 Ee Qian Lee1
Ee Qian Lee1 Chin-King Looi2
Chin-King Looi2 Lu Ping Tan3
Lu Ping Tan3 Yik Ling Chew1
Yik Ling Chew1 Wei-Meng Lim4
Wei-Meng Lim4 Lian-Chee Foong5
Lian-Chee Foong5 Chee-Onn Leong6Kok Whye Cheong1
Chee-Onn Leong6Kok Whye Cheong1 Chun-Wai Mai2*
Chun-Wai Mai2*- 1Faculty of Pharmaceutical Sciences, UCSI University, Cheras, Kuala Lumpur, Malaysia
- 2Centre for Cancer and Stem Cell Research, Institute for Research, Development and Innovation (IRDI), IMU University, Bukit Jalil, Kuala Lumpur, Malaysia
- 3Molecular Pathology Unit, Cancer Research Centre, Institute for Medical Research, National Institutes of Health, Ministry of Health Malaysia, Shah Alam, Selangor, Malaysia
- 4School of Pharmacy, Monash University Malaysia, Jalan Lagoon Selatan, Bandar Sunway, Subang Jaya, Selangor, Malaysia
- 5State Key Laboratory of Systems Medicine for Cancer, Renji-Med X Clinical Stem Cell Research Center, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Pudong New District, Shanghai, China
- 6AGTC Genomics Sdn. Bhd, Kuala Lumpur, Malaysia
Radiotherapy (RT) is the first-line treatment for more than 50% of newly diagnosed cancer patients and remains a cornerstone of cancer therapy, particularly for tumors that are inoperable, recurrent, or incompletely resected. Despite advancements in RT techniques, locoregional recurrence and distant metastasis remain critical clinical challenges, contributing significantly to cancer-related inflammation and mortality. Emerging evidence suggests that RT may inadvertently promote metastasis through inflammation-related immune modulations, such as the dysregulation of signaling cascades, such as focal adhesion kinase (FAK), phosphoinositide 3-kinases (PI3K)/protein kinase B (AKT), p38 mitogen-activated protein kinase (p38 MAPK), and nuclear factor-kappa B (NF-κB) signaling cascades. Targeting these pro-metastatic pathways using specific inflammatory inhibitors and clinically available repurposed drugs has shown promise in numerous preclinical models, offering a rational approach to mitigate radiation-induced inflammation in metastatic progression. With the rapid advancements of high-throughput sequencing and medical imaging technologies, radiogenomics, which incorporates medical imaging and genomic data, offers great promise for cancer diagnosis, tumor classification, treatment selection, and disease monitoring through the identification of predictive and prognostic biomarkers. This review critically unravels the immune modulation underlying radiation-induced inflammation in cancer metastasis and highlights the need for comprehensive studies combining radiogenomics with RT and targeted therapies. Such approaches hold potential to improve therapeutic outcomes and reduce metastatic burden, paving the way for more effective and personalized cancer treatments.
1 Introduction
Radiotherapy (RT) has remained a crucial treatment modality for patients with incompletely resected tumors, recurrent disease, or tumors located in inoperable areas since its development in the late 19th century, following Wilhelm Conrad Roentgen’s discovery of X-rays in 1895 (1–4). Over the years, RT has advanced significantly through continuous technological innovation aimed at improving treatment efficacy while reducing the severe acute and late toxicities associated with conventional two-dimensional RT (2D-CRT) (5). Modern RT techniques, such as intensity-modulated radiotherapy (IMRT), stereotactic body radiotherapy (SBRT), and image-guided radiation therapy (IGRT), enable the precise delivery of high-dose radiation to tumor sites, typically ranging from 60-100 Gy, depending on the modality and fractionation schedule, thereby enhancing tumor control while minimizing damage to surrounding healthy tissues (6–8). To further enhance its therapeutic efficacy, recent studies have explored combining RT with other therapies, including chemotherapy, targeted therapy, immunotherapy, and radionuclide therapy (9, 10). Today, more than 50% of all cancer patients receive RT at some point during their illness, underscoring its critical role in cancer management (11).
While RT is well known for activating cytotoxic signaling pathways and mediating immunostimulatory effects to promote cancer cell death, cancer recurrence, and distant metastasis continue to pose a major challenge in the medical field (2, 12). Studies indicate that nearly 40% of head and neck cancer patients treated with RT experience locoregional recurrence, which is a leading cause of cancer-related mortality (2). Raab et al. (2024) analyzed data from the National Cancer Database (2004–2018) and reported a 90-day mortality rate of 3.5% among head and neck cancer patients undergoing RT (13). One key factor driving recurrence is the intrinsic biological properties of the tumor cells, including inherent sensitivity to RT and the development of acquired resistance (14). A previous study evaluating recurrence and metastasis patterns in nasopharyngeal carcinoma (NPC) patients treated with IMRT found that most patients who experienced local recurrence had received high-dose RT (70–74 Gy), compared to those who received low doses (54–66 Gy) (14). These findings suggest that increasing the radiation dose does not effectively prevent recurrence; instead, it may potentially lead to the development of tumor resistance, further complicating therapeutic outcomes (14). In addition, genomic instability, epigenetic modifications, and variations in the tumor microenvironment (TME) can collectively contribute to tumor heterogeneity, which plays a significant role in mediating RT resistance, enabling tumor cells to adapt and survive (15).
Accumulating evidence suggests that RT may also inadvertently promote the recruitment of tumor cells into the circulation by damaging tumor vasculature, thereby facilitating distant metastasis (16). Metastasis typically begins when tumor cells acquire an invasive and migratory phenotype via the activation of epithelial–mesenchymal transition (EMT), enabling them to detach from their primary site and invade surrounding tissues (4). These cells then enter the bloodstream and travel to distant organs (4). Once in circulation, circulating tumor cells can either metastasize to other sites or reinfiltrate their tumor of origin (4). Although von Essen has outlined four possible mechanisms influencing the rate of metastasis following RT since 1991: radiation-induced biological and behavioral changes in surviving tumor cells; effects on distant normal tissue that may support metastasis; radiation-induced release of tumor cells into the circulation; and the prolonged release of metastatic cells into the circulation due to delayed tumor progression, the molecular mechanisms underlying RT-induced metastasis remain poorly understood (17).
Therefore, gaining a deeper understanding of the mechanisms underlying treatment resistance and RT-induced metastasis is essential for overcoming these significant obstacles in RT. By investigating the molecular and cellular pathways involved, it holds promise for uncovering new therapeutic targets that could enhance the effectiveness of RT and ultimately improve patient outcomes (18). However, translating these experimental findings into clinical applications remains a major challenge, particularly in identifying biomarkers that can predict therapeutic responses and developing targeted interventions capable of preventing or reversing radiation-induced metastasis 19.
Although biopsy is the standard research approach, it provides only a localized representation of the tumor, failing to account for intratumoral heterogeneity and the broader TME, both of which play critical roles in disease progression (19). Furthermore, the identification of clinically relevant biomarkers is complicated by genetic variability, necessitating large-scale studies to ensure their predictive reliability (19). These limitations underscore the urgent need for accurate and cost-effective predictive models to enhance cancer prognosis and optimize treatment strategies.
This review critically discusses the inflammation in metastasis pathways induced by RT, focusing on how these immune-related inflammations contribute to cancer progression. It also explores the use of repurposed and investigational drugs in combination therapies designed to counteract radiation-induced metastasis. In addition, emerging trends in radiomics and radiogenomics are discussed, with a focus on their potential to identify predictive biomarkers for cancer diagnosis and treatment optimization. The review aims to support the development of personalized treatments for cancer patients.
2 Radiation-induced inflammation in metastasis
Inevitably, radiation could induce inflammation, leading to immune modulation via four key inflammatory signaling pathways, namely (i) focal adhesion mechanism, (ii) phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) activation, (iii) p38 mitogen-activated protein kinase (p38 MAPK) activation, and (iv) nuclear factor-kappa B (NF-κB) activation. Radiation-induced focal adhesion activation has been observed in prostate, breast, and colon cancers, while radiation-induced PI3K/AKT activation has been reported in NPC (20–23). Radiation-induced p38 MAPK activation has been reported in hepatocellular carcinoma (HCC), glioma, lung, and cervical cancers, while the radiation-induced NF-κB inflammatory activation has been demonstrated in esophageal, lung, and non-small-cell lung cancers (NSCLC) (24–28). A recent study further suggests that RT can alter the TME, promoting resistance to therapy primarily through the upregulation of inflammatory cytokines such as transforming growth factor-beta (TGF-β) and activation of the AKT, NF-κB, and signal transducer and activator of transcription (STAT) signaling pathways (29). These changes enhance cell proliferation, oxidative stress, M2 macrophage polarization, and angiogenesis, collectively contributing to tumor progression. 30.
2.1 Radiation-induced inflammatory focal adhesion mechanism
The extracellular matrix (ECM) is a non-cellular, crosslinked network of macromolecules, including collagens, fibronectins, proteoglycans, and glycoproteins, that provide structural and biochemical support to surrounding cells (30). However, tumor cells can hijack ECM remodeling by altering its composition and organization to facilitate invasion and migration (31). This process is tightly regulated by focal adhesion proteins (31). Focal adhesions are dynamic multi-protein complexes that function as mechanosensors, linking the intracellular actin cytoskeleton to the ECM via integrins (32). Integrins are key cell adhesion proteins that mediate both outside-in and inside-out signaling, and an increase in ECM stiffness activates integrin-mediated mechanosignaling, promoting cell proliferation and local tumor cell invasion (31, 33). Furthermore, integrin binding to ECM exerts mechanical forces that alter ECM conformation, influence matrix degradation and deposition, and promote cancer cell migration even in the absence of proteolytic activity (31). Thus, the natural biomechanisms of ECM remodeling and integrin-mediated focal adhesions provide the basis for tumor invasion, wherein radiation can further modulate these processes.
Over the years, numerous studies have reported that RT can activate focal adhesion mechanisms, leading to the upregulation of integrins and Ras homolog family member A (RhoA), both of which are key regulators of cell adhesion and migration (20–22, 34). For instance, You and co-workers (2021) demonstrated that cell motility mechanisms, particularly focal adhesion, are key pathways in promoting tumor invasion and radioresistance in head and neck cancer (35). Notably, integrin alpha-6 (ITGA6), a heterodimeric component of the integrin receptor protein, was significantly upregulated in tumor cells (35). Subsequent shRNA-mediated knockdown of ITGA6 expression led to reduced migration and invasion, as well as increased reactive oxygen species (ROS) production (35). These findings suggest that tumor cells may upregulate ITGA6 to promote cell migration and invasion through ECM integrity modulation and facilitate radioresistance by reducing intracellular ROS levels (35). Therefore, the inhibition of ITGA6 warrants further studies to determine whether its inhibition could synergize with RT to suppress the migration and invasion of radioresistant cells, as well as re-sensitize tumors to irradiation.
Furthermore, RhoA, the small guanosine triphosphate-binding protein (GTPase), is closely associated with the regulation of cytoskeletal reorganization and vascular permeability after radiation exposure (36). Under normal physiological conditions, RhoA functions as a molecular switch, cycling between an inactive guanosine diphosphate (GDP)-bound state and an active guanosine triphosphate (GTP)-bound form (37). Activation of RhoA is tightly regulated by guanine nucleotide exchange factors (GEFs), which promote the release of GDP from inactive RhoA, allowing GTP to bind and activate it (37). Conversely, Rho GTPase-activating proteins (RhoGAPs) promote the hydrolysis of GTP to GDP, returning RhoA to its inactive state (37). Upon irradiation, DNA double-strand breaks (DSBs) are induced, triggering the DNA damage response (DDR) (38). This signaling cascade, particularly through the epidermal growth factor (EGF) or c-Jun N-terminal kinase (JNK) pathways, promotes the chromosome region maintenance 1 (CRM1)-dependent nuclear export and cytoplasmic translocation of neuroepithelial cell transforming gene 1 (NET1) (39). NET1, a RhoA-specific GEF, catalyzes the exchange of GDP for GTP, converting RhoA into its active form (39). Activated RhoA subsequently interacts with Rho-associated coiled-coil containing kinases (ROCK; comprising ROCK1 and ROCK2) by binding to their Rho-binding domain (RBD) (40). This binding induces a conformational change that relieves autoinhibition, leading to ROCK activation (40). Once activated, ROCK phosphorylates the myosin light chain (MLC), thereby enhancing myosin II (Myo II) contractility (40). Additionally, ROCK activates LIM domain kinase (LIMK), which phosphorylates and inactivates cofilin, leading to the stabilization of actin filament (37). In parallel, RhoA also activates formin proteins such as the mammalian homolog of diaphanous (mDia), facilitating linear actin polymerization and stabilization (37). The coordinated activities of ROCK and mDia promote the formation of filamentous-actin stress fiber and the assembly of focal adhesion proteins, potentially enhancing the invasiveness of tumor cells following radiation exposure (37). Taken together, these findings position radiation-induced RhoA activation as a critical molecular link between DDR signaling and cytoskeletal remodeling (38). Figure 1 provides an overview of how irradiation-induced integrin activation and RhoA-mediated cytoskeletal remodeling promote tumor cell invasion and migration.
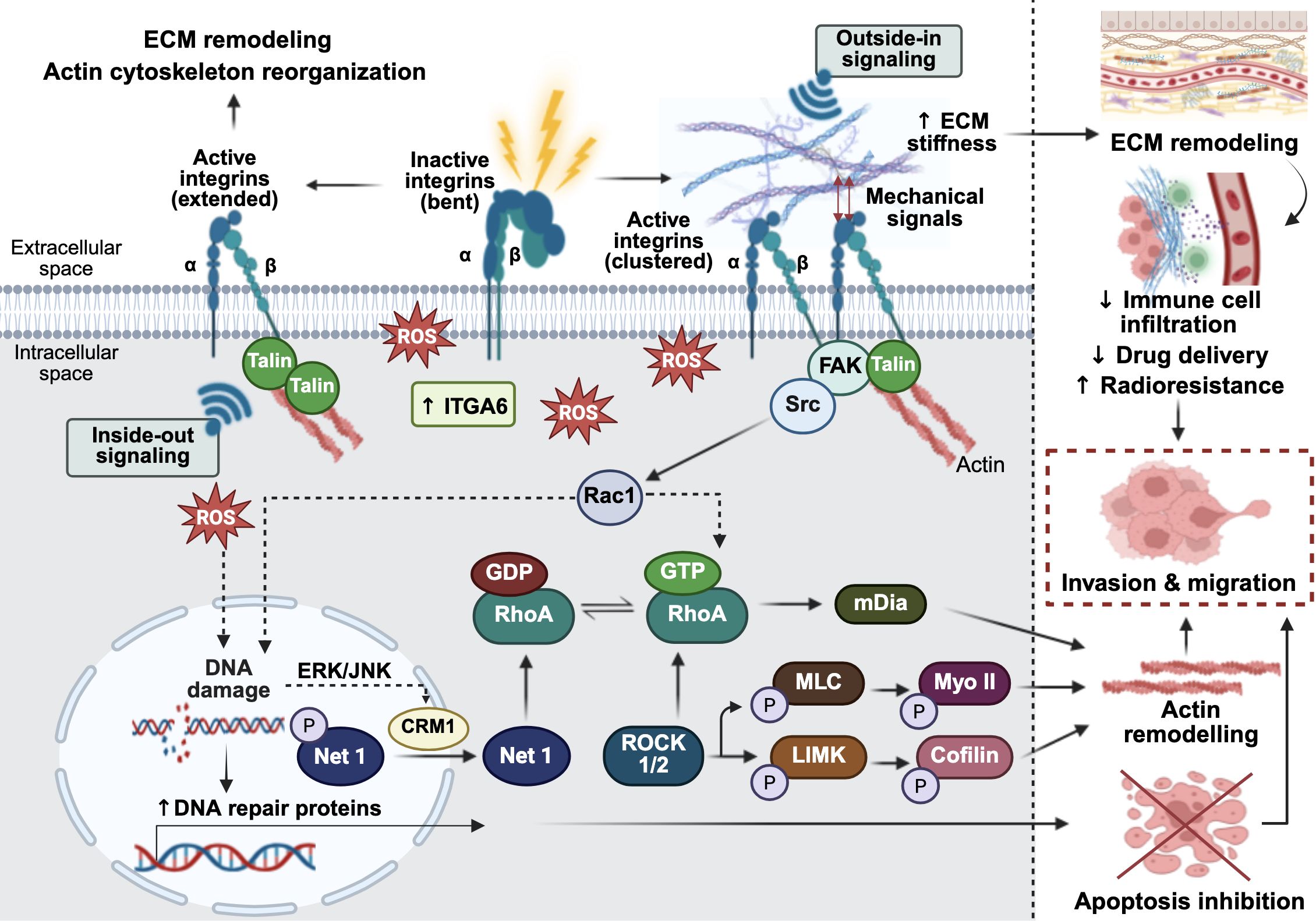
Figure 1. Radiation-induced extracellular matrix (ECM) remodeling, cell migration through focal adhesion signaling and cytoskeletal remodeling leading to cancer immune modulation. In the absence of stimulation, integrins adopt an inactive, closed, bent conformation. Upon exposure to ionizing radiation, reactive oxygen species (ROS) are generated, stimulating inside-out integrin signaling through talin, which reorganizes the actin cytoskeleton and contributes to ECM remodeling. Radiation also upregulates integrin alpha-6 (ITGA6), altering ECM integrity to promote cell invasion. Concurrently, the outside-in binding of ECM ligands to cell surface integrins triggers mechanosignaling that activates focal adhesion kinase (FAK). FAK then associates with proto-oncogene tyrosine-protein kinase Src, leading to activation of the RhoA/ROCK signaling axis. In tumor cells, radiation-induced ROS can cause DNA damage while simultaneously upregulating DNA repair proteins, thereby inhibiting apoptotic cell death and promoting cell survival. The ROS-induced DNA damage response also leads to the dephosphorylation of neuroepithelial cell transforming gene 1 (Net1), a RhoA-specific guanine nucleotide exchange factor (GEF), resulting in its nuclear export via extracellular signal-regulated kinase/c-Jun N-terminal kinase (ERK/JNK)-mediated chromosome region maintenance 1 (CRM1) signaling. Once in the cytoplasm, NET1 activates RhoA, which in turn activates Rho-associated coiled-coil containing protein kinase ½ (ROCK1/2) and the mammalian diaphanous-related formin (mDia). This cascade promotes myosin II contractility and actin filament stabilization, leading to cytoskeletal remodeling. Collectively, these radiation-induced signaling events drive tumor cell migration and invasion. The figure was created using BioRender (https://BioRender.com).
2.2 Radiation-induced inflammatory PI3K/AKT via PNUTS
Protein phosphatase 1 nuclear-targeting subunit (PNUTS), originally identified as a nuclear protein, has been found to modulate the PI3K/AKT pathway, a signaling cascade frequently implicated in cancer progression and cell survival (23, 41). Under normal cellular conditions, PNUTS forms a stable complex with protein phosphatase 1 (PP1), thereby regulating its catalytic activity (42). As a key modulator of PP1, PNUTS participates in various PP1-mediated cellular processes, including transcriptional regulation, cell cycle, apoptosis, and DNA repair (42). Upon exposure to cellular stress, such as hypoxia or radiation, PNUTS dissociates from PP1, allowing PP1 to dephosphorylate the retinoblastoma (Rb) protein, an essential regulator of cell proliferation and apoptosis (43). This dephosphorylation promotes apoptosis, inhibits irradiation-induced EMT, reduces cell viability, and ultimately contributes to tumor suppression (43).
In contrast, overexpression of PNUTS following high-dose irradiation may paradoxically reverse its tumor-suppressive function and potentially contribute to tumor progression, as demonstrated in vitro studies conducted by Yu and colleagues (2019) using a 4 Gy dose (23). The study demonstrated that radiation significantly upregulated PNUTS expression in the CNE-2, NPC cell line, and this increase was significantly associated with enhanced migratory and invasive capacities, suggesting that PNUTS overexpression may promote more aggressive tumor behavior following irradiation (23). Further mechanistic studies revealed that PNUTS induces EMT to accelerate CNE-2 cell progression through the PI3K/AKT signaling pathway (23). However, how PNUTS activates the PI3K/AKT pathway was not elucidated in the study.
Previous studies reported that PNUTS binds directly to the lipid-binding C2 domain of phosphatase and tensin homolog (PTEN) via its N-terminal TF2S domain, resulting in the sequestration of PTEN in the nucleus and thereby preventing it from inhibiting PI3K/AKT pathway activation (44). In general, PTEN functions by dephosphorylating phosphatidylinositol 3,4,5-trisphosphate (PIP3), a critical docking site for 3-phosphoinositide-dependent protein kinase-1 (PDK1) and AKT, converting it back to phosphatidylinositol 4,5-bisphosphate (PIP2) (45, 46). This results in reduced PIP3 levels, preventing AKT activation, and thereby inhibiting PI3K/AKT downstream signaling (45, 46). Notably, fractionated ionizing radiation (IR) has been shown to suppress PTEN expression and activate PDK1 and AKT signaling, leading to elevated levels of EMT-related proteins in human esophageal cancer cells (47). Thus, it is suggested that the PNUTS overexpression in response to radiation exposure stimulates PI3K/AKT pathway activation to induce EMT, primarily through the regulation of PTEN activity (23). Figure 2 illustrates the dual functions of PNUTS in regulating cancer development and progression, highlighting its roles in both tumor suppression and promotion.
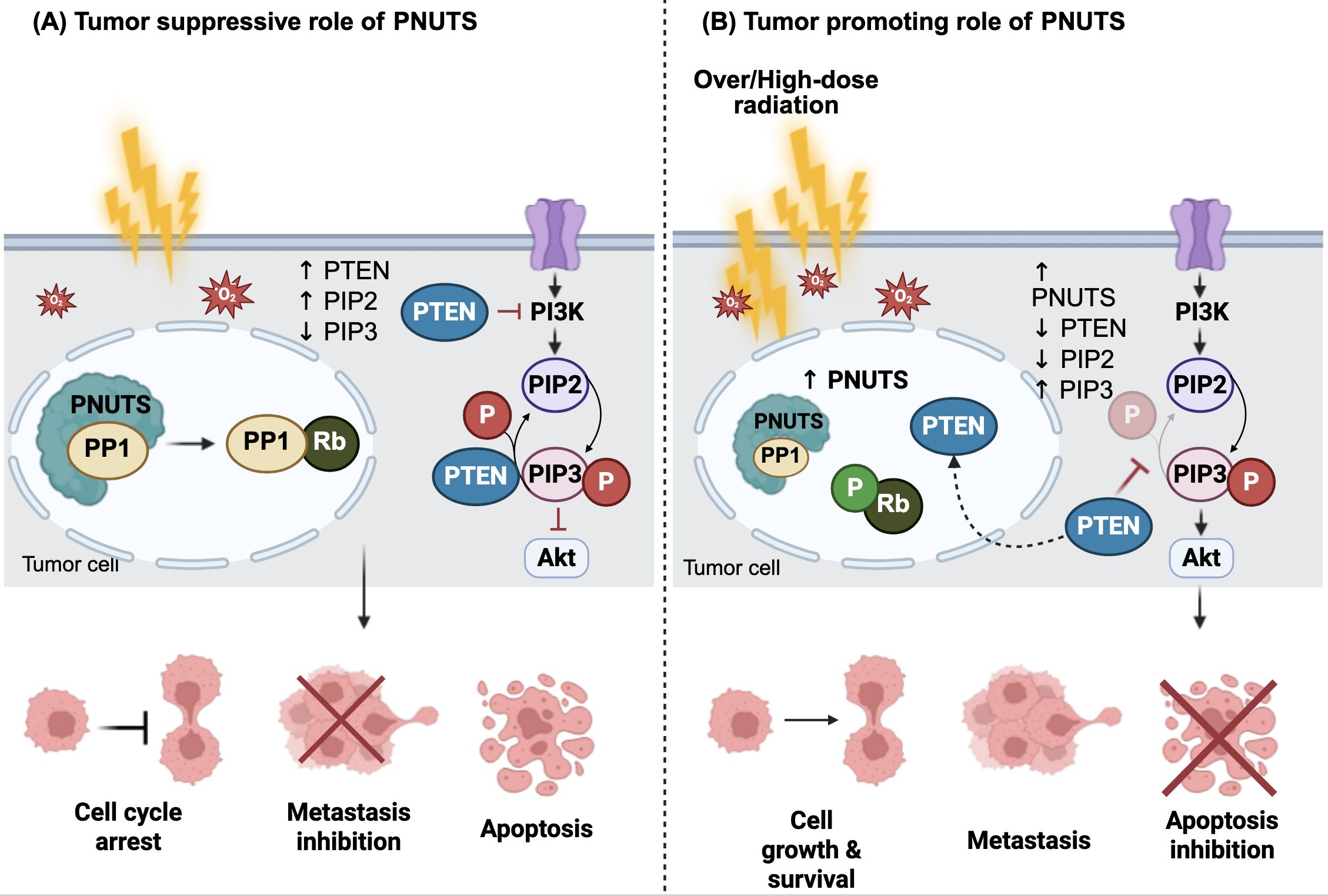
Figure 2. The dual roles of PNUTS in cancers. (A) Under basal conditions, protein phosphatase 1 nuclear-targeting subunit (PNUTS) is predominantly localized in the nucleus, where it associates with protein phosphatase 1 (PP1) to regulate its activity. In response to irradiation-induced reactive oxygen species (ROS), PNUTS dissociates from PP1, allowing PP1 to dephosphorylate the retinoblastoma tumor suppressor protein (Rb). This dephosphorylation induces cell cycle arrest and promotes apoptosis, thereby suppressing metastasis. (B) In contrast, high-dose irradiation leads to PNUTS upregulation and increased binding to PP1, maintaining Rb in a phosphorylated (inactive) state. Additionally, PNUTS binds directly to phosphatase and tensin homolog (PTEN), sequestering it in the nucleus. This prevents PTEN from dephosphorylating phosphatidylinositol 3,4,5-trisphosphate (PIP3) to phosphatidylinositol 4,5-bisphosphate (PIP2), resulting in cytoplasmic accumulation of PIP3 and constitutive activation of the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) signaling pathway. This, in turn, promotes tumor cell growth and metastasis. The figure was created using BioRender (https://BioRender.com).
2.3 Radiation-induced inflammatory p38 MAPK pathway
MAPKs are a family of serine/threonine protein kinases that mediate signal transduction from the cell surface to the nucleus in response to various extracellular stimuli. The conventional MAPK superfamily comprises extracellular signal-regulated kinases (ERK), JNK, and p38 MAPK (48), playing a pivotal role in regulating a wide range of cellular processes, including immune activation, proliferation, differentiation, apoptosis, motility, and stress adaptation (48). Increasing evidence implicates these canonical MAPK pathways as central regulators of cancer cell migration and metastasis. For example, external stimuli such as chemotherapeutic agents and IR can activate the rat sarcoma virus oncogene (Ras)/rapidly accelerated fibrosarcoma (Raf)/mitogen-activated protein kinase (MEK)/ERK cascade, thereby enhancing the migratory capacity of HCC cells (49). Similarly, activation of the JNK/cellular jun proto-oncogene (c-JUN)/matrix metalloproteinase 9 (MMP9) signaling axis facilitates ECM degradation, promoting metastasis in HCC (50). In radioresistant cervical cancer, Kirsten rat sarcoma viral oncogene homolog (KRAS)–driven cell migration has been linked to the activation of the cellular-Raf (c-Raf)/p38 MAPK pathway (51). Collectively, these findings underscore the roles of ERK, JNK, and p38 as key mediators of radiation-enhanced tumor cell migration and invasion.
Among these, p38 MAPK has emerged as a particularly important mediator of radiation-induced tumor aggressiveness. In HCC, residual cancer cells following radiation exhibit increased p38 MAPK activity, which promotes EMT, migration, and invasion. Notably, this effect appears to be driven by hydrogen sulfide (H2S), a gaseous neurotransmitter (24). H2S is endogenously produced via the transsulfuration pathway, primarily by the catalytic activity of enzymes cystathionine β-synthase (CBS) or cystathionine γ-lyase (CSE), and has been shown to modulate p38 MAPK activity and contribute to tumor cell proliferation (24, 52). Inhibition of H2S production effectively reverses these effects, resulting in reduced tumor growth, increased radiosensitivity, and downregulated EMT-associated protein expression, such as Snail and N-cadherin (24). These findings suggest that H2S-driven activation of p38 MAPK is a crucial mechanism of radiation-induced aggressiveness, and targeting H2S metabolism may potentially enhance radiosensitivity.
Moreover, p38 MAPK can exerts its pro-tumorigenic effects through its downstream effectors such as MAPK-activated protein kinase 2 (MK2), which is known to promote the phosphorylation of heat shock protein 27 (HSP27) (53). This modification subsequently activates the NF-κB signaling cascade, leading to increased expression of pro-inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α), interleukin-1 alpha (IL-1α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6), thereby facilitating tumor progression (53). Berggren et al. (2019) demonstrated that MK2 inhibition following RT significantly reduced cytokine induction and delayed tumor regrowth in a murine model of head and neck squamous cell carcinoma (HNSCC), positioning MK2 as a potential therapeutic target to mitigate radiation-induced metastasis (53).
Interestingly, even low-dose radiation can activate p38 MAPK pathway, which in turn facilitates tumor migration (25). For instance, in glioma, lung, and cervical cancer cell lines, exposure to low-dose γ-radiation (10–20 cGy) selectively activates p38 MAPK-mediated invasion via the upregulation of connexin-43 (Cx43), a gap junction protein (25). Notably, knockdown of Cx43 effectively prevents radiation-induced p38 MAPK phosphorylation and suppresses the associated cell migration and invasion, whereas it does not affect ERK1/2 activity or cell proliferation (25). Although the precise mechanism of their direct interaction is not fully understood, Cx43 may promote p38 MAPK signaling by forming hemichannels that release pro-migratory factors (54). These findings suggest that targeting Cx43 could be a potential strategy to limit invasion induced by low-dose radiation.
Overall, both high-dose and low-dose irradiation converge on the p38 MAPK pathway through distinct but interconnected mechanisms, including H2S signaling, MK2-driven inflammation, and Cx43-mediated invasion, to promote tumor aggressiveness. These pathways highlight multiple therapeutic opportunities to enhance radiosensitivity and inhibit radiation-induced metastasis. Figure 3 illustrates the mechanisms by which irradiation activates the p38 MAPK pathway to promote cancer metastasis.
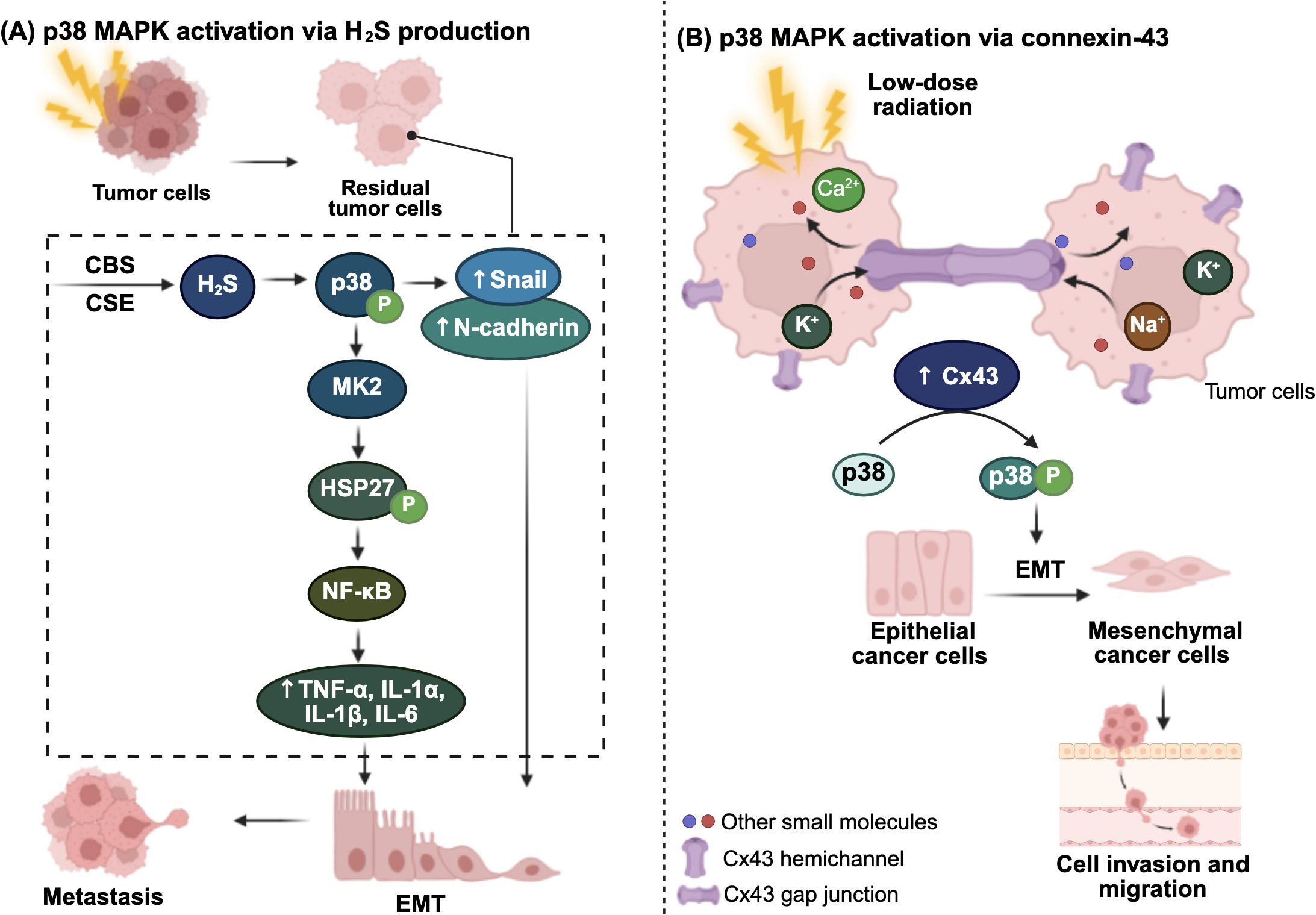
Figure 3. Radiation-induced inflammatory p38 MAPK activation via (A) hydrogen sulfide (H2S) and (B) connexin-43 (Cx43). (A) In residual tumor cells, endogenous H2S production is constitutively upregulated through increased expression of the two key H2S-producing enzymes, cystathionine-γ-lyase (CSE) and cystathionine-β-synthase (CBS). Elevated H2S levels activate and phosphorylate p38 mitogen-activated protein kinase (MAPK), which promotes epithelial-mesenchymal transition (EMT) by upregulating EMT markers, such as Snail and N-cadherin. Activated p38 also stimulates MAPK-activated protein kinase 2 (MK2; MAPKAPK2), which phosphorylates heat shock protein 27 (HSP27), ultimately leading to NF-κB activation. This results in increased secretion of immune modulating inflammatory cytokines, further driving tumor progression and metastasis. (B) Low-dose γ-radiation induces p38 MAPK activation through upregulation of Cx43. Cx43 forms hemichannel between neighboring tumor cells, facilitating the release of signaling molecules or pro-migratory factors, thereby promoting EMT and enhancing cell invasion and migration. The figure was created using BioRender (https://BioRender.com).
2.4 Radiation-induced inflammatory NF-κB signaling
NF-κB is a family of inducible transcription factors that play a pivotal role in regulating genes involved in inflammation, innate and adaptive immunity, cell proliferation, apoptosis, and oncogenesis. The NF-κB family comprises five structurally related members: RelA (p65), RelB, c-Rel, NF-κB1 (p50), and NF-κB2 (p52) (55). In unstimulated cells, NF-κB dimers, especially p50/RelA are sequestered in the cytoplasm by NF-κB protein inhibitors (IκB) (55). Upon stimulation by pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), such as TNF-α, ROS, or IR, the IκB kinase (IKK) complex becomes activated and triggers IκB degradation (55). This releases NF-κB, allowing its translocation into the nucleus to activate transcription of target genes (55). However, dysregulated or constitutive activation of NF-κB can lead to chronic inflammation and uncontrolled immune modulation, which drives cancer initiation and progression(56).
Notably, IR-induced inflammatory NF-κB activation has been linked to cancer metastasis in various tumor types (26–28, 56, 57). For instance, irradiated T cell-derived exosomes promoted esophageal cancer cells metastasis by upregulating β-catenin and the NF-κB/Snail pathway, facilitating EMT (26). In lung cancer, IR has been shown to promote tumor cell invasion and migration through the upregulation of IL-1β and its receptor, interleukin-1 receptor type I (IL-1RI) or II (IL-1RII) (27). Although IL-1RII is traditionally viewed as a decoy receptor that competes with IL-1RI to bind IL-1β and prevent NF-κB activation, Liu et al. (2022) demonstrated that IL-1RII can also promote oncogenic signaling by activating the Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) pathway in clear cell renal cell carcinoma (ccRCC) (58, 59). In HCC, activation of JAK2/STAT3 has been shown to increase matrix metalloproteinase 2 (MMP2), MMP9, and vascular endothelial growth factor (VEGF), promoting cell migration and invasion (60).
Furthermore, siRNA-mediated silencing and pharmacological inhibition of receptor-interacting protein kinase 1 (RIP1) and NF-κB significantly reduced IL-1β secretion, while direct inhibition of IL-1β suppressed IR-induced EMT and cell invasion (27). These findings suggest that IR may concurrently activate NF-κB to promote IL-1β release, thereby enhancing metastasis through EMT (27). Similarly, low-dose radiation (4 Gy) activates NF-κB signaling through the upregulation of chemokine (C-X-C motif) ligand 1 (CXCL1) expression in NSCLC, facilitating EMT and invasion (28). In addition, NF-κB also mediates IR-induced EMT through the RIP1/proto-oncogene tyrosine-protein kinase Src (Src kinase)/STAT3 pathway, which enhances the activity of ECM-degrading enzymes, such as MMP-2 and MMP-9 (56). Targeting these pathways with inhibitors that suppress NF-κB activity and MMP-9 expression may demonstrate promising immune modulation that could result in inhibiting IR-induced metastasis (57).
Collectively, these findings clearly demonstrate the role NF-κB as a central player in radiation-induced metastasis, promoting immune overactivation, EMT, migration, and invasion through multiple interconnected signaling cascades (Figure 4). These insights underline the importance of developing targeted therapies to mitigate the pro-metastatic effects of RT.
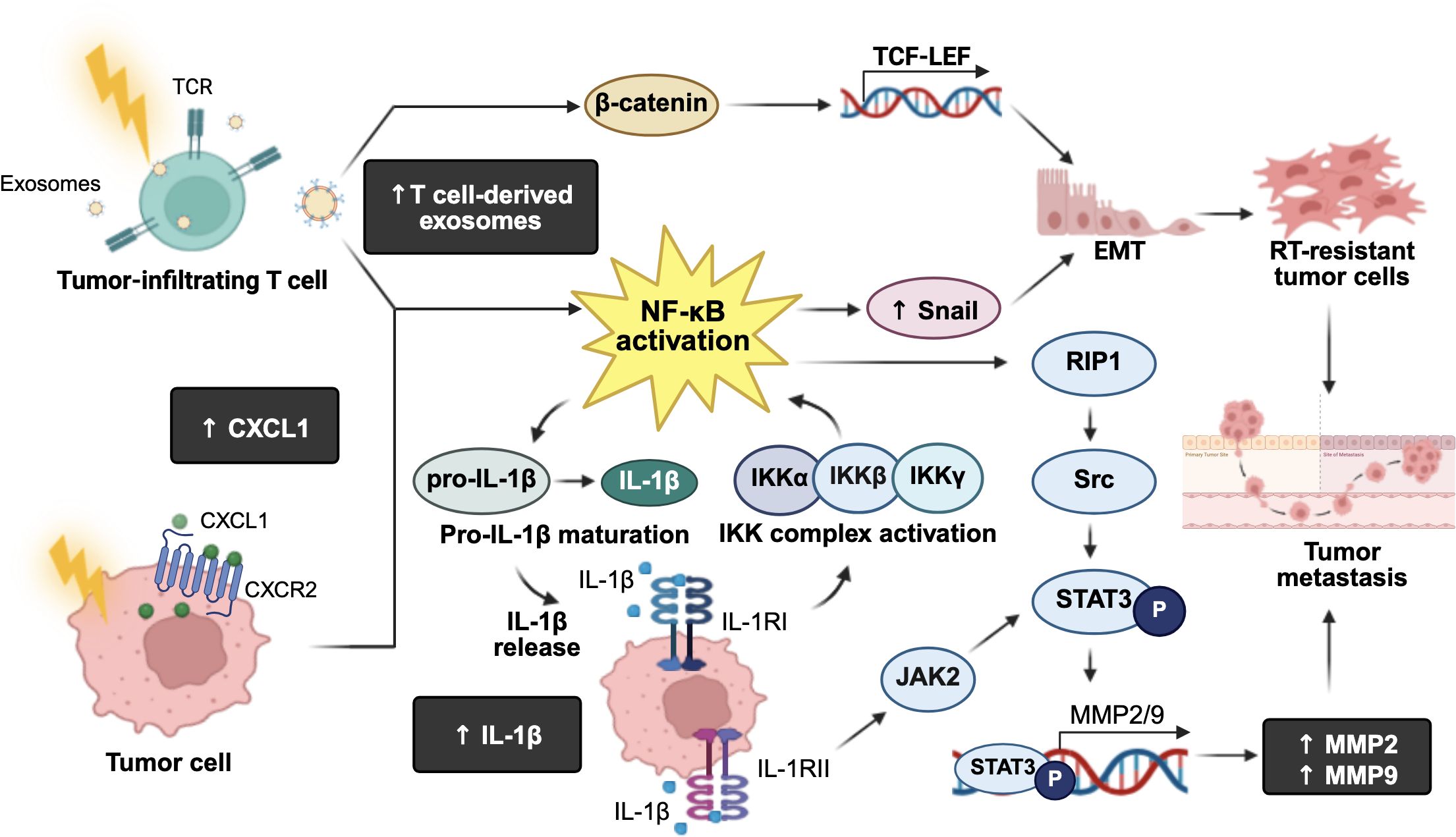
Figure 4. Roles of radiation-induced NF-κB immune modulation leading to cancer metastasis. Exposure to ionizing radiation activates the IκB kinase (IKK) complex, which phosphorylates inhibitor of NF-κB (IκB) proteins, leading to their proteasomal degradation and the subsequent activation of NF-κB. Activated NF-κB triggers the maturation and production of interleukin-1β (IL-1β), and its receptors, IL-1 receptor type I (IL-1RI) and type II (IL-1RII). IL-1RI further activates the IKK complex, creating a positive feedback loop that amplifies NF-κB signaling. On the other hand, IL-1RII activates the Janus kinase 2 (JAK)/signal transducer and activator of transcription 3 (STAT3) pathway, leading to the transcription of matrix metalloproteinase (MMP)-2 and MMP9, which promote tumor metastasis. Additionally, activated NF-κB upregulates MMP2/9 via the receptor-interacting protein kinase 1 (RIP1)/proto-oncogene tyrosine-protein kinase Src/STAT3 signaling pathway, further enhancing tumor metastatic potential. Irradiated tumor-infiltrating T cells-derived exosomes can upregulate β-catenin and NF-κB, inducing Snail expression and transcriptional activation of T cell factor-lymphoid enhancer factor proteins (TCF-LEF) target genes. This results in an upregulation of epithelial-mesenchymal transition (EMT), supporting radioresistant tumor cell survival. Moreover, low-dose radiation increases chemokine C-X-C motif ligand 1 (CXCL1) expression and activates NF-κB signaling to promote EMT, contributing to tumor invasion and metastasis. The figure was created using BioRender (https://BioRender.com).
3 Drug repurposing and clinical trial studies on radiation-induced inflammation in cancer metastasis
While RT remains a cornerstone of cancer management due to its effectiveness in reducing primary tumor burden, it often falls short in preventing post-treatment metastatic progression (17). In parallel, tumor cells can detect radiation-induced DNA damage and activate a cascade of immune signaling pathways to repair the damage, protecting them from therapy-induced cytotoxicity (38). Consequently, strategies have been developed to interfere with DDR pathways in tumor cells, and accumulating evidence suggests that the combination of RT with target agents may enhance the therapeutic efficacy and tumor control. Table 1 provides an overview of FAK, PI3K/AKT pathway, p38 MAPK, and NF-κB inhibitors that have been evaluated in preclinical studies and phase I/II clinical trials for their potential to improve outcomes when used in combination with RT (53, 61–80). However, acquired resistance to these targeted therapies may develop over time, and the risk of excessive adverse events associated with combination regimens remains a concern (81). In light of this, drug repurposing—a strategy that identifies new applications for existing therapeutics—offers a cost-effective and expedited approach to enhance the efficacy of RT (82). By leveraging known pharmacodynamics and safety profiles, repurposed drugs may serve as promising candidates to target key molecular pathways involved in IR-induced metastasis (Table 2) (82). Table 2 summarizes repurposed drugs with potential activity as FAK, PI3K/AKT pathway, p38 MAPK, and NF-κB inhibitors in cancer treatment. (84–100).
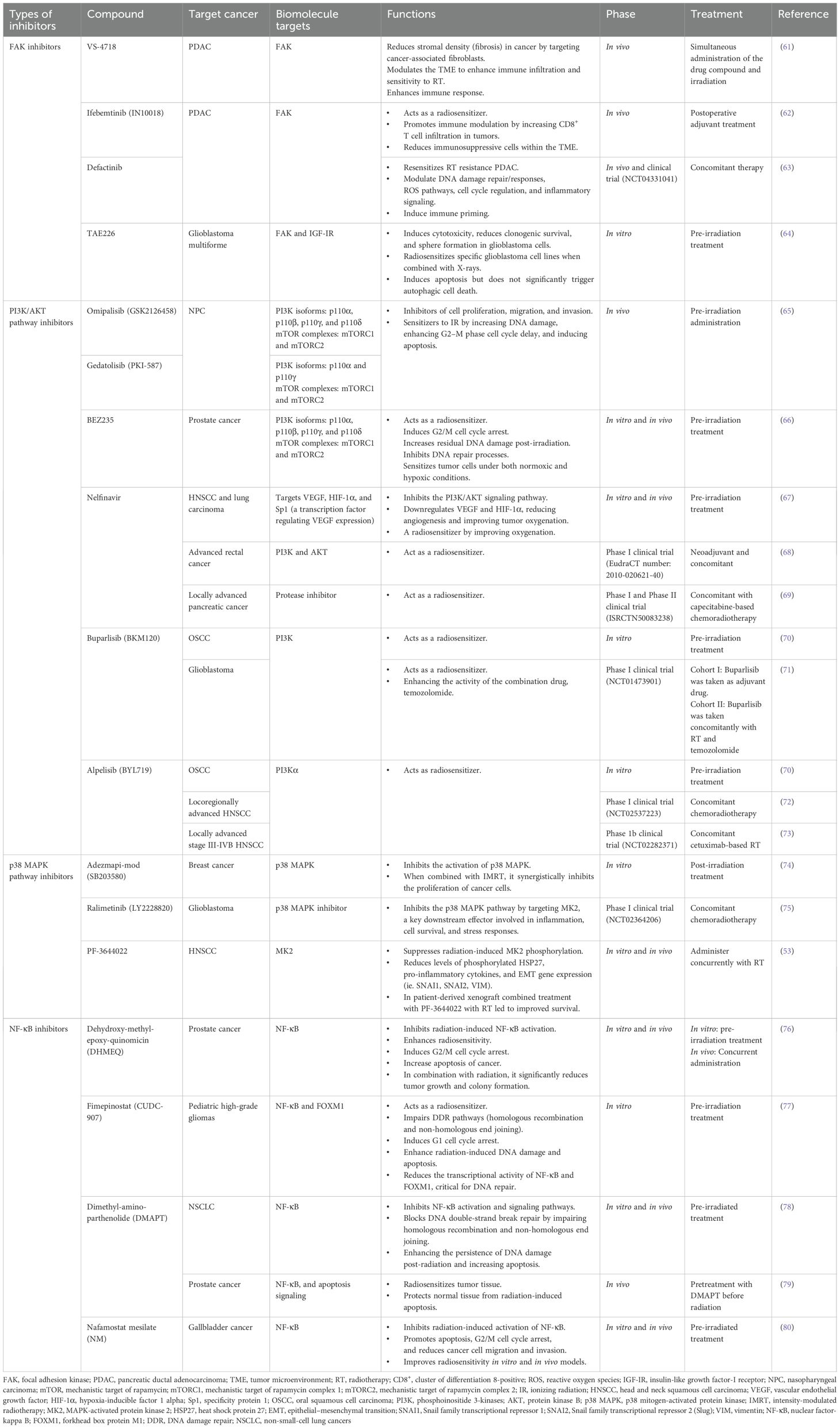
Table 1. Drugs combined with radiotherapy for cancer treatment.
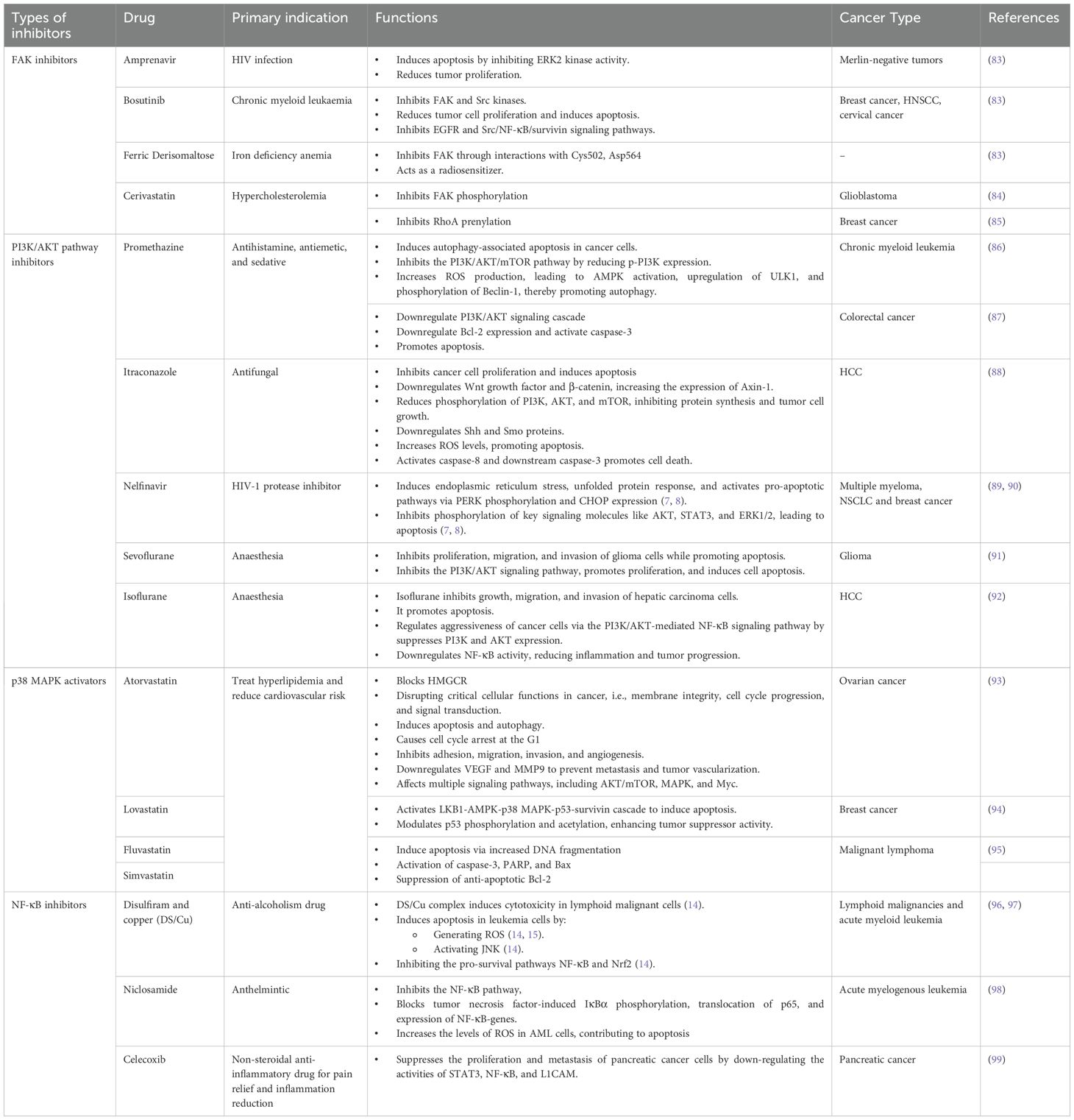
Table 2. List of repurposed drugs for cancer treatment evaluated in preclinical studies.
3.1 Targeting FAK to remodel the TME and enhance radiosensitivity
As discussed earlier, IR-induced focal adhesion kinase (FAK) activation initiates a cascade of intracellular signaling pathways that promote cell survival, migration, and proliferation (34, 100). Given FAK’s central role in these oncogenic pathways, it has emerged as a promising therapeutic target for overcoming radiation-induced cancer progression.
One of the most notable roles of FAK inhibitors is their ability to modulate the TME by altering the composition and behavior of immune and stromal cells within the TME (61). For instance, in the pancreatic cancer mouse model, FAK inhibition by VS-4718 in combination with low-dose radiation (10 Gy) has been shown to reduce stromal density and tumor hypoxia, thereby facilitating immune cell infiltration and enhancing the radiosensitivity of tumor cells (61). Similarly, Osipov and coworkers demonstrated that the adjuvant use of ifebemtinib (IN10018), a FAK inhibitor, improved survival in RT-treated murine pancreatic ductal adenocarcinoma (PDAC) model in a cluster of differentiation 8-positive (CD8+) T cell-dependent manner (62). Although the combination regimen increased the proportions of tumor-associated macrophages (TAMs) and regulatory T cells (Tregs), these immunosuppressive effects were offset by a robust infiltration of CD8+ T cells (62). These findings suggest that targeting FAK may potentially enhance the efficacy of RT by modulating the TME to favor antitumor immune cell infiltration (62). TAE226 (2-(5-chloro-2-(2-methoxy-4-(4-morpholinyl)phenylamino)pyrimidin-4-ylamino)-N-methylbenzamide), a potent adenosine triphosphate (ATP)-competitive FAK inhibitor, has also demonstrated significant antiproliferative and antitumor effects in glioblastoma cells, markedly reducing cell survival in a concentration- and time-dependent manner (64) (64).However, TAE226 exhibited minimal to no effect on enhancing radiosensitivity, suggesting that its efficacy may be tumor type-dependent and highlighting the importance of selecting inhibitors based on tumor-specific biology (64). Despite these promising preclinical findings, no FAK inhibitors have yet received clinical approval, and their efficacy in combination with RT remains to be fully elucidated. A phase II clinical study (NCT04331041) is ongoing to evaluate the efficacy and safety profile of combining FAK inhibitor defacitinib and SBRT in patients with advanced PDAC (63).
Meanwhile, several repurposed drugs have shown potential in inhibiting FAK-mediated signaling pathways and are currently in early-stage preclinical evaluation. For instance, an in silico study by Siyah (2024) identified amprenavir, originally approved for Human Immunodeficiency Virus (HIV) treatment; ferric derisomaltose, used for iron deficiency anemia; and bosutinib, a tyrosine kinase inhibitor, as compounds with high predicted binding affinity for the FAK protein (83). These findings suggest that these agents may directly bind to FAK and effectively inhibit the activation of its downstream oncogenic signaling pathways (83). Cerivastatin, a statin commonly used to lower cholesterol, has been shown to impair actin stress fiber formation and inhibit focal adhesion assembly by downregulating FAK phosphorylation (84). This results in the loss of cell shape and tension, detachment from the ECM, and ultimately reduces migration and invasion of glioblastoma cells (84). In breast tumor cells, cerivastatin exerts its anticancer effects by inhibiting RhoA prenylation, thereby suppressing the RhoA/ROCK pathway and inducing cytoskeletal destabilization, which leads to a loss of traction forces essential for cell migration (85). Additionally, it disrupts the RhoA/FAK/AKT signaling pathway, which regulates gene transcription involved in proliferation and invasion (85). Downstream pathways involving NF-κB, β-catenin, and activator protein-1 (AP-1) are also further suppressed by cerivastatin, collectively contributing to reduced tumor cell proliferation, invasion, and survival (85). These findings suggest that, beyond direct FAK inhibition, agents like statins can indirectly target FAK-related signaling to reduce tumor invasiveness and potentially improve radiosensitivity, expanding therapeutic options.
Taken together, these findings underscore the promising potential of targeting the FAK signaling cascade to counteract metastasis and enhance radiosensitivity. Moving forward, the integration of FAK inhibitors with RT warrants rigorous clinical validation through well-designed trials to confirm their therapeutic benefit while minimizing toxicity and adverse effects (11).
3.2 Targeting PI3K/AKT pathway for radiosensitization
Dysregulation of the PI3K/AKT pathway in cancer cells is well known to contribute to drug and treatment resistance (101). Given the differential activation of this pathway in tumor cells compared to normal cells, targeting it has been proposed as a promising strategy to enhance tumor cell sensitivity and improve therapeutic efficacy (101). As such, investigating inhibitors of the PI3K/AKT pathway represents a worthwhile strategy to overcome treatment resistance in cancer 102.
To date, multiple ongoing clinical trials are evaluating the safety and efficacy of drugs targeting the PI3K/mechanistic target of rapamycin (mTOR) pathways (66, 71, 73, 102). Emerging evidence suggests that combining these agents with RT enhances antitumor efficacy (66, 71, 73, 102). For instance, dual PI3K/mTOR inhibitors such as omipalisib (GSK2126458/GSK458), gedatolisib (PKI-587), and dactolisib (BEZ235) have been shown to effectively suppress the phosphorylation of mTOR, AKT, and eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) in NPC and prostate cancer cells. This suppression leads to improved antitumor activity and radiosensitization, primarily through inhibition of DNA repair, induction of G2/M cell cycle arrest, and increased apoptosis (66, 67).
In a phase I clinical study (NCT00972686), omipalisib demonstrated tolerability at a dose of 2.5 mg/day in patients with breast, renal cell, bladder, or endometrial cancers, without any life-threatening adverse events (102). However, durable objective responses were observed in only 5% of patients, with two patients achieving sustained responses lasting over four years. Notably, PIK3CA expression did not correlate with clinical outcomes in this study (102) (102).In contrast, a phase Ib clinical trial (NCT02282371) evaluating alpelisib (BYL719), an oral class I α-specific PI3K inhibitor, in combination with cetuximab and IMRT, reported promising outcomes in patients with locally advanced HNSCC, particularly among those harboring PIK3CA mutations (73). This differential clinical efficacy may be attributed to the distinct targeting profiles of the two agents. Alpelisib primarily inhibits both the wild-type and mutant PI3Kα isoforms, whereas omipalisib targets a broader range of PI3K isoforms, including mutant forms of PIK3CA and mTOR complexes (73, 102). Despite this broader targeting capacity, omipalisib has been shown to primarily target the mTOR signaling pathway, with its downstream proteins being significantly downregulated in a dose-dependent manner (103, 104). The findings suggest that distinct mechanisms of action of PI3K inhibitors may contribute to variations in clinical efficacy across diverse cancer types. Nonetheless, these results highlight the urgent need for more reliable biomarkers to identify patients who are likely to respond to PI3K inhibitors, in order to improve therapeutic efficacy and clinical outcomes.
Besides that, buparlisib (BKM120), a pan-class I PI3K inhibitor, has demonstrated synergistic effects with radiation and mTOR inhibitors in preclinical models of oral squamous cell carcinoma (OSCC) (70). However, its clinical application has been hindered by toxicity (70, 71). In a phase I study (NCT01473901) evaluating buparlisib in combination with temozolomide and RT in patients with newly diagnosed glioblastoma, the trial was terminated early due to the inability to determine the maximum tolerated dose, coupled with a high incidence of adverse events (71). This, in turn, raises concerns about the viability of buparlisib for future development 72.
Given the limitations of current targeted therapies, several non-oncogenic drugs, such as promethazine, itraconazole, and nelfinavir, have shown promising anticancer activity by targeting the PI3K signaling pathway. For instance, promethazine, a first-generation antihistamine with sedative and antiemetic properties, has been shown to inhibit the PI3K/AKT/mTOR signaling in chronic myeloid leukemia by downregulating phosphorylated PI3K (p-PI3K) (86). This relieves autophagy suppression and activates AMP-activated protein kinase (AMPK), which further inhibits mTOR and downregulates the anti-apoptotic protein myeloid cell leukemia 1 (MCL-1), thereby promoting autophagy. (86) Similarly, in colorectal cancer models, promethazine has been shown to reduce p-PI3K and phosphorylated AKT (p-AKT) levels, suppress cell proliferation, and induce apoptosis by downregulating B-cell lymphoma 2 (Bcl-2) and activating caspase-3 (87). Itraconazole, an antifungal agent, suppresses HCC cell growth and induces apoptosis via multiple mechanisms, including inhibition of the Hedgehog, Wnt/β-catenin, and AKT/mTOR/ribosomal protein S6 kinase (S6K) signaling pathways, as well as by inducing ROS production and activating death receptor signaling 89.
Notably, nelfinavir, originally developed as an HIV protease inhibitor, has been extensively studied for its antitumor activity due to its ability to inhibit the PI3K/AKT/mTOR signaling pathways and function as a radiosensitizer (105, 106). In multiple myeloma, nelfinavir induces apoptotic cell death by activating caspase 3 and downregulating the phosphorylation of AKT, STAT3, and ERK1/2, which leads to activation of the pro-apoptotic unfolded protein response pathway (89). Beyond its pro-apoptotic effects, nelfinavir also modulates the TME by downregulating hypoxia-inducible factor 1 alpha (HIF-1α) and VEGF, thereby improving tumor oxygenation and suppressing angiogenesis (67). This effect is mediated through inhibition of AKT signaling and reduced specificity protein 1 (Sp1) phosphorylation (67). The therapeutic efficacy of nelfinavir has been evaluated in a non-randomized, open-label clinical trial, SONATINA (European Union Drug Regulating Authorities Clinical Trial Database, EudraCT number: 2010-020621-40), in combination with RT in patients with advanced rectal cancer (68). This combination regimen was well tolerated and associated with increased tumor perfusion (68). Notably, the median tumor cell density decreased from 24.3% at baseline to 9.2% seven days post-treatment, supporting its potential as a radiosensitizer for targeting metastatic disease (68). However, the therapeutic efficacy of nelfinavir appears to be cancer-type dependent. Despite evidence that nelfinavir induces AKT inhibition in both in vitro and in vivo models of prostate cancer, it did not lead to improvements in local tumor control and radiosensitivity (107). Similarly, in the multi-center, two-stage SCALOP-2 trial (ISRCTN50083238), the combination of nelfinavir with capecitabine-based chemoradiotherapy was well tolerated in patients with locally advanced pancreatic cancer, but it failed to improve progression-free survival (PFS) (69, 108). These findings suggest that tumor types may potentially play critical roles in shaping the cytotoxicity of nelfinavir, underscoring the need for cancer-specific therapeutic strategies and more reliable predictive biomarkers.
Overall, clinical outcomes indicate that PI3K/AKT pathway inhibitors may benefit only a subset of patients whose tumors are highly dependent on this signaling axis. Despite encouraging preclinical results, their translation into clinical success has been limited, primarily due to challenges in patient stratification and the management of treatment-related toxicities. Continued investigation is warranted, particularly in identifying robust predictive biomarkers, such as PNUTS overexpression and PTEN loss, which could help refine patient selection and enhance the therapeutic efficacy of PI3K/AKT/mTOR-targeted therapies (109).
3.3 p38 MAPK pathway in therapy implications
Activation of p38 MAPK in response to stressors such as IR has been implicated in promoting metastasis by driving inflammation, EMT, and increased cellular motility (110). Therefore, targeting p38 MAPK represents a promising strategy for radiosensitization. Selective p38 inhibitors such as adezmapimod (SB203580) and ralimetinib (LY2228820) have demonstrated synergistic effects with RT in preclinical and early clinical studies by enhancing apoptosis and impairing DNA repair mechanisms (74, 75). For instance, Zhao et al. (2018) showed that adezmapimod (SB203580), a selective p38α and p38β MAPK inhibitor, enhanced tumor suppression and reduced proliferation in MCF-7 breast cancer cells when combined with RT (74). Similarly, encouraging results were reported in a phase I clinical trial evaluating ralimetinib (LY2228820), a selective, ATP-competitive p38 MAPK inhibitor, in combination with standard chemoradiotherapy (temozolomide + RT) for glioblastoma patients (75). The trial showed a median PFS of 12.8 months and an overall survival (OS) of 24.5 months, outperforming historical benchmarks (75). These early-phase clinical trial results illustrated the therapeutic potential of p38 MAPK inhibition for radiosensitization. Nevertheless, larger-scale studies and biomarker-driven trials are needed to determine whether p38 inhibition can be effectively integrated into standard cancer therapies.
Despite numerous p38 MAPK inhibitors being investigated, none have received regulatory approval for clinical use (98). This is largely due to the pathway multifaceted roles, limited sustained efficacy, and significant adverse effects such as hepatotoxicity, cardiotoxicity, and tachyphylaxis, all of which collectively complicate the assessment of their therapeutic benefits in cancer treatment (111). To address these limitations, statins have emerged as potential modulators of the p38 MAPK pathway. Statins such as atorvastatin, lovastatin, fluvastatin, and simvastatin have been shown to induce apoptosis and G1 cell cycle arrest in ovarian, breast, and lymphoma cells via AKT/mTOR inhibition and p38 MAPK activation (93–95). However, this mechanism appears to conflict with the strategies proposed by Zhao et al. (2018) and Biau et al. (2021), where p38 MAPK inhibition suppressed IR-induced tumor metastasis (74, 75). These contrasting findings suggest that p38 MAPK may act as either a tumor suppressor or promoter, depending on the cancer type and TME, further complicating its utility as a therapeutic target in oncology.
Given these complexities, recent cancer research has shifted toward targeting MK2, the downstream effector of p38 MAPK (111). MK2 is a kinase involved primarily in regulating inflammatory responses, cell cycle progression, apoptosis, and cell motility (112). Its activation has been shown to upregulate the production of proinflammatory cytokines and has been implicated in various inflammatory diseases (113, 114). Given the established link between inflammation and cancer, MK2 is likely to play a significant role in cancer development and progression (113, 114). Indeed, MK2 inhibition has shown promise in combination with RT to suppress tumor cell growth and reduce metastatic potential. For instance, shRNA-mediated silencing of MK2 in bladder cancer cells has been shown to significantly enhance apoptotic cell death following RT via the upregulation of caspase 3 activity and cytochrome c release (116). Berggren et al. (2019) demonstrated that pharmacologic inhibition of MK2 using PF-3644022 and genetic silencing significantly reduced RT-induced MK2 phosphorylation and proinflammatory cytokine expression (53). Moreover, the combination of PF-3644022 with RT markedly reduced tumor growth and improved survival in patient-derived xenograft models of HNSCC (53). Collectively, these findings suggest that MK2 pathway activation may potentially contribute to RT resistance and that its inhibition could enhance tumor radiosensitivity.
In summary, while both p38 MAPK and its downstream effector MK2 have emerged as promising targets in cancer therapy, their clinical potential remains underexplored. Given their significant roles in regulating inflammation, metastasis, and treatment resistance, further investigation is warranted to validate the therapeutic benefit of targeting these kinases, particularly in combination with RT.
3.4 Targeting NF-κB signaling to curb inflammation in cancer
Given the crucial roles of NF-κB in cancer development and progression, targeting it has been extensively explored over the past few decades (27). A few compounds, such as dehydroxy-methyl-epoxy-quinomicin (DHMEQ), fimepinostat (CUDC-907), and dimethylaminoparthenolide (DMAPT), have been proposed as potential radiosensitizers (76–78, 80). These NF-κB inhibitors primarily exert their cytotoxic effects through inducing G2/M cell cycle arrest and inhibiting DNA repair pathways, thereby sensitizing tumor cells to radiation and reducing cell proliferation (76–78, 80). For instance, the combination of DHMEQ, a compound derived from a natural product, with 4 Gy of irradiation produced a synergistic antitumor effect in LNCaP and PC-3 prostate cancer cells by inhibiting radiation-induced NF-κB and upregulating the expression of cell cycle-related proteins, such as p53 and p21 in LNCaP, and 14-3-3σ in PC-3 cells (98). This collectively leads to enhanced G2/M cell cycle arrest (76). In vivo, the combination of DHMEQ with IR significantly reduced tumor volume in PC-3 xenograft models compared to either treatment alone (76). Similarly, combining CUDC-907, a dual PI3K/HDAC inhibitor, or DMAPT, an orally active NF-κB inhibitor, with irradiation sensitized glioma and NSCLC cells, respectively, by inhibiting DNA repair (77, 78). In glioma cells, this combination treatment markedly increased p21 expression and reduced phosphorylated CDK1 levels, further enhancing radiosensitivity (77). Collectively, targeting NF-κB, alone or in combination with PI3K/HDAC inhibition, effectively enhances radiosensitivity across multiple cancer types by disrupting DNA repair and promoting G2/M cell cycle arrest, supporting further translational efforts to validate these compounds in clinical settings (77).
Although current evidence supporting the use of NF-κB inhibitors to prevent radiation-induced metastasis is largely confined to preclinical models, several compounds have demonstrated clinical promise based on early-phase safety evaluations. For example, fimepinostat has demonstrated antitumor activity by downregulating p-PI3K downstream targets, increasing histone H3 acetylation, reducing cellular myelocytomatosis (Myc) protein levels, upregulating pro-apoptotic proteins, and inducing G1 cell cycle arrest (115, 116). These effects have been shown to associate with an inhibition of NF-κB signaling (115, 116). Fimepinostat has been investigated in phase I (NCT01742988) and II (NCT02674750) clinical trials in patients with relapsed or refractory diffuse large B-cell lymphoma and high-grade B-cell lymphoma, respectively, demonstrating a durable antitumor response with an acceptable safety profile at 60 mg, administered daily for 5 consecutive days followed by a 2-day break (117, 118). Notably, patients with Myc-altered disease demonstrated an encouraging objective response rate of 64% (7 out of 11 patients), compared to only 29% (2 out of 7 patients) in those with unaltered Myc (119). It is anticipated that the therapeutic efficacy of fimepinostat may be enhanced through combination with other conventional therapies or by selecting patients with Myc overexpression, warranting further investigation.
DMAPT, a water-soluble analog of the natural sesquiterpene lactone parthenolide, has been shown to inhibit NF-κB signaling and eradicate tumor cells in various cancers, including acute myelogenous leukemia, NSCLC, prostate cancer, and PDAC (120–123). Preclinical studies have demonstrated that DMAPT-mediated NF-κB inhibition induces apoptotic cell death by impairing DSB repair, increasing c-Jun phosphorylation, promoting oxidative stress responses, and activating the tumor suppressor p53 (120, 121, 123). Importantly, DMAPT has also been shown to selectively target the tumor cells while protecting neighboring healthy tissues from cancer therapy-induced damage. Morel and co-workers (2017) reported that treatment with DMAPT in a prostate cancer mouse model, administered prior to a combination of low (10 mGy) and high doses (6 Gy) whole-body X-ray, doubled radiation-induced apoptosis in prostate tumor tissue while reducing apoptosis in normal prostate tissues and distant tissues, such as the spleen tissues and colorectal tissues, by 71.7%, 48.2%, and 38.0%, respectively (79). In addition, DMAPT markedly reduces RT-induced fibrosis in normal tissues following long-term exposure to RT, as demonstrated in a preclinical, fractionated irradiation mouse model (124). These findings suggest that combining DMAPT with RT not only enhances the tumor radiosensitivity but may also serve as a radioprotective strategy to minimize RT-induced side effects in normal tissues. Despite its promise, clinical data on DMAPT remain very limited and unpublished to date. A phase I clinical trial conducted by Cardiff University in patients with hematological malignancies demonstrated promising pharmacokinetics with no reported normal tissue toxicity induced by DMAPT (122). Further clinical studies are warranted to validate the therapeutic potential of DMAPT as a radiosensitizer and/or radioprotective agents in well-designed trials, thereby advancing its clinical application.
Another NF-κB inhibitor, DHMEQ, has demonstrated strong anticancer and anti-inflammatory potential in animal models without signs of toxicity (125). Treatment with DHMEQ in both in vitro and in vivo models of OSCC have been shown to suppress tumor growth and survival in a dose-dependent manner by inducing DNA fragmentation and downregulating anti-apoptotic proteins such as survivin (126). Similarly, DHMEQ exhibited dose-dependent anti-proliferative and anti-invasive effects against NPC cells by inducing G2/M cell cycle arrest and inhibiting TNF-α-induced NF-κB activity (127). Furthermore, the combination of DHMEQ with RT effectively suppressed prostate cancer cell growth and induced G2/M cell cycle arrest compared to RT alone (76). These findings suggest that DHMEQ could potentially overcome radioresistance and RT-induced metastasis, warranting further investigation in early-phase clinical trials to establish a safe and efficacious dose in patients.
In addition to conventional NF-κB inhibitors, several promising repurposed drugs targeting the NF-κB pathway have been identified, including nafamostat mesylate, a synthetic serine protease inhibitor; disulfiram-copper complex, used to treat alcohol use disorder; niclosamide, an anti-helminthic agent; and celecoxib, a non-steroidal anti-inflammatory drug. These agents warrant further investigation for their potential clinical benefits in NF-κB-mediated cancer development (96, 97). For instance, nafamostat mesylate has been shown to inhibit radiation-induced NF-κB activity and exert antitumor effects in gallbladder cancer by promoting apoptosis and enhancing cell cycle arrest (80). In vivo, the combination treatment of nafamostat and X-ray (5 Gy) significantly reduced tumor burden without causing body weight loss, indicating a favorable toxicity profile for potential clinical translation (80). Overall, NF-κB inhibitors show promising radiosensitizing potential, particularly by inducing cell cycle arrest and enhancing DNA damage.
4 The future with radiogenomics
Despite promising results from preclinical and early-phase clinical studies targeting these pathways in cancers, only a limited number of agents have been successfully translated into clinical applications due to their unclear therapeutic effects and undesirable side effects (70, 71, 83, 111). Furthermore, our understanding of the precise mechanisms underlying radiation-induced inflammation in metastasis and radioresistance remains limited. Since these events are typically mediated by multiple regulatory pathways that vary across cancer types, tailored molecular interventions or combination therapies are likely required for different tumor subtypes. Additionally, tumor heterogeneity further complicates the development of effective and personalized cancer treatments, posing a major challenge to advancing therapeutic strategies (15). Despite the well-established role of online databases on cancer studies, overcoming these challenges will require the integration of advanced technologies coupled with comprehensive databases to decode the complexity of radioresistance and enable the development of safer and more effective therapeutic strategies.
Recent advances in genomic sequencing technologies and image acquisition have already begun to support this effort. These tools enable the identification of prognostic and predictive biomarkers, enhancing early detection, disease prognosis, and treatment selection. In particular, radiogenomics, an emerging tool in precision medicine that leverages artificial intelligence (AI) to correlate genomic alterations (e.g., gene mutations and expression patterns) and imaging features, has gained significant attention in precision oncology (128). By integrating molecular and radiological data, radiogenomics offers deeper insights into tumor biology and tumor heterogeneity, potentially leading to the discovery of optimal biomarkers to improve cancer prognosis and treatment outcomes (128).
In general, the workflow of radiogenomics involves image acquisition, region of interest (ROI) identification and segmentation, quantitative image feature extraction, data mining and informatics analysis, and modeling (128). It relies on high-throughput computing as well as machine learning and deep learning algorithms to manage and analyze a large amount of imaging datasets (129). Compared to traditional imaging approaches, radiogenomics offers a non-invasive and less time-consuming alternative to assist clinicians in early diagnosis and disease monitoring, potentially reducing the risk of recurrence and cancer metastasis. In clinical practice, follow-up imaging after RT is essential for assessing treatment response. Hence, integrating radiomic or radiogenomic analysis into routine imaging workflows can aid in the detection of residual cancer cells and offer insights into resistance mechanisms and therapeutic adaptation (128).
Moreover, the development of AI-powered models enables more accurate characterization of tumor heterogeneity, assesses metastatic potential, and helps uncover clinically relevant imaging signatures that may be imperceptible to the human eye (129). By analyzing radiomic signatures and differentially expressed genes, researchers can gain insight into molecular mechanisms underlying cancer development and progression. For example, Zeng et al. (2021) developed a radiogenomic predictive model to study molecular characteristics and forecast OS of patients with ccRCC by integrating gene expression data from The Cancer Genome Atlas (TCGA) with matched contrast-enhanced computed tomography images from The Cancer Imaging Archive (TCIA) (130). The model demonstrated strong predictive capability in identifying several genetic mutations and mRNA-based molecular subtypes in ccRCC patients, achieving an area under the curve (AUC) ranging from 0.949 to 0.973 (130). By incorporating multi-omics data, such as genomic, transcriptomic, and proteomic information into the radiomic analysis, the model resulted in accurate predictions of OS in ccRCC patients with an AUC of 0.846 compared to 0.755 for the radiomics model alone (130). These findings support the potential of radiogenomics as a non-invasive tool for personalized prognosis and treatment planning, reducing the reliance on invasive tissue biopsies in clinical practice.
Furthermore, recent advancements in computational methods have positioned the Connectivity Map (CMap), Reactome or Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) (131) as some powerful in silico platforms that match disease-specific gene expression signatures with drug-induced transcriptional profiles (132), enabling the identification of compounds capable of reversing pathological expression patterns (131, 132). The integration of radiogenomic approaches with in silico platforms is anticipated to offer a cost-effective and scalable strategy for discovering candidate therapeutics targeting radiation-induced metastatic pathways (129, 131). This integration may further advance personalized medicine by aligning targeted therapies with patient-specific molecular profiles, ultimately maximizing therapeutic efficacy (133). Figure 5 illustrates the integration of radiomic and other omics data (134, 135), which supports the development of predictive models to assess treatment response and metastatic risk following RT.
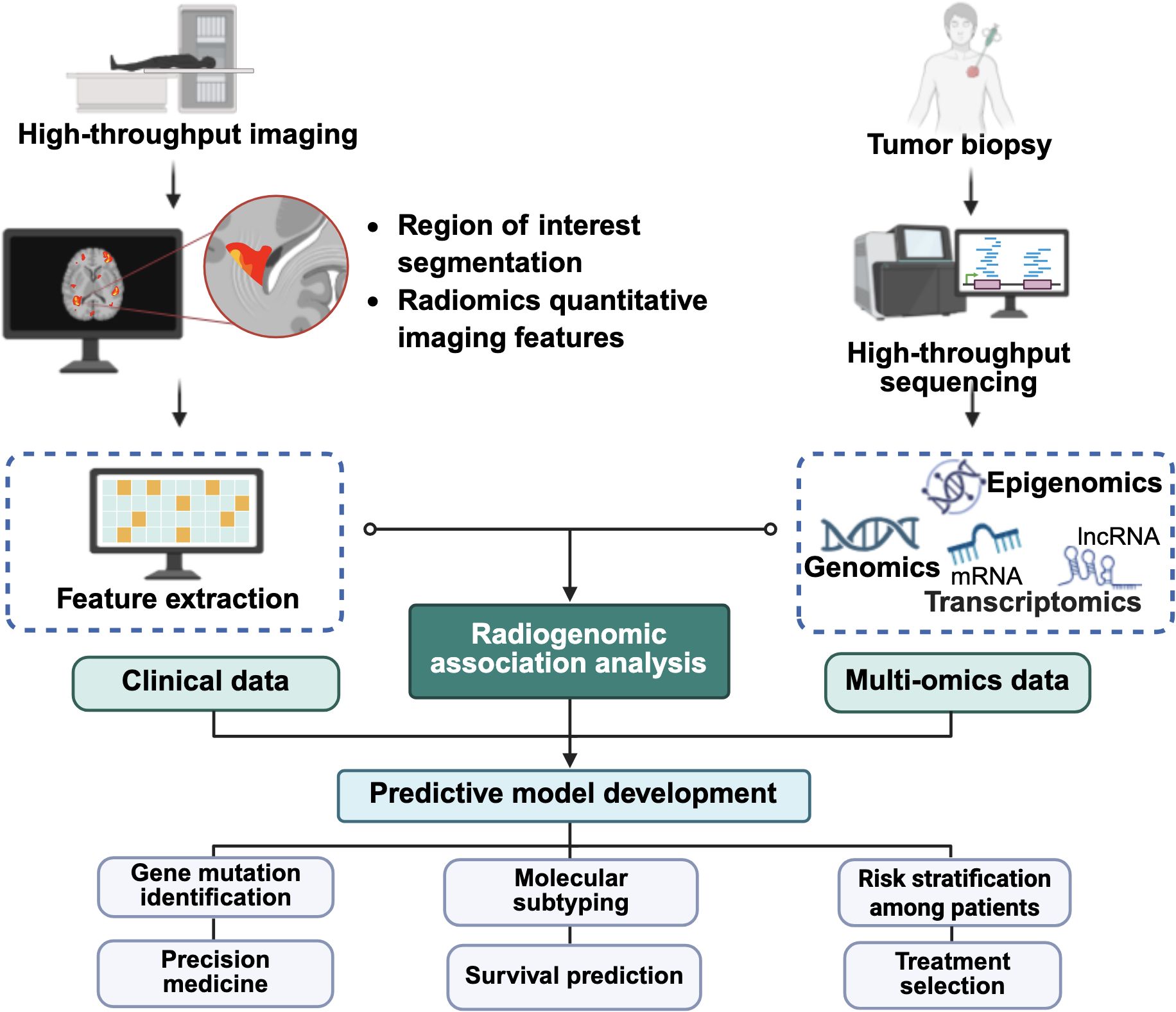
Figure 5. Integration of multi-omics analysis and artificial intelligence in cancer management. The schematic illustrates how medical imaging, clinical, genomic, transcriptomic, and other omics data are integrated and fed into artificial intelligence-driven radiogenomic analysis to develop predictive models with potential applications in gene mutation prediction, survival prognosis, molecular subtyping, treatment risk stratification, and treatment selection. The figure was created using BioRender (https://BioRender.com).
5 Conclusion
RT can inadvertently promote tumor cell survival and metastatic potential and thus a deeper understanding of these mechanisms is important to develop more effective treatments. With the increasing demand for effective cancer treatments, drug repurposing along with the advances in gene sequencing technologies now enable comprehensive tumor profiling, to repositioning drug candidate in cancer treatment. Radiogenomics further offers a non-invasive, cost-effective approach to assess tumor heterogeneity, holding great potential to revolutionize diagnosis, treatment planning, and prognosis. Nevertheless, further research is needed to refine AI algorithms, enhance multi-omics data integration, discover reliable imaging biomarkers, and validate the clinical applicability of radiogenomics across diverse cancer types. Ultimately, these advancements will pave the way for more personalized, efficient, and cost-effective cancer treatment strategies.
Author contributions
EL: Writing – original draft, Formal Analysis, Visualization, Data curation. C-KL: Formal Analysis, Visualization, Data curation, Writing – original draft. LT: Supervision, Writing – review & editing. YC: Supervision, Writing – review & editing. W-ML: Supervision, Writing – review & editing. LF: Validation, Writing – review & editing. C-OL: Resources, Visualization, Formal Analysis, Writing – review & editing. KC: Supervision, Resources, Writing – review & editing. C-WM: Conceptualization, Resources, Supervision, Visualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The authors would like to express their sincere gratitude to research grant provided by the Malaysia Ministry of Higher Education through the Fundamental Research Grant Scheme (Grant Number: FRGS/1/2023/SKK10/UCSI/02/1).
Acknowledgments
The authors would like to express their sincere gratitude to IMU University, Monash University and UCSI University for their kind support and library resources for completing this review. All figures in this review paper were created by the authors using BioRender.com.
Conflict of interest
Author C-OL was employed by company AGTC Genomics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript in proofreading any potential grammatical or typo-errors only. FAK, focal adhesion kinase; HIV, Human Immunodeficiency Virus; ERK2, extracellular signal-regulated kinases 2; Src, Src kinase; EGFR, epidermal growth factor receptor; NF-κB, nuclear factor-kappa B; HNSCC, head and neck squamous cell carcinoma; RhoA, Ras homolog family member A; PI3K, phosphoinositide 3-kinases; AKT, protein kinase B; mTOR, mechanistic target of rapamycin; p-PI3K, phosphorylated phosphoinositide 3-kinases; ROS, reactive oxygen species; AMPK, AMP-activated protein kinase; ULK1, Unc-51 like autophagy activating kinase 1; Bcl-2, B-cell lymphoma 2; Axin-1, axis inhibition protein 1; Shh, Sonic hedgehog proteins; Smo, Smoothened proteins; PERK, protein kinase RNA-like endoplasmic reticulum kinase; CHOP, C/EBP homologous protein; STAT3, signal transducer and activator of transcription 3; NSCLC, non-small-cell lung cancers; HCC, hepatocellular carcinoma; p38 MAPK, p38 mitogen-activated protein kinase; HMGCR, HMG-CoA reductase; VEGF, vascular endothelial growth factor; MMP9, matrix metalloproteinase 9; Myc, cellular myelocytomatosis; LKB1, liver kinase B1; PARP, Poly (adenosine diphosphate-ribose) polymerase; Bax: Bcl-2 associated X protein; JNK, c-Jun N-terminal kinase; Nrf2, Nuclear factor erythroid 2–related factor 2; IκBα, NF-κB protein inhibitors alpha; LICAM, L1 cell adhesion molecule.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Latrèche A, Bourbonne V, and Lucia F. Unrecognized thoracic radiotherapy toxicity: A review of literature. Cancer Radiotherapie: J la Societe Francaise Radiotherapie Oncologique. (2022) 26:616–21. doi: 10.1016/j.canrad.2021.10.008
2. Zhang S, Zeng N, Yang J, He J, Zhu F, Liao W, et al. Advancements of radiotherapy for recurrent head and neck cancer in modern era. . Radiat Oncol (London England). (2023) 18. doi: 10.1186/s13014-023-02342-0
3. Gianfaldoni S, Gianfaldoni R, Wollina U, Lotti J, Tchernev G, and Lotti T. An overview on radiotherapy: from its history to its current applications in dermatology. Open Access Macedonian J Med Sci. (2017) 5:521. doi: 10.3889/oamjms.2017.122
4. Vilalta M, Rafat M, and Graves EE. Effects of radiation on metastasis and tumor cell migration. Cell Mol Life Sciences: CMLS. (2016) 73:2999. doi: 10.1007/s00018-016-2210-5
5. Du T, Xiao J, Qiu Z, and Wu K. The effectiveness of intensity-modulated radiation therapy versus 2D-RT for the treatment of nasopharyngeal carcinoma: A systematic review and meta-analysis. PloS One. (2019) 14. doi: 10.1371/journal.pone.0219611
6. Zhang W, Xie Q, Zhu B, Wang X, He L, and Zhang Y. Intensity-modulated radiotherapy with more than 60 Gy improved the survival of inoperable patients with locally advanced esophageal squamous cell carcinoma: A population-based real-world study. Medicine. (2022) 101:e29166. doi: 10.1097/MD.0000000000029166
7. Tateishi Y, Takeda A, Horita N, Tsurugai Y, Eriguchi T, Kibe Y, et al. Stereotactic body radiation therapy with a high maximum dose improves local control, cancer-specific death, and overall survival in peripheral early-stage non-small cell lung cancer. Int J Radiat Oncol Biol Physics. (2021) 111:143–51. doi: 10.1016/j.ijrobp.2021.04.014
8. Huang SY, Wu CT, Liu DW, Wang TH, Liao YH, Chen YW, et al. Dose escalation (81 gy) with image-guided radiation therapy and volumetric-modulated arc therapy for localized prostate cancer: A retrospective preliminary result. Tzu-Chi Med J. (2019) 32:75. doi: 10.4103/tcmj.tcmj_2_19
9. Zhao L, Pang Y, Zhou Y, Chen J, Fu H, Guo W, et al. Antitumor efficacy and potential mechanism of FAP-targeted radioligand therapy combined with immune checkpoint blockade. Signal Transduction Targeted Ther. (2024) 9:1–17. doi: 10.1038/s41392-024-01853-w
10. Wu L, Li B, Wan G, Wang Y, Zhu J, Liang L, et al. Toripalimab plus chemotherapy and radiotherapy for treatment-naive advanced esophageal squamous cell carcinoma: A single-arm phase 2 trial. Nat Commun. (2024) 15:1–12. doi: 10.1038/s41467-024-51105-2
11. Baskar R, Lee KA, Yeo R, and Yeoh KW. Cancer and radiation therapy: current advances and future directions. Int J Med Sci. (2012) 9:193. doi: 10.7150/ijms.3635
12. Petroni G, Cantley LC, Santambrogio L, Formenti SC, and Galluzzi L. Radiotherapy as a tool to elicit clinically actionable signalling pathways in cancer. Nat Rev Clin Oncol. (2021) 19:114. doi: 10.1038/s41571-021-00579-w
13. Raab G, Yu Y, Sherman E, Wong R, Mell LK, Lee NY, et al. Nomogram to predict risk of early mortality following definitive or adjuvant radiation and systemic therapy for head and neck cancer. Clin Trans Radiat Oncol. (2024) 45:100725. doi: 10.1016/j.ctro.2024.100725
14. Chen S, Yang D, Liao X, Lu Y, Yu B, Xu M, et al. Failure patterns of recurrence and metastasis after intensity-modulated radiotherapy in patients with nasopharyngeal carcinoma: results of a multicentric clinical study. Front Oncol. (2022) 11:693199. doi: 10.3389/fonc.2021.693199
15. Zhu L, Jiang M, Wang H, Sun H, Zhu J, Zhao W, et al. A narrative review of tumor heterogeneity and challenges to tumor drug therapy. Ann Trans Med. (2021) 9:1351. doi: 10.21037/atm-21-1948
16. Blyth BJ, Cole AJ, MacManus MP, and Martin OA. Radiation therapy-induced metastasis: radiobiology and clinical implications. Clin Exp Metastasis. (2018) 35:223–36. doi: 10.1007/s10585-017-9867-5
17. von Essen CF. Radiation enhancement of metastasis: A review. Clin Exp Metastasis. (1991) 9:77–104. doi: 10.1007/BF01756381
18. Liu C, Li X, Huang Q, Zhang M, Lei T, Wang F, et al. Single-cell RNA-sequencing reveals radiochemotherapy-induced innate immune activation and MHC-II upregulation in cervical cancer. Signal Transduction Targeted Ther. (2023) 8. doi: 10.1038/s41392-022-01264-9
19. Chang L, Liu J, Zhu J, Guo S, Wang Y, Zhou Z, et al. Advancing precision medicine: the transformative role of artificial intelligence in immunogenomics, radiomics, and pathomics for biomarker discovery and immunotherapy optimization. Cancer Biol Med. (2025) 22:33–47. doi: 10.20892/j.issn.2095-3941.2024.0376
20. Lee SH, Cheng H, Yuan Y, and Wu S. Regulation of ionizing radiation-induced adhesion of breast cancer cells to fibronectin by alpha5beta1 integrin. Radiat Res. (2014) 181:650–8. doi: 10.1667/RR13543.1
21. Eke I, Makinde AY, Aryankalayil MJ, Reedy JL, Citrin DE, Chopra S, et al. Long-term tumor adaptation after radiotherapy: therapeutic implications for targeting integrins in prostate cancer. Mol Cancer Research : MCR. (2018) 16:1855–64. doi: 10.1158/1541-7786.MCR-18-0232
22. Lee M, Lee HJ, Seo WD, Park KH, and Lee YS. Sialylation of integrin beta1 is involved in radiation-induced adhesion and migration in human colon cancer cells. Int J Radiat Oncology Biology Physics. (2010) 76:1528–36. doi: 10.1016/j.ijrobp.2009.11.022
23. Yu D, An X, Fan W, Wang X, He Y, and Li B. PNUTS mediates ionizing radiation-induced CNE-2 nasopharyngeal carcinoma cell migration, invasion, and epithelial–mesenchymal transition via the PI3K/AKT signaling pathway. OncoTargets Ther. (2019) 12:1205. doi: 10.2147/OTT.S188571
24. Zhang H, Song Y, Zhou C, Bai Y, Yuan D, Pan Y, et al. Blocking endogenous H2S signaling attenuated radiation-induced long-term metastasis of residual hepG2 cells through inhibition of EMT. Radiat Res. (2018) 190:374–84. doi: 10.1667/RR15074.1
25. Ghosh S, Kumar A, Tripathi RP, and Chandna S. Connexin-43 regulates p38-mediated cell migration and invasion induced selectively in tumour cells by low doses of γ-radiation in an ERK-1/2-independent manner. Carcinogenesis. (2014) 35:383–95. doi: 10.1093/carcin/bgt303
26. Min H, Sun X, Yang X, Zhu H, Liu J, Wang Y, et al. Exosomes derived from irradiated esophageal carcinoma-infiltrating T cells promote metastasis by inducing the epithelial-mesenchymal transition in esophageal cancer cells. Pathol Oncol Research : POR. (2018) 24:11–8. doi: 10.1007/s12253-016-0185-z
27. Kang AR, Cho JH, Lee NG, Kwon JH, Song JY, Hwang SG, et al. Radiation-induced IL-1β Expression and secretion promote cancer cell migration/invasion via activation of the NF-κB-RIP1 pathway. Biochem Biophys Res Commun. (2021) 534:973–9. doi: 10.1016/j.bbrc.2020.10.057
28. Li J, Wu DM, Han R, Deng YYSH, Liu T, Zhang T, et al. Low-dose radiation promotes invasion and migration of A549 cells by activating the CXCL1/NF-κB signaling pathway. OncoTargets Ther. (2020) 13:3619–29. doi: 10.2147/OTT.S243914
29. Li D, Wang J, Li X, Wang Z, Yu Q, Koh SB, et al. Interactions between radiotherapy resistance mechanisms and the tumor microenvironment. Crit Rev Oncology/Hematology. (2025) 210:104705. doi: 10.1016/j.critrevonc.2025.104705
30. Elgundi Z, Papanicolaou M, Major G, Cox TR, Melrose J, Whitelock JM, et al. Cancer metastasis: the role of the extracellular matrix and the heparan sulfate proteoglycan perlecan. Front Oncol. (2020) 9:1482. doi: 10.3389/fonc.2019.01482
31. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, and Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. (2020) 11:1–19. doi: 10.1038/s41467-020-18794-x
32. Legerstee K and Houtsmuller AB. A layered view on focal adhesions. Biology. (2021) 10:1189. doi: 10.3390/biology10111189
33. Gkretsi V and Stylianopoulos T. Cell adhesion and matrix stiffness: coordinating Cancer cell invasion and metastasis. Front Oncol. (2018) 8:342205. doi: 10.3389/fonc.2018.00145
34. Lacombe J and Zenhausern F. Effect of mechanical forces on cellular response to radiation. Radiotherapy Oncol. (2022) 176:187–98. doi: 10.1016/j.radonc.2022.10.006
35. You GR, Chang JT, Li YL, Chen YJ, Huang YC, Fan KH, et al. Molecular interplays between cell invasion and radioresistance that lead to poor prognosis in head-neck cancer. Front Oncol. (2021) 11:681717. doi: 10.3389/fonc.2021.681717
36. Huang Q, Zhou Z, Yan F, Dong Q, Wang L, Sha W, et al. Low-dose X-ray irradiation induces morphological changes and cytoskeleton reorganization in osteoblasts. Exp Ther Med. (2020) 20:283. doi: 10.3892/etm.2020.9413
37. Lessey EC, Guilluy C, and Burridge K. From mechanical force to rhoA activation. Biochemistry. (2012) 51:7420. doi: 10.1021/bi300758e
38. Magalhaes YT, Boell VK, Cardella GD, and Forti FL. Downregulation of the rho GTPase pathway abrogates resistance to ionizing radiation in wild-type P53 glioblastoma by suppressing DNA repair mechanisms. Cell Death Dis. (2023) 14:1–16. doi: 10.1038/s41419-023-05812-1
39. Ulu A, Oh W, Zuo Y, and Frost JA. Stress-activated MAPKs and CRM1 regulate the subcellular localization of net1A to control cell motility and invasion. J Cell Science. (2018) 131. doi: 10.1242/jcs.204644
40. Julian L and Olson MF. Rho-associated coiled-coil containing kinases (ROCK): structure, regulation, and functions. Small GTPases. (2014) 5:e29846. doi: 10.4161/sgtp.29846
41. He Y, Sun MM, Zhang GG, Yang J, Chen KS, Xu WW, et al. Targeting PI3K/akt signal transduction for cancer therapy. Signal Transduction Targeted Ther. (2021) 6:1–17. doi: 10.1038/s41392-021-00828-5
42. Choy MS, Hieke M, Kumar GS, Lewis GR, Gonzalez-DeWhitt KR, Kessler RP, et al. Understanding the antagonism of retinoblastoma protein dephosphorylation by PNUTS provides insights into the PP1 regulatory code. Proc Natl Acad Sci United States America. (2014) 111:4097–102. doi: 10.1073/pnas.1317395111
43. Landsverk HB, Mora-Bermúdez F, Landsverk OJB, Hasvold G, Naderi S, Bakke O, et al. The protein phosphatase 1 regulator PNUTS is a new component of the DNA damage response. EMBO Rep. (2010) 11:868. doi: 10.1038/embor.2010.134
44. Kavela S, Shinde SR, Ratheesh R, Viswakalyan K, Bashyam MD, Gowrishankar S, et al. PNUTS functions as a proto-oncogene by sequestering PTEN. Cancer Res. (2012) 73:205–14. doi: 10.1158/0008-5472.CAN-12-1394
45. Kang Y, He W, Qiao J, Guo Q, Ren C, Hu J, et al. Advances in targeted therapy mainly based on signal pathways for nasopharyngeal carcinoma. Signal Transduction Targeted Ther. (2020) 5:1–20. doi: 10.1038/s41392-020-00340-2
46. Engelman JA, Luo J, and Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. (2006) 7:606. doi: 10.1038/nrg1879
47. He E, Pan F, Li G, and Li J. Fractionated ionizing radiation promotes epithelial-mesenchymal transition in human esophageal cancer cells through PTEN deficiency-mediated akt activation. PloS One. (2015) 10. doi: 10.1371/journal.pone.0133097
48. Cargnello M and Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Reviews : MMBR. (2011) 75:50. doi: 10.1128/MMBR.00031-10
49. Li L, Zhao GD, Shi Z, Qi LL, Zhou LY, and Fu ZX. The ras/raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC. Oncol Letters. (2016) 12:3045. doi: 10.3892/ol.2016.5110
50. Song F, Zhang W, Li X, Chen X, Yuan X, Jiang M, et al. FSBP suppresses tumor cell migration by inhibiting the JNK pathway. IScience. (2023) 26:106440. doi: 10.1016/j.isci.2023.106440
51. Su WH, Chuang PC, Huang EY, and Yang KD. Radiation-induced increase in cell migration and metastatic potential of cervical cancer cells operates via the K-ras pathway. Am J Pathology. (2012) 180:862–71. doi: 10.1016/j.ajpath.2011.10.018
52. Chen HJ, Li K, Qin YZ, Zhou JJ, Li T, Qian L, et al. Recent advances in the role of endogenous hydrogen sulphide in cancer cells. Cell Proliferation. (2023) 56:e13449. doi: 10.1111/cpr.13449
53. Berggren KL, Restrepo Cruz S, Hixon MD, Cowan AT, Keysar SB, Craig S, et al. MAPKAPK2 (MK2) inhibition mediates radiation-induced inflammatory cytokine production and tumor growth in head and neck squamous cell carcinoma. Oncogene. (2019) 38:7329. doi: 10.1038/s41388-019-0945-9
54. Behrens J, Kameritsch P, Wallner S, Pohl U, and Pogoda K. The carboxyl tail of cx43 augments p38 mediated cell migration in a gap junction-independent manner. Eur J Cell Biol. (2010) 89:828–38. doi: 10.1016/j.ejcb.2010.06.003
55. Zinatizadeh MR, Schock B, Chalbatani GM, Zarandi PK, Jalali SA, Miri SR, et al. The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Genes Diseases. (2021) 8:287–97. doi: 10.1016/j.gendis.2020.06.005
56. Kang AR, Cho JH, Lee NG, Song JY, Hwang SG, Lee DH, et al. RIP1 is a novel component of γ-ionizing radiation-induced invasion of non-small cell lung cancer cells. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21134584
57. Jung CH, Han AR, Chung HJ, Ha IH, and Um HD. Linarin inhibits radiation-induced cancer invasion by downregulating MMP-9 expression via the suppression of NF-κB activation in human non-small-cell lung cancer A549. Natural Product Res. (2019) 33:3582–6. doi: 10.1080/14786419.2018.1484460
58. Mantovani A, Barajon I, and Garlanda C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol Rev. (2018) 281:57. doi: 10.1111/imr.12614
59. Liu Y, Xing Z, Yuan M, Xu B, Chen L, Zhang D, et al. IL1R2 promotes tumor progression via JAK2/STAT3 pathway in human clear cell renal cell carcinoma. Pathol - Res Practice. (2022) 238:154069. doi: 10.1016/j.prp.2022.154069
60. Lei RE, Shi C, Zhang PL, Hu BL, Jiang HX, and Qin SY. IL-9 promotes proliferation and metastasis of hepatocellular cancer cells by activating JAK2/STAT3 pathway. Int J Clin Exp Pathology. (2017) 10:7940–46.
61. Chen H, Tu W, Lu Y, Zhang Y, Xu Y, Chen X, et al. Low-dose X-ray irradiation combined with FAK inhibitors improves the immune microenvironment and confers sensitivity to radiotherapy in pancreatic cancer. Biomedicine Pharmacotherapy. (2022) 151:113114. doi: 10.1016/j.biopha.2022.113114
62. Osipov A, Blair AB, Liberto J, Wang J, Li K, Herbst B, et al. Inhibition of focal adhesion kinase enhances antitumor response of radiation therapy in pancreatic cancer through CD8+ T cells. Cancer Biol Med. (2021) 18:206–14. doi: 10.20892/j.issn.2095-3941.2020.0273
63. Lander VE, Belle JI, Kingston NL, Herndon JM, Hogg GD, Liu X, et al. Stromal reprogramming by FAK inhibition overcomes radiation resistance to allow for immune priming and response to checkpoint blockade. Cancer Discovery. (2022) 12:2774. doi: 10.1158/2159-8290.CD-22-0192
64. Storch K, Sagerer A, and Cordes N. Cytotoxic and radiosensitizing effects of FAK targeting in human glioblastoma cells in vitro. Oncol Rep. (2015) 33:2009–16. doi: 10.3892/or.2015.3753
65. Liu T, Sun Q, Li Q, Yang H, Zhang Y, Wang R, et al. Dual PI3K/mTOR inhibitors, GSK2126458 and PKI-587, suppress tumor progression and increase radiosensitivity in nasopharyngeal carcinoma. Mol Cancer Ther. (2015) 14:429–39. doi: 10.1158/1535-7163.MCT-14-0548
66. Potiron VA, Abderrhamani R, Giang E, Chiavassa S, Di Tomaso E, Maira SM, et al. Radiosensitization of prostate cancer cells by the dual PI3K/mTOR inhibitor BEZ235 under normoxic and hypoxic conditions. Radiotherapy Oncology : J Eur Soc Ther Radiol Oncol. (2013) 106:138–46. doi: 10.1016/j.radonc.2012.11.014
67. Pore N, Gupta AK, Cerniglia GJ, Jiang Z, Bernhard EJ, Evans SM, et al. Nelfinavir down-regulates hypoxia-inducible factor 1α and VEGF expression and increases tumor oxygenation: implications for radiotherapy. Cancer Res. (2006) 66:9252–9. doi: 10.1158/0008-5472.CAN-06-1239
68. Hill EJ, Roberts C, Franklin JM, Enescu M, West N, MacGregor TP, et al. Clinical Trial of Oral Nelfinavir before and during Radiation Therapy for Advanced Rectal Cancer. Clin Cancer Research : Off J Am Assoc Cancer Res. (2016) 22:1922–31. doi: 10.1158/1078-0432.CCR-15-1489
69. Mukherjee S, Qi C, Shaw R, Jones CM, Bridgewater JA, Radhakrishna G, et al. Standard or high dose chemoradiotherapy, with or without the protease inhibitor nelfinavir, in patients with locally advanced pancreatic cancer: the phase 1/randomised phase 2 SCALOP-2 trial. Eur J Cancer (Oxford England : 1990). (2024) 209. doi: 10.1016/j.ejca.2024.114236
70. Chuang FC, Wang CC, Chen JH, Hwang TZ, Yeh SA, and Su YC. PI3k inhibitors (BKM120 and BYL719) as radiosensitizers for head and neck squamous cell carcinoma during radiotherapy. PloS One. (2021) 16:e0245715. doi: 10.1371/journal.pone.0245715
71. Wen PY, Rodon JA, Mason W, Beck JT, Degroot J, Donnet V, et al. Open-label, multicentre study of buparlisib in combination with temozolomide or with concomitant radiation therapy and temozolomide in patients with newly diagnosed glioblastoma. ESMO Open. (2020) 5. doi: 10.1136/esmoopen-2020-000673
72. Day D, Prawira A, Spreafico A, Waldron J, Karithanam R, Giuliani M, et al. Phase I trial of alpelisib in combination with concurrent cisplatin-based chemoradiotherapy in patients with locoregionally advanced squamous cell carcinoma of the head and neck. Oral Oncol. (2020) 108. doi: 10.1016/j.oraloncology.2020.104753
73. Dunn LA, Riaz N, Fury MG, McBride SM, Michel L, Lee NY, et al. A phase 1b study of cetuximab and BYL719 (Alpelisib) concurrent with intensity modulated radiation therapy in stage III-IVB head and neck squamous cell carcinoma. Int J Radiat Oncology Biology Physics. (2020) 106:564–70. doi: 10.1016/j.ijrobp.2019.09.050
74. Zhao J, Cheng G, and Liu J. Combination of intensity modulated radiotherapy followed treatment with p38 MAPK activation inhibitor inhibits the proliferation of MCF-7 breast cancer cells. Saudi J Biol Sci. (2018) 25:10–4. doi: 10.1016/j.sjbs.2017.01.061
75. Biau J, Thivat E, Chautard E, Stefan D, Boone M, Chauffert B, et al. Phase 1 trial of ralimetinib (LY2228820) with radiotherapy plus concomitant temozolomide in the treatment of newly diagnosed glioblastoma. Radiotherapy Oncology : J Eur Soc Ther Radiol Oncol. (2021) 154:227–34. doi: 10.1016/j.radonc.2020.09.036
76. Kozakai N, Kikuchi E, Hasegawa M, Suzuki E, Ide H, Miyajima A, et al. Enhancement of radiosensitivity by a unique novel NF-κB inhibitor, DHMEQ, in prostate cancer. Br J Cancer. (2012) 107:652–7. doi: 10.1038/bjc.2012.321
77. Pal S, Kozono D, Yang X, Fendler W, Fitts W, Ni J, et al. Dual HDAC and PI3K inhibition abrogates NFκB- and FOXM1-mediated DNA damage response to radiosensitize pediatric high-grade gliomas. Cancer Res. (2018) 78:4007–21. doi: 10.1158/0008-5472.CAN-17-3691
78. Deraska PV, O’Leary C, Reavis HD, Labe S, Dinh TK, Lazaro JB, et al. NF-κB inhibition by dimethylaminoparthenolide radiosensitizes non-small-cell lung carcinoma by blocking DNA double-strand break repair. Cell Death Discov. (2018) ;4:1–12. doi: 10.1038/s41420-017-0008-3
79. Morel KL, Ormsby RJ, Bezak E, Sweeney CJ, and Sykes PJ. Parthenolide selectively sensitizes prostate tumor tissue to radiotherapy while protecting healthy tissues in vivo. Radiat Res. (2017) 187:501–12. doi: 10.1667/RR14710.1
80. Takada N, Sugano H, Shirai Y, Saito N, Hamura R, Taniai T, et al. Nafamostat mesilate, a nuclear factor kappa B inhibitor, enhances the antitumor action of radiotherapy on gallbladder cancer cells. PloS One. (2021) 16:e0257019. doi: 10.1371/journal.pone.0257019
81. Wrona A, Dziadziuszko R, and Jassem J. Combining radiotherapy with targeted therapies in non-small cell lung cancer: focus on anti-EGFR, anti-ALK and anti-angiogenic agents. Trans Lung Cancer Res. (2021) 10:2032. doi: 10.21037/tlcr-20-552
82. Katukam LP, Padakanti AP, and Chella N. Drug repurposing: history, significance, benefits, approaches, and challenges. In: Chella N, Ranjan OP, and Alexander A, editors. Drug repurposing. Singapore. Gateway East, Singapore: Springer (2024). p. 1–11.
83. Siyah P. Advanced computational pipeline for FAK inhibitor discovery: combining multiple docking methods with MD and QSAR for cancer therapy. Computation. (2024) 12:222. doi: 10.3390/computation12110222
84. Obara S, Nakata M, Takeshima H, Kuratsu JI, Maruyama I, and Kitajima I. Inhibition of migration of human glioblastoma cells by cerivastatin in association with focal adhesion kinase (FAK). Cancer Letters. (2002) 185:153–61. doi: 10.1016/s0304-383502)00278-1
85. Denoyelle C, Albanese P, Uzan G, Hong L, Vannier JP, Soria J, et al. Molecular mechanism of the anti-cancer activity of cerivastatin, an inhibitor of HMG-coA reductase, on aggressive human breast cancer cells. Cell Signalling. (2003) 63:1684–95. doi: 10.1016/S0898-6568(02)00124-9
86. Medeiros HCD, Colturato-Kido C, Ferraz LS, Costa CA, Moraes VWR, Paredes-Gamero EJ, et al. AMPK activation induced by promethazine increases NOXA expression and beclin-1 phosphorylation and drives autophagy-associated apoptosis in chronic myeloid leukemia. Chemico-Biological Interactions. (2020) 315:108888. doi: 10.1016/j.cbi.2019.108888
87. Tan X, Gong L, Li X, Zhang X, Sun J, Luo X, et al. Promethazine inhibits proliferation and promotes apoptosis in colorectal cancer cells by suppressing the PI3K/AKT pathway. Biomedicine Pharmacotherapy. (2021) 143:112174. doi: 10.1016/j.biopha.2021.112174
88. Wang W, Dong XX, Liu Y, Ni B, Sai N, You L, et al. Itraconazole exerts anti-liver cancer potential through the Wnt, PI3K/AKT/mTOR, and ROS pathways. Biomedicine Pharmacotherapy. (2020) 131:110661. doi: 10.1016/j.biopha.2020.110661
89. Bono C, Karlin L, Harel S, Mouly E, Labaume S, Galicier L, et al. The human immunodeficiency virus-1 protease inhibitor nelfinavir impairs proteasome activity and inhibits the proliferation of multiple myeloma cells in vitro and in vivo. Haematologica. (2012) 97:1101–9. doi: 10.3324/haematol.2011.049981
90. Gills JJ, LoPiccolo J, Tsurutani J, Shoemaker RH, Best CJM, Abu-Asab MS, et al. Is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res. (2007) 13:5183–94. doi: 10.1158/1078-0432.CCR-07-0161
91. Gao C, He XF, Xu QR, Xu YJ, and Shen J. Sevoflurane downregulates insulin-like growth factor-1 to inhibit cell proliferation, invasion and trigger apoptosis in glioma through the PI3K/AKT signaling pathway. Anti-Cancer Drugs. (2019) 30:670–6. doi: 10.1097/CAD.0000000000000744
92. Hu J, Hu J, Jiao H, and Li Q. Anesthetic effects of isoflurane and the molecular mechanism underlying isoflurane-inhibited aggressiveness of hepatic carcinoma. Mol Med Rep. (2018) 18:184–92. doi: 10.3892/mmr.2018.8945
93. Jones HM, Fang Z, Sun W, Clark LH, Stine JE, Tran AQ, et al. Atorvastatin exhibits anti-tumorigenic and anti-metastatic effects in ovarian cancer in vitro. Am J Cancer Res. (2017) 7:2478–90.
94. Huang SW, Chyuan IT, Shiue C, Yu MC, Hsu YF, and Hsu MJ. Lovastatin-mediated MCF-7 cancer cell death involves LKB1-AMPK-p38MAPK-P53-survivin signalling cascade. J Cell Mol Med. (2020) 24:1822–36. doi: 10.1111/jcmm.14879
95. Qi XF, Zheng L, Lee KJ, Kim DH, Kim CS, Cai DQ, et al. HMG-coA reductase inhibitors induce apoptosis of lymphoma cells by promoting ROS generation and regulating akt, erk and p38 signals via suppression of mevalonate pathway. Cell Death Dis. (2013) 4:e518–8. doi: 10.1038/cddis.2013.44
96. Xu B, Wang S, Li R, Chen K, He L, Deng M, et al. Disulfiram/copper selectively eradicates AML leukemia stem cells in vitro and in vivo by simultaneous induction of ROS-JNK and inhibition of NF-κB and nrf2. Cell Death Disease. (2017) 8:e2797. doi: 10.1038/cddis.2017.176
97. Zha J, Chen F, Dong H, Shi P, Yao Y, Zhang Y, et al. Disulfiram targeting lymphoid Malignant cell lines via ROS-JNK activation as well as nrf2 and NF-KB pathway inhibition. J Trans Med. (2014) 12. doi: 10.1186/1479-5876-12-163
98. Jin Y, Lu Z, Ding K, Li J, Du X, Chen C, et al. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: inactivation of the NF-κB pathway and generation of reactive oxygen species. Cancer Res. (2010) 70:2516–27. doi: 10.1158/0008-5472.CAN-09-3950
99. Zuo C, Hong Y, Qiu X, Yang D, Liu N, Sheng X, et al. Celecoxib suppresses proliferation and metastasis of pancreatic cancer cells by down-regulating STAT3/NF-kB and L1CAM activities. Pancreatology. (2018) 18:328–33. doi: 10.1016/j.pan.2018.02.006
100. Tan X, Yan Y, Song B, Zhu S, Mei Q, and Wu K. Focal adhesion kinase: from biological functions to therapeutic strategies. Exp Hematol Oncol. (2023) 12:83. doi: 10.1186/s40164-023-00446-7
101. Glaviano A, Foo ASC, Lam HY, Yap KCH, Jacot W, Jones RH, et al. PI3K/AKT/MTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer. (2023) 22:1–37. doi: 10.1186/s12943-023-01827-6
102. Munster P, Aggarwal R, Hong D, Schellens JHM, van der Noll R, Specht J, et al. First-in-human phase I study of GSK2126458, an oral pan-class I phosphatidylinositol-3-kinase inhibitor, in patients with advanced solid tumor Malignancies. Clin Cancer Research : Off J Am Assoc Cancer Res. (2016) 22:1932–9. doi: 10.1158/1078-0432.CCR-15-1665
103. Zhu DS, Dong JY, Xu YY, Zhang XT, Fu SB, and Liu W. Omipalisib inhibits esophageal squamous cell carcinoma growth through inactivation of phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (MTOR) and ERK signaling. Med Sci Monitor : Int Med J Exp Clin Res. (2020) 26:e927106–1. doi: 10.12659/MSM.927106
104. Knight SD, Adams ND, Burgess JL, Chaudhari AM, Darcy MG, Donatelli CA, et al. Discovery of GSK2126458, a highly potent inhibitor of PI3K and the mammalian target of rapamycin. ACS Medicinal Chem Letters. (2010) 1:39. doi: 10.1021/ml900028r
105. Gupta AK, Cerniglia GJ, Mick R, McKenna WG, and Muschel RJ. HIV protease inhibitors block akt signaling and radiosensitize tumor cells both in vitro and in vivo. Cancer Res. (2005) 65:8256–65. doi: 10.1158/0008-5472.CAN-05-1220
106. Zeng J, See AP, Aziz K, Thiyagarajan S, Salih T, Gajula RP, et al. Nelfinavir induces radiation sensitization in pituitary adenoma cells. Cancer Biol Ther. (2011) 12:657–63. doi: 10.4161/cbt.12.7.17172
107. Liebscher S, Koi L, Löck S, Muders MH, and Krause M. The HIV protease and PI3K/akt inhibitor nelfinavir does not improve the curative effect of fractionated irradiation in PC-3 prostate cancer in vitro and in vivo. Clin Trans Radiat Oncol. (2017) 2:7. doi: 10.1016/j.ctro.2016.12.002
108. Hirata E and Sahai E. Tumor microenvironment and differential responses to therapy. Cold Spring Harbor Perspect Med. (2017) 7:a026781. doi: 10.1101/cshperspect.a026781
109. Josephs DH and Sarker D. Pharmacodynamic biomarker development for PI3K pathway therapeutics. Trans Oncogenomics. (2016) 7:33. doi: 10.4137/TOG.S30529
110. Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, and Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. (2020) 19:1997. doi: 10.3892/etm.2020.8454
111. Ganguly P, Macleod T, Wong C, Harland M, and McGonagle D. Revisiting p38 mitogen-activated protein kinases (MAPK) in inflammatory arthritis: A narrative of the emergence of MAPK-activated protein kinase inhibitors (MK2i). Pharmaceuticals. (2023) 16:1286. doi: 10.3390/ph16091286
112. Alimbetov D, Umbayev B, Tsoy A, Begimbetova D, Davis T, Kipling D, et al. Small molecule targeting of the p38/Mk2 stress signaling pathways to improve cancer treatment. BMC Cancer. (2023) 23:1–13. doi: 10.1186/s12885-023-11319-x
113. Gaur R, Mensah KA, Stricker J, Adams M, Parton A, Cedzik D, et al. CC-99677, a novel, oral, selective covalent MK2 inhibitor, sustainably reduces pro-inflammatory cytokine production. Arthritis Res Ther. (2022) 24:1–15. doi: 10.1186/s13075-022-02850-6
114. Morgan D, Berggren KL, Spiess CD, Smith HM, Tejwani A, Weir SJ, et al. Mitogen-activated protein kinase-activated protein kinase-2 (MK2) and its role in cell survival, inflammatory signaling, and migration in promoting cancer. Mol Carcinogenesis. (2022) 61:173. doi: 10.1002/mc.23348
115. Mondello P, Derenzini E, Asgari Z, Philip J, Brea EJ, Seshan V, et al. Dual inhibition of histone deacetylases and phosphoinositide 3-kinase enhances therapeutic activity against B cell lymphoma. Oncotarget. (2017) 8:14017–28. doi: 10.18632/oncotarget.14876
116. Ishikawa C and Mori N. The role of CUDC-907, a dual phosphoinositide-3 kinase and histone deacetylase inhibitor, in inhibiting proliferation of adult T-cell leukemia. Eur J Haematology. (2020) 105:763–72. doi: 10.1111/ejh.13508
117. Younes A, Berdeja JG, Patel MR, Kelly KR, Flinn IW, Gerecitano JF, et al. Phase 1 trial testing single agent CUDC-907, a novel, oral dual inhibitor of histone deacetylase (HDAC) and PI3K: initial assessment of patients with relapsed or refractory (RR) diffuse large B-cell lymphoma (DLBCL), including double expressor (DE) lymphoma. Blood. (2015) 126:257. doi: 10.1182/blood.V126.23.257.257
118. Landsburg DJ, Barta SK, Ramchandren R, Batlevi C, Iyer S, Kelly K, et al. Fimepinostat (CUDC-907) in patients with relapsed/refractory diffuse large B cell and high-grade B-cell lymphoma: report of a phase 2 trial and exploratory biomarker analyses. Br J Haematology. (2021) 195:201–9. doi: 10.1111/bjh.17730
119. Oki Y, Kelly KR, Flinn I, Patel MR, Gharavi R, Ma A, et al. CUDC-907 in relapsed/refractory diffuse large B-cell lymphoma, including patients with MYC-alterations: results from an expanded phase I trial. Haematologica. (2017) 102:1923–30. doi: 10.3324/haematol.2017.172882
120. Guzman ML, Rossi RM, Neelakantan S, Li X, Corbett CA, Hassane DC, et al. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood. (2007) 110:4427. doi: 10.1182/blood-2007-05-090621
121. Estabrook NC, Chin-Sinex H, Borgmann AJ, Dhaemers RM, Shapiro RH, Gilley D, et al. Inhibition of NF-κB and DNA double-strand break repair by DMAPT sensitizes non-small-cell lung cancers to X-rays. Free Radical Biol Med. (2011) 51:2249–58. doi: 10.1016/j.freeradbiomed.2011.09.029
122. Morel KL, Hamid AA, Clohessy JG, Pandell N, Ellis L, and Sweeney CJ. NF-κB blockade with oral administration of dimethylaminoparthenolide (DMAPT), delays prostate cancer resistance to androgen receptor (AR) inhibition and inhibits AR variants. Mol Cancer Research : MCR. (2021) 19:1137. doi: 10.1158/1541-7786.MCR-21-0099
123. Lamture G, Crooks PA, and Borrelli MJ. Actinomycin-D and dimethylamino-parthenolide (DMAPT) synergism in treating human pancreatic cancer cells. Drug Dev Res. (2018) 79:287. doi: 10.1002/ddr.21441
124. Morel KL, Ormsby RJ, Klebe S, Sweeney CJ, and Sykes PJ. DMAPT is an effective radioprotector from long-term radiation-induced damage to normal mouse tissues in vivo. Radiat Res. (2019) 192:231–9. doi: 10.1667/RR15404.1
125. Umezawa K, Breborowicz A, and Gantsev S. Anticancer activity of novel NF-κB inhibitor DHMEQ by intraperitoneal administration. Oncol Res. (2020) 28:541. doi: 10.3727/096504020X15929100013698
126. Yasuda A, Kondo S, Nagumo T, Tsukamoto H, Mukudai Y, Umezawa K, et al. Anti-tumor activity of dehydroxymethylepoxyquinomicin against human oral squamous cell carcinoma cell lines in vitro and in vivo. Oral Oncol. (2011) 47:334–9. doi: 10.1016/j.oraloncology.2011.03.001
127. Wong JHT, Lui VWY, Umezawa K, Ho Y, Wong EYL, Ng MHL, et al. A small molecule inhibitor of NF-κB, dehydroxymethylepoxyquinomicin (DHMEQ), suppresses growth and invasion of nasopharyngeal carcinoma (NPC) cells. Cancer Letters. (2010) 287:23–32. doi: 10.1016/j.canlet.2009.05.022
128. Shui L, Ren H, Yang X, Li J, Chen Z, Yi C, et al. The era of radiogenomics in precision medicine: an emerging approach to support diagnosis, treatment decisions, and prognostication in oncology. Front Oncol. (2021) 10:570465. doi: 10.3389/fonc.2020.570465
129. He W, Huang W, Zhang L, Wu X, Zhang S, and Zhang B. Radiogenomics: bridging the gap between imaging and genomics for precision oncology. MedComm. (2024) 5:e722. doi: 10.1002/mco2.722
130. Zeng H, Chen L, Wang M, Luo Y, Huang Y, and Ma X. Integrative radiogenomics analysis for predicting molecular features and survival in clear cell renal cell carcinoma. Aging (Albany NY). (2021) 13:9960–75. doi: 10.18632/aging.202752
131. Liew K, Yu GQS, Wei Pua LJ, Wong LZ, Tham SY, Hii LW, et al. Parallel genome-wide RNAi screens identify lymphocyte-specific protein tyrosine kinase (LCK) as a targetab le vulnerability of cell proliferation and chemoresistance in nasopharyngeal carcinoma. Cancer Lett. (2021) 504:81–90. doi: 10.1016/j.canlet.2021.02.006
132. Looi CK, Foong LC, Chung FF, Khoo AS, Loo EM, Leong CO, et al. Targeting the crosstalk of epigenetic modifications and immune evasion in nasopharyngeal cancer. Cell Biol Toxicol. (2023) 39:2501–26. doi: 10.1007/s10565-023-09830-9
133. Mai CW, Sridhar SB, Karattuthodi MS, Ganesan PM, Shareef J, Lee EL, et al. Scoping review of enablers and challenges of implementing pharmacogenomics testing in the primary care settings. BMJ Open. (2024) 14:e087064. doi: 10.1136/bmjopen-2024-087064
134. Li WZ, Song JL, Wu G, Dong MY, Liao YT, Huang ZJ, et al. Dynamic contrast-enhanced magnetic resonance imaging-based radiomics for predicting programmed death ligand 1 expression in nasopharyngeal carcinoma. BMC Med Imaging. (2025) 25:377. doi: 10.1186/s12880-025-01917-5
Keywords: radiation, inflammation, immune, therapeutics, radiogenomics
Citation: Lee EQ, Looi C-K, Tan LP, Chew YL, Lim W-M, Foong L-C, Leong C-O, Cheong KW and Mai C-W (2025) Targeting radiotherapy-induced inflammation in cancer metastasis: insights into immune modulation, therapeutic opportunities and radiogenomics. Front. Oncol. 15:1682522. doi: 10.3389/fonc.2025.1682522
Received: 12 August 2025; Accepted: 08 October 2025;
Published: 23 October 2025.
Edited by:
Andrei Fodor, IRCCS San Raffaele Scientific Institute, ItalyReviewed by:
Chiara Lucrezia Deantoni, San Raffaele Hospital (IRCCS), ItalyDengxiong Li, The First Affiliated Hospital of Zhejiang Chinese Medical University (Zhejiang Provincial Hospital of Traditional Chinese Medicine), China
Copyright © 2025 Lee, Looi, Tan, Chew, Lim, Foong, Leong, Cheong and Mai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chun-Wai Mai, Y2h1bndhaV9tYWlAaW11LmVkdS5teQ==