 Zihao Ye
Zihao Ye Hao Wu
Hao Wu Zhanhao Li1
Zhanhao Li1 Baoshan Gao
Baoshan Gao- 1Department of Urology II, The First Hospital of Jilin University, Changchun, China
- 2School of Basic Medicine, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3Department of Urology, China-Japan Hospital of Jilin University, Changchun, China
Ammonium metabolism represents a critically understudied yet pivotal driver of prostate tumorigenesis and tumor microenvironment (TME) remodeling. The interplay between tumor metabolic reprogramming and the tumor microenvironment has emerged as a critical frontier in oncology research. While previous studies on prostate cancer metabolism have predominantly focused on lipid metabolism and the Warburg effect, the role of ammonium metabolism, particularly the urea cycle in tumor immune regulation remains insufficiently explored. This metabolic reprogramming constitutes a central node connecting catabolic nutrient breakdown to anabolic biosynthesis by integrating upstream amino acid deamination and transamination reactions with downstream pathways, generating key intermediates including α-ketoglutarate, coenzyme A, and citrate that concurrently fuel the tricarboxylic acid cycle and macromolecular synthesis. Crucially, oncogenic drivers such as Myc and p53 modulate this flux through epigenetic regulation of core enzymes such as glutaminase, glutamine synthetase and ornithine transcarbamylase, thereby channeling metabolism toward tumor progression. The immunomodulatory consequences manifest through dual mechanisms including TME immunosuppression driven by M2 macrophage polarization and immune evasion mediated via glutathione dependent redox homeostasis disruption. Beyond its established role in modulating redox homeostasis, ammonium metabolic reprogramming may additionally trigger novel cell death modalities such as ferroptosis by GSH/GPX4 axis. This emerging pathway offers promising therapeutic avenues for prostate cancer intervention. Synthesizing mechanistically validated insights from in vivo or in vitro models and clinical trials of ammonium-targeting inhibitors, this review proposes novel therapeutic strategies and candidate biomarkers. Moreover, the unique citrate and polyamine metabolism characteristics of prostate cancer may be impacted by these processes, offering promising avenues for future treatments.
1 Introduction
Prostate cancer (PCa) is the second most common solid tumor in men worldwide and the fifth leading cause of cancer-related death. Its incidence and mortality rates vary by region (1). In 2020, there were more than 1.4 million new cases and more than 375,000 deaths globally. Prostate cancer can be classified into androgen-sensitive and androgen-insensitive types, which influence treatment options. Common treatments include active surveillance, chemotherapy, radiotherapy, hormone therapy, surgery, and focal therapy (2). Surgery and radiotherapy are effective for localized prostate cancer, whereas androgen deprivation therapy is the standard treatment for advanced or metastatic cases (3). Although 80% of prostate cancer cases are localized, 20% to 40% of patients experience recurrence within five years, and approximately 20% of localized cases progress to metastatic hormone-sensitive prostate cancer (mHSPC), which can further evolve into metastatic castration-resistant prostate cancer (mCRPC) (4). Therefore, there is an urgent need to develop new treatment strategies, especially for CRPC.
Metabolic reprogramming, characterized by alterations in lipid, glucose, and amino acid metabolism, is a hallmark of cancer and enables malignant cells to adapt to the unique features of the tumor microenvironment (TME) (5). In prostate cancer, a shift from lipid-dominated to glucose-dependent metabolism occurs during disease progression, particularly from early to metastatic stages, which may contribute to therapeutic resistance. Metastatic prostate cancer uses glycolysis to maintain an acidic tumor microenvironment, which suppresses immune cell activity and promotes immune evasion (6, 7). In bone metastasis, accompanied with adipocyte, prostate cancer cells exhibit high glycolytic rates and increased HIF-1α expression, which regulates Warburg effect genes, leading to enhanced lactate production and inhibition of oxidative phosphorylation (8). These findings confirm that glycolysis is a crucial metabolic pathway in prostate cancer metastasis and progression. These metabolic changes may lead to drug resistance in cancer cells, enabling them to evade therapeutic treatments (9). While extensive studies have explored lipid and glucose pathways, limited therapeutic opportunities have emerged due to metabolic heterogeneity and redundancy (10, 11). Specifically, in prostate cancer, PI3K activation and MYC-induced lipid metabolism promote glycolysis and contribute to metabolic heterogeneity (12). Additionally, FASN inhibitors can be bypassed by exogenous fatty acids, and cancer cells regulate acetyl-CoA sources like acetate and glutamine to adapt to metabolic stress (13, 14). Recently, ammonium metabolism, a key node integrating amino acid turnover, nitrogen balance, and intracellular pH regulation, has attracted growing interest (15, 16). Under nutrient-deprived conditions, Intermediates such as glutamine and glutamate not only serve as nitrogen and carbon sources for anabolic processes but also participate in TME remodeling and immunoregulation, particularly under nutrient deprivation, relevant to prostate cancer biology (17).
This review summarizes recent studies on the role of ammonium metabolism in the prostate cancer microenvironment, discusses its potential as a therapeutic target, and explores its application as a diagnostic biomarker for prostate cancer to improve both detection and prognosis.
2 Ammonium metabolism in tumor cells: sources, fates, and functional implications
2.1 Metabolic reprogram and crosstalk of ammonium metabolism in prostate cancers
PCa cells exhibit distinct ammonium metabolism characterized by dysregulated upstream amino acid catabolism and coordinated crosstalk with glucose and lipid metabolic pathways. Accumulating evidence establishes asparagine synthetase (ASNS) as a critical driver of malignant progression in solid tumors including prostate cancer, where it sustains tumorigenesis through dual metabolic reprogramming (18–21). In CRPC, TP53 mutation or deletion drives ASNS upregulation via the mTOR/ATF4 axis, enhancing cellular reliance on ASNS-mediated asparagine biosynthesis. This metabolic plasticity enables tumor survival in androgen-deprived microenvironments (22). This regulation typically arises due to the disruption of the balance between the recruited corepressor and coactivator following TP53 gene mutation. Another mechanism involves the mutant CRCP of TP53 predominantly binding to ATF4, thereby exerting an inhibitory effect, in contrast to the wild-type CRPC of TP53. TP53 mutation transcriptionally suppresses ASNS expression, thereby disrupting asparagine-aspartate homeostasis. Crucially, this regulatory circuit operates bidirectionally, asparagine and aspartate reciprocally modulate AMPK-mediated p53 activation through allosteric binding to LKB1 that bidirectionally regulates its kinase activity (23). Recent studies reveal ASNS gene amplification with concomitant mRNA overexpression, augmenting asparagine production (19). Aspartate fuels tumor progression by integrating into glycolysis and lipogenesis pathways while suppressing apoptosis under glucose deprivation via JNK/SAPK activation (24). Previous studies have suggested that TP53-mutant CRPC exhibits metabolic vulnerability as an adaptive response to the nutrient-deprived tumor microenvironment (23). Therapeutic strategies targeting asparagine metabolism have shown promising efficacy in this context.
In PCa, glutamine serves as a critical upstream hub in ammonium metabolism, with tumors exhibiting profound metabolic rewiring to sustain proliferative and stress-adaptive demands. Glutamine metabolism serves as a metabolic intersection in prostate cancer ammonium reprogramming, where this conditionally essential amino acid propels tumor proliferation through multifaceted biosynthetic and bioenergetic contributions including its catabolic flux furnishes precursors for de novo purine/pyrimidine biosynthesis and hexosamine pathway activation, drives reductive carboxylation-dependent lipogenesis, sustains redox homeostasis via glutathione synthesis and NADPH regeneration, generates non-essential amino acids, and fuels mitochondrial oxidative phosphorylation through α-KG (α-ketoglutarate) mediated anaplerosis, collectively establishing glutaminolysis as an indispensable axis supporting prostate cancer malignancy (9, 25). Under hypoxia, PCa cells divert excess nitrogen, derived from upstream amino acids and accumulated ammonium, toward dihydroorotate synthesis rather than uridine monophosphate (UMP) production, effectively circumventing ammonium toxicity (26). Mechanistically, hypoxia-induced NADH accumulation, rather than canonical HIF1 signaling, drives this glutamine metabolic reprogramming (27). Unlike normal cells, which utilize glutamine for basal nitrogen/energy homeostasis, PCa cells elevate glutamine uptake and catabolism to meet biosynthetic demands (28, 29).Glutamine-derived carbon undergoes reductive carboxylation to generate acetyl-CoA for fatty acid synthesis, while its nitrogen is channeled into nucleotide biosynthesis (30). Concurrently, glutamine metabolism sustains redox balance via glutathione synthesis, enabling adaptation to oxidative stress in hypoxic, nutrient-deprived microenvironments (31). This metabolic dependency positions glutaminolysis as a therapeutic strategy. From the perspective of signaling pathways, mainstream research indicates that glutamine addiction is associated with the progression and metastasis of prostate cancer cells through three distinct pathways: the AR pathway, the MYC pathway, and the PTEN/PI3K/mTOR pathway. ASCT2 (SLC1A5, solute carrier transporters) is a critical transporter responsible for glutamine uptake in prostate cancer cells with its expression is elevated in tumor tissues and further increased in CRPC. ASCT2 is directly regulated by AR signaling (32). In AR-sensitive cells such as LNCaP, androgens like dihydrotestosterone (DHT) significantly upregulate ASCT2 expression and enhance glutamine uptake. In contrast, AR-insensitive cell lines (e.g., DU-145, PC-3) also rely on ASCT2-mediated glutamine transport but are not directly modulated by AR signaling (33, 34). GLS, the rate-limiting enzyme in glutamine catabolism, exhibits markedly higher expression in AR-insensitive cells, particularly DU-145, contributing to their increased glutamine dependency. Inhibition of GLS, using agents such as BPTES or siRNA, effectively reduces viability across all PCa cell lines, with AR-independent cells showing heightened sensitivity (35). From a therapeutic perspective, combined inhibition of AR (androgen receptor) signaling and GLS activity yields synergistic anti-tumor effects in AR-sensitive cells. In contrast, targeting glutamine metabolism alone, especially through GLS inhibition, represents a more effective strategy for AR-independent tumors. These findings support a tailored therapeutic approach based on the AR status of prostate cancer.
MYC functions as a central regulator of glutamine metabolism in prostate cancer by repressing miR-23a/b, thereby relieving the inhibition of mitochondrial GLS and promoting glutaminolysis, particularly in androgen-independent PC-3 cells (36, 37). The antitumor efficacy of the GLS inhibitor CB-839 is strongly MYC-dependent (38). In AR-positive cells such as LNCaP, MYC also upregulates ASCT2 expression and enhances glutamine uptake in an androgen-responsive manner (39). The metabolic role of MYC is further modulated by PTEN/PI3K signaling, where concurrent mTORC1 activation in PTEN-deficient cells amplifies glutamine metabolism (40). Glutamine metabolic reprogramming not only sustains tumor proliferation but also contributes to therapy resistance; inhibition of GLS or MYC sensitizes prostate cancer cells to radiotherapy, while autophagy enables glutamine-independent cells to withstand metabolic stress. Additional regulatory mechanisms include SRC-2–mediated reductive carboxylation, compensatory glutamine addiction induced by PDHA1 loss, Guanine Monophosphate Synthetase(GMPS) driven purine biosynthesis, reciprocal regulation between GLS and CAD, epigenetic modulation via MeCP2/DNMT, and C5a complement signaling that promotes glutamine consumption in castration-resistant prostate cancer (41–43). Notably, PC-3M (prostate cancer cell lines) subpopulations with stem-like features exhibit heightened glutamine dependency characterized by elevated GLS1 expression and enhanced reductive carboxylation activity, forming a bidirectional regulatory loop between glutamine metabolism and epigenetic reprogramming (44). Collectively, these findings underscore the essential role of glutamine metabolism in prostate cancer progression and therapy resistance, and support the development of targeted combinatorial strategies.
Metabolomic profiling reveals elevated alanine levels in PCa versus normal prostate tissues, potentially reflecting heightened membrane biosynthesis requirements (45, 46). Alanine transamination supports glutamate oxidation to α-KG, providing an alternative lipogenic carbon source distinct from pyruvate/lactate flux, while excess alanine may directly fuel protein synthesis (47, 48). Studies in PCa suggest that arginine derived metabolites promote tumor proliferation by suppressing apoptosis through redox modulation (49). Mechanistically, nitric oxide synthase (NOS) converts arginine, NADPH, and oxygen into nitric oxide, which inhibits apoptosis by altering cellular redox states (50, 51). PCa cells exhibit upregulated tryptophan-hydroxylating enzymes and aromatic L-amino acid decarboxylases, key drivers of tryptophan metabolic reprogramming. These enzymes orchestrate tryptophan catabolism, with tryptophan hydroxylase (TPH) catalyzing the conversion of tryptophan (Trp) to serotonin. Elevated expression of tryptophan-2,3-dioxygenase 2 (TDO2) and indoleamine 2,3-dioxygenase 1 (IDO1) enhances kynurenine (Kyn) production, which fosters PCa progression through tumor-associated immunosuppression and direct pro-survival signaling (52–54). Methionine addiction characterizes PCa ammonium metabolism, with SNHG3 overexpression identified as a key upstream regulator through its interaction with the miR-152-3p/SLC7A11 axis (55, 56). Functional validation confirms SLC7A11 as a direct miR-152-3p target whose overexpression rescues methionine dependency in SNHG3-deficient PCa cells, establishing the SNHG3/miR-152-3p/SLC7A11 regulatory axis (Figure 1). Proline metabolism, regulated by oncogenic c-MYC and PI3K pathways, modulates AR transcriptional activity through cyclin D3/CDK11p58-mediated serine phosphorylation, a mechanism requiring further mechanistic exploration (37, 55, 57, 58).
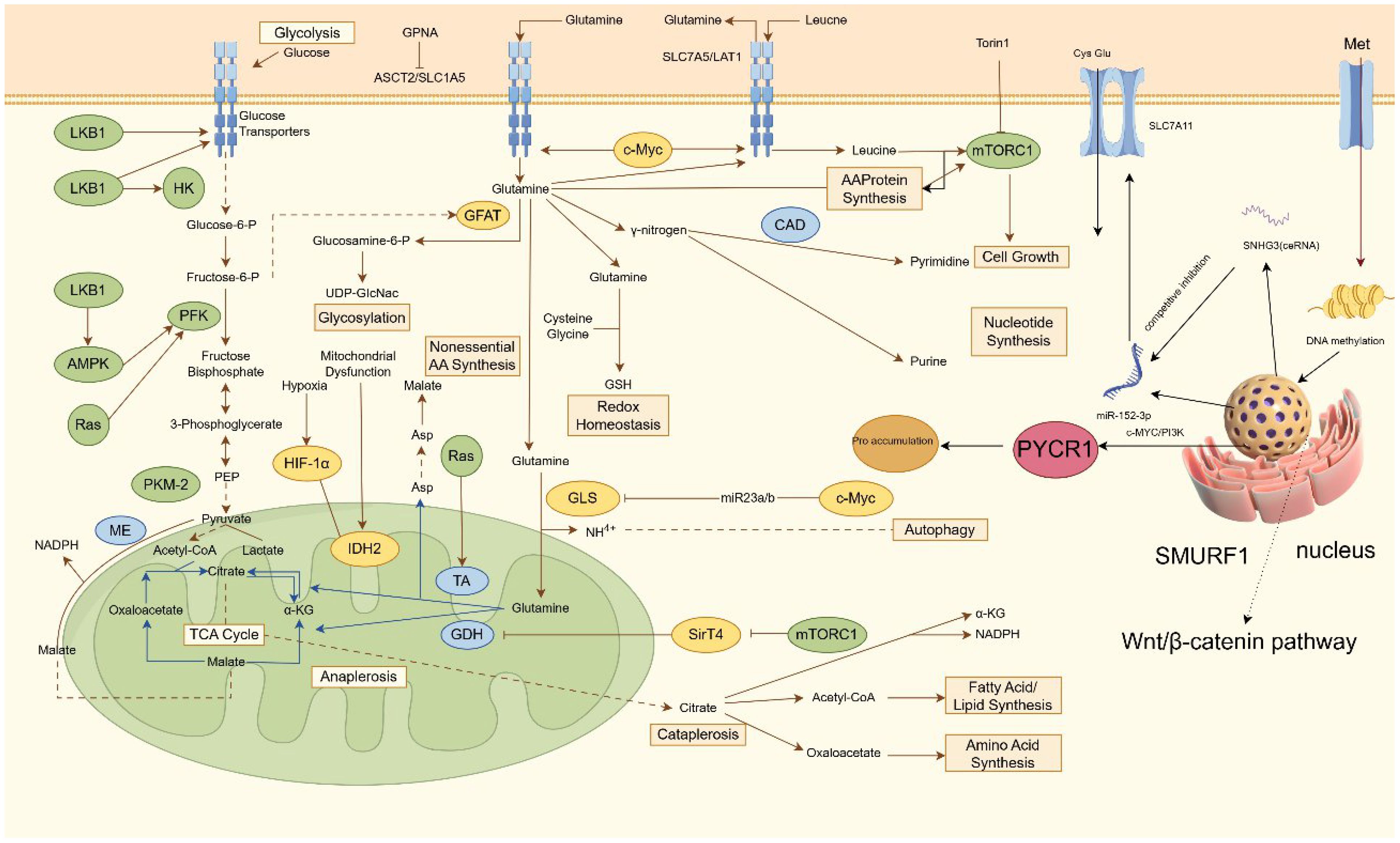
Figure 1. Regulatory mechanism of ammonium metabolism in PCa. This diagram outlines a coordinated network of cellular nutrient metabolism and signaling pathways that support growth and survival. Glucose enters via transporters and undergoes glycolysis, regulated by HK, PFK, and the LKB1-AMPK-Ras axis, yielding pyruvate. Amino acid transporters ASCT2 and SLC7A5 import glutamine and leucine; leucine activates mTORC1 to promote protein synthesis. Glutamine metabolism diverges into multiple pathways: it supports hexosamine biosynthesis (via GFAT), nucleotide synthesis (via CAD for pyrimidines), non-essential amino acid production, and glutathione-mediated redox balance. Converted to glutamate by GLS, it also fuels the TCA cycle, where enzymes including IDH2 and GDH sustain anaplerosis and intermediate turnover, influenced under hypoxia by HIF-1α. Signaling through mTORC1, SirT4, and the Wnt/β-catenin-SMURF1 pathway regulates gene expression and epigenetic modifications. Additionally, non-coding RNAs such as miR-152-3p and SNHG3 fine-tune processes including autophagy and proline metabolism via targets like c-Myc/PI3K, ensuring integrated control of metabolic and proliferative functions. HK, hexokinase; PFK, phosphofructokinase, UDP-GlcNac, Uridine Diphosphate N-Acetylglucosamine; GSH, glutathione; SNHG3, Small Nucleolar RNA Host Gene; ceRNA, competing endogenous RNAs.
The downstream metabolites of ammonium metabolism in prostate cancer cells, notably citrate and polyamines, exert significant functional influences on prostate cancer development. The prostate gland contains the highest polyamine concentrations in the human body, with spermine constituting the predominant polyamine species. Androgens coordinately induce the enzymatic activities of key polyamine biosynthetic enzymes, ornithine decarboxylase (ODC), S-adenosylmethionine decarboxylase (SAMDC), and spermidine synthase (SDS), with predominant expression localized to prostatic glandular epithelial cells (59–61). Murine ODC gene studies reveal an androgen response element-like sequence within the ODC promoter that binds androgen receptor in vitro. The functions of ODC and polyamines in prostate tissue are mechanistically linked to cellular proliferation and secretory activity (62). Recent investigations demonstrate overexpression of ODC, the rate-limiting enzyme in polyamine metabolism, in prostate cancer cells, accompanied by elevated ODC protein and mRNA levels (63, 64). Comparative analyses of polyamine levels in human normal, benign, and malignant prostatic tissues reveal elevated spermine concentrations in normal and benign hyperplastic prostates, contrasting with reduced spermine levels in tumor tissues, particularly metastatic prostate cancer (65). This marked depletion of prostatic spermine may signify the phenotypic transition from benign to malignant states. Consequently, ODC has been proposed as a proto-oncogene expression product in prostate carcinogenesis. Notably, enzymes governing polyamine biosynthesis, including glutamic oxaloacetic transaminase 2 (GOT2), aminoacylase-1 (ACY1), ODC, and SDS, exhibit overexpression in prostate cancer, while ornithine aminotransferase (OAT) demonstrates insufficient expression (66). The net effect redirects substrate flux toward polyamine biosynthesis while diverting it from proline synthesis, potentially contributing to oncogenic transformation. Emerging evidence suggests these elevated enzymatic levels are sustained through combinatorial mechanisms: enhanced biosynthesis, increased transporter activity, and reduced catabolism. Multiple oncogenes, including MYC, JUN, FOS, KRAS, and BRAF, are associated with ODC and SAMDC expression, with ODC and SAMDC being particularly linked to MYC activation (67, 68) (Figure 2).
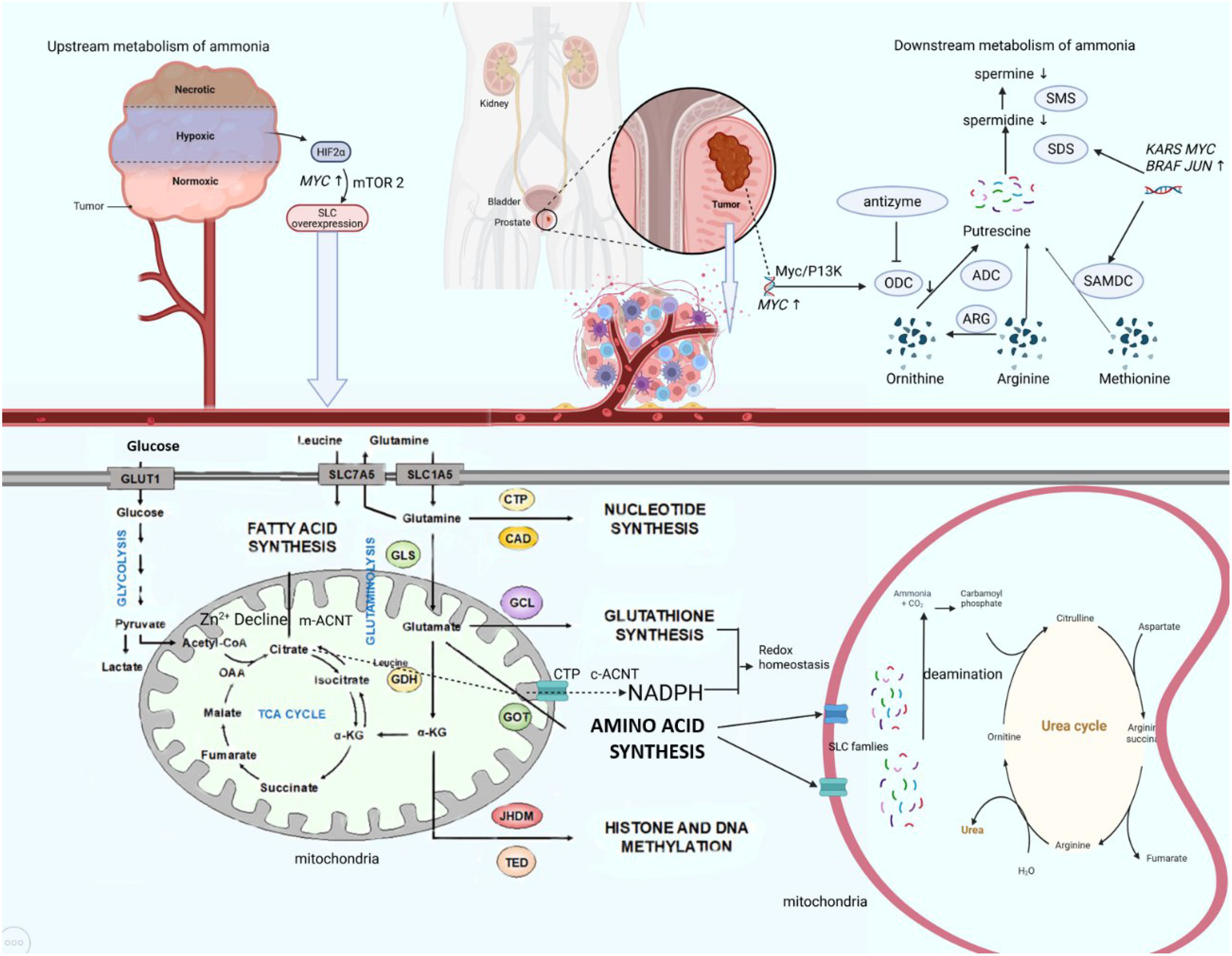
Figure 2. Ammonium metabolic reprogram axis in PCa. In the tumor microenvironment, hypoxia activates HIF-2α, which upregulates MYC and subsequently enhances mTORC2 signaling to promote expression of SLC transporters. This leads to increased uptake of glucose and amino acids such as glutamine. Glutamine is converted to glutamate by GLS, supporting biosynthesis of lipids, nucleotides, and glutathione. Concurrently, the ornithine–arginine–methionine pathway drives polyamine synthesis via ODC and SAMDC, regulated by MYC, JUN, and BRAF. The urea cycle mitigates ammonia toxicity. Oncogene activation disrupts these interconnected metabolic networks, sustaining tumor growth. HIF, hypoxia inducible factor; GLS, glutaminase; CTP, cytidine triphosphate; CAD, Caspase-activated deoxyribonulease; ACNT, aconitase; ODC, ornithine decarboxylase; ARG, arginase; ADC, arginine decarboxylase; SMS, spermine synthase; SDS, spermidine synthase.
Mechanistically, PCa cells exhibit impaired ODC antizyme regulation (69, 70). In spermine-insensitive cells, antizyme levels fail to upregulate, resulting in unabated ODC activity due to insufficient inhibition and degradation. Reduced antizyme levels observed across multiple cancer types suggest tumor-suppressive properties of ODC antizyme. Paradoxically, while most cancers demonstrate elevated intracellular polyamines correlating with proliferative metabolism, prostate cancer exhibits an inverse polyamine profile, warranting further mechanistic investigation. The prostate gland exhibits unique metabolic features characterized by extraordinary citrate production (up to 180 mM in prostatic fluid) to support sperm energetics, sustained through zinc-mediated inhibition of mitochondrial aconitase(m-ACNT), which blocks citrate-to-isocitrate conversion (71). In prostatic cells, citrate within the mitochondrial TCA (tricarboxylic acid) cycle undergoes compartment-specific metabolic partitioning. Mitochondrial citrate is either exported to the cytosol via solute carrier family 25 transporters, notably the citrate transport protein (CTP) embedded in the inner mitochondrial membrane, to fuel cytosolic fatty acid and sterol biosynthesis, or retained intramitochondrially to contribute to ATP generation (72). Additionally, extracellular citrate derived from systemic circulation can be imported into prostatic cells through SLC13 family transporters, a class of citrate carriers ubiquitously expressed across multiple organ systems (73, 74). This dual citrate sourcing, endogenous mitochondrial production and exogenous uptake, underscores the metabolic adaptability of prostatic cells in maintaining citrate homeostasis for both bioenergetic and biosynthetic demands. In PCa, metabolic reprogramming disrupts this homeostasis through sequential and interconnected mechanisms. First, dramatic Zn²+ depletion relieves m-ACNT suppression, enabling mitochondrial citrate catabolism. Concurrently, a metabolic shift to “citrate oxidation” splits citrate into acetyl-CoA, fueling lipogenesis via overexpressed fatty acid synthase (FAS), and α-KG which sustains TCA cycle activity to support proliferation. Despite compensatory upregulation of sodium-dependent citrate transporters (NaCitT) in advanced tumors, accelerated citrate consumption consistently outpaces extracellular uptake (75). Furthermore, elevated cytosolic aconitase (c-ACNT) and isocitrate dehydrogenase (ICD2), synergistically enhanced by iron accumulation, drive citrate flux toward NADPH and α-KG generation, thereby exacerbating intracellular citrate depletion. Paradoxically, while normal prostate physiology prioritizes polyamine synthesis (e.g., spermine), regulated by androgen-dependent enzymes like ODC to support secretory functions, PCa progression exhibits a contradictory polyamine landscape: spermine levels decline sharply despite ODC overexpression, a phenomenon contrasting with polyamine accumulation patterns in other cancers. Collectively, this dual metabolic identity, preserving physiological specialization in citrate production while hijacking polyamine and citrate pathways for oncogenic rewiring, not only distinguishes PCa from other malignancies but also unveils targetable vulnerabilities for diagnostic and therapeutic innovation.
2.2 Metabolic reprogramming of ammonium metabolism as pivotal part of tumor metabolism
Ammonium metabolism in tumor cells exhibits distinct features compared to normal cells, serving dual roles in energy production and substrate provision for proliferation. Similar to normal cells, tumor cells generate ammonium primarily via amino acid deamination, notably through the glutamine cycle, where glutamate dehydrogenase (GDH) catalyzes glutamate deamination to produce free ammonium and α-KG. GDH produces α-KG and ammonia through oxidative deamination, while transaminases convert α-KG into glutamate, enabling flexible regulation of nitrogen metabolism (76). Further studies indicate that glutamine serves as both a nitrogen and carbon source in prostate cancer, with higher expression of glutamine transporters (ASCT2), GDH1, AST1, and GLUL (Glutamine synthetase). This elevated enzyme expression is linked to tumor metabolic reprogramming and increased glutamine dependency, emphasizing the central role of glutamine metabolism in ammonia metabolism in prostate cancer (77, 78). In normal physiology, ammonium derived from deamination is transported as glutamine or alanine to hepatocyte mitochondria, where it undergoes enzymatic conversion to urea via the urea cycle (Details in Table 1). This cycle involves sequential reactions that ultimately produce urea, which is excreted renally to prevent neurotoxic ammonium accumulation. To meet heightened metabolic demands, tumor cells exhibit elevated glutamine consumption. Animal studies reveal that colorectal cancer (CRC) cells accumulate ammonium due to downregulation of transcription factors HNF4-α and ornithine transcarbamylase (OTC), a phenomenon also observed in CRC patients. Concurrent overexpression of SLC4A11, glutaminase (GLS), and GDH further amplifies glutamine derived ammonium production (79). Intriguingly, tumor-associated hyperammonemia (elevated glutamate and NH4+) promotes lipogenesis via glucose-dependent mechanisms. Specifically, ammonium stimulates sterol regulatory element-binding protein (SREBP) cleavage, enhancing nuclear translocation and upregulating fatty acid synthase (FASN) and stearoyl-CoA desaturase 1(SCD1). Conversely, glucose deprivation suppresses SCAP (SREBP cleavage-activating protein) N-glycosylation, blocking SREBP activation, suggesting synergistic regulation of lipogenesis by glucose and ammonium (80). Beyond energy metabolism, glutamine-derived ammonium contributes to nucleotide synthesis, though its downstream nitrogen utilization pathways remain poorly defined (81, 82). In estrogen receptor (ER)-positive breast cancer, Carbamoyl Phosphate Synthetase 1(CPS 1) (normally absent in hepatic cells), GS (glutamine synthetase), and GDH are overexpressed. Isotopic tracing with 15N-labeled glutamine revealed that 57% of ammonium is recycled via GDH-mediated reductive amination, generating 15N-labeled amino acids (excluding proline and glutathione, which are synthesized directly from glutamate) (83). Spinelli et al. demonstrated that tumors produce ammonia through glutamine metabolism, which is converted into glutamate and downstream amino acids via reductive amination to support cellular nitrogen requirements. Using 15N-labeled glutamine and HILIC-MS, the study tracked ammonia metabolism, revealing its recycling in tumor cells into glutamate and its derivatives. Although conducted in breast cancer cell lines, recent studies have confirmed similar metabolic processes in prostate cancer (77).These findings highlight tumor-specific adaptations in ammonium metabolism, emphasizing its role in sustaining proliferation and metabolic flexibility. Aside from glutamine, proline is also a key type of amino acid which is active in cancer cells. Proline metabolism not only supports collagen synthesis for the extracellular matrix (ECM), but also enables tumor cells to recycle collagen-derived proline under nutrient stress in the tumor microenvironment, promoting tumor growth (84, 85). Key enzymes such as P4H are activated by HIF-α (Hypoxia inducible factor-α), enhancing collagen deposition and tumor invasiveness (86, 87).
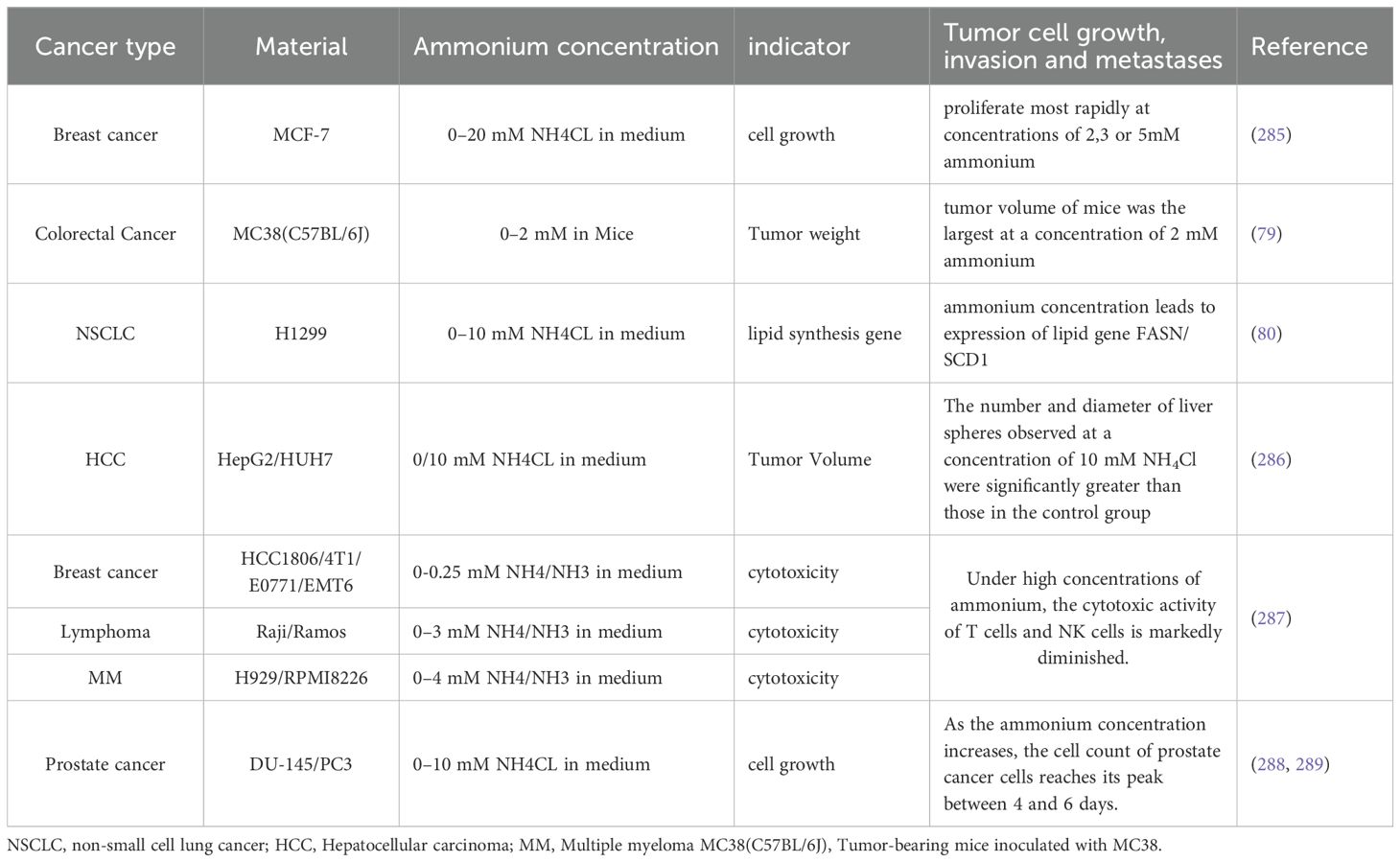
Table 1. The impact of ammonium accumulation in tumor cells.
Emerging evidence highlights the oncogenic rewiring of glutamine metabolism through multifaceted mechanisms. The Myc oncoprotein directly stimulates glutamine uptake by binding to promoters of glutamine metabolism genes (e.g., the glutamine transporter SLC1A5) and indirectly enhances glutaminolysis by repressing microRNA miR-23a/b, a negative regulator of GLS1 (36, 88). Conversely, the tumor suppressor p53 upregulates the glutaminase isoform GLS2 (89). Moreover, the transcription factor c-Jun, encoded by the proto-oncogene JUN, upregulates GLS through coordinated transcriptional and post-translational mechanisms. Directly, c-Jun binds to the GLS promoter at a v-Jun-homologous response element, enhancing GLS transcription. Indirectly, oncogenic Rho GTPase signaling activates JNK (c-Jun N-terminal kinase), which phosphorylates and stabilizes c-Jun, forming a coherent JNK/c–Jun/GLS promoter axis that amplifies GLS expression and promotes glutamine metabolism in breast cancer cells (90). Additional oncogenic drivers, including IDH1/2 mutations, STAT1, ERK, and KRAS, further modulate glutamine metabolic abundance (91–94). However, current studies on oncogene-driven metabolic reprogramming remain disproportionately focused on carbohydrates and lipids, with limited exploration of ammonium metabolism, a critical gap warranting systematic investigation.
The tumor microenvironment exerts profound bidirectional crosstalk with cancer cell metabolism. Composed of tumor cells, stromal cells, immune populations, and bioactive molecules, the TME imposes nutrient constraints that drive adaptive metabolic responses. HIF-1 orchestrates this adaptation under low oxygen tension, transcriptionally activating glycolysis-associated genes such as glucose transporters (GLUT1/3), glycolytic enzymes (HK1/2, ENO1, PGK1, PKM2), and lactate dehydrogenase A (LDHA), thereby shifting energy production from oxidative phosphorylation to aerobic glycolysis (94–96). This metabolic shift elevates cytosolic lactate concentrations from physiological levels (1.5–3 mM) to pathological levels (10–30 mM) in cancer cells. Proton-coupled monocarboxylate transporters (MCTs) facilitate lactate and proton extrusion, alleviating pH-dependent inhibition of phosphofructokinase 1 (PFK1) to sustain glycolytic abundance (97).
Concomitantly, HIF-1 suppresses mitochondrial fatty acid oxidation by downregulating c-Myc and its targets, medium-chain and long-chain acyl-CoA dehydrogenases(MCAD/LCAD) and inhibiting the PTEN pathway (98, 99). To maintain oxidation equilibrium, HIF-1α activation under hypoxia suppresses mitochondrial respiration by inhibiting oxygen consumption and fatty acid oxidation via HIG2-mediated suppression of lipolysis. It also reprograms glucose and glutamine metabolism, while impairing electron transport chain activity, leading to ROS accumulation and altered energy production (100). This dual mechanism preserves redox homeostasis and ensures lipid availability for membrane biosynthesis. The resulting lactate-rich, acidic TME further reinforces tumor progression via histone lactylation, an epigenetic modification linking metabolic byproducts to transcriptional reprogramming. However, the precise mechanisms underlying lactate-mediated transcriptional regulation and immune suppression remain elusive (101, 102).
Ammonium metabolic reprogramming in prostate cancer constitutes a pathogenic cornerstone that drives tumor initiation, progression, and therapeutic resistance through its dual role as a metabolic integrator and microenvironmental modulator (103). This reprogramming directly responds to coordinated oncogenic alterations—including MYC amplification, androgen receptor signaling hyperactivity, and mTOR pathway activation, which dysregulate core ammonium-metabolizing enzymes as GS, GLS and SLC (104, 105). Concurrently, ammonium accumulation remodels the tumor microenvironment by skewing immune cell differentiation toward immunosuppressive phenotypes and activating cancer-associated fibroblasts (106, 107). Metabolic intermediates including α-ketoglutarate and acetyl-CoA fulfill biosynthetic demands for nucleotide/lipid production while enabling epigenetic reprogramming via TET (Ten-eleven translocation enzymes) dioxygenase-mediated DNA demethylation (108, 109). Furthermore, glutathione synthesized from glutamine-derived precursors confers treatment resistance by scavenging therapy-induced reactive oxygen species, thereby suppressing radiation- and chemotherapy-triggered autophagic cell death and establishing a self-perpetuating oncogenic circuit (110, 111). These metabolic alterations not only intrinsically reprogram tumor cells but also profoundly remodel the TME and associated immune regulation through ammonium metabolic reprogramming.
2.3 Ammonium metabolism in the TME: impact on immune cells
The role of ammonium metabolism in tumor immune regulation has drawn significant attention. Research indicates that the metabolic reprogramming of tumor cells leads to the overproduction and accumulation of ammonium, which directly disrupts immune cell functions, inhibiting T cell proliferation and cytotoxicity (112). Moreover, ammonium accumulation alters the metabolic state of immune cells, further weakening their anti-tumor responses (113). These findings highlight ammonia’s critical role in tumor-induced immune suppression and offer a theoretical foundation for developing novel immunotherapy strategies.
Ammonium affects immune cells through several mechanisms. Firstly, the abnormal accumulation of ammonium in immune cells causes metabolic disruptions, impairing the function of T cells, NK cells, and other immune cells. Recent studies highlight that the effectiveness of anti-tumor T cell responses depends on nutrient availability and the metabolic flexibility between cancer cells and immune cells. Tumor cells compete with T cells for essential nutrients in the TME, particularly glucose and amino acids involved in nucleotide synthesis, such as glutamine, glycine, and serine (114–116). These molecules are crucial for both cancer and immune cells to meet biosynthetic and energy demands. Deficiencies or excessive consumption of Arg, Glu, and Branched-chain amino acids (BCAAs) can compromise the ability of tumor-infiltrating lymphocytes (TILs) to clear cancer cells (117). For example, plasma arginine levels have been found to decrease in various cancer types, suggesting that tumors may deplete this amino acid. Arginine depletion via arginase activity can restrict T cell activation and contribute to the establishment of an immunosuppressive environment (118). This is mainly due to the similar amino acid requirements of TILs and cancer cells, with TILs often at a disadvantage in this competition. Arginine plays an essential role in immune responses, particularly in patients with severe trauma, immune suppression, or cancer cachexia, where arginine demand exceeds endogenous production (119–121). Immune cells, particularly CD4+ and CD8+ T cells, depend on sufficient arginine concentrations to maintain effector functions. Arginine synthesis defects, often linked to ASS1 deficiency, are common in cancer cells. Similarly, glutamine is another vital nutrient for both cancer and immune cells. Cancer cells have a high dependence on glutamine, and excess glutamine can stimulate tumor growth while also supporting immune cell function. BCAAs are critical for T cell activation, and their absence impedes T cell expansion and effector differentiation (122).
In the TME, T cell activation is closely associated with ammonium metabolism. Specific mechanisms may involve glutaminase activity promoting effector functions in TH1 cells and cytotoxic CD8+ T cells while inhibiting TH17 cells (123). Additionally, SLC1A5 plays a critical role in the polarization and inflammatory activity of TH1 and TH17 cells (124). Immune checkpoint molecules like PD-1 (programmed death-1) and CTLA-4 (Cytotoxic T lymphocyte-associated antigen-4) function as negative regulators of immune activation, and when triggered by tumor or tumor-associated cells, they suppress immune responses (105, 125–128). A strong relationship exists between checkpoint pathways and cellular metabolism. Immune checkpoint blockade (ICB) therapy can directly influence the metabolism of both immune cells and cancer cells, particularly through the upregulation of PD-1 expression (129). Although CTLA-4 and PD-1 are part of separate pathways, they exert similar inhibitory effects on effector T cell metabolism, including downregulation of AKT phosphorylation, reduced amino acid uptake, and general suppression of metabolic activity. Depletion of tryptophan within the TME can impair anti-tumor immune function of infiltrating lymphocytes. Cells within the TME facilitate immune evasion by reprogramming ammonium metabolism, thereby inhibiting the functions of infiltrating immune cells. Tumor cells, tumor-associated macrophages, and certain dendritic cells can decrease local tryptophan levels by provoking metabolic enzymes, including indoleamine 2,3-dioxygenase (IDO1) and tryptophan 2,3-dioxygenase (TDO2) (130). Increased activity of tryptophan catabolic enzymes promotes the deposition of tryptophan to its metabolite kynurenine (131–133). The depletion of tryptophan in the TME impairs effector T cell function, while kynurenine degradation products can suppress tumor immunity by inducing the generation of Foxp3+ regulatory T cells (Tregs) (134). Recent studies indicate that alanine is crucial for early T cell activation and the re-stimulation of memory CD8+ T cells. However, due to the limited expression of ALT1/2 (or GPT1/2) and low transaminase activity, which restricts alanine biosynthesis, T cells need extracellular alanine for protein synthesis (135). This suggests that during nutrient deprivation, the consumption of extracellular alanine by cancer cells negatively impacts T cell function. The metabolic composition of the TME significantly influences both tumor cells and infiltrating T cells. Glutamine-derived nitrogen is essential for the clonal expansion and differentiation of activated T cells into effector cells (122, 136). However, limiting glutamine consumption through SLC1A5 deficiency or restricted local glutamine availability can promote the expression of Foxp3, a key transcription factor for Treg lineage specification (124, 137). Decreased β-lactam degradation releases α-KG-dependent demethylation at the Foxp3 locus, which promotes the generation of suppressive Tregs and inhibits TH1 differentiation (138). Another study suggests that the accumulation of 2-HG is likely due to increased transamination activity mediated by glutamate Oxaloacetate Transaminase(GOT1), resulting in promoter methylation at the Foxp3 locus and reduced TcB induction (139). Furthermore, glutamine is a precursor for glucosamine synthesis, which is essential for protein glycosylation and has been shown to be critical for activated T cell function (140, 141). Therefore, increased glutamine consumption by cancer cells can modulate anti-tumor immunity by depleting the local glutamine pool required for effector T cell responses, while promoting the development of suppressive Treg populations. A recent research showed that while cancer cells are sensitive to glutamine antagonism, effector T cells can redirect their metabolism towards a more oxidative, long-lived activation phenotype (142). Additionally, ammonium accumulation is closely associated with the redox balance of immune cells, as redox imbalance further hampers their functions, particularly the antigen-presenting capability of dendritic cells (143). By inhibiting dendritic cell maturation from the immature to the mature form, ammonium weakens their ability to initiate T cell responses, thus reducing the immune system’s ability to surveil and combat tumor cells. Furthermore, ammonium alters the types of cytokines secreted by dendritic cells, diminishing their ability to activate T cells and further limiting anti-tumor immune responses (144).
Ammonium TME acidification mainly results from lactate accumulation via anaerobic glycolysis. Ammonium contributes by suppressing aerobic respiration and promoting glycolysis through ammonia-mediated metabolic reprogramming, thereby exacerbating lactate accumulation and acidosis. Reported inhibition of TCA enzymes (e.g., pyruvate dehydrogenase) by ammonia enhances anaerobic glycolysis, further accelerating acidification. Research has shown that ammonium’s inhibitory effects on immune cells, such as T cells and macrophages, are closely associated with TME acidification. This acidification decreases immune cell activity and facilitates tumor cell immune escape. For example, ammonium inhibits perforin maturation, leading to reduced levels of mature perforin in NK cells and consequently diminishing NK cell cytotoxicity. Specifically, ammonium impairs the conversion of perforin from its precursor to the mature form, disrupting its normal function. Perforin maturation and function are pH-dependent within the lysosome. Ammonium raises the lysosomal pH, interfering with perforin maturation. Perforin typically transitions from its precursor to its active form in a low-pH environment. Ammonium accumulation weakens the acidic environment of the lysosome, preventing effective perforin maturation or causing its degradation, thus diminishing its cytotoxic activity. Ammonium not only reduces the level of mature perforin in NK cells but also alters the distribution of other lysosomal markers (e.g., LAMP-1). Ammonium suppresses NK cell cytotoxicity primarily through a dose-dependent reduction in perforin protein levels, without altering its intrinsic activity. This rapid and reversible effect which is independent of transcriptional regulation suggests post-translational impairment of perforin maturation. In vivo studies show that mature perforin decreases at 1 mM ammonium and becomes undetectable at 4–5 mM (145). Mechanistically, ammonium disrupts acidic compartments, as indicated by reduced LysoTracker staining, likely impairing the pH-dependent maturation of perforin (79).
Furthermore, ammonium metabolism suppresses immune cell effector functions, including cytotoxicity, by altering their energy metabolic pathways, thus providing a favorable growth environment for tumor cells. 1.5 ng ug-1 protein ammonium exposure can induce apoptosis in CD8+ T cell (146). Ammonium may promote immune cell apoptosis through several mechanisms: 1) Ammonium activates the TNF-α/TNFR1 signaling pathway, triggering the death receptor-mediated apoptosis. The binding of ammonium to TNF-α activates its death domain, recruits TRADD (Tumor Necrosis factor receptor-associated death domain protein) and FADD (Fas-associating protein with a novel death domain) proteins, forms a complex, activates Caspase-8, and subsequently Caspase-3, leading to apoptosis (147). 2) Ammonia induces apoptosis via the mitochondrial pathway. It downregulates mitochondrial membrane potential, causing pro-apoptotic factors such as Bax, Bid, and Bak to bind to the mitochondrial membrane, disrupt it, release Cytc, and activate Caspase-9, which then activates Caspase-3, inducing cell death (148). 3) Ammonium reduces miR-27b-3p expression, enhancing apoptosis-related gene expression (e.g., TRADD, FADD), thus promoting both death receptor and mitochondrial pathway activation (112, 149). 4) Ammonium exposure alters immune cell cytokine secretion, causing an imbalance in immune responses. Specifically, cytokines secreted by Th1 and Treg cells (e.g., IFN-γ, IL-2) decrease, while those from Th2 and Th17 cells (endocrine IL-1β, IL-4, IL-6) increase, suppressing immune function and promoting apoptosis (150). 5) Ammonium exposure activates heat shock proteins (HSPs) like HSP 25, HSP 40, and HSP 70, which regulate immune factor expression and may play a key role in immune cell apoptosis and immune suppression (151, 152). More details can be found in Figure 3. While ammonium accumulation-mediated acidification has been experimentally validated (in vitro and in vivo) to drive immunosuppression in the prostate cancer TME, primarily through impairing T cell and NK cell cytotoxicity and promoting M2 macrophage polarization. Mechanisms of ammonium metabolic reprogramming orchestrates immunosuppression via cytokine networks and stromal crosstalk remain mechanistically unresolved and demand urgent investigation. To harness potential ammonium metabolism reprogramming targets for the treatment of solid tumors, such as prostate cancer, it is essential to investigate the regulatory genes and associated metabolic enzymes involved in the ammonium metabolism reprogramming process.
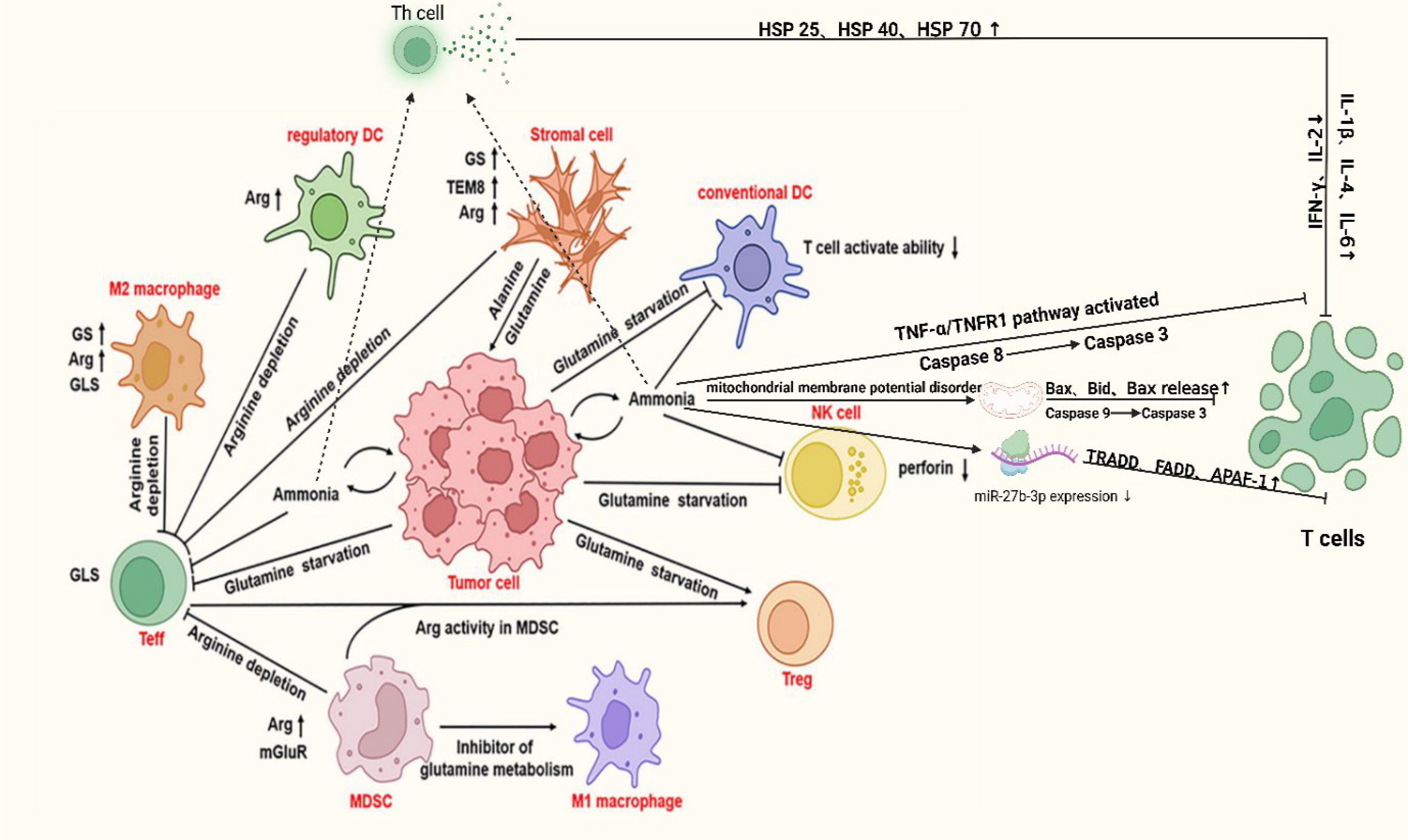
Figure 3. Mechanism of immune cell been suppressed in PCa. In the tumor microenvironment, tumor cells drive immune dysfunction by competitively depleting key metabolites such as glutamine and arginine. Tumor and stromal cells upregulate GS and tumor endothelial marker 8 (TEM8), promoting tumor proliferation and angiogenesis while exhausting extracellular glutamine, thereby impairing T cell activation. M2 macrophages and regulatory dendritic cells enhance anti-inflammatory functions via arginine metabolism, whereas myeloid-derived suppressor cells (MDSCs) suppress immune responses through autophagy and inhibition of glutamine metabolism. Additionally, TNF-α/TNFR1 pathway activation and mitochondrial membrane potential disruption induce apoptosis in immune cells (e.g., NK cells) via Caspase cascades, coupled with reduced perforin levels and dysregulated miR-27b-9p expression. These interconnected mechanisms, metabolic competition, pro-survival adaptations, and apoptotic signaling, collectively establish a highly immunosuppressive niche, enabling tumor immune evasion. DC, dendritic cell; Arg, Arginine; Bax, BCL-2-associated; X, protein; Bid, BH3-interaction domain death agonist; TRADD, Tumor Necrosis factor receptor-associated death domain protein; FADD, Fas-associating protein with a novel death domain; APAF-1, Apoptotic protease activating factor-1; TEM8, tumor endothelial marker 8 Created in https://BioRender.com.
2.4 Molecular regulation of ammonium metabolic reprogramming in tumors
Tumor cells exhibit distinct ammonium metabolism compared to normal cells, characterized by aberrant expression and activity of SLC, metabolic enzymes, and signal transduction pathway rewiring. The SLC family, comprising 455 members across 66 subfamilies, serves as a critical node in tumor ammonium metabolism. Key transporters such as SLC1A5 (glutamine transporter), SLC7A5(LAT1, A heterodimer of SLC7A11 and SLC3A2), and SLC43A1, facilitate glutamine and leucine uptake, activating the mTOR pathway to drive glutaminolysis (153, 154). This process converts glutamine into glutamate, which is further metabolized to α-ketoglutarate for TCA cycle, fueling tumor proliferation (155). SLC6A14 and SLC43A1 are also implicated in leucine transport, with SLC43A1 overexpression correlating with prostate cancer aggressiveness (156–159). Mechanistically, SLC transporters promote glutamine efflux and leucine influx, sustaining mTOR hyperactivation in tumors (160). SLC7A5 expression is regulated by ATF4 (ATF4 is a known transcriptional activator of ASNS expression) under low leucine conditions via the GAAC pathway and by c-Myc through direct promoter binding, while HIF-2α enhances SLC7A5-mediated mTORC1 phosphorylation (161). Integrative omics analyses reveal that MYC orchestrates ammonium metabolism reprogramming in prostate cancer through direct transcriptional control of GLS, with TCGA-PRAD data confirming co-amplification of MYC and GLS (q < 0.001) (162). Parallel mechanisms involve mutant oncogene-mediated dysregulation of metabolic carriers, notably citrin-dependent upregulation of the aspartate-glutamate transporter SLC25A13. These coordinated alterations drive pathological ammonium redistribution, creating a therapeutically exploitable metabolic vulnerability.
Enzyme dysregulation further defines tumor ammonium metabolism. Urea cycle enzymes, such as CPS1, are downregulated in tumors but paradoxically enhance proliferation via S-adenosylmethionine dependent m6A modification of the aspartate transporter SLC1A3, elevating intracellular aspartate (163). GLUL, overexpressed in pancreatic and liver cancers due to c-Myc driven promoter demethylation, supports tumor growth (105). Glutaminase isoforms (GLS1/2), transcriptionally upregulated by c-Myc and hypoxia, catalyze glutamine-to-glutamate conversion, with glutamate dehydrogenase (GLUD) channeling glutamate into α-KG for the TCA cycle and glutathione synthesis, sustaining redox balance and mTOR-driven anabolism. Adaptive expression of urea cycle enzymes ARG1/2, OTC, ASL and regulators like NAT10, which stabilizes ATF4 mRNA via ac4C modification to upregulate asparagine synthetase (ASNS) optimizing nitrogen utilization for tumor proliferation (164). Collectively, these alterations highlight ammonium’s dual role as a metabolic byproduct and biosynthetic precursor, offering therapeutic targets to disrupt tumor metabolic plasticity (as shown in Figure 2).
Current research on targeted therapy for prostate cancer primarily focuses on identifying potential inhibitory mechanisms. Massive researches have concentrated on SLC family, with SLC7A11, the SLC3 family, and SLC7A5 emerging as promising therapeutic targets (165). SLC7A11 activity may be influenced by environmental metal elements such as antimony, which modulate the Nrf2/SLC7A11/GPX4 axis and inhibit ferroptosis in PCa cells (166). The SLC3 family has been implicated in epithelial–mesenchymal transition (EMT) and cell cycle–related apoptosis, while SLC7A5 is speculated to be associated with distant metastasis (167, 168). Several inhibitors targeting SLC transporters are currently in preclinical trials. In addition to transporters, enzymes involved in ammonium metabolism have also drawn attention as potential therapeutic targets. Current research primarily focuses on blocking glutamine utilization in cancer cells using GLS inhibitors such as CB-839 and JHU083. Some of these agents have already progressed to phase III clinical trials, providing new hope for improved therapeutic strategies against prostate cancer.
3 Metabolic reprogramming of ammonium metabolism in prostate cancer microenvironment
Metabolic characteristics, metabolic reprogramming, immune microenvironment, and immune evasion are intricately interconnected, forming a complex network that drives the initiation and progression of prostate cancer. Under high concentration of ammonium, prostate cancer cell stays active and keep cell viability via several mechanism. Recent studies show that aspartate levels are elevated in the prostate cancer TME, suggesting partial activation of the urea cycle (169). Higher CPS enzyme activity in prostate cancer tissues compared to controls indicates that the active urea cycle helps mitigate ammonia toxicity. Additionally, excessive urea cycle activation promotes CPS expression, converting ammonia-derived nitrogen into pyrimidine metabolites, supporting tumor cell proliferation (170). Modulating ammonium metabolism holds significant potential in the treatment of prostate cancer, offering deeper insights into its biological characteristics and providing novel therapeutic strategies aimed at improving survival and quality of life for patients with advanced prostate cancer.
3.1 Metabolic crosstalk in prostate cancer microenvironment: acidification and its role in immunosuppression TME
The TME in PCa is a complex and dynamic system that plays a critical role in tumor progression, immune evasion, and therapy resistance. Immunosuppressive factors such as HIF-1α, CD73, and PGE2 (Prostaglandin E), along with immunosuppressive cell populations including tumor-associated macrophages (TAMs), regulatory T cells (Tregs), and myeloid-derived suppressor cells (MDSCs), have been shown to be present in the prostate cancer immune microenvironment and may contribute to immune evasion (171–175). TAMs and Tregs are pivotal in shaping the immunosuppressive landscape. TAMs promote immune evasion by secreting immunosuppressive cytokines, including TGF-β and IL-10, which inhibit the function of cytotoxic T cells (particularly CD8+ T cells) and impair antigen presentation. Tregs further contribute to immune suppression by dampening the effect of T-cell responses, thereby enabling tumor cells to evade immune surveillance (176).
Hypoxia alters the metabolic profiles of immune cells, impairing their anti-tumor functions and facilitating immune evasion. Tumor cells exploit these metabolic changes to suppress immune cell activity, for example, by increasing lactate production and accumulating immunosuppressive metabolites, which further inhibit T cell proliferation and cytotoxicity (177, 178). Previous studies have established that metabolic reprogramming in solid tumors like prostate cancer promotes immunosuppression through TME acidification (179). Elevated lactate concentrations (>20 mM) impair immune cell function by competitively inhibiting monocarboxylate transporters (MCTs), MCT1 in T cells and MCT4 in NK cells, leading to metabolic paralysis or apoptosis (180) (Figure 4). Simultaneously, lactate drives histone lactylation (Kla), a novel epigenetic modification identified via HPLC-MS/MS analyses, revealing 28 conserved Kla sites. This modification directly reprograms gene expression, exemplified by its capacity to polarize macrophages toward M2 phenotypes through non-inflammatory pathways (181). Notably, M2 macrophages exhibit superior survival under acidic pH compared to M1 counterparts (6). Furthermore, lactate activates oncogenic signaling via G-protein-coupled receptor GPR81, upregulating immune checkpoints like PD-L1 (182, 183). While ammonium metabolic rewiring likely exacerbates TME acidification through glycolytic-lipogenic crosstalk, further in vivo/in vitro validation is required to delineate these indirect mechanisms. (Figure 4).
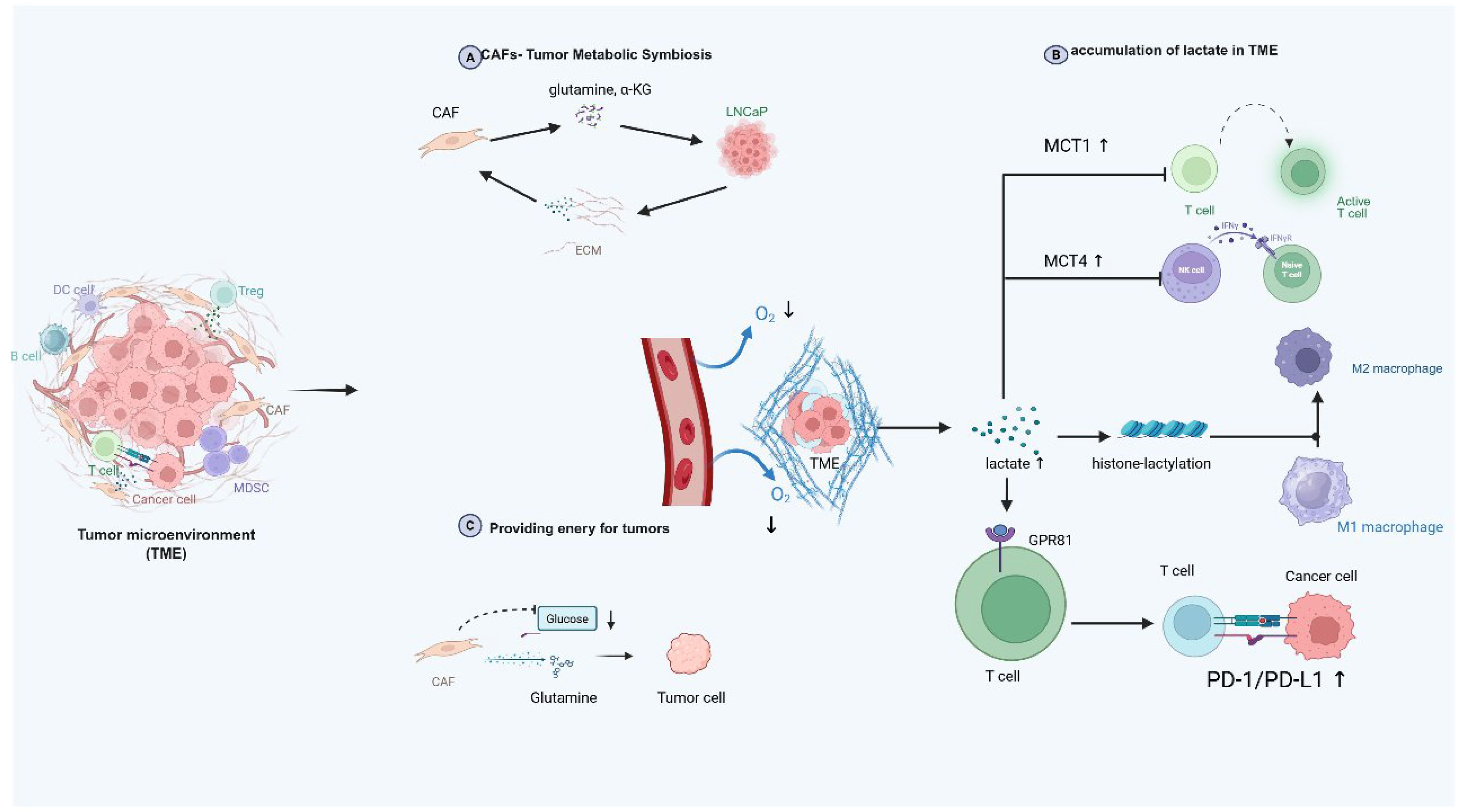
Figure 4. Ammonium metabolism in TME and possible interplay with TME components. This diagram vividly depicts the intricate dynamics within the TME. It showcases the metabolic symbiosis between cancer - associated fibroblasts (CAFs) and tumor cells like LNCaP, where CAFs secrete glutamine and α-ketoglutarate for tumor cells, with reciprocal exchanges via the extracellular matrix. In the oxygen-deprived TME, tumor cells generate lactate, which accumulates and triggers the upregulation of MCT1 and MCT4 transporters. This lactate not only induces histone-lactylation to potentially modify gene expression but also exerts a profound impact on immune cells, such as inhibiting the function of T and NK cells, skewing macrophage polarization towards the M2 phenotype, and engaging with GPR81 on T cells to upregulate the PD-1/PD-L1 pathway for immune evasion. Additionally, CAFs utilize glucose and glutamine, potentially funneling metabolites to fuel tumor cell growth, highlighting the complex interplay of metabolism and immune regulation in the TME that supports tumor progression. TME, tumor microenvironment; CAF, cancer-associated fibroblast; MCT, monocarboxylate transporter; GPR81, G protein-coupled receptor 81, PD-1, programmed death-1, PD-L1, programmed death ligand 1.
Beyond ammonia-driven TME acidification that broadly suppresses antitumor immunity, competitive depletion of arginine, a key ammonium metabolism related substrate, represents an underappreciated axis of immune evasion in prostate cancer (184). Androgen signaling as evidenced in castration therapy studies, manipulates this immunosuppressive network by inhibiting arginine metabolism via two synergistic pathways: 1) Myeloid-derived suppressor cells (e.g., CD11b+ cells) overexpressing ARG1 deplete extracellular L-arginine, crippling T-cell function through CD3ζ chain downregulation, impaired antigen recognition, and dual activation of the GCN2-eIF2α stress pathway with concurrent mTOR suppression (185–187). 2) Nitric oxide (NO) generated by nitric oxide synthase (NOS) disrupts IL-2 signaling via inhibition of JAK-STAT, ERK, and AKT phosphorylation, destabilizing IL-2 and inducing T-cell apoptosis (188–191). Critically, arginine scarcity triggers a pathological shift in NOS activity, from NO production to superoxide (O2-) and reactive nitrogen oxide species (RNOS) generation, which amplifies T-cell suppression through combined oxidative damage and signal blockade (192, 193). These mechanisms are exacerbated in castration-resistant prostate cancer (CRPC), where TAMs exhibit elevated ARG I/II activity and heightened susceptibility to low arginine environments. Targeting this metabolic-immune crosstalk may thus yield novel immunotherapeutic strategies for CRPC.
Recent studies reveal that prostate TME regulates tumor progression by modulating ammonium metabolism substrates. In vitro studies demonstrate that cancer-associated fibroblasts (CAFs) support hormone-sensitive prostate cancer (HSPC) proliferation via glutamine and α-KG secretion, fueling energy and biosynthetic demands (194, 195). This metabolic symbiosis exhibits biphasic regulation: LNCaP cells upregulate Gln catabolic pathways (e.g., GLS1-dependent glutaminolysis), while CAFs activate extracellular matrix (ECM) remodeling pathways. Notably, GLS1 expression serves as a dynamic biomarker of this metabolic interplay. Broader solid tumor studies corroborate that pharmacological inhibition of CAF-mediated Gln synthesis disrupts tumor metabolic fitness, suggesting combinatorial targeting of tumoral and stromal glutamine metabolism, particularly through dual GLS1/ECM pathway inhibition, may offer promising therapeutic avenues for advanced prostate cancer (196).
3.2 New insight of cellular programmed death associated with metabolic reprogram in PCa
Emerging research underscores the intricate crosstalk between metabolic reprogramming and regulated cell death pathways in PCa, where hypoxia and nutrient deprivation within the TME drive adaptive survival mechanisms (197). PCa cells and stromal components sustain a hypermetabolic state characterized by heightened dependence on glutamine and mTORC1-mediated suppression of autophagy, a process critical for oncogenesis, as evidenced by dysregulated autophagy-related genes (e.g., STK11/LKB1) upstream of mTORC signaling (198). The tumor suppressor STK11/LKB1 activates AMPK, a central metabolic sensor that integrates energy stress signals through cross-talk with PI3K, mTOR, and MAPK pathways (199). In LKB1-deficient non-small cell lung cancer (NSCLC) models, AMPK inactivation derepresses CPS1, whose silencing induces nucleotide pool imbalance (reduced pyrimidine/purine ratio), S-phase arrest, and DNA damage, mechanisms hypothesized to operate in PCa, where ammonium metabolism inhibitors (e.g., targeting glutaminase) combined with autophagy modulators (e.g., ULK1 activators) could exploit metabolic vulnerabilities targeting tumor cells (200). Concurrently, ammonium metabolites exhibit mitochondrial toxicity, collapsing membrane potential, and depleting ATP, while nutrient stress activates AMPK to phosphorylate ULK1, initiating pro-survival autophagy (201, 202).
Paradoxically, phosphoserine phosphatase suppresses hepatocellular carcinoma autophagy via the AMPK/mTOR/ULK1 axis, analogous regulatory networks remain uncharacterized in PCa.
PCa also engages ferroptosis, an iron-dependent death pathway driven by glutathione peroxidase 4 (GPX4) downregulation and lipid peroxidation (202). Key regulators include SLC7A11 and SLC3A2 (LAT 1), components of the cystine/glutamate antiporter system Xc-. Erastin-induced inhibition of system Xc- depletes glutathione (GSH), disrupting redox balance and triggering ferroptosis (203, 204). Intriguingly, ferroptosis inducers activate non-canonical endoplasmic reticulum (ER) stress cascades, including the ATF4-CHOP/ERK-eIF2 axis, while bypassing apoptosis, a resilience mechanism attenuated in LNCaP-AI cells with elevated ATF6 expression. ATF6-mediated transcriptional activation of PLA2G4A confers ferroptosis resistance, whereas pharmacological inhibition of ATF6 (e.g., Ceapin-A7) synergizes with enzalutamide to suppress CRPC growth (205). Detailed mechanisms are portrayed in Figure 5. These findings implicate ATF transcription factors as critical nodes linking ammonium metabolic rewiring (e.g., glutaminolysis) to ferroptosis sensitivity. Collectively, co-targeting metabolic dependencies (e.g., glutaminase inhibitors, ammonium scavengers) and death evasion mechanisms (e.g., ferroptosis inducers, ATF6 inhibitors) may overcome therapeutic resistance, with preclinical validation urgently needed for strategies combining autophagy modulation (AMPK/mTOR/ULK1 targeting), ferroptosis potentiation (system Xc− blockade), and stromal disruption (CAF-derived glutamine inhibition) (205, 206). Unresolved questions include the tissue-specific regulation of ULK1 complexes in PCa autophagy, role of CPS1 in nucleotide metabolism, and spatiotemporal dynamics of ATF6-mediated therapy resistance, all critical for advancing precision therapies in this metabolic-death nexus.
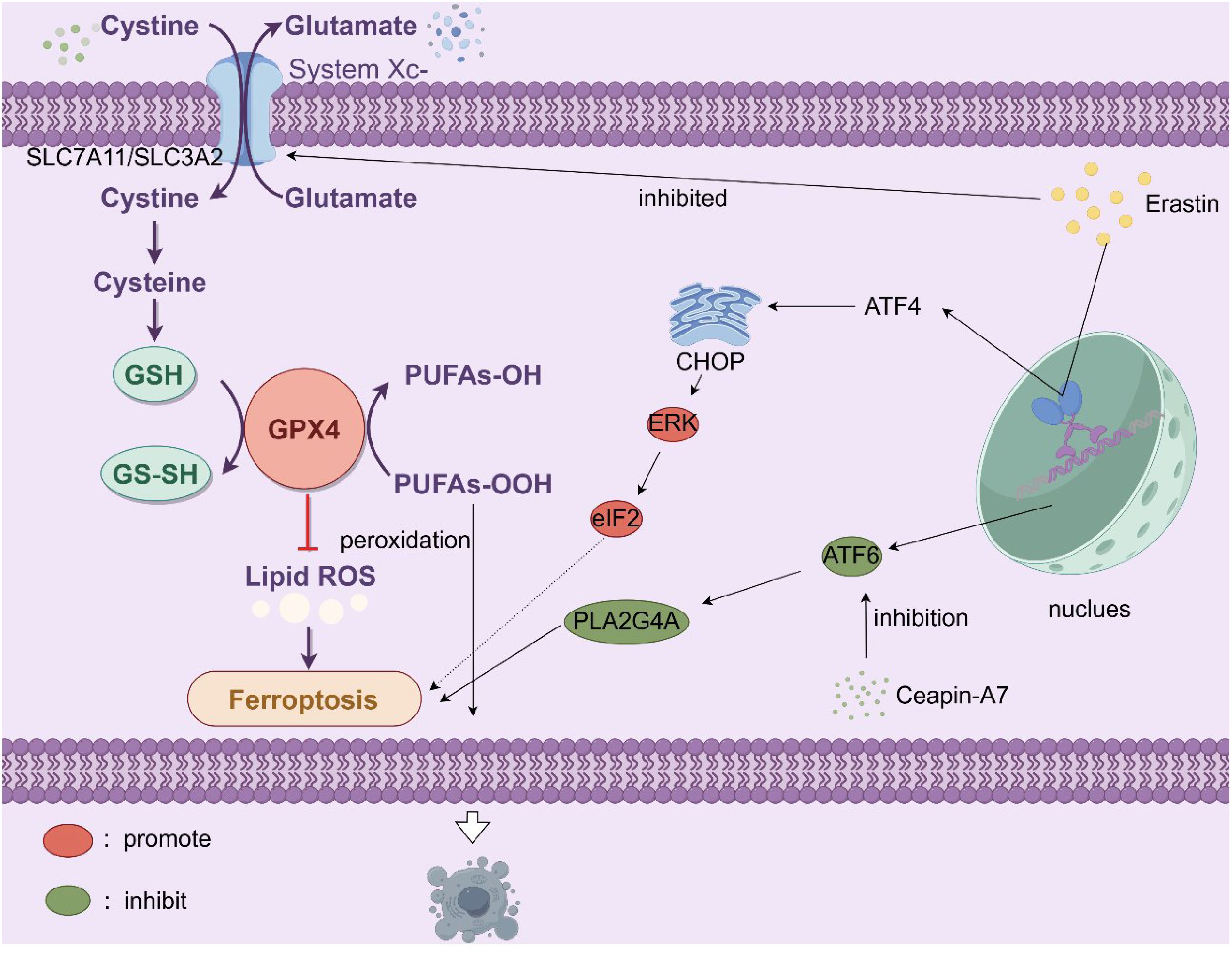
Figure 5. The cell membrane System Xc- (SLC7A11/SLC3A2) facilitates the exchange of extracellular cystine for intracellular glutamate. Cystine is subsequently converted to cysteine, a key component in the synthesis of GSH. GSH is vital as it supplies the reducing power for GPX4 to convert PUFAs-OOH into PUFAs-OH, thereby curbing lipid reactive oxygen species (lipid ROS) accumulation and preventing ferroptosis. However, the compound erastin inhibits System Xc-, diminishing cystine uptake and consequently reducing GSH levels. This undermines GPX4’s function, allowing lipid ROS to build up and triggering ferroptosis. Additionally, stress-related signaling pathways play a role. Activating transcription factor 4 (ATF4) is upregulated under stress, leading to the induction of C/EBP homologous protein (CHOP), which in turn activates extracellular signal-regulated kinase (ERK), promoting ferroptosis. Activating transcription factor 6 (ATF6), whose inhibition can be mediated by Ceapin-A7, influences the activity of phospholipase A2 group IVA (PLA2G4A), which can contribute to lipid peroxidation and the onset of ferroptosis. In essence, disruptions in cystine-glutamate transport, antioxidant defenses, and activation of specific signaling cascades all converge to initiate ferroptosis, characterized by iron-dependent lipid peroxidation. Erastin, ferroptosis activator; GPX4, glutathione peroxidase 4; PUFA, polyunsaturated fatty acid; ATF, activating transcription factor, C/EBP, CCAAT/enhancer-binding protein; ERK, extracellular signal-regulated kinase; PLA2G4A, phospholipase A2 group IVA.
4 Application prospects of ammonium metabolism in prostate cancer
4.1 Key therapeutic targets in ammonium metabolism for prostate cancer
Ammonium metabolism involves key enzymes that regulate metabolic processes, including ASNS, GS, and GLS. By modulating these enzymes, the concentrations of ammonium metabolism substrates and products are altered, which in turn affects PCa behaviors such as proliferation, differentiation, and invasion (207). Specific enzymes and their targeted drugs, along with clinical trial information, are outlined in the Table 2.
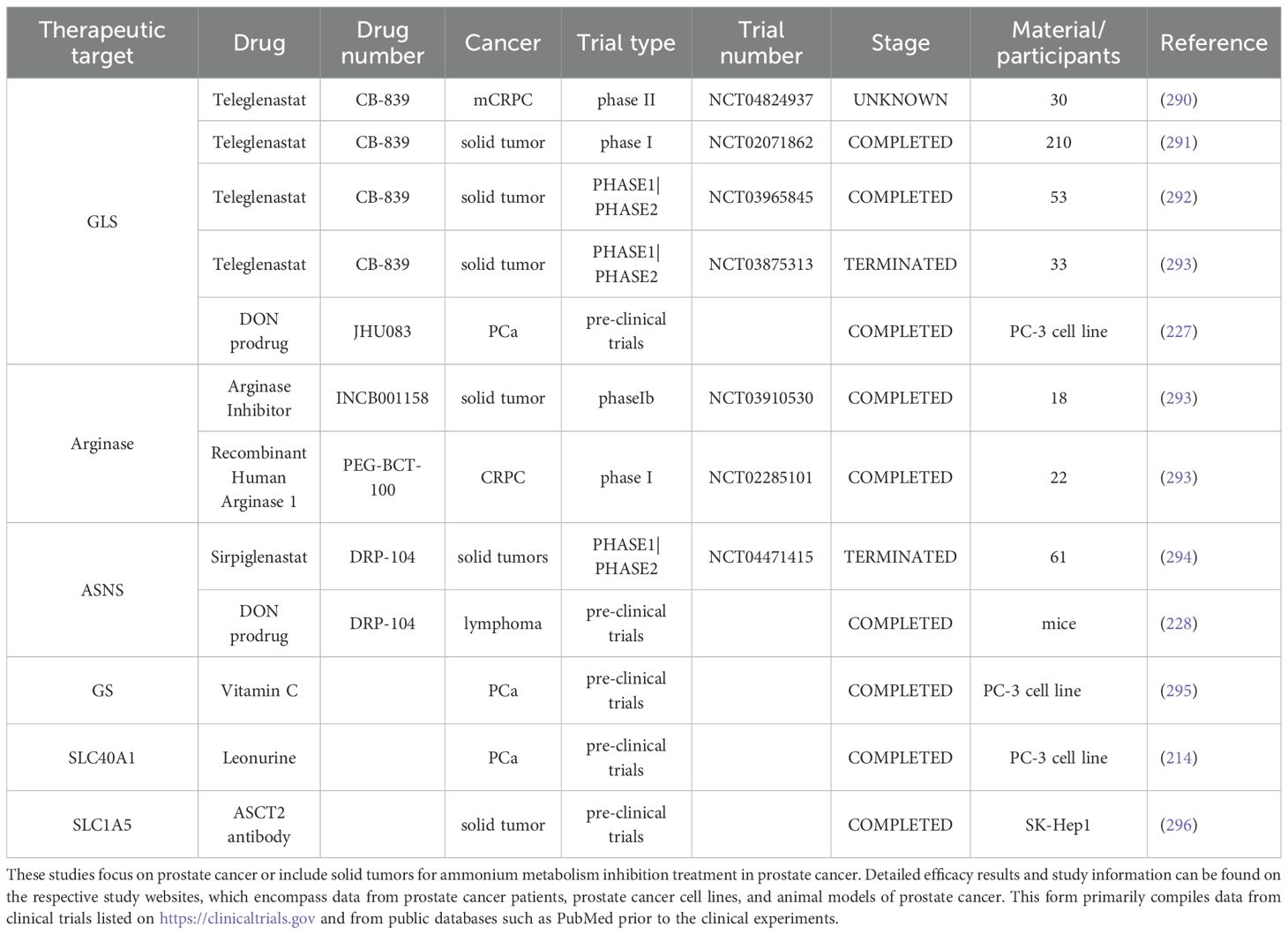
Table 2. researches focus on the utilization of ammonia metabolism inhibitor.
These drugs show significant promise for metabolic therapy in prostate cancer. This section highlights new research demonstrating that CB-839, a drug that targets GLS, inhibits prostate cancer proliferation through several mechanisms (90). GLS inhibition prevents the conversion of glutamine to glutamate in cancer cells (208). This process generates α-ketoglutarate, a key fuel for the TCA cycle, and inhibiting this step deprives cancer cells of energy for proliferation. Furthermore, glutamate not only serves as an essential TCA cycle intermediate but also helps maintain the cellular redox balance (209, 210). In cancer cells, GLS supports the production of glutamate, which is vital for the synthesis of GSH, a major antioxidant that scavenges ROS, triggering oxidative stress and eventually leading to apoptosis (211). Another mechanism involves GLS inhibition mediated activation of apoptosis pathways in prostate cancer cells, evidenced by the upregulation of apoptosis-related genes. This process may involve the p53 pathway (a known tumor suppressor) or the activation of other proapoptotic genes, resulting in programmed cell death (212). Moreover, GLS inhibition impairs the metabolic adaptability of cancer cell, potentially triggering excessive autophagy, resulting in apoptosis. Inhibition of other ammonium metabolism enzymes works through similar mechanisms, halting cancer cell proliferation via the coupling of ammonium metabolism to the TCA cycle. Research on ASNS reveals that inhibiting ASNS in mCRPC may reduce resistance to androgen therapy (19). ASNS also helps cancer cells survive through anti-apoptotic mechanisms. In addition, ASNS inhibition promotes aspartate accumulation, activates p53, and suppresses prostate cancer growth by regulating metabolic pathways, DNA repair, cell cycle progression, and apoptosis (22). Combining CB-839, a well-tolerated GLS inhibitor, with ASNase effectively limits asparagine synthesis and inhibits the growth of CRPC tumors driven by TP53 mutations. These findings open a new therapeutic avenue for prostate cancer and other solid tumors. Inhibition of arginase suppresses prostate cancer cells, as these cells often rely on external arginine for rapid proliferation. Arginine deprivation thus becomes an important therapeutic target (213). Additionally, arginine depletion inhibits T cell proliferation and promotes the recruitment of TAMs and myeloid-derived suppressor cells (MDSCs), facilitating immune evasion. Nitric oxide (NO) signaling plays dual roles in tumor immune evasion and tumorigenesis. Arginine depletion reduces NO synthesis and may alter its protumor and antitumor effects.
New research has focused on the SLC transporter family, with findings showing that SLC proteins facilitate the transport of substances such as ammonium and glutamine across lipid bilayers. One study revealed that leopurine inhibits SLC40A1, reducing its transport efficiency (214). Furthermore, SLC40A1 expression is linked to microRNA-18a-5P, a noncoding RNA that can regulate gene expression by binding to specific genes. In this context, SLC40A1 inhibition leads to the binding of microRNA-18a-5P with RUNX1, affecting the transcription process and influencing prostate cancer cell proliferation.
4.2 Ammonium metabolism-related biomarkers in prostate cancer
Prostate specific antigen (PSA) has been the gold standard for diagnosing PCa in hospitals and research institutes (215). However, while PSA has high sensitivity, its specificity is low, and factors such as prostate inflammation or other noncancerous diseases can also lead to elevated PSA levels (216–218). Furthermore, improvements such as PSA velocity, PSA density, and the free-to-total PSA ratio have shown only marginal improvements in specificity. Consequently, new biomarkers are urgently needed to aid in the early detection and treatment of PCa. Biochemical changes in cancer cells occur earlier than cytological, imaging, or functional changes. Metabolic profiling of bodily fluids is a promising method for identifying clinically valuable non-invasive biomarkers. A study revealed significantly elevated concentrations of Asp, Tyr, Val, Arg, Cit, Gly, Gln, and His (P < 0.05) in the blood samples of PCa patients, whereas Glu, Trp, Orn, and Ser levels were reduced. Among these, the Glu/Gln ratio is the most valuable for research, with specific AUC, sensitivity, and specificity data provided in the table (219). Metabolic changes in urinary vesicles may also serve as potential diagnostic biomarkers (220). Recent studies have shown that specific amino acids in these vesicles can accurately differentiate PCa from BPH and classify PCa into different stages, as detailed in the Table 3.
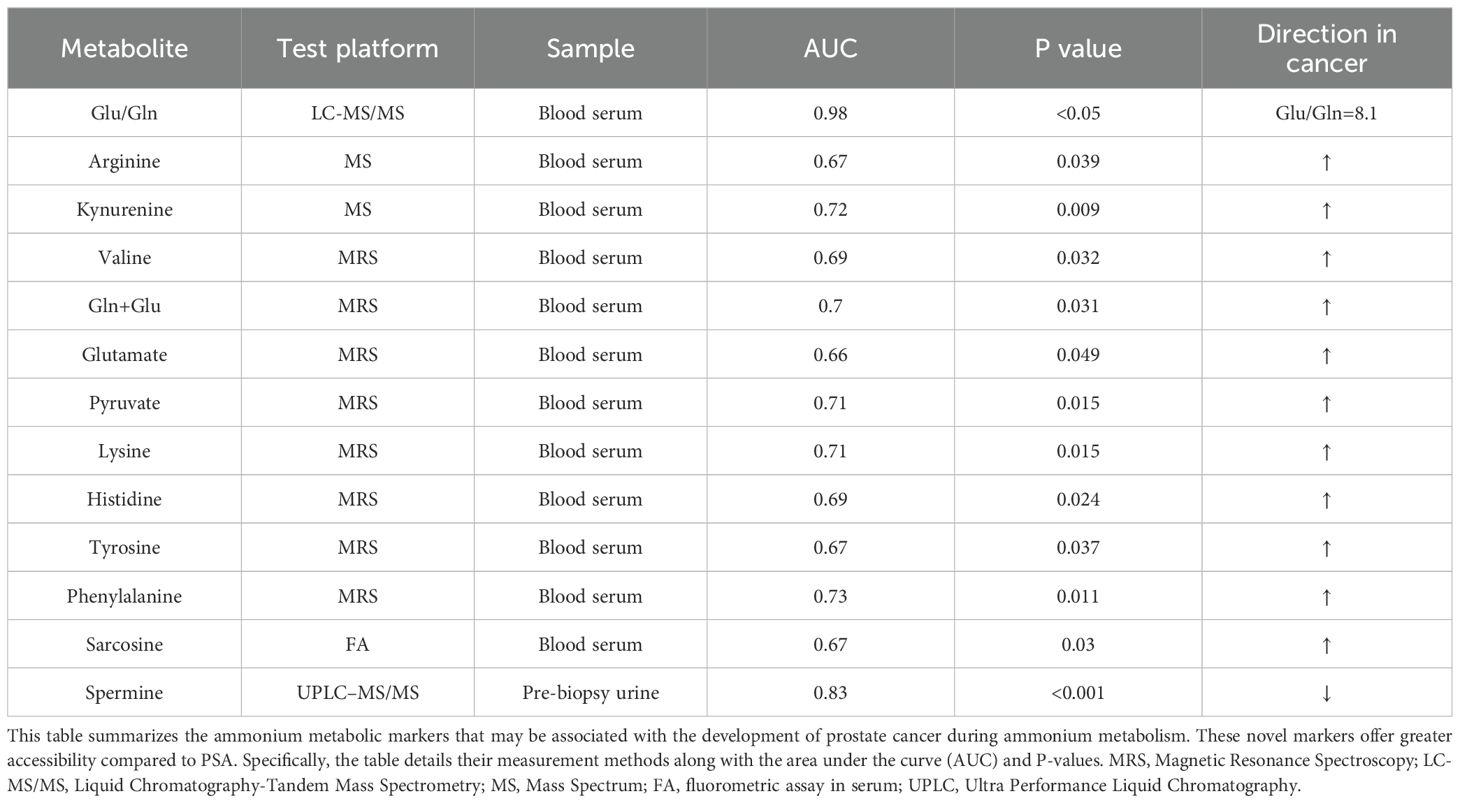
Table 3. New metabolic markers of prostate cancer related to ammonium metabolism.
4.3 Ammonium metabolism-related treatment combined with other therapies
4.3.1 Combining ammonium metabolism inhibition with targeted therapy
TP53-targeted therapy combined with metabolism inhibitors exploits the metabolic vulnerability caused by TP53 deletions, particularly the reliance on the asparagine synthesis pathway. Recent in vitro experiments have shown that tumor cells lacking TP53 produce a large amount of aspartic acid when cultured in vitro (22). TP53 deletion activates the ATF4/ASNS pathway, which enhances de novo synthesis of asparagine and supports tumor cell survival and proliferation in androgen-deprived or nutrient-limited environments. ATF4, a transcriptional activator of ASNS, is upregulated when TP53 deleted, leading to increased ASNS expression and increased intracellular asparagine synthesis (22).
GLS1 (GAC, glutaminase C), such as CB-839 and DONs, suppress glutamine metabolism, mainly by affecting GDH in the active site of GAC reducing its conversion to glutamate and thus limiting the synthesis of asparagine. Since asparagine synthesis depends on glutamate as a precursor, GLS inhibitors further suppress asparagine synthesis by reducing glutamine availability. Combining GLS inhibitors with targeted therapy can suppress glutamine production and asparagine synthesis via their respective metabolic pathways, restoring tumor cell sensitivity to ASNase and significantly reducing asparagine levels, thereby increasing therapeutic efficacy (221).
This combination therapy not only effectively disrupts the metabolic adaptation caused by TP53 mutations but also provides a new direction for the treatment of CRPC and other prostate cancers by targeting key metabolic pathways. The combination of GLS inhibitors and ASNase has shown promising therapeutic potential against TP53-mutated CRPC tumors in experimental studies and may offer new approaches for the personalized treatment of these cancers. Further treatment strategies are needed to confirm which targets GLS should be combined with for PCa treatment.
4.3.2 Combining ammonium metabolism targeting with chemotherapy
Paclitaxel resistance in mCRPC is associated with multiple cellular and metabolic mechanisms (222). Resistant cells often exhibit increased invasiveness, motility, and tumorigenic potential, along with metabolic reprogramming. Specifically, there is an increase in oxidative phosphorylation (OXPHOS), GSH synthesis, and reactive oxygen species (ROS) scavenging, which are closely linked to glutamine metabolism. Gln metabolism supports OXPHOS by providing energy, promotes GSH synthesis, and enhances ROS scavenging, helping to counteract paclitaxel-induced oxidative stress (223). Additionally, paclitaxel-resistant PCa cells often upregulate the expression of the antiapoptotic protein Bcl-2 and increase GSH levels, further enhancing antioxidant defense and resistance.
Furthermore, Gln metabolism contributes to chemotherapy resistance in prostate cancer cells by modulating drug efflux, particularly through the upregulation of the ATP-binding cassette transporter ABCB1 (224). ABCB1 reduces the intracellular concentration of chemotherapy drugs, thereby diminishing their therapeutic efficacy (225, 226). The activation of other pro-survival signaling pathways, such as the PI3K/Akt, MAPK, and NF-kB pathways, as well as the regulation of DNA repair mechanisms (e.g., ATM/ATR and Chk1/Chk2 pathways), also play critical roles in paclitaxel resistance. Therefore, Gln metabolism is essential for the cellular energy supply and aids tumor cells in adapting to paclitaxel treatment stress by regulating redox balance and drug efflux, thereby providing a survival advantage for resistance.
4.3.3 Integration of ammonium metabolism inhibition with immune checkpoint blockade
The inhibition of glutamine utilization by DON (Gln antagonist) and its prodrugs, such as JHU 083 and DRP-104, has been shown to enhance antitumor immune responses (142, 227, 228), suggesting a potential strategy when combined with immune checkpoint inhibitors. DRP-104 has been tested in the NCT 04471415 clinical trial to assess its preliminary safety and efficacy as a monotherapy or in combination with anti-PD-L1 immunotherapy (atezolizumab) in patients with advanced solid tumors such as PCa. However, this study remains unfinished, and the results have not yet been made available.
DON is considered a promising anticancer agent, but its clinical application has been limited by dose-limiting toxicity. DON exerts its effects by inhibiting several enzymes that utilize glutamine in both tumor and normal tissues mainly digestive tract. To minimize toxicity to normal tissues, DRP-104, a peptide prodrug of DON, was developed to preferentially convert DON in tumor tissues. Studies have demonstrated that DON and its prodrugs, such as JHU 083 and DRP-104, enhance antitumor immune responses by inhibiting glutamine utilization and that their combination with immune checkpoint inhibitors could improve therapeutic outcomes. Furthermore, DRP-104 effectively inhibits the carbon and nitrogen glutamine pathways in CRPC cells, thereby suppressing their growth. Currently, DRP-104 is being evaluated int he NCT 04471415 clinical trial for its safety and efficacy as a monotherapy or in combination with anti-PD-L1 immunotherapy (atezolizumab) in patients with advanced solid tumors, however, the study was recently terminated, and the results has not yet been released.
4.3.4 Synergistic combinations of ammonium metabolism inhibitors
CB-839, when combined with ASNS, reduces asparagine levels. Studies have shown that this combination effectively limits the synthesis of asparagine and inhibits the growth of CRPC tumors induced by TP53 mutations. This strategy offers a new therapeutic approach for solid tumors, such as prostate cancer. The combination of GLS inhibitors, such as CB-839, with ASNase, works synergistically by not only inhibiting asparagine synthesis but also enhancing its degradation. CB-839 inhibits the metabolic conversion of glutamine, thereby reducing asparagine synthesis and increasing tumor cell dependency on asparagine. ASNase further degrades asparagine, limiting the availability of essential amino acids to cancer cells and thereby inhibiting their growth. This combination therapy has demonstrated promising therapeutic potential in prostate cancer and other types of tumors. Moreover, while the use of ASNS alone can cause adverse effects such as pancreatitis and thrombosis, its combination with CB-839 effectively mitigates these risks (19).
4.3.5 Combination of ammonium metabolism drugs and radiotherapy
Excessive ROS production leads to an imbalance between ROS and the cell’s antioxidant defense, resulting in DNA damage, a key mechanism in radiation therapy-mediated tumor treatment (40). Radio-resistance in prostate cancer is mediated by enhanced glutamine metabolism, whose downstream product GSH confers therapeutic resistance. Targeting glutamine metabolism suppresses GSH synthesis and restores radiosensitivity, offering a promising therapeutic strategy. Additionally, GLS inhibition modifies the cellular redox state, altering the cell’s response to radiation therapy. Cancer stem cells (CSCs) constitute a unique population within tumors with self-renewal and differentiation potential. These cells are more resistant to radiation and chemotherapy than regular cancer cells and are typically located in the core of tumors, making them difficult to eliminate completely with conventional treatments. Studies have shown that glutamine metabolism is crucial not only for the energy and nitrogen supply of cells but also for the maintenance of CSCs. Glutamine regulates CSCs through signaling pathways such as the mTOR and Notch. In this study, GLS inhibition reduced glutamine metabolism in prostate cancer cells, significantly decreasing the expression of stem cell markers (such as ALDH, OCT4, and Sox2), with the cells exhibiting reduced stemness characteristics. By decreasing the stemness of cancer cells, GLS inhibition decreases their resistance to radiation, thus enhancing the radiosensitivity of prostate cancer.
Chemical inhibition of key points of glutamine metabolism, such as GLS and MYC, or genetic knockdown of these genes and glutamine transporters, suppresses glutamine decomposition, thereby inhibiting crucial CSC-driving pathways (WNT/β-catenin, the oxidative stress response, and the DNA damage response), resulting in CSC depletion both in vitro and in vivo and enhancing tumor radiosensitivity. ALDH-positive (ALDH+) PCa CSC populations exhibit radio-resistance, tumor initiation, and metastatic traits (229–231). Inhibition of GLS with BPTES or genetic silencing of GLS/GLS2 genes increases radiation sensitivity in lung and prostate cancer cell lines.
4.3.6 Combination of ammonium metabolism and endocrine therapy
Prostate cancer cells adapt to extreme conditions, such as hypoxia in the TME, through metabolic reprogramming to sustain growth and proliferation, as discussed earlier. Specifically, androgen treatment induces excessive oxidative stress in prostate cancer cells, leading to cell death. To maintain rapid proliferation, these cells require efficient protein synthesis and amino acid metabolism (232). This may lead to the accumulation of ammonium, which in turn activates intracellular antioxidant responses and other metabolic pathways, alleviating the oxidative stress induced by endocrine therapy. Ammonium accumulation enhances the cellular antioxidant capacity by activating a series of enzymes and pathways, such as glutamine synthetase, leading to the production of antioxidant compounds such as GSH.
Additionally, recent studies have shown that the transporter SLC1A5 (ASCT-2), which is involved in glutamine metabolism is regulated by androgens (233). In prostate tumor samples, ASCT-2 expression is significantly higher than that in normal prostate tissue, especially in castration-resistant tumors, and is associated with the growth and metastatic potential of prostate cancer cells. Prostate cancer cells regulate glutamine metabolism through ASCT2 (SLC1A5) and LAT1/3 transporters, which support amino acid uptake for proliferation and metastasis. ASCT2 is highly expressed in prostate cancer cells, facilitating glutamine uptake, cell cycle progression, mTORC1 activation, fatty acid synthesis, and energy metabolism. Inhibition of ASCT2 significantly reduces glutamine uptake, cell proliferation, tumor growth, and metastasis by lowering E2F pathway proteins. Glutamine metabolism in prostate cancer is also regulated by oncogenes like MYC, androgen receptors (AR), and mTOR, with metabolic tracing studies highlighting glutamine’s role in energy and precursor synthesis (234). Targeting ASCT2 and glutamine metabolism is a potential therapeutic strategy for prostate cancer (235). DU-145 cells, in particular, show marked glutamine dependency, with prominent GLS gene and protein expression. GLS inhibition (e.g., via BPTES) significantly reduces cell viability, demonstrating potent antitumor effects, especially in AR-independent cell lines (29, 236, 237). A combined therapy targeting both AR and glutamine metabolism could be more advantageous. In conclusion, AR plays a crucial role in PCa progression by regulating the glutamine metabolism network. Although AR-targeted therapies have been effective in the early stages, the emergence of CRPC is associated with adaptive changes, including increased glutamine utilization. Advanced PCa cells shift towards glutamine metabolism to meet energy demands, positioning it as a central hub in the PCa metabolic network. Therefore, targeting glutamine metabolism is a promising strategy, particularly since PCa cells (including CSCs) are highly dependent on glutamine during disease progression. Combining therapies targeting both AR and glutamine metabolism may play a greater role in combating advanced PCa and overcoming treatment resistance.
Metformin, an antidiabetic agent, promotes glutamine metabolic dependency in PCa cells by restricting glucose-derived carbon entry into the TCA cycle, thereby forcing PCa cells to utilize glutamine reductive carboxylation for citrate production. Pharmacological inhibition of GLS via CB-839 or BPTES amplifies PCa cell sensitivity to metformin (29). Compared with single-agent approaches, combined glutamine depletion and metformin treatment synergistically increase radiosensitivity in autophagy-deficient PCa models (40). These findings indicate a synergistic interaction between glutamine metabolism suppression and metformin in PCa cells.
4.3.7 Limitations of drugs targeting ammonium metabolism
Although systematic investigations into the adverse effects of ammonium metabolism-targeting agents in prostate cancer remain limited, their toxicological profiles have been relatively well characterized in other solid tumors, such as breast, pancreatic, and gastrointestinal cancers (211, 222, 238). Representative glutamine metabolism inhibitors, including DON, CB-839, and BPTES, have exhibited varying degrees of toxicity during development, with gastrointestinal side effects being particularly prominent and constituting a major barrier to clinical translation (239–242). These adverse events are likely due to the broad inhibition of multiple glutamine metabolism-related enzymes. Additional toxicities such as hypocalcemia and reversible myelosuppression have also been reported (243–245). In terms of pharmacological properties, DON and BPTES show moderate potency, poor metabolic stability, low water solubility, and limited selectivity for GLS, resulting in inferior antitumor efficacy compared to CB-839 (246, 247). By contrast, CB-839 is a highly selective GLS inhibitor that has demonstrated promising therapeutic efficacy across multiple tumor models. Several studies have identified potential biomarkers predictive of CB-839 sensitivity, including the intracellular glutamate/glutamine ratio, expression levels of the GLS isoform GAC, and GLS enzymatic activity (90, 93, 248, 249). These indicators could facilitate the development of predictive tools and support personalized treatment strategies, although further validation is warranted.
Nevertheless, both intrinsic and acquired resistance to CB-839 remains a critical challenge for widespread application. Tumor cells can bypass glutamine metabolism by activating alternative pathways; for instance, the upregulation of pyruvate carboxylation to generate oxaloacetate (OAA) has been observed in breast and pancreatic ductal adenocarcinomas (250). Furthermore, fatty acid oxidation (FAO) is markedly upregulated in resistant tumor cells, providing additional substrates such as acetyl-CoA and propionyl-CoA to fuel the TCA cycle via citrate synthase and succinyl-CoA synthesis (251, 252). Interestingly, the mTORC1 signaling pathway plays a pivotal role in this adaptive metabolic reprogramming, especially under glutamine-restricted conditions. Notably, combinatorial treatment with CB-839 and mTOR inhibitors has shown synergistic effects in preclinical studies, offering a compelling rationale for dual-targeted strategies and highlighting their potential for future clinical translation (253, 254).
5 Future outlook and direction
The metabolic reprogramming of prostate cancer cells has garnered significant attention, and drugs targeting metabolic processes are constantly being developed. However, there studies have notable limitations. Focusing solely on the metabolism of prostate cancer cells is insufficient, as the TME operates as a dynamic and balanced system. The interactions among immune cells, cytokine networks, and ammonium metabolism within the TME still requires further investigation. Additionally, the role of ammonium metabolism in prostate cancer remains underexplored, with insufficient experimental validation of its underlying mechanisms. Only by elucidating the upstream and downstream regulatory mechanisms can new targeted therapies be developed. Additionally, we have identified varying degrees of interference in experimental studies on ammonia metabolic reprogramming. As pointed out by De Berardinis et al., the changes in tumor ammonia metabolism between in vivo and in vitro experiments may differ significantly (255). For example, in in vitro studies, shRNA is often used to silence the expression of metabolic enzymes to observe their effects on ammonia metabolism, whereas in vivo, the affected sites may be more numerous and complex. This discrepancy suggests a potential direction for future research: whether the conclusions regarding the impact of ammonium metabolism on tumor progression are reliable. Furthermore, relying solely on metabolic flux is insufficient to determine the complete picture of ammonia metabolic reprogramming in tumor cells and its relative contribution compared to other metabolic pathways. Therefore, the most suitable approach currently is to provide a snapshot of metabolism rather than a reliable measurement of metabolic flux. More sensitive methods, such as using hyperpolarized 13C-labeled probes, may ultimately support in vivo flux analysis (256).
5.1 Potential effects of ammonium metabolism on various components in the TME of prostate cancer
As integral components of the TME, tumor-infiltrating immune cells (TIICs) can significantly alter the immune landscape of tumors (25, 257–259). Previous studies have demonstrated that the interactions between TIICs and prostate cancer cells can serve as therapeutic targets, with experimental evidence supporting these relationships (260, 261). However, immune checkpoint-based therapies have not yielded satisfactory outcomes in prostate cancer patients. Consequently, the impact of metabolic reprogramming, particularly ammonium metabolism, on TIICs within the TME warrants further investigation.
Recent studies have highlighted the detrimental effects of ammonium metabolism on nonspecific immune cells within the TME, particularly NK cells. Ammonium accumulation disrupts NK cell metabolism by impairing glycolysis and OXPHOS, both of which are essential for ATP production and cytotoxic activity (142, 227). Ammonium accumulation specifically inhibits hexokinase and lactate dehydrogenase in glycolysis, while sparing other enzymes (262). It may modulate TCA cycle activity via secondary messengers like cAMP, though further validation is needed (263). Additionally, ammonium impairs mitochondrial function, further inhibiting OXPHOS and NK cell activation (142). In addition to energy metabolism, ammonium accumulation affects lipid metabolism, altering membrane fluidity and receptor activation, which compromises NK cell efficiency in target cell recognition and elimination. Moreover, ammonium contributes to metabolic acidosis in the TME by forming ammonium ions (NH4+), lowering the pH, disrupting redox balance, and further suppressing NK cell function. These studies suggest that ammonium accumulation may induce programmed cell death of immune cells within TME, such as NK cells, through oxidative imbalance and mitochondria-mediated autophagy, thereby promoting immune evasion by prostate cancer cells. However, the specific mechanisms underlying this process remain to be elucidated. Moreover, additional therapeutic targets within this pathway are yet to be identified, and combination strategies involving immune checkpoint blockade (ICB) may offer enhanced therapeutic efficacy. This approach has demonstrated efficacy in the treatment of other solid tumors. For instance, the V9032 inhibitor has been shown to enhance tumor cell sensitivity to PD-1-targeted therapy in breast cancer (223, 264).
Ammonium also promotes tumor immune escape by modulating immune regulatory pathways. It enhances the immunosuppressive activity of Tregs, promoting their expansion and increasing the secretion of TGF-β and IL-10, which dampens antitumor immunity. Additionally, ammonium upregulates immune checkpoint molecules such as PD-L1 on tumor cells, directly inhibiting NK cell activation. By altering metabolic pathways, including glycolysis and fatty acid oxidation, ammonium promotes immune tolerance and weakens NK cell effector functions, particularly in solid tumors such as PCa. Macrophages, especially those in the M1 and M2 phenotypes, play crucial roles in tumor progression, particularly in PCa (265). M1 macrophages are involved in antitumor immunity, whereas M2 macrophages promote tumor growth, metastasis, and immune suppression. M2 macrophage presence is linked to worse clinical outcomes in several cancers, including PCa, and may cooperate with Tregs to create an immunosuppressive environment that assists the tumor to evade immune surveillance. Targeting M2 macrophages and their pathways, in addition to regulating Tregs, could offer a potential strategy for treating lethal PCa.
Matrix of TME are also influenced by the consumption of glutamine leading to a decrease in energy supply required for T cells to perform immune functions, thereby suppressing their activity (266). Arginine plays a vital role in T cell immunity by synthesizing nitric oxide, and its deficiency inhibits T cell proliferation and function. Tryptophan metabolism via the IDO enzyme pathway suppresses T-cell proliferation and promotes immune tolerance. Nonessential amino acids produced during amino acid metabolism are critical for the metabolic reprogramming of T cells. After activation, T-cells shift from glycolysis to amino acid synthesis and utilization to support proliferation and differentiation, which competes with tumor cells, leading to T-cell suppression. The uptake of glutamine depends on amino acid transporters such as ASCT2, which play a key role in T cell differentiation. Arginine enters T cells via the CAT-1 transporter, and its deficiency impacts T-cell proliferation, differentiation, and cytokine secretion. Asparagine aids in protein synthesis and restores mTOR activity, helping tumor cells proliferate and maintaining T-cell function (267, 268). Joint deprivation of arginine and asparagine leads to extensive T-cell death, indicating their complementary role in maintaining T-cell activity. Additionally, serine synthesis is essential for T-cell proliferation and tumor growth. Limiting serine suppresses T-cell proliferation, although some tumors adapt by increasing phosphoglycerate dehydrogenase (PHGDH) activity. Although, targeting PHGDH may inhibit tumor growth, but its impact on the immune response remains unclear.
According to previous studies, ammonium metabolism not only affects TIICs in the TME but also influences cytokines secreted by immune cells or other immunoreactive substances upstream of genes. These noncellular components may serve as potential targets for future drug therapies and can function as prognostic indicators. Recent research has demonstrated that pH levels in the TME significantly impact immune cell function, cytokine secretion, and ammonium metabolism reprogramming, which may contribute to the decreased pH value in the TME (269). A novel immunoconjugate compound, L-DOS47, was utilized to increase the pH of an acidic medium in vitro to evaluate its effects on Jurkat, a human T lymphoblast-like cell line.
5.2 Influence of ammonium metabolism on other major therapeutic modalities
Current prostate cancer treatments primarily include surgical and nonsurgical approaches. Among nonsurgical treatments, castration is currently the most attractive option; however, in androgen-insensitive prostate cancers such as CRPC, androgen therapy may be ineffective (232, 270, 271). The long-term use of ADT can lead to metabolic disorders in patients. Drugs that target metabolic reprogramming may synergize with ADT to some extent. Metabolic reprogramming, as an emerging research direction, has shown considerable efficacy in combination with other treatments in various solid tumors. However, in prostate cancer treatment, particularly regarding the combined treatment of ammonium metabolism and androgens, effective studies remain scarce, with most focusing on glycolysis. Ammonium metabolism is also a primary metabolic mode after tumor cell metabolic reprogramming, potentially representing a future research direction. Previous studies have demonstrated that AR signaling (DHT, 5-alpha-dihydrotestosterone) primarily enhances glutamine (Gln) uptake by regulating the expression of various Gln transporters, such as SLC1A4 (ASCT1), SLC1A5 (ASCT2), SLC3A2 (4F2hc), SLC7A5 (LAT1), and SLC43A1 (LAT3), to meet the proliferative and differentiative demands of PCa cells (39, 233, 236). ADT alone typically induces the development of CRPC, during which CRPC cells often undergo metabolic reprogramming toward enhanced ammonium metabolism, characterized by a higher Gln utilization rate. This metabolic shift in CRPC is reflected in increased Gln uptake and upregulation of key regulatory genes involved in glutaminolysis (272–274). The elevated demand for Gln is further evidenced by the overexpression of Gln transporters such as ASCT2 and LAT1, as well as the increased expression of the androgen-independent GLS isoform GAC. As PCa progresses to more aggressive stages, both PCa cells and prostate cancer CSCs exhibit Gln addiction (274, 275). Therefore, targeting the Gln metabolic network represents a unique opportunity to overcome therapeutic resistance and sensitize tumor cells to anticancer treatments, potentially offering greater benefit than AR-targeted monotherapy (39, 168). CRPC cells generally rely on glutamine metabolism for energy supply, providing a mechanistic explanation for the effectiveness of ammonium metabolism inhibitors, especially glutamine metabolism inhibitors, in combination with second-generation androgen receptor antagonists (276). Moreover, the AR signaling axis can activate GLS activity, promoting the conversion of glutamine to glutamate and providing energy substrates for the tricarboxylic acid cycle, another potential mechanism for the use of metabolic inhibitors in combination with ADT. Further studies comparing LNCaP cells (androgen-sensitive prostate cancer) with mCRPC cells revealed a stronger metabolic dependence on glutamine in LNCaP cells. The androgen signaling pathway not only drives prostate cancer proliferation and survival but also provides metabolic advantages through ammonium metabolic reprogramming. Combined targeting of AR signaling and glutamine metabolic pathways holds promise as a new strategy to improve CRPC treatment. Advanced metabolomics techniques can assist in accurately screening precise metabolic targets to optimize treatment. Although ADT is effective, the metabolic reprogramming it induces drives CRPC development. Therapies incorporating metabolic targets, such as those inhabit amino acid metabolism, are expected to significantly improve outcomes in CRPC patients.
In addition to developing therapeutic agents that target the reprogramming of ammonium metabolism in prostate cancer, it is essential to identify reliable indicators for evaluating treatment efficacy, such as sensitivity-related gene signatures and metabolomic profiling (277). While previous imaging techniques have mainly focused on magnetic resonance spectroscopy (MRS), which detects localized concentrations of metabolites such as choline, citrate, and various amino acids, these metabolic profiles reflect tumor metabolic intensity and may indicate responsiveness to ammonium-targeted therapies (278, 279). When integrated with conventional imaging and biopsy, MRS can aid in treatment assessment; however, its clinical application remains limited due to the low concentration of metabolites relative to water, necessitating high-field magnets, long acquisition times, and specialized expertise in spectral analysis. To overcome these limitations, novel metabolic PET tracers have been developed, offering improved sensitivity and specificity in detecting early metabolic changes in prostate cancer (280–282). Additionally, advanced hyperpolarized 13C-labeled spectral MRI techniques provide dynamic and real-time evaluation of tumor metabolism (283). Metabolomics-based research may further reveal valuable biomarkers for therapy monitoring, as the reprogramming of ammonium metabolism in prostate cancer generates a range of intermediate metabolites, such as polyamines, citrate, and various amino acids, as well as key metabolic enzymes, which hold promise as indicators of therapeutic response (284). Further experimental validation and clinical investigation are needed to establish their clinical utility.
6 Conclusion
Overall, ammonium metabolism, particularly downstream glutamine metabolism, is crucial for the metabolic treatment of prostate cancer. Corresponding metabolic inhibitors can be targeted, and products from this metabolic pathway can serve as diagnostic or prognostic markers for prostate cancer. Additionally, ammonium metabolism inhibitors can enhance immune cell function by affecting the immune microenvironment in prostate cancer, achieving therapeutic effects. Therefore, the role of ammonium metabolism in the immune microenvironment of prostate cancer remains worth exploring, along with its potential application value in diagnosis and treatment.
Author contributions
ZY: Writing – review & editing, Writing – original draft, Conceptualization, Data curation. HW: Investigation, Writing – review & editing, Supervision. ZL: Writing – review & editing, Methodology. RY: Investigation, Methodology, Writing – review & editing. YR: Writing – review & editing, Investigation. BL: Data curation, Writing – review & editing, Formal analysis. BG: Software, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
mCRPC, metastatic castration-resistant prostate cancer; mHSPC, metastatic hormone-sensitive prostate cancer; ADT, androgen-deprivation therapy; CPS, carbamoyl phosphate synthetase I; OTC, ornithine transcarbamylase; LDH, Lactate Dehydrogenase; GDH, Glutamate Dehydrogenase; SREBP, sterol regulatory element-binding protein; GLS, glutaminase; TCA, Tricarboxylic Acid; SLC, solute carrier family; GABA, aminobutyric acid; TIL, tumor-infiltrating lymphocytes; Tregs, regulatory T cells; TAMs, tumor-associated macrophages; PSA, Prostate Specific Antigen; PD-1, programmed death-1; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; GSH, glutathione; TIGIT, T cell immunoreceptor with Ig and ITIM domain.
References
1. Bergengren O, Pekala KR, Matsoukas K, Fainberg J, Mungovan SF, Bratt O, et al. 2022 Update on prostate cancer epidemiology and risk factors—A systematic review. Eur Urol. (2023) 84:191–206. doi: 10.1016/j.eururo.2023.04.021
2. Sekhoacha M, Riet K, Motloung P, Gumenku L, Adegoke A, and Mashele S. Prostate cancer review: genetics, diagnosis, treatment options, and alternative approaches. Molecules. (2022) 27:5730. doi: 10.3390/molecules27175730
3. Hamdy FC, Donovan JL, Lane JA, Mason M, Metcalfe C, Holding P, et al. 10-year outcomes after monitoring, surgery, or radiotherapy for localized prostate cancer. N Engl J Med. (2016) 375:1415–24. doi: 10.1056/NEJMoa1606220
4. Gong J, Janes JL, Trustram Eve C, Stock S, Waller J, De Hoedt AM, et al. Epidemiology, treatment patterns, and clinical outcomes in de novo oligometastatic hormone-sensitive prostate cancer. Cancer. (2024) 130:3815–25. doi: 10.1002/cncr.35466
5. Furukawa T, Tabata S, Minami K, Yamamoto M, Kawahara K, and Tanimoto A. Metabolic reprograming of cancer as a therapeutic target. Biochim Biophys Acta (BBA) - Gen Subj. (2023) 1867:130301. doi: 10.1016/j.bbagen.2022.130301
6. Colegio OR, Chu N-Q, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. (2014) 513:559–63. doi: 10.1038/nature13490
7. Chetta P, Sriram R, and Zadra G. Lactate as key metabolite in prostate cancer progression: what are the clinical implications? Cancers. (2023) 15:3473. doi: 10.3390/cancers15133473
8. Diedrich JD, Rajagurubandara E, Herroon MK, Mahapatra G, Hüttemann M, and Podgorski I. Bone marrow adipocytes promote the Warburg phenotype in metastatic prostate tumors via HIF-1α activation. Oncotarget. (2016) 7:64854–77. doi: 10.18632/oncotarget.11712
9. Ahmad F, Cherukuri MK, and Choyke PL. Metabolic reprogramming in prostate cancer. Br J Cancer. (2021) 125:1185–96. doi: 10.1038/s41416-021-01435-5
10. DiNardo CD, Stein EM, De Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable remissions with ivosidenib in IDH1 -mutated relapsed or refractory AML. N Engl J Med. (2018) 378:2386–98. doi: 10.1056/NEJMoa1716984
11. Gonçalves AC, Richiardone E, Jorge J, Polónia B, Xavier CPR, Salaroglio IC, et al. Impact of cancer metabolism on therapy resistance - Clinical implications. Drug Resist Update. (2021) 59:100797. doi: 10.1016/j.drup.2021.100797
12. Priolo C, Pyne S, Rose J, Regan ER, Zadra G, Photopoulos C, et al. AKT1 and MYC induce distinctive metabolic fingerprints in human prostate cancer. Cancer Res. (2014) 74:7198–204. doi: 10.1158/0008-5472.CAN-14-1490
13. Röhrig F and Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer. (2016) 16:732–49. doi: 10.1038/nrc.2016.89
14. Sadowski MC, Pouwer RH, Gunter JH, Lubik AA, Quinn RJ, and Nelson CC. The fatty acid synthase inhibitor triclosan: repurposing an anti-microbial agent for targeting prostate cancer. Oncotarget. (2014) 5:9362–81. doi: 10.18632/oncotarget.2433
15. Hindes MT, McElligott AM, Best OG, Ward MP, Selemidis S, Miles MA, et al. Metabolic reprogramming, Malignant transformation and metastasis: Lessons from chronic lymphocytic leukaemia and prostate cancer. Cancer Lett. (2025) 611:217441. doi: 10.1016/j.canlet.2025.217441
16. Liberti MV and Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci. (2016) 41:211–8. doi: 10.1016/j.tibs.2015.12.001
17. Liu P-S, Wang H, Li X, Chao T, Teav T, Christen S, et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. (2017) 18:985–94. doi: 10.1038/ni.3796
18. Knott SRV, Wagenblast E, Khan S, Kim SY, Soto M, Wagner M, et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature. (2018) 554:378–81. doi: 10.1038/nature25465
19. Sircar K, Huang H, Hu L, Cogdell D, Dhillon J, Tzelepi V, et al. Integrative molecular profiling reveals asparagine synthetase is a target in castration-resistant prostate cancer. Am J Pathol. (2012) 180:895–903. doi: 10.1016/j.ajpath.2011.11.030
20. Zhang B, Dong L-W, Tan Y-X, Zhang J, Pan Y-F, Yang C, et al. Asparagine synthetase is an independent predictor of surgical survival and a potential therapeutic target in hepatocellular carcinoma. Br J Cancer. (2013) 109:14–23. doi: 10.1038/bjc.2013.293
21. Xu Y, Lv F, Zhu X, Wu Y, and Shen X. Loss of asparagine synthetase suppresses the growth of human lung cancer cells by arresting cell cycle at G0/G1 phase. Cancer Gene Ther. (2016) 23:287–94. doi: 10.1038/cgt.2016.28
22. Yoo YA, Quan S, Yang W, Guo Q, Rodríguez Y, Chalmers ZR, et al. Asparagine dependency is a targetable metabolic vulnerability in TP53 -altered castration-resistant prostate cancer. Cancer Res. (2024) 84:3004–22. doi: 10.1158/0008-5472.CAN-23-2910
23. Deng L, Yao P, Li L, Ji F, Zhao S, Xu C, et al. p53-mediated control of aspartate-asparagine homeostasis dictates LKB1 activity and modulates cell survival. Nat Commun. (2020) 11:1755. doi: 10.1038/s41467-020-15573-6
24. Cui H, Darmanin S, Natsuisaka M, Kondo T, Asaka M, Shindoh M, et al. Enhanced expression of asparagine synthetase under glucose-deprived conditions protects pancreatic cancer cells from apoptosis induced by glucose deprivation and cisplatin. Cancer Res. (2007) 67:3345–55. doi: 10.1158/0008-5472.CAN-06-2519
25. Bader DA and McGuire SE. Tumour metabolism and its unique properties in prostate adenocarcinoma. Nat Rev Urol. (2020) 17:214–31. doi: 10.1038/s41585-020-0288-x
26. Rajagopalan KN, Marian CO, Shay JW, and DeBerardinis RJ. Abstract 1248: Enhanced glutamine metabolism contributes to docetaxel resistance in DU145 prostate cancer cells. Cancer Res. (2011) 71:1248–8. doi: 10.1158/1538-7445.AM2011-1248
27. Menendez-Montes I, Solmonson A, MAcedo-Pereira AH, Cardoso AC, Nakada Y, Thet S, et al. Abstract P3193: cardiac glutamine metabolism as NAD+ Generating pathway during chronic hypoxia. Circ Res. (2023) 133:AP3193–AP3193. doi: 10.1161/res.133.suppl_1.P3193
28. DeBerardinis RJ and Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. (2010) 29:313–24. doi: 10.1038/onc.2009.358
29. Fendt S-M, Bell EL, Keibler MA, Davidson SM, Wirth GJ, Fiske B, et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. (2013) 73:4429–38. doi: 10.1158/0008-5472.CAN-13-0080
30. Carracedo A, Cantley LC, and Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. (2013) 13:227–32. doi: 10.1038/nrc3483
31. Canada AT, Roberson KM, Vessella RL, Trump DL, Robertson CN, and Fine RL. Glutathione and glutathione S-transferase in benign and Malignant prostate cell lines and prostate tissues. Biochem Pharmacol. (1996) 51:87–90. doi: 10.1016/0006-2952(95)02157-4
32. Okudaira H, Oka S, Ono M, Nakanishi T, Schuster DM, Kobayashi M, et al. Accumulation of trans-1-amino-3-[(18)F]fluorocyclobutanecarboxylic acid in prostate cancer due to androgen-induced expression of amino acid transporters. Mol Imaging Biol. (2014) 16:756–64. doi: 10.1007/s11307-014-0756-x
33. Canapè C, Catanzaro G, Terreno E, Karlsson M, Lerche MH, and Jensen PR. Probing treatment response of glutaminolytic prostate cancer cells to natural drugs with hyperpolarized [5-(13) C]glutamine. Magn Reson Med. (2015) 73:2296–305. doi: 10.1002/mrm.25360
34. Zhang J, Mao S, Guo Y, Wu Y, Yao X, and Huang Y. Inhibition of GLS suppresses proliferation and promotes apoptosis in prostate cancer. Biosci Rep. (2019) 39:BSR20181826. doi: 10.1042/BSR20181826
35. Jin J, Byun J-K, Choi Y-K, and Park K-G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp Mol Med. (2023) 55:706–15. doi: 10.1038/s12276-023-00971-9
36. Gao P, Tchernyshyov I, Chang T-C, Lee Y-S, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. (2009) 458:762–5. doi: 10.1038/nature07823
37. Liu W, Le A, Hancock C, Lane AN, Dang CV, Fan TW-M, et al. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc Natl Acad Sci U.S.A. (2012) 109:8983–8. doi: 10.1073/pnas.1203244109
38. Qu X, Sun J, Zhang Y, Li J, Hu J, Li K, et al. c-Myc-driven glycolysis via TXNIP suppression is dependent on glutaminase-MondoA axis in prostate cancer. Biochem Biophys Res Commun. (2018) 504:415–21. doi: 10.1016/j.bbrc.2018.08.069
39. White MA, Lin C, Rajapakshe K, Dong J, Shi Y, Tsouko E, et al. Glutamine transporters are targets of multiple oncogenic signaling pathways in prostate cancer. Mol Cancer Res. (2017) 15:1017–28. doi: 10.1158/1541-7786.MCR-16-0480
40. Mukha A, Kahya U, Linge A, Chen O, Löck S, Lukiyanchuk V, et al. GLS-driven glutamine catabolism contributes to prostate cancer radiosensitivity by regulating the redox state, stemness and ATG5-mediated autophagy. Theranostics. (2021) 11:7844–68. doi: 10.7150/thno.58655
41. Dasgupta S, Putluri N, Long W, Zhang B, Wang J, Kaushik AK, et al. Coactivator SRC-2-dependent metabolic reprogramming mediates prostate cancer survival and metastasis. J Clin Invest. (2015) 125:1174–88. doi: 10.1172/JCI76029
42. Li Y, Li X, Li X, Zhong Y, Ji Y, Yu D, et al. PDHA1 gene knockout in prostate cancer cells results in metabolic reprogramming towards greater glutamine dependence. Oncotarget. (2016) 7:53837–52. doi: 10.18632/oncotarget.10782
43. Wang Q, Guan YF, Hancock SE, Wahi K, van Geldermalsen M, Zhang BK, et al. Inhibition of guanosine monophosphate synthetase (GMPS) blocks glutamine metabolism and prostate cancer growth. J Pathol. (2021) 254:135–46. doi: 10.1002/path.5665
44. Xu L, Zhao B, Butler W, Xu H, Song N, Chen X, et al. Targeting glutamine metabolism network for the treatment of therapy-resistant prostate cancer. Oncogene. (2022) 41:1140–54. doi: 10.1038/s41388-021-02155-z
45. Albers MJ, Bok R, Chen AP, Cunningham CH, Zierhut ML, Zhang VY, et al. Hyperpolarized 13C lactate, pyruvate, and alanine: noninvasive biomarkers for prostate cancer detection and grading. Cancer Res. (2008) 68:8607–15. doi: 10.1158/0008-5472.CAN-08-0749
46. Costello LC and Franklin RB. Why do tumour cells glycolyse?”: from glycolysis through citrate to lipogenesis. Mol Cell Biochem. (2005) 280:1–8. doi: 10.1007/s11010-005-8841-8
47. Moreadith RW and Lehninger AL. The pathways of glutamate and glutamine oxidation by tumor cell mitochondria. Role of mitochondrial NAD(P)+-dependent Malic enzyme. J Biol Chem. (1984) 259:6215–21. doi: 10.1016/S0021-9258(20)82128-0
48. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U.S.A. (2007) 104:19345–50. doi: 10.1073/pnas.0709747104
49. El Gaafary M, Morad SAF, Schmiech M, Syrovets T, and Simmet T. Arglabin, an EGFR receptor tyrosine kinase inhibitor, suppresses proliferation and induces apoptosis in prostate cancer cells. Biomedicine Pharmacotherapy. (2022) 156:113873. doi: 10.1016/j.biopha.2022.113873
50. Lala PK and Chakraborty C. Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncol. (2001) 2:149–56. doi: 10.1016/S1470-2045(00)00256-4
51. Chen L, Xu Y-X, Wang Y-S, and Zhou J-L. Lipid metabolism, amino acid metabolism, and prostate cancer: a crucial metabolic journey. Asian J Andrology. (2024) 26:123–34. doi: 10.4103/aja202363
52. Zhong T, Zhang W, Guo H, Pan X, Chen X, He Q, et al. The regulatory and modulatory roles of TRP family channels in Malignant tumors and relevant therapeutic strategies. Acta Pharm Sin B. (2022) 12:1761–80. doi: 10.1016/j.apsb.2021.11.001
53. Shinka T, Onodera D, Tanaka T, Shoji N, Miyazaki T, Moriuchi T, et al. Serotonin synthesis and metabolism-related molecules in a human prostate cancer cell line. Oncol Lett. (2011) 2:211–5. doi: 10.3892/ol.2011.244
54. Rodríguez-Blanco G, Burgers PC, Dekker LJM, Vredenbregt-van Den Berg MS, Ijzermans JNM, Schenk-Braat EAM, et al. Serum kynurenine/tryptophan ratio is not a potential marker for detecting prostate cancer. Clin Biochem. (2014) 47:1347–8. doi: 10.1016/j.clinbiochem.2014.05.001
55. Wang X, Song Y, Shi Y, Yang D, Li J, and Yin B. SNHG3 could promote prostate cancer progression through reducing methionine dependence of PCa cells. Cell Mol Biol Lett. (2022) 27:13. doi: 10.1186/s11658-022-00313-z
56. Han Q, Tan Y, and Hoffman RM. Oral dosing of recombinant methioninase is associated with a 70% Drop in PSA in a patient with bone-metastatic prostate cancer and 50% Reduction in circulating methionine in a high-stage ovarian cancer patient. Anticancer Res. (2020) 40:2813–9. doi: 10.21873/anticanres.14254
57. Wang H, Zhang Q, Wen Q, Zheng Y, Lazarovici P, Jiang H, et al. Proline-rich Akt substrate of 40kDa (PRAS40): a novel downstream target of PI3k/Akt signaling pathway. Cell Signal. (2012) 24:17–24. doi: 10.1016/j.cellsig.2011.08.010
58. Funderburk SF, Shatkina L, Mink S, Weis Q, Weg-Remers S, and Cato ACB. Specific N-terminal mutations in the human androgen receptor induce cytotoxicity. Neurobiol Aging. (2009) 30:1851–64. doi: 10.1016/j.neurobiolaging.2007.12.023
59. Fjösne HE, Strand H, and Sunde A. Dose-dependent induction of ornithine decarboxylase and S-adenosyl-methionine decarboxylase activity by testosterone in the accessory sex organs of male rats. Prostate. (1992) 21:239–45. doi: 10.1002/pros.2990210307
60. Blackshear PJ, Manzella JM, Stumpo DJ, Wen L, Huang JK, Oyen O, et al. High level, cell-specific expression of ornithine decarboxylase transcripts in rat genitourinary tissues. Mol Endocrinol. (1989) 3:68–78. doi: 10.1210/mend-3-1-68
61. Cyriac J, Haleem R, Cai X, and Wang Z. Androgen regulation of spermidine synthase expression in the rat prostate. Prostate. (2002) 50:252–61. doi: 10.1002/pros.10052
62. Crozat A, Palvimo JJ, Julkunen M, and Jänne OA. Comparison of androgen regulation of ornithine decarboxylase and S-adenosylmethionine decarboxylase gene expression in rodent kidney and accessory sex organs. Endocrinology. (1992) 130:1131–44. doi: 10.1210/endo.130.3.1537280
63. Vujcic S, Liang P, Diegelman P, Kramer DL, and Porter CW. Genomic identification and biochemical characterization of the mammalian polyamine oxidase involved in polyamine back-conversion. Biochem J. (2003) 370:19–28. doi: 10.1042/BJ20021779
64. Schipper RG, Romijn JC, Cuijpers VMJI, and Verhofstad AAJ. Polyamines and prostatic cancer. Biochem Soc Trans. (2003) 31:375–80. doi: 10.1042/bst0310375
65. Smith RC, Litwin MS, Lu Y, and Zetter BR. Identification of an endogenous inhibitor of prostatic carcinoma cell growth. Nat Med. (1995) 1:1040–5. doi: 10.1038/nm1095-1040
66. Wallace HM, Fraser AV, and Hughes A. A perspective of polyamine metabolism. Biochem J. (2003) 376:1–14. doi: 10.1042/BJ20031327
67. Bello-Fernandez C, Packham G, and Cleveland JL. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc Natl Acad Sci U.S.A. (1993) 90:7804–8. doi: 10.1073/pnas.90.16.7804
68. Peters MC, Minton A, Phanstiel Iv O, Gilmour SK, and Novel A. Polyamine-targeted therapy for BRAF mutant melanoma tumors. Med Sci (Basel). (2018) 6:3. doi: 10.3390/medsci6010003
69. Ramos-Molina B, Lambertos A, and Peñafiel R. Antizyme inhibitors in polyamine metabolism and beyond: physiopathological implications. Med Sci. (2018) 6:89. doi: 10.3390/medsci6040089
70. Pietilä M, Lampinen A, Pellinen R, and Alhonen L. Inducible expression of antizyme 1 in prostate cancer cell lines after lentivirus mediated gene transfer. Amino Acids. (2012) 42:559–64. doi: 10.1007/s00726-011-1033-9
71. Kavanagh JP. Isocitric and citric acid in human prostatic and seminal fluid: implications for prostatic metabolism and secretion. Prostate. (1994) 24:139–42. doi: 10.1002/pros.2990240307
72. Palmieri F, Bisaccia F, Iacobazzi V, Indiveri C, and Zara V. Mitochondrial substrate carriers. Biochim Biophys Acta. (1992) 1101:223–7. doi: 10.1016/S0005-2728(05)80026-X
73. Pajor AM. Sequence and functional characterization of a renal sodium/dicarboxylate cotransporter. J Biol Chem. (1995) 270:5779–85. doi: 10.1074/jbc.270.11.5779
74. Chen XZ, Shayakul C, Berger UV, Tian W, and Hediger MA. Characterization of a rat Na+-dicarboxylate cotransporter. J Biol Chem. (1998) 273:20972–81. doi: 10.1074/jbc.273.33.20972
75. Mycielska ME, Patel A, Rizaner N, Mazurek MP, Keun H, Patel A, et al. Citrate transport and metabolism in mammalian cells: Prostate epithelial cells and prostate cancer. BioEssays. (2009) 31:10–20. doi: 10.1002/bies.080137
76. Coloff JL, Murphy JP, Braun CR, Harris IS, Shelton LM, Kami K, et al. Differential glutamate metabolism in proliferating and quiescent mammary epithelial cells. Cell Metab. (2016) 23:867–80. doi: 10.1016/j.cmet.2016.03.016
77. Hodo Y, Tressler CM, Ghaemi B, Thomas R, Webster AS, Bains Williams KN, et al. Imaging the uptake and metabolism of glutamine in prostate tumor models using CEST MRI. NPJ Imaging. (2025) 3:34. doi: 10.1038/s44303-025-00100-3
78. Zou W, Han Z, Wang Z, and Liu Q. Targeting glutamine metabolism as a potential target for cancer treatment. J Exp Clin Cancer Res. (2025) 44:180. doi: 10.1186/s13046-025-03430-7
79. Bell HN, Huber AK, Singhal R, Korimerla N, Rebernick RJ, Kumar R, et al. Microenvironmental ammonia enhances T cell exhaustion in colorectal cancer. Cell Metab. (2023) 35:134–149.e6. doi: 10.1016/j.cmet.2022.11.013
80. Cheng C, Geng F, Li Z, Zhong Y, Wang H, Cheng X, et al. Ammonia stimulates SCAP/Insig dissociation and SREBP-1 activation to promote lipogenesis and tumour growth. Nat Metab. (2022) 4:575–88. doi: 10.1038/s42255-022-00568-y
81. Altman BJ, Stine ZE, and Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. (2016) 16:749. doi: 10.1038/nrc.2016.114
82. Tardito S, Oudin A, Ahmed SU, Fack F, Keunen O, Zheng L, et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat Cell Biol. (2015) 17:1556–68. doi: 10.1038/ncb3272
83. Spinelli JB, Yoon H, Ringel AE, Jeanfavre S, Clish CB, and Haigis MC. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science. (2017) 358:941–6. doi: 10.1126/science.aam9305
84. Page-McCaw A, Ewald AJ, and Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. (2007) 8:221–33. doi: 10.1038/nrm2125
85. D’Aniello C, Patriarca EJ, Phang JM, and Minchiotti G. Proline metabolism in tumor growth and metastatic progression, front. Oncol. (2020) 10:776. doi: 10.3389/fonc.2020.00776
86. Goggins E, Kakkad S, Mironchik Y, Jacob D, Wildes F, Krishnamachary B, et al. Hypoxia inducible factors modify collagen I fibers in MDA-MB-231 triple negative breast cancer xenografts. Neoplasia. (2018) 20:131–9. doi: 10.1016/j.neo.2017.11.010
87. Xiong G, Stewart RL, Chen J, Gao T, Scott TL, Samayoa LM, et al. Collagen prolyl 4-hydroxylase 1 is essential for HIF-1α stabilization and TNBC chemoresistance. Nat Commun. (2018) 9:4456. doi: 10.1038/s41467-018-06893-9
88. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang X-Y, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction, Proc. Natl Acad Sci USA. (2008) 105:18782–7. doi: 10.1073/pnas.0810199105
89. Hu W, Zhang C, Wu R, Sun Y, Levine A, and Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA. (2010) 107:7455–60. doi: 10.1073/pnas.1001006107
90. Wang J-B, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. (2010) 18:207–19. doi: 10.1016/j.ccr.2010.08.009
91. Borodovsky A, Seltzer MJ, and Riggins GJ. Altered cancer cell metabolism in gliomas with mutant IDH1 or IDH2. Curr Opin Oncol. (2012) 24:83–9. doi: 10.1097/CCO.0b013e32834d816a
92. Zhao L, Huang Y, and Zheng J. STAT1 regulates human glutaminase 1 promoter activity through multiple binding sites in HIV-1 infected macrophages. PloS One. (2013) 8:e76581. doi: 10.1371/journal.pone.0076581
93. Thangavelu K, Pan CQ, Karlberg T, Balaji G, Uttamchandani M, Suresh V, et al. Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc Natl Acad Sci USA. (2012) 109:7705–10. doi: 10.1073/pnas.1116573109
94. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. (2013) 496:101–5. doi: 10.1038/nature12040
95. Nakazawa MS, Keith B, and Simon MC. Oxygen availability and metabolic adaptations. Nat Rev Cancer. (2016) 16:663–73. doi: 10.1038/nrc.2016.84
96. Xie H and Simon MC. Oxygen availability and metabolic reprogramming in cancer. J Biol Chem. (2017) 292:16825–32. doi: 10.1074/jbc.R117.799973
97. Soni VK, Mehta A, Ratre YK, Chandra V, Shukla D, Kumar A, et al. Counteracting action of curcumin on high glucose-induced chemoresistance in hepatic carcinoma cells. Front Oncol. (2021) 11:738961. doi: 10.3389/fonc.2021.738961
98. Huang D, Li T, Li X, Zhang L, Sun L, He X, et al. HIF-1-mediated suppression of acyl-coA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. (2014) 8:1930–42. doi: 10.1016/j.celrep.2014.08.028
99. Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, et al. Fatty acid uptake and lipid storage induced by HIF-1α Contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. (2014) 9:349–65. doi: 10.1016/j.celrep.2014.08.056
100. Zhang X, Saarinen AM, Hitosugi T, Wang Z, Wang L, Ho TH, et al. Inhibition of intracellular lipolysis promotes human cancer cell adaptation to hypoxia. eLife. (2017) 6:e31132. doi: 10.7554/eLife.31132
101. Wang X, Fan W, Li N, Ma Y, Yao M, Wang G, et al. YY1 lactylation in microglia promotes angiogenesis through transcription activation-mediated upregulation of FGF2. Genome Biol. (2023) 24:87. doi: 10.1186/s13059-023-02931-y
102. Yu J, Chai P, Xie M, Ge S, Ruan J, Fan X, et al. Histone lactylation drives oncogenesis by facilitating m6A reader protein YTHDF2 expression in ocular melanoma. Genome Biol. (2021) 22:85. doi: 10.1186/s13059-021-02308-z
103. Peitzsch C, Gorodetska I, Klusa D, Shi Q, Alves TC, Pantel K, et al. Metabolic regulation of prostate cancer heterogeneity and plasticity. Semin Cancer Biol. (2022) 82:94–119. doi: 10.1016/j.semcancer.2020.12.002
104. Wang T, Cai B, Ding M, Su Z, Liu Y, and Shen L. c-myc overexpression promotes oral cancer cell proliferation and migration by enhancing glutaminase and glutamine synthetase activity. Am J Med Sci. (2019) 358:235–42. doi: 10.1016/j.amjms.2019.05.014
105. Bott AJ, Peng I-C, Fan Y, Faubert B, Zhao L, Li J, et al. Oncogenic myc induces expression of glutamine synthetase through promoter demethylation. Cell Metab. (2015) 22:1068–77. doi: 10.1016/j.cmet.2015.09.025
106. Chen G, Wang C, Huang S, Yang S, Su Q, Wang Y, et al. Novel roles of ammonia in physiology and cancer. J Mol Cell Biol. (2025) 17:mjaf007. doi: 10.1093/jmcb/mjaf007
107. Li X, Zhu H, Sun W, Yang X, Nie Q, and Fang X. Role of glutamine and its metabolite ammonia in crosstalk of cancer-associated fibroblasts and cancer cells. Cancer Cell Int. (2021) 21:479. doi: 10.1186/s12935-021-02121-5
108. Etchegaray J-P and Mostoslavsky R. Interplay between metabolism and epigenetics: A nuclear adaptation to environmental changes. Mol Cell. (2016) 62:695–711. doi: 10.1016/j.molcel.2016.05.029
109. Carey BW, Finley LWS, Cross JR, Allis CD, and Thompson CB. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. (2015) 518:413–6. doi: 10.1038/nature13981
110. Amores-Sánchez MI and Medina MA. Glutamine, as a precursor of glutathione, and oxidative stress. Mol Genet Metab. (1999) 67:100–5. doi: 10.1006/mgme.1999.2857
111. Han L, Meng L, Liu J, Xie Y, Kang R, Klionsky DJ, et al. Macroautophagy/autophagy promotes resistance to KRASG12D-targeted therapy through glutathione synthesis. Cancer Lett. (2024) 604:217258. doi: 10.1016/j.canlet.2024.217258
112. Zhang J, Cui J, Wang Y, Lin X, Teng X, and Tang Y. Complex molecular mechanism of ammonia-induced apoptosis in chicken peripheral blood lymphocytes: miR-27b-3p, heat shock proteins, immunosuppression, death receptor pathway, and mitochondrial pathway. Ecotoxicology Environ Saf. (2022) 236:113471. doi: 10.1016/j.ecoenv.2022.113471
113. Ye Q, Li D, Zou Y, and Yuan Y. The role and treatment strategies of ammonia-related metabolism inTumor microenvironment. CGT. (2024) 25(3):199–209. doi: 10.2174/0115665232301222240603100840
114. Lim AR, Rathmell WK, and Rathmell JC. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. eLife. (2020) 9:e55185. doi: 10.7554/eLife.55185
115. Delgoffe GM. Filling the tank: keeping antitumor T cells metabolically fit for the long haul. Cancer Immunol Res. (2016) 4:1001–6. doi: 10.1158/2326-6066.CIR-16-0244
116. DePeaux K and Delgoffe GM. Metabolic barriers to cancer immunotherapy. Nat Rev Immunol. (2021) 21:785–97. doi: 10.1038/s41577-021-00541-y
117. Wetzel TJ, Erfan SC, Figueroa LD, Wheeler LM, and Ananieva EA. Crosstalk between arginine, glutamine, and the branched chain amino acid metabolism in the tumor microenvironment, Front. Oncol. (2023) 13:1186539. doi: 10.3389/fonc.2023.1186539
118. Líndez A-AM, Dunand-Sauthier I, Conti M, Gobet F, Núñez N, Hannich JT, et al. Mitochondrial arginase-2 is a cell−autonomous regulator of CD8+ T cell function and antitumor efficacy. JCI Insight. (2019) 4:e132975. doi: 10.1172/jci.insight.132975
119. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. (2016) 167:829–842.e13. doi: 10.1016/j.cell.2016.09.031
120. Poillet-Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu ZS, et al. Autophagy maintains tumour growth through circulating arginine. Nature. (2018) 563:569–73. doi: 10.1038/s41586-018-0697-7
121. Riess C, Shokraie F, Classen CF, Kreikemeyer B, Fiedler T, Junghanss C, et al. Arginine-depleting enzymes – an increasingly recognized treatment strategy for therapy-refractory Malignancies. Cell Physiol Biochem. (2018) 51:854–70. doi: 10.1159/000495382
122. Sinclair LV, Rolf J, Emslie E, Shi Y-B, Taylor PM, and Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. (2013) 14:500–8. doi: 10.1038/ni.2556
123. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct regulation of th17 and th1 cell differentiation by glutaminase-dependent metabolism. Cell. (2018) 175:1780–1795.e19. doi: 10.1016/j.cell.2018.10.001
124. Nakaya M, Xiao Y, Zhou X, Chang J-H, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. (2014) 40:692–705. doi: 10.1016/j.immuni.2014.04.007
125. Chamoto K, Chowdhury PS, Kumar A, Sonomura K, Matsuda F, Fagarasan S, et al. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc Natl Acad Sci U.S.A. (2017) 114(5):E761–70. doi: 10.1073/pnas.1620433114
126. Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
127. Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing CD8+ T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell. (2017) 32:377–391.e9. doi: 10.1016/j.ccell.2017.08.004
128. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. (2015) 6:6692. doi: 10.1038/ncomms7692
129. Chang C-H, Curtis JD, Maggi LB, Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
130. Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor ζ-chain and induce a regulatory phenotype in naive T cells. J Immunol. (2006) 176:6752–61. doi: 10.4049/jimmunol.176.11.6752
131. Matés JM, Di Paola FJ, Campos-Sandoval JA, Mazurek S, and Márquez J. Therapeutic targeting of glutaminolysis as an essential strategy to combat cancer. Semin Cell Dev Biol. (2020) 98:34–43. doi: 10.1016/j.semcdb.2019.05.012
132. Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science. (2002) 297:1867–70. doi: 10.1126/science.1073514
133. Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. (2003) 9:1269–74. doi: 10.1038/nm934
134. Sharma MD, Baban B, Chandler P, Hou D-Y, Singh N, Yagita H, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase, J. Clin Invest. (2007) 117:2570–82. doi: 10.1172/JCI31911
135. Ron-Harel N, Ghergurovich JM, Notarangelo G, LaFleur MW, Tsubosaka Y, Sharpe AH, et al. T cell activation depends on extracellular alanine. Cell Rep. (2019) 28:3011–3021.e4. doi: 10.1016/j.celrep.2019.08.034
136. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
137. Metzler B, Gfeller P, and Guinet E. Restricting glutamine or glutamine-dependent purine and pyrimidine syntheses promotes human T cells with high FOXP3 expression and regulatory properties. J Immunol. (2016) 196:3618–30. doi: 10.4049/jimmunol.1501756
138. Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, et al. Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal. (2015) 8(396):ra97. doi: 10.1126/scisignal.aab2610
139. Xu T, Stewart KM, Wang X, Liu K, Xie M, Ryu JK, et al. Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature. (2017) 548:228–33. doi: 10.1038/nature23475
140. Swamy M, Pathak S, Grzes KM, Damerow S, Sinclair LV, Van Aalten DMF, et al. Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and Malignancy. Nat Immunol. (2016) 17:712–20. doi: 10.1038/ni.3439
141. Araujo L, Khim P, Mkhikian H, Mortales C-L, and Demetriou M. Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. eLife. (2017) 6:e21330. doi: 10.7554/eLife.21330
142. Leone RD, Zhao L, Englert JM, Sun I-M, Oh M-H, Sun I-H, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. (2019) 366:1013–21. doi: 10.1126/science.aav2588
143. Luo C, Shen G, Liu N, Gong F, Wei X, Yao S, et al. Ammonia drives dendritic cells into dysfunction. J Immunol. (2014) 193:1080–9. doi: 10.4049/jimmunol.1303218
144. Norian LA, Rodriguez PC, O’Mara LA, Zabaleta J, Ochoa AC, Cella M, et al. Tumor-infiltrating regulatory dendritic cells inhibit CD8+ T cell function via L-arginine metabolism. Cancer Res. (2009) 69:3086–94. doi: 10.1158/0008-5472.CAN-08-2826
145. Kumar A, Welch N, Mishra S, Bellar A, Silva RN, Li L, et al. Metabolic reprogramming during hyperammonemia targets mitochondrial function and postmitotic senescence. JCI Insight. (2021) 6:e154089. doi: 10.1172/jci.insight.154089
146. Zhang H, Liu J, Yuan W, Zhang Q, Luo X, Li Y, et al. Ammonia-induced lysosomal and mitochondrial damage causes cell death of effector CD8+ T cells. Nat Cell Biol. (2024) 26:1892–902. doi: 10.1038/s41556-024-01503-x
147. Wang S, Wang W, Li X, Zhao X, Wang Y, Zhang H, et al. Cooperative application of transcriptomics and ceRNA hypothesis: LncRNA-107052630/miR-205a/G0S2 crosstalk is involved in ammonia-induced intestinal apoptotic injury in chicken. J Hazardous Materials. (2020) 396:122605. doi: 10.1016/j.jhazmat.2020.122605
148. Lin W, Tsai BC, Day CH, Chiu P, Chen R, Chen MY, et al. Arecoline induces heart injure via Fas/Fas ligand apoptotic pathway in heart of Sprague–Dawley rat. Environ Toxicol. (2021) 36:1567–75. doi: 10.1002/tox.23153
149. Chen D, Yu F, Wu F, Bai M, Lou F, Liao X, et al. The role of Wnt7B in the mediation of dentinogenesis via the ERK1/2 pathway. Arch Oral Biol. (2019) 104:123–32. doi: 10.1016/j.archoralbio.2019.05.009
150. Lee N and Kim W-U. Microbiota in T-cell homeostasis and inflammatory diseases. Exp Mol Med. (2017) 49:e340–0. doi: 10.1038/emm.2017.36
151. Magouz FI, Mahmoud SA, El-Morsy RAA, Paray BA, Soliman AA, Zaineldin AI, et al. Dietary menthol essential oil enhanced the growth performance, digestive enzyme activity, immune-related genes, and resistance against acute ammonia exposure in Nile tilapia (Oreochromis niloticus). Aquaculture. (2021) 530:735944. doi: 10.1016/j.aquaculture.2020.735944
152. Liu Y, Yu M, Cui J, Du Y, Teng X, and Zhang Z. Heat shock proteins took part in oxidative stress-mediated inflammatory injury via NF-κB pathway in excess manganese-treated chicken livers. Ecotoxicology Environ Saf. (2021) 226:112833. doi: 10.1016/j.ecoenv.2021.112833
153. Pizzagalli MD, Bensimon A, and Superti-Furga G. A guide to plasma membrane solute carrier proteins. FEBS J. (2021) 288:2784–835. doi: 10.1111/febs.15531
154. Hediger MA, Romero MF, Peng J-B, Rolfs A, Takanaga H, and Bruford EA. The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteins. Pflugers Archiv Eur J Physiol. (2004) 447:465–8. doi: 10.1007/s00424-003-1192-y
155. Bhutia YD and Ganapathy V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim Biophys Acta. (2016) 1863:2531–9. doi: 10.1016/j.bbamcr.2015.12.017
156. Ganapathy V, Thangaraju M, and Prasad PD. Nutrient transporters in cancer: relevance to Warburg hypothesis and beyond. Pharmacol Ther. (2009) 121:29–40. doi: 10.1016/j.pharmthera.2008.09.005
157. Wang Q and Holst J. L-type amino acid transport and cancer: targeting the mTORC1 pathway to inhibit neoplasia. Am J Cancer Res. (2015) 5(4):1281–94.
158. Bodoy S, Fotiadis D, Stoeger C, Kanai Y, and Palacín M. The small SLC43 family: facilitator system l amino acid transporters and the orphan EEG1. Mol Aspects Med. (2013) 34:638–45. doi: 10.1016/j.mam.2012.12.006
159. Wang Q, Bailey CG, Ng C, Tiffen J, Thoeng A, Minhas V, et al. Androgen receptor and nutrient signaling pathways coordinate the demand for increased amino acid transport during prostate cancer progression. Cancer Res. (2011) 71:7525–36. doi: 10.1158/0008-5472.CAN-11-1821
160. Cha Y, Kim E-S, and Koo J. Amino acid transporters and glutamine metabolism in breast cancer. IJMS. (2018) 19:907. doi: 10.3390/ijms19030907
161. Chen R, Zou Y, Mao D, Sun D, Gao G, Shi J, et al. The general amino acid control pathway regulates mTOR and autophagy during serum/glutamine starvation. J Cell Biol. (2014) 206:173–82. doi: 10.1083/jcb.201403009
162. Owusu-Ansah M, Guptan N, Alindogan D, Morizono M, and Caldovic L. NAGS, CPS1, and SLC25A13 (Citrin) at the crossroads of arginine and pyrimidines metabolism in tumor cells. Int J Mol Sci. (2023) 24:6754. doi: 10.3390/ijms24076754
163. Chen S, Tang Q, Hu M, Song S, Wu X, Zhou Y, et al. Loss of carbamoyl phosphate synthetase 1 potentiates hepatocellular carcinoma metastasis by reducing aspartate level. Adv Sci (Weinh). (2024) 11:e2402703. doi: 10.1002/advs.202402703
164. Zou Y, Guo S, Wen L, Lv D, Tu J, Liao Y, et al. Targeting NAT10 inhibits osteosarcoma progression via ATF4/ASNS-mediated asparagine biosynthesis. Cell Rep Med. (2024) 5:101728. doi: 10.1016/j.xcrm.2024.101728
165. Hushmandi K, Einollahi B, Saadat SH, Lee EHC, Farani MR, Okina E, et al. Amino acid transporters within the solute carrier superfamily: Underappreciated proteins and novel opportunities for cancer therapy. Mol Metab. (2024) 84:101952. doi: 10.1016/j.molmet.2024.101952
166. Shi J, Ma C, Zheng Z, Zhang T, Li Z, Sun X, et al. Low-dose antimony exposure promotes prostate cancer proliferation by inhibiting ferroptosis via activation of the Nrf2-SLC7A11-GPX4 pathway. Chemosphere. (2023) 339:139716. doi: 10.1016/j.chemosphere.2023.139716
167. Sugiura M, Sato H, Okabe A, Fukuyo M, Mano Y, Shinohara K, et al. Identification of AR-V7 downstream genes commonly targeted by AR/AR-V7 and specifically targeted by AR-V7 in castration resistant prostate cancer. Trans Oncol. (2021) 14:100915. doi: 10.1016/j.tranon.2020.100915
168. Xu M, Sakamoto S, Matsushima J, Kimura T, Ueda T, Mizokami A, et al. Up-regulation of LAT1 during antiandrogen therapy contributes to progression in prostate cancer cells. J Urol. (2016) 195:1588–97. doi: 10.1016/j.juro.2015.11.071
169. Franko A, Shao Y, Heni M, Hennenlotter J, Hoene M, Hu C, et al. Human prostate cancer is characterized by an increase in urea cycle metabolites. Cancers (Basel). (2020) 12:1814. doi: 10.3390/cancers12071814
170. Shou Y, Liu R, Xiong H, Xu K, Chen X, Huang L, et al. Urea cycle dysregulation: a new frontier in cancer metabolism and immune evasion. Cell Commun Signal. (2025) 23:307. doi: 10.1186/s12964-025-02328-3
171. Karpisheh V, Fakkari Afjadi J, Nabi Afjadi M, Haeri MS, Abdpoor Sough TS, Heydarzadeh Asl S, et al. Inhibition of HIF-1α/EP4 axis by hyaluronate-trimethyl chitosan-SPION nanoparticles markedly suppresses the growth and development of cancer cells. Int J Biol Macromol. (2021) 167:1006–19. doi: 10.1016/j.ijbiomac.2020.11.056
172. Jadidi-Niaragh F, Atyabi F, Rastegari A, Kheshtchin N, Arab S, Hassannia H, et al. CD73 specific siRNA loaded chitosan lactate nanoparticles potentiate the antitumor effect of a dendritic cell vaccine in 4T1 breast cancer bearing mice. J Controlled Release. (2017) 246:46–59. doi: 10.1016/j.jconrel.2016.12.012
173. Karpisheh V, Nikkhoo A, Hojjat-Farsangi M, Namdar A, Azizi G, Ghalamfarsa G, et al. Prostaglandin E2 as a potent therapeutic target for treatment of colon cancer. Prostaglandins Other Lipid Mediators. (2019) 144:106338. doi: 10.1016/j.prostaglandins.2019.106338
174. Karpisheh V, Joshi N, Zekiy AO, Beyzai B, Hojjat-Farsangi M, Namdar A, et al. EP4 receptor as a novel promising therapeutic target in colon cancer. Pathol - Res Pract. (2020) 216:153247. doi: 10.1016/j.prp.2020.153247
175. Yazdani Y, Mohammadnia-Afrouzi M, Yousefi M, Anvari E, Ghalamfarsa G, Hasannia H, et al. Myeloid-derived suppressor cells in B cell Malignancies. Tumour Biol. (2015) 36:7339–53. doi: 10.1007/s13277-015-4004-z
176. Novysedlak R, Guney M, Al Khouri M, Bartolini R, Koumbas Foley L, Benesova I, et al. The immune microenvironment in prostate cancer: A comprehensive review. Oncology. (2024) 103(6):521–45. doi: 10.1159/000541881
177. Dai J, Lu Y, Roca H, Keller JM, Zhang J, McCauley LK, et al. Immune mediators in the tumor microenvironment of prostate cancer. Chin J Cancer. (2017) 36:29. doi: 10.1186/s40880-017-0198-3
178. Sung J-Y and Cheong J-H. New immunometabolic strategy based on cell type-specific metabolic reprogramming in the tumor immune microenvironment. Cells. (2022) 11:768. doi: 10.3390/cells11050768
179. Boufaied N, Chetta P, Hallal T, Cacciatore S, Lalli D, Luthold C, et al. Obesogenic high-fat diet and MYC cooperate to promote lactate accumulation and tumor microenvironment remodeling in prostate cancer. Cancer Res. (2024) 84:1834–55. doi: 10.1158/0008-5472.CAN-23-0519
180. Long Y, Gao Z, Hu X, Xiang F, Wu Z, Zhang J, et al. Downregulation of MCT 4 for lactate exchange promotes the cytotoxicity of NK cells in breast carcinoma. Cancer Med. (2018) 7:4690–700. doi: 10.1002/cam4.1713
181. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. (2019) 574:575–80. doi: 10.1038/s41586-019-1678-1
182. Feng J, Yang H, Zhang Y, Wei H, Zhu Z, Zhu B, et al. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene. (2017) 36:5829–39. doi: 10.1038/onc.2017.188
183. Lee YJ, Shin KJ, Park S-A, Park KS, Park S, Heo K, et al. G-protein-coupled receptor 81 promotes a Malignant phenotype in breast cancer through angiogenic factor secretion. Oncotarget. (2016) 7:70898–911. doi: 10.18632/oncotarget.12286
184. Gannon PO, Godin-Ethier J, Hassler M, Delvoye N, Aversa M, Poisson AO, et al. Androgen-regulated expression of arginase 1, arginase 2 and interleukin-8 in human prostate cancer. PloS One. (2010) 5:e12107. doi: 10.1371/journal.pone.0012107
185. Zhang P, McGrath BC, Reinert J, Olsen DS, Lei L, Gill S, et al. The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol. (2002) 22:6681–8. doi: 10.1128/MCB.22.19.6681-6688.2002
186. Lee J, Ryu H, Ferrante RJ, Morris SM, and Ratan RR. Translational control of inducible nitric oxide synthase expression by arginine can explain the arginine paradox. Proc Natl Acad Sci U.S.A. (2003) 100:4843–8. doi: 10.1073/pnas.0735876100
187. El-Gayar S, Thüring-Nahler H, Pfeilschifter J, Röllinghoff M, and Bogdan C. Translational control of inducible nitric oxide synthase by IL-13 and arginine availability in inflammatory macrophages. J Immunol. (2003) 171:4561–8. doi: 10.4049/jimmunol.171.9.4561
188. Bingisser RM, Tilbrook PA, Holt PG, and Kees UR. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J Immunol. (1998) 160:5729–34. doi: 10.4049/jimmunol.160.12.5729
189. Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. (2002) 168:689–95. doi: 10.4049/jimmunol.168.2.689
190. Fischer TA, Palmetshofer A, Gambaryan S, Butt E, Jassoy C, Walter U, et al. Activation of cGMP-dependent protein kinase Ibeta inhibits interleukin 2 release and proliferation of T cell receptor-stimulated human peripheral T cells. J Biol Chem. (2001) 276:5967–74. doi: 10.1074/jbc.M009781200
191. Macphail SE, Gibney CA, Brooks BM, Booth CG, Flanagan BF, and Coleman JW. Nitric oxide regulation of human peripheral blood mononuclear cells: critical time dependence and selectivity for cytokine versus chemokine expression. J Immunol. (2003) 171:4809–15. doi: 10.4049/jimmunol.171.9.4809
192. Bronte V and Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. (2005) 5:641–54. doi: 10.1038/nri1668
193. Munder M. Arginase: an emerging key player in the mammalian immune system. Br J Pharmacol. (2009) 158:638–51. doi: 10.1111/j.1476-5381.2009.00291.x
194. Hönscheid PV, Baretton GB, Puhr M, Siciliano T, Israel JS, Stope MB, et al. Prostate cancer’s silent partners: fibroblasts and their influence on glutamine metabolism manipulation. IJMS. (2024) 25:9275. doi: 10.3390/ijms25179275
195. Bedeschi M, Marino N, Cavassi E, Piccinini F, and Tesei A. Cancer-associated fibroblast: role in prostate cancer progression to metastatic disease and therapeutic resistance. Cells. (2023) 12:802. doi: 10.3390/cells12050802
196. Yang L, Achreja A, Yeung T-L, Mangala LS, Jiang D, Han C, et al. Targeting stromal glutamine synthetase in tumors disrupts tumor microenvironment-regulated cancer cell growth. Cell Metab. (2016) 24:685–700. doi: 10.1016/j.cmet.2016.10.011
197. Guo JY and White E. Autophagy, metabolism, and cancer. Cold Spring Harb Symp Quant Biol. (2016) 81:73–8. doi: 10.1101/sqb.2016.81.030981
198. Menon S, Yecies JL, Zhang HH, Howell JJ, Nicholatos J, Harputlugil E, et al. Chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Sci Signal. (2012) 5(217):ra24. doi: 10.1126/scisignal.2002739
199. Chiacchiera F and Simone C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle. (2010) 9:1091–6. doi: 10.4161/cc.9.6.11035
200. Kim J, Hu Z, Cai L, Li K, Choi E, Faubert B, et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature. (2017) 546:168–72. doi: 10.1038/nature22359
201. Niknahad H, Jamshidzadeh A, Heidari R, Zarei M, and Ommati MM. Ammonia-induced mitochondrial dysfunction and energy metabolism disturbances in isolated brain and liver mitochondria, and the effect of taurine administration: relevance to hepatic encephalopathy treatment. Ceh. (2017) 3:141–51. doi: 10.5114/ceh.2017.68833
202. Zhang J, Wang E, Zhang L, and Zhou B. PSPH induces cell autophagy and promotes cell proliferation and invasion in the hepatocellular carcinoma cell line Huh7 via the AMPK/mTOR/ULK1 signaling pathway. Cell Biol Int. (2021) 45:305–19. doi: 10.1002/cbin.11489
203. Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, and Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. (2020) 66:89–100. doi: 10.1016/j.semcancer.2019.03.002
204. Lu J, Yang J, Zheng Y, Chen X, and Fang S. Extracellular vesicles from endothelial progenitor cells prevent steroid-induced osteoporosis by suppressing the ferroptotic pathway in mouse osteoblasts based on bioinformatics evidence. Sci Rep. (2019) 9:16130. doi: 10.1038/s41598-019-52513-x
205. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. (2014) 3:e02523. doi: 10.7554/eLife.02523
206. Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. (2007) 447:864–8. doi: 10.1038/nature05859
207. Twum-Ampofo J, Fu D-X, Passaniti A, Hussain A, and Siddiqui MM. Metabolic targets for potential prostate cancer therapeutics. Curr Opin Oncol. (2016) 28:241–7. doi: 10.1097/CCO.0000000000000276
208. Xu X, Meng Y, Li L, Xu P, Wang J, Li Z, et al. Overview of the development of glutaminase inhibitors: achievements and future directions. J Med Chem. (2019) 62:1096–115. doi: 10.1021/acs.jmedchem.8b00961
209. Curthoys NP and Watford M. Regulation of glutaminase activity and glutamine metabolism. Annu Rev Nutr. (1995) 15:133–59. doi: 10.1146/annurev.nu.15.070195.001025
210. Hensley CT, Wasti AT, and DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. (2013) 123:3678–84. doi: 10.1172/JCI69600
211. Yang W-H, Qiu Y, Stamatatos O, Janowitz T, and Lukey MJ. Enhancing the efficacy of glutamine metabolism inhibitors in cancer therapy. Trends Cancer. (2021) 7:790–804. doi: 10.1016/j.trecan.2021.04.003
212. Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA. (2010) 107:7461–6. doi: 10.1073/pnas.1002459107
213. Wilder CS, Chen Z, and DiGiovanni J. Pharmacologic approaches to amino acid depletion for cancer therapy. Mol Carcinogenesis. (2022) 61:127–52. doi: 10.1002/mc.23349
214. Liang B, Cui S, and Zou S. Leonurine Suppresses Prostate Cancer Growth in vitro and in vivo by Regulating miR-18a-5p/SLC40A1 Axis. Chin J Physiol. (2022) 65:319–27. doi: 10.4103/0304-4920.365459
215. Saxby H, Mikropoulos C, and Boussios S. An update on the prognostic and predictive serum biomarkers in metastatic prostate cancer. Diagnostics. (2020) 10:549. doi: 10.3390/diagnostics10080549
216. Sato H, Narita S, Tsuchiya N, Koizumi A, Nara T, Kanda S, et al. Impact of early changes in serum biomarkers following androgen deprivation therapy on clinical outcomes in metastatic hormone-sensitive prostate cancer. BMC Urol. (2018) 18:32. doi: 10.1186/s12894-018-0353-4
217. Thoms JW, Dal Pra A, Anborgh PH, Christensen E, Fleshner N, Menard C, et al. Plasma osteopontin as a biomarker of prostate cancer aggression: relationship to risk category and treatment response. Br J Cancer. (2012) 107:840–6. doi: 10.1038/bjc.2012.345
218. Litwin MS and Tan H-J. The diagnosis and treatment of prostate cancer: A review. JAMA. (2017) 317:2532. doi: 10.1001/jama.2017.7248
219. Yu C, Niu L, Li L, Li T, Duan L, He Z, et al. Identification of the metabolic signatures of prostate cancer by mass spectrometry-based plasma and urine metabolomics analysis. Prostate. (2021) 81:1320–8. doi: 10.1002/pros.24229
220. Clos-Garcia M, Loizaga-Iriarte A, Zuñiga-Garcia P, Sánchez-Mosquera P, Rosa Cortazar A, González E, et al. Metabolic alterations in urine extracellular vesicles are associated to prostate cancer pathogenesis and progression. J Extracellular Vesicle. (2018) 7:1470442. doi: 10.1080/20013078.2018.1470442
221. Lemberg KM, Gori SS, Tsukamoto T, Rais R, and Slusher BS. Clinical development of metabolic inhibitors for oncology. J Clin Invest. (2022) 132:e148550. doi: 10.1172/JCI148550
222. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. (2014) 13:890–901. doi: 10.1158/1535-7163.MCT-13-0870
223. Erb HHH, Polishchuk N, Stasyk O, Kahya U, Weigel MM, and Dubrovska A. Glutamine metabolism and prostate cancer. Cancers. (2024) 16:2871. doi: 10.3390/cancers16162871
224. Lee ACK, Lau PM, Kwan YW, and Kong SK. Mitochondrial fuel dependence on glutamine drives chemo-resistance in the cancer stem cells of hepatocellular carcinoma. IJMS. (2021) 22:3315. doi: 10.3390/ijms22073315
225. Vasiliou V, Vasiliou K, and Nebert DW. Human ATP-binding cassette (ABC) transporter family. Hum Genomics. (2008) 3:281. doi: 10.1186/1479-7364-3-3-281
226. Zhu Y, Liu C, Nadiminty N, Lou W, Tummala R, Evans CP, et al. Inhibition of ABCB1 expression overcomes acquired docetaxel resistance in prostate cancer. Mol Cancer Ther. (2013) 12:1829–36. doi: 10.1158/1535-7163.MCT-13-0208
227. Praharaj M, Shen F, Lee AJ, Zhao L, Nirschl TR, Theodros D, et al. Metabolic reprogramming of tumor-associated macrophages using glutamine antagonist JHU083 drives tumor immunity in myeloid-rich prostate and bladder cancers. Cancer Immunol Res. (2024) 12:854–75. doi: 10.1158/2326-6066.CIR-23-1105
228. Rais R, Lemberg KM, Tenora L, Arwood ML, Pal A, Alt J, et al. Discovery of DRP-104, a tumor-targeted metabolic inhibitor prodrug. Sci Adv. (2022) 8:eabq5925. doi: 10.1126/sciadv.abq5925
229. Cojoc M, Peitzsch C, Kurth I, Trautmann F, Kunz-Schughart LA, Telegeev GD, et al. Aldehyde dehydrogenase is regulated by β-catenin/TCF and promotes radioresistance in prostate cancer progenitor cells. Cancer Res. (2015) 75:1482–94. doi: 10.1158/0008-5472.CAN-14-1924
230. Gorodetska I, Offermann A, Püschel J, Lukiyanchuk V, Gaete D, Kurzyukova A, et al. ALDH1A1 drives prostate cancer metastases and radioresistance by interplay with AR- and RAR-dependent transcription. Theranostics. (2024) 14:714–37. doi: 10.7150/thno.88057
231. Püschel J, Dubrovska A, and Gorodetska I. The multifaceted role of aldehyde dehydrogenases in prostate cancer stem cells. Cancers. (2021) 13:4703. doi: 10.3390/cancers13184703
232. Chetta P and Zadra G. Metabolic reprogramming as an emerging mechanism of resistance to endocrine therapies in prostate cancer. CDR. (2021) 4(1):143–62. doi: 10.20517/cdr.2020.54
233. Wang Q, Tiffen J, Bailey CG, Lehman ML, Ritchie W, Fazli L, et al. Targeting amino acid transport in metastatic castration-resistant prostate cancer: effects on cell cycle, cell growth, and tumor development. JNCI: J Natl Cancer Institute. (2013) 105:1463–73. doi: 10.1093/jnci/djt241
234. Fidelito G, De Souza DP, Niranjan B, De Nardo W, Keerthikumar S, Brown K, et al. Multi-substrate metabolic tracing reveals marked heterogeneity and dependency on fatty acid metabolism in human prostate cancer. Mol Cancer Res. (2023) 21:359–73. doi: 10.1158/1541-7786.MCR-22-0796
235. Fidelito G, Watt MJ, and Taylor RA. Personalized medicine for prostate cancer: is targeting metabolism a reality? Front Oncol. (2021) 11:778761. doi: 10.3389/fonc.2021.778761
236. Kahya U, Köseer AS, and Dubrovska A. Amino acid transporters on the guard of cell genome and epigenome. Cancers. (2021) 13:125. doi: 10.3390/cancers13010125
237. Cardoso HJ, Figueira MI, Vaz CV, Carvalho TMA, Brás LA, Madureira PA, et al. Glutaminolysis is a metabolic route essential for survival and growth of prostate cancer cells and a target of 5α-dihydrotestosterone regulation. Cell Oncol. (2021) 44(2):385–403. doi: 10.1007/s13402-020-00575-9
238. Lemberg KM, Vornov JJ, Rais R, and Slusher BS. We’re not “DON” Yet: optimal dosing and prodrug delivery of 6-diazo-5-oxo-L-norleucine. Mol Cancer Ther. (2018) 17:1824–32. doi: 10.1158/1535-7163.MCT-17-1148
239. Magill GB, Myers WPL, Reilly HC, Putnam RC, Magill JW, Sykes MP, et al. Pharmacological and initial therapeutic observations on 6-Diazo-5-Oxo-L-Norleucine (Don) in human neoplastic disease. Cancer. (1957) 10:1138–50. doi: 10.1002/1097-0142(195711/12)10:6%253C1138::AID-CNCR2820100608%253E3.0.CO;2-K
240. Krantz S, Rivers S, Dwight RW, Corbus HF, Wolf J, Green I, Spear PW, et al. A CLINICAL study of the comparative effect of nitrogen mustard and DON in patients with bronchogenic carcinoma, Hodgkin’s disease, lymphosarcoma, and melanoma. J Natl Cancer Inst. (1959) 22:433–9. doi: 10.1093/jnci/22.2.433
241. Li MC. Effects of combined drug therapy on metastatic cancer of the testis. JAMA. (1960) 174:1291. doi: 10.1001/jama.1960.03030100059013
242. Sullivan MP, Beatty EC, Hyman CB, Murphy ML, Pierce MI, and Severo NC. A comparison of the effectiveness of standard dose 6-mercaptopurine, combination 6-mercaptopurine and DON, and high-loading 6-mercaptopurine therapies in treatment of the acute leukemias of childhood: results of a coperative study. Cancer Chemother Rep. (1962) 18:83–95.
243. Sullivan MP, Nelson JA, Feldman S, and Van Nguyen B. Pharmacokinetic and phase I study of intravenous DON (6-diazo-5-oxo-L-norleucine) in children, Cancer Chemother. Pharmacol. (1988) 21:78–84. doi: 10.1007/BF00262746
244. Earhart RH, Koeller JM, and Davis HL. Phase I trial of 6-diazo-5-oxo-L-norleucine (DON) administered by 5-day courses. Cancer Treat Rep. (1982) 66:1215–7.
245. Sklaroff RB, Casper ES, Magill GB, and Young CW. Phase I study of 6-diazo-5-oxo-L-norleucine (DON). Cancer Treat Rep. (1980) 64:1247–51.
246. Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK, et al. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem J. (2007) 406:407–14. doi: 10.1042/BJ20070039
247. Hartwick EW and Curthoys NP. BPTES inhibition of hGA(124-551), a truncated form of human kidney-type glutaminase. J Enzyme Inhib Med Chem. (2012) 27:861–7. doi: 10.3109/14756366.2011.622272
248. Cassago A, Ferreira APS, Ferreira IM, Fornezari C, Gomes ERM, Greene KS, et al. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism, Proc. Natl Acad Sci USA. (2012) 109:1092–7. doi: 10.1073/pnas.1112495109
249. DeLaBarre B, Gross S, Fang C, Gao Y, Jha A, Jiang F, et al. Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry. (2011) 50:10764–70. doi: 10.1021/bi201613d
250. Méndez-Lucas A, Lin W, Driscoll PC, Legrave N, Novellasdemunt L, Xie C, et al. Identifying strategies to target the metabolic flexibility of tumours. Nat Metab. (2020) 2:335–50. doi: 10.1038/s42255-020-0195-8
251. Biancur DE, Paulo JA, Małachowska B, Quiles Del Rey M, Sousa CM, Wang X, et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun. (2017) 8:15965. doi: 10.1038/ncomms15965
252. Reis LMD, Adamoski D, Ornitz Oliveira Souza R, Rodrigues Ascenção CF, Sousa De Oliveira KR, Corrêa-da-Silva F, et al. Dual inhibition of glutaminase and carnitine palmitoyltransferase decreases growth and migration of glutaminase inhibition–resistant triple-negative breast cancer cells. J Biol Chem. (2019) 294:9342–57. doi: 10.1074/jbc.RA119.008180
253. Momcilovic M, Bailey ST, Lee JT, Fishbein MC, Braas D, Go J, et al. The GSK3 signaling axis regulates adaptive glutamine metabolism in lung squamous cell carcinoma. Cancer Cell. (2018) 33:905–921.e5. doi: 10.1016/j.ccell.2018.04.002
254. Tanaka K, Sasayama T, Irino Y, Takata K, Nagashima H, Satoh N, et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J Clin Invest. (2015) 125:1591–602. doi: 10.1172/JCI78239
255. Deberardinis RJ. Is cancer a disease of abnormal cellular metabolism? New angles on an old idea. Genet Med. (2008) 10:767–77. doi: 10.1097/GIM.0b013e31818b0d9b
256. Parsons DW, Jones S, Zhang X, Lin JC-H, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. (2008) 321:1807–12. doi: 10.1126/science.1164382
257. Pertega-Gomes N, Felisbino S, Massie CE, Vizcaino JR, Coelho R, Sandi C, et al. A glycolytic phenotype is associated with prostate cancer progression and aggressiveness: a role for monocarboxylate transporters as metabolic targets for therapy. J Pathol. (2015) 236:517–30. doi: 10.1002/path.4547
258. Eidelman E, Twum-Ampofo J, Ansari J, and Siddiqui MM. The metabolic phenotype of prostate cancer. Front Oncol. (2017) 7:131. doi: 10.3389/fonc.2017.00131
259. Cutruzzolà F, Giardina G, Marani M, Macone A, Paiardini A, Rinaldo S, et al. Glucose metabolism in the progression of prostate cancer. Front Physiol. (2017) 8:97. doi: 10.3389/fphys.2017.00097
260. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
261. Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. (2015) 125:3335–7. doi: 10.1172/JCI83871
262. Ratnakumari L and Murthy CHRK. Response of rat cerebral glycolytic enzymes to hyperammonemic states. Neurosci Lett. (1993) 161:37–40. doi: 10.1016/0304-3940(93)90134-7
263. Cooper AJ and Plum F. Biochemistry and physiology of brain ammonia. Physiol Rev. (1987) 67:440–519. doi: 10.1152/physrev.1987.67.2.440
264. Li Q, Zhong X, Yao W, Yu J, Wang C, Li Z, et al. Inhibitor of glutamine metabolism V9302 promotes ROS-induced autophagic degradation of B7H3 to enhance antitumor immunity. J Biol Chem. (2022) 298:101753. doi: 10.1016/j.jbc.2022.101753
265. Erlandsson A, Carlsson J, Lundholm M, Fält A, Andersson S, Andrén O, et al. M2 macrophages and regulatory T cells in lethal prostate cancer. Prostate. (2019) 79:363–9. doi: 10.1002/pros.23742
266. Hope HC and Salmond RJ. The role of non-essential amino acids in T cell function and anti-tumour immunity, arch. Immunol Ther Exp. (2021) 69:29. doi: 10.1007/s00005-021-00633-6
267. Yan C, Yang J, Saleh N, Chen S-C, Ayers GD, Abramson VG, et al. Inhibition of the PI3K/mTOR pathway in breast cancer to enhance response to immune checkpoint inhibitors in breast cancer. IJMS. (2021) 22:5207. doi: 10.3390/ijms22105207
268. Martinez-Outschoorn UE, Lisanti MP, and Sotgia F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol. (2014) 25:47–60. doi: 10.1016/j.semcancer.2014.01.005
269. Jardim-Perassi BV, Irrera P, Oluwatola OE, Abrahams D, Estrella VC, Ordway B, et al. L-DOS47 elevates pancreatic cancer tumor pH and enhances response to immunotherapy. Biomedicines. (2024) 12:461. doi: 10.3390/biomedicines12020461
270. Shih J-W, Wang L-Y, Hung C-L, Kung H-J, and Hsieh C-L. Non-coding RNAs in castration-resistant prostate cancer: regulation of androgen receptor signaling and cancer metabolism. IJMS. (2015) 16:28943–78. doi: 10.3390/ijms161226138
271. Lloyd SM, Arnold J, and Sreekumar A. Metabolomic profiling of hormone-dependent cancers: a bird’s eye view. Trends Endocrinol Metab. (2015) 26:477–85. doi: 10.1016/j.tem.2015.07.001
272. Xu L, Yin Y, Li Y, Chen X, Chang Y, Zhang H, et al. A glutaminase isoform switch drives therapeutic resistance and disease progression of prostate cancer. Proc Natl Acad Sci USA. (2021) 118:e2012748118. doi: 10.1073/pnas.2012748118
273. Sun J, Bok RA, DeLos Santos J, Upadhyay D, DeLos Santos R, Agarwal S, et al. Resistance to androgen deprivation leads to altered metabolism in human and murine prostate cancer cell and tumor models. Metabolites. (2021) 11:139. doi: 10.3390/metabo11030139
274. Kaushik AK, Vareed SK, Basu S, Putluri V, Putluri N, Panzitt K, et al. Metabolomic profiling identifies biochemical pathways associated with castration-resistant prostate cancer. J Proteome Res. (2014) 13:1088–100. doi: 10.1021/pr401106h
275. Mukha A, Kahya U, and Dubrovska A. Targeting glutamine metabolism and autophagy: the combination for prostate cancer radiosensitization. Autophagy. (2021) 17:3879–81. doi: 10.1080/15548627.2021.1962682
276. Mitsuzuka K and Arai Y. Metabolic changes in patients with prostate cancer during androgen deprivation therapy. Int J Urol. (2018) 25:45–53. doi: 10.1111/iju.13473
277. Retter A, Gong F, Syer T, Singh S, Adeleke S, and Punwani S. Emerging methods for prostate cancer imaging: evaluating cancer structure and metabolic alterations more clearly. Mol Oncol. (2021) 15:2565–79. doi: 10.1002/1878-0261.13071
278. Turkbey B, Pinto PA, Mani H, Bernardo M, Pang Y, McKinney YL, et al. Prostate cancer: value of multiparametric MR imaging at 3 T for detection—Histopathologifc correlation. Radiology. (2010) 255:89–99. doi: 10.1148/radiol.09090475
279. García-Figueiras R, Baleato-González S, Padhani AR, Luna-Alcalá A, Vallejo-Casas JA, Sala E, et al. How clinical imaging can assess cancer biology. Insights Imaging. (2019) 10:28. doi: 10.1186/s13244-019-0703-0
280. Kung B, Yong T, and Tong C. The pearl of FDG PET/CT in preoperative assessment of patients with potentially operable non-small-cell lung cancer and its clinical impact. World J Nucl Med. (2017) 16:21–5. doi: 10.4103/1450-1147.176882
281. De Wever W, Ceyssens S, Mortelmans L, Stroobants S, Marchal G, Bogaert J, et al. Additional value of PET-CT in the staging of lung cancer: comparison with CT alone, PET alone and visual correlation of PET and CT. Eur Radiol. (2007) 17:23–32. doi: 10.1007/s00330-006-0284-4
282. Im H-J, Pak K, Cheon GJ, Kang KW, Kim S-J, Kim I-J, et al. Prognostic value of volumetric parameters of 18F-FDG PET in non-small-cell lung cancer: a meta-analysis. Eur J Nucl Med Mol Imaging. (2015) 42:241–51. doi: 10.1007/s00259-014-2903-7
283. Wang ZJ, Ohliger MA, Larson PEZ, Gordon JW, Bok RA, Slater J, et al. Hyperpolarized13 C MRI: state of the art and future directions. Radiology. (2019) 291:273–84. doi: 10.1148/radiol.2019182391
284. Al-Daffaie FM, Al-Mudhafar SF, Alhomsi A, Tarazi H, Almehdi AM, El-Huneidi W, et al. Metabolomics and proteomics in prostate cancer research: overview, analytical techniques, data analysis, and recent clinical applications. Int J Mol Sci. (2024) 25:5071. doi: 10.3390/ijms25105071
285. Merhi A, Delrée P, and Marini AM. The metabolic waste ammonium regulates mTORC2 and mTORC1 signaling. Sci Rep. (2017) 7:44602. doi: 10.1038/srep44602
286. Elaimy AL, El-Derany MO, James J, Wang Z, Pearson AN, Holcomb EA, et al. SLC4A11 mediates ammonia import and promotes cancer stemness in hepatocellular carcinoma. JCI Insight. (2024) 9:e184826. doi: 10.1172/jci.insight.184826
287. Domagala J, Grzywa TM, Baranowska I, Justyniarska M, Tannir R, Graczyk-Jarzynka A, et al. Ammonia suppresses the antitumor activity of natural killer cells and T cells by decreasing mature perforin. Cancer Res. (2025) 85:2448–67. doi: 10.1158/0008-5472.CAN-24-0749
288. Won JH, Parekattil SJ, Davidson SD, Luddy JS, Choudhury MS, Mallouh C, et al. Ammonium-chloride-induced prostatic hypertrophy in vitro: urinary ammonia as a potential risk factor for benign prostatic hyperplasia. Urological Res. (1999) 27:376–81. doi: 10.1007/s002400050166
289. Moreno-Sánchez R, Marín-Hernández Á., Gallardo-Pérez JC, Pacheco-Velázquez SC, Robledo-Cadena DX, Padilla-Flores JA, et al. Physiological role of glutamate dehydrogenase in cancer cells, front. Oncol. (2020) 10:429. doi: 10.3389/fonc.2020.00429
290. A phase II Study of the Glutaminase Inhibitor Telaglenastat (CB-839) in Combination With the PARP Inhibitor Talazoparib in Participants With Metastatic Castration-Resistant Prostate Cancer (2021). Available online at: https://clinicaltrials.gov/study/NCT04824937 (Accessed July 1, 2021).
291. Ph1 Study of the Safety, PK, and PDn of Escalating Oral Doses of the Glutaminase Inhibitor CB-839, as a Single Agent and in Combination With Standard Chemotherapy in Patients With Advanced and/or Treatment-Refractory Solid Tumors (2014). Available online at: https://clinicaltrials.gov/study/NCT02071862 (Accessed February 1, 2014).
292. A Phase 1b/2, Open Label, Dose Escalation and Expansion Study of the Glutaminase Inhibitor Telaglenastat (CB-839) in Combination With CDK4/6 Inhibitor Palbociclib in Patients With Advanced or Metastatic Solid Tumors (2019). Available online at: https://clinicaltrials.gov/study/NCT03965845 (Accessed June 25, 2019).
293. A Phase 1b/2 Open Label, Dose Escalation and Expansion Study of the Glutaminase Inhibitor CB-839 in Combination With the PARP Inhibitor Talazoparib in Patients With Advanced or Metastatic Solid Tumors (2019). Available online at: https://clinicaltrials.gov/study/NCT03875313 (Accessed May 20, 2019).
294. Phase 1 and Phase 2a, First-in-human Study of DRP-104, a Glutamine Antagonist, in Adult Patients With Advanced Solid Tumors (2020). Available online at: https://clinicaltrials.gov/study/NCT04471415 (Accessed August 31, 2020).
295. Long Y, Qiu J, Zhang B, He P, Shi X, He Q, et al. Pharmacological vitamin C treatment impedes the growth of endogenous glutamine-dependent cancers by targeting glutamine synthetase. Front Pharmacol. (2021) 12:671902. doi: 10.3389/fphar.2021.671902
Keywords: ammonium metabolism, prostate cancer, ADT, TME, SLC
Citation: Ye Z, Wu H, Li Z, Ye R, Rao Y, Liu B and Gao B (2025) Ammonium metabolism rewiring in the prostate cancer microenvironment: Mechanisms and clinical prospects. Front. Oncol. 15:1673513. doi: 10.3389/fonc.2025.1673513
Received: 26 July 2025; Accepted: 02 October 2025;
Published: 16 October 2025.
Edited by:
Rafael Moreno-Sánchez, National Autonomous University of Mexico, MexicoReviewed by:
Sara Rodriguez-Enriquez, Facultad de Estudios Superiores Iztacala, MexicoGio Fidelito, University of Melbourne, Australia
Farida Usman, Bayero University Faculty of Basic Medical Sciences, Nigeria
Copyright © 2025 Ye, Wu, Li, Ye, Rao, Liu and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baoshan Gao, Z2FvYnNAamx1LmVkdS5jbg==