Int-2 risk PMF, the NCCN guidelines provide a Category 1 recommendation for JAK1/2 inhibitor therapy. Therefore, the initiation of ruxolitinib was a guideline-directed decision to address his significant disease burden, including massive splenomegaly and progressive anemia. A further diagnostic consideration was the confirmation of HS. We acknowledge that targeted gene sequencing of red cell membrane protein genes (ANK1, SPTB, SLC4A1, SPTA1, EPB42) is the gold standard for a definitive molecular diagnosis. This testing was recommended to the patient to confirm the diagnosis and clarify the inheritance pattern. However, the patient declined to undergo this investigation due to prohibitive out-of-pocket costs, a decision that was formally documented. The clinical diagnosis of HS remains robustly supported by the presence of 20% spherocytes on the peripheral blood smear and a definitively positive eosin-5’-maleimide (EMA) binding test. While the absence of genetic data is a limitation of this report, it did not alter the immediate therapeutic management, which was driven by his symptomatic, Intermediate-2 risk PMF. We plan to seek opportunities for this genetic analysis for the patient during future follow-up. Case reports have suggested that patients with HS may develop secondary myelofibrosis and acute monocytic leukemia (M5) following splenectomy (21). PMF is a hematopoietic clonal disorder of unknown etiology, and the relationship between PMF and HS remains to be elucidated. This hypothesis posits that defects in red blood cell membranes of HS result in elevated ATP consumption. This raises the question of whether prolonged ATP deficiency in the bone marrow predisposes individuals to driver mutations in JAK2, CALR, and MPL, thereby contributing to the development of myelofibrosis. The potential correlation between HS and PMF necessitates further investigation to determine whether this association is coincidental or indicative of an underlying relationship. When a singular theoretical framework fails to account for all observed phenomena, a comprehensive approach should be adopted to enhance diagnostic precision and prevent misdiagnosis or oversight. By emphasizing the morphology of peripheral blood smears, diagnostic and differential capabilities can be maximized, thereby significantly advancing hematological morphological diagnostics.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Ethics Committee of Northeast Yunnan Central Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
C-EQ: Writing – original draft. LL: Data curation, Writing – original draft. GJ: Writing – review & editing. XH: Writing – original draft. YL: Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
The authors express gratitude to the medical staff and research participants who contributed to the diagnosis and treatment of this case.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Suttorp M and Classen CF. Splenomegaly in children and adolescents. Front Pediatr. (2021) 9:704635. doi: 10.3389/fped.2021.704635
2. Bai L, Zheng L, Li B, Huang H, Shi X, Yi Y, et al. Clinical and genetic diagnosis for 26 paitents with hereditary spherocytosis. Zhong Nan Da Xue Xue Bao Yi Xue Ban. (2023) 48:565–74. doi: 10.11817/j.issn.1672-7347.2023.220390
3. Da Costa L, Galimand J, Fenneteau O, and Mohandas N. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. (2013) 27:167–78. doi: 10.1016/j.blre.2013.04.003
4. Ma H, Liu J, Li Z, Xiong H, Zhang Y, Song Y, et al. Expression profile analysis reveals hub genes that are associated with immune system dysregulation in primary myelofibrosis. Hematology. (2021) 26:478–90. doi: 10.1080/16078454.2021.1945237
5. Bhagya D and Chamindri W. Review: JAK2V617F allele burden in diagnosis and therapeutic monitoring of myeloproliferative neoplasms. Eur J Med Health Sci. (2023) 5:35–40. doi: 10.24018/ejmed.2023.5.1.1587
6. Şahin E, Yönal-Hindilerden İ, Hindilerden F, Aday A, and Nalçacı M. The impact of JAK2V617F mutation on Philadelphia-negative myeloproliferative neoplasms. Turkish J Med Sci. (2022) 52:150–65. doi: 10.3906/sag-2103-247
7. Cvetković Z, Pantić N, Cvetković M, Virijević M, Sabljić N, Marinković G, et al. The role of the spleen and the place of splenectomy in autoimmune hemolytic anemia-A review of current knowledge. Diagn (Basel Switzerland). (2023) 13:2891. doi: 10.3390/diagnostics13182891
8. Zabihi MR, Zangooie A, Piroozkhah M, Harirchian MH, and Salehi Z. From multiple sclerosis to organ-specific autoimmune disorders: insights into the molecular and clinical implications of comorbidity. Mol Neurobiol. (2025) 62:3396–411. doi: 10.1007/s12035-024-04458-0
9. Vi TP and Lavanya NS. 8194 Extramedullary hematopoiesis involving bilateral adrenal glands in a patient with sickle cell anemia. J Endocrine Soc. (2024) 8. doi: 10.1210/jendso/bvae163.186
10. Ma S, Deng X, Liao L, Deng Z, Qiu Y, Wei H, et al. Analysis of the causes of the misdiagnosis of hereditary spherocytosis. Oncol Rep. (2018) 40:1451–8. doi: 10.3892/or.2018.6578
11. Navya KT, Keerthana P, and Kumar SBM. Classification of blood cells into white blood cells and red blood cells from blood smear images using machine learning techniques. (2021). 2nd Global Conference for Advancement in Technology (GCAT), Bangalore, India, 2021, pp. 1–4. doi: 10.1109/GCAT52182.2021.9587524
12. Iolascon A, Andolfo I, and Russo R. Advances in understanding the pathogenesis of red cell membrane disorders. Br J Jaematol. (2019) 187:13–24. doi: 10.1111/bjh.16126
13. Karlsson LK, Mottelson MN, Helby J, Petersen J, and Glenthøj A. Acquired spherocytosis in the setting of myelodysplasia. Leukemia Res Rep. (2022) 17:100332. doi: 10.1016/j.lrr.2022.100332
14. Jimut Bahan P, Aniket B, Debasis B, Br. Tamal M, et al. Advancing instance segmentation and WBC classification in peripheral blood smear through domain adaptation: A study on PBC and the novel RV-PBS datasets. Expert Syst Appl. (2024) 249. Article 123660. doi: 10.1016/j.eswa.2024.123660
15. Ciepiela O. Old and new insights into the diagnosis of hereditary spherocytosis. Ann Trans Med. (2018) 6:339. doi: 10.21037/atm.2018.07.35
16. Nakai T and Okamoto Y. A megakaryocyte in a peripheral blood smear. Int J Hematol. (2019) 110:647. doi: 10.1007/s12185-019-02752-5
17. Lewis JE and Pozdnyakova O. Digital assessment of peripheral blood and bone marrow aspirate smears. Int J Lab Hematol. (2023) 45 Suppl 2:50–8. doi: 10.1111/ijlh.14082
18. Sonam P and Attilio O. How I diagnose primary myelofibrosis. Am J Clin Pathol. (2022) 157:518–30. doi: 10.1093/ajcp/aqac016
19. Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. (2022) 140:1200–28. doi: 10.1182/blood.2022015850
20. Gerds AT, Gotlib J, Ali H, Bose P, Dunbar A, Elshoury A, et al. Myeloproliferative neoplasms, version 3.2022, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2022) 20:1033–62. doi: 10.6004/jnccn.2022.0046
Keywords: hereditary spherocytosis, primary myelofibrosis, JAK2V617F, comorbidity, myeloproliferative neoplasm
Citation: Qiu C-E, Lei L, Jiang G, Huang X and Li Y (2025) Hereditary spherocytosis concomitant with JAK2V617F-positive primary myelofibrosis: a case report. Front. Oncol. 15:1665179. doi: 10.3389/fonc.2025.1665179
Received: 13 July 2025; Accepted: 03 October 2025;
Published: 17 October 2025.
Edited by:
Cyrus Khandanpour, Universitätsklinikum Schleswig-Holstein, GermanyReviewed by:
Elena Beatriz Moiraghi, Hospital Ramos Mejía, ArgentinaJianfei Fu, Tongji Hospital Affiliated to Tongji University, China
Copyright © 2025 Qiu, Lei, Jiang, Huang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chi-E Qiu, MTM1MDg3MDIwNjlAMTM5LmNvbQ==
†These authors have contributed equally to this work and share first authorship
‡ORCID: Chi-E Qiu, orcid.org/0009-0008-4423-2766