 Francesco Cuccia1*
Francesco Cuccia1* Salvatore D’Alessandro1Marco Messina2
Salvatore D’Alessandro1Marco Messina2 Giovanni Tringali3Giuseppe Craparo4Livio Blasi2Francesco Azzarello5
Giovanni Tringali3Giuseppe Craparo4Livio Blasi2Francesco Azzarello5 Giuseppe Carruba6
Giuseppe Carruba6 Giuseppe Ferrera1
Giuseppe Ferrera1- 1Radiation Oncology, Azienda ospedaliera di Riferimento Nazionale e di Alta Specializzazione (ARNAS) Civico Hospital, Palermo, Italy
- 2Medical Oncology, ARNAS Civico Hospital, Palermo, Italy
- 3Neurosurgery Unit, ARNAS Civico Hospital, Palermo, Italy
- 4Neuroradiology Unit, ARNAS Civico Hospital, Palermo, Italy
- 5Medical Physics, ARNAS Civico Hospital, Palermo, Italy
- 6Division of Internationalization and Health Research (SIRS), ARNAS Civico Hospital, Palermo, Italy
Background: Chordoid glioma is a rare World Health Organization (WHO) Grade II brain tumor located near the third ventricle. Fewer than 100 cases have been reported in the literature, and surgery represents the main treatment option. Due to its typical location, complete surgical resection is uncommon, and the role of adjuvant radiotherapy remains controversial.
Methods: Based on a case report, we performed a literature review focused on the potential role of adjuvant radiotherapy for chordoid glioma. We summarized data on patient characteristics, surgical approach and extent of resection, radiotherapy technique and dose, and clinical outcomes.
Results: A total of 18 patients from 14 studies were identified. Stereotactic treatments were used more frequently than conventional external beam radiotherapy, with doses ranging from 11.5–18 Gy and 45–59.4 Gy, respectively, and a median local control time of 26 months. Five patients developed disease recurrence after a median of 22.4 months. In the present case, the patient received adjuvant conventional radiotherapy (59.4 Gy/33 fractions) and remains alive after 12 months of follow-up with no major side effects.
Conclusions: From the available evidence, modern radiotherapy may be considered a therapeutic tool that combines less invasive surgical procedures with improved local control, potentially reducing the risk of severe postoperative complications. Larger studies with longer follow-up periods are needed.
1 Introduction
In 1998, Brat et al. first described chordoid glioma (CG) as a brain tumor typically located near the third ventricle, characterized histologically by glial fibrillary acidic protein (GFAP)-positive tumor cells surrounded by a myxoid matrix (1, 2).
The most recent World Health Organization (WHO, 2021) classification of central nervous system (CNS) tumors lists CG as an astrocytic Grade II tumor due to its relatively slow growth pattern (3). Nonetheless, its peculiar location is closely associated with the sudden onset of neurological symptoms, including hydrocephalus (due to cerebrospinal fluid obstruction), headache, behavioral disorders, visual impairment, ataxia, and memory issues (4).
From a radiological perspective, CG usually appears on MRI as a well-defined, midline, suprasellar, contrast-enhancing mass, with infrequent invasion of the surrounding brain tissue (5). Maximal safe resection remains the mainstay of treatment, although the critical location often precludes complete resection, resulting in a higher risk of disease relapse or the need for adjuvant treatment. Moreover, the postoperative course is frequently complicated by endocrine and metabolic sequelae, such as hypothalamic dysfunction, diabetes, and other endocrinological disorders (6–8).
Fewer than 100 cases have been reported in the literature, with variable outcomes and limited understanding of the disease behavior in terms of recurrence patterns, adjuvant treatment, and prognosis. Due to the rarity of the disease, there is currently no standardized treatment approach. In particular, no consensus has been established regarding the optimal dose and fractionation schedule for radiotherapy.
In this article, we describe the clinical course of a young adult woman with a third ventricle CG treated with surgery followed by adjuvant radiotherapy and discuss this case in the context of published literature, with a specific focus on the potential role of adjuvant radiotherapy.
2 Methods
A PubMed and Embase literature search was conducted in December 2024 using the following MeSH terms: “chordoid” AND “glioma” AND “radiotherapy,” covering publications from 2000 to 2024. Additional hand searching of reference lists from the retrieved articles was also performed.
For this review, all articles published in English were included. Reviews or papers focusing exclusively on the histopathological features of the tumor, without reporting clinical outcomes, were excluded. Case reports and case series were appraised using the JBI Critical Appraisal Checklist (28).
Two independent reviewers (FC and SDA) performed the data collection, and a third reviewer (MM) was involved in cases of disagreement. Data were extracted on baseline patient characteristics (age, sex, presenting symptoms, and tumor size), surgical approach and extent of resection, radiotherapy technique and dose (where available), and clinical outcomes. Because of the rarity of the disease, studies without radiotherapy details were also included.
3 Results
3.1 Case description
A 42-year-old woman was admitted to our hospital due to the onset of severe headache, ataxia, visual impairment, and speech disorders.
Her medical history was positive for smoking, overweight, Raynaud syndrome, hypertension, and a previous cervical conization for a high-grade squamous intraepithelial lesion.
A contrast-enhanced brain computed tomography (CT) scan revealed an ovoid solid mass measuring 2.9 × 2.6 cm. These findings were confirmed on contrast-enhanced brain MRI, which showed a homogeneously enhancing, polylobulated mass of 2.7 × 2.4 cm in the hypothalamic–chiasmatic region, causing compression of the left optic nerve, which appeared enlarged in the prechiasmatic portion. Moreover, bilateral involvement of the optic tracts was observed, along with anterior obstruction of the third ventricle and bilateral Monro foramina, although without hydrocephalus (Figure 1A).
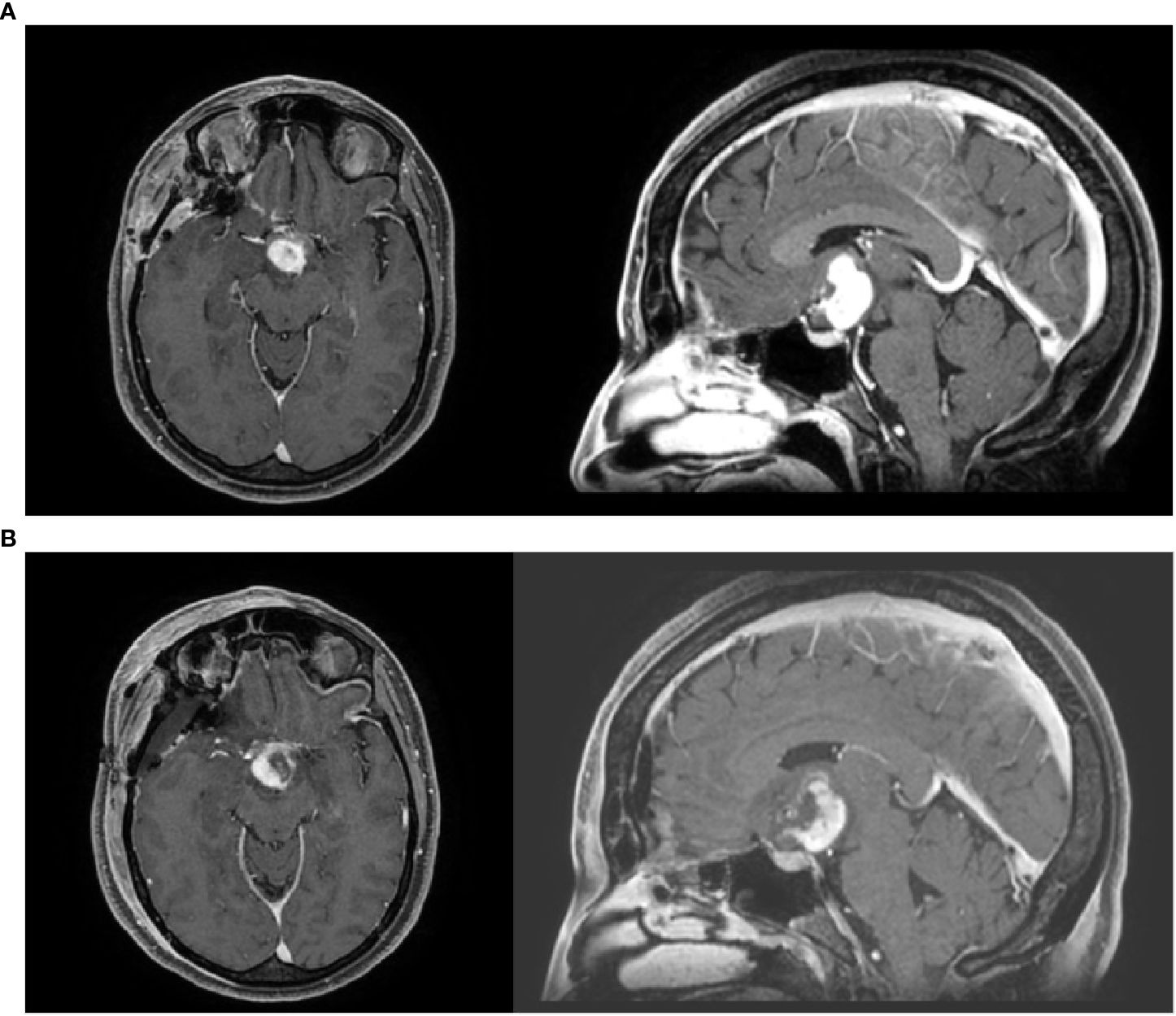
Figure 1. (A, B) Pre-operative and post-operative MRI imaging.
In January 2024, the patient underwent surgical excision of the tumor via a pterional approach. Histological examination revealed strong positivity for GFAP, synaptophysin, and thyroid transcription factor 1 (TTF-1), with focal expression of CKPan and a Ki-67 index of 2%. These findings confirmed a diagnosis of WHO Grade II chordoid glioma (Figure 2).
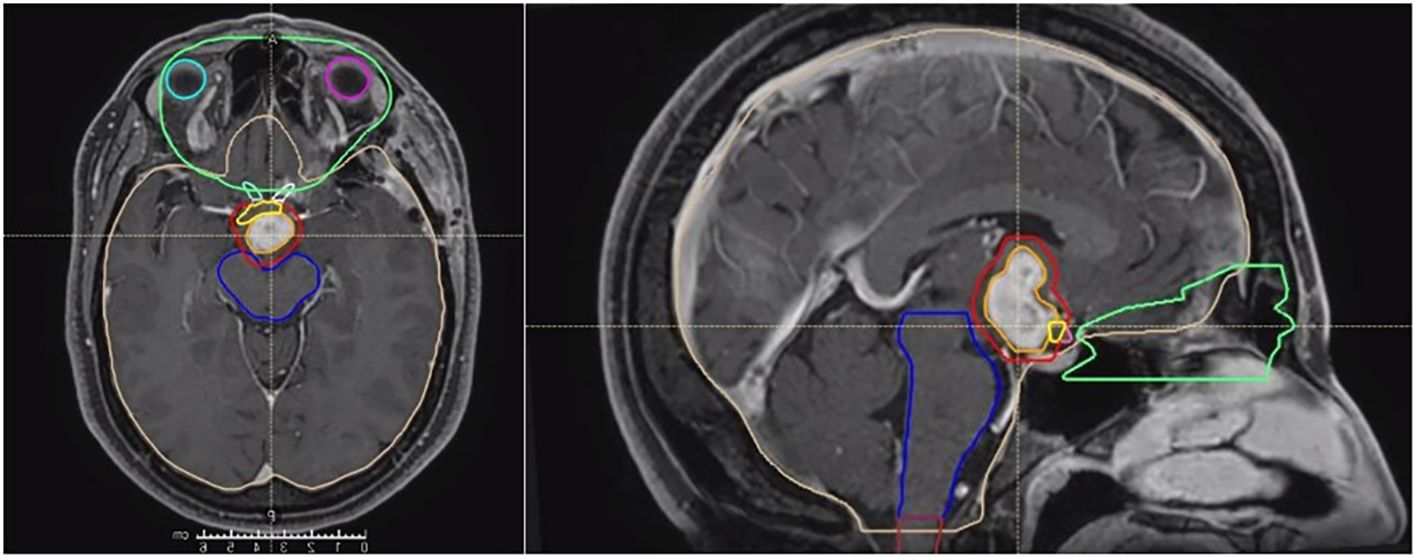
Figure 2. MRI imaging after surgery.
One month later, postoperative MRI revealed residual or relapsed disease measuring 1.9 × 2.7 cm. After multidisciplinary discussion, the patient was considered a candidate for adjuvant radiotherapy (Figure 1B).
At the initial evaluation in the Radiotherapy Department, the patient was in good general condition, with significant improvement in visual symptoms after surgery and persistent headache as the main residual symptom. No other neurological deficits were observed at baseline assessment.
Radiotherapy was performed using Radixact Helical TomoTherapy (Accuray, Sunnyvale, CA, USA), following a conventional fractionation schedule of 59.4 Gy delivered in 33 daily fractions. Target volume delineation was based on T1-weighted contrast-enhanced sequences to define the gross tumor volume (GTV). The planning target volume (PTV) consisted of the GTV plus an isotropic margin of 3 mm. An overlap structure was delineated in the intersection between the PTV and optic chiasm, with a prescription dose of 56.4 Gy to comply with dose constraints for optic structures (Figure 3).
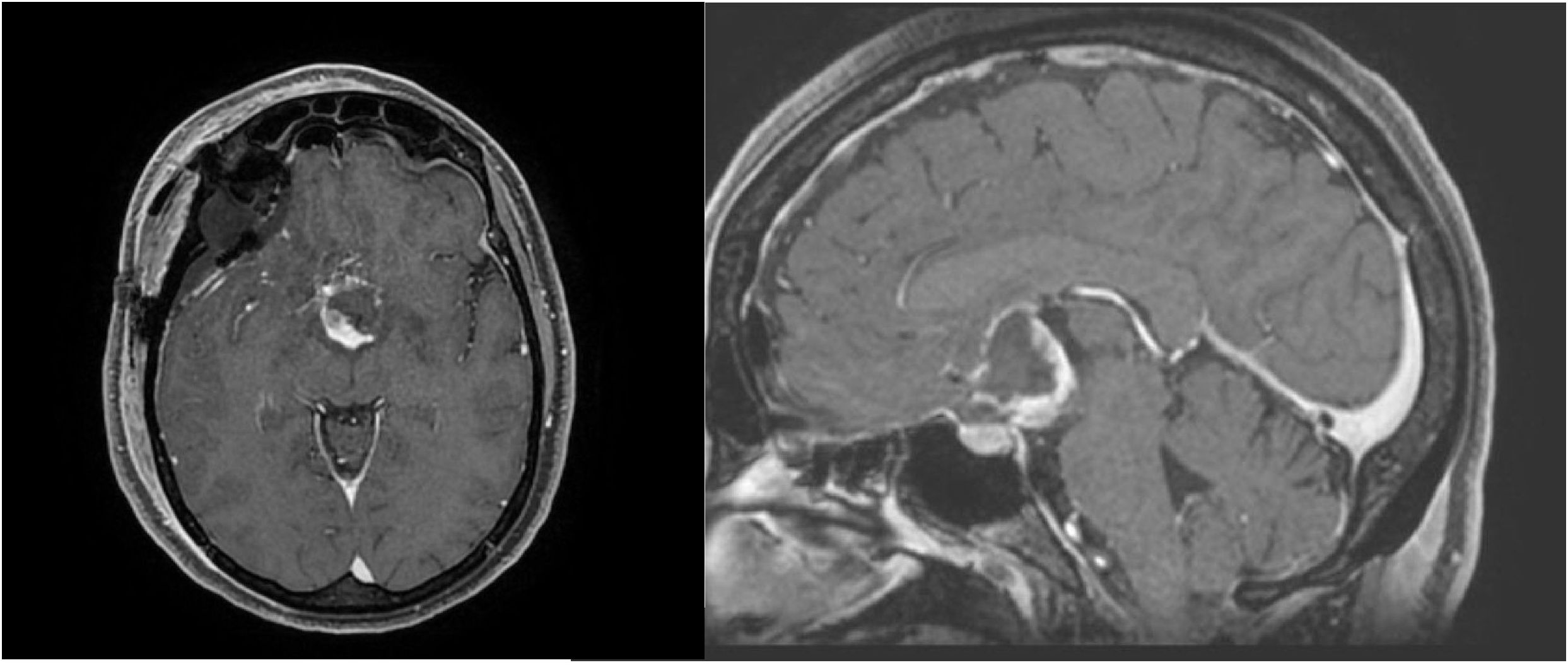
Figure 3. Target volume delineation of the clinical case.
Treatment was well tolerated. The patient received prophylactic low-dose steroids (dexamethasone 2 mg/day for the entire treatment duration), and no treatment interruptions occurred. No major side effects were observed, and the headache symptoms resolved. One month after completing radiotherapy, the patient continued steroid therapy for two weeks due to persistent headache, which led to suspected iatrogenic Cushing syndrome that was further investigated by the hospital’s Endocrinology and Rheumatology teams.
Headache was fully resolved at the first follow-up visit, 45 days after the end of treatment. Three months later, the patient developed asthenia and depression; laboratory tests and clinical evaluation led to a diagnosis of systemic sclerosis.
At 12 months of follow-up, serial MRI examinations showed no change in the lesion, indicating stable disease after radiotherapy. The patient remains in good general condition, fully recovered from metabolic disturbances, and is under rheumatologic surveillance, with no major neurological disorders or relevant radiotherapy-related side effects at the last follow-up visit.
3.2 Literature review
After the initial search identified 18 studies, additional hand searching of bibliographies yielded a final total of 14 studies (Table 1) (1, 9–21).
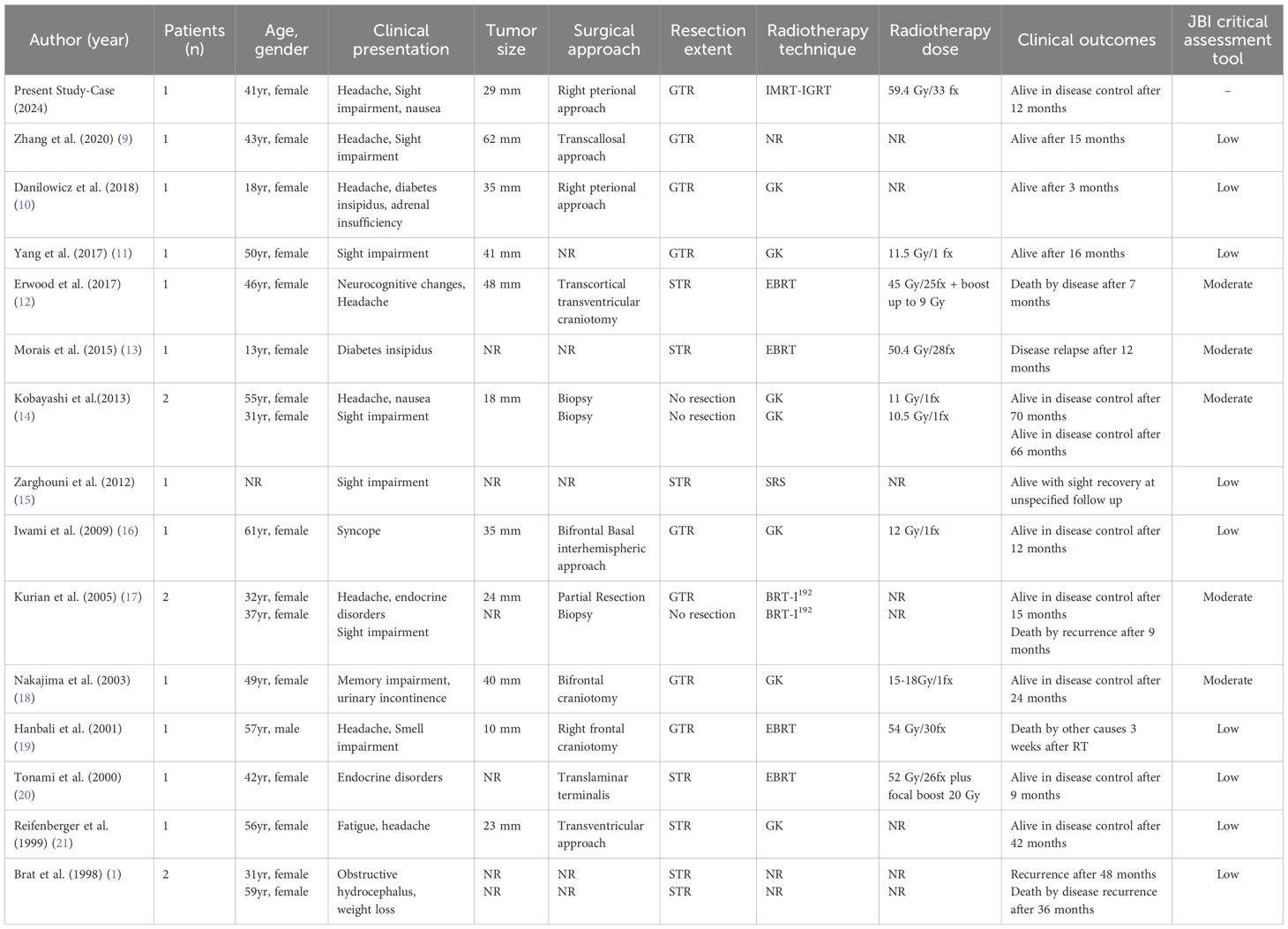
Table 1. Literature experiences of radiotherapy for chordoid glioma.
In total, 18 patients (including the present case) received radiotherapy for chordoid glioma, mainly in the form of Gamma Knife radiosurgery, with doses ranging from 11.5–18 Gy in patients with postoperative residual disease or who were unfit for surgery (five and two patients, respectively). Details regarding the radiotherapy technique were not reported for four patients, whereas brachytherapy with I192-seeds was described in two patients in the study by Kurian et al. (17)
Conventional radiotherapy was reported in only five patients, including our case, with doses ranging from 45–59.4 Gy in 1.8–2 Gy per fraction (12, 13, 19, 20).
Most patients treated with adjuvant radiotherapy were female (16 cases), with one male and one unreported, and a median age at diagnosis of 40 years (range, 13–57 years). The most common presenting symptom was headache (44% of cases), followed by visual deficits (13.6%).
Radiotherapy was performed after subtotal resection in 33% of cases and after gross total resection in 44% of cases. The remaining patients received radiotherapy as definitive treatment after stereotactic biopsy.
Three patients died due to disease recurrence at 7, 9, and 36 months, respectively. Recurrence after radiotherapy was also described in two other cases, at 12 and 48 months. In the remaining cases, the median local control time was 26 months. No patient in this series received chemotherapy.
4 Discussion
Chordoid glioma is a relatively newly characterized glial neoplasm, first described by Brat et al. as typically located near the third ventricle. From a pathological standpoint, the histogenesis of this tumor remains unclear, although strong GFAP positivity supports a likely glial origin (22).
Fewer than 100 cases have been reported in the literature. Radiologically, chordoid glioma is often difficult to distinguish from craniopharyngioma, as both present as well-defined, T1-isointense, ovoid masses with consistent and homogeneous contrast enhancement. Clinically, presentation is heterogeneous—ranging from incidental, asymptomatic cases to a broad spectrum of symptoms including headache, memory deficits, visual impairment, endocrine or metabolic disorders, and, in some cases, psychosis (23).
Although categorized as a WHO Grade II glioma, the prognosis for chordoid glioma is often poor, primarily due to the low likelihood of achieving complete surgical resection regardless of the surgical approach. The extent of resection remains one of the main predictors of improved survival, as tumor recurrence is more likely when gross total resection is not achieved, with a reported 5-year progression-free survival rate of 35.5%.
Nonetheless, a systematic review by Ampie et al. highlighted that surgical resection remains the cornerstone of treatment, while the role of adjuvant therapy remains controversial. Similarly, Huo et al. emphasized the uncertain benefit of adjuvant therapy for chordoid glioma (24).
However, severe postoperative complications are frequently reported, including cerebrospinal fluid leakage, hypothalamic dysfunction, sinonasal morbidity, and pulmonary thromboembolism. Therefore, several authors have proposed a potential role for adjuvant radiotherapy as a therapeutic strategy that may combine less invasive surgery with improved local disease control (25).
As chemotherapy has not been reported in the literature, radiotherapy remains the most common adjuvant treatment, with a slight preference for radiosurgery—likely due to the tumor’s critical location and the need to minimize exposure of nearby healthy structures.
Some authors have also reported the use of conventional radiotherapy, although with generally poor outcomes, possibly related to the relatively low total doses delivered. In the present case, after multidisciplinary discussion, we decided to perform conventional radiotherapy with the aim of delivering a higher biological dose to the target compared with previously reported schedules, given the unfavorable prognosis of this neoplasm.
Our patient received 59.4 Gy in 33 fractions—the highest conventional radiotherapy dose reported to date—derived from prior institutional experience in treating rare brain tumors. This approach was based on a similar adjuvant radiotherapy schedule applied to a young adult patient with papillary pineal tumor, which achieved favorable results.
After 12 months of follow-up, no evidence of local progression was observed on serial MRI scans, and the patient fully recovered from the initial persistent headache. The overlapping diagnosis of systemic sclerosis immediately after radiotherapy slowed the recovery process but did not affect oncological outcomes, reinforcing limited existing evidence supporting the feasibility of radiotherapy in patients with connective tissue diseases or immunosuppression (26, 27).
Consistently, a recent study by Rzepka et al. (29) assessed chromosomal radiosensitivity in oncological and non-oncological patients, showing higher radiosensitivity rates—defined as the average number of DNA breaks per metaphase—in oncological patients with connective tissue disorders (29).
This study has several limitations, beginning with its case-report design, which inherently restricts the level of evidence. Moreover, the data available in the literature and summarized here remain limited and carry a low level of evidence, being mainly composed of case reports and small case series. Many of these studies also lack key information regarding treatment parameters and clinical outcomes, thereby preventing a direct comparison between stereotactic and conventional radiotherapy approaches.
5 Conclusions
Considering the rarity of this disease, the management of chordoid glioma remains controversial. The use of modern adjuvant radiotherapy techniques may represent a favorable option following surgery, potentially reducing the incidence of severe postoperative sequelae and improving local tumor control. Larger databases and national registries with longer follow-up periods are necessary to gain more insights into recurrence patterns and optimal therapeutic strategies.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
Ethical approval was not required for the study involving humans in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
FC: Conceptualization, Writing – original draft, Writing – review & editing. SD: Data curation, Investigation, Writing – original draft, Writing – review & editing. MM: Data curation, Formal Analysis, Methodology, Writing – original draft, Writing – review & editing. GT: Conceptualization, Data curation, Supervision, Writing – original draft, Writing – review & editing. GCr: Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing. LB: Methodology, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing. FA: Formal Analysis, Software, Supervision, Writing – original draft, Writing – review & editing. GCa: Conceptualization, Methodology, Project administration, Visualization, Writing – original draft, Writing – review & editing. GF: Conceptualization, Data curation, Project administration, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Brat DJ, Scheithauer BW, Staugaitis SM, Cortez SC, Brecher K, and Burger PC. Third ventricular chordoid glioma: a distinct clinicopathologic entity. J Neuropathol Exp Neurol. (1998) 57:283–90. doi: 10.1097/00005072-199803000-00009
2. Cenacchi G, Roncaroli F, Cerasoli S, Ficarra G, Merli GA, and Giangaspero F. Chordoid glioma of the third ventricle: an ultrastructural study of three cases with a histogenetic hypothesis. Am J Surg Pathol. (2001) 25:401–5. doi: 10.1097/00000478-200103000-00016
3. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. (2021) 23:1231–51. doi: 10.1093/neuonc/noab106
4. Pomper MG, Passe TJ, Burger PC, Scheithauer BW, and Brat DJ. Chordoid glioma: a neoplasm unique to the hypothalamus and anterior third ventricle. AJNR Am J Neuroradiol. (2001) 22:464–9.
5. Muneer MS, Mohamed AS, Vizcaino MA, Raghunathan A, Naidich TP, and Vibhute PG. Chordoid glioma: a rare old foe but a new pathological and radiological presentation. Clin Imaging. (2021) 78:160–4. doi: 10.1016/j.clinimag.2021.03.008
6. Bongetta D, Risso A, Morbini P, Butti G, and Gaetani P. Chordoid glioma: a rare radiologically, histologically, and clinically mystifying lesion. World J Surg Oncol. (2015) 13:188. doi: 10.1186/s12957-015-0603-9
7. Estronza S, Saavedra FM, De Jesus O, and Pastrana EA. Chordoid glioma with psychosis: case report. P R Health Sci J. (2018) 37:174–6.
8. Dziurzynski K, Delashaw JB, Gultekin SH, Yedinak CG, and Fleseriu M. Diabetes insipidus, panhypopituitarism, and severe mental status deterioration in a patient with chordoid glioma: case report and literature review. Endocr Pract. (2009) 15:240–5. doi: 10.4158/EP.15.3.240
9. Zhang GB, Huang HW, Li HY, Zhang XK, Wang YG, and Lin S. Intracranial chordoid glioma: A clinical, radiological and pathological study of 14 cases. J Clin Neurosci. (2020) 80:267–73. doi: 10.1016/j.jocn.2020.09.019
10. Danilowicz K, Abbati SG, Sosa S, Witis FL, and Sevlever G. Suprasellar chordoid glioma: a report of two cases. Arch Endocrinol Metab. (2018) 62:648–54. doi: 10.20945/2359-3997000000092
11. Yang D, Xu Z, Qian Z, Wang L, Nie Q, Ge J, et al. Chordoid glioma: A neoplasm found in the anterior part of the third ventricle. J Craniofac Surg. (2021) 32:e311–3. doi: 10.1097/SCS.0000000000002514
12. Erwood AA, Velazquez-Vega JE, Neill S, Solomon DA, Butowski N, Nowlan A, et al. Chordoid glioma of the third ventricle: report of a rapidly progressive case. J Neurooncol. (2017) 132:487–95. doi: 10.1007/s11060-017-2399-7
13. Morais BA, Menendez DF, Medeiros RS, Teixeira MJ, and Lepski GA. Chordoid glioma: Case report and review of the literature. Int J Surg Case Rep. (2015) 7C:168–71. doi: 10.1016/j.ijscr.2015.01.027
14. Kobayashi T, Tsugawa T, Hashizume C, Arita N, Hatano H, Iwami K, et al. Therapeutic approach to chordoid glioma of the third ventricle. Neurol Med Chir (Tokyo). (2013) 53:249–55. doi: 10.2176/nmc.53.249
15. Zarghouni M, Vandergriff C, Layton KF, McGowan JB, Coimbra C, Bhakti A, et al. Chordoid glioma of the third ventricle. Proc (Bayl Univ Med Cent). (2012). doi: 10.1080/08998280.2012.11928853
16. Iwami K, Arima T, Oooka F, Fukumoto M, Takagi T, and Takayasu M. Chordoid glioma with calcification and neurofilament expression: case report and review of the literature. Surg Neurol. (2009) 71:115–20. doi: 10.1016/j.surneu.2007.07.032
17. Kurian KM, Summers DM, Statham PF, Smith C, Bell JE, and Ironside JW. Third ventricular chordoid glioma: clinicopathological study of two cases with evidence for a poor clinical outcome despite low grade histological features. Neuropathol Appl Neurobiol. (2005) 31:354–61. doi: 10.1111/j.1365-2990.2005.00551.x
18. Nakajima M, Nakasu S, Hatsuda N, Takeichi Y, Watanabe K, and Matsuda M. Third ventricular chordoid glioma: case report and review of the literature. Surg Neurol. (2003) 59:424–8. doi: 10.1016/s0090-3019(03)00066-1
19. Hanbali F, Fuller GN, Leeds NE, and Sawaya R. Choroid plexus cyst and chordoid glioma. Rep two cases. Neurosurg Focus. (2001) 10:E5. doi: 10.3171/foc.2001.10.6.6
20. Tonami H, Kamehiro M, Oguchi M, Higashi K, Yamamoto I, Njima T, et al. Chordoid glioma of the third ventricle: CT and MR findings. J Comput Assist Tomogr. (2000) 24:336–8. doi: 10.1097/00004728-200003000-00029
21. Reifenberger G, Weber T, Weber RG, Wolter M, Brandis A, Kuchelmeister K, et al. Chordoid glioma of the third ventricle: immunohistochemical and molecular genetic characterization of a novel tumor entity. Brain Pathol. (1999) 9:617–26. doi: 10.1111/j.1750-3639.1999.tb00543.x
22. Bhardwaj N, Salunke P, Singla N, Ahuja C, Gendle C, and Gupta K. Suprasellar masquerader: Chordoid glioma. Clin Neuropathol. (2024) 43:23–8. doi: 10.5414/NP301577
23. Galloway M, Afshar F, and Geddes JF. Chordoid glioma: an uncommon tumour of the third ventricle. Br J Neurosurg. (2001) 15:147–50. doi: 10.1080/02688690120036865
24. Ampie L, Choy W, Lamano JB, Kesavabhotla K, Mao Q, Parsa AT, et al. Prognostic factors for recurrence and complications in the surgical management of primary chordoid gliomas: A systematic review of literature. Clin Neurol Neurosurg. (2015) 138:129–36. doi: 10.1016/j.clineuro.2015.08.011
25. Calanchini M, Cudlip S, Hofer M, Byrne J, Fabbri A, and Grossman A. Chordoid glioma of the third ventricle: a patient presenting with SIADH and a review of this rare tumor. Pituitary. (2016) 19:356–61. doi: 10.1007/s11102-016-0711-8
26. Mazzola R, Cuccia F, Bertani A, Tubin S, Conaldi PG, Corradini S, et al. The role of radiotherapy in patients with solid tumours after solid organ transplantation: a systematic review. Lancet Oncol. (2021) 22:e93–e104. doi: 10.1016/S1470-2045(20)30590-8
27. Giaj-Levra N, Sciascia S, Fiorentino A, Fersino S, Mazzola R, Ricchetti F, et al. Radiotherapy in patients with connective tissue diseases. Lancet Oncol. (2016) 17:e109–17. doi: 10.1016/S1470-2045(15)00417-9
28. Moola S, Munn Z, Tufanaru C, Aromataris E, Sears K, Sfetcu R, et al. Chapter 7: Systematic reviews of etiology and risk. In: Aromataris E and Munn Z, editors. Joanna Briggs Institute Reviewer’s Manual. The Joanna Briggs Institute (2017). Available online at: https://reviewersmanual.joannabriggs.org/ (Accessed September 15, 2025).
Keywords: chordoid glioma, radiotherapy, adjuvant treatment, rare tumors, CNS tumors
Citation: Cuccia F, D’Alessandro S, Messina M, Tringali G, Craparo G, Blasi L, Azzarello F, Carruba G and Ferrera G (2025) Adjuvant radiotherapy treatment of chordoid glioma: a case report with a literature review. Front. Oncol. 15:1680822. doi: 10.3389/fonc.2025.1680822
Received: 06 August 2025; Accepted: 24 October 2025;
Published: 17 November 2025.
Edited by:
Raphael Bertani, University of São Paulo, BrazilReviewed by:
Parisa Javadnia, Iran University of Medical Sciences, IranPeng Cao, The General Hospital of Northern Command, China
Copyright © 2025 Cuccia, D’Alessandro, Messina, Tringali, Craparo, Blasi, Azzarello, Carruba and Ferrera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Cuccia, ZnJhbmNlc2NvLmN1Y2NpYUBhcm5hc2Npdmljby5pdA==