 Renata Polaniak1*
Renata Polaniak1* Aleksander Kwiatkowski2
Aleksander Kwiatkowski2 Michał Górski3Elżbieta Grochowska-Niedworok4Małgorzata Latocha5
Michał Górski3Elżbieta Grochowska-Niedworok4Małgorzata Latocha5- 1Department of Dietetics, Division of Human Nutrition Faculty of Public Health in Bytom Medical University of Silesia in Katowice, Bytom, Poland
- 2Department of Human Anatomy, Faculty of Medical Sciences in Katowice, Medical University of Silesia in Katowice, Katowice, Poland
- 3Department of Chronic Diseases and Civilization-Related Hazards, Faculty of Public Health in Bytom, Medical University of Silesia in Katowice, Katowice, Poland
- 4Health Public School in Nysa, Nysa, Poland
- 5Department of Cell Biology, Faculty of Pharmacy with Division of Laboratory Medicine in Sosnowiec, Medical University of Silesia in Katowice, Sosnowiec, Poland
Cytostatic drugs are widely applied in cancer therapy. Among the most commonly used agents are anthracyclines, such as doxorubicin, and platinum (II) complexes, including cisplatin, carboplatin, and oxaliplatin. Treatment with cytostatic drugs has been shown to enhance the mitochondrial production of reactive oxygen species (ROS). Cells regulate redox homeostasis through scavenging systems, with antioxidant enzymes playing a crucial role in neutralizing ROS. Key enzymes involved in this defense include superoxide dismutase, catalase, and glutathione S-transferase, whose activity may be modulated under oxidative stress conditions. Previous research has documented the effects of cytostatic drugs on cancer cell cultures in vitro, as well as the corresponding alterations in antioxidant enzyme activity observed under these conditions.
Introduction
In healthy cells, oxidative stress is tightly regulated by compartmentalized antioxidant systems-glutathione, thioredoxin/peroxiredoxin cycles, catalase in peroxisomes, and SOD isoforms in cytosol, mitochondria, and extracellular space -maintaining ROS at low, signaling-competent levels (1, 2). In cancer cells, oncogenic signaling (e.g., MYC, RAS), mitochondrial dysfunction, rapid proliferation, and hypoxia/reoxygenation events cumulatively elevate ROS. This persistent redox shift fuels genomic instability and malignant progression, yet also creates therapeutic liabilities by lowering the threshold for ROS-mediated cell death (1).
Key differences include: (a) increased basal mitochondrial ROS and NADPH oxidase activity; (b) altered redox buffering (high glutathione turnover, reliance on pentose phosphate pathway for NADPH); (c) adaptive upregulation of antioxidant enzymes (e.g., MnSOD, GPx) in drug-resistant phenotypes; and (d) microenvironmental factors (hypoxia, inflammatory cytokines, metal ions) that modulate oxidative fluxes. These distinctions are central to interpreting the effects of cytostatics that further perturb redox homeostasis (2, 3).
This review synthesizes enzymatic and non-enzymatic antioxidant responses to major cytostatics and explicitly contrasts outcomes in normal versus cancer cells. We integrate scattered in vitro findings, highlight translational gaps, and outline therapeutic opportunities and pitfalls in targeting antioxidant networks.
Oxidative stress arises within a cell due to an imbalance between the formation of free radicals (FR) – specifically, reactive oxygen species (ROS) – and the cell’s ability to eliminate them and repair the damage they cause. Reactive oxygen species interact with nucleic acids, causing DNA mutations, which disrupt replication and transcription. Consequently, repair mechanisms are activated, or cellular death occurs. Additionally, the accumulation of mutations in genetic material often initiates carcinogenesis through uncontrolled proliferation of cells that are unresponsive to pro-apoptotic signals (1, 2, 4) (Figure 1).
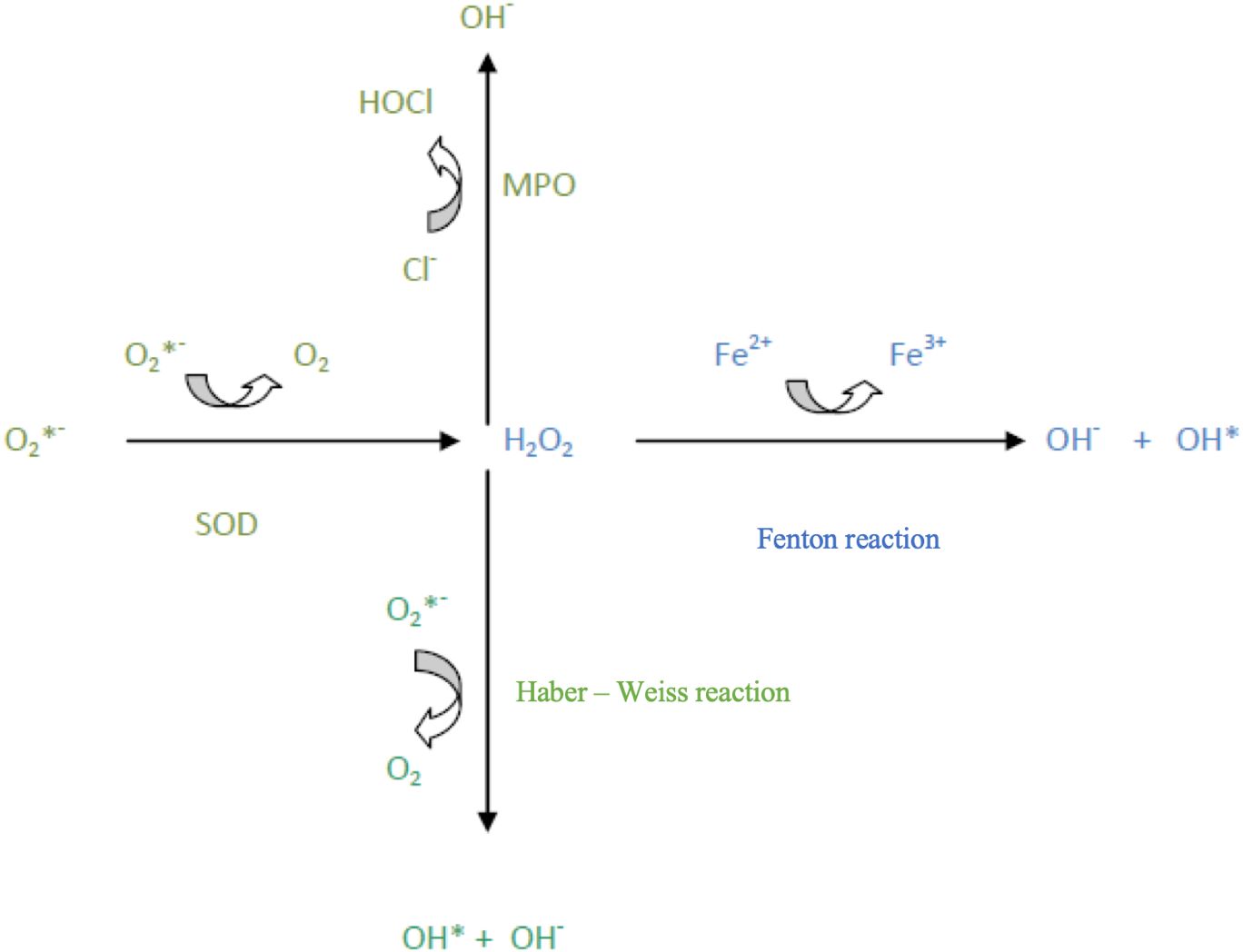
Figure 1. Reactive oxygen species (ROS) transformations.
A well-known free radical process is lipid peroxidation, which involves the oxidation of polyunsaturated fatty acids. This may affect phospholipids of cell, nuclear, mitochondrial, and peroxisomal membranes (2). Lipid peroxides form during this process, participating in further lipid peroxidation, with the final reaction products being phospholipid dimers and aldehydes. An example of a lipid peroxidation marker is malondialdehyde (MDA) (2). Free radicals also react with proteins, often resulting in altered or lost enzymatic functions (5). Thus, free radicals damage specific cellular structures and may lead to cell death. Through their action, they may also affect cell proliferation, differentiation, or interfere with signal transduction (2, 6).
In cells, free radicals primarily arise from oxygen-dependent processes in mitochondria, producing reactive oxygen species (ROS) such as the superoxide anion (O2-), hydroxyl radical (·OH), and hydroperoxide radical (HO2.) (3). This necessitates cellular defense mechanisms to counteract free radical attacks and repair induced damage (4). The cell has multiple defense mechanisms, both non-enzymatic, involving antioxidants such as vitamins C, E, and A, and enzymatic, involving antioxidant enzymes (2, 7, 8).
The role of cytostatics in oxidative stress reactions
Antioxidant enzymes
Key antioxidant enzymes in the cell include superoxide dismutase (SOD) [EC 1.15.1.1]; the zinc-copper isoenzyme of superoxide dismutase, SOD1 (Cu/ZnSOD), which is primarily found in the cytoplasm of liver, testes, kidney, nervous system, erythrocytes and manganese-dependent superoxide dismutase, SOD2 (MnSOD), located in the mitochondrial matrix, peroxisomes, and to a lesser extent, extracellularly. The third SOD isoenzyme is extracellular superoxide dismutase, SOD3 (EC-SOD), predominantly found in the extracellular space, with activity observed in blood, lymph, interstitial fluid, and cerebrospinal fluid (9).
These enzymes are metalloenzymes, containing a metal ion in their active center that alternately reduces or oxidizes, catalyzing the two-step dismutation of the superoxide anion, a highly active free radical, into oxygen and hydrogen peroxide (9). To illustrate the critical role of manganese-dependent superoxide dismutase, we can refer to research by Zhang et al. (10), who examined biochemical transformations in SOD2-/- mouse fibroblasts, cells lacking this enzyme. In vivo, such a phenotype is lethal, and researchers found that the absence of mitochondrial SOD activity disrupts cellular signaling pathways, slowing growth and proliferation (10) (Table 1).
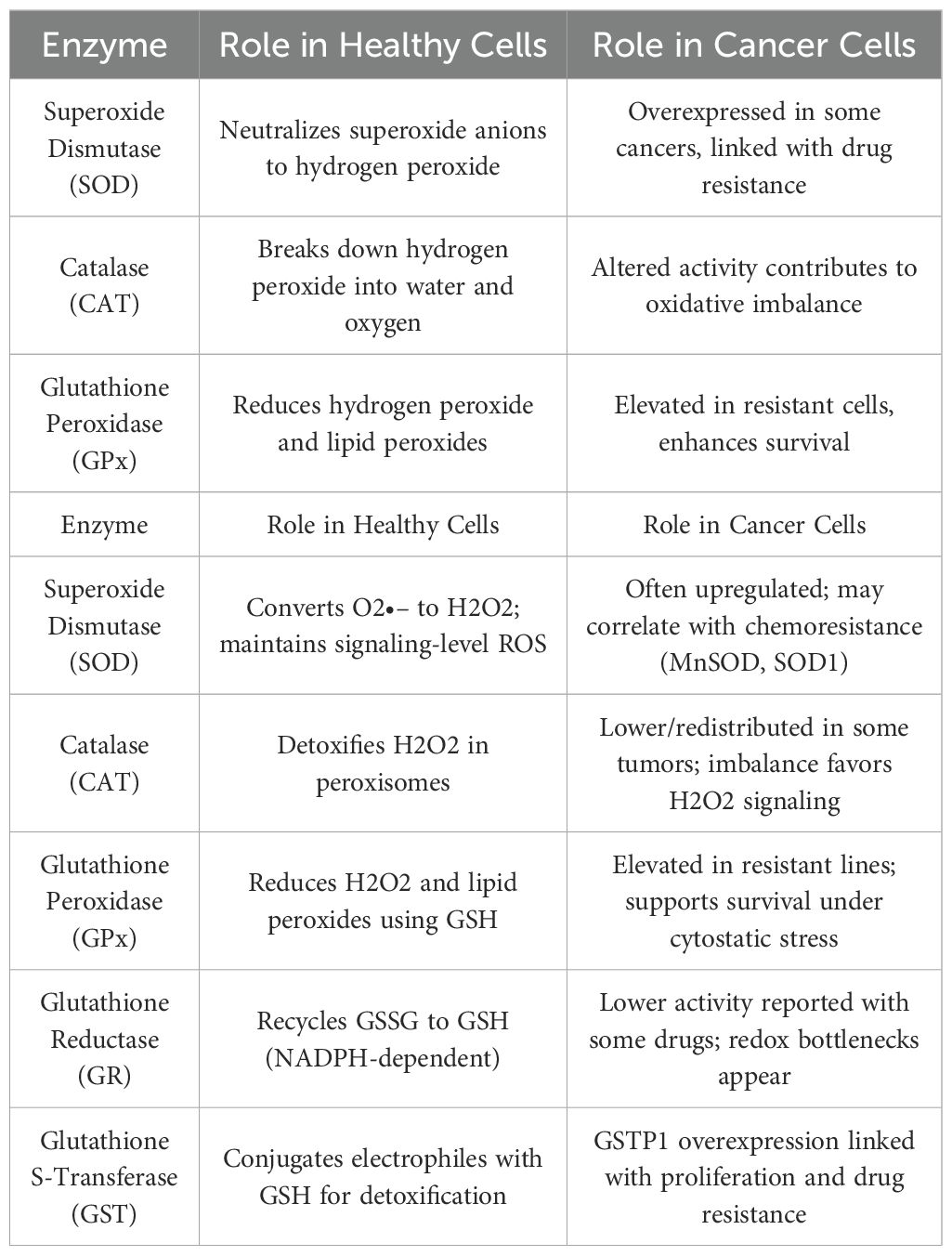
Another antioxidant enzyme is catalase (CAT) [EC 1.11.1.6], which catalyses the dismutation of hydrogen peroxide into water and oxygen. At high hydrogen peroxide concentrations, catalase primarily exhibits catalase activity; at low concentrations H2O2, it displays peroxidase activity (9).
It is possible to encapsulate superoxide dismutase or catalase molecules in large unilamellar liposomes of about 110 nm in size. These resulting enzymosomes are used to deliver antioxidant enzymes directly to target cells, increasing their biodistribution. Studies in rats have shown that enzymosomes reduce oxidative stress induced by radiotherapy and positively impact retinal cells in new-born rats (11).
The third group of antioxidant enzymes, selenoperoxidases, plays a significant role in defense mechanisms against free radicals and contains selenocysteine in the enzyme’s active center. The main representative of this group is glutathione peroxidase (GPx) [EC 1.11.1.9], which reduces hydrogen peroxide and organic peroxides. It exists in several isoforms: cytosolic (cGPx), gastrointestinal (giGPx), plasmatic (pGPx), nuclear (spGPx), and as phospholipid hydroperoxide peroxidase (phGPx) (9). A recently described glutathione peroxidase (snGPx) protects sperm DNA from oxidative damage and is involved in chromatin condensation (9). Glutathione peroxidase activity depends on cellular glutathione, which is consumed in the reaction and can be regenerated by glutathione reductase (GR) [EC 1.6.4.2] with NADPH+H+ (9).
Another enzyme in this group, glutathione S-transferase (GST) [EC 2.5.1.18], catalyzes the conjugation of electrophilic compounds with glutathione, including harmful metabolites like bilirubin, fatty acid peroxides, and xenobiotics such as cytostatic drugs. Conjugation makes these compounds water-soluble, allowing safe excretion via urine (9). GST has multiple isoforms, with GSTP1 receiving significant attention in literature due to its overexpression in various cancer cell types (12). Researches (12, 13) demonstrated that elevated GSTP1 activity in human HCT 116 colon cancer cells contributes to their uncontrolled proliferation.
Reactive oxygen species (ROS) are continuously generated in cells, accompanied by antioxidant processes that maintain a balance. Oxidative stress, a disruption of this balance, represents the cell’s response to physical, chemical, and biological factors (2, 7). Studies have shown that irritants, such as asbestos (14) or tobacco smoke (15), increase free radical production within cells. This also occurs in response to certain drugs. This article examines changes in antioxidant enzyme activity under the influence of cytostatic drugs on cells (16).
Therapeutic applications and challenges in targeting antioxidant enzymes
Targeting antioxidant enzymes (e.g., SOD2, GPx, GST) can sensitize tumors to cytostatics by pushing ROS beyond cytotoxic thresholds. Examples include GST inhibitors that prevent drug conjugation and efflux, or modulation of GSH synthesis to transiently lower cellular buffering capacity (17).
Certain challenges associated with this have also been identified: (a) therapeutic window—systemic suppression of antioxidants risks normal-tissue toxicity (cardiotoxicity, neurotoxicity); (b) compensatory rewiring—cancer cells upregulate parallel redox pathways; (c) pharmacokinetics—achieving tumor-selective delivery; (d) biomarker selection—lack of standardized, clinically actionable ROS/redox biomarkers to guide patient selection (17, 18).
Despite these limitations, it is also worth pointing out the significant opportunities: (a) liposomal and pegylated formulations (e.g., PLD) and enzymosome carriers may co-deliver cytostatics and redox modulators; (b) radiochemotherapy regimens can exploit ROS bursts; and (c) adaptive dosing based on early redox readouts (e.g., MDA, 4-HNE adducts) may optimize efficacy while limiting harm (17–19).
Cytostatic drugs
Cytostatics are a chemically diverse group of drugs with anticancer activity. Chemotherapy regimens are based on multi-center clinical trials (20, 21). Types of chemotherapy include induction chemotherapy to reduce tumor mass before planned surgery, postoperative adjuvant chemotherapy, and palliative chemotherapy for inoperable cancers (22). Numerous studies have investigated the impact of cytostatics on cellular mechanisms and their induction of cell death (23).
Among clinically significant cytostatic groups, this work focuses on cisplatin and its derivatives, carboplatin and oxaliplatin.
Cisplatin (CIS) is a fundamental chemotherapeutic used to treat various stages of testicular and ovarian cancers, as well as bladder, esophageal, advanced head and neck cancers, and both small-cell and non-small-cell lung cancer. It is a Platinum (II) compound with two chloride ligands and two NH3 residues in a cis configuration, acting by alkylating DNA and forming intra- and inter-strand bonds, thus inhibiting DNA replication and transcription to RNA (24, 25). Notably, the trans-isomer of this compound lacks anti-cancer activity, though certain trans-platinum derivatives exhibit anti-carcinogenic properties (26) (Figure 2).
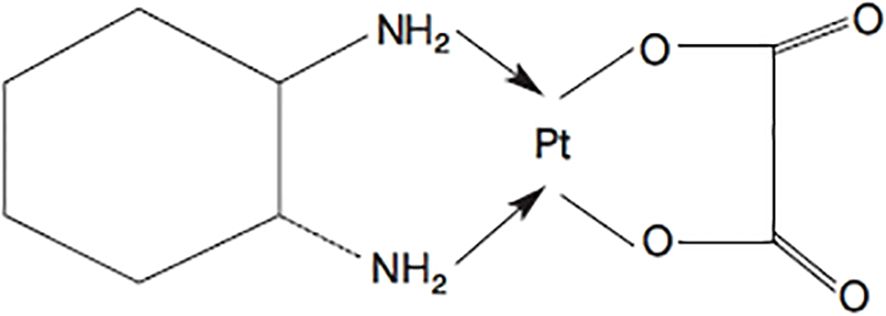
Figure 2. Ryc. I. Cisplatyna (cis-diaminodichloroplatyna).
With the increasing resistance of cancer cells to cisplatin, it is necessary to search for its new derivatives (16). Among the various platinum compounds, carboplatin and oxaliplatin are mainly used in treatment. Like cisplatin, these are alkylating compounds but exhibit different pharmacokinetics and tend to cause fewer side effects (27). Both maintain the cis configuration in their structure, differing in substituents (24).
Carboplatin contains two NH3 ligands in a cis conformation and a cyclobutane-1,1-dicarboxylic acid residue. This drug is used in treating ovarian cancer, cervical cancer, testicular cancer, small-cell and non-small-cell lung cancer, as well as squamous cell carcinoma of the head and neck (24, 25, 28). There are numerous reports of cross-resistance of cancer cells to cisplatin and carboplatin (25).
Oxaliplatin is distinguished in its structure by 1,2-diaminocyclohexane and an oxalate group (24). It is a cisplatin derivative used to treat rectal and colon cancer at various stages, typically combined with 5-fluorouracil and folic acid in therapeutic regimens (25, 29). It is also present in chemoradiotherapy protocols for rectal cancer (30). The cytotoxic properties of oxaliplatin against various cancer types, including those resistant to cisplatin, have been frequently described in the scientific literature (25).
Another group of cytotoxic drugs includes anthracycline antibiotics. These are used in cancer treatment due to their cytotoxic properties. Their mechanism involves preventing DNA transcription by embedding into its helix and inhibiting the action of topoisomerase II (24, 31).
The main representative of anthracycline antibiotics is doxorubicin (DOX), also known as adriamycin (Figure 3).
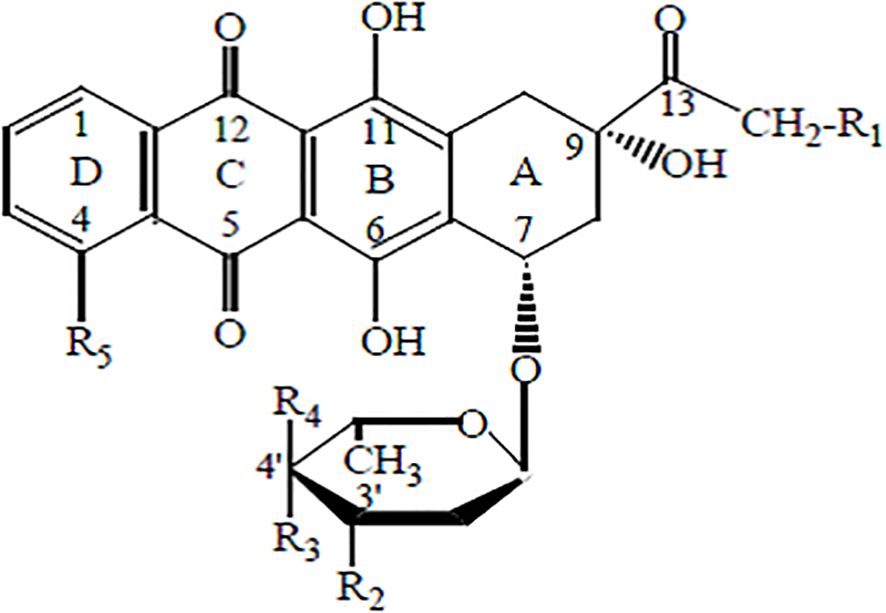
Figure 3. Ryc. II. Chemical structure of anthracyclines.
Its molecule consists of a four-ring aglycone structure and a sugar moiety. The substituents include hydroxyl, carbonyl, and amino groups. This antibiotic is widely used in chemotherapy for cancers of the breast, ovary, endometrium, thyroid, bladder, stomach, prostate, liver, as well as leukemias, immunological malignancies, pediatric cancers such as Wilms’ tumor and neuroblastoma, and AIDS-related cancers, including Kaposi’s sarcoma (32, 33). The drug is typically administered intravenously as part of the so-called “red chemotherapy.” Intravesical administration of doxorubicin is also possible for the treatment of bladder cancer (34).
Numerous studies highlight the significant cardiotoxicity of doxorubicin and the frequent development of cellular resistance (35–37). As a result, its liposomal formulation—liposomal doxorubicin—is increasingly used in therapy, proving more advantageous for treating patients with cancer recurrence (31). Additionally, pegylated liposomal doxorubicin (PLD) is available and utilized in the treatment of metastatic breast cancer, ovarian cancer, and highly vascularized tumors such as Kaposi’s sarcoma. This modification extends the drug’s circulation time and enhances its bioavailability (28, 32, 38). Doxorubicin is used both in monotherapy and in combination with other cytostatic agents, such as cyclophosphamide (39). Currently, numerous doxorubicin analogs with varying toxicity profiles and pharmacokinetics are employed in treatment (24).
The literature contains extensive research on the effects of cytostatic drugs on cancer cells in in vitro cultures. Studies on the mechanisms of doxorubicin action on cancer cells have been conducted using human lung adenocarcinoma cell line A549 (40), as well as human ovarian teratoma cell line PA-1 and ovarian cancer cell line CA5171 (41), and human breast adenocarcinoma cell line MCF-7 (36). In vitro investigations have also explored the metabolism of human colorectal cancer cell line HCT-116, T-cell acute lymphoblastic leukemia cell line Jurkat, and human promyelocytic leukemia cell line HL-60 under the influence of doxorubicin and cisplatin (42). Georgakis et al. (43) examined the apoptosis mechanisms in Hodgkin lymphoma cell lines HD-MyZ, HD-LM-2, L-428, and KM-H2 in response to doxorubicin. Kachadourian et al. (42) studied the effects of cisplatin on human lung adenocarcinoma cell line A549 in in vitro culture (42). Similarly, studies on the effects of carboplatin on cancer cells in in vitro cultures were conducted on human melanoma cell line Me45 (44) and human glioblastoma cell line SNB19 (45) (Table 2).
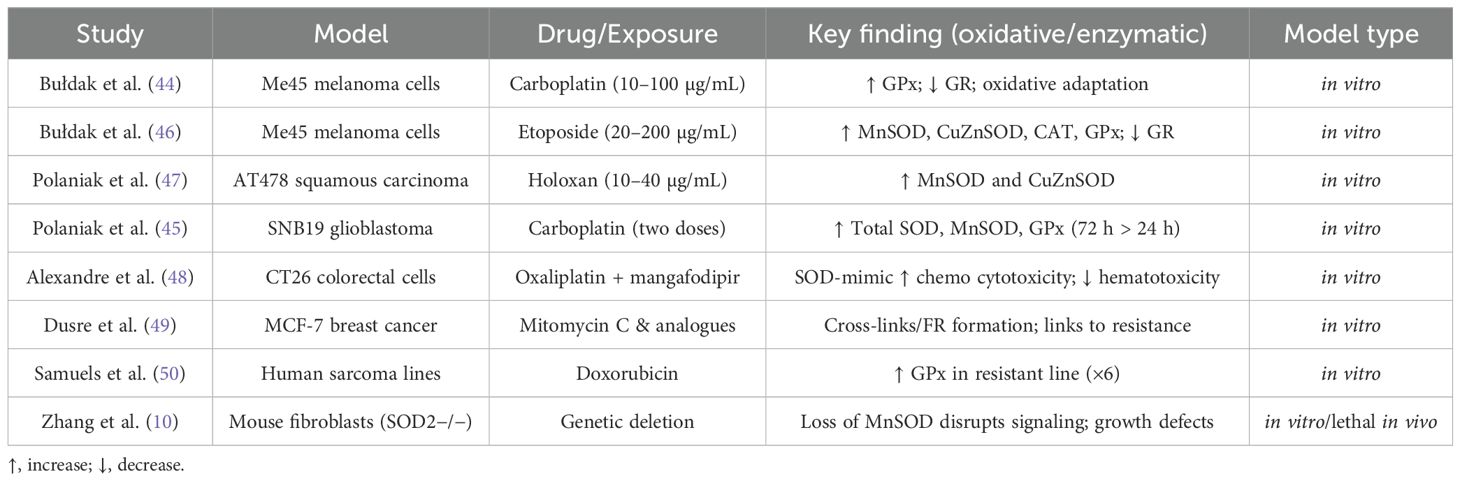
Table 2. Selected studies on cytostatics and oxidative stress (models, exposures, findings) (10, 44–50).
In the presence of cytostatic drugs, including platinum derivatives (51) and doxorubicin (33), the levels of reactive oxygen species (ROS) increase within cells (29). Depending on the cell type, drug concentration, or experimental conditions, the cellular response to oxidative stress varies (29, 51).
Cancer cells are characterized by an inherently higher baseline level of reactive oxygen species, making them more susceptible to oxidative stress (15, 52). By studying changes in the activity of superoxide dismutase isoenzymes, catalase, and glutathione peroxidase in cells cultured in vitro under the influence of a cytostatic drug, the extent of oxidative stress induced by the drug can be measured. Numerous reports in the literature indicate that an increase in antioxidant enzyme activity contributes to greater cellular resistance to cytostatic drugs. L’Ecuyer et al. (53) examined the effects of anthracycline antibiotics on rat cardiomyocytes of the H9C2 cell line, suggesting that selective overexpression of antioxidant enzymes in cardiomyocytes could reduce the cardiotoxicity of chemotherapy (53).
Zhong et al. (54) investigated the effects of antioxidants in vitro on human prostate adenocarcinoma RWPE-2 cells, depending on the expression of manganese superoxide dismutase (MnSOD). Their study demonstrated that cells overexpressing MnSOD exhibited greater sensitivity to the cytotoxic effects of buthionine sulfoximine, a compound that reduces intracellular glutathione levels, and vitamin C. Conversely, these cells showed reduced sensitivity to selenium compounds. The same effect was observed when the cells were treated with an exogenous MnSOD analog. Considering that glutathione peroxidase is a selenium-dependent enzyme, the authors concluded that MnSOD overexpression exerts both pro-oxidative and antioxidative effects, depending on the activity of other antioxidant enzymes. This finding underscores the need for further research into the activity of antioxidant enzymes in cancer cells and their interrelations, as this may have implications for the outcomes of anticancer therapies (54).
The literature contains numerous reports of higher antioxidant enzyme activity, such as GPx and MnSOD, in cancer cells resistant to cytostatic drugs (49, 50, 55, 56). As early as the 1990s, Dusre et al. (49) demonstrated that in vitro doxorubicin-resistant MCF-7 breast cancer cells exhibited higher glutathione peroxidase activity compared to cells that were not resistant to the cytostatic. Similarly, Samuels et al. (50), investigating GPx activity in two human sarcoma cell lines with differing resistance to doxorubicin, found that GPx activity was six times higher in the cell line less sensitive to apoptosis induced by doxorubicin treatment. This highlights the role of antioxidant enzymes in the development of drug resistance in cancer cells (49, 50).
Bułdak et al. (44), investigating the effects of carboplatin on antioxidant activity in malignant melanoma Me45 cells, demonstrated an increase in glutathione peroxidase (GSH-Px) activity following carboplatin treatment. Specifically, GSH-Px activity increased to 125.2 ± 12.1 IU/L and 99.1 ± 13.3 IU/L after 24 hours of exposure to carboplatin at concentrations of 10 µg/mL and 100 µg/mL, respectively, compared to 91.6 ± 12.1 IU/L in the control group. Additionally, a reduction in glutathione reductase (GR) activity was observed, with GR activity decreasing to 8.49 ± 0.31 IU/L after 24 hours of treatment with carboplatin at a concentration of 10 µg/mL, compared to 9.49 ± 0.49 IU/L in the control group.
In a related study by Bułdak et al. (46), examining the increase in antioxidant enzyme activity in malignant melanoma Me45 cells cultured in vitro following treatment with etoposide—a cytotoxic drug from the podophyllotoxin derivatives group—a significant increase in the activity of MnSOD, CuZnSOD, CAT, and GSH-Px was observed after 24 hours of exposure to etoposide at concentrations of 20 µg/mL and 200 µg/mL, compared to the control group. Simultaneously, GR activity decreased in both experimental groups. These results suggest an adaptive response of the cells to oxidative stress induced by the cytostatics.
There are scientific reports on the effects of other cytostatic drugs on antioxidant enzyme activity in cells. In the study by Polaniak et al. (47), an increase in MnSOD isoenzyme activity was observed in AT478 squamous carcinoma cells treated with holoxan at concentrations of 10 µg/mL and 40 µg/mL for 24 hours, compared to the control group, with increases of 9.2 NU/mL and 14.69 NU/mL, respectively, versus 1.2 NU/mL in the control group. Additionally, an increase in Cu/ZnSOD isoenzyme activity was noted in these cells, with levels of 3.7 NU/mL and 4.1 NU/mL versus 1.4 NU/mL in the control group.
In another study on changes in the activity of the pro-oxidant/antioxidant enzyme system in human glioblastoma SNB19 cells under the influence of carboplatin, Polaniak et al. (45) found that the total SOD activity in cells treated with carboplatin at two different concentrations was higher compared to controls at both 24 and 72 hours. The highest activity of SOD, MnSOD, and GSH-Px was observed in samples treated with the higher concentration of carboplatin after 72 hours. The activity of the SOD2 isoenzyme and GSH-Px was higher in all experimental groups at 72 hours compared to 24 hours. In contrast, the activity of the copper-zinc superoxide dismutase isoenzyme (Cu/ZnSOD) in samples exposed to carboplatin at both concentrations was higher than in the control at both 24 and 72 hours. However, in the lower-concentration group, SOD1 activity was lowest at 72 hours (45).
Alexandre et al. (48) investigated the effects of mangafodipir, a substance used as a contrast in magnetic resonance imaging, which acts as a superoxide dismutase (SOD) mimetic and exhibits catalase and glutathione reductase activity, on the cytotoxicity of anticancer drugs. They demonstrated that the use of this oxidative stress modulator increased the cytotoxicity of oxaliplatin and paclitaxel in CT26 colorectal cancer cells in in vitro cultures while reducing hematotoxicity. They hypothesized that this effect was due to the antioxidant activity of mangafodipir.
Current challenges in research
One of the main difficulties is the dual nature of ROS, which can both induce carcinogenesis and promote apoptosis depending on context. Another challenge is tumor heterogeneity: ROS levels and enzyme activities vary significantly across cancer types, complicating universal strategies. Moreover, translating promising in vitro findings to in vivo and clinical settings remains problematic (57).
Methodological caveats include assay selection (e.g., DCF-DA vs. mitochondria-specific probes), artifact-prone measurements, and endpoint timing (24 h vs. 72 h) that can invert interpretations. Standardized protocols and reference controls are needed to improve reproducibility across labs (58).
Clinical translation hurdles comprise patient heterogeneity, prior therapy exposure reshaping tumor redox landscapes, and difficulty in serially sampling tumors. Liquid biomarkers (lipid peroxidation products, oxidized nucleotides) and imaging surrogates may partially bridge this gap (57–60).
Numerous studies on the mechanisms of action of cytotoxic drugs and their efficacy have focused on exploring the mechanisms of cell resistance to these agents. For years, we have wanted to understand the underlying causes of cancer, its origins, and methods of prevention. In the literature, there is significant interest in studying changes in antioxidant enzyme activity in cancer cell lines during in vitro cultures under the influence of cytostatic drugs.
Future perspectives
Future work should integrate nanotechnology, gene therapy, and ROS-modulating strategies with conventional cytostatics. Personalized medicine approaches based on tumor-specific ROS profiles could improve therapeutic selectivity. Combination therapies, integrating cytostatics with antioxidants or pro-oxidants, may offer synergistic benefits and reduce side effects.
Precision redox oncology will likely rely on composite biomarkers (enzyme activities, GSH/GSSG ratio, redox-sensitive transcriptional signatures) to stratify patients. Adaptive trials can test cytostatics ± redox modulators with early stopping based on toxicity/efficacy readouts.
Engineering advances (stimuli-responsive nanoparticles releasing payloads in high-ROS niches, tumor-penetrating peptides, and mitochondrial-targeted carriers) could widen the therapeutic window, enhancing tumor selectivity while sparing normal tissues.
Integration with immunotherapy is also promising. ROS can remodel antigen presentation and the tumor microenvironment. Rational scheduling may synergize redox modulation with checkpoint blockade or adoptive cell therapies.
Conclusion
Research on oxidative stress enzyme activity changes provides essential insights into the mechanisms of cytostatic action in cancer cells. A deeper understanding of ROS-antioxidant interactions may enable the design of selective therapies with reduced toxicity. With advancements in gene therapy, targeted drug delivery, and nanomedicine, chemotherapy effectiveness could be significantly improved. Translating these findings into clinical applications will be key to overcoming resistance and improving patient survival.
Author contributions
RP: Formal Analysis, Resources, Methodology, Conceptualization, Writing – original draft, Supervision. AK: Writing – review & editing, Investigation, Project administration. MG: Software, Supervision, Funding acquisition, Writing – review & editing, Formal Analysis. EG-N: Project administration, Visualization, Writing – review & editing, Investigation. ML: Project administration, Methodology, Visualization, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Carocho M and Ferreira IC. A review on antioxidants, prooxidants and related controversy: natural and synthetic compounds, screening and analysis methodologies and future perspectives. Food Chem Toxicol. (2013) 51:15–25. doi: 10.1016/j.fct.2012.09.021
2. Abdelazim AM and Abomughaid MM. Oxidative stress: an overview of past research and future insights. All Life. (2024) 17. doi: 10.1080/26895293.2024.2316092
3. Sies H, Belousov VV, Chandel NS, Davies MJ, Jones DP, Mann GE, et al. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat Rev Mol Cell Biol. (2022) 23:499–515. doi: 10.1038/s41580-022-00456-z
4. Murotomi K, Umeno A, Shichiri M, Tanito M, and Yoshida Y. Significance of singlet oxygen molecule in pathologies. Int J Mol Sci. (2023) 24:2739. doi: 10.3390/ijms24032739
5. Fu C, Li Y, Xi H, Niu Z, Chen N, Wang R, et al. Benzo(a)pyrene and cardiovascular diseases: An overview of pre-clinical studies focused on the underlying molecular mechanism. Front Nutr. (2022) 9:978475. doi: 10.3389/fnut.2022.978475
6. Tsiara E, Economou M, Stamatopoulou S, Kourti M, Voulgarelis M, and Vogiatzoglou A. Redox biomarker levels in patients with myelodysplastic syndrome. Biomed Rep. (2025) 23:1923. doi: 10.3892/br.2025.1923
7. Didier AJ, Stiene J, Fang L, Watkins D, Dworkin LD, and Creeden JF. Antioxidant and anti-tumor effects of dietary vitamins A, C and E. Antioxidants. (2023) 12:632. doi: 10.3390/antiox12030632
8. Martemucci G, Costagliola C, Mariano M, D’Andrea L, Napolitano P, and D’Alessandro AG. Free radical properties, source and targets, antioxidant consumption and health. Oxygen. (2022) 2:48–78. doi: 10.3390/oxygen2020006
9. Roy Z, Bansal R, Siddiqui L, and Chaudhary N. Understanding the role of free radicals and antioxidant enzymes in human diseases. Curr Pharm Biotechnol. (2023) 24:1265–76. doi: 10.2174/1389201024666221121160822
10. Zhang Y, Zhang HM, Shi Y, Lustgarten M, Li Y, Qi W, et al. Loss of manganese superoxide dismutase leads to abnormal growth and signal transduction in mouse embryonic fibroblasts. Free Radic Biol Med. (2010) 49:1255–62. doi: 10.1016/j.freeradbiomed.2010.07.006
11. Skólmowska M and Kmieć M. Enzymosomy antyoksydacyjne - właściwości i zastosowanie. Postępy Hig. Med Dośw. (2011) 65:640–4.
12. Aliya S, Reddanna P, and Thyagaraju K. Does glutathione S-transferase Pi (GST-Pi) a marker protein for cancer? Mol Cell Biochem. (2003) 253:319–27.
13. Wang F, Zhang C, Zhu X, Zhang D, Zhang Z, Ni S, et al. Overexpression of GSTP1 promotes colorectal cancer cell proliferation, invasion and metastasis by upregulating STAT3. Adv Clin Exp Med. (2022) 31:139–49. doi: 10.17219/acem/142461
14. Wu K, El Zowalaty AE, Sayin VI, and Papagiannakopoulos T. The pleiotropic functions of reactive oxygen species in cancer. Nat Cancer. (2024) 5:384–99. doi: 10.1038/s43018-024-00738-9
15. Sahoo BM, Banik BK, Borah P, and Jain A. Reactive oxygen species (ROS): key components in cancer therapies. Anti-Cancer Agents Medicinal Chem. (2022) 22:215–22. doi: 10.2174/1871520621666210608095512
16. Romani AM. Cisplatin in cancer treatment. Biochem Pharmacol. (2022) 206:115323. doi: 10.1016/j.bcp.2022.115323
17. Meng X, Wu Z, Yuan Y, Liu J, Wu S, Shi W, et al. Redox-manipulating nanocarriers for anticancer drug delivery. J Nanobiotechnol. (2024) 22:859. doi: 10.1186/s12951-024-02859-w
18. Sha H, Zheng X, Xu J, Liu Z, Li M, and Sun H. NBDHEX re-sensitizes adriamycin-resistant breast cancer by inhibiting GSTP1 activity. Cancer Med. (2023) 12:5370. doi: 10.1002/cam4.5370
19. Roy N, Saha B, Ghosh S, Mukherjee S, and Dasgupta S. Glutathione depletion and stalwart anticancer activity of small-molecule modulators. ACS Omega. (2024) 9:8890–902. doi: 10.1021/acsomega.3c08890
20. National Collaborating Centre for Cancer (UK). Colorectal cancer: the diagnosis and management of colorectal cancer. In: Management of local disease, vol. 3. National Collaborating Centre for Cancer (UK, Cardiff (2011). Available online at: http://www.ncbi.nlm.nih.gov/books/NBK116630/. (NICE Clinical Guidelines, No. 131.).
21. National Collaborating Centre for Cancer (UK). Ovarian cancer: the recognition and initial management of ovarian cancer. In: Management of advanced stage (II-IV) ovarian cancer. National Collaborating Centre for Cancer (UK, Cardiff (UK (2011). Available online at: http://www.ncbi.nlm.nih.gov/books/NBK83844/. (NICE Clinical Guidelines, No. 122.)Chapter 5.
22. Fokas E, Appelt A, Glynne-Jones R, Beets G, Perez R, Garcia-Aguilar J, et al. International consensus recommendations on key outcome measures for organ preservation after (chemo)radiotherapy in patients with rectal cancer. Nat Rev Clin Oncol. (2021) 18:805–16. doi: 10.1038/s41571-021-00538-5
23. Ježek P, Jabůrek M, Holendová B, and Plecitá-Hlavatá L. Pitfalls of mitochondrial redox signalling research. Antioxidants. (2023) 12:1696. doi: 10.3390/antiox12091696
24. Trynda-Lemiesz L and Śliwińska-Hill U. Metal complexes in anticancer therapy. Present future. (2011) 6:465–74.
25. Ali MM and Aziz TA. Toxic effect of platinum compounds: molecular mechanisms of toxicity. Al-Rafidain J Med Sci. (2021) 1:81–8. doi: 10.54133/ajms.v1i.32
26. Bakalova A, Ruseva N, and Cherneva E. Non-classical” Platinum complexes: A concise review. Int J Mol Sci. (2025) 26:6270. doi: 10.3390/ijms26136270
27. Kopacz-Bednarska A and Król T. Selected platinum complexes in standard and modern anti-cancer therapies. Biuletyn Polskiego Towarzystwa Onkologicznego Nowotwory. (2022) 7:106–15. doi: 10.5603/NJO.a2022.0011
28. Khemapech N, Oranratanaphan S, Termrungruanglert W, Lertkhachonsuk R, and Vasurattana A. Salvage chemotherapy in recurrent platinum-resistant or refractory epithelial ovarian cancer with Carboplatin and distearoylphosphatidylcholine pegylated liposomal Doxorubicin (lipo-dox®). Asian Pac. J Cancer Prev. (2013) 14:2131–5. doi: 10.7314/APJCP.2013.14.3.2131
29. Llesuy S. Reduction in the toxicity of doxorubicin (Adriamycin). In: Vitamin E in Health and Disease: Biochemistry and Clinical Applications, CRC Press,vol. 417. (2023).
30. Rödel C, Liersch T, Hermann R, Arnold D, Reese T, and Hipp M. Multicenter phase II trial of chemoradiation with oxaliplatin for rectal cancer. J Clin Oncol. (2007) 25:110–7. doi: 10.1200/JCO.2006.08.3675
31. Tacar O, Sriamornsak P, and Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. (2013) 65(2):157–70. doi: 10.1111/j.2042-7158.2012.01567.x
32. Gibson JM, Alzghari S, Ahn C, Trantham H, and La-Beck NM. The role of pegylated liposomal doxorubicin in ovarian cancer: a meta-analysis of randomized clinical trials. Oncologist. (2013) 18:1022–31. doi: 10.1634/theoncologist.2013-0126
33. Mancardi D, Mezzanotte M, Arrigo E, Barinotti A, and Roetto A. Iron overload, oxidative stress, and ferroptosis in the failing heart and liver. Antioxidants. (2021) 10:1864. doi: 10.3390/antiox10121864
34. Stephenson A, Bass EB, Bixler BR, Daneshmand S, Kirkby E, Marianes A, et al. Diagnosis and treatment of early-stage testicular cancer: AUA guideline amendment 2023. J Urol. (2024) 211:20–5. doi: 10.1097/JU.0000000000003694
35. Ramani S and Park S. HSP27 role in cardioprotection by modulating chemotherapeutic doxorubicin-induced cell death. J Mol Med. (2021) 99:771–84. doi: 10.1007/s00109-021-02048-4
36. Wang S, Konorev EA, Kotamraju S, Joseph J, Kalivendi S, and Kalyanaraman B. Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms. intermediacy of H(2)O(2)- and p53-dependent pathways. J Biol Chem. (2004) 279:25535–43. doi: 10.1074/jbc.M400944200
37. Patintingan CG, Louisa M, Juniantito V, Arozal W, Hanifah S, Wanandi SI, et al. Moringa oleifera leaves extract ameliorates doxorubicin-induced cardiotoxicity via its mitochondrial biogenesis modulatory activity in rats. J Exp Pharmacol. (2023), 307–19. doi: 10.2147/JEP.S413256
38. Renu K, Pureti LP, Vellingiri B, and Valsala Gopalakrishnan A. Toxic effects and molecular mechanism of doxorubicin on different organs–an update. Toxin Rev. (2022) 41:650–74. doi: 10.1080/15569543.2021.1912099
39. Jones SE, Savin MA, Holmes FA, O’Shaughnessy JA, and Blum JL. et all: Phase III trial comparing doxorubicin plus cyclophosphamide with docetaxel plus cyclophosphamide as adjuvant therapy for operable breast cancer. J Clin Oncol. (2006) 24:5381–7. doi: 10.1200/JCO.2006.06.5391
40. Poornima P, Kumar VB, Weng CF, and Padma W. Doxorubicin induced apoptosis was potentiated by neferine in human lung adenocarcima, A549 cells. Food Chem.Toxicol. (2014) 68:87–98.
41. Chen YA, Lu CY, Cheng WF, Kuo KT, Yu CW, Ho HN, et al. An experimental model for ovarian cancer: propagation of ovarian cancer initiating cells and generation of ovarian cancer organoids. BMC Cancer. (2022) 22:967. doi: 10.1186/s12885-022-10042-3
42. KaChadourian R, Leitner HM, and Day BJ. Selected flavonoids potentiate the toxicity of cisplatin in human lung adenocarcinoma cells: a role for glutathione depletion. Int J Oncol. (2007) 31:161–8. doi: 10.3892/ijo.31.1.161
43. Georgakis GV, Li Y, Humphreys R, Andreeff M, O’Brien S, Younes M, et al. Activity of selective fully human agonistic antibodies to the TRAIL death receptors TRAIL-R1 and TRAIL-R2 in primary and cultured lymphoma cells: induction of apoptosis and enhancement of doxorubicin- and bortezomib-induced cell death. Brit.J. Haematology. (2005) 130:501–10. doi: 10.1111/j.1365-2141.2005.05656.x
44. Bułdak RJ, Polaniak R, Kukla M, Kubina R, Skonieczna M, Bułdak M, et al. Wpływ karboplatyny na aktywność peroksydazy glutationu (GSH-Px), reduktazy glutationu (GR) oraz stężenie dialdehydu malonowego (MDA) w mediach hodowlanych ludzkich komórek czerniaka złośliwego linii Me45 in vitro. Ann Acad Med Siles. (2011) 65:7–15.
45. Polaniak R, Bułdak RJ, Latocha M, Romuk E, Karbowska D, Ochocki K, et al. Zaburzenia układu prooksydacyjno/antyoksydacyjnego komórek glejaka mózgu pod wpływem karboplatyny–in vitro. Brom.Chem. Toksykol. (2013), 461–5.
46. Bułdak RJ, Polaniak R, and Kukla M. Wpływ etopozydu na aktywność enzymów stresu oksydacyjnego oraz stężenie dialdehydu malonowego (MDA) w hodowli ludzkich komórek czerniaka złośliwego linii Me45 in vitro. Farm. Przegl. Nauk. (2010) 11:11–9. i wsp.
47. Polaniak R, Wideł M, Beck B, Chwalińska E, Gruca- Mamczar E, and Birkner E. Wpływ holoksanu na aktywność izoenzymów dysmutazy ponadtlenkowej i stężenie dialdehydu malonowego w hodowli megakolonii komórek raka płaskonabłonkowego in vitro. Bromat. Chem Toksykol. (2008) 1:89–94.
48. Alexandre J, Nicco C, Chéreau C, Laurent A, Weill B, Goldwasser F, et al. Improvement of the therapeutic index of anticancer drugs by the superoxide dismutase mimic mangafodipir. J Natl Cancer Institute. (2006) 98:236–44. doi: 10.1093/jnci/djj049
49. Dusre L, Rajagopalan S, Eliot HM, Covey JM, and Sinha BK. DNA interstrand cross-link and free radical formation in a human multidrug-resistant cell line from mitomycin C and its analogues. Cancer Res. (1990) 50:648–52.
50. Samuels BL, Murray JL, Cohen MB, Safa AR, Sinha BK, Townsend AJ, et al. Increased glutathione peroxidase activity in a human sarcoma cell line with inherent doxorubicin resistance. Cancer Res. (1991) 51:521–7.
51. Mozafari F, Rashidzadeh H, Ghaffarlou M, Salehiabar M, Ertas YN, Ramazani A, et al. ROS-based cancer radiotherapy. In: Harnessing Materials for X-ray Based Cancer Therapy and Imaging. Cham: Springer International Publishing (2022) p. 265–309.
52. Engwa GA, Nweke FN, and Nkeh-Chungag BN. Free radicals, oxidative stress-related diseases and antioxidant supplementation. Altern Therapies Health Med. (2022) 28.
53. L’Ecuyer T, Horenstein MS, Thomas R, and Vander Heide R. Anthracycline-induced cardiac injury using a cardiac cell line: potential for gene therapy studies. Mol Genet Metab. (2001) 74:370–9. doi: 10.1006/mgme.2001.3243
54. Zhong W, Yan T, Webber MM, and Oberley TD. Alteration of cellular phenotype and responses to oxidative stress by manganese superoxide dismutase and a superoxide dismutase mimic in RWPE-2 human prostate adenocarcinoma cells. Antioxid Redox Signal. (2004) 6:513–22. doi: 10.1089/152308604773934279
55. Dong C, Zhang NJ, and Zhang LJ. Oxidative stress in leukemia and antioxidant treatment. Chin Med J. (2021) 134:1897–907. doi: 10.1097/CM9.0000000000001628
56. Li H, Wang H, Li Z, Kelley N, Ouyang M, Wu JW, et al. Anti-proliferative and anti-invasive effects of exogenous thermostable MnSOD in gastric cancer associated with p53 and ZEB1 expression. J Cancer. (2025) 16:2062. doi: 10.7150/jca.102600
57. Murphy MP, Bayir H, Belousov V, Chang CJ, Davies KJA, Davies MJ, et al. Guidelines for measuring reactive oxygen species and oxidative damage. Nat Metab. (2022) 4:651–62. doi: 10.1038/s42255-022-00591-Z
58. An X, Yu W, Liu J, Tang D, Yang L, Chen X, et al. Oxidative cell death in cancer: mechanisms and therapeutic opportunities. Cell Death Dis. (2024) 15:412. doi: 10.1038/s41419-024-06939-5
59. Zhou X, An B, Lin Y, Ni Y, Zhao X, and Liang X. Molecular mechanisms of ROS-modulated cancer drug resistance. Cancer Lett. (2023) 562:216223. doi: 10.1016/j.canlet.2023.216223
Keywords: cytostatic, cisplatin, doxorubicin, reactive oxygen species (ROS), cancer
Citation: Polaniak R, Kwiatkowski A, Górski M, Grochowska-Niedworok E and Latocha M (2025) The role of cytostatic in oxidative stress reactions. Front. Oncol. 15:1667522. doi: 10.3389/fonc.2025.1667522
Received: 16 July 2025; Accepted: 22 September 2025;
Published: 13 October 2025.
Edited by:
Qingbin Cui, University of Toledo College of Medicine and Life Sciences, United StatesReviewed by:
Sivapar V. Mathan, University of Delhi, IndiaHardeep Singh Tuli, Maharishi Markandeshwar University, India
Copyright © 2025 Polaniak, Kwiatkowski, Górski, Grochowska-Niedworok and Latocha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renata Polaniak, cnBvbGFuaWFrQHN1bS5lZHUucGw=