 Yifan Luo1
Yifan Luo1 Jun Gu
Jun Gu- 1School of Medicine, Southeast University, Nanjing, China
- 2State Key Laboratory of Bioelectronics, School of Biological Science and Medical Engineering, Southeast University, Nanjing, China
Breast cancer, as the most common cancer in women, is a highly heterogeneous and complex tumor. One of the important reasons for the poor prognosis and high mortality of breast cancer patients is drug resistance. More and more evidence shows that epithelial-to-mesenchymal transition (EMT) is a key driver of malignant behavior of breast cancer, and also the core promoter of drug resistance. Multiple EMT-related signaling pathways activate EMT-transcription factors (EMT-TFs) and interact with each other, ultimately inducing drug resistance. The role of EMT in promoting invasion and metastasis has been studied in detail and systematically summarized, but its role in drug resistance of breast cancer has not been elucidated comprehensively. The purpose of this review is to clarify the EMT-related regulatory network in breast cancer and the possible mechanisms of EMT-induced drug resistance. Moreover, we have discussed the potential therapeutic advantages of reversing EMT and drug resistance by effectively targeting key elements of the regulatory network, with particular emphasis on EMT-related signaling pathways and microRNAs. This review summarizes the drug resistance of breast cancer induced by EMT systematically, which is of great significance for solving the drug resistance problem of breast cancer and improving the prognosis of patients.
1 Introduction
In recent years, breast cancer, surpassing lung cancer, has become the most common cancer among women and the primary cause of cancer death (1). Breast cancer causes 685,000 deaths annually among cancer patients, far exceeding the data from 2018, which indicates that the burden of breast cancer incidence and mortality is increasing rapidly worldwide (2). Breast cancer is highly heterogenous and has various subtypes, thus it is destined to be a complicated disease to treat, especially triple negative breast cancer (TNBC) which lacks the expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) (3). According to the guideline, effective therapeutic methods currently used in clinic include chemotherapy, endocrine therapy, targeted therapy, and so on (4). Although tumors are initially sensitive to the anti-tumor drugs, they may develop resistance through various pathways with the continued use of these drugs (5). Widely utilized chemotherapy agents like doxorubicin and paclitaxel combat cancer through their cytotoxic actions. Although TNBC is initially sensitive to chemotherapy than subtypes, most patients gradually develop drug resistance with continued drug administration (6). Tamoxifen is a common endocrine therapy, which can control the progression of ER-positive breast cancer. However, about half of patients with advanced ER-positive breast cancer and almost all patients with metastatic disease have no response to first-line tamoxifen treatment, in addition (7). Trastuzumab, another commonly used drug, has an effective response rate of only 26% as a single first-line treatment in patients with HER2-positive metastatic breast carcinomas (8). Statistical data shows that over ninety percent of cancer deaths are attributed to drug resistance (9, 10). It has always been a persistent factor restricting the therapeutic effect of breast cancer patients (11). Therefore, it is of far-reaching significance to explore the mechanism and treatment strategy of drug resistance in breast cancer.
Drug resistance is frequently complicated and multifactorial owing to the dynamic tumor microenvironment (9). Various intertwined mechanisms interact with each other and signal pathways interfere, ultimately leading to drug resistance (5, 9, 11). In recent years, the important role of epithelial-to-mesenchymal transition (EMT) in drug resistance has gradually been discovered. EMT is a cellular program that involves the transition of cells from epithelial phenotype to mesenchymal phenotype, accompanied by the acquisition of migration ability (12). EMT plays a role in various physiological and pathological processes, including tumor malignant progression and drug resistance (13). Although most of the reports in the past decades have focused on the crucial role of EMT in breast cancer metastasis, increasing evidence shows that EMT is an important mechanism leading to an increased likelihood of drug resistance in breast cancer populations (14). According to the clinical analysis between the chemotherapy response of breast cancer patients and the gene expression profiles of tumor samples, the expression activation of EMT related genes is strongly related to the occurrence of treatment resistance (15, 16). Moreover, Inayatullah et al. used MAST test to compare gene expression profiles and found that chemotherapy resistance in TNBC is related to the activation of EMT program at the beginning of treatment (17). Many studies have reported that the activation of EMT in breast cancer cell lines makes them unresponsive or less sensitive to the treatment of tamoxifen, paclitaxel and doxorubicin (18–20). However, the relationship between EMT and the metastasis of breast cancer has been widely discussed, but the drug resistance caused by EMT has rarely been systematically and completely sorted out, specifically for breast cancer. This review aims to fill this gap and concludes with a discussion on the relationship between EMT and drug resistance, as well as therapeutic strategies targeting EMT for breast cancer.
2 EMT: an overview
EMT is a cellular process that allows polarized epithelial cells to exhibit a mesenchymal phenotype by undergoing various biochemical changes, accompanied with the loss of cell-cell adhesive properties and apical–basal polarity, as well as the acquisition of mesenchymal characteristics, including upregulation of vimentin and loss of cell adhesion (21). (Figure 1) During the EMT process, it can be observed that the use of intermediate filaments shifts from cytokeratin to vimentin, and cells achieve higher mobility and invasiveness (12, 22). Cancer cells undergoing EMT can be identified by detecting changes in the expression of EMT-related genes or proteins (23, 24). In the past few decades, research related to EMT markers has been continuously emerging, but the most closely related to the phenotypic changes of EMT are still the loss of cell-cell adhesion and its associated protein expression abnormalities (23). E-cadherin, as a calcium-dependent cell-cell adhesion molecule, is currently the most convincing marker for assessing EMT status, which is also used in clinical diagnosis of cancer progression (24). The downregulation of epithelial marker E-cadherin, accompanied by simultaneous upregulation of mesenchymal marker N-cadherin, is established as a hallmark of EMT. Furthermore, the tight junction components, such as claudins, occludins, zonula occlusions‐1(ZO-1), exhibit downregulation of expression or weakened function when EMT occurs (23, 25). Apart from loss of cell-cell adhesion, change in motility and invasiveness is another major hallmark of EMT. Expression of cytokeratin and vimentin can be used to monitor the status of EMT in breast cancer (25, 26). Additionally, mesenchymal-related cytoskeletal markers(vimentin, FSP1, α-SMA), ECM proteins (fibronectin, type I/III collagen) and matrix metalloproteinases (MMPs) were also significantly upregulated during EMT (25).
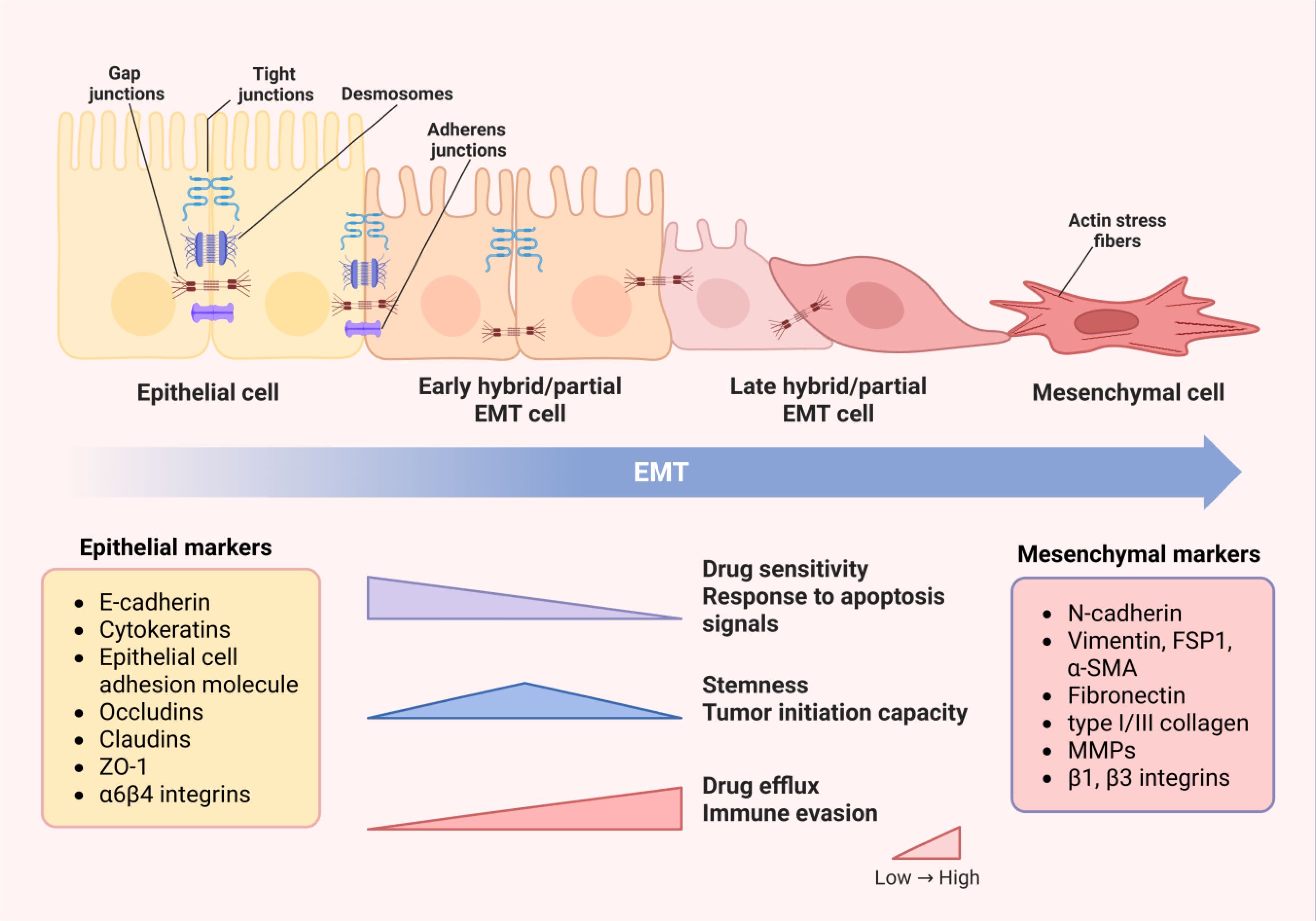
Figure 1. Changes in cell phenotypes and properties during EMT process. With the occurrence of EMT, intercellular junctions dissolve and cell polarity is lost, leading to cytoskeleton remodeling. Therefore, the transformation of cell morphology from epithelial to mesenchymal phenotype and changes in protein expression can be observed. Epithelial markers, including E-cadherin, cytokines, epithelial cell adhesion molecules and tight junction proteins, gradually decrease in expression levels or become inactive, while mesenchymal markers, such as N-cadherin, mesenchymal related cytoskeletal markers, ECM proteins and MMPs, are significantly upregulated. The changes in cell structure and phenotype determine the alterations in properties and functions. The drug sensitivity and response to apoptosis signals of cells decccteristics of drug efflux and immune evasion become increasingly prominent. Cells in the partial EMT state have the highest levels of stemness and tumor initiation ability. Image created using Bio-Render.com software.
An important feature of EMT is reversibility. Mesenchymal–epithelial transition (MET) is the reverse process of EMT, and both of them contribute to the tumor metastasis (27, 28). Another notable feature of EMT is that the transition from epithelial to mesenchymal status in adult tissues is often incomplete, and few cells can complete the entire EMT process (12, 27). Therefore, most cancer cells undergoing EMT will remain in the epithelial/mesenchymal intermediate state, also known as hybrid E/M or partial EMT phenotype. Due to varying degrees of activation of the EMT program, a spectrum of partial EMT cells is generated, which is considered to have higher plasticity than complete mesenchymal cells (29).
3 EMT-related regulatory networks in breast cancer
The initiation of EMT programs is co-regulated by multiple signaling pathways. The cooperation and crosstalk of these signaling pathways ultimately stimulate the activation of EMT-transcription factors (EMT-TFs), whose transcription targets are aforementioned hallmarks of EMT. (Figure 2) In addition to signaling pathways, the activation of EMT is also regulated by epigenetic modifications. In summary, EMT is a multifaceted program activated by a regulatory network formed by the combined action of multiple factors.
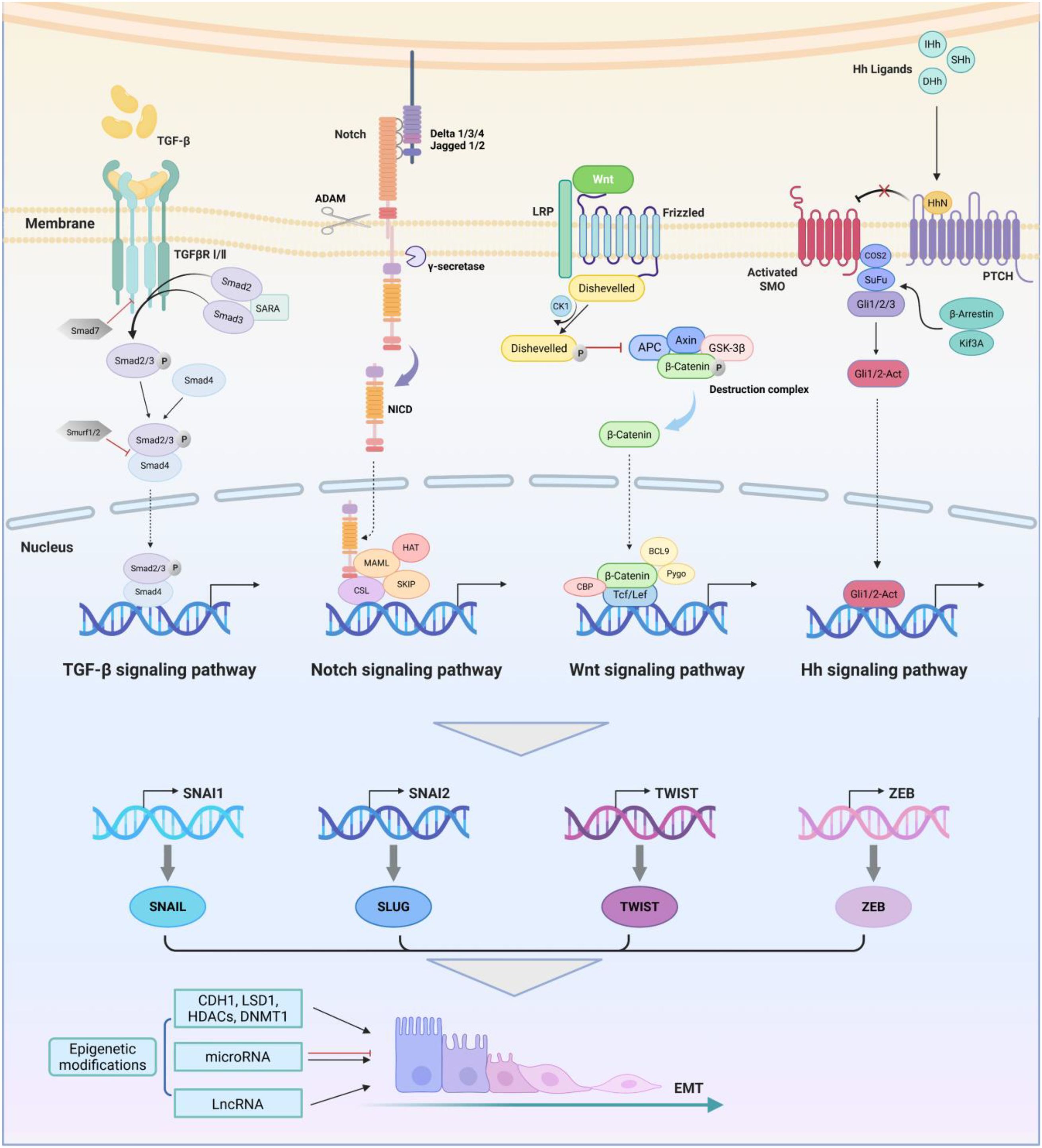
Figure 2. EMT-related regulatory networks in breast cancer. The TGF-β receptor is a complex composed of two types of transmembrane serine threonine kinase receptors. The activation of the canonical TGF-β signaling pathway is mediated by SMAD transcription factors. Activated TGF-β ligands bind to TβRII, inducing the recruitment, phosphorylation, and activation of TβRI, thereby phosphorylating downstream mediators SMAD2 and SMAD3, which then form the trimer with Smad4. After entering the nucleus, the trimer binds to EMT-TFs and initiates transcription. The Notch signaling pathway is mediated by four receptors (Notch-1, -2, -3, -4) and five ligands (Delta-like-1, -3, -4 and Jagged-1, -2). The Notch receptor is activated by interaction with its ligand. The gamma-secretase is responsible for cleaving intracellular C-terminal fragments of the Notch receptor and a disintegrin and metalloproteases (ADAM) is responsible for cleaving the extracellular part, leading to the release of NICD and formation of the complex with CSL. Wnt binds to the LRP5/6-Frizzled receptor to form functional receptor complex and induce recruitment and activation of Disheveled receptor, thereby inhibiting the phosphorylation and ubiquitination of β-catenin by the destruction complex. Dissociative β-catenin in the cytoplasm enters the nucleus and initiates transcription of downstream target genes. There are three types of Hh ligands, including Sonic Hedgehog (SHh), Indian Hedgehog (IHh), and Desert Hedgehog (DHh). Hedgehog protein forms a signal active N-terminal fragment HhN via self cleavage, which binds to PTCH to activate SMO. The activated SMO binds to the COS2/Sufu/Gali trimer, causing Gli to dissociate from the trimer and become the active Gli1/2-Act, which leads to upregulation of Hh target genes. The synergistic effect of these EMT-related signaling pathways collectively activates SNAIL, TWIST, ZEB and other EMT-related transcription factors. They regulate downstream transcriptional networks together with epigenetic factors, further mediating the biological effects of EMT. Image created using Bio-Render.com software.
3.1 EMT-related signaling pathways
EMT is regulated and affected by multiple signaling pathways, among which the critical pathways that regulate EMT initiation in breast cancer include transforming growth factor-beta (TGF-β), Notch, Wnt and Hedgehog (Hh) pathways (30). Growing evidence suggests that these signaling pathways dominate the activation of EMT, tumor development, and the occurrence of drug resistance. We summarize the process of regulating EMT by the four critical signaling pathways in breast cancer concisely, providing a theoretical basis for subsequent targeted therapy strategies.
The TGF-β pathway is one of the earliest discovered and the most extensively studied signaling cascade in the induction of EMT (31). Activated TGF-β ligands phosphorylate downstream mediator small mothers against decapentaplegics (SMADs) by binding to TGF-β receptors and form a trimer (32). After entering the nucleus, the trimer binds to EMT-TFs and initiates transcription (33). It is reported that the Notch receptor Notch-4 plays a primary role in EMT signaling in breast cancer cells and could be a potential target (34). The Noth intracellular domain (NICD) is released through a cascade of proteolytic cleavage, which forms a complex with RBP-jk (i.e. CSL) and triggers EMT-TFs (35). The Wnt signaling pathway has been gradually discovered to play an indispensable role in regulating EMT, stemness and drug resistance of various cancers, including breast cancer (36, 37). Wnt binds to Frizzled (Fzd) receptor on the cell membrane to form Disheveled (Dvl)-Fzd complexes, and inhibits β-catenin phosphorylation that originally occurred in the construction complex, allowing it to enter the nucleus and ultimately activate EMT-TFs (38). Hh binds to its receptor Patched (PTCH) to activate Smoothened (SMO) that was originally suppressed during transport and allow the Hh signaling to be transmitted downstream to glioma (GLI) family, leading to upregulation of Hh target genes, such as EMT-TFs (39).
In addition to the pathways mentioned above, there are other signals involved with EMT in breast cancer, including Nuclear factor-kappaB (NF-κB), Phosphatidylinositol 3-Kinase (PI3K)/AKT, Mitogen-Activated Protein Kinase (MAPK), Hypoxia‐Induced Factor (HIF), Epidermal Growth Factor (EGF) and so on. These pathways crosstalk and interact with each other, forming an EMT-related signal network. For example, hypoxia activated HIF-1α promotes EMT by enhancing the Notch signaling pathway (35, 40). NF-κB signal can enhance the Hh pathway, which is also influenced by TGF-β/Smad, HIF‐1α, RAS/MAPK or PI3K/AKT signaling (41–45). EGF receptors can also regulate downstream signaling molecules, including β-catenin, RAS, MAPK, and PI3K/AKT, to regulate EMT associated events (46).
3.2 EMT-related transcription factors
The combined effect of signaling pathways leads to the activation of EMT-TFs. The transition of cancer cells from epithelial to mesenchymal states is regulated by EMT-TFs, including SNAIL, TWIST, ZEB, FOX, SOX, PRRX, etc (47). SNAIL, TWIST, and ZEB family are currently recognized as the main regulatory factors driving the transcription pathways of EMT, which can regulate cellular characterization, such as intercellular adhesion, cell polarity, and motility (48, 49). SNAIL family (SNAI1, SNAI2, SNAI3) and basic helix loop helix (BHLH) family (TWIST1, TWIST2) regulate EMT by downregulating epithelial gene expression and upregulating mesenchymal gene expression (50, 51). The E-box elements in the E-cadherin (CDH1) promoter play a critical negative regulatory role in gene transcription (52). ZEB family of zinc finger (ZEB1, ZEB2) regulate gene sequences by binding to E-boxes, ultimately activating transcription (53). The Snail family can also induce EMT via binding to the elements and inhibiting CDH1 (54, 55). EMT-TFs typically control expression with each other and collaborate functionally on target genes to achieve the activation of the EMT program (56). For example, TWIST and SNAIL are frequently co-expressed in breast cancer, and the expression of SNAI2 (SLUG) directly depends on TWIST1 (18).
To date, all established EMT processes involve at least one member of these EMT-TFs. However, the occurrence of EMT cannot be inferred solely from the expression of EMT-TFs in cells, as these transcription factors are also involved in other cellular processes such as proliferation and apoptosis (12). Additionally, it should also be noted that the signaling pathways have more than a unidirectional effect on EMT-TFs. Although numerous reports have shown that various signaling pathways activate EMT-TFs to induce the occurrence of EMT, there are also many studies demonstrating that EMT-TFs can stimulate the transmission of signaling pathways. For example, Snail and Slug have been shown to activate TGF-β and MAPK pathways (57, 58). The positive feedback between signaling pathways and EMT seems to be a vicious cycle, which may explain why tumor progression is often so rapid and difficult to cure.
3.3 Epigenetic modifications
The course of EMT is related to epigenetic alterations, which are achieved by regulating the function of EMT-TFs (59). Epigenetic modifications, including DNA methylation, histone modifications and non-coding RNA regulation, have been confirmed to be largely involved in the EMT process. It is reported that CDH1 promoter metabolism plays an important role in EMT of various human tumors, including breast cancer (60, 61). EMT-TFs, such as SNAIL and ZEB, bind to E-box on CDH1 promoter, directly leading to inhibition of E-cadherin expression (62). In addition, it was discovered that demethylases play an important role in EMT (63). The histone demethylase lysine-specific demethylase 1 (LSD1) interacts with SNAIL and is recruited into the CDH1 promoter, leading to histone demethylation (64, 65). Exosomal piRNA-17560 enhances the stable expression of ZEB1 by reducing N6-methyladenosine RNA methylation, thereby inducing EMT and chemoresistance (66).
The image of non-coding RNA as a key factor that can affect the EMT process has become increasingly prominent. MicroRNAs (miRNAs) are a group of non‐coding single‐stranded small RNAs. They bind to the 3’-UTR region of downstream target mRNA through base pairing, which can cause the degradation of target mRNA or inhibit its translation, thus they are considered as the main post-transcriptional regulators of gene expression (67, 68). Research has shown that miRNAs affect the EMT process by mediating the expression of EMT-TFs. The miR-200 family is believed to inhibit the expression of ZEB, and interestingly, ZEB simultaneously suppresses the expression of the miR-200 family through a double negative feedback loop in breast cancer (69–71). Similar double negative feedback loops can also be observed between other miRNAs and EMT-TFs, including miR-203/SNAIL1, miR-129-5p/SOX4 and miR-30a/SOX4 (72–74). Mounting evidence has indicated that long non-coding RNAs (lncRNAs) contribute to malignant behaviors of breast cancer, including EMT and drug resistance (75). It is reported that LincK can regulate the expression of ZEB1 and the function of miR-200 in breast cancer (76). Similarly, lncRNA LINC00460 has been shown to promote Cancer stem cell (CSC)/EMT-like characteristics and resistance to doxorubicin by forming a positive feedback loop with c-MYC (77). Interestingly, high expression of lncRNA SNHG6 can promote EMT initiation and tamoxifen resistance in ER-positive breast cancer by inhibiting miR-101 (78). Another new study have revealed that lncRNA PTENP1 regulates EMT and drug resistance of breast cancer through isolating miR-21 by constructing a dynamic Boolean network model (79). In murine or human breast cancer, there are other examples of lncRNA-miRNA interactions like this, and some studies have referred to these pathways as lncRNA/miRNA axis, which can exert the oncogenic effect and promote drug resistance by regulating EMT process (80).
In conclusion, the elements involved in the EMT process are not independent, nor are they acting unilaterally. These factors and signaling pathways interact and entangle with each other, synergistically regulating EMT. The same factor can affect every step of the EMT process, and downstream factors can also have a reverse impact on upstream signals. New evidence suggests that the presence of TGFβ2/Smad‐Snail1/EZH2‐miRNAs loop in TNBC can maintain EMT phenotype and induce drug resistance, which is a good example of the interaction of multiple regulatory factors (81). There are countless interplays and feedback loops like this between various elements. Therefore, the EMT-related regulatory network is a complex and dynamic system that deserves further research.
4 Mechanisms of EMT-induced drug resistance in breast cancer
Insufficient drug dosage during chemotherapy not only fails to eradicate cancer cells far from blood vessels, but also accelerates the EMT procedure, ultimately inducing drug resistance (82). Luo et al. identified that the molecular biomarkers of HR+/HER2 metastatic breast cancer patients resistant to standard treatment were related to EMT through single-cell RNA sequencing (83). Although there have been new advances in the study of the significance of EMT in drug resistance, the molecular mechanism is incomplete due to the lack of suitable in vivo models and limited human samples for comprehensive research (84). Next, we will provide a summary of several widely discussed potential mechanisms underlying EMT-induced drug resistance in breast cancer (Figure 3).
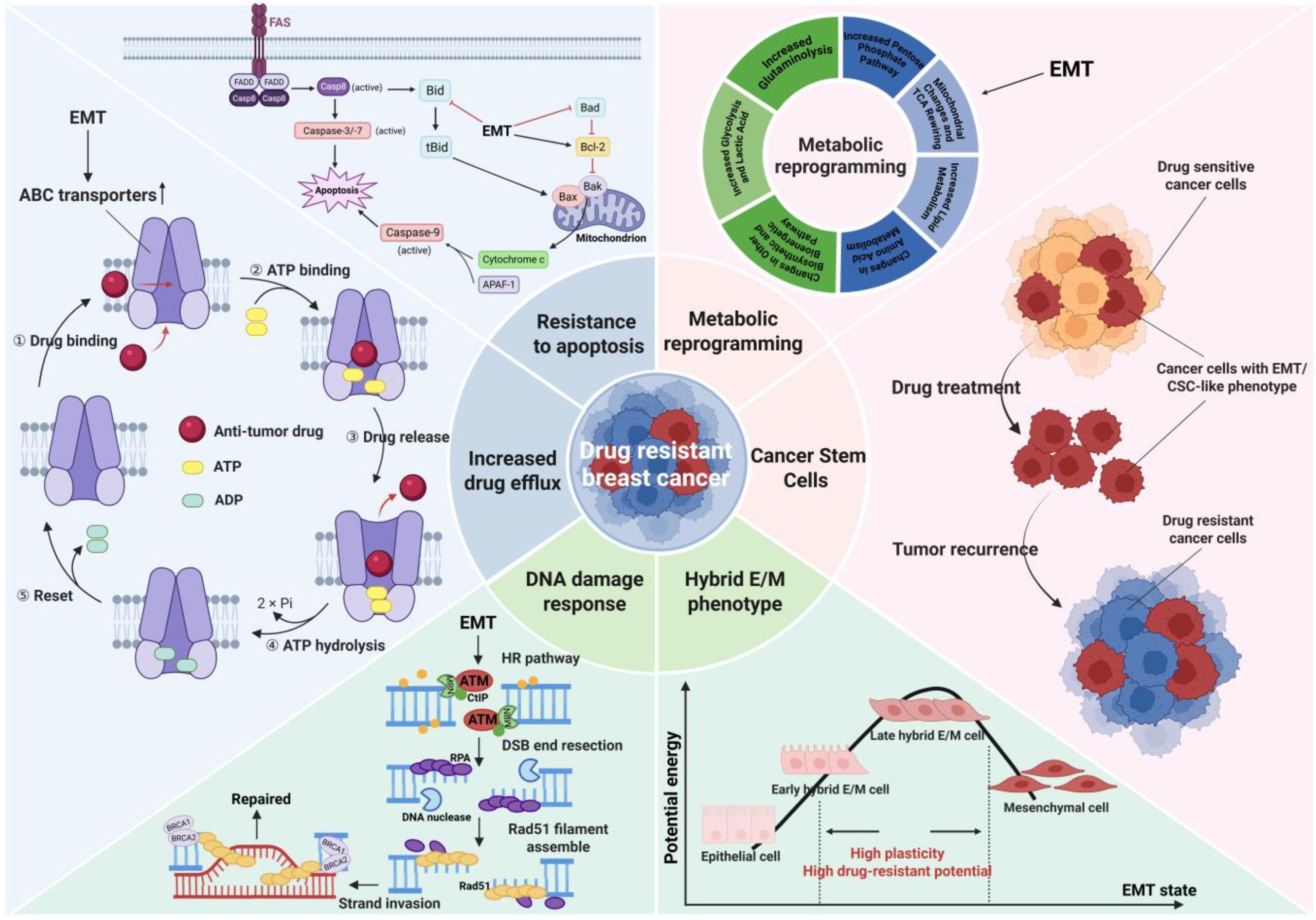
Figure 3. Potential mechanisms underlying EMT-induced drug resistance in breast cancer. EMT cells are highly similar in phenotype and function to CSCs, and EMT process is recognized to promote the production of CSCs. After killing drug sensitive cancer cells, residual cancer cells with EMT/CSC-like phenotype may trigger tumor recurrence and develop drug resistance. Most cancer cells that undergo EMT will remain in the hybrid E/M state, which exhibits the most prominent phenotypic heterogeneity and plasticity, enabling better adaptation to the microenvironment and resistance to drug action. The drug and ATP molecules enter the ABC transporter and bind to corresponding binding sites, forming an outward-open conformation that causes the drug to flow out of the cell. EMT promotes the expression and activation of ABC transporters, increasing drug efflux and leading to drug resistance. Cells that receive apoptotic signals activate pro-apoptotic protein and caspase through intrinsic and extrinsic apoptotic pathways to conduct apoptosis. EMT can upregulate the anti-apoptotic protein Bcl-2 and inhibit pro-apoptotic proteins, thereby resisting drug-induced apoptosis. The HR pathway is a classic DDR pathway for repairing DNA damage such as double strand breaks. EMT activates the HR pathway (such as activating the ATM promoter) to reverse the cell damage caused by platinum-based drugs, resulting in the tumor exhibiting a drug-resistant phenotype. EMT can induce metabolic reprogramming in cancer cells, resulting in marked changes such as increased glycolysis, increased lipid metabolism, mitochondrial respiratory inhibition and so on, which in turn induce tumor drug resistance. Image created using software.
4.1 Acquisition of CSC-like characteristics
Thanks to the rapid development of research related to CSCs, researchers have gained a new understanding of the mechanism of drug resistance caused by EMT. CSCs are a small subset of malignant cells that possess self-renewal ability and tumorigenesis potential (85, 86). There are studies reporting a high degree of similarity between EMT cells and CSCs (87). EMT cells can also serve as effective seeds for both primary and metastatic tumors, making it challenging to distinguish them from CSCs in function (88). In addition, the signaling pathways related to self-renewal and maintenance of CSCs highly overlap with the pathways regulating EMT, such as Wnt, Notch and Hh signaling pathways (89, 90). Moreover, CSCs isolated from breast tissue express a number of typical EMT markers (91). Most importantly, there are publications proving that breast epithelial cells or breast cancer cells can obtain pluripotent stem cell-like phenotype (CD44high, CD24low) through EMT induction, which further indicates that EMT is closely related to the production of CSCs (91, 92). EMT-TFs have been identified as key factors involved in the stemness regulation of CSCs (87). EMT-TFs, represented by SNAIL, TWIST and SOX, can induce the emergence of breast cancer stem cells (BCSCs) exhibiting CSC-like phenotypes while driving the EMT process (91, 93–95). In summary, induction of EMT can promote the production of BCSCs, which can form mammospheres and express EMT markers. Therefore, the emergence of CSC-like phenotype will be regarded as evidence and result of the occurrence of EMT process in this review.
It is generally believed that CSCs are inherently resistant to anti-tumor drugs and cause the relapse, or they can acquire resistance under the influence of the tumor microenvironment (96). It has been found that cells with CSC/EMT-like characteristics in breast cancer are resistant to neoadjuvant chemotherapy (97). Further research shows that breast cancer cells with CSC/EMT-like properties still survive after receiving neoadjuvant chemotherapy or pharmacological inhibition targeting HER2, which underscore that these cells encode drug resistance (97, 98). Therefore, cancer cells treated with anti-tumor drugs may achieve drug resistance by undergoing EMT to obtain CSC-like features (99). It may be one of the possible mechanisms by which EMT induces drug resistance.
4.2 Enhanced plasticity of hybrid epithelial/mesenchymal phenotype
As mentioned above, most cancer cells that undergo EMT will eventually maintain a hybrid epithelial/mesenchymal (or partial EMT) phenotype, which is also one of the reasons why EMT tends to cause drug resistance. After 8 days of TGF-β induction treatment of human mammary epithelial cells, single-cell RNA sequencing analysis was used to identify ten different EMT subgroups, most of which were in the partial EMT state rather than the complete epithelial or mesenchymal state (100). More importantly, the hybrid E/M cells possess higher metastatic and drug-resistant potential in breast cancer as compared to cells on either end of the EMT spectrum (15, 101). This is because during the entire process of epithelial mesenchymal transition along the E-to-M spectrum, individual cells generate extensive phenotypic heterogeneity, and cells in the hybrid E/M state exhibit high plasticity, which provide greater adaptability and resistance for cancer cells (12). Consistently, through multi-modal translational data-bulk, single-cell, and spatial transcriptomics, it can be concluded that breast cancer cells can obtain higher heterogeneity along EMT spectrum, thus limiting the drug efficacy (102). EMT-driven cell plasticity makes breast cancer cells resistant to paclitaxel by promoting the formation of primary cilia (103). In stage III breast cancer patients, EMT and the accompanying epithelial-mesenchymal heterogeneity serve as prognostic indicators for survival outcomes (104). In summary, most cancer cells that undergo the EMT process will finally remain in a highly plastic E/M intermediate state and acquire the ability to resist the effects of chemotherapy drugs and adapt to this microenvironment, exhibiting a drug-resistant phenotype in clinical practice. However, it is worth noting that the latest evidence claims that higher plasticity may not be directly related to partial EMT state, which is a direction that needs further exploration in the future (105).
4.3 Increased drug efflux and reduced drug intake
Like other types of drug resistance, an important mechanism by which EMT induces drug resistance is through the regulation of the ATP binding cassette (ABC) transporter family of proteins, which increases drug efflux and reduces drug efficacy in cancer cells (106). Currently, there are up to 16 ABC transporters in the ABC transporter family related to multidrug resistance (MDR), including P-glycoprotein (P-gp, also known as ABCB1 or MDR1), Multi-drug Resistance associated Protein-1 (MRP-1; also known as ABCC1) and Breast Cancer Resistance Protein (BCRP) (107). They actively efflux a series of commonly used anti-tumor drugs, including mitoxantrone, anthracyclines, vinca alkaloids, taxanes and other drugs suitable for breast cancer treatment (107). It is reported that EMT induction upregulates the expression of ABC transporters and exacerbates drug resistance in breast cancer (108). It has been proven that EMT-TFs are the regulators in directly modulating ABC transporters, and promoters of ABC transporter genes contain the binding sites for EMT-TFs, such as TWIST, SNAIL and FOXC2 (108). Taken together, EMT-TFs can augment the activity of ABC transporters in various breast cancer cell lines by directly binding to their promoters, thereby leading to enhanced drug efflux, which constitutes a pivotal molecular mechanism of EMT-induced drug resistance.
4.4 Insensitivity and resistance to apoptosis mechanisms
In addition to increased drug efflux, avoidance of drug-induced apoptosis and necrosis is another possible mechanism of EMT-induced drug resistance (109). Growing evidence suggests that EMT can induce cell apoptosis resistance by upregulating the anti-apoptotic protein Bcl-2, downregulating pro-apoptotic proteins (such as Bad, Bax, Bim, p53, Noxa), activating PI3-K/Akt pathways, or interfering with cell cycle (110–112). EMT-TFs, especially SNAIL family, play a master role in increasing resistance to apoptosis. SNAIL stimulates the PI3-K/Akt pathway and inhibits pro-apoptotic protein Bad by inhibiting PTEN transcription, thereby promoting apoptosis resistance (113). SNAIL has also been reported to interfere with the function of pro-apoptotic proteins p53 to prevent cell apoptosis and lead to drug resistance (114). Moreover, SNAIL can inhibit the transcription of cyclin D2 and block cell cycle progression, making cells resistant to apoptosis (115). In addition to activating the PI3K/Akt pathway, TWIST also promotes EMT and upregulates anti-apoptotic protein Bcl-2 to contribute to apoptosis resistance (116, 117).
EMT can also protect cells from drug-induced apoptosis by activating autophagy. The EMT-related signaling pathways, including TGF-β, RAS, WNT, and NF-κB, not only activate EMT, but also closely associate with autophagy (118). EMT-related signaling triggers, such as TGF-β and hypoxia, can effectively induce autophagy under different environmental conditions (119, 120). TGF-β induces autophagosome formation and upregulates the expression of autophagy-related genes in MDA-MB-231 cells (121). Although autophagy and apoptosis are activated by multiple overlapping upstream signals, they mostly cross-regulate each other in an inhibitory manner. Therefore, EMT-induced autophagy reduces the tendency of cells to undergo apoptosis, which manifests as drug resistance in clinical practice (122). It is reported that autophagy induces resistance of breast cells to epirubicin and pacitaxel (123–125). In summary, EMT can combat drug-induced apoptosis of mammary carcinoma cells through various pathways, leading to drug resistance.
4.5 Activation of DNA damage response pathways
DNA damage response/repair (DDR) is a mechanism within cells to resist DNA damage induced by external or internal factors, which monitors and transmits damage signals and makes appropriate responses (126, 127). Homologous recombination (HR) pathway is a classic DDR pathway used to repair DNA damage like double-strand break (128, 129). Some chemotherapeutic agents exert anti-tumor effects by damaging the nuclear or mitochondrial DNA of cancer cells, driving their direct or indirect death (130). For example, cisplatin is an efficient DNA-damaging agent with significant anti-cancer effects. However, some cancer cells can reverse the damage induced by anti-tumor drugs, including DNA-damaging agents, by enhancing their ability of DNA damage repair, thus exhibiting a drug-resistant phenotype (5). The resistance of platinum-based drugs is closely related to DDR. Nucleotide exit repair and HR pathway are the main DDR pathways for reversing platinum damage in cancer cells (131, 132).
A pioneering report reveals the connection between EMT and DDR. ZEB1 has been identified as a response target for Ataxia‐telangiectasia‐mutated (ATM), which is a key protein kinase in HR signaling (133). Furthermore, phosphorylated ZEB1 can interact with USP7 deubiquitylating enzyme and promote HR-dependent DDR pathways. In breast cancer, ZEB1 can also activate the ATM promoter by binding to p300/pCAFj, forming a positive feedback loop that promotes DNA repair and resists DNA damage caused by chemotherapy (134). In addition, ubiquitinated TWIST1 can modulate DDR pathway and upregulate HR gene expression (135). In summary, EMT-TFs, especially ZEB1, can promote DDR and increase HR pathway activity. As mentioned earlier, HR pathway is the main DNA repair mechanism for reversing platinum damage, which may explain why EMT promotes tumor resistance to platinum-based drugs.
4.6 Other mechanisms
There is mutual influence between tumor microenvironment (TME) and EMT. The stromal cells that constitute TME, including cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs) and T lymphocytes, interact with neighboring cancer cells by secreting cytokines or other means, activating their EMT program (30). Carcinoma cells undergoing EMT also have an impact on various cells in TME, thereby affecting tumor progression and drug resistance (30, 96). EMT cells exert immunosuppressive effects to regulate immune cells in TME. For example, when MCF-7 cells overexpress SNAIL, the function of co-cultured T cells is severely reduced (136). In addition, the EMT cells secrete or indirectly activate immunosuppressive factors, such as TGF-β, TNF and CCL5, which affect the activity of various immune cells in TME (137, 138). Spatial colorization analysis reveals that at the tumor boundary characterized by EMT, CAFs and M2-like TAMs interact to promote immune exclusion and drug resistance (139). A new research has developed a biomimetic codelivery system that can reverse the EMT and CSC-like characteristics of TNBC cells to reshape the immunosuppressive microenvironment, thereby enhancing the sensitivity of TNBC to paclitaxel (140). Therefore, EMT-induced immune reconstitution may be a possible mechanism of drug resistance in breast cancer.
Metabolic reprogramming is another potential mechanism of EMT-induced drug resistance. It is reported that overexpression of SNAIL can induce aberrant glucose metabolism in cancer cells, including increased glucose uptake and lactate production, decreased oxygen consumption by mitochondria, and so on, thus altering TME and enhancing chemoresistance (141–143). The rate of glycolysis in EMT cells is significantly increased, resulting in the production of more ATP to meet the energy requirements for wound repair and resistance to attacks, which is manifested as refractory breast cancer (144). Meanwhile, acidification of TME can induce breast cancer cells to become resistant to mitoxantrone (145). Growing evidence shows that the reprogramming of lipid metabolism is conducive to the development of drug resistance in breast cancer (146). Fatty acid synthase (FASN), as a kind of metabolic oncogene involved in neoplastic lipogenesis, has been found to induce drug resistance (147). EMT was also found to be involved in this process. New evidence indicates that CD36-mediated fatty acid uptake makes HER2-positive breast cancer cells obtain drug resistance by regulating the EMT-like phenotype (148). EMT, accompanied by changes in lipid metabolism, limits the entry of drugs into cells through the plasma membrane and prevents drug accumulation (149, 150). In addition, the occurrence of EMT also causes the upregulation of P-gp, which can remove the lipid peroxidation products induced by the application of doxorubicin, thus leading to doxorubicin resistance in breast cancer (151).
EMT-induced drug resistance is usually multifactorial and complex, resulting from the combined effects of various cells and molecules. The above only summarizes the relatively important mechanisms mentioned in recent research, and more influencing factors and comprehensive mechanisms need to be explored in the future.
5 Therapeutic strategies by targeting EMT program
The drug resistance is not only an important focus in the development of traditional chemotherapy drugs, but also a focal point worthy of attention in emerging targeted therapies. Since EMT has been established as a fundamental mechanism that endows breast cancer cells with drug resistance and CSC-like traits, targeting EMT to control drug resistance represents a promising therapeutic strategy for breast cancer (152). In recent years, numerous studies have delved into promoting the drug sensitivity of breast cancer by blocking the EMT process and reversing the CSC/EMT-like phenotype of tumor cells. Blocking the occurrence of EMT involves disrupting the regulatory network associated with EMT, which prevents the initiation of EMT program. For example, the latest evidence shows that pentagalloyl glucose can reverse the resistance of breast cancer to doxorubicin by targeting EMT and the expression of miRNAs (153). Targeting EMT includes blocking upstream signaling pathways, directly targeting EMT-TFs, regulating epigenetic modifications (especially non-coding RNAs), and blocking the possible mechanisms of EMT-induced drug resistance. (Figure 4) Among them, some agents in the last strategy directly target key factors in the mechanism of resistance, such as ABC transporter family and Bcl-2, rather than overcoming drug resistance by affecting the EMT process, thus they are not included in the discussion here. Some of these molecules targeting the EMT process, especially signaling pathways, are already in the clinical trial phase and have great potential for application in future clinical treatments (Table 1).
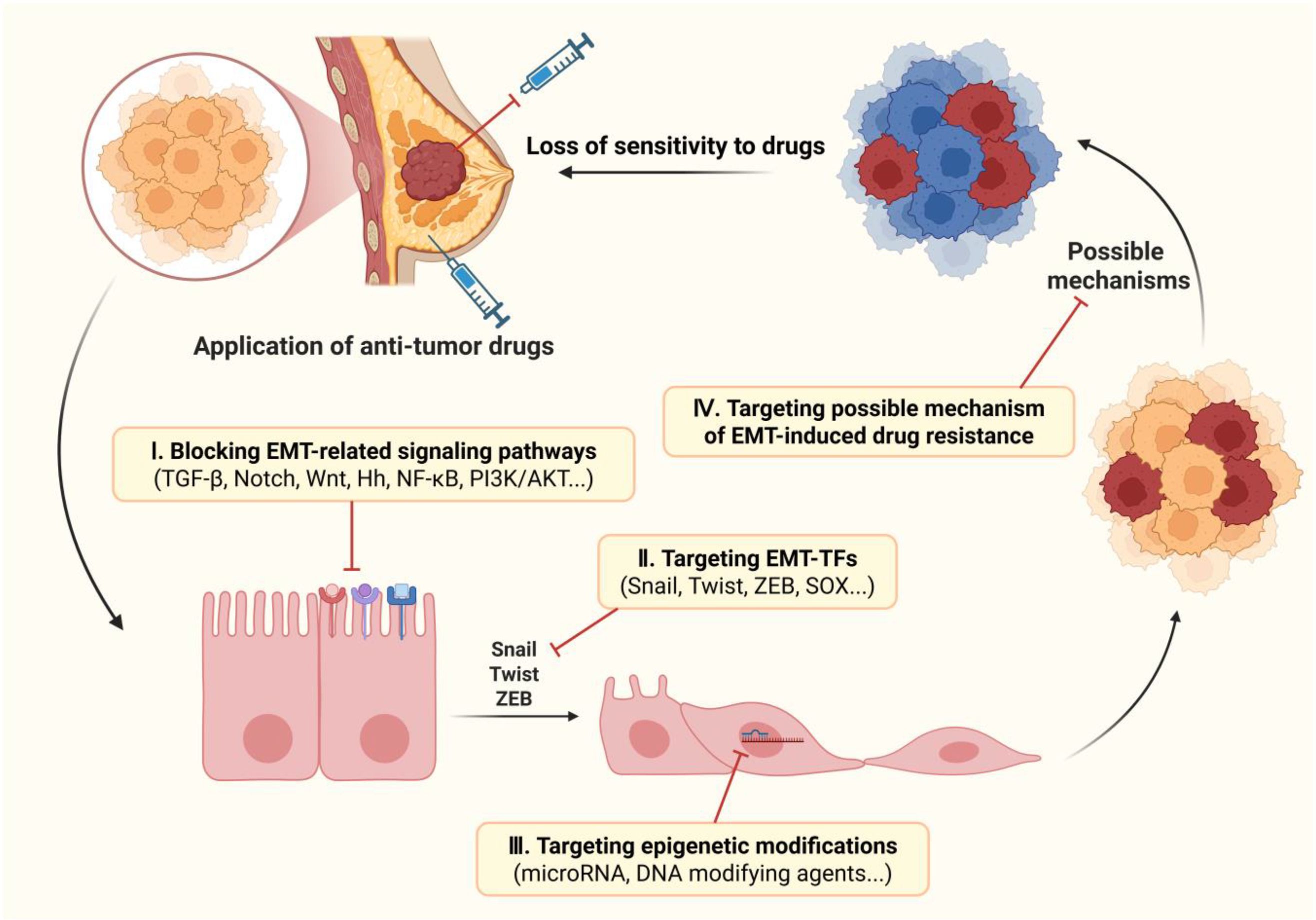
Figure 4. Therapeutic strategies to overcome EMT-induced drug resistance in breast cancer. There are four possible strategies for targeting EMT to overcome drug resistance in breast cancer: 1) inhibiting the occurrence of EMT by blocking the upstream signaling pathways; 2) blocking transcription of EMT by targeting EMT-TFs; 3) inhibiting EMT progression by regulating post-transcriptional epigenetic modifications; 4) maintaining sensitivity to drugs by targeting the possible mechanisms of EMT-induced drug resistance. Image created using Bio-Render.com software.
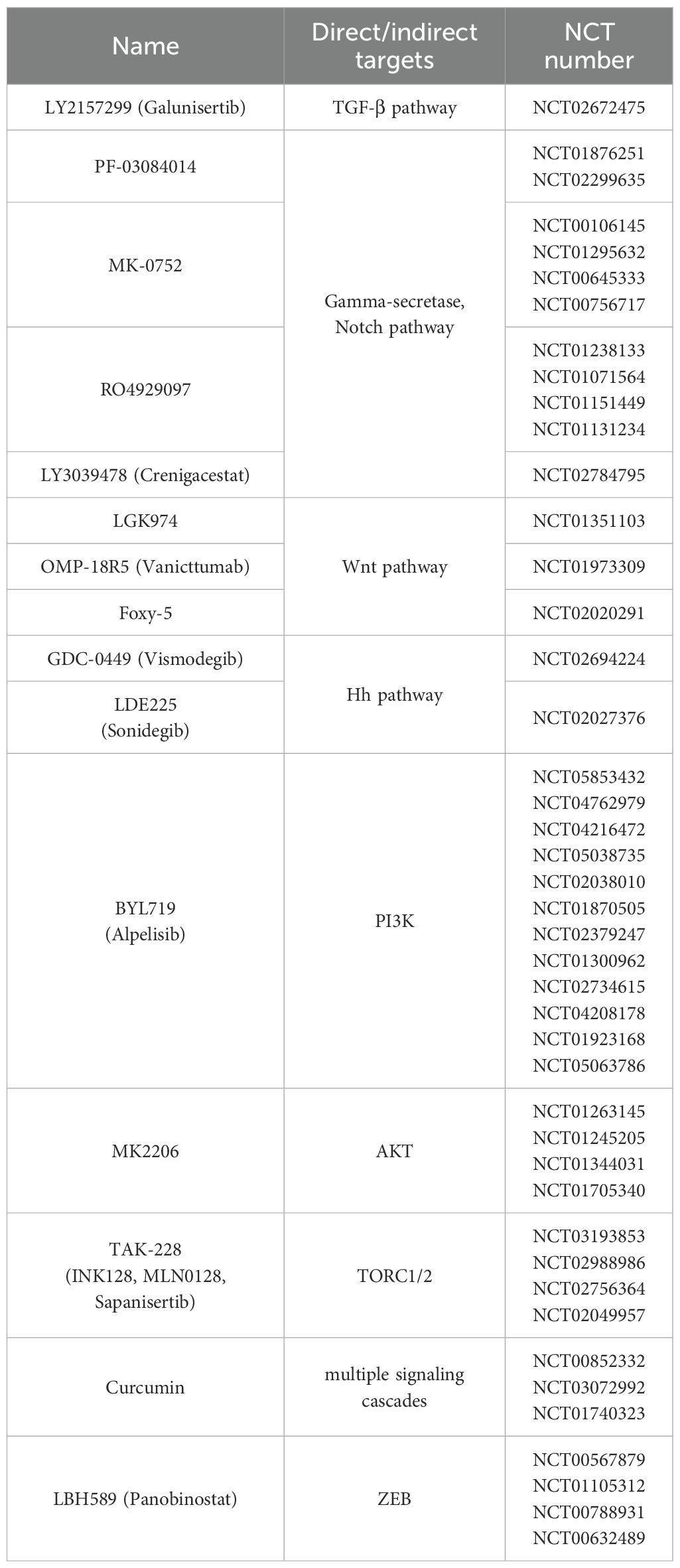
Table 1. The molecules that have undergone/completed clinical trials and their NCT numbers of clinical projects. All information is sourced from ClinicalTrials.gov.
5.1 Inhibitors of EMT-related signaling pathways
5.1.1 TGF-β signaling pathway
Insufficient chemotherapy may induce the initiation of EMT program by activating TGF-β signaling (154). Doxorubicin, cisplatin, paclitaxel and other anti-tumor drugs have been shown to induce the expression of TGF-β1 and the occurrence of EMT in various malignancies (154–156). For example, it has been reported that MDA-MB-231 cells treated with cisplatin have elevated levels of TGF-β, making themselves resistant to the cytotoxic effects of cisplatin (157). Another research shows that application of epirubicin can activate the TGF-β pathway in TNBC cells and regulates EMT-related markers, ultimately leading to drug resistance (158). TGF-β induces breast cancer cells to transform into partial EMT phenotype and exhibit CSC-like characteristics, which may explain why TGF-β can induce drug resistance through EMT (100). More and more similar studies have focused on the relationship between TGF-β signaling pathway and drug resistance, reminding us that the pathway may be a therapeutic target for drug resistance in breast cancer.
There is a study reporting that neutrophil extracellular traps activate the TGF-β signaling pathway by inducing SMAD2 phosphorylation in breast cancer, leading to EMT and chemotherapy resistance (159). However, TGF-β type I receptor inhibitor (TβRI), which inhibits SMAD2 phosphorylation, can block the activation of TGF-β signaling and reduce the expression of EMT-related genes, thereby improving and reversing the resistance response caused by chemotherapy (159). Another research has also reported that chemotherapy drugs lead to drug resistance through inducing the occurrence of EMT, however, TβRI kinase inhibitor (TβRI-KI) can reverse the EMT program and combined treatment with doxorubicin may improve efficacy and reduce the dosage of doxorubicin (155). Similarly, TβRI-KI LY2157299 (Galunisertib) can block the conduction of the TGF-β pathway and inhibit the development of drug-resistant CSCs and tumor recurrence induced by paclitaxel (160). TβRI/TβRII inhibitor LY2109761 can also reverse EMT and enhance chemosensitivity by inhibiting the TGF-β signaling pathway (161, 162). Curcumin, as a natural agent, is closely related to the occurrence and development of cancer (163). Curcumin can suppress doxorubicin-induced EMT and improve the efficacy of chemotherapy by inhibiting TGF-β/Smad and PI3K/AKT signaling cascades (164).
In addition to the aforementioned, there are also many TGF-β inhibitors that have been provided with compelling ability to block the TGF-β pathway and reverse EMT, including Ki26894, LY364947, IN-1130, SM16, SB-431542, YR-290 etc (165–170). Unfortunately, TGF-β pathway is more considered to be related to tumor growth and metastasis, so these inhibitors are mostly used to control breast cancer growth and metastasis, rather than reduce drug resistance. Thus, whether they can also reverse drug resistance necessitates more comprehensive and in-depth investigation.
5.1.2 Notch signaling pathway
The importance of the Notch signaling pathway in EMT program and drug resistance has been recognized, and regulating the Notch pathway may be a good approach to overcome drug resistance. The latest report has shown that imatinib, a tyrosine kinase inhibitor, has been proven to have the ability to reverse EMT by inhibiting the Notch pathway and significantly reduce the stemness of TNBC cells and induce apoptosis (171). In the past few decades, many methods have been developed and reported to regulate cancer drug resistance by inhibiting the activation of the Notch pathway, including gamma-secretase inhibitors (GSIs), ADAM inhibitors, monoclonal antibodies, etc (172). Among them, the development prospect of GSIs in reversing drug resistance is gratifying. GSIs, as the first proposed and successfully developed Notch pathway inhibitors, have shown potential in increasing tumor sensitivity to chemotherapy, whereas GSIs combined with conventional therapy have been reported to have better efficacy than using GSIs alone (173). The mechanism of GSIs reversing drug resistance in breast cancer is multifactorial. GSIs block the Notch signaling pathway by inhibiting gamma-secretase, thereby regulating CSCs, EMT, ABC transporters, and affecting crosstalk between the Notch pathway and other pathways (172). GSIs have been reported to be able to reverse drug resistance in breast cancer through the above mechanisms and achieve better therapeutic effects when combined with conventional drugs in clinical trials.
Doxorubicin can induce overexpression of MRP-1, an ABC transporter, in breast cancer cells by activating Notch pathway (174). DAPT, as a GSI, can reverse this process, reducing doxorubicin efflux and enhancing doxorubicin-induced apoptosis program. High expression of Nicastrin and Notch4 in breast cancer cells can induce the acquisition of EMT phenotype and tamoxifen resistance. Anti-Nicastrin mAbs and GSI PF03084014 can inhibit the expression of EMT-related molecules and partially alleviate drug resistance (175). Docetaxel induces the initiation of EMT program and drug resistance of breast cancer cells by activating Notch pathway, and PF-03084014 combined with docetaxel can reverse the above process (176). The combination therapy of MK-0752 and docetaxel can reduce BCSCs in breast tumor transplants by inhibiting the Notch pathway and enhance the efficacy of docetaxel (177). In addition, biopsy results of clinical trials showed a reduction in CSC-like phenotype cells and a decreased mammosphere after the combined therapy, which can be seen as a reversal of EMT. A classic GSI, RO4929097, has been proven safe and effective when used in combination with other conventional drugs in various clinical and preclinical trials (178, 179). However, there are limited reports on whether RO4929097 can reverse EMT and drug resistance by inhibiting the Notch pathway. It is reported that short-term treatment of breast cancer with tamoxifen or fulvestrant will increase the activity of BCSCs by upregulating Noth4 target gene, while RO4929097 can inhibit BCSCs and alleviate drug resistance (180). Another Notch inhibitor, LY3039478 (Crenigacestat) has been reported to have poor tolerability and results in disappointing clinical survival rates for breast cancer patients (181).
5.1.3 Wnt signaling pathway
Wnt signaling pathway plays a role in promoting tamoxifen acquired drug resistance in breast cancer. Won et al. have reported that the tamoxifen-resistant MCF-7 cell line possesses mesenchymal phenotype and significantly increased level of β-catenin (182). After treatment with classical WNT inhibitor ICG-001 or β-catenin siRNA, the expression of active β-catenin was inhibited, and viability of the drug-resistant cell line was also reduced (182). Another Wnt inhibitor, IWP-2, has also been proved to improve the sensitivity of breast cancer cells to tamoxifen by inhibiting EMT (183). Similarly, pyridine derivatives reverse the resistance of MCF-7 breast cancer cells to tamoxifen by inhibiting the activation of Wnt/β-catenin and NF-κB pathways (184). It can be inferred that the Wnt signaling pathway may act as a potential therapeutic regimen for alleviating resistance.
After treating TNBC cell lines with Wnt inhibitor FH535, the expression of EMT-related markers (E-cadherin) and EMT-TFs (Snail and Twist1) was significantly downregulated, indicating partial reversal of the EMT process (185). Activation of Wnt/β-catenin pathway will induce trastuzumab resistance in breast cancer cells with HER2 overexpression, thus, knocking out Wnt3 by siRNA can result in downregulation of EMT-related expression and restoration of trastuzumab’s inhibitory effect on cell growth (186). Porcupine is a key factor regulating the release of Wnt ligands, and LGK974 is a specific inhibitor of it (187). It is reported that enhanced activity of the Wnt/β-catenin pathway induces drug resistance in TBNC cells and enhances the expression of CSC-like markers, which can be reversed by LGK974 (188, 189).
As a key receptor for Wnt/β-catenin signaling, Frizzled-7 (Fzd7) is abnormally expressed in TNBC, which is associated with poor prognosis and resistance to chemotherapy (190, 191). Fzd2 promotes the maintenance of mesenchymal phenotype in breast cancer cells, and endows cells with stemness and drug resistance by combining Wnt5a/b and Wnt3 (192). Knockout of Fzd2 significantly reduced the expression of ABC transporter subfamily G isoform 2 (ABCG2) and IC50 of paclitaxel, indicating that knockout of Fzd2 enhanced the sensitivity of breast cancer cells to paclitaxel. Bevacizumab, as a first-line combination drug for various cancers, has limited therapeutic effect on TNBC, because it simultaneously activates Wnt/β-catenin signaling to induce EMT process and stemness of breast cancer cells (190, 193, 194). A novel humanized antibody, SHH002-hu1, can specifically target Fzd7-positive cells and block the Wnt/β-catenin pathway, thereby inhibiting EMT and enhancing the anti-breast cancer effect of bevacizumab (195). Another Fzd receptor inhibitor OMP-18R5 (Vanicttumab), which has entered the clinical trial phase, combined with paclitaxel has more excellent anti-tumor effect than paclitaxel alone in the treatment of breast cancer (194).
Salinomycin has been proved to be an inhibitor of Wnt/β-catenin signaling pathway in breast cancer (196). A study first identified that salinomycin had specific toxicity against BCSCs and reversed the general resistance of breast cancer to multiple drug therapies, but the mechanism by which salinomycin inhibited CSCs was not explained in this research (197). Over a decade later, another study confirms that the combination of salinomycin and doxorubicin can reverse the resistance of adriamycin-resistant breast cancer cells (198). In addition, the genes related to the Wnt/β-catenin pathway and EMT was found to be downregulated, which suggested that this combination therapy can suppress CSCs by inhibiting the Wnt pathway and inverting the EMT process, ultimately reversing the drug resistance of breast cancer cells. Salinomycin also can remove markers on the surface of breast cancer cells by inhibiting Wnt signaling transduction, such as CD44 and ABCG2, a drug resistance marker (199).
The role of Wnt signaling pathway in breast cancer has been very clear and inhibitors of Wnt pathways are constantly being developed and entering clinical trials (199). Endogenous agents and pharmacological inhibitors targeting various elements of Wnt pathway have also been widely studied and reported, such as Foxy-5 (Wnt5a mimetic), DKK3, ZFP57 and so on (200). However, these Wnt inhibitors are mainly reported to prevent cell proliferation and tumor metastasis by inhibiting Wnt signaling. It is unclear whether they can improve the drug sensitivity of breast cancer by reversing the Wnt/EMT/drug resistance axis, which is also the direction we can explore and study in the future.
5.1.4 Hh signaling pathway
Overexpression of Hh pathway has been proved to regulate the proliferation and self-renewal of CSCs in different cancers, including breast cancer, and can induce chemotherapy resistance by activating multiple pathways (201). Analysis of embryonic pathways showed that EMT markers were significantly increased in TNBC cells resistant to paclitaxel and doxorubicin, accompanied by activation of the Hh pathway and Notch receptor expression (202). Moreover, cilengitide is reported to overcome the resistance of HER2-positive breast cancer to trastuzumab by targeting ITGβ3 to inhibit the activity of Hh pathway and the transcription of EMT-TFs (203). Therefore, targeting Hh signaling pathway is a potential therapeutic direction to alleviate chemotherapy resistance and improve prognosis of breast cancer.
Cyclopamine (11deoxojervine) is the prototype of Hh inhibitor and is currently widely studied and used as an agent for preclinical studies (204). It achieves the effect of blocking the Hh signaling by binding to SMO signaling elements and inactivating them. Nitidine chloride (NC), a natural bioactive alkaloid, has been proved to have anti-cancer effect and enhance the inhibitory effect of doxorubicin on breast cancer (205). NC regulates the expression of EMT-related markers and reverses EMT by inhibiting Hh pathway, while reducing the CSC/EMT-like properties of breast cancer cells by regulating the pathway (206). Moreover, the combination of cycloamine and NC can enhance the above effect, which indicates that cycloamine can also enhance the sensitivity of breast cancer to anti-cancer drugs by inhibiting Hh pathway (206). The consistent research results show that another non-canonical Hh inhibitor GANT61 (Gli1 inhibitor) can effectively increase the expression of E-cadherin in breast cancer cells and down-regulate CSC/EMT-like phenotype, thereby promoting cells apoptosis (207–209). Moreover, its combination therapy with paclitaxel enhances the efficacy of chemotherapy drugs for anti-cell growth and anti-CSC activities (207). Some SMO inhibitors, such as IPI-926, GDC-0449 (Vismodegib) and LDE225 (Sonidegib), have undergone clinical trials for breast cancer treatment (210–212). TNBC cells treated with GDC-0449 or LDE225 showed downregulation of both Hh target genes and genes regulating CSC-like phenotype (212). Furthermore, the combination of LDE225 and docetaxel can improve the sensitivity of tumors to chemotherapy, and its safety and efficacy have been demonstrated (211, 212).
5.1.5 Other signaling pathways
Although there are many signaling pathways proven to be associated with EMT or drug resistance, the exact description of signaling pathways that can induce drug resistance through EMT is limited, and there are relatively few reports on corresponding inhibitors (Figure 5). Among them, there are comparatively more discussions on the NF-κB and PI3K/AKT/mTOR signaling pathways. Tannic acid can inhibit the activation of EMT and NF-κB signaling pathway in MCF-7 cells, thereby suppressing the formation of drug-resistant BCSCs and the expression of stemness markers (213). Similarly, the co-delivery system of NF-κB inhibitor PDTC and doxorubicin can alleviate the multidrug resistance of breast cancer (214). Garcinol has been reported to inhibit NF-κB/Twist1 signaling activated by paclitaxel and downregulate the expression level of EMT-TFs, thus significantly improving the efficacy of paclitaxel in breast cancer in the orthotopic breast cancer model (215). Recently, an increasing number of PI3K inhibitors have entered the clinical trial stage and been proven to be effective in combination with other traditional chemotherapy drugs for advanced breast cancer (216). Among them, the isoform-specific PI3K inhibitor BYL719 (Alpelisib) has been shown to overcome eribulin resistance by inhibiting EMT and stemness of BCSCs (217). In addition to classic PI3K inhibitors, Cx43t is also proved to inhibit the activation of PI3K/Akt signaling by reducing Akt phosphorylation, thereby suppressing EMT and increasing tamoxifen sensitivity (218). Luo et al. reported that 14, 15-EET induces the occurrence of EMT and cisplatin resistance by activating the FAK/PI3K/AKT pathway, thus the antagonist of 14, 15-EET, 14, 15-EEZE, can reverse EMT and cisplatin resistance in breast cancer (219). mTOR has been reported to interact with EMT-related signaling pathways, including PI3K/Akt, Notch, TGF-β, and non-coding RNAs to maintain BCSC-like characteristics of cancer cells and mediate drug resistance (220). The transmembrane protein-45A (TMEM45A) induces glycolysis and EMT program by activating AKT/mTOR signaling pathway, therefore the siRNA targeting TMEM45A can reverse the above pathway and improve the sensitivity of breast cancer to palbocilib (221). MK2206 (AKT kinase inhibitor), TAK-228 (formerly INK128 or MLN0128, dual TORC1/2 inhibitor) and RapaLink-1 (mTOR inhibitor) can alleviate drug resistance by regulating the stability of CSCs phenotype and inhibiting cell viability (222). Stevens LE et al. found that pSTAT3 regulates EMT-related genes in inflammatory breast cancer cell lines resistant to paclitaxel and doxorubicin (223). Meanwhile, the combination therapy of paclitaxel and JAK2/STAT3 inhibitor, AZD1480, can prevent the occurrence of the drug-resistant subpopulation with EMT-like characteristics (223). DUSP4 (MKP-2) can block the initiation of EMT program by inhibiting the activation of JNK signaling pathway, and restore the sensitivity of breast cancer cells to doxorubicin (224).
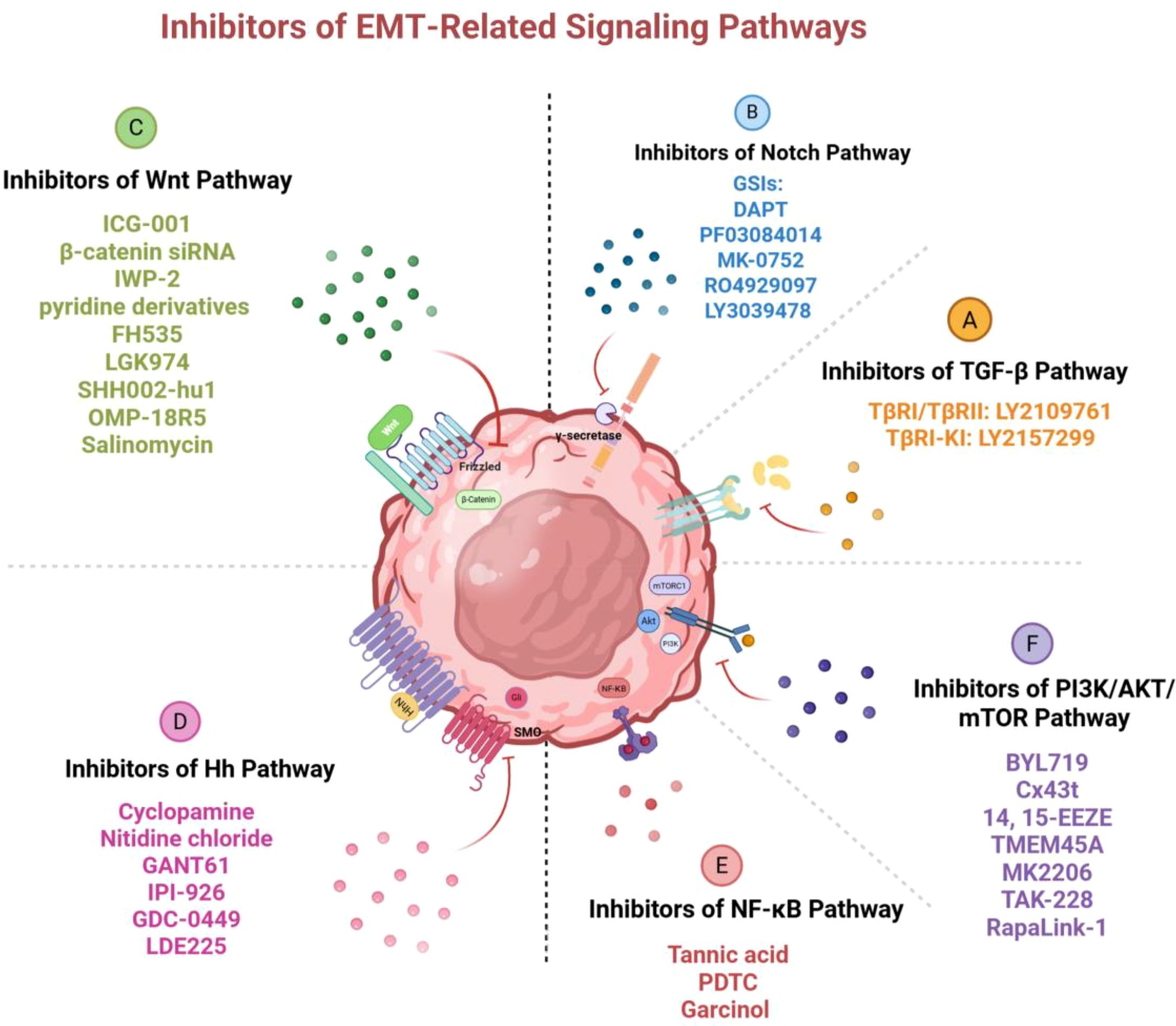
Figure 5. Inhibitors of EMT-related signaling pathways. Various inhibitory molecules block the activation of these pathways and the occurrence of EMT by targeting key factors in the signaling pathways, thus reversing drug resistance in breast cancer. Image created using Bio-Render.com software.
Due to the crosstalk and interference between signaling pathways, which affect and interact with each other, a drug or inhibitor may induce the participation and alteration of multiple factors and signaling pathways simultaneously. For example, besides suppressing TGF-β/Smad and PI3K/AKT signaling, curcumin is also reported to be associated with multiple signaling cascades, such as Notch, NF-κB and Wnt/β-catenin pathway (163). Especially in the Wnt pathway, curcumin hampers activation of Slug and suppresses CSCs by blocking nuclear translocation of β-catenin (225). Similarly, in addition to targeting the Wnt pathway, salinomycin in combination with budesonide may suppress stemness of TNBC cells and activate apoptosis by inhibiting AKT/mTOR pathway and EMT (226). Pentadecanoic acid can inhibit multiple survival signaling pathways (MAPK, ERK1/2, mTOR and EGFR) and EMT, resulting in reversal of tamoxifen resistance (227). Consistently, BYL-719, as a PI3K inhibitor, can not only block the PI3K/AKT/mTOR pathway, but also inhibit the Notch, JAK/STAT, and MAPK/ERK pathways, ultimately inhibiting EMT and overcoming drug resistance (217). Therefore, the action of a drug or inhibitor may involve more than one signaling pathway, and overcoming drug resistance may be the result of the synergy and interaction of multiple pathways.
5.2 Inhibitors of EMT-TFs
Targeting EMT-TFs is another therapeutic strategy to overcome drug resistance in breast cancer. It is reported that overexpression of Snail in MCF-7 cells increases the level of P-gp and shows a tendency to develop resistance to adriamycin, therefore, Snail is a promising target (228). SNAI1 enhancer RNA depletion inhibits the EMT process and chemoresistance of breast cancer cells (229). MCF-7 cells transfected with pCDNA3.1-Snail can promote EMT characterization, which leads to an increased expression of BCRP and drug resistance to mitoxantrone (230). AMP-activated protein kinase (AMPK) agonists can enhance the sensitivity of TNBC cells to chemotherapy by phosphorylating Snail1 (231). The interaction between N-terminal SNAG repressor domain and LSD1 plays an important role in Slug activating EMT (65, 232), and LSD1 can maintain the CSC-like phenotype and induce doxorubicin resistance in breast cancer (233). The application of LSD1 inhibitors, 2-PCPA and GSK-LSD1, can significantly reduce CSCs, and the combination therapy with doxorubicin improves the sensitivity of cancer cells (233).
The overexpression of TWIST1 in palbociclib-resistant luminal breast cancer activates EMT (234). Moreover, the survival time of mice treated with pSilencer-twist and adriamycin was significantly prolonged compared to mice treated with adriamycin alone, suggesting that inhibiting Twist may be a possible method to enhance chemotherapy efficacy and reverse drug resistance (20). Similarly, Twist1 siRNA can reverse the high expression of EMT markers induced by adriamycin, and the anti-cancer efficacy in combination with adriamycin is significantly better than that of monotherapy (20). The CDK1 inhibitor RO-3306 can significantly inhibit the CSC/EMT-like phenotype and increase the sensitivity of TNBC cells to cisplatin and paclitaxel by downregulating the protein level of TWIST1 (235).
Growing evidence shows that ZEB influences the sensitivity of cancer cells to chemotherapy by regulating EMT (236). LBH589 (Panobinostat) is reported to mediate the inhibition of EMT by targeting ZEB expression, and inhibit BCSCs and enhance the apoptosis of TNBC cells by regulating EMT when combined with salinomycin (237, 238). Eribulin reverses EMT progression by disrupting the interaction between ZEB1 and SWI/SNF, thereby preventively increasing the sensitivity of TNBC cells to drugs (239). PEG10-siRNA has been reported to inhibit EMT and overcome drug resistance by activating SIAH1, the post-translational degrader of ZEB1 (240).
Although EMT-TFs usually exert effects by directly affecting EMT, there is evidence to suggest that EMT-TFs can also induce drug resistance through other pathways without affecting the EMT process. It is reported that Snail or Slug induces tamoxifen resistance in breast cancer by activating EGFR/ERK pathway independent of EMT, and inhibitors of EGFR/ERK pathway can restore the sensitivity of cancer cells with high expression of Snail/Slug to tamoxifen, without reversing the EMT phenotype of these cells (241). This further confirms what was previously mentioned, that regulation of EMT-TFs may be one of the necessary conditions for the occurrence of EMT, but EMT is not the inevitable result of abnormal expression of EMT-TFs (12). As mentioned above, EMT-TFs can further activate related signaling pathways through positive feedback to enhance EMT, so targeting EMT-TFs can also inhibit activation of signaling pathways. It is reported that after Sox4 expression of breast cancer cells is knocked down by siRNA, Wnt/β-catenin signaling is also observed to be synchronously inhibited through feedback loop, and subsequently, EMT, CSC-like features and cisplatin resistance are reversed (242).
5.3 MicroRNA
In recent years, many studies have delved into and reported on the relationship between miRNAs and EMT-induced drug resistance, and the roles of miRNAs are gradually clarified (243). Some miRNAs can reverse drug resistance by targeting the signaling pathways or EMT-TFs mentioned above, while others can exert their effects through other key molecules related to EMT. Herein, we only introduce the tumor suppressor miRNAs that are prominent in breast cancer and the critical processes to function. Other miRNAs are listed in Table 2.
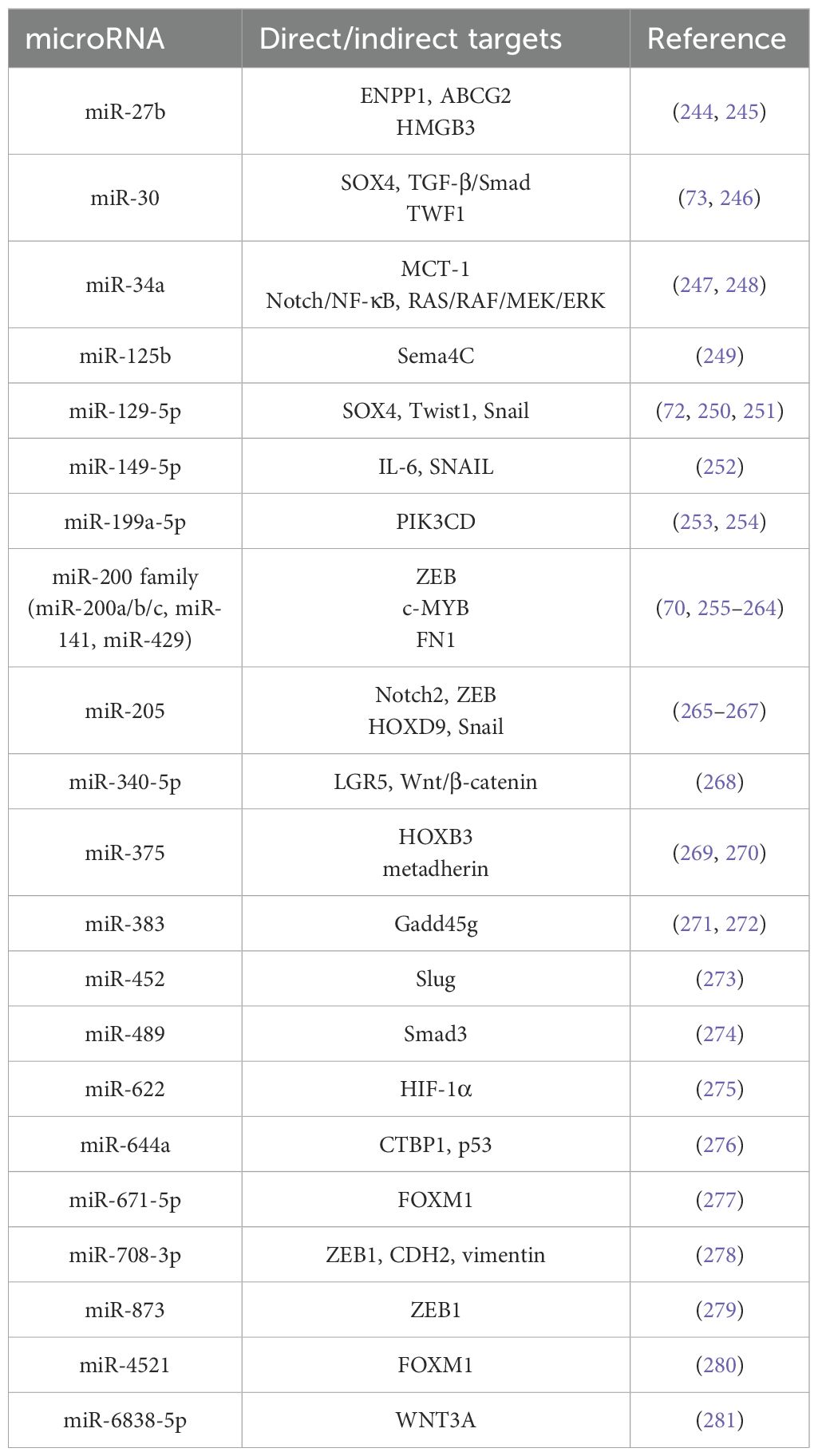
Table 2. EMT‐associated tumor suppresser miRNAs and their targets for drug resistance in breast cancer.
The miR-200 family, as one of the most widely studied EMT-related miRNA, consists of five members, namely miR-200a, miR-200b, miR-200c, miR-141, and miR-429 (282, 283). MiR-200 family members directly target ZEB to exert inhibitory effects and reverse EMT processes by upregulating E-cadherin expression (70, 255–258). For TNBC cells in the mesenchymal state, tamoxifen can promote upregulation of miR-200c through demethylating its promoter, thereby reversing EMT and increasing sensitivity to traditional chemotherapeutic agents (259). MiR-200c restored the sensitivity of HER2-positive breast cancer to trastuzumab by targeting ZNF217/TGF-β/ZEB1 axis and suppressing CSC-like phenotype (260, 261). MiR-200b/c regulates E-cadherin by targeting ZEB, thereby inhibiting EMT and increasing sensitivity of breast cancer cells to doxorubicin (262). The overexpression of miR-200b/c has also been found to downregulate c-MYB, thereby reversing EMT-induced tamoxifen resistance in ER-positive breast cancer (263). In addition, miR-200b can also reverse EMT-induced chemoresistance by targeting FN1 (264).
The miR-30 family is regarded as an important group of microRNAs that negatively regulates the malignant behaviors of tumors. It has been demonstrated that the downregulation of miR-30a facilitates EMT process and metastasis by modulating EMT-TFs (284–288). Moreover, miR-30a inhibit the initiation of EMT program and drug-resistant CSC-like phenotype by forming a double negative feedback loop with SOX4 (73). MiR-622 can exert the inhibitory effect of miR-30a on EMT and drug resistance by increasing its expression (275). Another family member, miR-30c, has also been reported to directly target TWF1 to reverse EMT, thereby restoring sensitivity of cells to chemotherapy (246).
The miR-34 family, especially miR-34a, is one of most intensively studied miRNAs in breast cancer (289). The combination therapy of miR-34 and traditional anti-cancer agents can inhibit drug resistance in various types of cancer (290). MiR-34a is reported to be able to target EMT-TFs to inhibit the EMT process (291). In TNBC, miR-34a targets MCT-1 to control M2 macrophages polarization, thereby reprogramming EMT and inhibiting stemness closely associated with drug resistance (247). Furthermore, the combination treatment of miR-34a and doxorubicin can significantly downregulate the expression of Snail by inhibiting the Notch/NF-κB and RAS/RAF/MEK/ERK pathways, thereby preventing doxorubicin-resistant breast cancer progression (248).
Although miR-129 may play a dual role in the development of tumors, more evidence tends to suggest that it acts as a tumor suppressor to prevent the malignant progression of breast cancer. In MCF-7 cells treated with adriamycin, the enhanced miR-129-5p expression significantly downregulates the expression of mesenchymal markers (vimentin and N-cadherin), indicating inhibition of EMT (72). By negatively regulating the expression of SOX4, miR-129-5p significantly reduces the IC50 of several drugs, including adriamycin, which proves that breast cancer cells are more sensitive to drugs. In addition, miR-129-5p negatively regulates Twist1 and Snail, therefore reverse the EMT process and eliminate epirubicin resistance (250, 251).
The role of miR-205 in different types of cancer is controversial, with dual effects of inhibition or carcinogenesis. According to existing reports, miR-205 typically exhibits tumor suppression and chemosensitivity enhancement in breast cancer, even though the specific effects are variant in different subtypes (292). MiR-205-5p has been proved to enhance the sensitivity of breast cancer to chemotherapeutic agents, including doxorubicin and docetaxel (293, 294). MiR-205 can negatively regulate the phenotype and activity of drug-resistant BCSCs by inhibiting related elements of EMT (265, 295). There are many known targets of miR-205 in breast cancer, including transcription factors ZEB1, ZEB2 and SIP1, which are responsible for regulating EMT (69, 267, 292). The upstream regulators of miR-205, polycomb protein MEL-18 and ligand Jagged1 also suppress the initiation of EMT via this pathway (267). Moreover, miR-205-5p can inhibit chemoresistance of TNBC by regulating Snail (266). The miR-205 and miR-200 family have similar functions and share several target genes, such as ZEB1 and SIP1, which may suggest that they may share more commonalities (69, 296). The combination therapy of miRNA-200/205 has been increasingly reported for regulating EMT and overcoming drug resistance (297).
The inhibitory function of some microRNAs on EMT and their anti-tumor metastasis effects have been widely reported, but whether they can overcome drug resistance by suppressing EMT has not been clearly revealed, which will be an orientation worthy of further research (298). It is worth noting that the role of microRNA in breast cancer may not be unidirectional promotion or inhibition, but often dual action. MiR-125b has been proved to reverse EMT and prevent drug resistance in many cancers, including breast cancer and lung cancer (299, 300). However, other studies reported the opposite results simultaneously that high levels of miR-125b are more likely to induce drug resistance and lead to poor prognosis in breast cancer patients (301, 302). The miRNAs listed above with anti-tumor effects in breast cancer may simultaneously promote malignant progression in other tumors or subtypes, which further reflects that the tumor environment is a complex and multifactorial system. Thus, the therapeutic strategy targeting miRNA still requires further exploration and investigation for future clinical application.
5.4 Limitations of molecules targeting EMT
Although some EMT inhibitors have entered clinical trials and been proven to improve the efficacy of conventional therapies, the safety in the long-term use of them is currently unclear (303). Connolly EC et al. first reported that long-term use of LY2109761 may induce an increase in the levels of EMT-related markers, such as E-cadherin, and would lead to acquired resistance to LY2109761 (304). Subsequently, there is increasing evidence that although blocking TGF-β signaling may provide clinical benefits, treatment with TGF-β inhibitors alone may lead to serious adverse reaction (156). In addition, the toxicity of EMT inhibitors remains a potential risk. For example, small molecule TGF-β inhibitors have shown severe cardiac toxicity in preclinical animal models (305, 306). miRNA is another promising research field for overcoming drug resistance. miRNAs may play opposite roles in different cancers, and even their functions may vary in different subtypes of the same cancer. This guides us to make more precise distinctions about the roles of miRNAs in tumors. In the past 20 years, the number of identified miRNAs and their targets has been increasing at an incredible rate. However, there are still many questions that need to be answered before miRNA therapy can be widely applied in clinical practice. How to prevent microRNA degradation in vivo and how to efficiently targeted delivery are still unresolved issues (307). Epigenetic modifications are closely related to EMT, stemness and drug resistance, which has aroused the interest of researchers in this field and has been widely discussed. In addition to miRNAs, the corresponding epi-drugs, such as DNA modifying agents, inhibitors of histone acetyltransferase (or deacetylase or methyltransferase or demethyltransferase), are also gradually emerging, trying to be applied in the treatment of breast cancer (308).
6 Conclusion
EMT is a complex and dynamic biological program that typically occurs during embryonic development and tumor progression. It is essentially a major reprogramming involving gene expression, which can affect the macroscopic malignant development of tumors by regulating the fate and behavior of cells. EMT has been reported to induce invasion and metastasis of breast cancer, and its impact on drug resistance is also becoming clearer. Increasingly direct evidence shows that EMT-related markers are closely related to the resistance to therapy in breast cancer (309, 310). The present review summarizes the current knowledge regarding EMT-induced drug resistance in breast cancer. The EMT-related regulatory network constitutes a complex system, with TGF-β, Notch, Wnt, and Hh pathways being the most explored and clearly defined pathways in breast cancer. These signaling pathways crosstalk and interact with each other, collectively activating EMT-TFs, which target the hallmarks of EMT and initiate the EMT program. Currently, the SNAIL, TWIST, and ZEB families are widely acknowledged as the primary transcription factors driving EMT. Epigenetic modifications also play a role in the EMT process by influencing the function of EMT-TFs, with particular attention being paid to non-coding RNAs. None of the elements within the EMT-related regulatory network functions in isolation. They can synergize or antagonize with other elements, forming positive or negative feedback loops. The molecular mechanism underlying EMT-induced drug resistance in breast cancer is still unclear, yet several possible mechanisms have been extensively proposed. Among them, the close relationship and high phenotypic similarity between EMT cells and drug-resistant CSCs have garnered the most attention. Furthermore, the hybrid epithelial/mesenchymal state of EMT cells endows them with a high degree of plasticity, which makes them more prone to drug resistance. Additionally, increased drug efflux, resistance to apoptosis and activation of DDR pathways induced by EMT contribute to the decreasing responsiveness of breast cancer cells to anti-tumor drugs. The elucidation of the regulatory network related to EMT and the potential mechanisms of EMT-induced drug resistance may contribute to the design of better targeted therapies combined with conventional treatments. Reversing the EMT process by modifying TGF-β, Wnt, Notch, Hh or other signaling pathways is expected to overcome drug resistance in breast cancer. Numerous inhibitors of these signaling pathways have advanced to preclinical or clinical trial phase and have been proven to partially alleviate drug resistance by reversing EMT. Directly targeting EMT-TFs can also make breast cancer cells more susceptible to anti-tumor drugs. Furthermore, as the interplay between miRNAs and EMT becomes increasingly well-understood, it is also a promising direction of research to leverage tumor suppressor miRNAs to regulate EMT and restore the sensitivity of breast cancer to anti-cancer drugs.
In recent years, with the development of technologies such as tumor genomics, transcriptomics and proteomics, the association of EMT and tumor drug resistance have attracted great interest. In the future, interdisciplinary approaches should be adopted to EMT research to further confirm its correlation with drug resistance and clarify the dominant mechanisms. For instance, database-based bioinformatics analysis can be used to develop personalized and customized treatments to overcome tumor drug resistance. Although the mechanisms and influencing factors of drug resistance are becoming increasingly complex, new targets are emerging to offer new hope to cancer patients worldwide.
Author contributions
YL: Conceptualization, Investigation, Visualization, Writing – original draft, Writing – review & editing. RS: Writing – review & editing. XT: Visualization, Writing – review & editing. JG: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, and Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
3. Davey MG, Ryan EJ, Davey MS, Lowery AJ, Miller N, and Kerin MJ. Clinicopathological and prognostic significance of programmed cell death ligand 1 expression in patients diagnosed with breast cancer: meta-analysis. Br J Surg. (2021) 108:622–31. doi: 10.1093/bjs/znab103
4. Cardoso F, Paluch-Shimon S, Senkus E, Curigliano G, Aapro MS, André F, et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann Oncol. (2020) 31:1623–49. doi: 10.1016/j.annonc.2020.09.010
5. Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, et al. Drug resistance in cancer: an overview. Cancers (Basel). (2014) 6:1769–92. doi: 10.3390/cancers6031769
6. Wang X, Chen T, Li C, Li W, Zhou X, Li Y, et al. CircRNA-CREIT inhibits stress granule assembly and overcomes doxorubicin resistance in TNBC by destabilizing PKR. J Hematol Oncol. (2022) 15:122. doi: 10.1186/s13045-022-01345-w
7. Normanno N, Di Maio M, De Maio E, De Luca A, de Matteis A, Giordano A, et al. Mechanisms of endocrine resistance and novel therapeutic strategies in breast cancer. Endocr Relat Can. (2005) 12:721–47. doi: 10.1677/erc.1.00857
8. Martin-Castillo B, Oliveras-Ferraros C, Vazquez-Martin A, Cufí S, Moreno JM, Corominas-Faja B, et al. Basal/HER2 breast carcinomas: integrating molecular taxonomy with cancer stem cell dynamics to predict primary resistance to trastuzumab (Herceptin). Cell Cycle. (2013) 12:225–45. doi: 10.4161/cc.23274
9. Bukowski K, Kciuk M, and Kontek R. Mechanisms of multidrug resistance in cancer chemotherapy. Int J Mol Sci. (2020) 21:3233. doi: 10.3390/ijms21093233
10. Lukasiewicz S, Czeczelewski M, Forma A, Baj J, Sitarz R, and Stanislawek A. Breast cancer-epidemiology, risk factors, classification, prognostic markers, and current treatment strategies-an updated review. Cancers (Basel). (2021) 13:4287. doi: 10.3390/cancers13174287
11. Vasan N and Baselga J. Hyman DM. A view Drug Res Can Nat. (2019) 575:299–309. doi: 10.1038/s41586-019-1730-1
12. Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. (2020) 21:341–52. doi: 10.1038/s41580-020-0237-9
13. Chen T, You Y, Jiang H, and Wang ZZ. Epithelial-mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J Cell Physiol. (2017) 232:3261–72. doi: 10.1002/jcp.25797
14. Mallini P, Lennard T, Kirby J, and Meeson A. Epithelial-to-mesenchymal transition: what is the impact on breast cancer stem cells and drug resistance. Cancer Treat Rev. (2014) 40:341–8. doi: 10.1016/j.ctrv.2013.09.008
15. Shibue T and Weinberg RA. EMT. CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. (2017) 14:611–29. doi: 10.1038/nrclinonc.2017.44
16. Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, Becette V, et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med. (2009) 15:68–74. doi: 10.1038/nm.1908
17. Inayatullah M, Mahesh A, Turnbull AK, Dixon JM, Natrajan R, and Tiwari VK. Basal-epithelial subpopulations underlie and predict chemotherapy resistance in triple-negative breast cancer. EMBO Mol Med. (2024) 16:823–53. doi: 10.1038/s44321-024-00050-0
18. Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, and Wang LH. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. (2007) 67:1979–87. doi: 10.1158/0008-5472.CAN-06-1479
19. Hiscox S, Jiang WG, Obermeier K, Taylor K, Morgan L, Burmi R, et al. Tamoxifen resistance in MCF7 cells promotes EMT-like behaviour and involves modulation of beta-catenin phosphorylation. Int J Can. (2006) 118:290–301. doi: 10.1002/ijc.21355
20. Li QQ, Xu JD, Wang WJ, Cao XX, Chen Q, Tang F, et al. Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin Cancer Res. (2009) 15:2657–65. doi: 10.1158/1078-0432.CCR-08-2372
21. Kalluri R and Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. (2009) 119:1420–8. doi: 10.1172/JCI39104
22. Huang Y, Hong W, and Wei X. The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis. J Hematol Oncol. (2022) 15:129. doi: 10.1186/s13045-022-01347-8
23. Steinestel K, Eder S, Schrader AJ, and Steinestel J. Clinical significance of epithelial-mesenchymal transition. Clin Transl Med. (2014) 3:17. doi: 10.1186/2001-1326-3-17
24. Pasquier J, Abu-Kaoud N, Al Thani H, and Rafii A. Epithelial to mesenchymal transition in a clinical perspective. J Oncol. (2015) 2015:792182. doi: 10.1155/2015/792182
25. Zeisberg M and Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. (2009) 119:1429–37. doi: 10.1172/JCI36183
26. Satelli A and Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. (2011) 68:3033–46. doi: 10.1007/s00018-011-0735-1
27. Nieto MA, Huang RY, Jackson RA, and Thiery JP. Emt: 2016. Cell. (2016) 166:21–45. doi: 10.1016/j.cell.2016.06.028
28. Singh M, Yelle N, Venugopal C, and Singh SK. EMT: Mechanisms and therapeutic implications. Pharmacol Ther. (2018) 182:80–94. doi: 10.1016/j.pharmthera.2017.08.009
29. Akhmetkaliyev A, Alibrahim N, Shafiee D, and Tulchinsky E. EMT/MET plasticity in cancer and Go-or-Grow decisions in quiescence: the two sides of the same coin? Mol Cancer. (2023) 22:90. doi: 10.1186/s12943-023-01793-z
30. Dongre A and Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. (2019) 20:69–84. doi: 10.1038/s41580-018-0080-4
31. Xu J, Lamouille S, and Derynck R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. (2009) 19:156–72. doi: 10.1038/cr.2009.5
32. Derynck R and Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. (2003) 425:577–84. doi: 10.1038/nature02006
33. Katsuno Y, Lamouille S, and Derynck R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol. (2013) 25:76–84. doi: 10.1097/CCO.0b013e32835b6371
34. Bui QT, Im JH, Jeong SB, Kim YM, Lim SC, Kim B, et al. Essential role of Notch4/STAT3 signaling in epithelial-mesenchymal transition of tamoxifen-resistant human breast cancer. Cancer Lett. (2017) 390:115–25. doi: 10.1016/j.canlet.2017.01.014
35. De Francesco EM, Maggiolini M, and Musti AM. Crosstalk between notch, HIF-1α and GPER in breast cancer EMT. Int J Mol Sci. (2018) 19:2011. doi: 10.3390/ijms19072011
36. Hashemi M, Hasani S, Hajimazdarany S, Ghadyani F, Olyaee Y, Khodadadi M, et al. Biological functions and molecular interactions of Wnt/β-catenin in breast cancer: Revisiting signaling networks. Int J Biol Macromol. (2023) 232:123377. doi: 10.1016/j.ijbiomac.2023.123377
37. Zhang J, Guo H, Gong C, Shen J, Jiang G, Liu J, et al. Therapeutic targets in the Wnt signaling pathway: Treating cancer with specificity. Biochem Pharmacol. (2025) 236:116848. doi: 10.1016/j.bcp.2025.116848
38. Barker N and Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov. (2006) 5:997–1014. doi: 10.1038/nrd2154
39. Habib JG and O’Shaughnessy JA. The hedgehog pathway in triple-negative breast cancer. Cancer Med. (2016) 5:2989–3006. doi: 10.1002/cam4.833
40. Landor SK and Lendahl U. The interplay between the cellular hypoxic response and Notch signaling. Exp Cell Res. (2017) 356:146–51. doi: 10.1016/j.yexcr.2017.04.030
41. Lei J, Fan L, Wei G, Chen X, Duan W, Xu Q, et al. Gli-1 is crucial for hypoxia-induced epithelial-mesenchymal transition and invasion of breast cancer. Tumour Biol. (2015) 36:3119–26. doi: 10.1007/s13277-014-2948-z
42. Das S, Samant RS, and Shevde LA. Nonclassical activation of Hedgehog signaling enhances multidrug resistance and makes cancer cells refractory to Smoothened-targeting Hedgehog inhibition. J Biol Chem. (2013) 288:11824–33. doi: 10.1074/jbc.M112.432302
43. Johnson RW, Nguyen MP, Padalecki SS, Grubbs BG, Merkel AR, Oyajobi BO, et al. TGF-beta promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical Hedgehog signaling. Cancer Res. (2011) 71:822–31. doi: 10.1158/0008-5472.CAN-10-2993
44. Colavito SA, Zou MR, Yan Q, Nguyen DX, and Stern DF. Significance of glioma-associated oncogene homolog 1 (GLI1) expression in claudin-low breast cancer and crosstalk with the nuclear factor kappa-light-chain-enhancer of activated B cells (NFkappaB) pathway. Breast Cancer Res. (2014) 16:444. doi: 10.1186/s13058-014-0444-4
45. Goel HL, Pursell B, Chang C, Shaw LM, Mao J, Simin K, et al. GLI1 regulates a novel neuropilin-2/α6β1 integrin based autocrine pathway that contributes to breast cancer initiation. EMBO Mol Med. (2013) 5:488–508. doi: 10.1002/emmm.201202078
46. Barr S, Thomson S, Buck E, Russo S, Petti F, Sujka-Kwok I, et al. Bypassing cellular EGF receptor dependence through epithelial-to-mesenchymal-like transitions. Clin Exp Metastasis. (2008) 25:685–93. doi: 10.1007/s10585-007-9121-7
47. Lu W and Kang Y. Epithelial-mesenchymal plasticity in cancer progression and metastasis. Dev Cell. (2019) 49:361–74. doi: 10.1016/j.devcel.2019.04.010
48. Wilson MM, Weinberg RA, Lees JA, and Guen VJ. Emerging mechanisms by which EMT programs control stemness. Trends Can. (2020) 6:775–80. doi: 10.1016/j.trecan.2020.03.011
49. Ribatti D, Tamma R, and Annese T. Epithelial-mesenchymal transition in cancer: A historical overview. Transl Oncol. (2020) 13:100773. doi: 10.1016/j.tranon.2020.100773
50. Meng J, Chen S, Han JX, Qian B, Wang XR, Zhong WL, et al. Twist1 regulates vimentin through cul2 circular RNA to promote EMT in hepatocellular carcinoma. Cancer Res. (2018) 78:4150–62. doi: 10.1158/0008-5472.CAN-17-3009
51. Thiam HR, Wong SL, Qiu R, Kittisopikul M, Vahabikashi A, Goldman AE, et al. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc Natl Acad Sci U S A. (2020) 117:7326–37. doi: 10.1073/pnas.1909546117
52. Hennig G, Behrens J, Truss M, Frisch S, Reichmann E, and Birchmeier W. Progression of carcinoma cells is associated with alterations in chromatin structure and factor binding at the E-cadherin promoter in vivo. Oncogene. (1995) 11:475–84. doi: 10.1016/0378-1119(95)98166-D
53. Gheldof A, Hulpiau P, van Roy F, De Craene B, and Berx G. Evolutionary functional analysis and molecular regulation of the ZEB transcription factors. Cell Mol Life Sci. (2012) 69:2527–41. doi: 10.1007/s00018-012-0935-3
54. Hajra KM, Chen DY, and Fearon ER. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. (2002) 62:1613–8. doi: 10.1002/cncr.10401
55. Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. (2000) 2:76–83. doi: 10.1038/35000025
56. Peinado H, Olmeda D, and Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. (2007) 7:415–28. doi: 10.1038/nrc2131
57. Dhasarathy A, Phadke D, Mav D, Shah RR, and Wade PA. The transcription factors Snail and Slug activate the transforming growth factor-beta signaling pathway in breast cancer. PloS One. (2011) 6:e26514. doi: 10.1371/journal.pone.0026514
58. Smith BN, Burton LJ, Henderson V, Randle DD, Morton DJ, Smith BA, et al. Snail promotes epithelial mesenchymal transition in breast cancer cells in part via activation of nuclear ERK2. PloS One. (2014) 9:e104987. doi: 10.1371/journal.pone.0104987
59. Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell. (2010) 6:479–91. doi: 10.1016/j.stem.2010.03.018
60. Tamura G, Yin J, Wang S, Fleisher AS, Zou T, Abraham JM, et al. E-Cadherin gene promoter hypermethylation in primary human gastric carcinomas. J Natl Cancer Inst. (2000) 92:569–73. doi: 10.1093/jnci/92.7.569
61. Bornman DM, Mathew S, Alsruhe J, Herman JG, and Gabrielson E. Methylation of the E-cadherin gene in bladder neoplasia and in normal urothelial epithelium from elderly individuals. Am J Pathol. (2001) 159:831–5. doi: 10.1016/S0002-9440(10)61758-0
62. Puisieux A, Brabletz T, and Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. (2014) 16:488–94. doi: 10.1038/ncb2976
63. Shao P, Liu Q, Maina PK, Cui J, Bair TB, Li T, et al. Histone demethylase PHF8 promotes epithelial to mesenchymal transition and breast tumorigenesis. Nucleic Acids Res. (2017) 45:1687–702. doi: 10.1093/nar/gkw1093
64. Lin T, Ponn A, Hu X, Law BK, and Lu J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene. (2010) 29:4896–904. doi: 10.1038/onc.2010.234
65. Lin Y, Wu Y, Li J, Dong C, Ye X, Chi YI, et al. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. (2010) 29:1803–16. doi: 10.1038/emboj.2010.63
66. Ou B, Liu Y, Gao Z, Xu J, Yan Y, Li Y, et al. Senescent neutrophils-derived exosomal piRNA-17560 promotes chemoresistance and EMT of breast cancer via FTO-mediated m6A demethylation. Cell Death Dis. (2022) 13:905. doi: 10.1038/s41419-022-05317-3
67. Guo H, Ingolia NT, Weissman JS, and Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. (2010) 466:835–40. doi: 10.1038/nature09267
68. Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. (2005) 433:769–73. doi: 10.1038/nature03315
69. Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. (2008) 10:593–601. doi: 10.1038/ncb1722
70. Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. (2008) 9:582–9. doi: 10.1038/embor.2008.74
71. Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. (2008) 68:7846–54. doi: 10.1158/0008-5472.CAN-08-1942
72. Luan QX, Zhang BG, Li XJ, and Guo MY. MiR-129-5p is downregulated in breast cancer cells partly due to promoter H3K27m3 modification and regulates epithelial-mesenchymal transition and multi-drug resistance. Eur Rev Med Pharmacol Sci. (2016) 20:4257–65.
73. Liu Z, Mi M, Zheng X, Zhang C, Zhu F, Liu T, et al. miR-30a/SOX4 Double Negative Feedback Loop is modulated by Disulfiram and regulates EMT and Stem Cell-like properties in Breast Cancer. J Can. (2021) 12:5053–65. doi: 10.7150/jca.57752
74. Moes M, Le Béchec A, Crespo I, Laurini C, Halavatyi A, Vetter G, et al. A novel network integrating a miRNA-203/SNAI1 feedback loop which regulates epithelial to mesenchymal transition. PloS One. (2012) 7:e35440. doi: 10.1371/journal.pone.0035440
75. Su J, Deng L, and Wang YD. Roles and mechanisms of long non-coding RNAs in breast cancer. Int J Mol Sci. (2022) 24:89. doi: 10.3390/ijms24010089
76. Li J, Hao Y, Mao W, Xue X, Xu P, Liu L, et al. LincK contributes to breast tumorigenesis by promoting proliferation and epithelial-to-mesenchymal transition. J Hematol Oncol. (2019) 12:19. doi: 10.1186/s13045-019-0707-8
77. Yang L, Wang M, Wang Y, Zhu Y, Wang J, Wu M, et al. LINC00460-FUS-MYC feedback loop drives breast cancer metastasis and doxorubicin resistance. Oncogene. (2024) 43:1249–62. doi: 10.1038/s41388-024-02972-y
78. Khan MI and Ahmad A. LncRNA SNHG6 sponges miR-101 and induces tamoxifen resistance in breast cancer cells through induction of EMT. Front Oncol. (2022) 12:1015428. doi: 10.3389/fonc.2022.1015428
79. Gupta S, Silveira DA, Lorenzoni PR, Mombach JCM, and Hashimoto RF. LncRNA PTENP1/miR-21/PTEN axis modulates EMT and drug resistance in cancer: dynamic boolean modeling for cell fates in DNA damage response. Int J Mol Sci. (2024) 25:8264. doi: 10.3390/ijms25158264
80. Venkatesh J, Wasson MD, Brown JM, Fernando W, and Marcato P. LncRNA-miRNA axes in breast cancer: Novel points of interaction for strategic attack. Cancer Lett. (2021) 509:81–8. doi: 10.1016/j.canlet.2021.04.002
81. Luo L, Xu N, Fan W, Wu Y, Chen P, Li Z, et al. The TGFβ2-snail1-miRNA(TGFβ2) circuitry is critical for the development of aggressive functions in breast cancer. Clin Transl Med. (2024) 14:e1558. doi: 10.1002/ctm2.1558
82. Tannock IF, Lee CM, Tunggal JK, Cowan DS, and Egorin MJ. Limited penetration of anticancer drugs through tumor tissue: a potential cause of resistance of solid tumors to chemotherapy. Clin Cancer Res. (2002) 8:878–84. doi: 10.1159/000048589
83. Luo L, Yang P, Mastoraki S, Rao X, Wang Y, Kettner NM, et al. Single-cell RNA sequencing identifies molecular biomarkers predicting late progression to CDK4/6 inhibition in patients with HR+/HER2- metastatic breast cancer. Mol Can. (2025) 24:48. doi: 10.1186/s12943-025-02226-9
84. De Las Rivas J, Brozovic A, Izraely S, Casas-Pais A, Witz IP, and Figueroa A. Cancer drug resistance induced by EMT: novel therapeutic strategies. Arch Toxicol. (2021) 95:2279–97. doi: 10.1007/s00204-021-03063-7
85. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. (2004) 432:396–401. doi: 10.1038/nature03128
86. Bjerkvig R, Tysnes BB, Aboody KS, Najbauer J, and Terzis AJ. Opinion: the origin of the cancer stem cell: current controversies and new insights. Nat Rev Can. (2005) 5:899–904. doi: 10.1038/nrc1740
87. Battula VL, Evans KW, Hollier BG, Shi Y, Marini FC, Ayyanan A, et al. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells. (2010) 28:1435–45. doi: 10.1002/stem.467
88. Feng YX, Sokol ES, Del Vecchio CA, Sanduja S, Claessen JH, Proia TA, et al. Epithelial-to-mesenchymal transition activates PERK-eIF2α and sensitizes cells to endoplasmic reticulum stress. Cancer Discov. (2014) 4:702–15. doi: 10.1158/2159-8290.CD-13-0945
89. Chu X, Tian W, Ning J, Xiao G, Zhou Y, Wang Z, et al. Cancer stem cells: advances in knowledge and implications for cancer therapy. Signal Transduct Target Ther. (2024) 9:170. doi: 10.1038/s41392-024-01851-y
90. Malanchi I, Peinado H, Kassen D, Hussenet T, Metzger D, Chambon P, et al. Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature. (2008) 452:650–3. doi: 10.1038/nature06835
91. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. (2008) 133:704–15. doi: 10.1016/j.cell.2008.03.027
92. Santisteban M, Reiman JM, Asiedu MK, Behrens MD, Nassar A, Kalli KR, et al. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. (2009) 69:2887–95. doi: 10.1158/0008-5472.CAN-08-3343
93. Fang X, Cai Y, Liu J, Wang Z, Wu Q, Zhang Z, et al. Twist2 contributes to breast cancer progression by promoting an epithelial-mesenchymal transition and cancer stem-like cell self-renewal. Oncogene. (2011) 30:4707–20. doi: 10.1038/onc.2011.181
94. Vesuna F, Lisok A, Kimble B, and Raman V. Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia. (2009) 11:1318–28. doi: 10.1593/neo.91084
95. Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. (2012) 148:1015–28. doi: 10.1016/j.cell.2012.02.008
96. Nassar D and Blanpain C. Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol. (2016) 11:47–76. doi: 10.1146/annurev-pathol-012615-044438
97. Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. (2008) 100:672–9. doi: 10.1093/jnci/djn123
98. Blick T, Hugo H, Widodo E, Waltham M, Pinto C, Mani SA, et al. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi/)CD24 (lo/-) stem cell phenotype in human breast cancer. J Mammar Gland Biol Neoplasia. (2010) 15:235–52. doi: 10.1007/s10911-010-9175-z
99. Singh A and Settleman JEMT. cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. (2010) 29:4741–51. doi: 10.1038/onc.2010.215
100. van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, et al. Recovering gene interactions from single-cell data using data diffusion. Cell. (2018) 174:716–29.e27. doi: 10.1016/j.cell.2018.05.061
101. Jolly MK, Somarelli JA, Sheth M, Biddle A, Tripathi SC, Armstrong AJ, et al. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol Ther. (2019) 194:161–84. doi: 10.1016/j.pharmthera.2018.09.007
102. Sahoo S, Ramu S, Nair MG, Pillai M, San Juan BP, Milioli HZ, et al. Increased prevalence of hybrid epithelial/mesenchymal state and enhanced phenotypic heterogeneity in basal breast cancer. iScience. (2024) 27:110116. doi: 10.1016/j.isci.2024.110116
103. Tessier CE, Derrien J, Dupuy AMM, Pelé T, Moquet M, Roul J, et al. EMT-ciliary signaling in quasi-mesenchymal-stem-like cells drives therapeutic resistance and is a druggable vulnerability in triple-negative breast cancer. EMBO Mol Med. (2025) 17:2536–61. doi: 10.1038/s44321-025-00289-1
104. Brown MS, Abdollahi B, Wilkins OM, Lu H, Chakraborty P, Ognjenovic NB, et al. Phenotypic heterogeneity driven by plasticity of the intermediate EMT state governs disease progression and metastasis in breast cancer. Sci Adv. (2022) 8:eabj8002. doi: 10.1126/sciadv.abj8002
105. Muller L, Fauvet F, Chassot C, Angileri F, Coutant A, Dégletagne C, et al. EMT-driven plasticity prospectively increases cell-cell variability to promote therapeutic adaptation in breast cancer. Cancer Cell Int. (2025) 25:32. doi: 10.1186/s12935-025-03637-w
106. Gottesman MM, Fojo T, and Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Can. (2002) 2:48–58. doi: 10.1038/nrc706
107. Fletcher JI, Haber M, Henderson MJ, and Norris MD. ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Can. (2010) 10:147–56. doi: 10.1038/nrc2789
108. Saxena M, Stephens MA, Pathak H, and Rangarajan A. Transcription factors that mediate epithelial-mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. (2011) 2:e179. doi: 10.1038/cddis.2011.61
109. Brabletz S, Schuhwerk H, Brabletz T, and Stemmler MP. Dynamic EMT: a multi-tool for tumor progression. EMBO J. (2021) 40:e108647. doi: 10.15252/embj.2021108647
110. Sánchez-Tilló E, Fanlo L, Siles L, Montes-Moreno S, Moros A, Chiva-Blanch G, et al. The EMT activator ZEB1 promotes tumor growth and determines differential response to chemotherapy in mantle cell lymphoma. Cell Death Differ. (2014) 21:247–57. doi: 10.1038/cdd.2013.123
111. Paoli P, Giannoni E, and Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta. (2013) 1833:3481–98. doi: 10.1016/j.bbamcr.2013.06.026
112. Wu Y and Zhou BP. Snail: more than EMT. Cell Adh Migr. (2010) 4:199–203. doi: 10.4161/cam.4.2.10943
113. Barrallo-Gimeno A and Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. (2005) 132:3151–61. doi: 10.1242/dev.01907
114. Kurrey NK, Jalgaonkar SP, Joglekar AV, Ghanate AD, Chaskar PD, Doiphode RY, et al. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells. (2009) 27:2059–68. doi: 10.1002/stem.154
115. Vega S, Morales AV, Ocaña OH, Valdés F, Fabregat I, and Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. (2004) 18:1131–43. doi: 10.1101/gad.294104
116. Xu W, Yang Z, and Lu N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh Migr. (2015) 9:317–24. doi: 10.1080/19336918.2015.1016686
117. Kwok WK, Ling MT, Lee TW, Lau TC, Zhou C, Zhang X, et al. Up-regulation of TWIST in prostate cancer and its implication as a therapeutic target. Cancer Res. (2005) 65:5153–62. doi: 10.1158/0008-5472.CAN-04-3785
118. Chen HT, Liu H, Mao MJ, Tan Y, Mo XQ, Meng XJ, et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol Can. (2019) 18:101. doi: 10.1186/s12943-019-1030-2
119. Ghavami S, Cunnington RH, Gupta S, Yeganeh B, Filomeno KL, Freed DH, et al. Autophagy is a regulator of TGF-β1-induced fibrogenesis in primary human atrial myofibroblasts. Cell Death Dis. (2015) 6:e1696. doi: 10.1038/cddis.2015.36
120. Heydarpour F, Sajadimajd S, Mirzarazi E, Haratipour P, Joshi T, Farzaei MH, et al. Involvement of TGF-β and autophagy pathways in pathogenesis of diabetes: A comprehensive review on biological and pharmacological insights. Front Pharmacol. (2020) 11:498758. doi: 10.3389/fphar.2020.498758
121. Kiyono K, Suzuki HI, Matsuyama H, Morishita Y, Komuro A, Kano MR, et al. Autophagy is activated by TGF-beta and potentiates TGF-beta-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. (2009) 69:8844–52. doi: 10.1158/0008-5472.CAN-08-4401
122. Mariño G, Niso-Santano M, Baehrecke EH, and Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. (2014) 15:81–94. doi: 10.1038/nrm3735
123. Chittaranjan S, Bortnik S, Dragowska WH, Xu J, Abeysundara N, Leung A, et al. Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clin Cancer Res. (2014) 20:3159–73. doi: 10.1158/1078-0432.CCR-13-2060
124. Sun WL, Chen J, Wang YP, and Zheng H. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy. (2011) 7:1035–44. doi: 10.4161/auto.7.9.16521
125. Ajabnoor GM, Crook T, and Coley HM. Paclitaxel resistance is associated with switch from apoptotic to autophagic cell death in MCF-7 breast cancer cells. Cell Death Dis. (2012) 3:e260. doi: 10.1038/cddis.2011.139
126. Jackson SP and Bartek J. The DNA-damage response in human biology and disease. Nature. (2009) 461:1071–8. doi: 10.1038/nature08467
127. Harper JW and Elledge SJ. The DNA damage response: ten years after. Mol Cell. (2007) 28:739–45. doi: 10.1016/j.molcel.2007.11.015
128. Moyret-Lalle C, Prodhomme MK, Burlet D, Kashiwagi A, Petrilli V, Puisieux A, et al. Role of EMT in the DNA damage response, double-strand break repair pathway choice and its implications in cancer treatment. Cancer Sci. (2022) 113:2214–23. doi: 10.1111/cas.15389
129. Chang HHY, Pannunzio NR, Adachi N, and Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol. (2017) 18:495–506. doi: 10.1038/nrm.2017.48
130. Jurkovicova D, Neophytou CM, Gašparović A, and Gonçalves AC. DNA damage response in cancer therapy and resistance: challenges and opportunities. Int J Mol Sci. (2022) 23:14672. doi: 10.3390/ijms232314672
131. Olaussen KA, Dunant A, Fouret P, Brambilla E, André F, Haddad V, et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med. (2006) 355:983–91. doi: 10.1056/NEJMoa060570
132. Bonanno L, Favaretto A, and Rosell R. Platinum drugs and DNA repair mechanisms in lung cancer. Anticancer Res. (2014) 34:493–501. doi: 10.1016/j.lungcan.2013.10.009
133. Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y, Zhang J, et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol. (2014) 16:864–75. doi: 10.1038/ncb3013
134. Zhang X, Zhang Z, Zhang Q, Zhang Q, Sun P, Xiang R, et al. ZEB1 confers chemotherapeutic resistance to breast cancer by activating ATM. Cell Death Dis. (2018) 9:57. doi: 10.1038/s41419-017-0087-3
135. Lee HJ, Li CF, Ruan D, Powers S, Thompson PA, Frohman MA, et al. The DNA damage transducer RNF8 facilitates cancer chemoresistance and progression through twist activation. Mol Cell. (2016) 63:1021–33. doi: 10.1016/j.molcel.2016.08.009
136. Akalay I, Janji B, Hasmim M, Noman MZ, Thiery JP, Mami-Chouaib F, et al. EMT impairs breast carcinoma cell susceptibility to CTL-mediated lysis through autophagy induction. Autophagy. (2013) 9:1104–6. doi: 10.4161/auto.24728
137. Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. (2011) 145:926–40. doi: 10.1016/j.cell.2011.04.029
138. Hsu DS, Wang HJ, Tai SK, Chou CH, Hsieh CH, Chiu PH, et al. Acetylation of snail modulates the cytokinome of cancer cells to enhance the recruitment of macrophages. Cancer Cell. (2014) 26:534–48. doi: 10.1016/j.ccell.2014.09.002
139. Wu Y, Shi Y, Luo Z, Zhou X, Chen Y, Song X, et al. Spatial multi-omics analysis of tumor-stroma boundary cell features for predicting breast cancer progression and therapy response. Front Cell Dev Biol. (2025) 13:1570696. doi: 10.3389/fcell.2025.1570696
140. Wu S, Tang Q, Fang W, Sun Z, Zhang M, Liu E, et al. Targeting stem-property and vasculogenic mimicry for sensitizing paclitaxel therapy of triple-negative breast cancer by biomimetic codelivery. Acta Pharm Sin B. (2025) 15:3226–42. doi: 10.1016/j.apsb.2025.04.006
141. Kim NH, Cha YH, Lee J, Lee SH, Yang JH, Yun JS, et al. Snail reprograms glucose metabolism by repressing phosphofructokinase PFKP allowing cancer cell survival under metabolic stress. Nat Commun. (2017) 8:14374. doi: 10.1038/ncomms14374
142. Lee SY, Jeon HM, Ju MK, Kim CH, Yoon G, Han SI, et al. Wnt/Snail signaling regulates cytochrome C oxidase and glucose metabolism. Cancer Res. (2012) 72:3607–17. doi: 10.1158/0008-5472.CAN-12-0006
143. Liu M, Hancock SE, Sultani G, Wilkins BP, Ding E, Osborne B, et al. Snail-overexpression induces epithelial-mesenchymal transition and metabolic reprogramming in human pancreatic ductal adenocarcinoma and non-tumorigenic ductal cells. J Clin Med. (2019) 8:822. doi: 10.3390/jcm8060822
144. Parker HN, Haberman KL, Ojo T, Watkins J, Nambiar A, Morales K, et al. Twist-induced epithelial-to-mesenchymal transition confers specific metabolic and mitochondrial alterations. Cells. (2025) 14:80. doi: 10.3390/cells14020080
145. Greijer AE, de Jong MC, Scheffer GL, Shvarts A, van Diest PJ, and van der Wall E. Hypoxia-induced acidification causes mitoxantrone resistance not mediated by drug transporters in human breast cancer cells. Cell Oncol. (2005) 27:43–9. doi: 10.1155/2005/236045
146. Leegwater H, Zhang Z, Zhang X, Wang X, Hankemeier T, Zweemer AJM, et al. Distinct lipidomic profiles in breast cancer cell lines relate to proliferation and EMT phenotypes. Biochim Biophys Acta Mol Cell Biol Lipids. (2025) 1870:159679. doi: 10.1016/j.bbalip.2025.159679
147. Flavin R, Peluso S, Nguyen PL, and Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. (2010) 6:551–62. doi: 10.2217/fon.10.11
148. Castagnoli L, Franceschini A, Cancila V, Dugo M, Bigliardi M, Chiodoni C, et al. CD36 enrichment in HER2-positive mesenchymal stem cells drives therapy refractoriness in breast cancer. J Exp Clin Cancer Res. (2025) 44:19. doi: 10.1186/s13046-025-03276-z
149. Babina IS, McSherry EA, Donatello S, Hill AD, and Hopkins AM. A novel mechanism of regulating breast cancer cell migration via palmitoylation-dependent alterations in the lipid raft affiliation of CD44. Breast Cancer Res. (2014) 16:R19. doi: 10.1186/bcr3614
150. Omran Z, Scaife P, Stewart S, and Rauch C. Physical and biological characteristics of multi drug resistance (MDR): An integral approach considering pH and drug resistance in cancer. Semin Cancer Biol. (2017) 43:42–8. doi: 10.1016/j.semcancer.2017.01.002
151. Szebényi K, Füredi A, Bajtai E, Sama SN, Csiszar A, Gombos B, et al. Effective targeting of breast cancer by the inhibition of P-glycoprotein mediated removal of toxic lipid peroxidation byproducts from drug tolerant persister cells. Drug Resist Updat. (2023) 71:101007. doi: 10.1016/j.drup.2023.101007
152. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Can. (2002) 2:442–54. doi: 10.1038/nrc822
153. Kantapan J, Innuan P, Viriyaadhammaa N, Anukul N, Chitapanarux I, and Dechsupa N. Pentagalloyl glucose reverses chemoresistance in triple-negative breast cancer via EMT inhibition and miRNA modulation. BioMed Pharmacother. (2025) 192:118572. doi: 10.1016/j.biopha.2025.118572
154. Fan JX, Zheng DW, Rong L, Zhu JY, Hong S, Li C, et al. Targeting epithelial-mesenchymal transition: Metal organic network nano-complexes for preventing tumor metastasis. Biomaterials. (2017) 139:116–26. doi: 10.1016/j.biomaterials.2017.06.007
155. Bandyopadhyay A, Wang L, Agyin J, Tang Y, Lin S, Yeh IT, et al. Doxorubicin in combination with a small TGFbeta inhibitor: a potential novel therapy for metastatic breast cancer in mouse models. PloS One. (2010) 5:e10365. doi: 10.1371/journal.pone.0010365
156. Chen J, Ding ZY, Li S, Liu S, Xiao C, Li Z, et al. Targeting transforming growth factor-β signaling for enhanced cancer chemotherapy. Theranostics. (2021) 11:1345–63. doi: 10.7150/thno.51383
157. Hirohashi S and Kanai Y. Cell adhesion system and human cancer morphogenesis. Cancer Sci. (2003) 94:575–81. doi: 10.1111/j.1349-7006.2003.tb01485.x
158. Xu X, Zhang L, He X, Zhang P, Sun C, Xu X, et al. TGF-β plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem Biophys Res Commun. (2018) 502:160–5. doi: 10.1016/j.bbrc.2018.05.139
159. Mousset A, Lecorgne E, Bourget I, Lopez P, Jenovai K, Cherfils-Vicini J, et al. Neutrophil extracellular traps formed during chemotherapy confer treatment resistance via TGF-beta activation. Cancer Cell. (2023) 41:757–75 e10. doi: 10.1016/j.ccell.2023.03.008
160. Bhola NE, Balko JM, Dugger TC, Kuba MG, Sánchez V, Sanders M, et al. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest. (2013) 123:1348–58. doi: 10.1172/JCI65416
161. Ganapathy V, Ge R, Grazioli A, Xie W, Banach-Petrosky W, Kang Y, et al. Targeting the Transforming Growth Factor-beta pathway inhibits human basal-like breast cancer metastasis. Mol Can. (2010) 9:122. doi: 10.1186/1476-4598-9-122
162. Bauer JA, Ye F, Marshall CB, Lehmann BD, Pendleton CS, Shyr Y, et al. RNA interference (RNAi) screening approach identifies agents that enhance paclitaxel activity in breast cancer cells. Breast Cancer Res. (2010) 12:R41. doi: 10.1186/bcr2595
163. Shanmugam MK, Rane G, Kanchi MM, Arfuso F, Chinnathambi A, Zayed ME, et al. The multifaceted role of curcumin in cancer prevention and treatment. Molecules. (2015) 20:2728–69. doi: 10.3390/molecules20022728
164. Chen WC, Lai YA, Lin YC, Ma JW, Huang LF, Yang NS, et al. Curcumin suppresses doxorubicin-induced epithelial-mesenchymal transition via the inhibition of TGF-β and PI3K/AKT signaling pathways in triple-negative breast cancer cells. J Agric Food Chem. (2013) 61:11817–24. doi: 10.1021/jf404092f
165. Ehata S, Hanyu A, Fujime M, Katsuno Y, Fukunaga E, Goto K, et al. Ki26894, a novel transforming growth factor-beta type I receptor kinase inhibitor, inhibits in vitro invasion and in vivo bone metastasis of a human breast cancer cell line. Cancer Sci. (2007) 98:127–33. doi: 10.1111/j.1349-7006.2006.00357.x
166. Bouquet F, Pal A, Pilones KA, Demaria S, Hann B, Akhurst RJ, et al. TGFβ1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin Cancer Res. (2011) 17:6754–65. doi: 10.1158/1078-0432.CCR-11-0544
167. Park CY, Min KN, Son JY, Park SY, Nam JS, Kim DK, et al. An novel inhibitor of TGF-β type I receptor, IN-1130, blocks breast cancer lung metastasis through inhibition of epithelial-mesenchymal transition. Cancer Lett. (2014) 351:72–80. doi: 10.1016/j.canlet.2014.05.006
168. Rausch MP, Hahn T, Ramanathapuram L, Bradley-Dunlop D, Mahadevan D, Mercado-Pimentel ME, et al. An orally active small molecule TGF-beta receptor I antagonist inhibits the growth of metastatic murine breast cancer. Anticancer Res. (2009) 29:2099–109.
169. Wiercinska E, Naber HP, Pardali E, van der Pluijm G, van Dam H, and ten Dijke P. The TGF-β/Smad pathway induces breast cancer cell invasion through the up-regulation of matrix metalloproteinase 2 and 9 in a spheroid invasion model system. Breast Cancer Res Treat. (2011) 128:657–66. doi: 10.1007/s10549-010-1147-x
170. Fang Y, Chen Y, Yu L, Zheng C, Qi Y, Li Z, et al. Inhibition of breast cancer metastases by a novel inhibitor of TGFβ receptor 1. J Natl Cancer Inst. (2013) 105:47–58. doi: 10.1093/jnci/djs485
171. Sarkar S, Kandasamy T, and Ghosh SS. Imatinib impedes EMT and notch signalling by inhibiting p300 acetyltransferase in breast cancer cells. Mol Carcinog. (2025) 64:344–56. doi: 10.1002/mc.23848
172. Feng M, Santhanam RK, Xing H, Zhou M, and Jia H. Inhibition of γ-secretase/Notch pathway as a potential therapy for reversing cancer drug resistance. Biochem Pharmacol. (2024) 220:115991. doi: 10.1016/j.bcp.2023.115991
173. Olsauskas-Kuprys R, Zlobin A, and Osipo C. Gamma secretase inhibitors of Notch signaling. Onco Targets Ther. (2013) 6:943–55. doi: 10.2147/OTT.S33766
174. Kim B, Stephen SL, Hanby AM, Horgan K, Perry SL, Richardson J, et al. Chemotherapy induces Notch1-dependent MRP1 up-regulation, inhibition of which sensitizes breast cancer cells to chemotherapy. BMC Can. (2015) 15:634. doi: 10.1186/s12885-015-1625-y
175. Lombardo Y, Faronato M, Filipovic A, Vircillo V, Magnani L, and Coombes RC. Nicastrin and Notch4 drive endocrine therapy resistance and epithelial to mesenchymal transition in MCF7 breast cancer cells. Breast Cancer Res. (2014) 16:R62. doi: 10.1186/bcr3675
176. Zhang CC, Yan Z, Zong Q, Fang DD, Painter C, Zhang Q, et al. Synergistic effect of the γ-secretase inhibitor PF-03084014 and docetaxel in breast cancer models. Stem Cells Transl Med. (2013) 2:233–42. doi: 10.5966/sctm.2012-0096
177. Schott AF, Landis MD, Dontu G, Griffith KA, Layman RM, Krop I, et al. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin Cancer Res. (2013) 19:1512–24. doi: 10.1158/1078-0432.CCR-11-3326
178. Means-Powell JA, Mayer IA, Ismail-Khan R, Del Valle L, Tonetti D, Abramson VG, et al. A phase ib dose escalation trial of RO4929097 (a γ-secretase inhibitor) in combination with exemestane in patients with ER + Metastatic breast cancer (MBC). Clin Breast Can. (2022) 22:103–14. doi: 10.1016/j.clbc.2021.10.013
179. Sardesai S, Badawi M, Mrozek E, Morgan E, Phelps M, Stephens J, et al. A phase I study of an oral selective gamma secretase (GS) inhibitor RO4929097 in combination with neoadjuvant paclitaxel and carboplatin in triple negative breast cancer. Invest New Drugs. (2020) 38:1400–10. doi: 10.1007/s10637-020-00895-5
180. Simões BM, O’Brien CS, Eyre R, Silva A, Yu L, Sarmiento-Castro A, et al. Anti-estrogen resistance in human breast tumors is driven by JAG1-NOTCH4-dependent cancer stem cell activity. Cell Rep. (2015) 12:1968–77. doi: 10.1016/j.celrep.2015.08.050
181. Massard C, Cassier PA, Azaro A, Anderson B, Yuen E, Yu D, et al. A phase 1b study of crenigacestat (LY3039478) in combination with gemcitabine and cisplatin or gemcitabine and carboplatin in patients with advanced or metastatic solid tumors. Cancer Chemother Pharmacol. (2022) 90:335–44. doi: 10.1007/s00280-022-04461-z
182. Won HS, Lee KM, Oh JE, Nam EM, and Lee KE. Inhibition of β-catenin to overcome endocrine resistance in tamoxifen-resistant breast cancer cell line. PloS One. (2016) 11:e0155983. doi: 10.1371/journal.pone.0155983
183. Loh YN, Hedditch EL, Baker LA, Jary E, Ward RL, and Ford CE. The Wnt signalling pathway is upregulated in an in vitro model of acquired tamoxifen resistant breast cancer. BMC Can. (2013) 13:174. doi: 10.1186/1471-2407-13-174
184. Yuan J, Wang G, Beeraka NM, Zhang H, Wang Q, Zhang D, et al. Dual action of pyrimidine derivatives: Targeting tamoxifen resistance in breast cancer. Transl Oncol. (2025) 58:102418. doi: 10.1016/j.tranon.2025.102418
185. Shome R, Sen P, Sarkar S, and Ghosh SS. Single-cell transcriptomics reveals the intra-tumoral heterogeneity and SQSTM1/P62 and Wnt/β-catenin mediated epithelial to mesenchymal transition and stemness of triple-negative breast cancer. Exp Cell Res. (2024) 438:114032. doi: 10.1016/j.yexcr.2024.114032
186. Wu Y, Ginther C, Kim J, Mosher N, Chung S, Slamon D, et al. Expression of Wnt3 activates Wnt/β-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol Cancer Res. (2012) 10:1597–606. doi: 10.1158/1541-7786.MCR-12-0155-T
187. Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci U S A. (2013) 110:20224–9. doi: 10.1073/pnas.1314239110
188. Tzeng HE, Yang L, Chen K, Wang Y, Liu YR, Pan SL, et al. The pan-PI3K inhibitor GDC-0941 activates canonical WNT signaling to confer resistance in TNBC cells: resistance reversal with WNT inhibitor. Oncotarget. (2015) 6:11061–73. doi: 10.18632/oncotarget.3568
189. Abreu de Oliveira WA, Moens S, El Laithy Y, van der Veer BK, Athanasouli P, Cortesi EE, et al. Wnt/β-catenin inhibition disrupts carboplatin resistance in isogenic models of triple-negative breast cancer. Front Oncol. (2021) 11:705384. doi: 10.3389/fonc.2021.705384
190. Linke F, Harenberg M, Nietert MM, Zaunig S, von Bonin F, Arlt A, et al. Microenvironmental interactions between endothelial and lymphoma cells: a role for the canonical WNT pathway in Hodgkin lymphoma. Leukemia. (2017) 31:361–72. doi: 10.1038/leu.2016.232
191. Pode-Shakked N, Harari-Steinberg O, Haberman-Ziv Y, Rom-Gross E, Bahar S, Omer D, et al. Resistance or sensitivity of Wilms’ tumor to anti-FZD7 antibody highlights the Wnt pathway as a possible therapeutic target. Oncogene. (2011) 30:1664–80. doi: 10.1038/onc.2010.549
192. Yin P, Wang W, Gao J, Bai Y, Wang Z, Na L, et al. Fzd2 contributes to breast cancer cell mesenchymal-like stemness and drug resistance. Oncol Res. (2020) 28:273–84. doi: 10.3727/096504020X15783052025051
193. Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci U S A. (2012) 109:2784–9. doi: 10.1073/pnas.1018866109
194. Gurney A, Axelrod F, Bond CJ, Cain J, Chartier C, Donigan L, et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci U S A. (2012) 109:11717–22. doi: 10.1073/pnas.1120068109
195. Xie W, Zhao H, Wang F, Wang Y, He Y, Wang T, et al. A novel humanized Frizzled-7-targeting antibody enhances antitumor effects of Bevacizumab against triple-negative breast cancer via blocking Wnt/β-catenin signaling pathway. J Exp Clin Cancer Res. (2021) 40:30. doi: 10.1186/s13046-020-01800-x
196. Lu W and Li Y. Salinomycin suppresses LRP6 expression and inhibits both Wnt/β-catenin and mTORC1 signaling in breast and prostate cancer cells. J Cell Biochem. (2014) 115:1799–807. doi: 10.1002/jcb.24850
197. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. (2009) 138:645–59. doi: 10.1016/j.cell.2009.06.034
198. Cui Y, Zhao M, Yang Y, Xu R, Tong L, Liang J, et al. Reversal of epithelial-mesenchymal transition and inhibition of tumor stemness of breast cancer cells through advanced combined chemotherapy. Acta Biomater. (2022) 152:380–92. doi: 10.1016/j.actbio.2022.08.024
199. Zhan T, Rindtorff N, and Boutros M. Wnt signaling in cancer. Oncogene. (2017) 36:1461–73. doi: 10.1038/onc.2016.304
200. Raut D, Vora A, and Bhatt LK. The Wnt/β-catenin pathway in breast cancer therapy: a pre-clinical perspective of its targeting for clinical translation. Expert Rev Anticancer Ther. (2022) 22:97–114. doi: 10.1080/14737140.2022.2016398
201. Dakal TC, Bhushan R, Xu C, Gadi BR, Cameotra SS, Yadav V, et al. Intricate relationship between cancer stemness, metastasis, and drug resistance. MedComm (2020). (2024) 5:e710. doi: 10.1002/mco2.710
202. Ruiz-Martínez S, Ribas X, Costas M, Landberg G, and Puig T. Characterization and targeting of chemoresistant triple-negative breast cancer subtypes using amino-pyridine compounds. Biochim Biophys Acta Mol Basis Dis. (2025) 1871:167899. doi: 10.1016/j.bbadis.2025.167899
203. Boz Er AB and Er I. Targeting ITGβ3 to overcome trastuzumab resistance through epithelial-mesenchymal transition regulation in HER2-positive breast cancer. Int J Mol Sci. (2024) 25:8640. doi: 10.3390/ijms25168640
204. Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. (2000) 406:1005–9. doi: 10.1038/35023008
205. Sun M, Zhang N, Wang X, Cai C, Cun J, Li Y, et al. Nitidine chloride induces apoptosis, cell cycle arrest, and synergistic cytotoxicity with doxorubicin in breast cancer cells. Tumour Biol. (2014) 35:10201–12. doi: 10.1007/s13277-014-2327-9
206. Sun M, Zhang N, Wang X, Li Y, Qi W, Zhang H, et al. Hedgehog pathway is involved in nitidine chloride induced inhibition of epithelial-mesenchymal transition and cancer stem cells-like properties in breast cancer cells. Cell Biosci. (2016) 6:44. doi: 10.1186/s13578-016-0104-8
207. Koike Y, Ohta Y, Saitoh W, Yamashita T, Kanomata N, Moriya T, et al. Anti-cell growth and anti-cancer stem cell activities of the non-canonical hedgehog inhibitor GANT61 in triple-negative breast cancer cells. Breast Can. (2017) 24:683–93. doi: 10.1007/s12282-017-0757-0
208. Neelakantan D, Zhou H, Oliphant MUJ, Zhang X, Simon LM, Henke DM, et al. EMT cells increase breast cancer metastasis via paracrine GLI activation in neighbouring tumour cells. Nat Commun. (2017) 8:15773. doi: 10.1038/ncomms15773
209. Riaz SK, Ke Y, Wang F, Kayani MA, and Malik MFA. Influence of SHH/GLI1 axis on EMT mediated migration and invasion of breast cancer cells. Sci Rep. (2019) 9:6620. doi: 10.1038/s41598-019-43093-x
210. Jimeno A, Weiss GJ, Miller WH Jr., Gettinger S, Eigl BJ, Chang AL, et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin Cancer Res. (2013) 19:2766–74. doi: 10.1158/1078-0432.CCR-12-3654
211. Ruiz-Borrego M, Jimenez B, Antolín S, García-Saenz JA, Corral J, Jerez Y, et al. A phase Ib study of sonidegib (LDE225), an oral small molecule inhibitor of smoothened or Hedgehog pathway, in combination with docetaxel in triple negative advanced breast cancer patients: GEICAM/2012-12 (EDALINE) study. Invest New Drugs. (2019) 37:98–108. doi: 10.1007/s10637-018-0614-9
212. Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan CL, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. (2018) 9:2897. doi: 10.1038/s41467-018-05220-6
213. Kim DA, Choi HS, Ryu ES, Ko J, Shin HS, Lee JM, et al. Tannic acid attenuates the formation of cancer stem cells by inhibiting NF-κB-mediated phenotype transition of breast cancer cells. Am J Cancer Res. (2019) 9:1664–81.
214. Cheng X, Li D, Sun M, He L, Zheng Y, Wang X, et al. Co-delivery of DOX and PDTC by pH-sensitive nanoparticles to overcome multidrug resistance in breast cancer. Colloids Surf B Biointerface. (2019) 181:185–97. doi: 10.1016/j.colsurfb.2019.05.042
215. Tu SH, Chiou YS, Kalyanam N, Ho CT, Chen LC, and Pan MH. Garcinol sensitizes breast cancer cells to Taxol through the suppression of caspase-3/iPLA(2) and NF-κB/Twist1 signaling pathways in a mouse 4T1 breast tumor model. Food Funct. (2017) 8:1067–79. doi: 10.1039/C6FO01588C
216. Verret B, Cortes J, Bachelot T, Andre F, and Arnedos M. Efficacy of PI3K inhibitors in advanced breast cancer. Ann Oncol. (2019) 30:x12–20. doi: 10.1093/annonc/mdz381
217. Yu L, Zang C, Ye Y, Liu H, and Eucker J. Effects of BYL-719 (alpelisib) on human breast cancer stem cells to overcome drug resistance in human breast cancer. Front Pharmacol. (2024) 15:1443422. doi: 10.3389/fphar.2024.1443422
218. Wu DP, Zhou Y, Hou LX, Zhu XX, Yi W, Yang SM, et al. Cx43 deficiency confers EMT-mediated tamoxifen resistance to breast cancer via c-Src/PI3K/Akt pathway. Int J Biol Sci. (2021) 17:2380–98. doi: 10.7150/ijbs.55453
219. Luo J, Yao JF, Deng XF, Zheng XD, Jia M, Wang YQ, et al. 14, 15-EET induces breast cancer cell EMT and cisplatin resistance by up-regulating integrin αvβ3 and activating FAK/PI3K/AKT signaling. J Exp Clin Cancer Res. (2018) 37:23. doi: 10.1186/s13046-018-0694-6
220. Zhang C, Xu S, Yin C, Hu S, and Liu P. The role of the mTOR pathway in breast cancer stem cells (BCSCs): mechanisms and therapeutic potentials. Stem Cell Res Ther. (2025) 16:156. doi: 10.1186/s13287-025-04218-4
221. Chen C, Chen Z, Zhao J, Wen X, Yao H, Weng Z, et al. TMEM45A enhances palbociclib resistance and cellular glycolysis by activating AKT/mTOR signaling pathway in HR+ breast cancer. Cell Death Discov. (2025) 11:47. doi: 10.1038/s41420-025-02336-9
222. Katsuno Y, Meyer DS, Zhang Z, Shokat KM, Akhurst RJ, Miyazono K, et al. Chronic TGF-β exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci Signal. (2019) 12:eaau8544. doi: 10.1126/scisignal.aau8544
223. Stevens LE, Peluffo G, Qiu X, Temko D, Fassl A, Li Z, et al. JAK-STAT signaling in inflammatory breast cancer enables chemotherapy-resistant cell states. Cancer Res. (2023) 83:264–84. doi: 10.1158/0008-5472.CAN-22-0423
224. Al-Mutairi MS and Habashy HO. DUSP4 silencing enhances the sensitivity of breast cancer cells to doxorubicin through the activation of the JNK/c-jun signalling pathway. Molecules. (2022) 27:6146. doi: 10.3390/molecules27196146
225. Mukherjee S, Mazumdar M, Chakraborty S, Manna A, Saha S, Khan P, et al. Curcumin inhibits breast cancer stem cell migration by amplifying the E-cadherin/β-catenin negative feedback loop. Stem Cell Res Ther. (2014) 5:116. doi: 10.1186/scrt506
226. Sarkar S and Ghosh SS. Synergistic effect of salinomycin with budesonide on TNBC regression via EMT reversal and autophagy induction. J Biochem Mol Toxicol. (2024) 38:e70045. doi: 10.1002/jbt.70045
227. To NB, Truong VN, Ediriweera MK, and Cho SK. Effects of combined pentadecanoic acid and tamoxifen treatment on tamoxifen resistance in MCF-7/SC breast cancer cells. Int J Mol Sci. (2022) 23:11340. doi: 10.3390/ijms231911340
228. Li W, Liu C, Tang Y, Li H, Zhou F, and Lv S. Overexpression of Snail accelerates adriamycin induction of multidrug resistance in breast cancer cells. Asian Pac J Cancer Prev. (2011) 12:2575–80. doi: 10.1007/978-3-642-16602-0_13
229. Fan C, Wang Q, Krijger PHL, Cats D, Selle M, Khorosjutina O, et al. Identification of a SNAI1 enhancer RNA that drives cancer cell plasticity. Nat Commun. (2025) 16:2890. doi: 10.1038/s41467-025-58032-w
230. Chen WJ, Wang H, Tang Y, Liu CL, Li HL, and Li WT. Multidrug resistance in breast cancer cells during epithelial-mesenchymal transition is modulated by breast cancer resistant protein. Chin J Can. (2010) 29:151–7. doi: 10.5732/cjc.009.10447
231. Li M, Zhang L, Guan T, Huang L, Zhu Y, Wen Y, et al. Energy stress-activated AMPK phosphorylates Snail1 and suppresses its stability and oncogenic function. Cancer Lett. (2024) 595:216987. doi: 10.1016/j.canlet.2024.216987
232. Baron R, Binda C, Tortorici M, McCammon JA, and Mattevi A. Molecular mimicry and ligand recognition in binding and catalysis by the histone demethylase LSD1-CoREST complex. Structure. (2011) 19:212–20. doi: 10.1016/j.str.2011.01.001
233. Verigos J, Karakaidos P, Kordias D, Papoudou-Bai A, Evangelou Z, Harissis HV, et al. The histone demethylase LSD1/κDM1A mediates chemoresistance in breast cancer via regulation of a stem cell program. Cancers (Basel). (2019) 11:1585. doi: 10.3390/cancers11101585
234. Cordani N, Mologni L, Piazza R, Tettamanti P, Cogliati V, Mauri M, et al. TWIST1 upregulation is a potential target for reversing resistance to the CDK4/6 inhibitor in metastatic luminal breast cancer cells. Int J Mol Sci. (2023) 24:16294. doi: 10.3390/ijms242216294
235. Guan T, Li M, Song Y, Chen J, Tang J, Zhang C, et al. Phosphorylation of USP29 by CDK1 governs TWIST1 stability and oncogenic functions. Adv Sci (Weinh). (2023) 10:e2205873. doi: 10.1002/advs.202205873
236. Doodmani SM, Safari MH, Akbari M, Farahani N, Alimohammadi M, Aref AR, et al. Metastasis and chemoresistance in breast cancer: Crucial function of ZEB1/2 proteins. Pathol Res Pract. (2025) 267:155838. doi: 10.1016/j.prp.2025.155838
237. Kai M, Kanaya N, Wu SV, Mendez C, Nguyen D, Luu T, et al. Targeting breast cancer stem cells in triple-negative breast cancer using a combination of LBH589 and salinomycin. Breast Cancer Res Treat. (2015) 151:281–94. doi: 10.1007/s10549-015-3376-5
238. Rhodes LV, Tate CR, Segar HC, Burks HE, Phamduy TB, Hoang V, et al. Suppression of triple-negative breast cancer metastasis by pan-DAC inhibitor panobinostat via inhibition of ZEB family of EMT master regulators. Breast Cancer Res Treat. (2014) 145:593–604. doi: 10.1007/s10549-014-2979-6
239. Bagheri M, Mohamed GA, Mohamed Saleem MA, Ognjenovic NB, Lu H, Kolling FW, et al. Pharmacological induction of chromatin remodeling drives chemosensitization in triple-negative breast cancer. Cell Rep Med. (2024) 5:101504. doi: 10.1016/j.xcrm.2024.101504
240. Katuwal NB, Kang MS, Ghosh M, Hong SD, Jeong YG, Park SM, et al. Targeting PEG10 as a novel therapeutic approach to overcome CDK4/6 inhibitor resistance in breast cancer. J Exp Clin Cancer Res. (2023) 42:325. doi: 10.1186/s13046-023-02903-x
241. Jiang Y, Zhao X, Xiao Q, Liu Q, Ding K, Yu F, et al. Snail and Slug mediate tamoxifen resistance in breast cancer cells through activation of EGFR-ERK independent of epithelial-mesenchymal transition. J Mol Cell Biol. (2014) 6:352–4. doi: 10.1093/jmcb/mju019
242. Hu MX, Li J, Fu YW, Xu ES, Li D, Huang SQ, et al. Establishment and characterization of cisplatin-resistant cell lines from canine mammary gland tumors. Theriogenology. (2024) 217:103–12. doi: 10.1016/j.theriogenology.2024.01.017
243. Pan G, Liu Y, Shang L, Zhou F, and Yang S. EMT-associated microRNAs and their roles in cancer stemness and drug resistance. Cancer Commun (Lond). (2021) 41:199–217. doi: 10.1002/cac2.12138
244. Takahashi RU, Miyazaki H, Takeshita F, Yamamoto Y, Minoura K, Ono M, et al. Loss of microRNA-27b contributes to breast cancer stem cell generation by activating ENPP1. Nat Commun. (2015) 6:7318. doi: 10.1038/ncomms8318
245. Li X, Wu Y, Liu A, and Tang X. MiR-27b is epigenetically downregulated in tamoxifen resistant breast cancer cells due to promoter methylation and regulates tamoxifen sensitivity by targeting HMGB3. Biochem Biophys Res Commun. (2016) 477:768–73. doi: 10.1016/j.bbrc.2016.06.133
246. Bockhorn J, Dalton R, Nwachukwu C, Huang S, Prat A, Yee K, et al. MicroRNA-30c inhibits human breast tumour chemotherapy resistance by regulating TWF1 and IL-11. Nat Commun. (2013) 4:1393. doi: 10.1038/ncomms2393
247. Weng YS, Tseng HY, Chen YA, Shen PC, Al Haq AT, Chen LM, et al. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol Can. (2019) 18:42. doi: 10.1186/s12943-019-0988-0
248. Yang X, Shang P, Yu B, Jin Q, Liao J, Wang L, et al. Combination therapy with miR34a and doxorubicin synergistically inhibits Dox-resistant breast cancer progression via down-regulation of Snail through suppressing Notch/NF-κB and RAS/RAF/MEK/ERK signaling pathway. Acta Pharm Sin B. (2021) 11:2819–34. doi: 10.1016/j.apsb.2021.06.003
249. Yang Q, Wang Y, Lu X, Zhao Z, Zhu L, Chen S, et al. MiR-125b regulates epithelial-mesenchymal transition via targeting Sema4C in paclitaxel-resistant breast cancer cells. Oncotarget. (2015) 6:3268–79. doi: 10.18632/oncotarget.3065
250. Yu Y, Zhao Y, Sun XH, Ge J, Zhang B, Wang X, et al. Down-regulation of miR-129-5p via the Twist1-Snail feedback loop stimulates the epithelial-mesenchymal transition and is associated with poor prognosis in breast cancer. Oncotarget. (2015) 6:34423–36. doi: 10.18632/oncotarget.5406
251. Yao N, Fu Y, Chen L, Liu Z, He J, Zhu Y, et al. Long non-coding RNA NONHSAT101069 promotes epirubicin resistance, migration, and invasion of breast cancer cells through NONHSAT101069/miR-129-5p/Twist1 axis. Oncogene. (2019) 38:7216–33. doi: 10.1038/s41388-019-0904-5
252. Tian D, Tian M, Ma ZM, Zhang LL, Cui YF, and Li JL. Anesthetic propofol epigenetically regulates breast cancer trastuzumab resistance through IL-6/miR-149-5p axis. Sci Rep. (2020) 10:8858. doi: 10.1038/s41598-020-65649-y
253. Chen J, Shin VY, Siu MT, Ho JC, Cheuk I, and Kwong A. miR-199a-5p confers tumor-suppressive role in triple-negative breast cancer. BMC Can. (2016) 16:887. doi: 10.1186/s12885-016-2916-7
254. Haghnavaz N, Asghari F, Shekari N, Shanehbandi D, Javadian M, Mohammadi A, et al. Paclitaxel may inhibit epithelial-mesenchymal transition properties of triple-negative breast cancer cell line via altering the expression of EMT-promoting and –inhibiting microRNAs. Midd East J Can. (2020) 11:42–9. doi: 10.30476/MEJC.2019.78716.0
255. Korpal M, Lee ES, Hu G, and Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. (2008) 283:14910–4. doi: 10.1074/jbc.C800074200
256. Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. (2009) 11:1487–95. doi: 10.1038/ncb1998
257. Li L, Li B, Chen D, Liu L, Huang C, Lu Z, et al. miR-139 and miR-200c regulate pancreatic cancer endothelial cell migration and angiogenesis. Oncol Rep. (2015) 34:51–8. doi: 10.3892/or.2015.3945
258. Park SM, Gaur AB, Lengyel E, and Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. (2008) 22:894–907. doi: 10.1101/gad.1640608
259. Wang Q, Cheng Y, Wang Y, Fan Y, Li C, Zhang Y, et al. Tamoxifen reverses epithelial–mesenchymal transition by demethylating miR-200c in triple-negative breast cancer cells. BMC Can. (2017) 17:492. doi: 10.1186/s12885-017-3457-4
260. Tang H, Song C, Ye F, Gao G, Ou X, Zhang L, et al. miR-200c suppresses stemness and increases cellular sensitivity to trastuzumab in HER2+ breast cancer. J Cell Mol Med. (2019) 23:8114–27. doi: 10.1111/jcmm.14681
261. Bai WD, Ye XM, Zhang MY, Zhu HY, Xi WJ, Huang X, et al. MiR-200c suppresses TGF-β signaling and counteracts trastuzumab resistance and metastasis by targeting ZNF217 and ZEB1 in breast cancer. Int J Can. (2014) 135:1356–68. doi: 10.1002/ijc.28782
262. Tryndyak VP, Beland FA, and Pogribny IP. E-cadherin transcriptional down-regulation by epigenetic and microRNA-200 family alterations is related to mesenchymal and drug-resistant phenotypes in human breast cancer cells. Int J Can. (2010) 126:2575–83. doi: 10.1002/ijc.24972
263. Gao Y, Zhang W, Liu C, and Li G. miR-200 affects tamoxifen resistance in breast cancer cells through regulation of MYB. Sci Rep. (2019) 9:18844. doi: 10.1038/s41598-019-54289-6
264. Yang X, Hu Q, Hu LX, Lin XR, Liu JQ, Lin X, et al. miR-200b regulates epithelial-mesenchymal transition of chemo-resistant breast cancer cells by targeting FN1. Discov Med. (2017) 24:75–85.
265. Chao CH, Chang CC, Wu MJ, Ko HW, Wang D, Hung MC, et al. MicroRNA-205 signaling regulates mammary stem cell fate and tumorigenesis. J Clin Invest. (2014) 124:3093–106. doi: 10.1172/JCI73351
266. Lin LF, Li YT, Han H, and Lin SG. MicroRNA-205-5p targets the HOXD9-Snail1 axis to inhibit triple negative breast cancer cell proliferation and chemoresistance. Aging (Albany NY). (2021) 13:3945–56. doi: 10.18632/aging.202363
267. Lee JY, Park MK, Park JH, Lee HJ, Shin DH, Kang Y, et al. Loss of the polycomb protein Mel-18 enhances the epithelial-mesenchymal transition by ZEB1 and ZEB2 expression through the downregulation of miR-205 in breast cancer. Oncogene. (2014) 33:1325–35. doi: 10.1038/onc.2013.53
268. Shi S, Chen X, Liu H, Yu K, Bao Y, Chai J, et al. LGR5 acts as a target of miR-340-5p in the suppression of cell progression and drug resistance in breast cancer via Wnt/β-catenin pathway. Gene. (2019) 683:47–53. doi: 10.1016/j.gene.2018.10.014
269. Fu H, Fu L, Xie C, Zuo WS, Liu YS, Zheng MZ, et al. miR-375 inhibits cancer stem cell phenotype and tamoxifen resistance by degrading HOXB3 in human ER-positive breast cancer. Oncol Rep. (2017) 37:1093–9. doi: 10.3892/or.2017.5360
270. Ward A, Balwierz A, Zhang JD, Küblbeck M, Pawitan Y, Hielscher T, et al. Re-expression of microRNA-375 reverses both tamoxifen resistance and accompanying EMT-like properties in breast cancer. Oncogene. (2013) 32:1173–82. doi: 10.1038/onc.2012.128
271. Jafarzadeh A, Noori M, Sarrafzadeh S, Tamehri Zadeh SS, Nemati M, Chatrabnous N, et al. MicroRNA-383: A tumor suppressor miRNA in human cancer. Front Cell Dev Biol. (2022) 10:955486. doi: 10.3389/fcell.2022.955486
272. Zhao L, Gu H, Chang J, Wu J, Wang D, Chen S, et al. MicroRNA-383 regulates the apoptosis of tumor cells through targeting Gadd45g. PloS One. (2014) 9:e110472. doi: 10.1371/journal.pone.0110472
273. Kim M, Jang K, Miller P, Picon-Ruiz M, Yeasky TM, El-Ashry D, et al. VEGFA links self-renewal and metastasis by inducing Sox2 to repress miR-452, driving Slug. Oncogene. (2017) 36:5199–211. doi: 10.1038/onc.2017.4
274. Jiang L, He D, Yang D, Chen Z, Pan Q, Mao A, et al. MiR-489 regulates chemoresistance in breast cancer via epithelial mesenchymal transition pathway. FEBS Lett. (2014) 588:2009–15. doi: 10.1016/j.febslet.2014.04.024
275. Cheng CW, Liu YF, Liao WL, Chen PM, Hung YT, Lee HJ, et al. miR-622 Increases miR-30a Expression through Inhibition of Hypoxia-Inducible Factor 1α to Improve Metastasis and Chemoresistance in Human Invasive Breast Cancer Cells. Cancers (Basel). (2024) 16:657. doi: 10.3390/cancers16030657
276. Raza U, Saatci Ö, Uhlmann S, Ansari SA, Eyüpoğlu E, Yurdusev E, et al. The miR-644a/CTBP1/p53 axis suppresses drug resistance by simultaneous inhibition of cell survival and epithelial-mesenchymal transition in breast cancer. Oncotarget. (2016) 7:49859–77. doi: 10.18632/oncotarget.10489
277. Tan X, Li Z, Ren S, Rezaei K, Pan Q, Goldstein AT, et al. Dynamically decreased miR-671-5p expression is associated with oncogenic transformation and radiochemoresistance in breast cancer. Breast Cancer Res. (2019) 21:89. doi: 10.1186/s13058-019-1173-5
278. Lee JW, Guan W, Han S, Hong DK, Kim LS, and Kim H. MicroRNA-708-3p mediates metastasis and chemoresistance through inhibition of epithelial-to-mesenchymal transition in breast cancer. Cancer Sci. (2018) 109:1404–13. doi: 10.1111/cas.13588
279. Wang G, Dong Y, Liu H, Ji N, Cao J, Liu A, et al. Loss of miR−873 contributes to gemcitabine resistance in triple−negative breast cancer via targeting ZEB1. Oncol Lett. (2019) 18:3837–44. doi: 10.3892/ol.2019.10697
280. Kuthethur R, Adiga D, Kandettu A, Jerome MS, Mallya S, Mumbrekar KD, et al. MiR-4521 perturbs FOXM1-mediated DNA damage response in breast cancer. Front Mol Biosci. (2023) 10:1131433. doi: 10.3389/fmolb.2023.1131433
281. Liu G, Wang P, and Zhang H. MiR-6838-5p suppresses cell metastasis and the EMT process in triple-negative breast cancer by targeting WNT3A to inhibit the Wnt pathway. J Gene Med. (2019) 21:e3129. doi: 10.1002/jgm.3129
282. Klicka K, Grzywa TM, Mielniczuk A, Klinke A, and Włodarski PK. The role of miR-200 family in the regulation of hallmarks of cancer. Front Oncol. (2022) 12:965231. doi: 10.3389/fonc.2022.965231
283. Howe EN, Cochrane DR, and Richer JK. The miR-200 and miR-221/222 microRNA families: opposing effects on epithelial identity. J Mammar Gland Biol Neoplasia. (2012) 17:65–77. doi: 10.1007/s10911-012-9244-6
284. Zhou Q, Yang M, Lan H, and Yu X. miR-30a negatively regulates TGF-β1-induced epithelial-mesenchymal transition and peritoneal fibrosis by targeting Snai1. Am J Pathol. (2013) 183:808–19. doi: 10.1016/j.ajpath.2013.05.019
285. di Gennaro A, Damiano V, Brisotto G, Armellin M, Perin T, Zucchetto A, et al. A p53/miR-30a/ZEB2 axis controls triple negative breast cancer aggressiveness. Cell Death Differ. (2018) 25:2165–80. doi: 10.1038/s41418-018-0103-x
286. Zhang N, Wang X, Huo Q, Sun M, Cai C, Liu Z, et al. MicroRNA-30a suppresses breast tumor growth and metastasis by targeting metadherin. Oncogene. (2014) 33:3119–28. doi: 10.1038/onc.2013.286
287. Chang CW, Yu JC, Hsieh YH, Yao CC, Chao JI, Chen PM, et al. MicroRNA-30a increases tight junction protein expression to suppress the epithelial-mesenchymal transition and metastasis by targeting Slug in breast cancer. Oncotarget. (2016) 7:16462–78. doi: 10.18632/oncotarget.7656
288. Liu Z, Tu K, and Liu Q. Effects of microRNA-30a on migration, invasion and prognosis of hepatocellular carcinoma. FEBS Lett. (2014) 588:3089–97. doi: 10.1016/j.febslet.2014.06.037
289. Imani S, Wu RC, and Fu J. MicroRNA-34 family in breast cancer: from research to therapeutic potential. J Can. (2018) 9:3765–75. doi: 10.7150/jca.25576
290. Naghizadeh S, Mohammadi A, Duijf PHG, Baradaran B, Safarzadeh E, Cho WC, et al. The role of miR-34 in cancer drug resistance. J Cell Physiol. (2020) 235:6424–40. doi: 10.1002/jcp.29640
291. Imani S, Wei C, Cheng J, Khan MA, Fu S, Yang L, et al. MicroRNA-34a targets epithelial to mesenchymal transition-inducing transcription factors (EMT-TFs) and inhibits breast cancer cell migration and invasion. Oncotarget. (2017) 8:21362–79. doi: 10.18632/oncotarget.15214
292. Xiao Y, Humphries B, Yang C, and Wang Z. MiR-205 dysregulations in breast cancer: the complexity and opportunities. Noncoding RNA. (2019) 5:53. doi: 10.3390/ncrna5040053
293. Hu Y, Qiu Y, Yagüe E, Ji W, Liu J, and Zhang J. miRNA-205 targets VEGFA and FGF2 and regulates resistance to chemotherapeutics in breast cancer. Cell Death Dis. (2016) 7:e2291. doi: 10.1038/cddis.2016.194
294. Cai Y, Yan X, Zhang G, Zhao W, and Jiao S. MicroRNA-205 increases the sensitivity of docetaxel in breast cancer. Oncol Lett. (2016) 11:1105–9. doi: 10.3892/ol.2015.4030
295. Zhang L, Liu L, Xu X, He X, Wang G, Fan C, et al. miR-205/RunX2 axis negatively regulates CD44(+)/CD24(-) breast cancer stem cell activity. Am J Cancer Res. (2020) 10:1871–87.
296. Berber U, Yilmaz I, Narli G, Haholu A, Kucukodaci Z, and Demirel D. miR-205 and miR-200c: predictive micro RNAs for lymph node metastasis in triple negative breast cancer. J Breast Can. (2014) 17:143–8. doi: 10.4048/jbc.2014.17.2.143
297. Ahmadi-Hadad A, de Queiroz PCC, Schettini F, and Giuliano M. Reawakening the master switches in triple-negative breast cancer: A strategic blueprint for confronting metastasis and chemoresistance via microRNA-200/205: A systematic review. Crit Rev Oncol Hematol. (2024) 204:104516. doi: 10.1016/j.critrevonc.2024.104516
298. Ghafouri-Fard S, Khanbabapour Sasi A, Abak A, Shoorei H, Khoshkar A, and Taheri M. Contribution of miRNAs in the pathogenesis of breast cancer. Front Oncol. (2021) 11:768949. doi: 10.3389/fonc.2021.768949
299. Hong L, Pan F, Jiang H, Zhang L, Liu Y, Cai C, et al. miR-125b inhibited epithelial-mesenchymal transition of triple-negative breast cancer by targeting MAP2K7. Onco Targets Ther. (2016) 9:2639–48. doi: 10.2147/OTT.S102713
300. Zhang Y and Huang S. Up-regulation of miR-125b reverses epithelial-mesenchymal transition in paclitaxel-resistant lung cancer cells. Biol Chem. (2015). doi: 10.1515/hsz-2015-0153
301. Wang HJ, Guo YQ, Tan G, Dong L, Cheng L, Li KJ, et al. miR-125b regulates side population in breast cancer and confers a chemoresistant phenotype. J Cell Biochem. (2013) 114:2248–57. doi: 10.1002/jcb.24574
302. Vilquin P, Donini CF, Villedieu M, Grisard E, Corbo L, Bachelot T, et al. MicroRNA-125b upregulation confers aromatase inhibitor resistance and is a novel marker of poor prognosis in breast cancer. Breast Cancer Res. (2015) 17:13. doi: 10.1186/s13058-015-0515-1
303. Connolly EC, Freimuth J, and Akhurst RJ. Complexities of TGF-β targeted cancer therapy. Int J Biol Sci. (2012) 8:964–78. doi: 10.7150/ijbs.4564
304. Connolly EC, Saunier EF, Quigley D, Luu MT, De Sapio A, Hann B, et al. Outgrowth of drug-resistant carcinomas expressing markers of tumor aggression after long-term TβRI/II kinase inhibition with LY2109761. Cancer Res. (2011) 71:2339–49. doi: 10.1158/0008-5472.CAN-10-2941
305. Anderton MJ, Mellor HR, Bell A, Sadler C, Pass M, Powell S, et al. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol Pathol. (2011) 39:916–24. doi: 10.1177/0192623311416259
306. Garber K. Companies waver in efforts to target transforming growth factor beta in cancer. J Natl Cancer Inst. (2009) 101:1664–7. doi: 10.1093/jnci/djp462
307. Simonson B and Das S. MicroRNA therapeutics: the next magic bullet? Mini Rev Med Chem. (2015) 15:467–74. doi: 10.2174/1389557515666150324123208
308. Sarkar S, Venkatesh D, Kandasamy T, and Ghosh SS. Epigenetic modulations in breast cancer: an emerging paradigm in therapeutic implications. Front Biosci (Landmark Ed). (2024) 29:287. doi: 10.31083/j.fbl2908287
309. Roger L, Jullien L, Gire V, and Roux P. Gain of oncogenic function of p53 mutants regulates E-cadherin expression uncoupled from cell invasion in colon cancer cells. J Cell Sci. (2010) 123(Pt 8):1295–305. doi: 10.1242/jcs.061002
Keywords: EMT, drug resistance, breast cancer, cancer stem cells, microRNA
Citation: Luo Y, Sheng R, Tan X and Gu J (2025) Role of EMT in drug resistance of breast cancer: molecular mechanisms and therapeutic strategies. Front. Oncol. 15:1680751. doi: 10.3389/fonc.2025.1680751
Received: 06 August 2025; Accepted: 09 October 2025;
Published: 23 October 2025.
Edited by:
Susmita Mondal, Presidency University, IndiaReviewed by:
Dr. Saptak Banerjee, Chittaranjan National Cancer Institute (CNCI), IndiaRittwika Bhattacharya, Netaji Subhas Chandra Bose Cancer Hospital, India
Copyright © 2025 Luo, Sheng, Tan and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Gu, Z3VqdW5qaWFuZ3N1QG91dGxvb2suY29t