 Hanan E. Alatwi1*
Hanan E. Alatwi1* Amnah A. Alharbi2
Amnah A. Alharbi2 Rashid Mir3
Rashid Mir3 Othman R. Alzahrani1
Othman R. Alzahrani1 Abdulrahman H. Alessa1
Abdulrahman H. Alessa1 Yousef M. Hawsawi4,5Mohammed Ali Arishi6Aziz Dhaher Albalawi6
Yousef M. Hawsawi4,5Mohammed Ali Arishi6Aziz Dhaher Albalawi6- 1Department of Biology, Faculty of Science, University of Tabuk, Tabuk, Saudi Arabia
- 2Department of Biochemistry, Faculty of Science, University of Tabuk, Tabuk, Saudi Arabia
- 3Department of Medical Lab Technology, Faculty of Applied Medical Sciences, and Prince Fahd Sultan Research Chair for Biomedical Research, University of Tabuk, Tabuk, Saudi Arabia
- 4Research Center, King Faisal Specialist Hospital and Research Center, Jeddah, Saudi Arabia
- 5Department of Biochemistry and Molecular Medicine, College of Medicine, Alfaisal University, Riyadh, Saudi Arabia
- 6Faculty of Laboratory, King Khaled Hospital, Ministry of Health, Tabuk, Saudi Arabia
Background: Colorectal cancer (CRC) shows significant inter-population heterogeneity in its genomic landscape, yet Middle Eastern populations are underrepresented in large-scale sequencing studies. This exploratory study aims to characterize somatic mutations and disrupted signaling pathways in Saudi Arabian CRC patients.
Methods: We performed whole-exome sequencing (WES) on tumor DNA from 24 Saudi CRC patients. Somatic variants were identified and analyzed in a curated panel of cancer-related genes. Comparative analysis was conducted against The Cancer Genome Atlas colorectal cancer dataset (TCGA-COADREAD), and pathway enrichment analysis was performed.
Results: Somatic variants were identified in 23 tumors, with recurrent mutations in BRCA2 (61%), TCF7L2 (52%), EGFR (43%), and SOS1 (43%). Compared to TCGA-COADREAD, mutation frequencies were significantly higher in BRCA2, EGFR, SLC25A5, and PIK3R2 (adjusted p < 0.0001). Among 258 total variants, 43% were novel, and 25 were classified as pathogenic, likely pathogenic, or deleterious, including 13 novel variants across nine genes. Pathway analysis revealed frequent disruptions in WNT/β-catenin (65%), homologous recombination (61%), PI3K (48%), and RTK/RAS (43%) signaling pathways.
Conclusion: Our results reveal a distinct mutational profile in Saudi CRC patients, characterized by novel and enriched somatic variants affecting key oncogenic pathways. These findings underscore the necessity of including underrepresented populations in cancer genomics to support globally equitable precision oncology.
1 Introduction
Colorectal cancer (CRC) is one of the most prevalent malignancies worldwide, ranking third in incidence and second in cancer-related mortality (1). While early detection and treatment programs in high-income countries have led to a decline in both incidence and mortality, CRC continues to pose a significant global health challenge. In contrast, many low- and middle-income countries, including those in the Arab region, are witnessing a steady rise in CRC incidence. Within the Gulf Cooperation Council (GCC), CRC is the second most common cancer among both men and women. In Saudi Arabia specifically, it accounts for approximately 10.1% of male and 9.3% of female cancer diagnoses (2, 3). These figures underscore the urgent need for region-specific research to better understand the molecular characteristics of CRC in Arab populations.
Large-scale genomic initiatives such as The Cancer Genome Atlas (TCGA) have identified key driver mutations, particularly in genes like APC, KRAS, TP53, and PIK3CA, that have shaped our understanding of CRC pathogenesis (4, 5). However, these studies predominantly involve Western populations, limiting their generalizability to more genetically diverse groups. Since genetic variability can influence cancer susceptibility, progression, and treatment response, population-specific genomic studies are essential for advancing precision oncology tailored to regional contexts.
CRC development is driven by the stepwise accumulation of genetic and epigenetic alterations affecting oncogenes, tumor suppressor genes, and DNA repair mechanisms. Key underlying processes include chromosomal instability (CIN), microsatellite instability (MSI), and the CpG island methylator phenotype (CIMP) (6, 7). The advent of high-throughput sequencing technologies, especially whole-exome sequencing (WES), now enables comprehensive profiling of somatic mutations, facilitating the identification of both well-known alterations and novel, population-specific variants (8). A recent Saudi-based study using targeted next-generation sequencing (NGS) reported high mutation frequencies in BRCA2, CHEK1, ATM, and PMS2, pointing to a potentially unique mutational landscape in this population. Although the study did not include some canonical CRC genes and found limited clinical correlations, it highlighted the critical role of molecular profiling in guiding personalized treatment strategies (2). Our previous studies have identified novel SNPs in Toll-like receptor 2 (TLR2) (9) and variants in the HER1 and HER2 genes (10) that associated with CRC in Saudi population.
In this context, the present study aimed to explore a targeted genomic analysis of CRC in a Saudi Arabian cohort using WES. The focus was on a curated panel of cancer-associated genes, encompassing well-established CRC drivers, BRCA2, EGFR, PIK3R2, PTEN, AXIN1, TGFB2, SOS1, BAX, and TCF7L2, as well as novel or understudied genes identified from our preliminary cohort-specific analyses. These established genes are involved in key signaling pathways such as Wnt/β-catenin, PI3K/AKT/mTOR, and MAPK, all of which are commonly dysregulated in CRC and contribute to tumorigenesis through impaired proliferation, apoptosis, and genomic stability (4, 8, 11–13).
In addition, the study explored a subset of novel or less-characterized genes, including KRT8, KRT18, TUBB6, SLC25A5, ELAVL1, and PRDX1, that were frequently mutated in this cohort. Though not traditionally associated with CRC, these genes may play important roles in tumor biology. For instance, KRT8 and KRT18 are cytoskeletal proteins that enhance motility and invasiveness in epithelial cancers (14–16), while PRDX1 is involved in redox regulation and may contribute to chemoresistance (17). TUBB6, a beta-tubulin isotype, is crucial for microtubule function during cell division, and its disruption may drive chromosomal instability, a hallmark of CRC (18, 19). SLC25A5 (also known as ANT2) facilitates ATP transport across the mitochondrial membrane and has been shown to reduce proliferation and promote apoptosis in colon cancer cells, partly through inhibition of MAPK signaling (20). Finally, ELAVL1, an RNA-binding protein that stabilizes mRNAs linked to cell growth and survival, is upregulated in multiple cancer types and has been implicated in tumor progression (21–24).
2 Materials and methods
2.1 Study population and ethical approval
This study included a cohort of 24 patients with clinically and pathologically confirmed CRC. Tissue specimens were collected from formalin-fixed paraffin-embedded (FFPE) blocks archived at the Division of Histopathology, King Khaled Hospitals, Tabuk, Saudi Arabia. Due to challenges in obtaining matched normal tissue samples from these archived FFPE blocks and existing budgetary constraints, this study adopted a tumor-only whole-exome sequencing approach for somatic variant discovery. All tumor specimens were collected at the time of diagnostic surgery, and none of the patients had received chemotherapy, radiotherapy, or targeted therapy before sample collection.
The research protocol was reviewed and approved by the Institutional Ethical Committee of the University of Tabuk (Protocol No. UT-115-13-2020). All procedures involving human participants were conducted in accordance with the ethical standards of the institutional and national research committees and with the 2013 revision of the Declaration of Helsinki.
2.1.1 Inclusion and exclusion criteria
Participants were selected based on the following inclusion criteria: a confirmed diagnosis of CRC through clinical, histopathological, and radiological assessments; Saudi Arabian nationality; and disease at any clinical stage. While the ethics-approved inclusion criteria permitted enrolment of patients with prior chemotherapy, radiotherapy, or hormone therapy, in practice, all patients included in this study were treatment-naïve at the time of tissue collection.
Exclusion criteria included: non-Saudi nationals, individuals of non-Arab descent, newly naturalized citizens, and patients presenting with multiple primary tumors. Patients unable to adhere to the study requirements or complete the consent process were also excluded. This ethnically focused recruitment strategy was designed to investigate population-specific genomic alterations in CRC.
2.1.2 Data collection
A standardized questionnaire was administered to each patient to collect demographic data, family medical history, and prior awareness of colorectal cancer. Additional clinical and laboratory information was obtained from medical records to create a comprehensive dataset for each participant. Informed consent was obtained in writing from all patients before inclusion, in compliance with the ethical guidelines of the University of Tabuk’s Research Ethics Committee.
2.2 DNA extraction, library preparation, and whole-exome sequencing
Genomic DNA was extracted from FFPE tissue blocks using the QIAamp DNA FFPE Tissue Kit (Cat. No. 56404), following the manufacturer’s (QIAGEN, Germany) protocol. The quality and quantity of the isolated DNA were assessed using both NanoDrop spectrophotometry for purity and Qubit fluorometry for accurate concentration measurement. DNA integrity was further confirmed by agarose gel electrophoresis.
Whole-exome libraries were constructed from 50 ng of high-quality genomic DNA using the Twist Bioscience Human Core Exome 2.0 Kit (Cat. No. 104207) manufactured by Twist Bioscience (USA). The library preparation process included DNA fragmentation, end repair, adapter ligation, and sample pooling, followed by targeted exome capture spanning approximately 10–50 Mb of coding regions. Library quality and fragment distribution were validated using the Agilent TapeStation system.
Final libraries were quantified and sequenced on an Illumina NovaSeq 6000 platform using paired-end reads. The sequencing was performed to achieve an average coverage depth of 500X, enabling high-confidence detection of variants within the captured exonic regions.
2.3 Sequencing read quality control, preprocessing, and alignment
The quality of the raw sequencing reads was initially assessed using FastQC software (v0.12.1), which provided comprehensive reports on key read quality metrics (25). To enhance the accuracy of downstream analyses, the raw reads underwent preprocessing to remove sequencing adapters and low-quality bases. This critical filtering step, which eliminates potential sequencing artifacts, was performed using Trimmomatic (v0.39) (26). After quality control and trimming, the resulting high-quality reads were aligned to the human reference genome (hg19) using the Burrows-Wheeler Aligner (BWA-MEM) algorithm, a robust tool for mapping short reads to large reference sequences (27). The alignment output, initially in SAM format, was converted to the more efficient BAM format using SAMtools, facilitating optimized storage and seamless integration into subsequent bioinformatics workflows (28). The sequencing data demonstrated high-quality metrics across all samples, with a Q30 score exceeding 90%. Alignment rates were consistently high, surpassing 99%, and the targeted panel coverage was robust, remaining above 95%.
2.4 Variant discovery and annotation
Variant discovery followed the GATK Best Practices workflow using GATK v4.3 (29). Somatic variants were identified using the Mutect2 caller with the Twist 2.0 target BED file, and further refined through a series of filtering steps using FilterMutectCalls and Mutect2 filters. These filters removed low-quality calls and germline variants flagged with labels such as contamination, germline, multiallelic, normal_artifact, weak_evidence, panel_of_normals, and clustered_events. Following variant calling, a comprehensive annotation was performed using ANNOVAR, leveraging the RefSeq database for gene-based annotation (30). Public databases, such as COSMIC and ClinVar, along with other resources, were used to annotate the identified variants (31). COSMIC was used to identify previously reported cancer somatic mutations, while ClinVar was leveraged to assess clinical significance and support ACMG/AMP classifications. To distinguish potentially pathogenic mutations from common polymorphisms, population frequency data were integrated from large-scale databases including the 1000 Genomes Project (32), ExAC (33), gnomAD (34), gnomAD-ME (34), and ESP (35). The functional impact of identified missense variants was predicted using multiple in silico tools, including SIFT (36), PolyPhen-2 (37), FATHMM (38), MutationTaster (39), and MutationAssessor (40). Additionally, variants were annotated with CADD scores to estimate their deleteriousness (41). While primarily developed for germline variant interpretation, relevant ACMG/AMP criteria were carefully applied using InterVar (42) to prioritize somatic variants for potential clinical significance within this study. Based on these criteria, all variants were categorized as benign (B), likely benign (LB), likely pathogenic (LP), pathogenic (P), or variants of uncertain significance (VUS).
2.5 Somatic variant filtering and classification
To mitigate the inherent challenges of tumor-only analysis, such as distinguishing somatic mutations from germline variants and reducing false positives, stringent bioinformatic filtering strategies were implemented. This approach, while not achieving the same precision as matched tumor-normal sequencing, allowed for the identification of potential somatic alterations in this unique cohort. Somatic variants were filtered and analyzed with a focused interest on a predefined panel based on established cancer genes, largely identified in diverse but often Western cohorts.: AXIN1, BAX, BRCA2, EGFR, ELAVL1, KRT18, KRT8, PIK3R2, PRDX1, PTEN, SLC25A5, SOS1, TCF7L2, TGFB2, and TUBB6. Initial filtering excluded synonymous and intronic variants, retaining only exonic non-synonymous and indel variants predicted to alter protein structure or function. To enrich for clinically and biologically significant alterations, only variants with a minor allele frequency (MAF) ≤1% in population databases (including Middle East population specific gnomAD-ME) were retained, avoiding common polymorphisms.
Variants were annotated as “known” if they were listed in dbSNP, gnomAD, COSMIC, or ClinVar, and labeled as “novel” if no such identifiers were present. dbSNP was primarily utilized to filter out common single nucleotide polymorphisms (SNPs) and other variants likely representing germline polymorphisms, thereby aiding in identifying novel variants not previously reported in the general population. Conversely, COSMIC served as the primary reference for identifying known cancer-associated somatic variants. Functional impact was predicted using multiple in silico tools, including SIFT (36), PolyPhen (37), FATHMM (38), MutationAssessor (40), MutationTaster (39), and CADD (41). Variants were classified as deleterious if they had a CADD score ≥20 and at least three of the other tools also predicted a deleterious effect.
The critical need to identify ‘actionable’ somatic mutations for precision oncology necessitates a standardized pathogenicity assessment approach, similar to germline variants. To improve clinical interpretation, variants were classified as Benign (B), Likely Benign (LB), Pathogenic (P), Likely Pathogenic (LP), or Variants of Uncertain Significance (VUS) using a combination of annotations from ClinVar, InterVar, and consensus in silico predictions. For those lacking ClinVar or InterVar annotations, classification as Deleterious (D) or Neutral (N) was based on CADD scores, Aloft predictions, and VEP impact scores, particularly in the case of indels and splice variants.
For comparative analyses, publicly available colorectal adenocarcinoma data was leveraged from The Cancer Genome Atlas (TCGA-COADREAD) via cBioPortal (accession ID: “coadread_tcga_pan_can_atlas_2018”) (43, 44). To ensure a direct and robust comparison with our Saudi cohort, this large dataset (n = 534) was stringently filtered to include only the identical gene set examined in our study. Furthermore, synonymous, intronic, and common polymorphic variants were excluded, allowing for a focused analysis of potentially pathogenic somatic alterations across both cohorts.
2.6 Statistical analysis and data visualization
All statistical analyses were performed using R software version 4.3.2 (45). Data processing and visualization were conducted primarily with the “tidyverse” (46), “ggplot2” (47), and “maftools” (48) packages within the R environment. Tables were generated using the “gt” (49) and “gtsummary” (50) packages for presentation. Specifically, Fisher’s Exact test was used to compare gene mutation frequencies between different groups. To account for multiple comparisons, p-values from these tests were adjusted using the Benjamini-Hochberg method to control the False Discovery Rate (FDR).
3 Results
3.1 Patient characteristics
A total of 24 patients were recruited for this preliminary study. The cohort predominantly comprised females (n=15, 62.5%), with males accounting for 9 patients (37.5%). The median age at diagnosis was 57 years (Interquartile Range: 42-63 years), with the majority of patients (n=15, 63%) being over 50 years old, while 9 patients were below 50. Tumor staging revealed that 46% of patients had T3 tumors and 42% had T4 tumors, indicating a predominance of locally advanced disease. Regarding lymph node involvement, 46% were classified as N2 and 42% as N3. Metastasis (M1) was observed in 88% of patients, confirming that the majority presented with advanced disease. Only 13% had early-stage cancer without evidence of metastasis. Tumor locations varied across the cohort, with 2 patients (8.3%) having tumors in the Ascending & Transverse colon, 3 (13%) in the Descending colon, 4 (17%) in Multicentric sites, and the largest proportion (n=15, 63%) in the Recto-sigmoid region. Regarding tumor differentiation, 17 patients (71%) had well-differentiated tumors, 4 (17%) had intermediate differentiation, and 3 (13%) had poorly differentiated tumors. Key characteristics of this cohort are further detailed in Table 1.
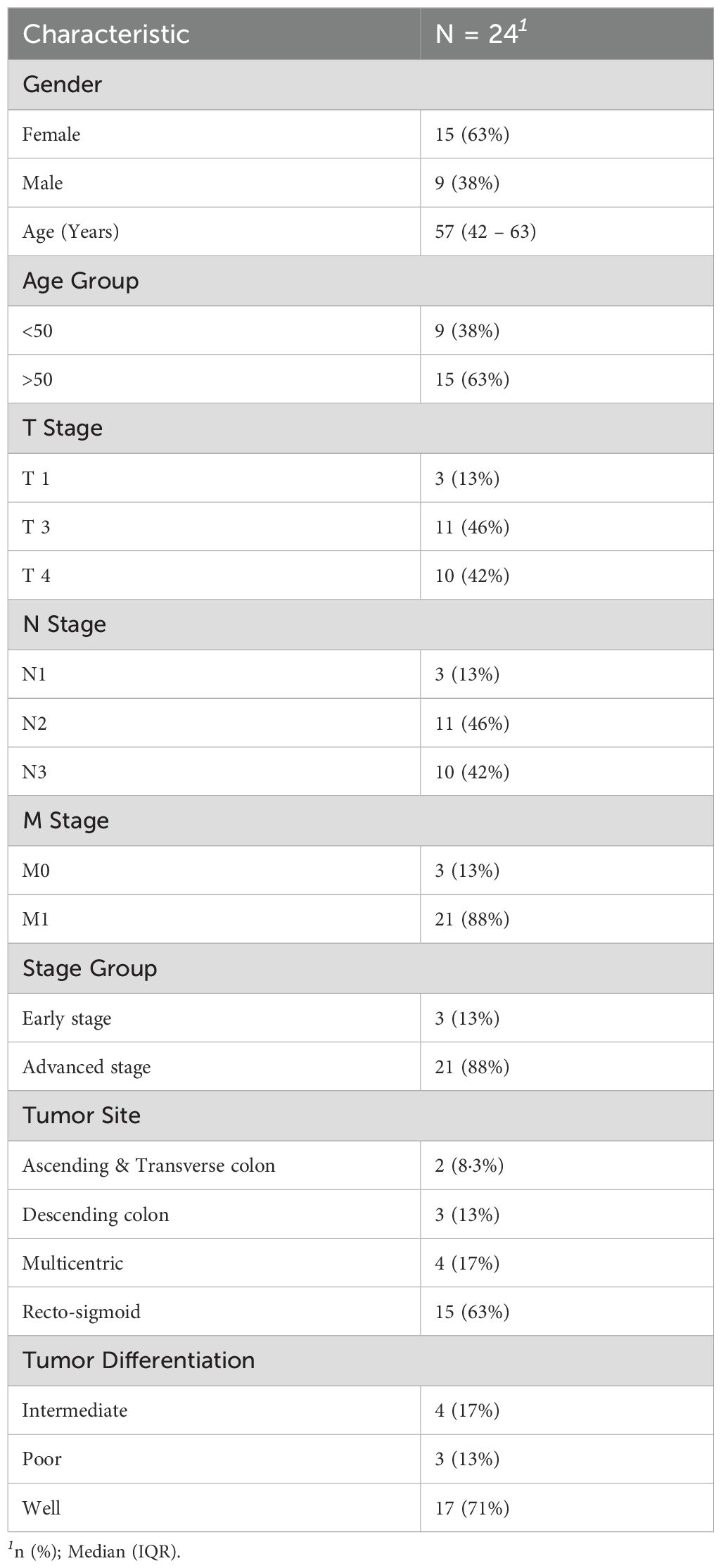
Table 1. Clinical characteristics of the patients included in the Saudi cohort.
The TCGA colorectal patient cohort comprised 64% colon adenocarcinoma, 26% rectal adenocarcinoma, and 10% mucinous adenocarcinoma cases. According to AJCC staging, the majority of patients in this cohort presented with no distant metastasis (M0; 74%), followed by distant metastasis (M1; 12%), undetermined distant metastasis (MX; 10%), and distant metastasis to a single organ or site (M1A; 2%).
3.2 Mutational landscape of selected genes
Examining the mutational landscape of the selected genes within the cohort revealed that 23 out of the 24 patients harbored at least one somatic variant. The most frequently mutated gene was BRCA2, with variants detected in 14 samples (61%). Following closely, the Beta-catenin pathway gene TCF7L2 was mutated in 52% of patients. Other frequently altered genes included EGFR (43%), SOS1 (43%), PIK3R2 (35%), and SLC25A5 (35%). Less frequent mutations were observed in KRT18 (30%), TUBB6 (26%), PTEN (22%), AXIN1 (17%), BAX (17%), TGFB2 (17%), KRT8 (13%), and ELAVL1 (9%). No mutations associated with PRDX1 were identified in any of the samples. The distribution of these mutations across individual samples is visually presented in Figure 1A. Further analysis of co-mutations identified key patterns of co-occurring and mutually exclusive variants. A statistically significant co-occurrence was noted between TGFB2 and EGFR variants (p < 0.05). Similarly, TUBB6 variants were found to co-occur significantly with KRT18, and KRT18 with SLC25A5, as well as PIK3R2 with SOS1. While not reaching statistical significance, TCF7L2 variants demonstrated a tendency towards mutual exclusivity with variants in BRCA2 and PTEN. Figure 1B depicts the mutation co-occurrence of the selected genes.
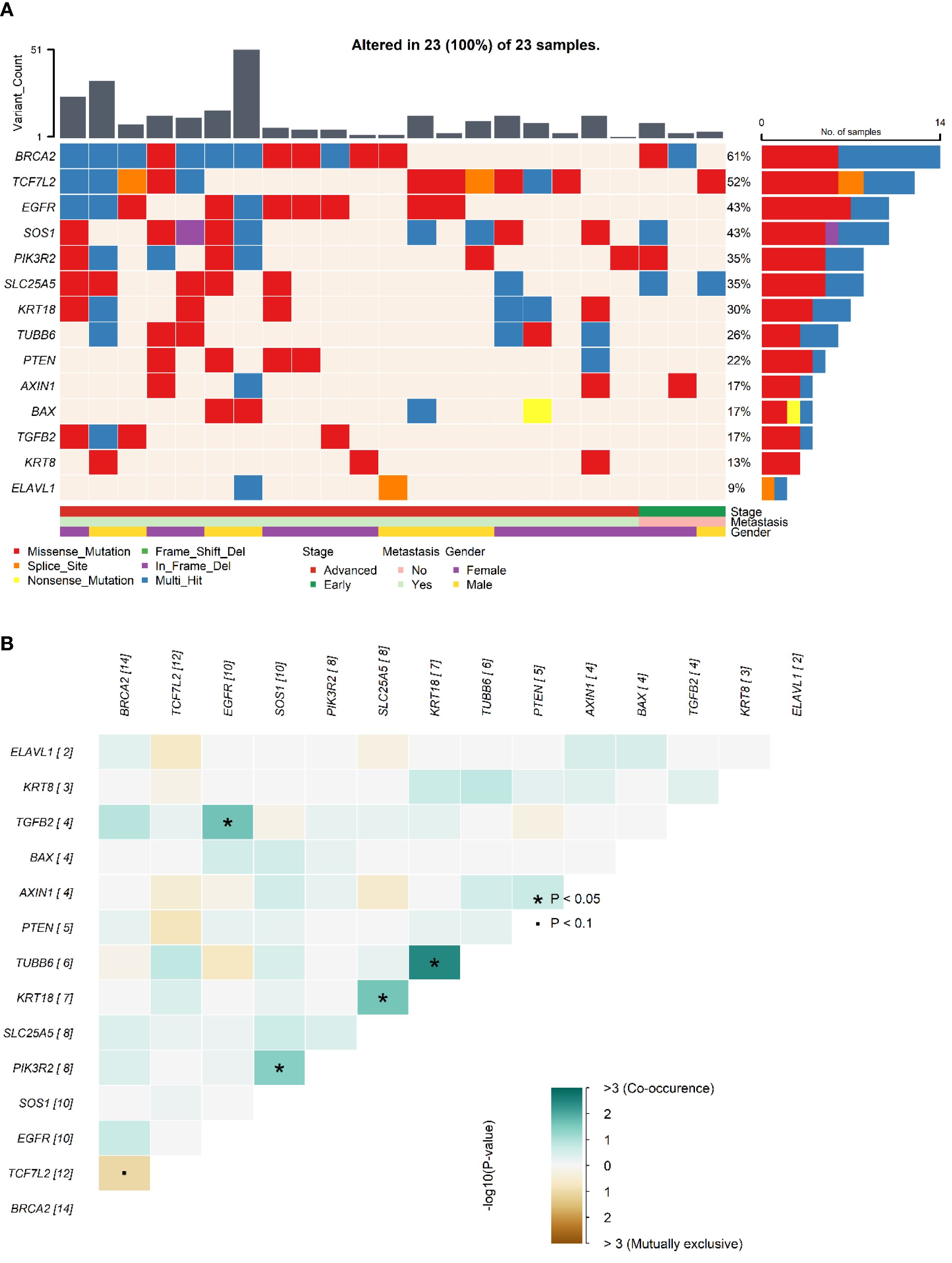
Figure 1. Somatic mutations and gene interaction patterns in Saudi CRC cohort. (A). Oncoplot showing the distribution of somatic mutations in the selected genes across individual samples in the Saudi cohort. Each row represents a gene, and each column represents a patient sample, indicating the presence or absence of mutations. (B). Plot depicting patterns of co-occurring and mutually exclusive gene variants observed across samples in the cohort.* indicates statistical significance (p < 0.05) for co-occurrence or mutual exclusivity. indicates marginal significance (p < 0.1).
3.3 Variant characteristics
Across the 15 selected genes, variants were identified in 14 genes, totaling 258 somatic variants across the cohort. The predominant variant type observed was missense mutations, accounting for the vast majority (n=241, 93%). Nonsense mutations constituted a smaller fraction (n=8, 3%), followed by splice site variants (n=5, 2%). Frameshift and inframe deletions each numbered 2. Overall, 254 variants (98%) were single nucleotide polymorphisms (SNPs), while 4 were insertions/deletions (indels). Among all identified variants across samples, 148 (57%) were classified as existing, meaning they had been previously reported in public databases. Conversely, 110 variants (43%) were considered novel. It is important to note that these counts reflect variant occurrences per sample and may include the same unique variant found in multiple patients. Examining the distribution at the gene level, SOS1 displayed the highest number of missense mutations (n=49), followed by BRCA2 (n=45), EGFR (n=31), TCF7L2 (n=25), and TUBB6 (n=20). For novel variants, the top five genes were SOS1 (n=33), TUBB6 (n=16), BAX (n=11), KRT18 (n=10), and PIK3R2 (n=7). In contrast, existing variants were most frequently found in BRCA2 (n=45), EGFR (n=29), TCF7L2 (n=22), SOS1 (n=18), and PTEN (n=7). Figure 2 visually represents these variant characteristics distributed across the genes. The Figure 2A illustrates the distribution of variants per gene, categorizing them as existing if they’re present in public databases or novel if they’re not. This distinction highlights the proportion of previously identified mutations versus newly discovered ones. The analysis is further refined in Figure 2B, which classifies the variants based on type—either SNPs or Indels (Insertions or Deletions). This classification provides insight into the underlying mutational events, whether they involve a single nucleotide change or a more extensive structural alteration. Finally, Figure 2C presents a functional classification of the variants, identifying their potential impact on protein function, such as missense, nonsense, splice site, in-frame deletion, and frameshift deletion mutations, offering a comprehensive view of the molecular consequences of the observed genetic variation.
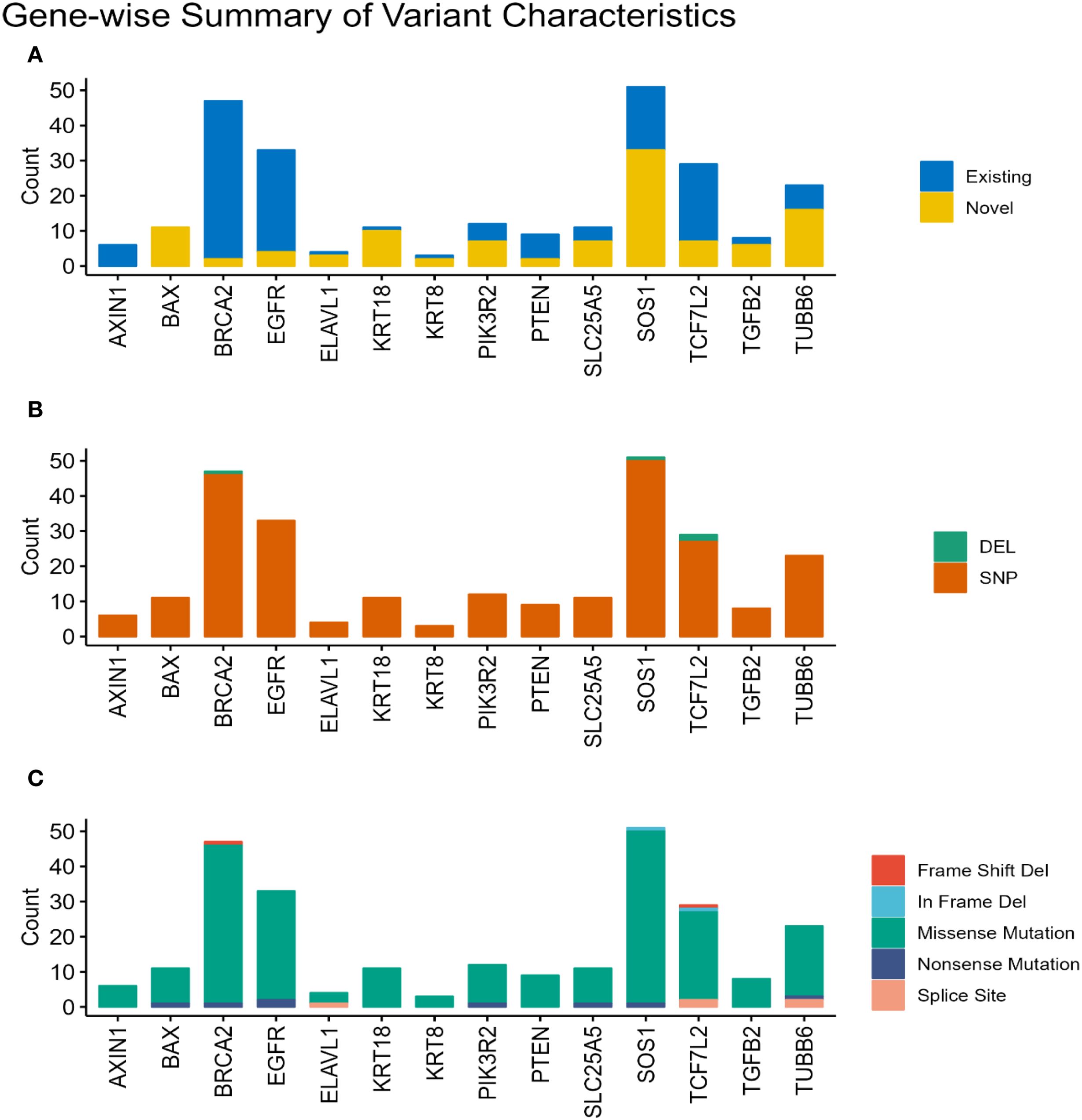
Figure 2. Characterization of somatic variants. Bar plots illustrating the characteristics of the identified somatic variants based on their status (A). (Existing or Novel), (B). type (Indel or SNP), and (C) functional classification (e.g., missense, nonsense, frameshift).
3.4 Differential distribution of somatic mutation: a comparison with TCGA cohort
To understand the uniqueness of the mutational landscape, somatic mutation frequencies of the selected genes in the Saudi cohort were compared with those in the TCGA-COADREAD cohort. This comparison revealed significant differential mutation frequencies across several genes, as detailed in Table 2.
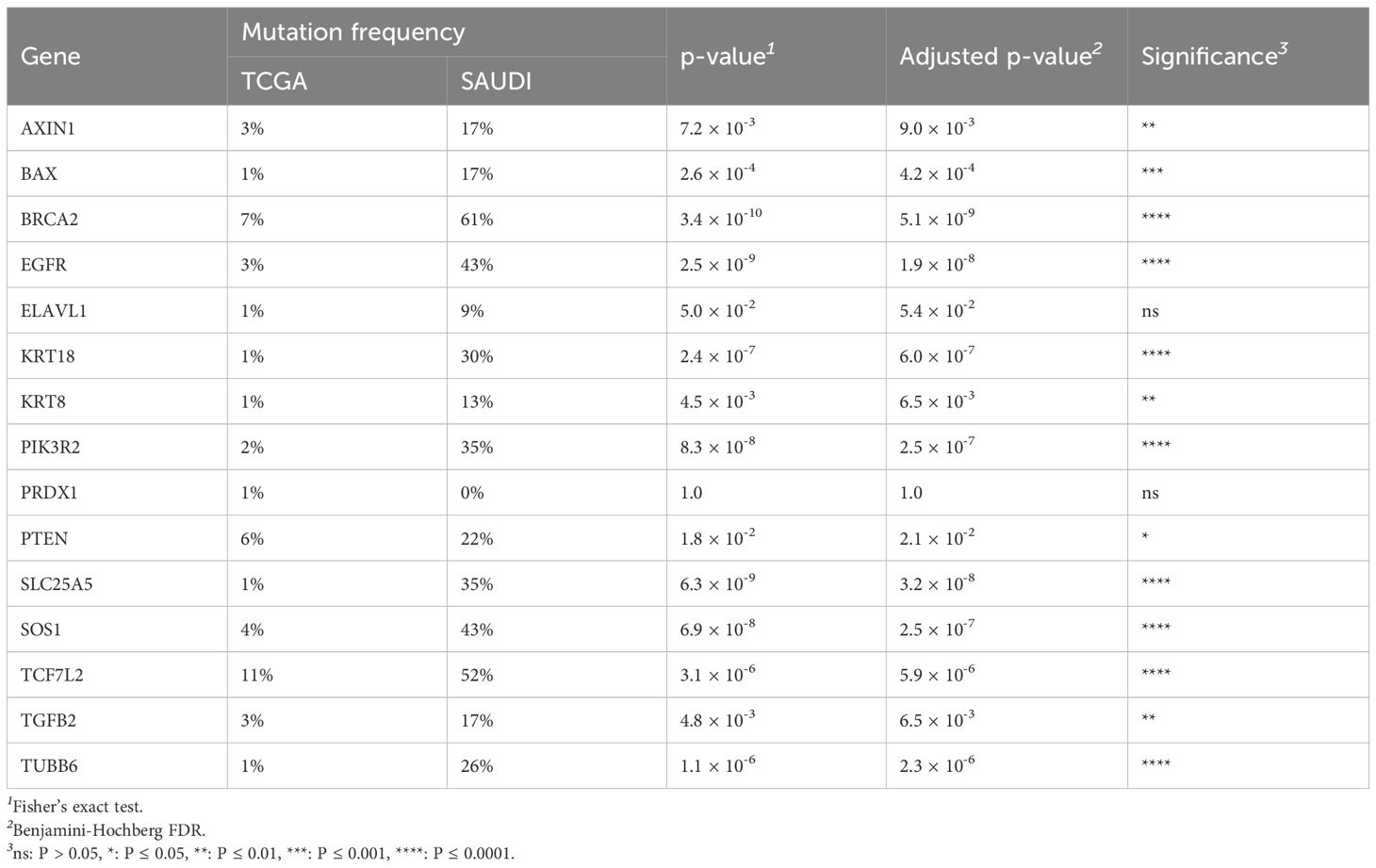
Table 2. Comparison of gene mutation frequencies between the TCGA-COADREAD cohort and the Saudi cohort, including statistical significance.
Notably, BRCA2 mutations were significantly enriched in the Saudi cohort compared to the TCGA cohort (61% vs 7%, p = 3.4×10−10). Similar significant enrichments in the Saudi cohort were observed for EGFR (43% vs 3%, p = 2.5×10−9), SLC25A5 (35% vs 1%, p = 6.3×10−9), SOS1 (43% vs 4%, p = 6.9×10−8), PIK3R2 (35% vs 2%, p = 8.3×10−8), KRT18 (30% vs 1%, p = 2.4×10−7), TUBB6 (26% vs 1%, p = 1.1×10−6), and TCF7L2 (52% vs 11%, p = 3.1×10−6). It is important to note that our cohort’s relatively small sample size (n=24) may contribute to these seemingly inflated frequencies, making rare events appear disproportionately common compared to large, heterogeneous datasets like TCGA. Therefore, these figures should be interpreted as observed patterns within our specific cohort rather than definitive population prevalences. These differential mutation patterns are visually presented in Figure 3A and Figure 4. Further investigation into the distribution of variants within individual genes did not reveal a universal pattern of specific domain enrichment. For BRCA2 and EGFR, the variant distribution across protein domains appeared similar in both cohorts. However, a contrasting pattern was observed for TCF7L2, where variants in the Saudi cohort were more enriched in the CTNNB1 binding domain, whereas those in the TCGA cohort showed greater enrichment in the SOX-TCF-HMGBOX domain. The domain-level distribution of variants for these genes in each cohort is depicted in Figures 3B–D.
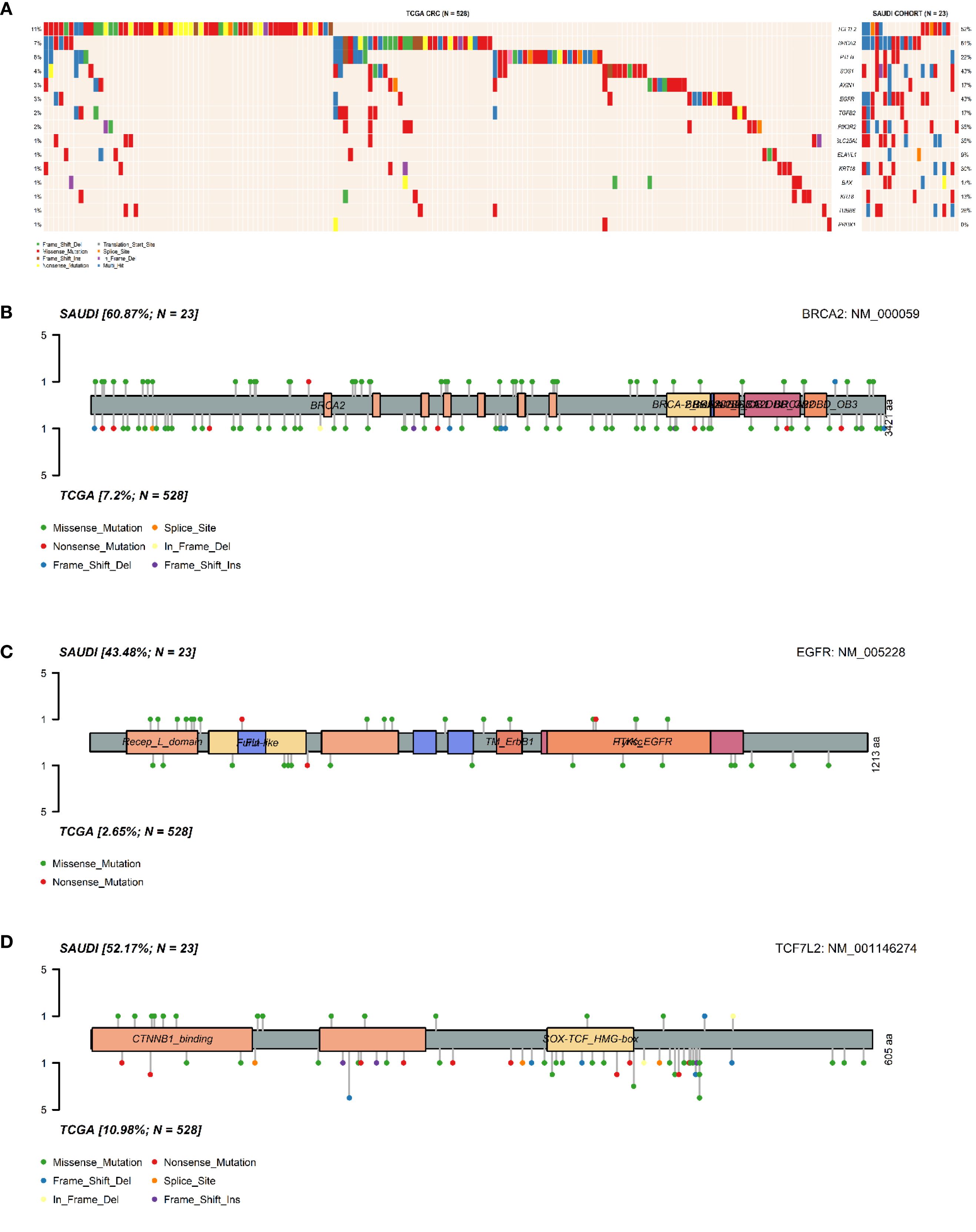
Figure 3. Comparative analysis of somatic mutations in Saudi and TCGA CRC cohorts. (A). Oncoplot comparing the distribution and frequency of mutations in the selected genes between the Saudi cohort and the TCGA-COADREAD cohort. Lolliplot illustrating the distribution of (B). BRCA2 variants, (C). EGFR variants, (D). TCF7L2 variants across protein domains, comparing patterns observed in the TCGA cohort and the Saudi cohort.
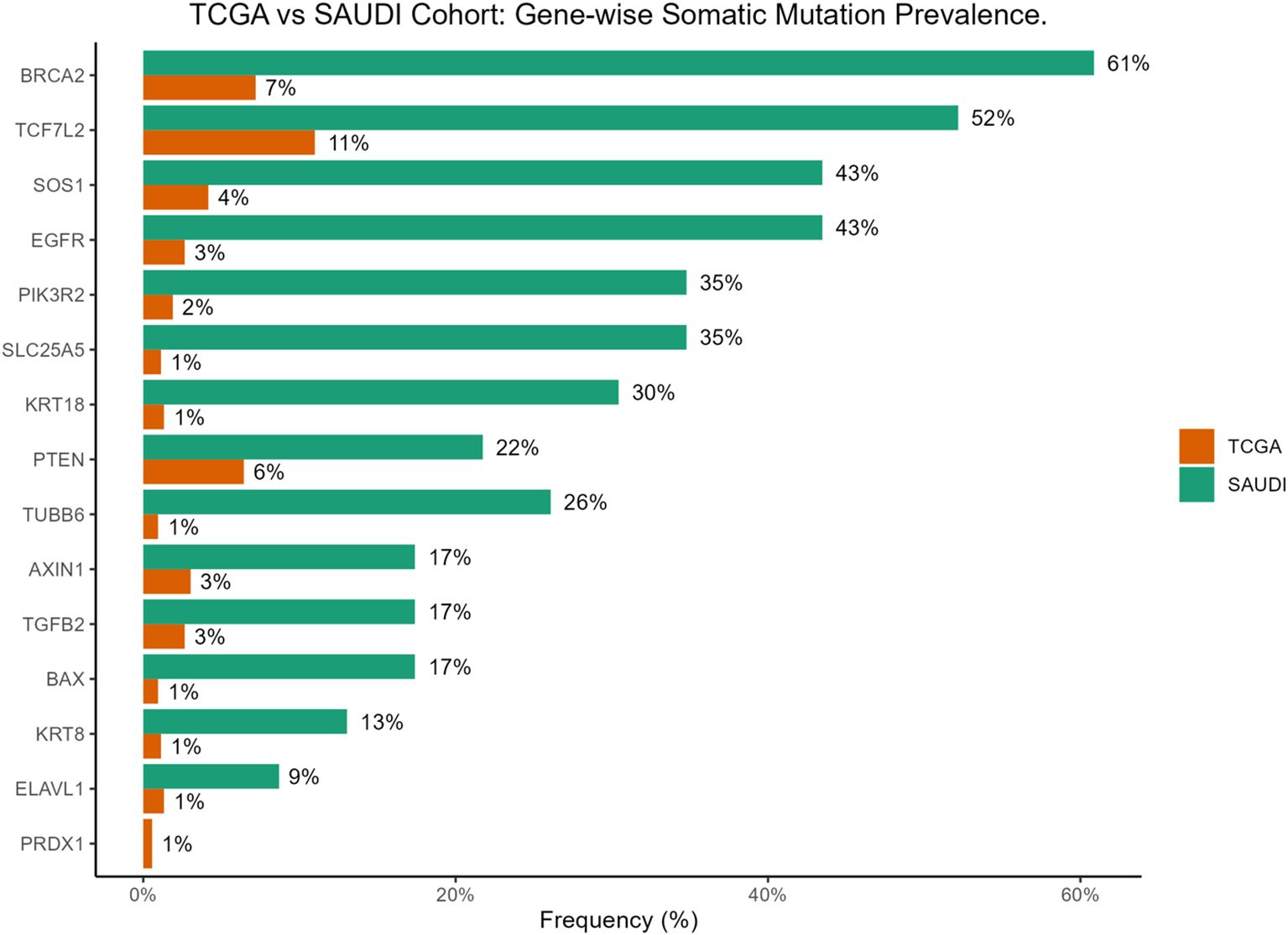
Figure 4. Bar plot comparing gene-wise somatic mutation prevalence between the TCGA-COADREAD and the Saudi cohorts.
3.5 Pathogenic variants identified in the cohort
Based on classification using ClinVar, InterVar, and functional prediction tools, a total of 25 variants were identified as Pathogenic (P), Likely Pathogenic (LP), or Deleterious (D) within the cohort. These potentially impactful variants were distributed across nine genes: EGFR (n=7), TCF7L2 (n=5), PIK3R2 (n=3), TGFB2 (n=3), BRCA2 (n=2), SOS1 (n=2), ELAVL1 (n=1), PTEN (n=1), and TUBB6 (n=1). The types of variants categorized as P/LP or D included 13 missense mutations, 6 nonsense mutations, 4 splice site variants, 1 frameshift deletion, and 1 in-frame deletion. Among these, 13 variants were novel, not previously reported in public databases, and were classified within this category. These novel pathogenic/deleterious variants included specific alterations in EGFR (NM_001346941.2:c.C2365T:p.Q789X), a splice variant in ELAVL1, multiple variants in PIK3R2 (NM_005027.4:c.C679T:p.Q227X; NM_005027.4:c.C374T:p.P125L; NM_005027.4:c.G415A:p.G139R), variants in SOS1 (NM_001382394.1:c.G2267A:p.W756X; NM_001382394.1:c.249_251del:p.Q84del), and several variants in TCF7L2 (Splice Variant; NM_001146285.1:c.1413_1435del:p.N475Rfs*7;NM_001349870.2:c.G101T:p.W34L; NM_001146285.1:c.1411_1412delinsTT:p.P471F), as well as a missense mutation in TGFB2 (NM_003238.6:c.G907A:p.A303T) and a splice variant in TUBB6. Detailed information on each of these pathogenic or deleterious variants is provided in Table 3.
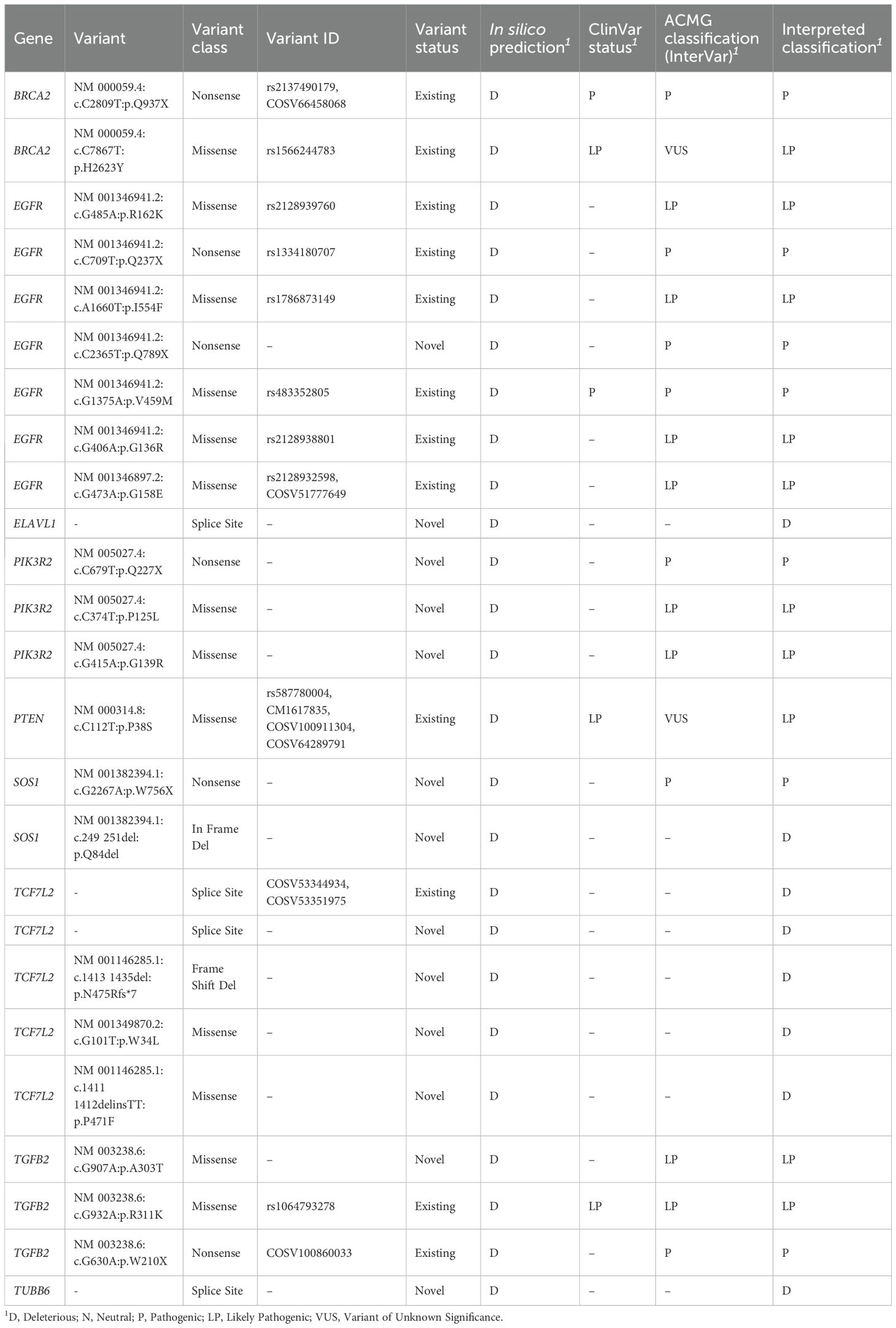
Table 3. List of Pathogenic (P), Likely Pathogenic (LP), and Deleterious (D) variants identified in the Saudi cohort, with associated details.
3.6 Genomic-based pathway alteration in the cohort
Building upon the observed somatic mutations, this study investigated the functional impact of these alterations by assessing the disruption of key cellular pathways (Figure 5). The analysis, based on the frequency of mutations in the selected genes, revealed widespread pathway alterations within the cohort. The Genomic Integrity pathway, specifically the Homologous Recombination Repair (HRR) mechanism, was found to be altered in a substantial proportion of patients (61%), primarily due to mutations identified in the BRCA2 gene. The critical Beta-catenin signaling pathway was disrupted in an even larger percentage of cases (65%), driven by mutations in both TCF7L2 and AXIN1. Downstream signaling cascades were also frequently affected; the PI3K signaling pathway showed alterations in 48% of patients, with the pathway being driven by a PIK3R2 mutation alone in six patients, a PTEN mutation in five patients, and mutations in both genes in two patients. Furthermore, Receptor Tyrosine Kinase (RTK) signaling, specifically involving the EGFR pathway, was altered in 43% of cases due to EGFR mutations. Another related signaling cascade, likely the RAS pathway, was also found to be altered in 43% of patients, attributable to mutations in the SOS1 gene.
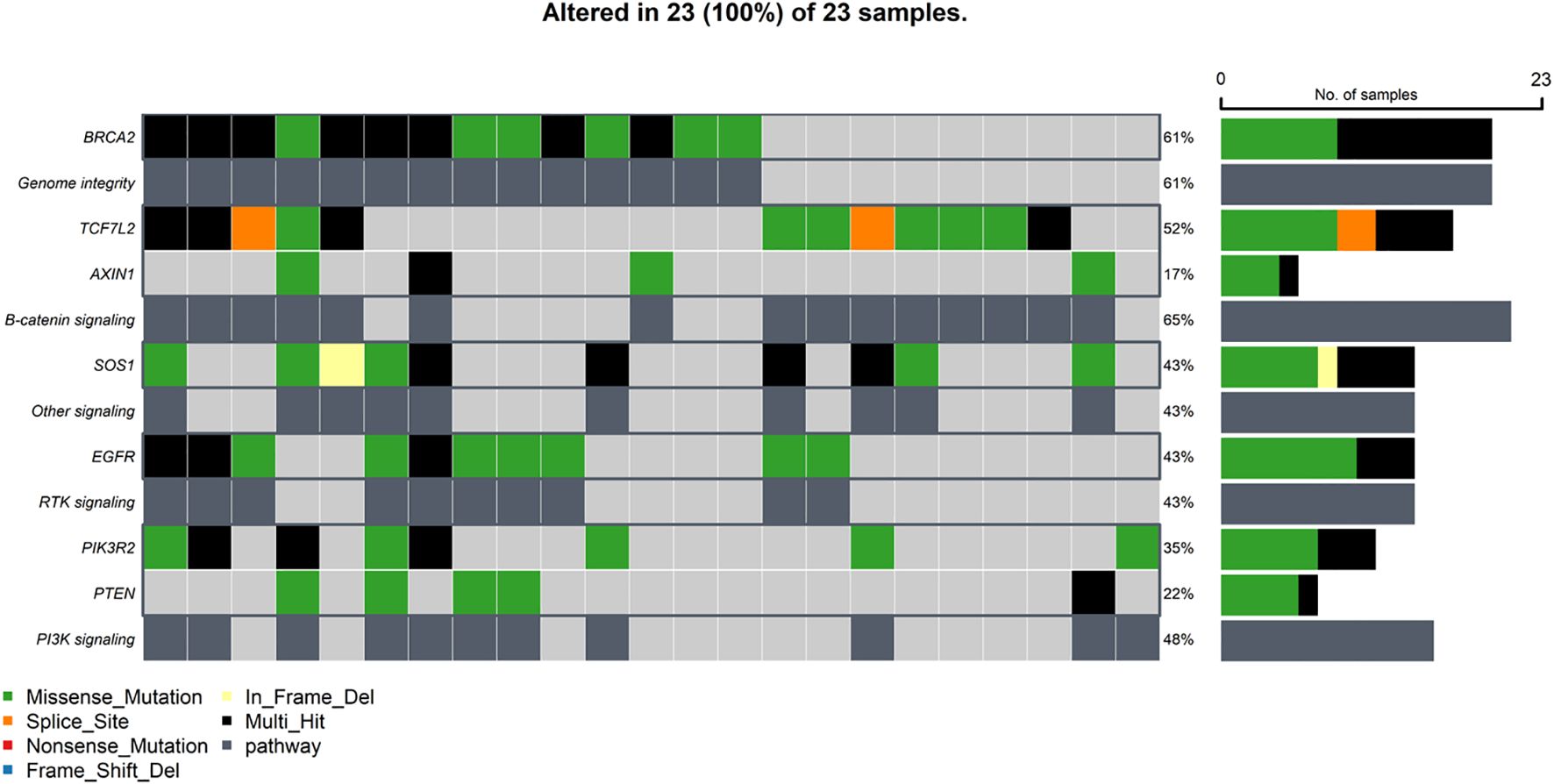
Figure 5. Oncoplot depicting the distribution of alterations in top genomic-based pathways across samples in the Saudi cohort.
4 Discussion
Our preliminary study offers an initial exploration of colorectal cancer within a Saudi Arabian patient cohort. By employing whole-exome sequencing, we’ve begun to examine a selected panel of cancer-associated genes. This targeted approach provides early insights into the mutation frequencies, pathogenic variants, and pathway disruptions that appear to be specific to this population whole-exome. This approach reveals molecular features that differ from international datasets, offering valuable information for precision oncology efforts in this region. Previously, we identified several variants associated with breast cancer (51) and iron deficiency (52) in the Saudi population.
Our cohort (Table 1) had a median age of diagnosis of 57 years, slightly younger than the global average, with 38% of patients diagnosed before age 50, aligning with global trends of rising early-onset CRC (53, 54). Alarmingly, 88% of patients presented with advanced-stage disease, including metastatic cases, suggesting delays in diagnosis and potentially limited access to early screening programs. This reinforces the urgent need to establish or enhance national CRC screening initiatives in Saudi Arabia (55).
Genomic analysis revealed a distinct mutational profile compared to the TCGA-COADREAD cohort (Table 2; Figures 3, 4). Among the most striking findings was the significantly higher frequency of BRCA2 mutations (61% vs. 7%), a gene canonically associated with DNA repair through homologous recombination (56–58). This observation is consistent with a recent NGS-based study of Saudi CRC patients, which reported BRCA2 mutations in 79% of cases (2), further supporting the potential significance of this gene in the regional disease profile. This notable enrichment may reflect population-specific genomic signatures or underlying hereditary predispositions. BRCA2 mutations were also a major contributor to the disruption of the Genomic Integrity pathway in over 60% of patients (Figure 5), indicating to defective DNA repair as a possible hallmark of CRC in this population. The high metastatic rate observed in our Saudi cohort (M1 in 88% of cases, compared to 22% in TCGA) could potentially be linked to these distinct mutational patterns, particularly alterations in genes like BRCA2, which are implicated in genomic instability and may promote more aggressive tumor behavior and metastatic progression.
Similarly, EGFR (43% vs. 3%) and TCF7L2 (52% vs. 11%) were more frequently mutated, both genes central to key oncogenic signalling cascades. EGFR is a well-established therapeutic target in CRC, particularly in KRAS wild-type tumors, and its high mutation rate may open avenues for targeted therapies such as tyrosine kinase inhibitors (59). It is important to clarify that EGFR expression, rather than EGFR mutations, serves as the key clinical biomarker for guiding anti-EGFR therapy in CRC. While EGFR mutations were observed in our study, they do not typically serve the same predictive role for response to these therapies as EGFR expression, especially in the context of RAS/BRAF wild-type tumors. As this study focused on genomic alterations, EGFR expression was not assessed.TCF7L2, a transcription factor in the Wnt/β-catenin pathway, showed not only a higher frequency of mutations but also a unique domain-level mutation distribution, with a preference for the CTNNB1-binding domain in the Saudi cohort (Figure 3D). This observation may suggest altered transcriptional regulation of Wnt target genes, contributing to uncontrolled proliferation (60, 61). Additionally, genes such as SOS1, PIK3R2, and SLC25A5 were frequently mutated, with SOS1 mutations contributing to both RTK and RAS pathway alterations in 43% of patients (Figure 5). The PI3K/AKT/mTOR pathway was also disrupted in 48% of cases (Figure 5), consistent with findings in aggressive or chemoresistant CRC subtypes (62, 63). Together, these results highlight the merging of multiple signalling pathways that may drive tumorigenesis through redundant or synergistic mechanisms in this population.
Notably, 25 pathogenic or likely pathogenic variants were identified and classified, including 13 novel variants not cataloged in public databases (Table 3), reinforcing the genetic uniqueness of the Saudi population and the importance of diverse representation in cancer genomics (64). Particularly, deleterious EGFR and BRCA2 variants suggest therapeutic vulnerability, as tumors harboring these mutations may be particularly susceptible to EGFR-targeted therapies and PARP inhibitors, respectively, due to their reliance on dysregulated signalling pathways or impaired DNA repair mechanisms (65, 66). Further, novel TCF7L2 and TGFB2 mutations may represent as-yet-undefined drivers of tumor progression. Notably, EGFR harbored the largest number of pathogenic variants, including several nonsense and missense mutations. Among them, NM_001346941.2:c.C2365T:p.Q789X, a novel truncating mutation, which may lead to loss of function or altered receptor dynamics, with potential implications for treatment resistance. Similarly, deleterious splice site and nonsense mutations in TCF7L2 and PIK3R2 may result in disrupted protein function, demonstrating a need for further experimental validation and possibly functional annotation through CRISPR-based models or transcriptome analysis.
Comparative analysis revealed unique mutational patterns in the Saudi cohort (Table 2; Figures 3, 4), including high mutation frequencies in KRT18 (30% vs. 1%) and TUBB6 (26% vs. 1%). While these genes are less well-characterized in CRC, their frequent mutation suggests a potential role in cytoskeletal regulation and cellular adhesion, processes critical to metastasis and tumor invasion (16, 67). Further, TUBB6 encodes a beta-tubulin isotype that is a critical component of the microtubule cytoskeleton, supporting cell division and intracellular transport (18). Disruptions in tubulin function may lead to defective cell division and contribute to chromosomal instability (CIN), a key factor in CRC progression. In contrast, the absence of PRDX1 mutations despite their presence in TCGA may reflect distinct selective pressures. Mutual exclusivity between TCF7L2 and BRCA2/PTEN mutations (Figure 1) further illustrates the complex interplay between tumor suppressors and signaling regulators, and points to potential functional redundancy or antagonism in tumor suppressor networks. Further mechanistic research is needed to understand how these genes interact. A crucial limitation of this study is the absence of functional assays to validate the impact of the identified mutations. While we have pinpointed specific pathogenic variants in different genes, our findings are based solely on genomic sequencing data and in-silico tools. The identification of these variants, however, provides a strong basis for future functional research to confirm their exact roles in CRC pathogenesis.
Our exploratory study reveals distinct mutational frequencies in the genes examined, yet a striking concordance with The Cancer Genome Atlas (TCGA) pan-cancer pathway analysis emerges upon converging these mutations onto their respective signaling networks. This suggests that while individual genes may exhibit varying mutation rates, the overall disruption of key oncogenic pathways remains remarkably consistent across different patient cohorts. Specifically, our cohort’s RTK-RAS signaling pathway was found to be altered in 69% of patients, primarily driven by mutations in EGFR or SOS1. This figure aligns closely with the TCGA’s findings, which report RTK-RAS pathway alterations in a wide range of colorectal cancer (CRC) subtypes, from 66% to 99% in chromosomal instability (CRC-CIN), genomically stable (CRC-GS), and microsatellite instability (CRC-MSI-POLE) subtypes (68). Similarly, the PI3K signaling pathway was altered in 48% of our patients, a frequency comparable to the 32–68% range observed in the TCGA cohort (68). This consistency highlights the central and conserved role of these pathways in colorectal carcinogenesis, regardless of the specific mutational drivers. In contrast, a notable divergence was observed in the Homologous Recombination Repair (HRR) pathway. Our cohort showed alterations in 61% of cases, a stark difference from the 21% reported in the TCGA CRC cohort (69). This discrepancy could reflect differences in the patient demographics, environmental exposures, or specific genetic backgrounds of our study population compared to the broader, more geographically diverse TCGA cohort. Alternatively, it might point to a unique, more frequent HRR deficiency in our patient group, which could have significant implications for therapeutic response to agents like PARP inhibitors. Further investigation is warranted to understand the factors underlying this significant variation and its clinical relevance.
While this exploratory study offers valuable initial insights into the molecular landscape of colorectal cancer in a Saudi Arabian cohort, several limitations warrant acknowledgment. Primarily, the small sample size significantly impacts the statistical power and generalizability of our findings. This restricted cohort also meant that the potential influence of confounding factors such as age, sex, and comorbidities on the observed molecular features could not be deeply explored; however, these will be critical considerations for larger-scale future investigations. Given that nine patients were diagnosed before the age of 50, the possibility of hereditary CRC cannot be excluded; however, as our study employed tumor-only sequencing without germline analysis, hereditary contributions could not be specifically addressed.
Furthermore, the reliance on a pre-defined gene panel for whole-exome sequencing, while enabling a targeted analysis, might introduce a degree of selection bias. This approach inherently limits the discovery of novel or population-specific driver mutations that fall outside the selected genes. To overcome this, future studies should consider employing broader sequencing strategies, such as whole-exome sequencing without a targeted panel or whole-genome sequencing, to provide a more comprehensive and unbiased view of the genomic landscape.
Another significant limitation arises when comparing our small cohort to large, ethnically diverse, yet predominantly European, datasets like The Cancer Genome Atlas (TCGA). These comparisons should be interpreted as preliminary, given the substantial differences in cohort size and ethnic composition. Moreover, methodological discrepancies in variant calling pipelines present a challenge for direct comparisons. Our study utilized a tumor-only whole-exome sequencing approach with a single variant caller (Mutect2), whereas large-scale initiatives like TCGA often employ matched tumor-normal sequencing and may integrate results from multiple variant callers. This difference in methodology can influence observed mutation frequencies and should be carefully considered when interpreting cross-dataset comparisons. Despite these limitations, our preliminary findings highlight the unique molecular features within this population, underscoring the necessity for further, more extensive, and methodologically harmonized research to advance precision oncology in Saudi Arabia.
5 Conclusion
Together, these initial findings underline the heterogeneity of CRC and the limitations of extrapolating Western-derived genomic data to other populations (70). The high frequency of actionable and novel mutations supports the implementation of population-specific precision oncology strategies. Expanding sequencing efforts and developing regional variant databases are essential steps toward integrating genomic data into clinical practice in Saudi Arabia and the broader Middle East. Future larger cohort studies are warranted to further validate and expand upon these initial findings.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA1149347.
Ethics statement
The studies involving humans were approved by The Ethics Committee of the University of Tabuk (protocol code UT-115-13-2020). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
HA: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. AAA: Methodology, Writing – original draft, Writing – review & editing. RM: Formal analysis, Methodology, Validation, Writing – original draft, Writing – review & editing. OA: Funding acquisition, Writing – original draft, Writing – review & editing. AHA: Writing – review & editing. YH: Writing – review & editing. MA: Resources, Writing – review & editing. ADA: Resources, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This project is funded by the Deanship of Research and graduate studies, University of Tabuk, Saudi Arabia through the project number (S-1441-0080).
Acknowledgments
The authors extend their appreciation to the Deanship of Research and graduate studies, University of Tabuk, Saudi Arabia, for funding this research through project number S-1441-0080. We would also like to thank all the CRC patients who participated in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Roshandel G, Ghasemi-Kebria F, and Malekzadeh R. Colorectal cancer: epidemiology, risk factors, and prevention. Cancers. (2024) 16:1530. doi: 10.3390/cancers16081530
2. Alsolme E, Alqahtani S, Fageeh M, Barakeh D, Sharma NK, Mangul S, et al. The genomic landscape of colorectal cancer in the Saudi Arabian population using a comprehensive genomic panel. Diagnostics. (2023) 13:2993. doi: 10.3390/diagnostics13182993
3. Almatroudi A. The incidence rate of colorectal cancer in Saudi Arabia: an observational descriptive epidemiological analysis. Int J Gen Med. (2020) 13:977–9. doi: 10.2147/IJGM.S277272
4. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. (2012) 487:330–7. doi: 10.1038/nature11252
5. Zhao Q, Wang F, Chen YX, Chen S, Yao YC, Zeng ZL, et al. Comprehensive profiling of 1015 patients’ exomes reveals genomic-clinical associations in colorectal cancer. Nat Commun. (2022) 13:2342. doi: 10.1038/s41467-022-30062-8
6. Pino MS and Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. (2010) 138:2059–72. doi: 10.1053/j.gastro.2009.12.065
7. Nazemalhosseini Mojarad E, Kuppen PJ, Aghdaei HA, and Zali MR. The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol Hepatol Bed Bench. (2013) 6:120–8. Available online at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4017514/
8. Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. (2018) 173:321–337.e10. doi: 10.1016/j.cell.2018.03.035
9. Semlali A, Parine NR, Al-Numair NS, Almutairi M, Hawsawi YM, Amri AA, et al. Potential role of Toll-like receptor 2 expression and polymorphisms in colon cancer susceptibility in the Saudi Arabian population. Onco Targets Ther. (2018) 11:8127–41. doi: 10.2147/OTT.S168478
10. Alanazi IO, Shaik JP, Parine NR, Azzam NA, Alharbi O, Hawsawi YM, et al. (2021) association of HER1 and HER2 gene variants in the predisposition of colorectal cancer. J Oncol. (2021) 2021:6180337. doi: 10.1155/2021/6180337
11. Nguyen LH, Goel A, and Chung DC. Pathways of Colorectal Carcinogenesis. Gastroenterology (2020) 158(2):291–302. doi: 10.1053/j.gastro.2019.08.059
12. Koveitypour Z, Panahi F, Vakilian M, Peymani M, Seyed Forootan F, Nasr Esfahani MH, et al. Signaling pathways involved in colorectal cancer progression. Cell Biosci. (2019) 9:97. doi: 10.1186/s13578-019-0361-4
13. Farooqi AA, de la Roche M, Djamgoz MBA, and Siddik ZH. Overview of the oncogenic signaling pathways in colorectal cancer: Mechanistic insights. Semin Cancer Biol. (2019) 58:65–79. doi: 10.1016/j.semcancer.2019.01.001
14. Makino T, Yamasaki M, Takeno A, Shirakawa M, Miyata H, Takiguchi S, et al. Cytokeratins 18 and 8 are poor prognostic markers in patients with squamous cell carcinoma of the oesophagus. Br J Cancer. (2009) 101:1298–306. doi: 10.1038/sj.bjc.6605313
15. Majumdar D, Tiernan JP, Lobo AJ, Evans CA, and Corfe BM. Keratins in colorectal epithelial function and disease. Int J Exp Pathol. (2012) 93:305–18. doi: 10.1111/j.1365-2613.00830.x
16. Fang J, Wang H, Liu Y, Ding F, Ni Y, and Shao S. High KRT8 expression promotes tumor progression and metastasis of gastric cancer. Cancer Sci. (2017) 108:178–86. doi: 10.1111/cas.13120
17. Lee YJ. Knockout mouse models for peroxiredoxins. Antioxidants (Basel). (2020) 9:182. doi: 10.3390/antiox9020182
18. Findeisen P, Mühlhausen S, Dempewolf S, Hertzog J, Zietlow A, Carlomagno T, et al. Six subgroups and extensive recent duplications characterize the evolution of the eukaryotic tubulin protein family. Genome Biol Evol. (2014) 6:2274–88. doi: 10.1093/gbe/evu187
19. Nami B and Wang Z. Genetics and expression profile of the tubulin gene superfamily in breast cancer subtypes and its relation to taxane resistance. Cancers (Basel). (2018) 10:274. doi: 10.3390/cancers10080274
20. Chen YJ, Hong WF, Liu ML, Guo X, Yu YY, Cui YH, et al. An integrated bioinformatic investigation of mitochondrial solute carrier family 25 (SLC25) in colon cancer followed by preliminary validation of member 5 (SLC25A5) in tumorigenesis. Cell Death Dis. (2022) 13:237. doi: 10.1038/s41419-022-04692-1
21. Dong R, Chen P, Polireddy K, Wu X, Wang T, Ramesh R, et al. An RNA-binding protein, hu-antigen r, in pancreatic cancer epithelial to mesenchymal transition, metastasis, and cancer stem cells. Mol Cancer Ther. (2020) 19:2267–77. doi: 10.1158/1535-7163.MCT-19-0822
22. Mao G, Mu Z, and Wu DA. Exosomal lncRNA FOXD3-AS1 upregulates ELAVL1 expression and activates PI3K/Akt pathway to enhance lung cancer cell proliferation, invasion, and 5-fluorouracil resistance. Acta Biochim Biophys Sin (Shanghai). (2021) 53:2267–77. doi: 10.1093/abbs/gmab129
23. Shi J, Guo C, and Ma J. CCAT2 enhances autophagy-related invasion and metastasis via regulating miR-4496 and ELAVL1 in hepatocellular carcinoma. J Cell Mol Med. (2021) 25:8985–96. doi: 10.1111/jcmm.16859
24. Cai Z, Xu H, Bai G, Hu H, Wang D, Li H, et al. ELAVL1 promotes prostate cancer progression by interacting with other m6A regulators. Front Oncol. (2022) 12:939784. doi: 10.3389/fonc.2022.939784
25. Andrews S. FastQC: A quality control tool for high throughput sequence data (2010). Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. (Accessed March 25, 2025).
26. Bolger AM, Lohse M, and Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. (2014) 30:2114–20. doi: 10.1093/bioinformatics/btu170
27. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv: Genomics. (2013) 1303.3997. Available online at: https://arxiv.org/abs/1303.3997
28. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinf (Oxford England). (2009) 25:2078–9. doi: 10.1093/bioinformatics/btp352
29. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinf. (2013) 43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43
30. Pruitt KD, Tatusova T, Brown GR, and Maglott DR. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. (2012) 40:D130–5. doi: 10.1093/nar/gkr1079
31. Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. (2014) 42:D980–5. doi: 10.1093/nar/gkt1113
32. 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. (2015) 526:68–74. doi: 10.1038/nature15393
33. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. (2016) 536:285–91. doi: 10.1038/nature19057
34. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581:434–43. doi: 10.1038/s41586-020-2308-7
36. Kumar P, Henikoff S, and Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. (2009) 4:1073–81. doi: 10.1038/nprot.2009.86
37. Adzhubei I, Jordan DM, and Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. In: Current protocols in human genetics. Hoboken, NJ, USA: John Wiley & Sons, Inc. (2013). doi: 10.1002/0471142905.hg0720s76
38. Shihab HA, Gough J, Mort M, Cooper DN, Day IN, and Gaunt TR. Ranking non-synonymous single nucleotide polymorphisms based on disease concepts. Hum Genomics. (2014) 8:11. doi: 10.1186/1479-7364-8-11
39. Schwarz JM, Rödelsperger C, Schuelke M, and Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods Aug. (2010) 7:575–6. doi: 10.1038/nmeth0810-575
40. Reva B, Antipin Y, and Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. (2011) 39:e118. doi: 10.1093/nar/gkr407
41. Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, and Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. (2014) 46:310–5. doi: 10.1038/ng.2892
42. Li Q and Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet. (2017) 100:267–80. doi: 10.1016/j.ajhg.2017.01.004
43. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
44. Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. (2018) 6:271–281.e7. doi: 10.1016/j.cels.2018.03.002
45. R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing (2023). Available online at: https://www.R-project.org.
46. Wickham H, Averick M, Bryan J, Chang W, McGowan LD, François R, et al. Welcome to the tidyverse. J Open Source Software. (2019) 4:1686. doi: 10.21105/joss.01686
48. Mayakonda A, Lin DC, Assenov Y, Plass C, and Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. (2018) 28(11):1747–56. Available online at: http://dx.doi.org/10.1101/gr.239244.118.
49. Iannone R, Cheng J, Schloerke B, Hughes E, Lauer A, and Seo J. gt:Easily Create Presentation-Ready Display Tables. (2023).
50. Sjoberg DD, Whiting K, Curry M, Lavery JA, and Larmarange J. Reproducible summary tables with the gtsummary package. R J. (2021) 13:570–80. doi: 10.32614/RJ-2021-053
51. Alzahrani OR, Mir R, Alatwi HE, Hawsawi YM, Alharbi AA, Alessa AH, et al. Potential Impact of PI3K-AKT Signaling Pathway Genes, KLF-14, MDM4, miRNAs 27a, miRNA-196a Genetic Alterations in the Predisposition and Progression of Breast Cancer Patients. Cancers (Basel). (2023) 15(4):1281. doi: 10.3390/cancers15041281
52. Al-Amer O, Hawasawi Y, Oyouni AAA, Alshehri M, Alasmari A, Alzahrani O, et al. Study the association of transmembrane serine protease 6 gene polymorphisms with iron deficiency status in Saudi Arabia. Gene. (2020) 751:144767. doi: 10.1016/j.gene.2020.144767
53. Siegel RL, Miller KD, Wagle NS, and Jemal A. Cancer statistics. CA Cancer J Clin. (2023) 73:17–48. doi: 10.3322/caac.21763
54. Araghi M, Soerjomataram I, Jenkins M, Brierley J, Morris E, Bray F, et al. Global trends in colorectal cancer mortality: projections to the year 2035. Int J Cancer. (2019) 144:2992–3000. doi: 10.1002/ijc.32055
55. Almadi MA and Basu P. Doing things right and doing the right things: Colorectal cancer screening in Saudi Arabia. Saudi J Gastroenterol. (2023) 29:67–70. doi: 10.4103/sjg.sjg_82_23
56. Roy R, Chun J, and Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. (2011) 12:68–78. doi: 10.1038/nrc3181
57. Le HP, Heyer WD, and Liu J. Guardians of the genome: BRCA2 and its partners. Genes (Basel). (2021) 12:1229. doi: 10.3390/genes12081229
58. Prakash R, Zhang Y, Feng W, and Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. (2015) 7:a016600. doi: 10.1101/cshperspect.a016600
59. Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. (2016) 27:1386–422. doi: 10.1093/annonc/mdw235
60. Wenzel J, Rose K, Haghighi EB, Lamprecht C, Rauen G, Freihen V, et al. Loss of the nuclear Wnt pathway effector TCF7L2 promotes migration and invasion of human colorectal cancer cells. Oncogene. (2020) 39:3893–909. doi: 10.1038/s41388-020-1259-7
61. Clevers H and Nusse R. Wnt/β-catenin signaling and disease. Cell. (2012) 149:1192–205. doi: 10.1016/j.cell.2012.05.012
62. Li Q, Geng S, Luo H, Wang W, Mo YQ, Luo Q, et al. Signaling pathways involved in colorectal cancer: pathogenesis and targeted therapy. Signal Transduct Target Ther. (2024) 9:266. doi: 10.1038/s41392-024-01953-7
63. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, and Abraham RT. The PI3K pathway in human disease. Cell. (2017) 170:605–35. doi: 10.1016/j.cell.2017.07.029
64. Popejoy A and Fullerton S. Genomics is failing on diversity. Nature. (2016) 538:161–4. doi: 10.1038/538161a
65. Dienstmann R, Rodon J, Barretina J, and Tabernero J. Genomic medicine frontier in human solid tumors: prospects and challenges. J Clin Oncol. (2013) 31:1874–84. doi: 10.1200/JCO.2012.45.2268
66. Lord CJ and Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science. (2017) 355:1152–8. doi: 10.1126/science.aam7344
67. Berx G and van Roy F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb Perspect Biol. (2009) 1:a003129. doi: 10.1101/cshperspect.a003129
68. Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic signaling pathways in the cancer genome atlas. Cell. (2018) 173:321–337.e10. doi: 10.1016/j.cell.2018.03.035
69. Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. (2018) 23:239–254.e6. doi: 10.1016/j.celrep.2018.03.076
Keywords: colorectal cancer, Saudi cohort, whole-exome sequencing, somatic mutation, pathway analysis, precision oncology
Citation: Alatwi HE, Alharbi AA, Mir R, Alzahrani OR, Alessa AH, Hawsawi YM, Arishi MA and Albalawi AD (2025) Whole-exome sequencing in Saudi colorectal cancer patients reveals distinct mutational patterns and population specific pathogenic variants. Front. Oncol. 15:1679528. doi: 10.3389/fonc.2025.1679528
Received: 04 August 2025; Accepted: 22 September 2025;
Published: 08 October 2025.
Edited by:
Jianxun J. Song, Texas A&M Health Science Center, United StatesReviewed by:
Sunilgowda Sunnagatta Nagaraja, Texas A&M University, United StatesRakesh Kumar, Texas A&M University, United States
Copyright © 2025 Alatwi, Alharbi, Mir, Alzahrani, Alessa, Hawsawi, Arishi and Albalawi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanan E. Alatwi, aF9hbGF0d2lAdXQuZWR1LnNh