 Andrei-Alexandru Tirpe1
Andrei-Alexandru Tirpe1 Andreea Nutu1Constantin Busuioc2,3
Andreea Nutu1Constantin Busuioc2,3 Ovidiu-Laurean Pop4
Ovidiu-Laurean Pop4 Ioana Berindan-Neagoe1,5,6*
Ioana Berindan-Neagoe1,5,6*- 1Department of Genomics, MEDFUTURE Institute for Biomedical Research, Iuliu Haţieganu University of Medicine and Pharmacy, Cluj-Napoca, Romania
- 2Department of Pathology, National Institute of Infectious Disease, Bucharest, Romania
- 3Department of Pathology, Onco Team Diagnostic, Bucharest, Romania
- 4Department of Morphological Sciences, Faculty of Medicine and Pharmacy, University of Oradea, Oradea, Romania
- 5Doctoral School, Iuliu Hatieganu University of Medicine and Pharmacy, Cluj-Napoca, Romania
- 6The Academy of Medical Sciences, Bucharest, Romania
Lung cancer is the most frequently diagnosed type of cancer worldwide, according to GLOBOCAN 2022 statistics. Key genetic alterations involve driver gene mutations that significantly enhance cancer aggressiveness. These include several EGFR mutations, ALK rearrangements, ROS1 rearrangements, RET translocations, MET alterations, NTRK fusions, BRAF mutations and KRAS mutations, such as the KRAS G12C mutation. Naturally, each of these is part of a larger signaling pathway that becomes dysregulated via genetic alterations. We highlight the transduction of EGFR: HER2 via RAS-RAF-MEK-MAPK pathway, PI3K-PTEN-AKT pathway and STAT pathway, of the ALK via PI3K/AKT, MAPK/ERK and JAK/STAT and of KRAS via effectors of the MAPK pathway and of the PI3K pathway. MicroRNAs (miRNAs) interfere at various levels with these pathways, either with pro-oncogenic effects or tumor suppressive effects. For instance, miR-33a is a tumor suppressive miRNA with a role in EGFR-tyrosine kinase inhibitor (TKI) resistance, miR-200c regulates the ALK pathway, and miR-22-3p regulates the MET pathway. The present paper also serves as an integrative work, highlighting the main cancer progression processes regulated by miRNAs, following these mutations. Specifically, we highlight the modulatory roles of miRNA in cancer cell survival and proliferation (miR-28, miR-30b/c), invasion and metastasis (miR-218, miR-182), neoangiogenesis (miR-29c), metabolic reprogramming (miR-124), and therapy resistance (miR-378, miR-328, miR-1244). The broad implications of miRNAs in lung cancer underline their potential real-world utility, as these entities can function as biomarkers for prognosis/diagnosis and even future therapeutic targets or agents.
1 Introduction
Lung cancer is an intricate malignancy, with an estimated 2.48 million new cases and 1.81 million deaths worldwide in 2022 alone (1). Many factors, including late diagnosis, patient characteristics and acquisition of cancer progression features can explain the poor prognosis (2). Comprehensive genomic characterization is increasingly recognized as essential in lung cancer management, as certain oncogene-driven lung cancers can benefit from targeted therapeutics. In specific cases, these targeted therapeutics can significantly prolong overall survival and/or progression-free survival compared to conventional chemotherapy and/or radiotherapy. For instance, in the PROFILE 1014 clinical trial, crizotinib, a first generation ALK tyrosine kinase inhibitor (TKI) significantly improved progression-free survival (PFS) in comparison to standard chemotherapy - median PFS (mPFS) = 10.9 months in the crizotinib group versus 7.0 months in the standard chemotherapy group; hazard ratio (HR) = 0.45, [95% CI 0.35-0.60], p < 0.001 for progression/death (3). Similarly, in patients with activating BRAF mutations, the combination of dabrafenib-trametinib in treatment-naive patients led to mPFS = 10.8 months [95% CI:7.0-14.5 months] and investigator-assessed median duration of response (mDoR) = 10.2 months [95% CI: 8.3-15.2 months] (4).
Histologically, lung cancer subtypes present significant differences that translate into prognostic differences, along with different treatment approaches. In general terms, lung cancers are classified as small cell lung cancers (SCLCs), which comprise about 15% of cases, and non-small cell lung cancers (NSCLCs), which comprise the rest of 85% of cases. NSCLC is further subclassified into lung adenocarcinoma (LUAD, with about 40% of total cases), lung squamous cell carcinoma (LUSC, 25-30%) and large cell carcinoma (LCC), with approximately 5-10% of all lung cancers (5, 6). Each of these lung cancer subtypes fosters diverse genomic and epigenomic alterations that develop and drive lung cancer from initiation to progression. For simplification purposes, the present paper will focus on NSCLC.
The human genome encompasses coding and non-coding regions, each playing distinct roles in cellular function (7). Coding regions, which make up about 2% of the genome, are responsible for producing proteins that perform various cellular functions. In contrast, non-coding regions, which constitute a large part of the genome, do not code for proteins. Instead, they are crucial in regulating gene expression and maintaining genomic stability (8). The interplay between the coding and non-coding regions is increasingly recognized as a fundamental aspect in the development and progression of various cancers, including lung cancer (9). Both the coding and non-coding genome are largely implicated in the carcinogenesis and progression processes of lung cancer. Specific genetic alterations in driver genes are the mainstay of targeted therapeutics in lung cancer, including subtypes such as LUAD. Some of the most well-known oncogenes and tumor suppressor genes associated with lung cancer include EGFR, KRAS, and TP53 (10). These genetic alterations of the coding genome are pivotal in the initiation and progression of lung cancer, making them primary targets for therapeutic interventions. By comparison, the non-coding genome encompasses various elements, including promoters, enhancers, microRNAs (miRNAs), and long non-coding RNAs (lncRNAs), which interact with coding regions to influence cancer development and progression. Non-coding RNAs can modulate the epithelial-to-mesenchymal transition (EMT)/invasion, angiogenesis, and other progression processes (11). The functions and mechanisms of action are variable between different classes of ncRNAs, as is their biogenesis (12). MiRNAs are small non-coding RNAs of various lengths (approximately 22 nucleotides) generated via primary miRNAs (pri-miRNA), derived from introns or other non-coding transcripts (13). Their discovery by the Victor Ambros’ group (14) and Gary Ruvkun’s group (15, 16) revolutionized the field of non-coding RNAs. The main function of miRNAs includes mRNA degradation or translational repression via binding the 3’-UTR of a target mRNA through the miRNA-induced silencing complex (miRISC) (17). Other mechanisms of action include binding to coding regions and thus inhibiting protein expression, or binding to the 5’-UTR of mRNAs (12). Alternatively, miRNAs can also directly upregulate expression (18).
The present study serves as an integrative and comprehensive review of the current roles of non-coding RNAs in lung cancer progression processes, including neoangiogenesis, metastasis, immune evasion, and the acquisition of therapeutic resistance, with a specific focus on miRNAs. It also consolidates the main genetically altered genes that drive lung cancer. The novelty of this paper is given by the translational overview of miRNAs in NSCLC. The updated information regarding miRNAs implicated in this malignancy, in parallel with the main pathway alterations and driver mutations reinforces the approach.
2 The mutational landscape in lung cancer: genetic alterations, signaling pathways, and relevant miRNAs
NSCLCs present a significant number of alterations within their coding genome. For didactic purposes, this section will examine the NSCLC mutational landscape, focusing on genes that encode essential oncogenic drivers in specific subtypes of NSCLC, as well as miRNAs that are involved in modulating these entities. Specific genetic alterations render driver genes in NSCLC that enhance the proliferation rate of cancer cells and cancer aggressiveness. However, driver genes offer multiple therapeutic strategies by exploiting these elements as druggable targets. This section will briefly discuss the main driver genes and targetable mutations in NSCLC, along with relevant modulatory miRNAs and their potential applications.
2.1 Epidermal growth factor receptor
EGFR is part of the HER/ErbB family of receptor tyrosine kinases (RTKs) (19) and is found in approximately 15-40% of non-squamous NSCLC tumors (20); when activated, physiological non-mutated EGFR can recruit HER2 in heterodimers, forming the EGFR::HER2 functional unit (21), leading to the downstream signaling pathways which promote cell survival, growth and migration (22). Intracellular signaling cascades pertain to the RAS-RAF-MEK-MAPK pathway, PI3K-PTEN-AKT pathway and STAT pathway (23), among others (22, 23). In lung cancer, the activated state is achieved through initial oncogenic mutations in exons 18-21, which encode the kinase domain (24). Two of the most frequent EGFR activating mutations are the L858R point mutation in exon 21 and the LREA in-frame deletion on exon 19 (19, 24). Activating EGFR mutations are druggable via EGFR TKIs, classified in first-generation EGFR TKIs, such as gefitinib and erlotinib and second-generation TKI afatinib (20). The exon 20 T790M “gatekeeper” mutation is considered one of the secondary EGFR point mutations in NSCLC. This mutation renders acquired resistance towards EGFR TKIs that do not specifically target this mutation (25). Third-generation EGFR TKI osimertinib is selective for the T790M resistance and for EGFR-TKI-sensitizing mutations. The FLAURA clinical trial showed that in patients with untreated EGFR mutation-positive (either L858R point mutation or exon 19 deletion) advanced NSCLC, the mPFS was significantly longer in patients treated with osimertinib than in the group treated with other standard EGFR-TKIs (gefitinib or erlotinib): 18.9 months versus 10.2 months, respectively, with HR = 0.46 [95% CI: 0.37-0.57, p < 0.001) for disease progression or death (26). This proves that osimertinib is superior to the other standard EGFR-TKIs. Further analysis in the FLAURA trial showed an improved overall survival (OS) of 38.6 months [95% CI: 34.5-41.8] in the group treated with osimertinib versus 31.8 months [95% CI: 26.6-36.0] in the group treated with other EGFR-TKIs, HR = 0.80 [95.05% CI: 0.64-1.00, p = 0.046] for death (27). Furthermore, recent clinical trial results show that in untreated EGFR-mutated (L858R mutation or exon 19 deletion) patients with advanced NSCLC, the addition of chemotherapy (pemetrexed + platinum-based agent in adjusted dosages) showed an increased investigator-assessed PFS, with HR = 0.62 [95% CI: 0.49-0.79, p < 0.001] for disease progression or death in comparison to the osimertinib monotherapy group. Moreover, 57% [95% CI: 50-63] of patients in the osimertinib-chemotherapy group were alive and disease progression-free at 24 months versus 41% [95% CI: 35-47] of patients receiving osimertinib monotherapy (28). The European Society for Medical Oncology Clinical Practice Guideline for Oncogene-Addicted Metastatic Non-Small-Cell Lung Cancer recommends the use of first-line EGFR TKIs in all patients with sensitizing EGFR mutation, regardless of performance status, gender, histology or tobacco exposure. In this case, osimertinib is the preferred first-line agent in patients with L858R/exon 21 or exon 19 deletions NSCLC and for patients with central nervous system metastases, as osimertinib can partly cross the blood-brain barrier (29).
Concomitantly, from a non-coding RNA perspective, there are several miRNAs associated with the modulation of EGFR-dependent processes in lung cancer. MiR-33a is involved in EGFR-TKI resistance. In gefitinib-resistant cells, resistance to TKIs is increased through HDAC1-dependent miR-33a suppression. This involves several processes, including cancer cell proliferation, migration, and apoptosis. Mechanistically, HDAC1 bound FOXK1 in gefitinib-resistant cells and silenced miR-33a. Conversely, miR-33a overexpression downregulated ABCB7 and p70S6K1 expression, leading to tumor-suppressive effects (30). Furthermore, miR-128b directly regulates EGFR. In tumor samples, loss of heterozygosity for miR-128b was frequent and correlated significantly with clinical response and survival after gefitinib treatment (31). In another study, let-7c expression led to increased cancer cell proliferation and invasion; high let-7c expression led to a reversal of EMT and increased cancer sell sensitivity to osimertinib via a WNT1- and TCF-4-dependent mechanism. From a more in-depth view, let-7c suppressed WNT1 and TCF-4 expression epigenetically via promoter methylation, leading to increased osimertinib activity upon the EGFR-mutated NSCLC cells (32).
2.2 ALK rearrangements
ALK rearrangement was first reported as a key driver in NSCLC by Soda et al. in 2007, when the authors identified the EML4-ALK fusion (33). Notably, other ALK fusions also exist (34). These alterations render constitutive ALK kinase activity (34). Usually, the ALK pathway includes downstream signaling via PI3K/AKT, MAPK/ERK and JAK/STAT pathways (35). Statistics indicate that rearrangements involving ALK are present in approximately 5% of NSCLCs (36, 37), while they are predominantly identified in adenocarcinomas (38). Tumors with ALK rearrangements exhibit aggressive behavior, including nodal metastasis and advanced stages at diagnosis (39). In this case, targeting these RTKs is a desideratum. First-line treatment options in NSCLC patients with ALK rearrangement include ALK-targeted TKIs such as crizotinib, alectinib, brigatinib and lorlatinib (29). Disease progression under treatment with crizotinib can be approached via newer-generation ALK TKIs, such as ceritinib (40) or alectinib (41), which have proven to have efficacy both intracranially and extracranially (40, 41). Furthermore, lorlatinib proved effective in subsequent lines of treatment in NSCLC patients previously treated with second-generation ALK TKIs, whilst presenting CNS activity (42).
In an in vitro and in silico study by Lai et al. on EML4-ALK mutant NSCLC cell lines, the authors found that miR-100-5p is a regulator of resistance to ALK TKIs. There was, however, no validation of the target. The in silico analysis identified the mTOR signaling pathway as a target (43). In a study by Fukuda et al., pretreatment with quisinostat, an HDAC inhibitor, resulted in the upregulation of miR-200c/141 promoter activity, thereby restoring miR-200c expression. This, in turn, reverted EMT. Next, administration of an ALK inhibitor can bypass EMT-induced therapy resistance (44). In a combined in vitro and in vivo study by Yun et al., treatment of cancer cells with panobinostat altered H3K27ac signal in promoters and enhancers, and led to activation of miRNAs with tumor suppressive effects, such as miR-449, followed by antiproliferative effects of ALK inhibitors upon resistant cancer cells, xenografts and EML4-ALK transgenic mice (45).
2.3 ROS1 rearrangements
ROS1 is a proto-oncogene that encodes a transmembrane protein with common structural features with ALK and insulin receptor families (46). ROS1 protein-tyrosine kinase protein fusions are rare, occurring in an estimated 1-2% of NSCLC (47), predominantly in adenocarcinomas, but other histology types have also been described (48). Although the literature reports numerous fusion partners, CD74 is the most common (47). Other fusion partners include SLC34A2, EZR and TMP3 (49). The ROS1 fusions lead to constitutive ROS1 kinase activity and increased cell survival, proliferation, and migration via the STAT, PI3K, RAS/RAF/MEK/ERK1/2 and Vav3 pathways (46). First-line treatment options in ROS1 translocation metastatic NSCLC include crizotinib, entrectinib and repotrectinib as an alternative option (29). Second-line options for NSCLC patients who had systemic progression and received first-line ROS1 TKI include alternative next-generation ROS1 TKI or platinum-based chemotherapy after rebiopsy (29).
One of the miRNAs that is in direct relation with ROS1 is miR-760. In a study by Yan et al., miR-760 levels were found to be downregulated in 71.4% of NSCLC tissues considered and in NSCLC cell lines. In addition, overexpressing miR-760 led to inhibition of cancer cell proliferation, migration and cell cycle. Mechanistically, miR-760 inhibited ROS1 expression in NSCLC cells and the miR-760 expression level was inversely correlated with ROS1 expression level in NSCLC tissues (50, 51).
2.4 RET translocations
Another chromosomal rearrangement that drives NSCLC is the fusion of the rearranged during transfection (RET) gene with other fusion partners, such as KIF5B and CCDC6. This leads in turn to RET protein overexpression (52–54). Mechanistically, the RET gene encodes a proto-oncogene RTK which transduces to downstream RAS/MAPK, PI3K/AKT and JNK (53). RET rearrangements lead to fusion proteins that present a ligand-independent constitutive activation of RET, downstream pathway activation and stimulatory effects upon cancer cell growth, survival and proliferation (47). RET translocations are encountered in approximately 1-2% of NSCLC, predominantly adenocarcinomas (53) and predispose the patient to the development of brain metastases (55). Targeted therapeutics that act on RET fusions include selpercatinib and pralsetinib (29) and are recommended in patients previously untreated with RET inhibitors. Both drugs present high intracranial response rates (29).
2.5 MET
The MET tyrosine kinase is expressed on epithelial cells in various localizations. The c-MET proto-oncogene encodes MET and is a member of the RTKs family, with the main ligand being hepatocyte growth factor (HGF) (56). The activation of the MET TK activates the downstream signaling cascades RAS/ERK/MAPK, PI3K/Akt, Wnt/β-catenin and STAT. This modulates cell proliferation, survival and migration, among other processes (57). In general terms, the main MET pathological alterations include MET exon 14 skipping mutation (58) through various mutations – insertions, deletions, point mutations and others (59), MET amplification that is reported in up to 5% of NSCLC patients (60), MET overexpression, and the formation of MET fusion products. The fusion counterparts include TPR, TRIM4 and HLA-DRB1 (59, 61). All these MET alterations lead to the activation of the MET tyrosine kinase with consecutive pro-oncogenic activity (56). In a clinical setting in NSCLC, capmatinib and tepotinib are FDA-approved for MET exon 14 skipping mutations but are not currently EMA-approved for first-line therapy (29). Capmatinib can also be used in patients with high MET amplification (>= 10 GCN) after immunotherapy and/or platinum-based chemotherapy, although this drug is currently not approved by the Food and Drug Administration (FDA) or European Medicines Agency (EMA) in this setting (29).
When considering a miRNA-dependent approach to lung cancers driven by MET alterations, several key points need to be addressed. First, in the Romano study, edited miR-411-5p (ed.miR-411-5p) induced EGFR TKIs sensitivity in gefitinib-resistant NSCLC cell lines that were only partially dependent on MET repression. Mechanistically, ed.miR-411-5p directly targeted MET and repressed the MAPK pathway (62). Moreover, in the Yang study, miR-22-3p was found to be downregulated in lung cancer tissues in comparison to normal lung tissues. MiR-22-3p mimics could reduce MET and STAT3 expression, leading to the induction of apoptosis (63). In the Migliore study, epigenetically induced miR-205 expression in NSCLC cells resistant to MET-TKIs resulted in the downregulation of ERRFI1, leading to EGFR activation and sustained resistance to MET-TKIs. Conversely, the in vivo transduction of anti-miR-205 reversed crizotinib resistance.
Furthermore, in the absence of EGFR alterations, the EGFR activation via miR-205/ERRFI1 led to sensitivity of MET-TKI-resistant cells to combined MET-EGFR inhibition (64). MiR-182 was found to be downregulated in metastatic NSCLC cells in comparison to primary tumor tissues. Furthermore, overexpression of miR-182 inhibited cancer cell migration and invasion, reduced Snail expression and increased E-cadherin expression. Additionally, miR-182 directly suppressed Met expression. As such, the authors concluded that miR-182 may inhibit EMT and metastasis via inactivation of the Met/AKT/Snail pathway in NSCLC cells (65), highlighting the significant implications of these non-coding RNAs in specific lung cancer settings.
2.6 NTRK
The NTRK genes encode tropomyosin receptor kinases (TRKs) such as TRKA, TRKB and TRKC (66). NSCLC NTRK fusions have a variable prevalence, depending on the study (66); however, ESMO guidelines suggest a prevalence of less than 0.1% (29). NTRK fusions are obtained through intra- and interchromosomal rearrangements, as the 3’-sequence of NTRK1, NTRK2 or NTRK3 is fused with the 5’-sequences of various other genes (67). The chimeric product obtained by the fusion presents constitutive activation that is independent of ligands (68). This leads to activation of the downstream signaling pathways such as MAPK, PI3K and PKC (68). Targeted therapeutics, including larotrectinib and entrectinib, are recommended for patients with NTRK fusion-positive NSCLC who have no satisfactory alternative treatments (29).
2.7 BRAF mutations
In NSCLC, BRAF mutations occur in approximately 3-5% of cases and, for the most part, are mutually exclusive with EGFR mutations and ALK and ROS1 rearrangements (29, 69). BRAF is a serine/threonine protein kinase of the RAF kinase family that can activate the MAPK signaling cascade via oncogenic mutations (70). Mechanistically, extracellular growth factors can activate RTK, activating the SOS family guanine nucleotide exchange factors (GEFs) and thus activating RAS. This cascade results in the activation and dimerization of RAF proteins via GTP-RAS, which further leads to the phosphorylation of MEK1/2, followed by the phosphorylation of ERK1/2. In its turn, ERK1/2 phosphorylates downstream effectors, which modulate cell survival, proliferation, differentiation, and cell motility (71). The classification of BRAF mutations has led to a better understanding of the impact of these alterations. Class I BRAF mutations include BRAF V600D/E/K/R mutants that generate an important BRAF kinase activity, with a constitutive MAPK signaling activation. Class II and III BRAF mutations are non-V600 BRAF mutations. These are identified in the activation segment or P-loop for class II BRAF mutants, leading to MAPK pathway activation, and P-loop, catalytic loop, DFG motif for class III mutants, which present a lower basal kinase activity compared to wild-type BRAF or that do not present kinase activity (72). According to ESMO guidelines, patients who present with BRAF V600E mutations benefit from the dual therapy of dabrafenib in combination with trametinib in the setting of advanced or metastatic V600-mutated NSCLC (29). Of note is that the clinical trial only included patients with the BRAF V600E mutation (4).
2.8 KRAS G12C mutation
Activating KRAS mutations are observed in approximately 30% of NSCLC-LUAD cases, with the KRAS G12C mutation being the most common (73, 74). Oncogenic mutations in the KRAS gene are most prevalent at codons 12, 13 and 61 (75). These mutations activate RAS signaling through impairment of intrinsic GTPase or by alteration of KRAS, leading to the inability of KRAS to respond to GTPase-activating proteins (75). In general terms, KRAS mutations are associated with poor prognosis (76). The activation of KRAS leads to interaction with the effectors of the MAPK pathway – RAF/MEK/ERK – and with effectors of the PI3K pathway – AKT/mTOR –, among others (77–79), leading to cell survival and cell proliferation (80, 81). Current ESMO guidelines for NSCLC patients with KRAS G12C mutation recommend classic first-line treatment algorithms according to the non-oncogene addicted metastatic NSCLC (29, 82). Targeted therapeutics include sotorasib, which is recommended for patients with NSCLC with failed prior therapy, as the phase III clinical trial tested sotorasib in patients who progressed under platinum-based chemotherapy or ICI-based therapy (83). Another targeted therapeutic for the KRAS G12C mutation is adagrasib, which is currently approved only by the FDA and not by the EMA. Adagrasib was also studied in patients who progressed under platinum-based chemotherapy or ICI-based therapy and demonstrated an mPFS = 6.5 months [95% CI: 4.7-8.4] (84).
When considering the implications of miRNAs, several KRAS-related studies warrant mention. First and foremost, in the Xie study, miR-148a-3p inhibited NSCLC cancer cell proliferation and EMT by reducing SOS2 expression, thereby decreasing RAS activation (85). In a study by Edmonds et al., miR-31 was found to be overexpressed in LUAD, overexpression that was independently correlated with reduced patient survival. In a transgenic mouse model, the induction of miR-31 led to lung hyperplasia, adenoma formation, and ultimately, the development of adenocarcinoma. In the Edmonds study, miR-31 promoted mutant KRAS-mediated oncogenesis by targeting and reducing the expression of negative regulators of RAS/MAPK cascade (86). In the Li study, METTL3 promoted the m6A methylation of circ_0000620, leading to increased expression and stability, which in turn modulated the miR-216b-5p/KRAS axis and influenced apoptosis and cisplatin sensitivity in NSCLC cells.
Furthermore, transfection with si-circ_0000620 or miR-216b-5p mimic led to decreased KRAS expression in LUAD cells compared to the control group (87). In the Yan study, miR-1205 was found to directly bind 3’-UTR of KRAS and downregulate its expression. Furthermore, in a A549 xenograft model in nude mice, miR-1205 inhibited tumor growth and decreased levels of KRAS, MDM4 and E2F1 in tumor tissues (88, 173).
Table 1 encompasses the targeted therapeutics discussed in this chapter.
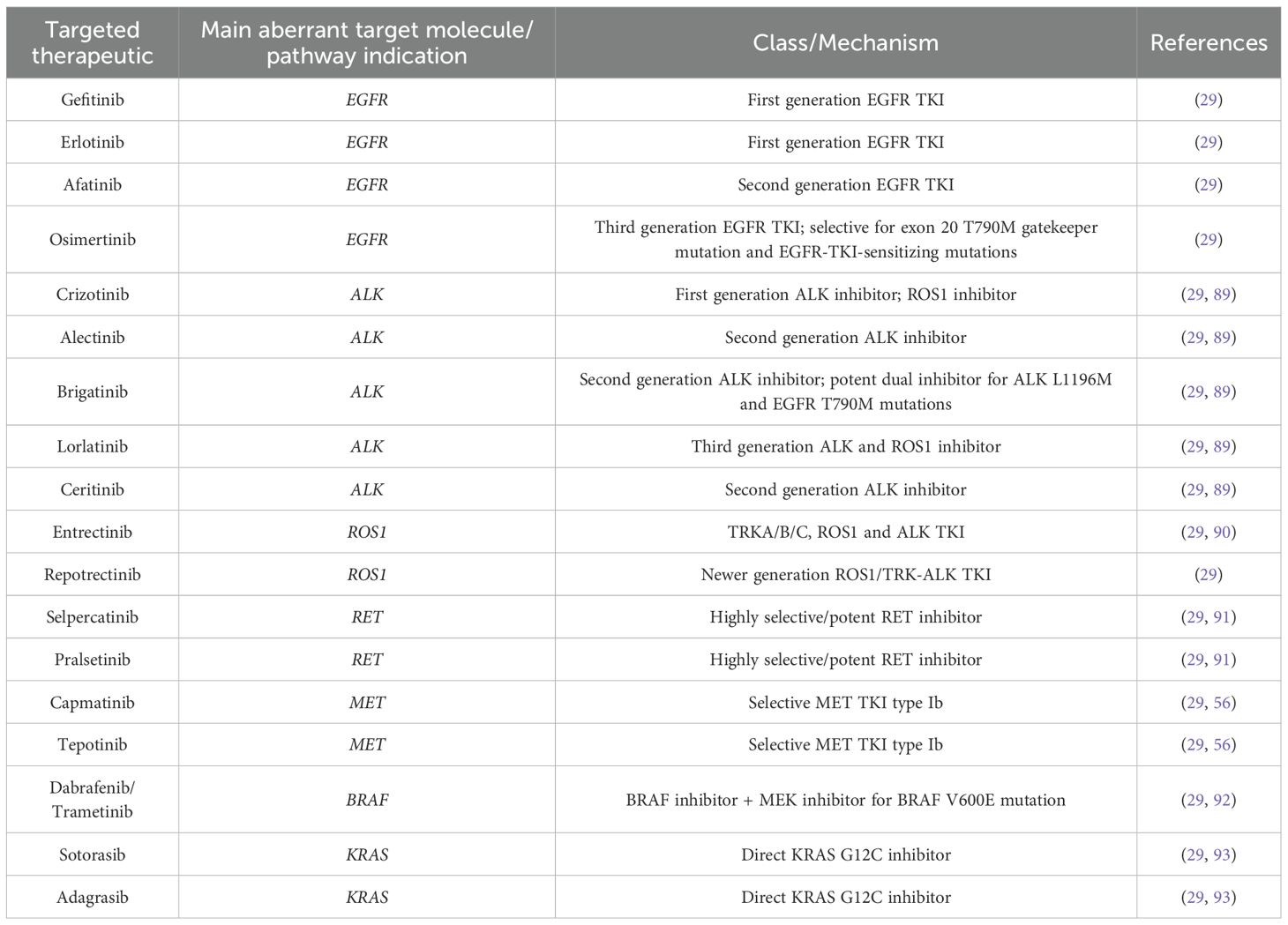
Table 1. Targeted therapeutics in lung cancer, their aberrant targets and class/mechanisms.
Figure 1 summarizes some of the specific molecular targets discussed in the present chapter and their signaling pathways in lung cancer, highlighting their modulatory role in several cancer development processes.
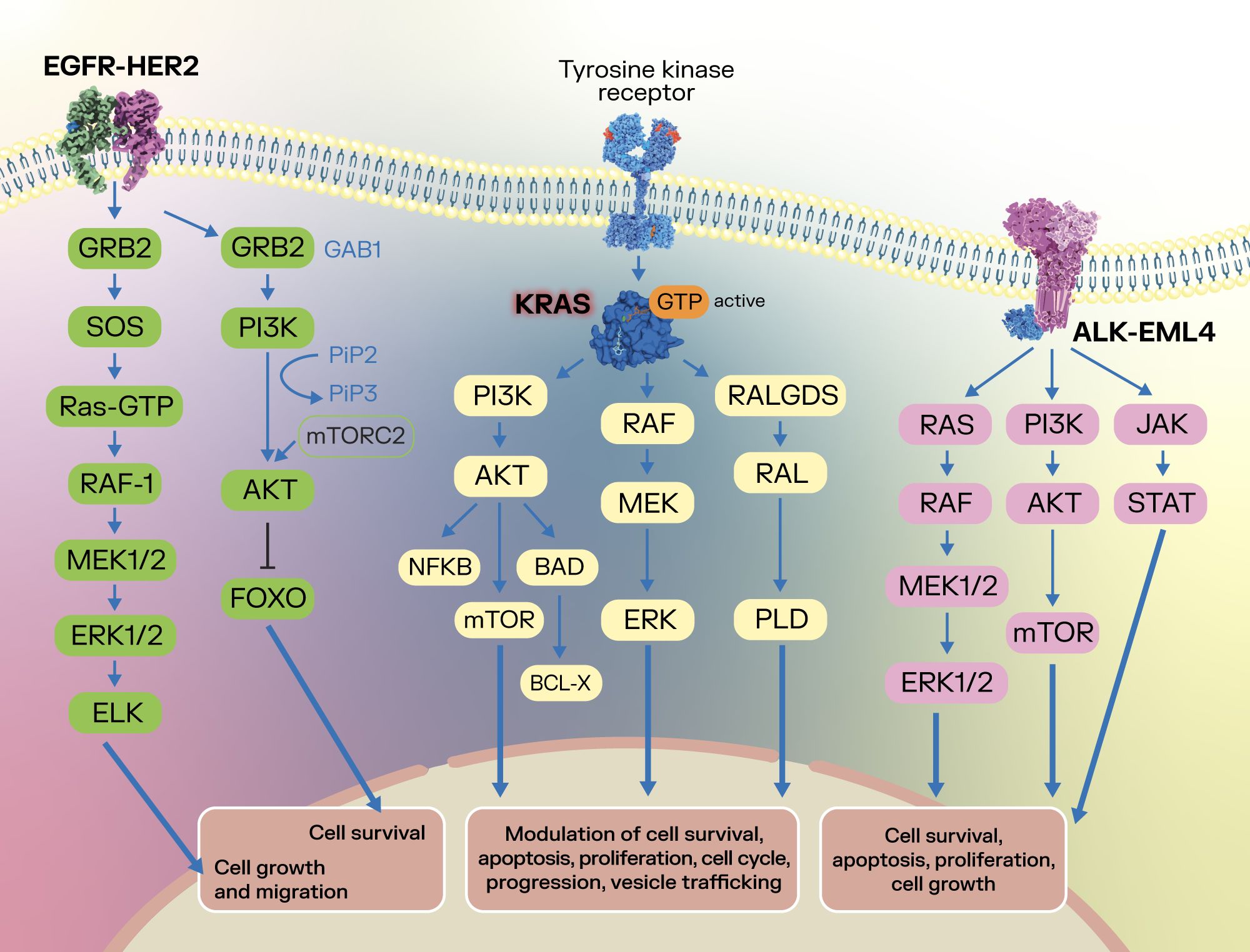
Figure 1. Summarization of several oncogenic targets and their signaling pathways in lung cancer. After binding with the corresponding ligands of the EGFR, the EGFR dimer activates downstream signaling pathways such as the RAS-RAF-MEK-ERK and PI3K-AKT, with an effect upon cell survival, cell growth and cell migration (19–21). In lung cancer, the activated state of EGFR is achieved through initial oncogenic mutations in exons 18-21, which encode the kinase domain (22). Concomitantly, KRAS plays a crucial role in signaling through the PI3K-AKT-mTOR, RAF-MEK-ERK and RALGDS-RAL-PLD pathways, modulating cell survival, apoptosis, cell proliferation, cell cycle, vesicle trafficking and progression (77–81), highlighting its oncogenic potential. In lung cancer, the ALK-EML4 fusion protein can form a dimer that does not require activation via ligand, which activates ALK and downstream RAS-RAF-MEK-ERK, PI3K-AKT-mTOR, and JAK-STAT signaling pathways, leading to effects upon cancer cell survival, apoptosis, proliferation and cell growth (33–35).
Furthermore, we sought to provide an integrative perspective on the current understanding of the modulation of these key oncogenes involved in NSCLC via non-coding RNAs, specifically miRNAs. As entities that are widely implicated in carcinogenesis and cancer progression, miRNAs can exert their oncogenic or tumor suppressive capabilities through silencing of molecules taking part in the signaling cascades of these specific oncogenes. Table 2 presents several key miRNAs that modulate specific target genes involved in these signaling cascades.
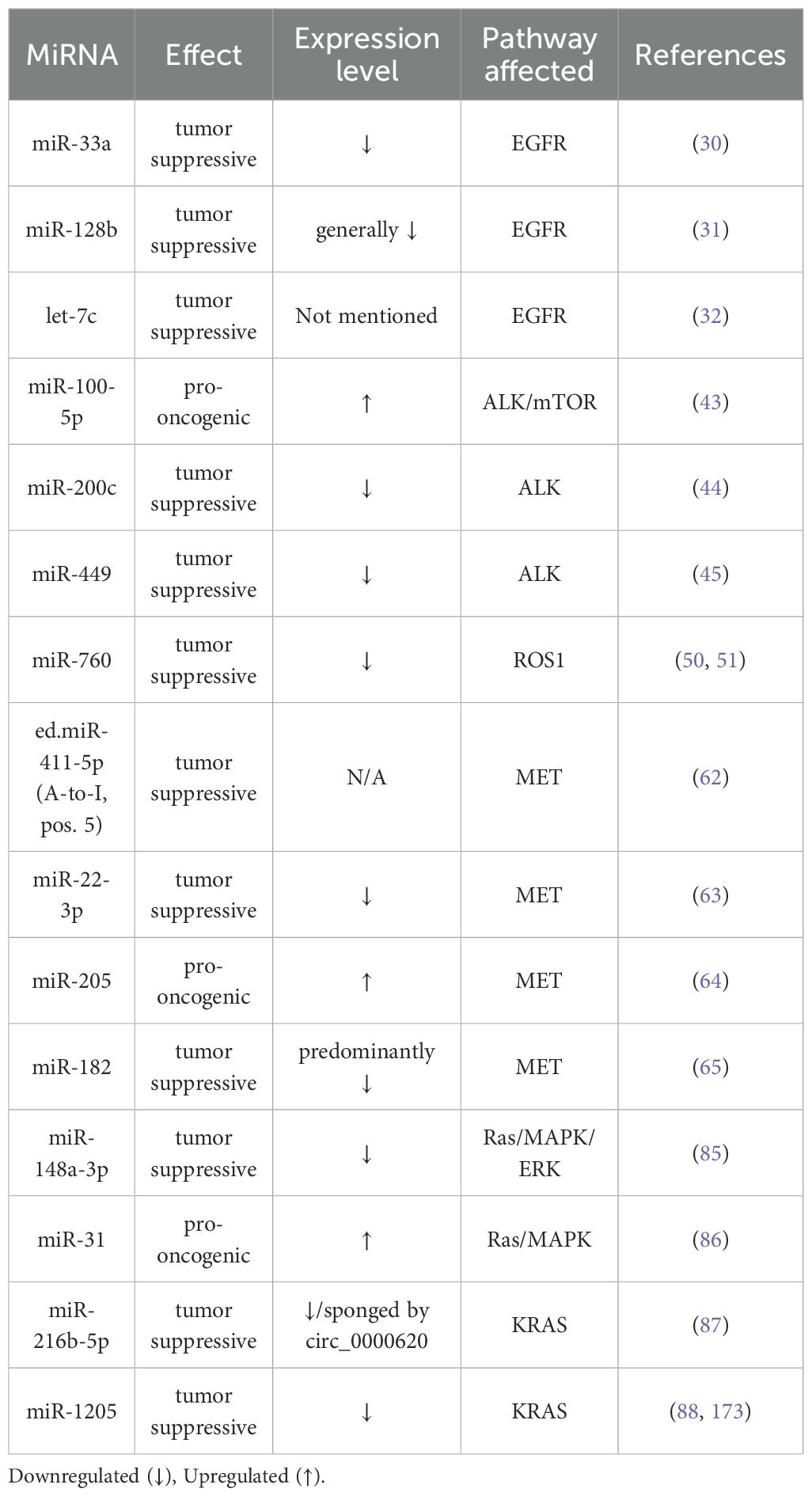
Table 2. A selection of miRNAs that target essential driver genes in lung cancer.
3 Dysregulation mechanisms of the non-coding genome in lung cancer
The non-coding genome describes the largest part of the human genome, as protein-coding sequences account for only approximately 1.5% of it (94). The non-coding genome is comprised of various entities, including non-coding regulatory regions such as non-coding RNAs (ncRNAs), promoters, enhancers and insulators, as well as untranslated regions (UTRs) (95). Advances in technology and high-throughput sequencing techniques led to the unravelment of this large part of the genome. As such, several types of ncRNAs have been described, some with a large number of evidence behind their proposed modulatory activity – miRNAs, long non-coding RNAs (lncRNAs), PIWI-interacting RNAs (piRNAs), small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs) and others (12). Although initially categorized as ncRNAs, recent reports suggest that some circRNAs may also have protein-coding abilities (96).
As miRNAs modulate numerous cellular processes, from cell growth and differentiation to development and apoptosis, it is abundantly clear that these entities play significant roles in malignancies (97). In cancer, miRNAs are categorized as oncogenes or tumor suppressors, depending on a given malignancy and the context (11); oncogenic miRNAs promote cancer traits and properties, while tumor suppressors have cancer suppressive abilities.
Regarding the practical interaction of miRNAs with the key updated hallmarks of cancer, as envisioned by Hanahan (98), it is comprehensible and realistic to expect the implication of modulatory miRNAs in each hallmark.
3.1 MiRNAs and the ECM
MiRNAs are significantly implicated in the regulation of the tumor microenvironment (TME), particularly in orchestrating and modulating the ECM. The dysregulated ECM has a significant role in lung cancer progression (99). Collagens are the most abundant proteins found within the ECM. Alteration in collagen expression influence TME cells (99). Discoidin domain receptor 1 (DDR1) is a RTK that binds collagen and activates downstream signaling cascades (100). DDR1 is largely implicated in cancer progression (101, 102). In a study by Ming et al., miR-199a-5p suppressed DDR1 expression. Circ_0087378 sponged miR-199a-5p and promoted malignant behavior through a DDR1-dependent mechanism (103). Concomitantly, integrins are proteins that intervene in the interaction between cells and ECM. This implies that integrins are involved in ECM remodeling and are largely implicated in cancer (104, 105). For instance, miR-338 may suppress lung cancer metastasis via integrin β3 (106). MiR-29c inhibits lung cancer metastasis and cancer cell adhesion to the ECM by integrin β1 and MMP2 expression inhibition (107). Moreover, exosomal miRNAs are involved in a variety of lung cancer processes, from cancer cell proliferation, apoptosis, epithelial-to-mesenchymal transition (EMT) and metastasis, to angiogenesis (108). Exosomes with low miR-34c-3p levels may promote NSCLC invasion and migration via upregulating integrin α2β1 (109).
3.2 MiRNAs and epigenetic regulation
MiRNAs are able to contribute to the epigenetic regulation via targeting several epigenetic regulators. MiR-101 interferes with lung cancer progression processes via the PTEN/AKT signaling cascade, by targeting the DNA-methyltransferase 3A (DNMT3A) (110). EZH2, a histone methyltransferase, functions as an epigenetic regulator by catalyzing the methylation of histone H3 - lysine 27 (111). In a study by Xia et al., anti-miR-21 led to a downregulation of EZH2 expression in lung cancer stem cells, proving the interaction between this miRNA and EZH2 (112). Another epigenetic modulators are histone deacetylases (HDACs), which interact with the chromatin structure and alter it, leading to transcriptional repression (113). In a study by Jeon et al., combined treatment with miR-449a and HDAC inhibitors in vitro led to a significant reduction in growth when compared to treatment with HDAC inhibitors alone (114). MiR-200b is involved in the therapeutic resistance of LUAD cells via E2F3; the suppression of HDAC1/4 increases miR-200b expression via histone-H3 acetylation upregulation, suggesting a crosstalk between miR-200b and HDAC1/4. In the Chen study, HDAC1/4 silencing led to G2/M cell cycle arrest, inhibited cancer cell proliferative abilities, increased cancer cell apoptosis, and counteracted the therapeutic resistance in docetaxel-resistant LUAD cells, in part via a miR-200b-controlled mechanism (115).
Furthermore, in the present chapter, we will briefly discuss some of the main NSCLC progression processes, providing several practical examples of modulatory miRNAs from the literature. Figure 2 presents these cancer processes and the associated miRNAs.
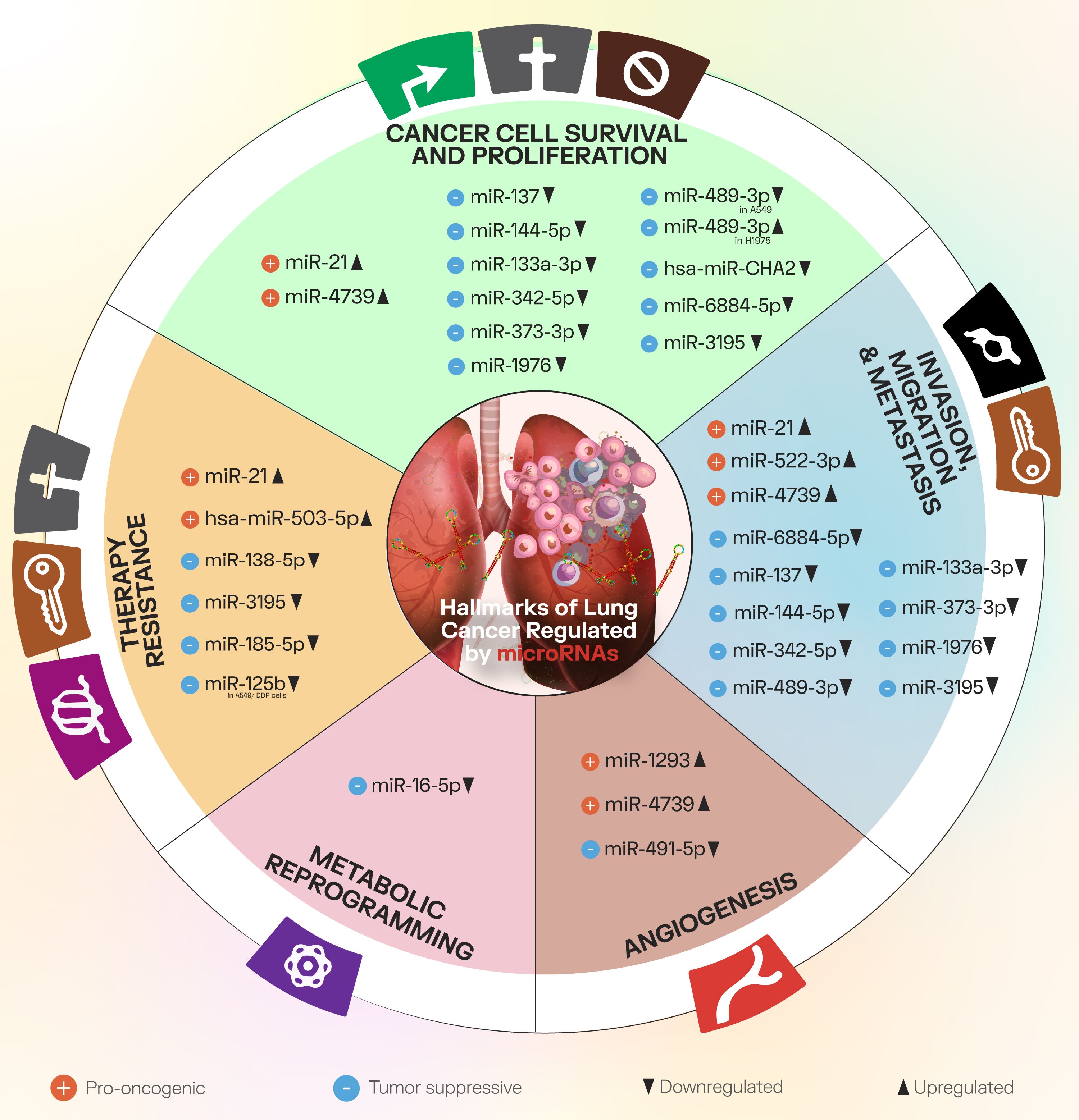
Figure 2. The main cancer progression processes discussed in the present paper, along with downregulated and upregulated miRNAs that modulate factors involved in these processes.
3.3 Survival and proliferation
MiRNAs can modulate NSCLC cell survival and proliferation. In a study by Gong et al., miR-20a promoted NSCLC cell proliferation through PTEN inhibition and upregulation of PD-L1 (116). In another study by Cui et al., the expression of miR-28 was significantly upregulated in NSCLC tissues and cell lines compared to adjacent normal tissue and control cell lines, respectively. Furthermore, miR-28 promoted cancer cell proliferation by directly targeting PTEN (117). When referring to miR-30b/c, Zhong et al. found that miR-30b/c levels were downregulated in NSCLC specimens in comparison to control – adjacent non-tumor tissues and that miR-30b/c targeted Rab18 in a direct manner leading to a downregulation in Rab18 expression and inhibition of NSCLC cell proliferation (118). Moreover, Peng et al. identified miR-19 as an oncogenic miRNA that is overexpressed in NSCLC tissues and several lung cancer cell lines. Herein, miR-19 inhibited CBX7 expression via CBX7 3’-UTR binding, leading to a decrease in CBX7 mRNA expression and CBX7 protein levels. Overexpression of miR-19 may significantly improve NSCLC cell proliferation and migration (119). Conversely, Liu et al. showed that miR-1253 was significantly downregulated in NSCLC tissues and associated with several clinical parameters such as advanced clinical stage, lymph node metastasis and even poor survival, highlighting the relevance of miRNAs in this malignancy. In the Liu study, overexpression of miR-1253 inhibited cancer cell proliferation, migration and invasion in vitro and identified the long isoform of WNT5A as a target for miR-1253 (120). Lastly, a study by Yoo et al. found that miR-CHA1 expression was downregulated in human lung cancer tissues (LUAD, SqCC) and cell lines. Overexpression of miR-CHA1 reduced XIAP mRNA expression and protein levels and thus inhibited NSCLC cell proliferation and induced apoptosis both in vitro and in vivo (121).
3.4 Invasion and metastasis
As versatile modulators of cancer progression processes, miRNAs control invasion and metastasis, among other factors from the local TME, such as hypoxia (122). In general terms, metastasis implies in its first phase the epithelial-to-mesenchymal transition, with consecutive loss of epithelial-like characteristics of cancer cells to a mesenchymal-like state. This leads to increased motility, invasiveness and the ability to degrade the ECM (123). Following EMT, cancer cells may regress back towards epithelial-like characteristics via mesenchymal-epithelial transition (MET) (123). In a study by Shi et al., miR-218 expression was found to be significantly downregulated in lung cancer tissues in comparison to control; the authors identified an association between miR-218 levels and lymph node metastasis and histological grade (124). Mechanistically, miR-218 suppressed invasion and metastatic spread via targeting Slug and ZEB2, key factors in the EMT process, and increased chemosensitivity to cisplatin in H1299 via Slug and ZEB2 (124). Moreover, Li et al. found that miR-182 is downregulated in metastatic NSCLC cells in comparison to primary tumor tissues; miR-182 overexpression reduced Snail expression and conversely promoted E-cadherin expression, leading to the inhibition of migration and invasion in lung cancer cells. In addition, miR-182 silenced MET expression, inhibited AKT phosphorylation and nuclear Snail accumulation. Li et al. concluded that miR-182 may inhibit NSCLC metastasis and EMT through the inactivation of MET/AKT/Snail signaling (65). Conversely, miRNAs can be regulated by a diverse number of factors. In the Chang et al. study, the authors found that miR-137 expression is induced by Slug and promoted metastasis via targeting transcription factor AP-2 gamma (TFAP2C) (125). MiR-137 knockdown inhibited Slug-induced NSCLC cell invasion and migration (125).
3.5 Neoangiogenesis
NSCLC progression is dependent on generating new vascularization via the neoangiogenic process, sustaining the relative oxygenation inside the TME. Our group has critically reviewed the neoangiogenesis process in NSCLC and the implication of miRNAs as potential neoangiogenesis-related therapeutic agents/targets in another paper (126). In general terms, neoangiogenesis is governed by pro-angiogenic factors, including VEGFA, FGF2, PDGFB, EGF, MMP2, and anti-angiogenic factors, such as THBS1 and TIMP1. Neoangiogenesis is driven by the imbalance between the pro-angiogenic and anti-angiogenic factors, termed the angiogenic switch (126). Intuitively, the miRNA-related modulation of neoangiogenesis can be achieved via miRNAs that target either pro-angiogenic or anti-angiogenic factors. In a lung cancer study by Liu et al., miR-29c was found to target VEGFA and act as a tumor suppressor by inhibiting cancer cell proliferation, migration and invasion, and angiogenesis in vitro. Furthermore, the authors identified a significant association between the downregulated miR-29c expression and poor prognosis in LUAD stage IIIA-N2 patients (127). In another study by Mao et al., tumor-derived miR-494 targeted and downregulated PTEN with a consecutive activation of the AKT/eNOS signaling pathway within human vascular endothelial cells, thus promoting neoangiogenesis (128).
3.6 Metabolic reprogramming
Cancer cells are able to reprogram their metabolism in order to increase the uptake of nutrients and promote their survival, growth and proliferation (129–131). One such alteration is the Warburg effect – a metabolism switch towards the use of glycolysis even in aerobic conditions (130, 131). Naturally, miRNAs can regulate factors that are in relation with metabolic rewiring as well. For instance, miR-124 overexpression inhibited NSCLC cell growth, energy metabolism, glucose consumption and lactate production via targeting glucose transporter 1 (GLUT1) and hexokinase II (HKII) and negatively regulating AKT1 and AKT2 (132).
3.7 Therapy resistance
Therapy resistance is an essential process in the evolution of NSCLC under treatment and represents a major challenge for clinicians, as it leads to therapeutic failure and cancer progression. In non-oncogene-addicted metastatic NSCLC, platinum-based chemotherapeutics remain a viable treatment option in various circumstances (82). Therapy resistance to cisplatin (cDDP) is multifaceted, as multiple mechanisms might be involved at a certain point. Such mechanisms include efflux transporters, EMT, autophagy, and modulation of different signaling cascades that pertain to cancer cell survival and apoptosis (133). For instance, miR-378 upregulation in cell lines A549/cDDP and Anip973/cDDP led to secreted form clusterin (sCLU) expression downregulation via direct targeting, sensitizing the NSCLC cells to cDDP. Concomitantly, patients sensitive to cDDP had higher levels of miR-378 and lower levels of sCLU in tumor tissues (134). In a study by Wang et al., miR-328 expression was significantly increased, and PTEN mRNA expression level was significantly decreased in tumor tissues from cDDP-resistant NSCLC patients in comparison to cDDP-sensitive NSCLC patients. Furthermore, the authors observed a higher miR-328 expression level and lower PTEN expression level in the cDDP-resistant A549 cell line (A549rCDDP) compared to the parental A549 cell line and confirmed that miR-328 targeted PTEN. Wang et al. showed that the inhibition of miR-328 in A549rCDDP cells treated with cDDP induced apoptosis and decreased cancer cell proliferation, highlighting the association of miR-328/PTEN in NSCLC cDDP resistance (135). In another study by Li et al., miR-589 and miR-1244 were downregulated in A549/cDDP cells in comparison to the parental A549 cell line, while the expression of miR-182 and miR-224 was found to be increased in the A549/cDDP cell line, with statistical significance. Transfection of the resistant A549/cDDP cells with miRNA mimics miR-589 or miR-1244 led to an improvement in cDDP sensitivity, reducing cancer cell invasion and apoptosis and underlining the implication of these miRNAs in cDDP chemosensitivity (136).
Figure 3 provides a visual representation of the implication of miRNAs in specific lung cancer processes.
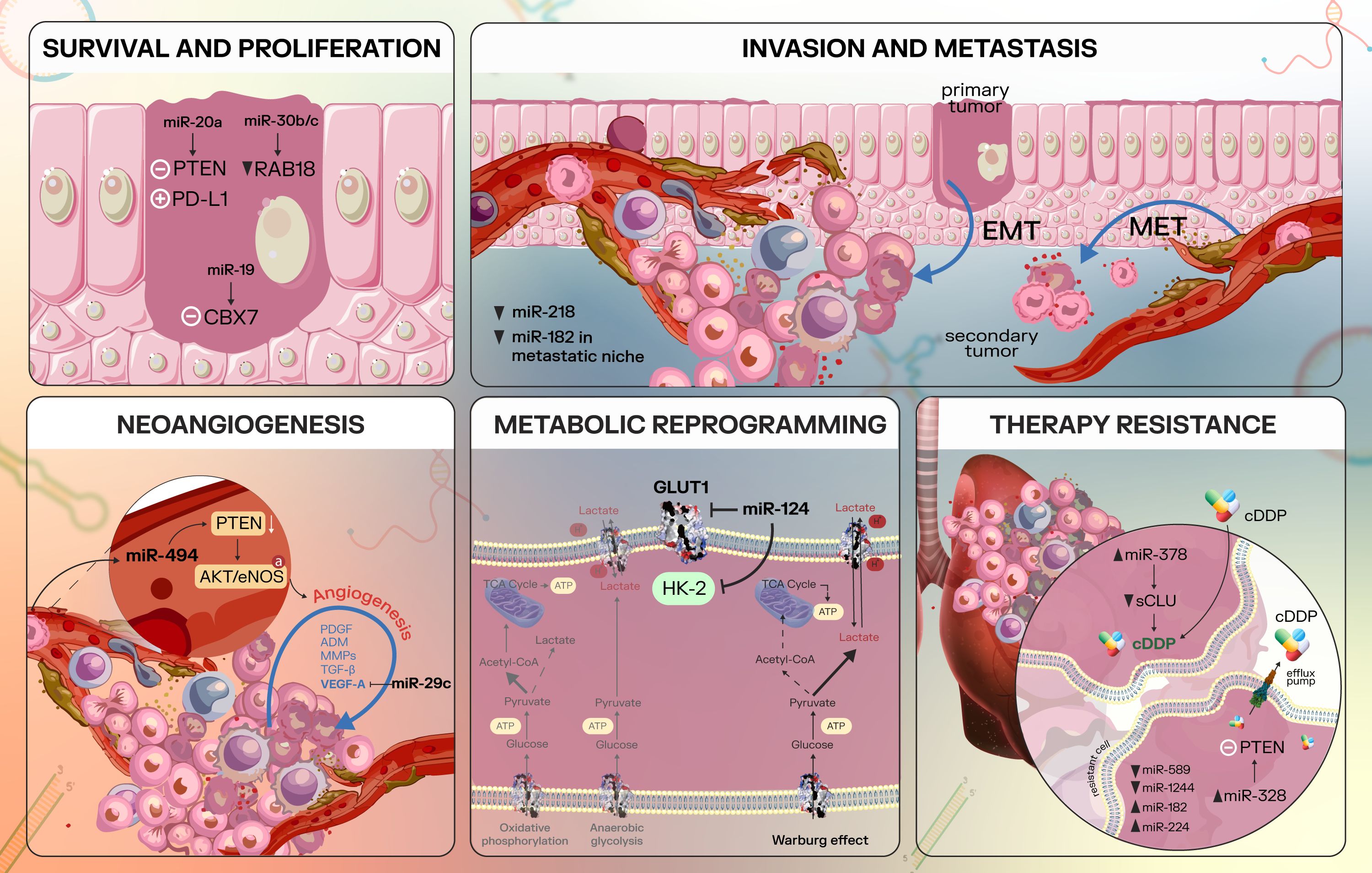
Figure 3. Overview of some cancer processes regulated by microRNAs in lung cancer. Survival and Proliferation: in lung cancer, miR-20a inhibits PTEN and upregulates PD-L1, promoting NSCLC cell proliferation (116); miR-30b/c is able to target Rab18 directly, leading to Rab18 repression and inhibition of NSCLC cell proliferation (118); miR-19 is an oncogenic miRNA that inhibits CBX7 expression (119). Invasion and metastasis: metastasis implies several essential processes, including epithelial-to-mesenchymal transition, with increased cancer cell invasiveness, motility and the ability to degrade the ECM. Furthermore, the mesenchymal-to-epithelial transition reverses these mesenchymal characteristics to epithelial-like characteristics (122, 123). In lung cancer, miR-218 is able to suppress invasion and metastatic spread by targeting key factors that modulate the EMT process - Slug and ZEB2 (124). In addition, a study found decreased miR-182 levels in metastatic NSCLC cells compared to the primary tumor and miR-182 may inhibit NSCLC EMT via inactivation of MET/AKT/Snail signaling pathway (65). Neoangiogenesis: essential pro-angiogenic factors include VEGFA, FGF2, PDGFB, EGF, MMP2 (126); miR-29c is able to target VEGFA and thus act as a tumor suppressor (127). MiR-494 targets and downregulates PTEN, with a consecutive activation of the AKT/eNOS pathway and stimulation of neoangiogenesis (128). Metabolic reprogramming: miR-124 interferes with cancer cell metabolism by targeting GLUT1 and HKII, negatively regulating AKT1 and AKT2 (132). Therapy resistance: miRNAs are largely implicated in therapy resistance in NSCLC, which is a multifaceted process. MiR-378 upregulation downregulates secreted form clusterin (sCLU) via direct targeting and led to sensitizing the NSCLC cells to cDDP (134). Other miRNAs implicated in NSCLC therapy resistance include the miR-328/PTEN duo (135), miR-589, miR-1244, miR-182, and miR-224 (136), with various expression levels of these non-coding RNAs.
Furthermore, Table 3 integrates a larger variety of miRNAs that modulate essential cancer processes, exemplifying their wide distribution in this malignancy.
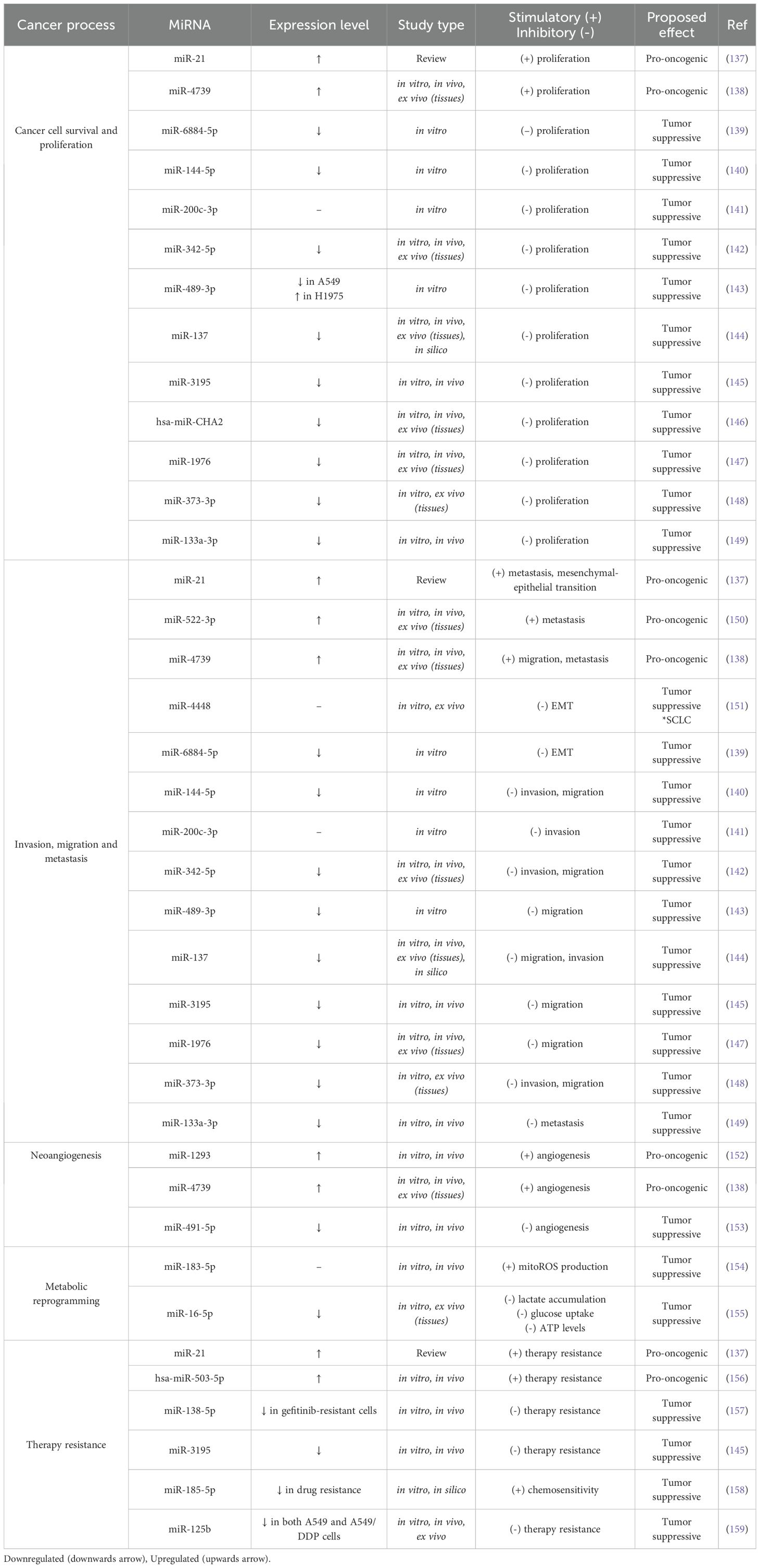
Table 3. A selection of miRNAs that modulate the main cancer processes.
4 Implications and future directions
As exemplified, lung cancer is an intricate malignancy with high incidence and poor overall prognosis. Recent developments in the field have brought a modest improvement in patient survival, with the addition of immunotherapy and targeted therapeutics in specific circumstances. Herein, we have critically reviewed the main driver mutations that may be identified in lung cancer, along with essential data regarding the associated signaling pathways and their modulation via non-coding RNAs, specifically miRNAs.
MiRNAs emerged as key players with modulatory roles across the spectra of cancer processes - from lung cancer development, to progression. Indeed, these entities are able to modulate cancer cell proliferation, invasion and metastasis, neoangiogenesis, metabolic effects, therapy resistance and other. Hence, considering their large implication in cancer, the interest in their use as therapeutic targets, agents, or even biomarkers for diagnosis/prognosis is increasing.
From a biomarker perspective, in a study by Liu et al. (160), the authors analyzed in 168 early-stage NSCLC patients, 100 healthy volunteers and 128 patients with benign lung nodules a number of clinical and biochemical parameters, along with miR-200 expression in peripheral blood-derived extracellular vesicles (EVs). The parameters taken into account included carbohydrate antigen 199 (CA199), carbohydrate antigen 242 (CA242), carcinoembryonic antigen (CEA), interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α). Interestingly, peripheral blood-derived miR-200 EVs displayed diagnostic value, with a sensitivity of 60.12%, specificity of 95.18%, area under the curve (AUC) = 0.855 [95% CI: 0.816-0.888], p < 0.001 for early-stage NSCLC. Furthermore, the addition of CA242, CEA and CA199 besides peripheral blood-derived miR-200 EVs displayed increased diagnostic efficacy, with a sensitivity of 89.88%, specificity of 98.68% and AUC = 0.942 [95% CI: 0.914-0.964], p < 0.001 for early-stage NSCLC, rendering the potential utility of miR-200 as a biomarker in this malignancy (160).
Furthermore, Wozniak et al. (161) sampled 100 NSCLC patients stages I to IIIA, marked as early-stage NSCLC and 100 non-cancer controls, screening 754 circulating miRNAs through qRT-PCR. This led to a model of 24-miRNA panel which can be consulted in the Wozniak study (161). Herein, logistic regression analyses showed diagnostic potential for the 24-miRNA panel with an AUC = 0.92 [95% CI: 0.87-0.95] in discriminating lung cancer cases from controls. Furthermore, when adjusted to sex, age and smoking status, the diagnostic efficacy of the combined 24-miRNA panel increased to an AUC = 0.94 [95% CI: 0.90-0.97]. Moreover, the 24-miRNA panel had similar performances across the different NSCLC subtypes - with an AUC = 0.94 for LUADs and AUC = 0.96 for LUSCs. In addition, subgroup analyses showed AUC = 0.96 for stage I (IA and IB), AUC = 0.98 for stage II (IIA and IIB) and AUC = 0.97 for stage IIIA NSCLC patients (161).
In a study by Wang et al. (162), the authors overlapped miRNA data set from miRNA sequencing data of 8 specimens that were collected from thoracic surgery of non-smoking female patients with LUAD and data extracted from the TCGA database. The authors identified hsa-miR-200a, hsa-miR-21 and hsa-miR-584 as miRNAs significantly associated with OS in this subset of patients, highlighting the potential of these miRNAs to be used as a prognostic model (162).
Abdipourbozorgbaghi et al. (163) analyzed plasma miRNA expression in a cohort of 122 patients - 78 NSCLC patients and 44 healthy controls. Although the authors found that miRNA expression levels in LUAD were independent of tumor stage, 2 miRNAs were identified as early-stage (stage I and II) biomarkers - hsa-miR-210-3p and hsa-miR-301a-5p, whilst hsa-miR-9-5p, hsa-miR-141-5p and hsa-miR-147b-3p were identified as late-stage (stage III and IV) biomarkers. Conversely, there was a higher variability between the stages, with only 6 miRNAs being common. Hsa-miR-210-3p and hsa-miR-301a-5p were found to be early-stage miRNAs in both LUAD and LUSC, and hsa-miR-9-5p was a late-stage biomarker in both LUAD and LUSC patients. The authors concluded with one miRNA diagnosis panel that included 7 miRNAs for LUAD diagnosis and a panel of 9 miRNAs for LUSC diagnosis. MiR-135b-5p, miR-196a-5p and miR-31-5p (LUAD) were found to be independent prognostic markers for survival in LUAD and miR-205 for LUSC (163).
From a practical perspective, the detection of miRNAs in liquid biopsy is constantly evolving and perfecting, as miRNAs are the most studied ncRNAs in liquid biopsies (164). According to Ma et al., several detection methods have been used for miRNAs, including qPCR, rolling circle amplification, strand displacement amplification and hybridization chain reaction (164). Although the detection of miRNAs in liquid biopsy holds great potential for future, current large-scale use is hampered by numerous factors. Limitations in moving from preclinical models to clinical applications include the need for validation in large-scale population (165), laboratory standardization (164), differences in sample processing techniques (166), and others.
The reproducibility of these markers/panel of miRNAs in clinical practice is seldom with great success. A major limitation is the reproducibility in independent cohorts due to variability in several parameters, including patient characteristics (e.g., age, sex), sample collection, analytical miRNA detection strategies, and others. Furthermore, miRNA expression is modulated via multiple methods, such as RNA-binding proteins (167), marking their potential variability between subjects. A future direction is also represented by the integration of miRNA panels with genomic and epigenomic data, leading to a more comprehensive characterization of the tumor. Yang et al. showed that there is a crosstalk between miRNA expression for immune regulation, DNA methylation and copy number variation in glioma (168).
When considering miRNAs in a therapeutic context, the agents can act either as miRNA mimics or miRNA inhibitors. Table 4 presents a selection of NSCLC preclinical studies that focus on miRNA mimics.
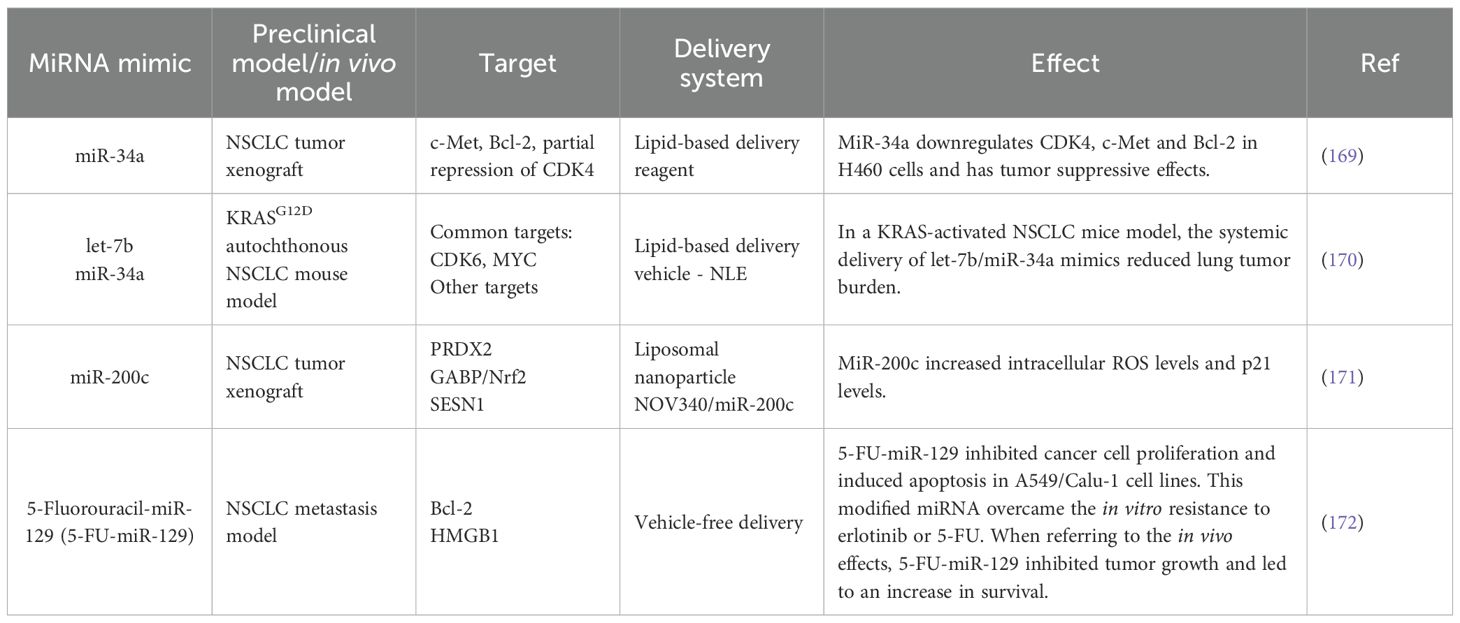
Table 4. Examples of miRNA mimics in NSCLC preclinical studies.
5 Conclusion
In the present paper, we have critically reviewed the main prospects pertaining to the effects of miRNAs in the personalized medicine of lung cancer.
Several mutated/genetically altered genes drive lung cancer to a more aggressive phenotype. For instance, EGFR L858R point mutation in exon 21 and the LREA in-frame deletion on exon 19 are two EGFR activating mutations, which are druggable via EGFR TKIs. ALK rearrangements lead to constitutive ALK kinase activity, with lung tumors exhibiting increased aggressive behavior. This includes nodal metastasis and advanced stages at diagnosis; these ALK rearrangements are also druggable via targeted therapeutics such as crizotinib. Furthermore, lung cancer tumors harboring KRAS mutations carry usually a poor prognosis; KRAS activation induces interactions with several key pathways, such as the RAF/MEK/ERK pathway and the PI3K/AKT pathway. Targeted therapeutics are also available for patients harboring KRAS mutation - sotorasib, or adagrasib for KRAS G12C mutation. Other well-known genetic alterations that may be encountered in lung cancers include ROS1 rearrangements, RET translocations, alterations in the MET gene, NTRK fusions and BRAF mutations. Herein, we also provide a selection of miRNAs that target these essential driver genes/signaling pathways in lung cancer - for instance, miR-33a and miR-128b that target the EGFR pathway, miR-200c and miR-449 that target the ALK signaling, miR-205 that interferes with MET, miR-148a-3p that is tumor suppressive on the RAS/MAPK/ERK signaling and miR-31 that has pro-oncogenic effect by modulating the RAS/MAPK pathway.
Furthermore, for a better integrative overview, our paper critically discusses the main cancer processes that govern lung cancer progression, along with essential prospects about miRNA modulation of these processes. We underline the implication of miR-28 as a promoter of cancer cell proliferation and miR-218 as a suppressor of invasion and metastatic spread via targeting Slug, ZEB2 and EMT. MiR-29c acts as a tumor suppressor via targeting VEGFA and several miRNAs have been described to be implicated in therapy resistance - miR-378, miR-328, miR-589, miR-1244.
When considering potential clinical applications and future directions in miRNA research in lung cancer, our paper highlights several developments in the field of lung cancer biomarkers - a combined peripheral blood-derived miR-200 EVs with CA242, CEA and CA199 that displays high diagnostic performance in early-stage NSCLC and also other miRNA diagnostic panels that may prove useful in early diagnosis or even in discriminating LUAD from LUSC. Although the liquid biopsy strategy is not entirely standardized and developed, it appears that the constant evolution of the field holds great potential.
The field of lung cancer is rapidly advancing, driven by the high mortality rates associated with this malignancy. The addition of ncRNAs and specifically of miRNAs into the complex molecular framework of lung cancer has the potential to improve patient care. Although there are still numerous aspects that need to be apprehended for a better integration of miRNA study in lung cancer, significant steps forward have already been achieved, as highlighted in this paper.
Author contributions
A-AT: Conceptualization, Investigation, Methodology, Writing – original draft, Writing – review & editing. AN: Visualization, Writing – review & editing. CB: Writing – original draft. O-LP: Writing – review & editing. IB-N: Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Acknowledgments
This work was supported by the internal grant received by A-AT, titled “The role of coding and non-coding genome in lung cancer progression and therapy resistance” - no. 777/44/13.01.2025 PCD from the “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania. This work was also supported by the Competitivity Operational Program, 2014–2020, entitled “Clinical and economical impact of personalized targeted anti-microRNA therapies in reconverting lung cancer chemoresistance”—CANTEMIR, number 35/01.09.2016, MySMIS 103375.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2024) 74:229–63. doi: 10.3322/caac.21834
2. Tirpe AA, Gulei D, Ciortea SM, Crivii C, and Berindan-Neagoe I. Hypoxia: overview on hypoxia-mediated mechanisms with a focus on the role of HIF genes. Int J Mol Sci. (2019) 20:6140. doi: 10.3390/ijms20246140
3. Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. (2014) 371:2167–77. doi: 10.1056/NEJMoa1408440
4. Planchard D, Besse B, Groen HJM, Hashemi SMS, Mazieres J, Kim TM, et al. Phase 2 study of dabrafenib plus trametinib in patients with BRAF V600E-mutant metastatic NSCLC: updated 5-year survival rates and genomic analysis. J Thorac Oncol. (2022) 17:103–15. doi: 10.1016/j.jtho.2021.08.011
5. Zappa C and Mousa SA. Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res. (2016) 5:288–300. doi: 10.21037/tlcr.2016.06.07
6. Duma N, Santana-Davila R, and Molina JR. Non-small cell lung cancer: epidemiology, screening, diagnosis, and treatment. Mayo Clin Proc. (2019) 94:1623–40. doi: 10.1016/j.mayocp.2019.01.013
7. Plank JL and Dean A. Enhancer function: mechanistic and genome-wide insights come together. Mol Cell. (2014) 55:5–14. doi: 10.1016/j.molcel.2014.06.015
8. Statello L, Guo CJ, Chen LL, and Huarte M. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol. (2021) 22:96–118. doi: 10.1038/s41580-020-00315-9
9. Kim SY, Na MJ, Yoon S, Shin E, Ha JW, Jeon S, et al. The roles and mechanisms of coding and noncoding RNA variations in cancer. Exp Mol Med. (2024) 56:1909–20. doi: 10.1038/s12276-024-01307-x
10. Blair LM, Juan JM, Sebastian L, Tran VB, Nie W, Wall GD, et al. Oncogenic context shapes the fitness landscape of tumor suppression. Nat Commun. (2023) 14:6422. doi: 10.1038/s41467-023-42156-y
11. Peng Y and Croce CM. The role of MicroRNAs in human cancer. Signal Transduct Target Ther. (2016) 1:15004. doi: 10.1038/sigtrans.2015.4
12. Nemeth K, Bayraktar R, Ferracin M, and Calin GA. Non-coding RNAs in disease: from mechanisms to therapeutics. Nat Rev Genet. (2024) 25:211–32. doi: 10.1038/s41576-023-00662-1
13. Shang R, Lee S, Senavirathne G, and Lai EC. microRNAs in action: biogenesis, function and regulation. Nat Rev Genet. (2023) 24:816–33. doi: 10.1038/s41576-023-00611-y
14. Lee RC, Feinbaum RL, and Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. (1993) 75:843–54. doi: 10.1016/0092-8674(93)90529-Y
15. Arasu P, Wightman B, and Ruvkun G. Temporal regulation of lin-14 by the antagonistic action of two other heterochronic genes, lin-4 and lin-28. Genes Dev. (1991) 5:1825–33. doi: 10.1101/gad.5.10.1825
16. Wightman B, Burglin TR, Gatto J, Arasu P, and Ruvkun G. Negative regulatory sequences in the lin-14 3'-untranslated region are necessary to generate a temporal switch during Caenorhabditis elegans development. Genes Dev. (1991) 5:1813–24. doi: 10.1101/gad.5.10.1813
17. Si W, Shen J, Zheng H, and Fan W. The role and mechanisms of action of microRNAs in cancer drug resistance. Clin epigenetics. (2019) 11:25. doi: 10.1186/s13148-018-0587-8
18. Jame-Chenarboo F, Ng HH, Macdonald D, and Mahal LK. High-Throughput Analysis Reveals miRNA Upregulating alpha-2,6-Sialic Acid through Direct miRNA-mRNA Interactions. ACS Cent Sci. (2022) 8:1527–36. doi: 10.1021/acscentsci.2c00748
19. da Cunha Santos G, Shepherd FA, and Tsao MS. EGFR mutations and lung cancer. Annu Rev Pathol. (2011) 6:49–69. doi: 10.1146/annurev-pathol-011110-130206
20. Diaz-Serrano A, Gella P, Jimenez E, Zugazagoitia J, and Paz-Ares Rodriguez L. Targeting EGFR in lung cancer: current standards and developments. Drugs. (2018) 78:893–911. doi: 10.1007/s40265-018-0916-4
21. Bai X, Sun P, Wang X, Long C, Liao S, Dang S, et al. Structure and dynamics of the EGFR/HER2 heterodimer. Cell Discov. (2023) 9:18. doi: 10.1038/s41421-023-00523-5
22. Uribe ML, Marrocco I, and Yarden Y. EGFR in cancer: signaling mechanisms, drugs, and acquired resistance. Cancers (Basel). (2021) 13:2748. doi: 10.3390/cancers13112748
23. Wee P and Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers (Basel). (2017) 9:52. doi: 10.3390/cancers9050052
24. Pao W and Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. (2010) 10:760–74. doi: 10.1038/nrc2947
25. Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci United States America. (2008) 105:2070–5. doi: 10.1073/pnas.0709662105
26. Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. New Engl J Med. (2018) 378:113–25. doi: 10.1056/NEJMoa1713137
27. Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. New Engl J Med. (2020) 382:41–50. doi: 10.1056/NEJMoa1913662
28. Planchard D, Janne PA, Cheng Y, Yang JC, Yanagitani N, Kim SW, et al. Osimertinib with or without chemotherapy in EGFR-mutated advanced NSCLC. New Engl J Med. (2023) 389:1935–48. doi: 10.1056/NEJMoa2306434
29. Hendriks LE, Kerr KM, Menis J, Mok TS, Nestle U, Passaro A, et al. Oncogene-addicted metastatic non-small-cell lung cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. (2023) 34:339–57. doi: 10.1016/j.annonc.2022.12.009
30. Liu J, Wang W, Wang K, Liu W, Zhao Y, Han X, et al. HDAC1 and FOXK1 mediate EGFR-TKI resistance of non-small cell lung cancer through miR-33a silencing. J Transl Med. (2024) 22:793. doi: 10.1186/s12967-024-05563-3
31. Weiss GJ, Bemis LT, Nakajima E, Sugita M, Birks DK, Robinson WA, et al. EGFR regulation by microRNA in lung cancer: correlation with clinical response and survival to gefitinib and EGFR expression in cell lines. Ann Oncol. (2008) 19:1053–9. doi: 10.1093/annonc/mdn006
32. Li XF, Shen WZ, Jin X, Ren P, and Zhang J. Let-7c regulated epithelial-mesenchymal transition leads to osimertinib resistance in NSCLC cells with EGFR T790M mutations. Sci Rep. (2020) 10:11236. doi: 10.1038/s41598-020-67908-4
33. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. (2007) 448:561–6. doi: 10.1038/nature05945
34. Du X, Shao Y, Qin HF, Tai YH, and Gao HJ. ALK-rearrangement in non-small-cell lung cancer (NSCLC). Thorac Cancer. (2018) 9:423–30. doi: 10.1111/tca.2018.9.issue-4
35. Holla VR, Elamin YY, Bailey AM, Johnson AM, Litzenburger BC, Khotskaya YB, et al. ALK: a tyrosine kinase target for cancer therapy. Cold Spring Harb Mol Case Stud. (2017) 3:a001115. doi: 10.1101/mcs.a001115
36. Addeo A, Tabbo F, Robinson T, Buffoni L, and Novello S. Precision medicine in ALK rearranged NSCLC: A rapidly evolving scenario. Crit Rev oncology/hematology. (2018) 122:150–6. doi: 10.1016/j.critrevonc.2017.12.015
37. Addeo A, Tabbo F, Robinson T, Buffoni L, and Novello S. Corrigendum to "Precision medicine in ALK rearranged NSCLC: A rapidly evolving scenario. Crit Rev oncology/hematology. (2019) 139:158. doi: 10.1016/j.critrevonc.2018.06.018
38. Pikor LA, Ramnarine VR, Lam S, and Lam WL. Genetic alterations defining NSCLC subtypes and their therapeutic implications. Lung cancer. (2013) 82:179–89. doi: 10.1016/j.lungcan.2013.07.025
39. Kim H, Jang SJ, Chung DH, Yoo SB, Sun P, Jin Y, et al. A comprehensive comparative analysis of the histomorphological features of ALK-rearranged lung adenocarcinoma based on driver oncogene mutations: frequent expression of epithelial-mesenchymal transition markers than other genotype. PloS One. (2013) 8:e76999. doi: 10.1371/journal.pone.0076999
40. Shaw AT, Kim TM, Crino L, Gridelli C, Kiura K, Liu G, et al. Ceritinib versus chemotherapy in patients with ALK-rearranged non-small-cell lung cancer previously given chemotherapy and crizotinib (ASCEND-5): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. (2017) 18:874–86. doi: 10.1016/S1470-2045(17)30339-X
41. Novello S, Mazieres J, Oh IJ, de Castro J, Migliorino MR, Helland A, et al. Alectinib versus chemotherapy in crizotinib-pretreated anaplastic lymphoma kinase (ALK)-positive non-small-cell lung cancer: results from the phase III ALUR study. Ann Oncol Off J Eur Soc Med Oncol. (2018) 29:1409–16. doi: 10.1093/annonc/mdy121
42. Solomon BJ, Besse B, Bauer TM, Felip E, Soo RA, Camidge DR, et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: results from a global phase 2 study. Lancet Oncol. (2018) 19:1654–67. doi: 10.1016/S1470-2045(18)30649-1
43. Lai Y, Kacal M, Kanony M, Stukan I, Jatta K, Kis L, et al. miR-100-5p confers resistance to ALK tyrosine kinase inhibitors Crizotinib and Lorlatinib in EML4-ALK positive NSCLC. Biochem Biophys Res Commun. (2019) 511:260–5. doi: 10.1016/j.bbrc.2019.02.016
44. Fukuda K, Takeuchi S, Arai S, Katayama R, Nanjo S, Tanimoto A, et al. Epithelial-to-mesenchymal transition is a mechanism of ALK inhibitor resistance in lung cancer independent of ALK mutation status. Cancer Res. (2019) 79:1658–70. doi: 10.1158/0008-5472.CAN-18-2052
45. Yun MR, Lim SM, Kim SK, Choi HM, Pyo KH, Kim SK, et al. Enhancer remodeling and microRNA alterations are associated with acquired resistance to ALK inhibitors. Cancer Res. (2018) 78:3350–62. doi: 10.1158/0008-5472.CAN-17-3146
46. Roskoski R Jr. ROS1 protein-tyrosine kinase inhibitors in the treatment of ROS1 fusion protein-driven non-small cell lung cancers. Pharmacol Res. (2017) 121:202–12. doi: 10.1016/j.phrs.2017.04.022
47. Gainor JF and Shaw AT. Novel targets in non-small cell lung cancer: ROS1 and RET fusions. oncologist. (2013) 18:865–75. doi: 10.1634/theoncologist.2013-0095
48. Lin JJ and Shaw AT. Recent advances in targeting ROS1 in lung cancer. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer. (2017) 12:1611–25. doi: 10.1016/j.jtho.2017.08.002
49. D'Angelo A, Sobhani N, Chapman R, Bagby S, Bortoletti C, Traversini M, et al. Focus on ROS1-positive non-small cell lung cancer (NSCLC): crizotinib, resistance mechanisms and the newer generation of targeted therapies. Cancers. (2020) 12:3293. doi: 10.3390/cancers12113293
50. Yan C, Zhang W, Shi X, Zheng J, Jin X, and Huo J. MiR-760 suppresses non-small cell lung cancer proliferation and metastasis by targeting ROS1. Environ Sci pollut Res Int. (2018) 25:18385–91. doi: 10.1007/s11356-017-1138-0
51. Yan C, Zhang W, Shi X, Zheng J, Jin X, and Huo J. Correction to: MiR-760 suppresses non-small cell lung cancer proliferation and metastasis by targeting ROS1. Environ Sci pollut Res Int. (2018) 25:18392. doi: 10.1007/s11356-018-2354-y
52. Matsubara D, Kanai Y, Ishikawa S, Ohara S, Yoshimoto T, Sakatani T, et al. Identification of CCDC6-RET fusion in the human lung adenocarcinoma cell line, LC-2/ad. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer. (2012) 7:1872–6. doi: 10.1097/JTO.0b013e3182721ed1
53. Drusbosky LM, Rodriguez E, Dawar R, and Ikpeazu CV. Therapeutic strategies in RET gene rearranged non-small cell lung cancer. J Hematol Oncol. (2021) 14:50. doi: 10.1186/s13045-021-01063-9
54. Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. (2012) 18:378–81. doi: 10.1038/nm.2658
55. Drilon A, Lin JJ, Filleron T, Ni A, Milia J, Bergagnini I, et al. Frequency of brain metastases and multikinase inhibitor outcomes in patients with RET-rearranged lung cancers. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer. (2018) 13:1595–601. doi: 10.1016/j.jtho.2018.07.004
56. Spagnolo CC, Ciappina G, Giovannetti E, Squeri A, Granata B, Lazzari C, et al. Targeting MET in non-small cell lung cancer (NSCLC): A new old story? Int J Mol Sci. (2023) 24:10119. doi: 10.3390/ijms241210119
57. Sakamoto M and Patil T. MET alterations in advanced non-small cell lung cancer. Lung cancer. (2023) 178:254–68. doi: 10.1016/j.lungcan.2023.02.018
58. Fujino T, Suda K, and Mitsudomi T. Lung cancer with MET exon 14 skipping mutation: genetic feature, current treatments, and future challenges. Lung Cancer (Auckl). (2021) 12:35–50. doi: 10.2147/LCTT.S269307
59. Liang H and Wang M. MET oncogene in non-small cell lung cancer: mechanism of MET dysregulation and agents targeting the HGF/c-met axis. OncoTargets Ther. (2020) 13:2491–510. doi: 10.2147/OTT.S231257
60. Kawakami H, Okamoto I, Okamoto W, Tanizaki J, Nakagawa K, and Nishio K. Targeting MET amplification as a new oncogenic driver. Cancers. (2014) 6:1540–52. doi: 10.3390/cancers6031540
61. Sun D, Wu W, Wang L, Qu J, Han Q, Wang H, et al. Identification of MET fusions as novel therapeutic targets sensitive to MET inhibitors in lung cancer. J Trans Med. (2023) 21:150. doi: 10.1186/s12967-023-03999-7
62. Romano G, Le P, Nigita G, Saviana M, Micalo L, Lovat F, et al. A-to-I edited miR-411-5p targets MET and promotes TKI response in NSCLC-resistant cells. Oncogene. (2023) 42:1597–606. doi: 10.1038/s41388-023-02673-y
63. Yang X, Su W, Li Y, Zhou Z, Zhou Y, Shan H, et al. MiR-22-3p suppresses cell growth via MET/STAT3 signaling in lung cancer. Am J Transl Res. (2021) 13:1221–32.
64. Migliore C, Morando E, Ghiso E, Anastasi S, Leoni VP, Apicella M, et al. miR-205 mediates adaptive resistance to MET inhibition via ERRFI1 targeting and raised EGFR signaling. EMBO Mol Med. (2018) 10. doi: 10.15252/emmm.201708746
65. Li Y, Zhang H, Li Y, Zhao C, Fan Y, Liu J, et al. MiR-182 inhibits the epithelial to mesenchymal transition and metastasis of lung cancer cells by targeting the Met gene. Mol Carcinog. (2018) 57:125–36. doi: 10.1002/mc.22741
66. Liu F, Wei Y, Zhang H, Jiang J, Zhang P, and Chu Q. NTRK fusion in non-small cell lung cancer: diagnosis, therapy, and TRK inhibitor resistance. Front Oncol. (2022) 12:864666. doi: 10.3389/fonc.2022.864666
67. Harada G, Santini FC, Wilhelm C, and Drilon A. NTRK fusions in lung cancer: From biology to therapy. Lung cancer. (2021) 161:108–13. doi: 10.1016/j.lungcan.2021.09.005
68. Cocco E, Scaltriti M, and Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. (2018) 15:731–47. doi: 10.1038/s41571-018-0113-0
69. Guaitoli G, Zullo L, Tiseo M, Dankner M, Rose AA, and Facchinetti F. Non-small-cell lung cancer: how to manage BRAF-mutated disease. Drugs context. (2023) 12:2022-11-3. doi: 10.7573/dic.2022-11-3
70. Hanrahan AJ, Chen Z, Rosen N, and Solit DB. BRAF - a tumour-agnostic drug target with lineage-specific dependencies. Nat Rev Clin Oncol. (2024) 21:224–47. doi: 10.1038/s41571-023-00852-0
71. Caunt CJ, Sale MJ, Smith PD, and Cook SJ. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer. (2015) 15:577–92. doi: 10.1038/nrc4000
72. Dankner M, Rose AAN, Rajkumar S, Siegel PM, and Watson IR. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene. (2018) 37:3183–99. doi: 10.1038/s41388-018-0171-x
73. Ricciuti B, Mira A, Andrini E, Scaparone P, Michelina SV, Pecci F, et al. How to manage KRAS G12C-mutated advanced non-small-cell lung cancer. Drugs context. (2022) 11:2022-7-4. doi: 10.7573/dic.2022-7-4
74. Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. (2014) 511:543–50. doi: 10.1038/nature13385
75. Luo J, Ostrem J, Pellini B, Imbody D, Stern Y, Solanki HS, et al. Overcoming KRAS-mutant lung cancer. Am Soc Clin Oncol Educ book Am Soc Clin Oncol Annu Meeting. (2022) 42:1–11. doi: 10.1200/EDBK_360354
76. Ricciuti B, Son J, Okoro JJ, Mira A, Patrucco E, Eum Y, et al. Comparative analysis and isoform-specific therapeutic vulnerabilities of KRAS mutations in non-small cell lung cancer. Clin Cancer research: an Off J Am Assoc Cancer Res. (2022) 28:1640–50. doi: 10.1158/1078-0432.CCR-21-2719
77. Huang L, Guo Z, Wang F, and Fu L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther. (2021) 6:386. doi: 10.1038/s41392-021-00780-4
78. Karimi N and Moghaddam SJ. KRAS-mutant lung cancer: targeting molecular and immunologic pathways, therapeutic advantages and restrictions. Cells. (2023) 12:749. doi: 10.3390/cells12050749
79. Westcott PM and To MD. The genetics and biology of KRAS in lung cancer. Chin J Cancer. (2013) 32:63–70. doi: 10.5732/cjc.012.10098
80. Liu J, Kang R, and Tang D. The KRAS-G12C inhibitor: activity and resistance. Cancer Gene Ther. (2022) 29:875–8. doi: 10.1038/s41417-021-00383-9
81. Liu J, Kang R, and Tang D. Correction: The KRAS-G12C inhibitor: activity and resistance. Cancer Gene Ther. (2023) 30:1715. doi: 10.1038/s41417-023-00692-1
82. Hendriks LE, Kerr KM, Menis J, Mok TS, Nestle U, Passaro A, et al. Non-oncogene-addicted metastatic non-small-cell lung cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. (2023) 34:358–76. doi: 10.1016/j.annonc.2022.12.013
83. de Langen AJ, Johnson ML, Mazieres J, Dingemans AC, Mountzios G, Pless M, et al. Sotorasib versus docetaxel for previously treated non-small-cell lung cancer with KRAS(G12C) mutation: a randomised, open-label, phase 3 trial. Lancet. (2023) 401:733–46. doi: 10.1016/S0140-6736(23)00221-0
84. Janne PA, Riely GJ, Gadgeel SM, Heist RS, Ou SI, Pacheco JM, et al. Adagrasib in non-small-cell lung cancer harboring a KRAS(G12C) mutation. New Engl J Med. (2022) 387:120–31. doi: 10.1056/NEJMoa2204619
85. Xie Q, Yu Z, Lu Y, Fan J, Ni Y, and Ma L. microRNA-148a-3p inhibited the proliferation and epithelial-mesenchymal transition progression of non-small-cell lung cancer via modulating Ras/MAPK/Erk signaling. J Cell Physiol. (2019) 234:12786–99. doi: 10.1002/jcp.v234.8
86. Edmonds MD, Boyd KL, Moyo T, Mitra R, Duszynski R, Arrate MP, et al. MicroRNA-31 initiates lung tumorigenesis and promotes mutant KRAS-driven lung cancer. J Clin Invest. (2016) 126:349–64. doi: 10.1172/JCI82720
87. Li X, Wang Y, Cheng J, Qiu L, Wang R, Zhang Y, et al. METTL3 -mediated m6A modification of circ_0000620 regulates cisplatin sensitivity and apoptosis in lung adenocarcinoma via the MiR-216b-5p/KRAS axis. Cell Signal. (2024) 123:111349. doi: 10.1016/j.cellsig.2024.111349
88. Yan H, Chen X, Li Y, Fan L, Tai Y, Zhou Y, et al. MiR-1205 functions as a tumor suppressor by disconnecting the synergy between KRAS and MDM4/E2F1 in non-small cell lung cancer. Am J Cancer Res. (2025) 15:2908–2910. doi: 10.62347/XVNE7528
89. Wu J, Savooji J, and Liu D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J Hematol Oncol. (2016) 9:19. doi: 10.1186/s13045-016-0251-8
90. Frampton JE. Entrectinib: A review in NTRK+ Solid tumours and ROS1+ NSCLC. Drugs. (2021) 81:697–708. doi: 10.1007/s40265-021-01503-3
91. Novello S, Califano R, Reinmuth N, Tamma A, and Puri T. RET fusion-positive non-small cell lung cancer: the evolving treatment landscape. Oncologist. (2023) 28:402–13. doi: 10.1093/oncolo/oyac264
92. Kelly RJ. Dabrafenib and trametinib for the treatment of non-small cell lung cancer. Expert Rev Anticancer Ther. (2018) 18:1063–8. doi: 10.1080/14737140.2018.1521272
93. O'Sullivan E, Keogh A, Henderson B, Finn SP, Gray SG, and Gately K. Treatment strategies for KRAS-mutated non-small-cell lung cancer. Cancers (Basel). (2023) 15. doi: 10.3390/cancers15061635
94. Lander ES. Initial impact of the sequencing of the human genome. Nature. (2011) 470:187–97. doi: 10.1038/nature09792
95. Zhang X and Meyerson M. Illuminating the noncoding genome in cancer. Nat cancer. (2020) 1:864–72. doi: 10.1038/s43018-020-00114-3
96. Lindner G, Takenaka K, Santucci K, Gao Y, and Janitz M. Protein-coding circular RNAs - mechanism, detection, and their role in cancer and neurodegenerative diseases. Biochem Biophys Res Commun. (2023) 678:68–77. doi: 10.1016/j.bbrc.2023.08.037
97. Saliminejad K, Khorram Khorshid HR, Soleymani Fard S, and Ghaffari SH. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J Cell Physiol. (2019) 234:5451–65. doi: 10.1002/jcp.v234.5
98. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. (2022) 12:31–46. doi: 10.1158/2159-8290.CD-21-1059
99. Gotte M and Kovalszky I. Extracellular matrix functions in lung cancer. Matrix Biol. (2018) 73:105–21. doi: 10.1016/j.matbio.2018.02.018
100. Wang J, Wang L, Qiang W, and Ge W. The role of DDR1 in cancer and the progress of its selective inhibitors. Bioorg Chem. (2025) 154:108018. doi: 10.1016/j.bioorg.2024.108018
101. Ambrogio C, Gomez-Lopez G, Falcone M, Vidal A, Nadal E, Crosetto N, et al. Combined inhibition of DDR1 and Notch signaling is a therapeutic strategy for KRAS-driven lung adenocarcinoma. Nat Med. (2016) 22:270–7. doi: 10.1038/nm.4041
102. Nokin MJ, Darbo E, Travert C, Drogat B, Lacouture A, San Jose S, et al. Inhibition of DDR1 enhances in vivo chemosensitivity in KRAS-mutant lung adenocarcinoma. JCI Insight. (2020) 5:e137869. doi: 10.1172/jci.insight.137869
103. Ming F, Li B, Yi S, and Pi G. Circ_0087378 intensifies the Malignant behavior of non-small cell lung cancer cells in vitro by facilitating DDR1 via sponging miR-199a-5p. Transl Lung Cancer Res. (2023) 12:770–85. doi: 10.21037/tlcr-23-88
104. Desgrosellier JS and Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. (2010) 10:9–22. doi: 10.1038/nrc2748
105. Ka M, Matsumoto Y, Ando T, Hinata M, Xi Q, Sugiura Y, et al. Integrin-alpha5 expression and its role in non-small cell lung cancer progression. Cancer Sci. (2025) 116:406–19. doi: 10.1111/cas.v116.2
106. Chen X, Wei L, and Zhao S. miR-338 inhibits the metastasis of lung cancer by targeting integrin beta3. Oncol Rep. (2016) 36:1467–74. doi: 10.3892/or.2016.4928
107. Wang H, Zhu Y, Zhao M, Wu C, Zhang P, Tang L, et al. miRNA-29c suppresses lung cancer cell adhesion to extracellular matrix and metastasis by targeting integrin beta1 and matrix metalloproteinase2 (MMP2). PloS One. (2013) 8:e70192. doi: 10.1371/journal.pone.0070192
108. Martinez-Espinosa I, Serrato JA, and Ortiz-Quintero B. The role of exosome-derived microRNA on lung cancer metastasis progression. Biomolecules. (2023) 13:1574. doi: 10.3390/biom13111574
109. Huang W, Yan Y, Liu Y, Lin M, Ma J, Zhang W, et al. Exosomes with low miR-34c-3p expression promote invasion and migration of non-small cell lung cancer by upregulating integrin alpha2beta1. Signal Transduct Target Ther. (2020) 5:39. doi: 10.1038/s41392-020-0133-y
110. Wang L, Yao J, Sun H, He K, Tong D, Song T, et al. MicroRNA-101 suppresses progression of lung cancer through the PTEN/AKT signaling pathway by targeting DNA methyltransferase 3A. Oncol Lett. (2017) 13:329–38. doi: 10.3892/ol.2016.5423
111. Entezari M, Taheriazam A, Paskeh MDA, Sabouni E, Zandieh MA, Aboutalebi M, et al. The pharmacological and biological importance of EZH2 signaling in lung cancer. BioMed Pharmacother. (2023) 160:114313. doi: 10.1016/j.biopha.2023.114313
112. Xia H, Zhang W, Zhang B, Zhao Y, Zhao Y, Li S, et al. miR-21 modulates the effect of EZH2 on the biological behavior of human lung cancer stem cells in vitro. Oncotarget. (2017) 8:85442–51. doi: 10.18632/oncotarget.20006
113. Cress WD and Seto E. Histone deacetylases, transcriptional control, and cancer. J Cell Physiol. (2000) 184:1–16. doi: 10.1002/(SICI)1097-4652(200007)184:1<1::AID-JCP1>3.0.CO;2-7
114. Jeon HS, Lee SY, Lee EJ, Yun SC, Cha EJ, Choi E, et al. Combining microRNA-449a/b with a HDAC inhibitor has a synergistic effect on growth arrest in lung cancer. Lung Cancer. (2012) 76:171–6. doi: 10.1016/j.lungcan.2011.10.012
115. Chen DQ, Pan BZ, Huang JY, Zhang K, Cui SY, De W, et al. HDAC 1/4-mediated silencing of microRNA-200b promotes chemoresistance in human lung adenocarcinoma cells. Oncotarget. (2014) 5:3333–49. doi: 10.18632/oncotarget.v5i10
116. Gong J, Shen Y, Jiang F, Wang Y, Chu L, Sun J, et al. MicroRNA-20a promotes non-small cell lung cancer proliferation by upregulating PD-L1 by targeting PTEN. Oncol Lett. (2022) 23:148. doi: 10.3892/ol.2022.13269
117. Cui F, Zhou Q, Xiao K, and Qian H. MicroRNA−28 promotes the proliferation of non−small−cell lung cancer cells by targeting PTEN. Mol Med Rep. (2020) 21:2589–96. doi: 10.3892/mmr.2020.11033
118. Zhong K, Chen K, Han L, and Li B. MicroRNA-30b/c inhibits non-small cell lung cancer cell proliferation by targeting Rab18. BMC Cancer. (2014) 14:703. doi: 10.1186/1471-2407-14-703
119. Peng X, Guan L, and Gao B. miRNA-19 promotes non-small-cell lung cancer cell proliferation via inhibiting CBX7 expression. Onco Targets Ther. (2018) 11:8865–74. doi: 10.2147/OTT.S181433
120. Liu M, Zhang Y, Zhang J, Cai H, Zhang C, Yang Z, et al. MicroRNA-1253 suppresses cell proliferation and invasion of non-small-cell lung carcinoma by targeting WNT5A. Cell Death disease. (2018) 9:189. doi: 10.1038/s41419-017-0218-x
121. Yoo JK, Lee JM, Kang SH, Jeon SH, Kim CM, Oh SH, et al. The novel microRNA hsa-miR-CHA1 regulates cell proliferation and apoptosis in human lung cancer by targeting XIAP. Lung cancer. (2019) 132:99–106. doi: 10.1016/j.lungcan.2018.04.011
122. Novikov NM, Zolotaryova SY, Gautreau AM, and Denisov EV. Mutational drivers of cancer cell migration and invasion. Br J Cancer. (2021) 124:102–14. doi: 10.1038/s41416-020-01149-0
123. Lambert AW, Pattabiraman DR, and Weinberg RA. Emerging biological principles of metastasis. Cell. (2017) 168:670–91. doi: 10.1016/j.cell.2016.11.037
124. Shi ZM, Wang L, Shen H, Jiang CF, Ge X, Li DM, et al. Downregulation of miR-218 contributes to epithelial-mesenchymal transition and tumor metastasis in lung cancer by targeting Slug/ZEB2 signaling. Oncogene. (2017) 36:2577–88. doi: 10.1038/onc.2016.414
125. Chang TH, Tsai MF, Gow CH, Wu SG, Liu YN, Chang YL, et al. Upregulation of microRNA-137 expression by Slug promotes tumor invasion and metastasis of non-small cell lung cancer cells through suppression of TFAP2C. Cancer letters. (2017) 402:190–202. doi: 10.1016/j.canlet.2017.06.002
126. Tirpe A, Gulei D, Tirpe GR, Nutu A, Irimie A, Campomenosi P, et al. Beyond conventional: the new horizon of anti-angiogenic microRNAs in non-small cell lung cancer therapy. Int J Mol Sci. (2020) 21:8002. doi: 10.3390/ijms21218002
127. Liu L, Bi N, Wu L, Ding X, Men Y, Zhou W, et al. MicroRNA-29c functions as a tumor suppressor by targeting VEGFA in lung adenocarcinoma. Mol Cancer. (2017) 16:50. doi: 10.1186/s12943-017-0620-0
128. Mao G, Liu Y, Fang X, Liu Y, Fang L, Lin L, et al. Tumor-derived microRNA-494 promotes angiogenesis in non-small cell lung cancer. Angiogenesis. (2015) 18:373–82. doi: 10.1007/s10456-015-9474-5
129. Safi A, Saberiyan M, Sanaei MJ, Adelian S, Davarani Asl F, Zeinaly M, et al. The role of noncoding RNAs in metabolic reprogramming of cancer cells. Cell Mol Biol letters. (2023) 28:37. doi: 10.1186/s11658-023-00447-8
130. Liberti MV and Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci. (2016) 41:211–8. doi: 10.1016/j.tibs.2015.12.001
131. Liberti MV and Locasale JW. Correction to: 'The Warburg Effect: How Does it Benefit Cancer Cells?'. Trends Biochem Sci. (2016) 41:287. doi: 10.1016/j.tibs.2016.01.004
132. Zhao X, Lu C, Chu W, Zhang B, Zhen Q, Wang R, et al. MicroRNA-124 suppresses proliferation and glycolysis in non-small cell lung cancer cells by targeting AKT-GLUT1/HKII. Tumour Biol. (2017) 39:1010428317706215. doi: 10.1177/1010428317706215
133. Cuttano R, Afanga MK, and Bianchi F. MicroRNAs and drug resistance in non-small cell lung cancer: where are we now and where are we going. Cancers. (2022) 14:5731. doi: 10.3390/cancers14235731
134. Chen X, Jiang Y, Huang Z, Li D, Chen X, Cao M, et al. miRNA-378 reverses chemoresistance to cisplatin in lung adenocarcinoma cells by targeting secreted clusterin. Sci Rep. (2016) 6:19455. doi: 10.1038/srep19455
135. Wang C, Wang S, Ma F, and Zhang W. miRNA−328 overexpression confers cisplatin resistance in non−small cell lung cancer via targeting of PTEN. Mol Med Rep. (2018) 18:4563–70. doi: 10.3892/mmr.2018.9478
136. Li W, Wang W, Ding M, Zheng X, Ma S, and Wang X. MiR-1244 sensitizes the resistance of non-small cell lung cancer A549 cell to cisplatin. Cancer Cell Int. (2016) 16:30. doi: 10.1186/s12935-016-0305-6
137. Silvia Lima RQD, Vasconcelos CFM, Gomes JPA, Bezerra de Menezes EDS, de Oliveira Silva B, Montenegro C, et al. miRNA-21, an oncomiR that regulates cell proliferation, migration, invasion and therapy response in lung cancer. Pathol Res Pract. (2024) 263:155601. doi: 10.1016/j.prp.2024.155601
138. Cen W, Yan Q, Zhou W, Mao M, Huang Q, Lin Y, et al. miR-4739 promotes epithelial-mesenchymal transition and angiogenesis in "driver gene-negative" non-small cell lung cancer via activating the Wnt/beta-catenin signaling. Cell Oncol (Dordr). (2023) 46:1821–35. doi: 10.1007/s13402-023-00848-z
139. Zhang L, Chi W, Wang X, Li J, Li F, Ma Y, et al. miR-6884-5p inhibits proliferation and epithelial-mesenchymal transition in non-small cell lung cancer cells. Heliyon. (2024) 10:e38428. doi: 10.1016/j.heliyon.2024.e38428
140. Tomioka Y, Seki N, Suetsugu T, Hagihara Y, Sanada H, Goto Y, et al. Identification of tumor suppressive miR-144-5p targets: FAM111B expression accelerates the Malignant phenotypes of lung adenocarcinoma. Int J Mol Sci. (2024) 25:9974. doi: 10.3390/ijms25189974
141. Yi X, Chen X, and Li Z. miR-200c targeting GLI3 inhibits cell proliferation and promotes apoptosis in non-small cell lung cancer cells. Med (Baltimore). (2024) 103:e39658. doi: 10.1097/MD.0000000000039658
142. Liu R, Wang J, Zhang L, Wang S, Li X, Liu Y, et al. GLIDR-mediated regulation of tumor Malignancy and cisplatin resistance in non-small cell lung cancer via the miR-342-5p/PPARGC1A axis. BMC Cancer. (2024) 24:1126. doi: 10.1186/s12885-024-12845-y
143. Cheng D, Liu Z, Sun R, Jiang Y, Zeng Z, Zhao R, et al. Overexpression of mir-489-3p inhibits proliferation and migration of non-small cell lung cancer cells by suppressing the HER2/PI3K/AKT/Snail signaling pathway. Heliyon. (2024) 10:e35832. doi: 10.1016/j.heliyon.2024.e35832
144. Wei Y, Wu R, Yang S, Cao Y, Li J, Ma H, et al. MiR-137 mediated high expression of TIGD1 promotes migration, invasion, and suppresses apoptosis of lung adenocarcinoma. Lung Cancer. (2024) 195:107918. doi: 10.1016/j.lungcan.2024.107918
145. Chen R, Gao X, and Shen Y. MiR-3195 inhibits non-small cell lung cancer Malignant behaviors along with cisplatin resistance through targeting PFKFB4. Cell Mol Biol (Noisy-le-grand). (2024) 70:73–8. doi: 10.14715/cmb/2024.70.7.10
146. Lee SJ, Jeon SH, Cho S, Kim CM, Yoo JK, Oh SH, et al. hsa-miR-CHA2, a novel microRNA, exhibits anticancer effects by suppressing cyclin E1 in human non-small cell lung cancer cells. Biochim Biophys Acta Mol Basis Dis. (2024) 1870:167250. doi: 10.1016/j.bbadis.2024.167250
147. Huang P, Zhao H, Sun R, Liu C, Wu L, Wang Y, et al. MiR-1976/NCAPH/P65 axis inhibits the Malignant phenotypes of lung adenocarcinoma. Sci Rep. (2024) 14:11211. doi: 10.1038/s41598-024-61261-6
148. Zhu X, Chen X, Zhang X, Zhao L, and Shen X. MicroRNA−373−3p inhibits the proliferation and invasion of non−small−cell lung cancer cells by targeting the GAB2/PI3K/AKT pathway. Oncol Lett. (2024) 27:221. doi: 10.3892/ol.2024.14353
149. Hu Q, Chen S, Li Y, Hu T, Hu J, Wang C, et al. ANGPTL4, a direct target of hsa-miR-133a-3p, accelerates lung adenocarcinoma lipid metabolism, proliferation and invasion. Aging (Albany NY). (2023) 16:8348–60. doi: 10.18632/aging.205313
150. Liu Q, Bao H, Zhang S, Li C, Sun G, Sun X, et al. MicroRNA-522-3p promotes brain metastasis in non-small cell lung cancer by targeting Tensin 1 and modulating blood-brain barrier permeability. Exp Cell Res. (2024) 442:114199. doi: 10.1016/j.yexcr.2024.114199
151. Koyama N, Ishikawa Y, Ohta H, Aoki T, Kyoyama H, Aoshiba K, et al. miR-4448/Girdin/Akt/AMPK axis inhibits EZH2-mediated EMT and tumorigenesis in small-cell lung cancer. Cancer Med. (2024) 13:e70093. doi: 10.1002/cam4.v13.19
152. Lou Y, Xu B, Huang K, Li X, Jin H, Ding L, et al. Knockdown of miR-1293 attenuates lung adenocarcinoma angiogenesis via Spry4 upregulation-mediated ERK1/2 signaling inhibition. Biochem Pharmacol. (2024) 226:116414. doi: 10.1016/j.bcp.2024.116414
153. Chen X, He L, Zhong H, Yan C, Ke B, and Shi L. The suppression of OTUD7B by miR-491-5p enhances the ubiquitination of VEGFA to suppress vascular mimicry in non-small cell lung cancer. J Gene Med. (2024) 26:e3743. doi: 10.1002/jgm.v26.10
154. Han P, Zhang B, Li Y, Gao R, Li X, Ren H, et al. MiR-183-5p inhibits lung squamous cell carcinoma survival through disrupting hypoxia adaptation mediated by HIF-1alpha/NDUFA4L2 axis. Oncogene. (2024) 43:2821–34. doi: 10.1038/s41388-024-03129-7
155. Arora S, Singh P, Tabassum G, Dohare R, and Syed MA. miR-16-5p regulates aerobic glycolysis and tumorigenesis of NSCLC cells via LDH-A/lactate/NF-kappaB signaling. Life Sci. (2022) 304:120722. doi: 10.1016/j.lfs.2022.120722
156. Han J and Wang Y. Hsa-miR-503-5p regulates CTDSPL to accelerate cisplatin resistance and angiogenesis of lung adenocarcinoma cells. Chem Biol Drug Des. (2023) 102:749–62. doi: 10.1111/cbdd.v102.4
157. Ding D, Shang W, Shi K, Ying J, Wang L, Chen Z, et al. FTO/m6A mediates miR-138-5p maturation and regulates gefitinib resistance of lung adenocarcinoma cells by miR-138-5p/LCN2 axis. BMC Cancer. (2024) 24:1270. doi: 10.1186/s12885-024-13036-5
158. Liang Y, Liang M, Yan T, Meng X, Zhou B, and Gao Y. miR-185-5p may modulate the chemosensitivity of LUSC to cisplatin via targeting PCDHA11: multi-omics analysis and experimental validation. Biochem Genet. (2024) 63(2):1734–51. doi: 10.1007/s10528-024-10795-5
159. Liu L, Guo NA, Li X, Xu Q, He R, Cheng L, et al. miR-125b reverses cisplatin resistance by regulating autophagy via targeting RORA/BNIP3L axis in lung adenocarcinoma. Oncol Res. (2024) 32:643–58. doi: 10.32604/or.2023.044491
160. Liu L, Zhang F, Niu D, Guo X, Lei T, and Liu H. Diagnostic value of microRNA-200 expression in peripheral blood-derived extracellular vesicles in early-stage non-small cell lung cancer. Clin Exp Med. (2024) 24:214. doi: 10.1007/s10238-024-01455-4
161. Wozniak MB, Scelo G, Muller DC, Mukeria A, Zaridze D, and Brennan P. Circulating microRNAs as non-invasive biomarkers for early detection of non-small-cell lung cancer. PloS One. (2015) 10:e0125026. doi: 10.1371/journal.pone.0125026
162. Wang H, Wang L, and Sun G. MiRNA and potential prognostic value in non-smoking females with lung adenocarcinoma by high-throughput sequencing. Int J Gen Med. (2023) 16:683–96. doi: 10.2147/IJGM.S401544
163. Abdipourbozorgbaghi M, Vancura A, Radpour R, and Haefliger S. Circulating miRNA panels as a novel non-invasive diagnostic, prognostic, and potential predictive biomarkers in non-small cell lung cancer (NSCLC). Br J Cancer. (2024) 131:1350–62. doi: 10.1038/s41416-024-02831-3
164. Ma L, Guo H, Zhao Y, Liu Z, Wang C, Bu J, et al. Liquid biopsy in cancer current: status, challenges and future prospects. Signal Transduct Target Ther. (2024) 9:336. doi: 10.1038/s41392-024-02021-w
165. Ochiya T, Hashimoto K, and Shimomura A. Prospects for liquid biopsy using microRNA and extracellular vesicles in breast cancer. Breast Cancer. (2025) 32:10–5. doi: 10.1007/s12282-024-01563-9
166. Wang K, Wang X, Pan Q, and Zhao B. Liquid biopsy techniques and pancreatic cancer: diagnosis, monitoring, and evaluation. Mol Cancer. (2023) 22:167. doi: 10.1186/s12943-023-01870-3
167. Jungers CF and Djuranovic S. Modulation of miRISC-mediated gene silencing in eukaryotes. Front Mol Biosci. (2022) 9:832916. doi: 10.3389/fmolb.2022.832916
168. Yang Z, Liu X, Xu H, Teschendorff AE, Xu L, Li J, et al. Integrative analysis of genomic and epigenomic regulation reveals miRNA mediated tumor heterogeneity and immune evasion in lower grade glioma. Commun Biol. (2024) 7:824. doi: 10.1038/s42003-024-06488-9
169. Wiggins JF, Ruffino L, Kelnar K, Omotola M, Patrawala L, Brown D, et al. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. (2010) 70:5923–30. doi: 10.1158/0008-5472.CAN-10-0655
170. Trang P, Wiggins JF, Daige CL, Cho C, Omotola M, Brown D, et al. Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol Ther. (2011) 19:1116–22. doi: 10.1038/mt.2011.48
171. Cortez MA, Valdecanas D, Zhang X, Zhan Y, Bhardwaj V, Calin GA, et al. Therapeutic delivery of miR-200c enhances radiosensitivity in lung cancer. Mol Ther. (2014) 22:1494–503. doi: 10.1038/mt.2014.79
172. Hwang GR, Yuen JG, Fesler A, Farley H, Haley JD, and Ju J. Development of a 5-FU modified miR-129 mimic as a therapeutic for non-small cell lung cancer. Mol Ther Oncolytics. (2023) 28:277–92. doi: 10.1016/j.omto.2023.02.007
Keywords: microRNA, lung cancer, signaling cascade, targeted therapy, cancer progression, miRNA
Citation: Tirpe A-A, Nutu A, Busuioc C, Pop O-L and Berindan-Neagoe I (2025) MicroRNAs and lung cancer: overview of essential pathways and somatic mutations in cancer progression. Front. Oncol. 15:1677471. doi: 10.3389/fonc.2025.1677471
Received: 31 July 2025; Accepted: 15 September 2025;
Published: 07 October 2025.
Edited by:
Matthew J. Marton, MSD, United StatesReviewed by:
Nikhil Samarth, National Centre for Cell Science, IndiaVenkatachalam Deepa Parvathi, Sri Ramachandra Institute of Higher Education and Research, India
Copyright © 2025 Tirpe, Nutu, Busuioc, Pop and Berindan-Neagoe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ioana Berindan-Neagoe, aW9hbmEubmVhZ29lQHVtZmNsdWoucm8=