 Leon Strzadala
Leon Strzadala- Hirszfeld Institute of Immunology and Experimental Therapy, Polish Academy of Sciences, Wroclaw, Poland
Resistance to programmed cell death is a defining hallmark of cancer and a persistent barrier to successful therapy. Dual-function proteins such as p53, Ras, HIF-1α, BNIP3, and NF-κB act as molecular switches that determine cell fate between apoptosis and survival. In tumors, these proteins are deregulated not only by intrinsic mutations but also by extrinsic signals from the tumor microenvironment (TME). This Mini Review critically analyzes previous therapeutic approaches, emphasizing overlooked mechanisms such as Ras-mediated suppression of p53. It proposes a sequential therapeutic strategy: first, dismantling TME adaptations (hypoxia, inflammation, protective autophagy); second, inhibiting oncogenic Ras signaling; and third, restoring p53 activity. The phased approach integrates biomarker-guided patient stratification, recognizes tumor–microenvironment co-evolution, and highlights how resistance evolves over time. Although the concept does not resolve all challenges, it outlines a rational framework for restoring apoptotic competence and provides a pathway for translational and clinical testing.
1 Introduction
Avoidance of programmed cell death is a hallmark of cancer and a fundamental challenge in oncology (1). This capability allows malignant cells to persist despite genomic instability, oncogenic signaling, and cytotoxic treatments. Crucially, apoptotic resistance emerges not only from cell-intrinsic genetic alterations but also from tumor microenvironment (TME) cues that collectively buffer pro-death signals (2). Hypoxia stabilizes hypoxia-inducible factor 1α (HIF-1α), promoting glycolysis, angiogenesis, and survival under metabolic stress (3). Inflammatory cytokines produced by stromal and immune cells chronically activate NF-κB, reinforcing survival, angiogenesis, and immune evasion (4). BNIP3, a BH3-only protein with pro-apoptotic potential, is frequently rewired to promote protective mitophagy and macroautophagy in stressed tumor cells (5). Finally, oncogenic Ras programs resistance by repressing p53 through multiple layers of signaling crosstalk, thereby raising the threshold for apoptosis (6). These mechanisms do not operate in isolation; rather, they co-evolve as transformed cells interact with and remodel their microenvironment, generating resilient ecological niches that adapt under therapeutic pressure (7). Addressing apoptosis resistance therefore requires a systems-level approach that integrates both intracellular wiring and microenvironmental context.
2 Dual-function proteins as molecular switches
2.1 p53
p53 coordinates genome-protective programs encompassing DNA damage repair, cell-cycle arrest, senescence, and apoptosis (8). TP53 represents one of the most frequently altered genes across cancers; missense, nonsense, and frameshift mutations can abolish transcriptional activity or confer dominant-negative and gain-of-function effects (9). Importantly, even when TP53 remains wild-type, p53 function is often suppressed in tumors. Oncogenic Ras activates PI3K/Akt, which stimulates the E3 ligase MDM2 to ubiquitinate and degrade p53, while MAPK/ERK signaling attenuates p53 transcriptional output (6). Ras-driven redox remodeling further elevates apoptotic thresholds: enhanced reactive oxygen species (ROS) generation elicits antioxidant responses that blunt p53-mediated death programs and favor survival under stress (10). In parallel, epigenetic mechanisms—histone acetylation/methylation dynamics and DNA methylation at p53 target promoters—can silence apoptotic effectors downstream of p53 (11). These multilayered constraints provide a mechanistic rationale for the limited efficacy of p53 reactivators such as APR-246 when deployed in isolation, before upstream Ras/TME repression is relieved (12). In a sequential paradigm, restoring p53 becomes effective only after dismantling microenvironmental buffering and inhibiting Ras-mediated suppression.
2.2 Ras
Ras GTPases integrate signals from receptor tyrosine kinases to drive proliferation, survival, and metabolic remodeling (13). Oncogenic mutations (e.g., KRAS G12C) lock the protein in an active, GTP-bound state, constitutively engaging PI3K/Akt and RAF/MEK/ERK cascades and thereby promoting angiogenesis, glycolysis, and resistance to apoptosis (14). Ras intersects with the p53 axis through several routes: activation of MDM2-mediated degradation, ERK-dependent repression of p53 transcriptional programs, redox adaptation that buffers p53-induced oxidative stress, and chromatin-level silencing of p53-responsive loci (6, 10, 11). Early attempts to inhibit Ras through farnesyltransferase blockade failed because KRAS and NRAS can undergo alternative prenylation, preserving membrane localization and signaling. By contrast, covalent inhibitors of KRAS G12C (e.g., sotorasib, adagrasib) have produced meaningful clinical responses in non–small cell lung cancer, validating the tractability of direct Ras inhibition and providing a critical lever for sequential strategies (14, 15).
2.3 HIF-1α
HIF-1α is a master regulator of the hypoxic response. Under normoxia, prolyl hydroxylases mark HIF-1α for von Hippel–Lindau (VHL)-mediated proteasomal degradation. Hypoxia inhibits hydroxylation, stabilizing HIF-1α and enabling transcription of angiogenic (e.g., VEGF) and glycolytic genes that sustain survival under low oxygen (3). Pharmacologic targeting of oxygen-sensing pathways has been clinically validated by HIF-2α inhibition with belzutifan in VHL-associated tumors (16, 17). Nevertheless, broader deployment of HIF-axis inhibitors in common solid tumors will likely require integration with complementary agents that relieve redundant survival circuits or sensitize tumors to apoptosis (Table 1).
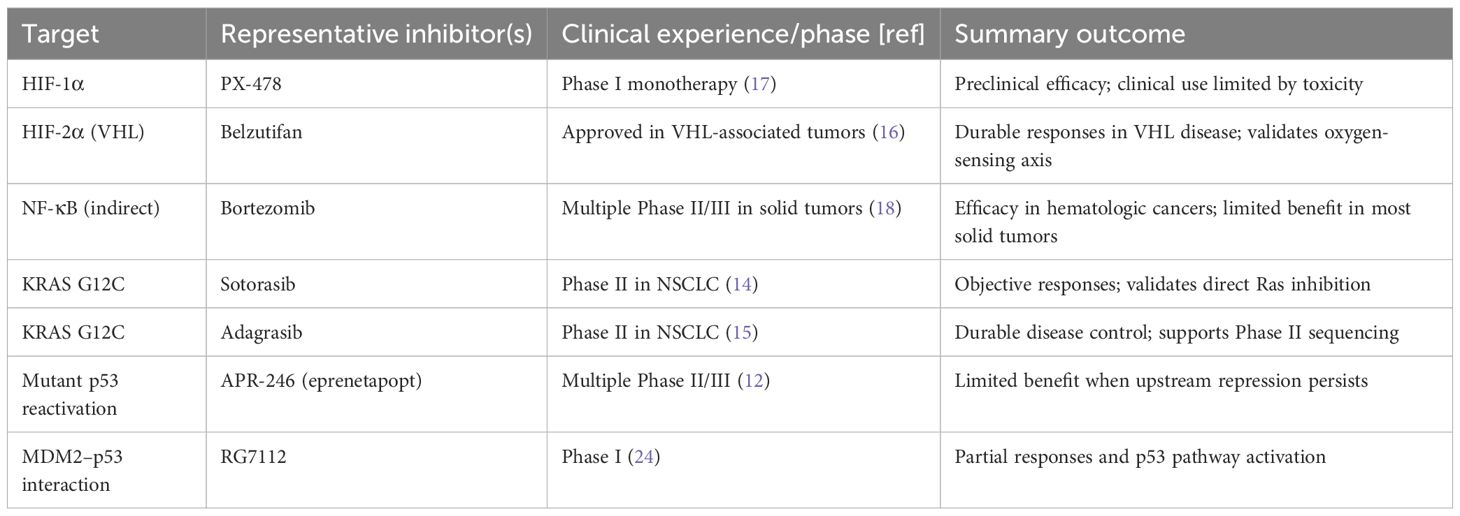
Table 1. Selected targets, representative inhibitors, and clinical experience.
2.4 BNIP3
BNIP3 illustrates the context dependence of dual-function proteins. While it can induce mitochondrial outer membrane permeabilization and apoptosis, in hypoxic tumors BNIP3 frequently drives mitophagy, removing dysfunctional mitochondria, limiting ROS accumulation, and thereby promoting survival (5). Therapeutic interventions must therefore tune BNIP3 activity toward pro-apoptotic outputs—either by inhibiting its mitophagic function during early sequencing or by combining it with agents that lower the survival advantage conferred by mitophagy.
2.5 NF-κB
NF-κB governs inflammation and innate immunity but is persistently activated in many cancers by TME-derived cytokines, leading to transcription of survival, angiogenic, and immunosuppressive programs (4). Canonical signaling proceeds through IKK-mediated phosphorylation of IκB, p65 nuclear translocation, and activation of target genes that can directly antagonize p53 functions. Clinical experience demonstrates context constraints: proteasome inhibitors such as bortezomib reduce NF-κB activity and are effective in several hematologic malignancies, yet they have generally not reproduced this benefit across solid tumors, reflecting pathway redundancy and microenvironmental buffering (18).
3 Sequential therapeutic strategy
A stepwise strategy can exploit the conditional nature of dual-function proteins. Phase I focuses on dismantling TME-driven adaptations by inhibiting hypoxia responses (e.g., HIF-1α/2α), tempering inflammatory signaling (NF-κB), and preventing survival-promoting mitophagy (BNIP3) (3–5). This reduces angiogenesis, lowers glycolytic buffering, and weakens survival circuits that would otherwise absorb pro-apoptotic stimuli. Phase II inhibits Ras, thereby relieving p53 repression via MDM2/ERK pathways and mitigating redox and epigenetic barriers (6, 10, 11, 13–15). Direct KRAS G12C inhibitors provide a clear entry point; in non-G12C settings, upstream RTK or downstream MEK/ERK and PI3K/Akt nodes may be targeted based on tumor dependencies. Phase III restores p53 activity—either by stabilizing wild-type p53 through MDM2 antagonism or by pharmacologically reactivating mutant p53—once upstream suppression has been alleviated (12, 24). Biomarker-guided decision points are essential: hypoxia imaging, cytokine profiles, Ras mutation status, and p53 integrity collectively inform where to begin and how to progress. Within this framework, premature activation of p53 is avoided, as it risks futility when Ras and the TME remain intact.
4 Lessons from past therapies
Past experiences underscore the importance of sequencing. APR-246 sought to reactivate mutant p53, but durable benefit was limited—consistent with a model in which p53 remains functionally constrained by Ras-driven and TME-mediated repression when these are not addressed first (12). Bortezomib effectively suppresses NF-κB in hematologic malignancies yet has delivered modest outcomes in most solid tumors, illustrating context dependency and pathway redundancy (18). PX-478, an HIF-1α inhibitor, produced strong preclinical activity but encountered dose-limiting toxicities clinically, limiting its standalone impact (17). Conversely, belzutifan’s success in VHL-associated tumors validates the oxygen-sensing axis as a drug target, while also highlighting that efficacy can be disease-context specific (16). Most notably, covalent KRAS G12C inhibitors achieved meaningful responses in lung cancer, conclusively demonstrating that direct Ras targeting is possible in patients and should be prioritized as the second phase of a sequential strategy where applicable (14, 15) (see Table 1).
5 Personalized application and biomarkers
Personalization is inherent to sequencing. Hypoxia-dominant tumors may warrant HIF-axis inhibition as Phase I, guided by imaging with radiotracers such as fluoromisonidazole (FMISO) positron emission tomography (19). Inflammation-driven phenotypes, evident from cytokine panels or TME transcriptional profiles, suggest prioritizing NF-κB blockade (20). Ras mutation status dictates feasibility of direct Ras inhibitors; in wild-type Ras tumors with amplified upstream signaling, RTK or pathway-node inhibitors may serve as surrogates (13–15). The status of p53—wild-type but suppressed versus mutant—can guide the use of MDM2 antagonists or reactivators, respectively, once upstream constraints are relieved (12, 24). Liquid biopsy approaches enable longitudinal monitoring of mutation dynamics and clonal selection during sequencing, informing when to switch phases or de-escalate therapy (21). Autophagy and mitophagy markers provide additional signals regarding BNIP3’s role and the degree of mitochondrial stress (5).
6 Challenges and TME co-evolution
Several challenges accompany sequential therapy. Tumor heterogeneity generates spatially distinct niches with variable hypoxia, inflammation, Ras pathway activity, and p53 status, complicating uniform biomarker thresholds (22). Combining multiple targeted agents sequentially (and sometimes in overlap windows) increases the risk of cumulative or synergistic toxicities; careful scheduling, dose optimization, and early safety stopping rules are essential (23). Most critically, resistance evolves dynamically. Therapy imposes selective pressures that reshape the TME—reprogramming fibroblasts, endothelial cells, and immune infiltrates—and foster expansion of resistant subclones with alternative survival strategies (7, 22). Adaptive clinical trial designs with predefined biomarker-driven transitions between phases, coupled with serial imaging and liquid biopsies, can operationalize the sequencing concept while minimizing exposure to ineffective regimens (23).
7 Conclusion
Dual-function proteins integrate context-dependent survival and death signals. Their rewiring by oncogenic mutations and the TME underlies the persistence of apoptosis-resistant cancer. Historical failures of single-node interventions can be reinterpreted through the lens of persistent Ras-mediated repression of p53 and microenvironmental buffering. A sequential framework—first dismantling TME adaptations, then inhibiting Ras to relieve p53 repression, and finally restoring p53—offers a biologically rational and testable path to restore apoptotic competence. This concept does not solve all resistance mechanisms but provides a clear direction for preclinical development and clinical trials.
Author contributions
LS: Conceptualization, Formal Analysis, Project administration, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Acknowledgments
The author thanks Prof. Janusz Rak (McGill University) for formative discussions.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. The author verifies and takes full responsibility for the use of generative AI in the preparation of this manuscript. Generative AI (ChatGPT, OpenAI) was used to assist in language editing.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
2. Junttila MR and de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. (2013) 501:346–54. doi: 10.1038/nature12626
3. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. (2003) 3:721–32. doi: 10.1038/nrc1187
4. Karin M. NF-κB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. (2009) 1:a000141.
5. Chourasia AH and Macleod KF. Tumor suppressor functions of BNIP3 and mitophagy regulation. Autophagy. (2015) 11:1937–8. doi: 10.1080/15548627.2015.1085136
6. Shaw AT and Settleman J. The molecular basis of the Ras–p53 connection. Oncogene. (2007) 26:6722–9.
7. Tabassum DP and Polyak K. Tumorigenesis: it takes a village. Nat Rev Cancer. (2015) 15:473–83. doi: 10.1038/nrc3971
8. Levine AJ and Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. (2009) 9:749–58. doi: 10.1038/nrc2723
9. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape across 12 major cancer types. Nature. (2013) 502:333–9. doi: 10.1038/nature12634
10. Trachootham D, Alexandre J, and Huang P. Redox regulation of cell survival: the role of ROS in cancer. Antioxid Redox Signal. (2008) 10:1343–74. doi: 10.1089/ars.2007.1957
11. Kouzarides T. Chromatin modifications and their function. Cell. (2007) 128:693–705. doi: 10.1016/j.cell.2007.02.005
12. Lehmann S, Bykov VJN, Ali D, Andrén O, Cherif H, Tidefelt U, et al. First-in-human clinical trial of APR-246 targeting p53. J Clin Oncol. (2012) 30:3633–9. doi: 10.1200/JCO.2011.40.7783
13. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. (2003) 3:11–22. doi: 10.1038/nrc969
14. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. (2019) 575:217–23. doi: 10.1038/s41586-019-1694-1
15. Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS G12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers. Cancer Discov. (2020) 10:54–71. doi: 10.1158/2159-8290.CD-19-1167
16. Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, et al. Belzutifan for renal cell carcinoma in von Hippel–Lindau disease. N Engl J Med. (2021) 385:2036–46. doi: 10.1056/NEJMoa2103425
17. Welsh SJ, Williams RR, Birmingham A, Newman DJ, Kirkpatrick DL, Powis G, et al. The thioredoxin inhibitor PX-12 and HIF-1α antagonist PX-478: preclinical and early clinical evaluation. Mol Cancer Ther. (2003) 2:235–43.
18. Richardson PG, Mitsiades C, Hideshima T, and Anderson KC. Proteasome inhibition in hematologic Malignancies and solid tumors. Cell. (2005) 122:571–82.
19. Yang DJ, LeBlanc M, Arbuckle M, Friedman HS, Coleman RE, Sampson JH, et al. Noninvasive assessment of tumor hypoxia by FMISO PET. J Nucl Med. (1999) 40:1993–2000.
20. Roxburgh CS and McMillan DC. Role of systemic inflammatory response in predicting survival in cancer. Future Oncol. (2010) 6:149–63. doi: 10.2217/fon.09.136
21. Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. (2017) 17:223–38. doi: 10.1038/nrc.2017.7
22. DagogoJack I and Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. (2018) 15:81–94. doi: 10.1038/nrclinonc.2017.166
23. Renfro LA and Mandrekar SJ. Clinical trial designs incorporating biomarkers and adaptive methods. Nat Rev Clin Oncol. (2018) 15:777–86.
Keywords: apoptosis, tumor microenvironment, p53, Ras, HIF-1α, BNIP3, NF-κB, sequencing
Citation: Strzadala L (2025) Context-dependent rewiring of dual-function proteins in cancer: a sequential strategy to restore apoptosis. Front. Oncol. 15:1675537. doi: 10.3389/fonc.2025.1675537
Received: 29 July 2025; Accepted: 09 September 2025;
Published: 07 October 2025.
Edited by:
Ciprian Tomuleasa, University of Medicine and Pharmacy Iuliu Hatieganu, RomaniaReviewed by:
Adrian Bogdan Tigu, University of Medicine and Pharmacy Iuliu Hatieganu, RomaniaBancos Anamaria, University of Medicine and Pharmacy Iuliu Hatieganu, Romania
Copyright © 2025 Strzadala. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leon Strzadala, bGVvbi5zdHJ6YWRhbGFAaGlyc3pmZWxkLnBs