 Zhuoran Tang
Zhuoran Tang Yanchun Xie2
Yanchun Xie2 Yanping Zeng
Yanping Zeng- 1Mudi Meng Honors College, China Pharmaceutical University, Nanjing, China
- 2Department of Neurology, Renmin Hospital of Wuhan University, Wuhan, China
Antibody–drug conjugates (ADCs) represent one of the most advanced drug configurations under current research, primarily composed of a monoclonal antibody (mAb), a highly potent cytotoxic payload, and a linker that connects the drug to the antibody. The mAb serves mainly as a targeting moiety, guiding the conjugate to specific cells. The cytotoxic payload is responsible for the anticancer activity, whereas the linker ensures stable attachment between the antibody and the payload during circulation. The core advantage of ADCs lies in their ability to leverage the specificity of antibodies to deliver highly potent cytotoxic agents precisely to tumor cells, thereby significantly improving the therapeutic index. However, they also face challenges such as systemic toxicity, drug resistance, tumor heterogeneity, and complex manufacturing processes. Currently, extensive research is focused on technological innovations, the development of novel ADCs, and the optimization of clinical combination therapies. This article provides a comprehensive review of the structure and mechanism of action of ADCs, their developmental history, current challenges, emerging novel agents, and combination strategies with immune checkpoint inhibitors (ICIs).
Introduction
Every year, cancer causes more than 8.2 million deaths worldwide, making it the second leading cause of death globally and a major public health threat (1, 2). For a long period of time, conventional treatments such as radiotherapy and chemotherapy have served as the basis for managing a wide range of cancers (3, 4). However, these approaches lack sufficient specificity and have a narrow therapeutic index, often leading to severe side effects due to non-specific drug exposure in healthy tissues (5). Thus, there is a critical need to develop novel cancer therapeutics with improved targeting capabilities.
The advent of monoclonal antibodies (mAbs) has reformed cancer treatment by enabling precise targeting of tumor antigens (6). However, mAb monotherapy has often shown limited efficacy compared with conventional chemotherapy in some cases. Advances in biopharmaceutical technology have led to the rise of antibody–drug conjugates (ADCs), a new class of anticancer agents that combine the specificity of antibodies with the efficiency of cytotoxic drugs. This innovation has opened a new era in oncology, yielding distinct clinical achievements. An ADC consists of three key parts: an mAb, a cytotoxic payload, and a chemical linker (Figure 1). The mAb directs the ADC to specific antigens on target cells. This mechanism enables the precise delivery of the cytotoxic agent with minimal off-target effects on healthy tissues. The payload is a potent cytotoxic drug that kills the targeted cells. The linker serves as a stable connection between the antibody and the payload, ensuring that the cytotoxic agent remains attached during circulation. Without a well-designed linker, the payload may be prematurely released, leading to systemic toxicity and damage to normal tissues (7).
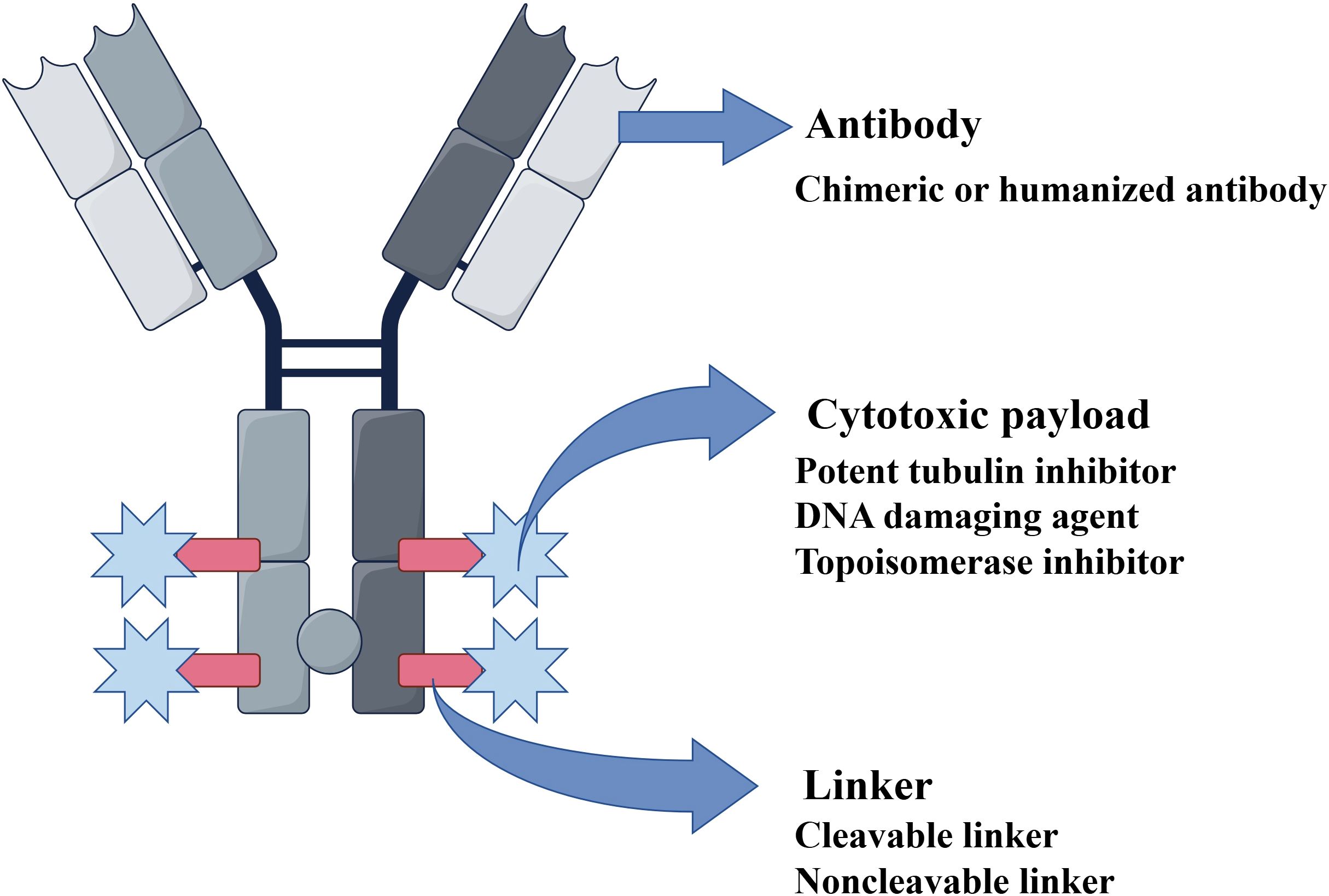
Figure 1. Structure of an antibody–drug conjugate (ADC). An ADC is structurally composed of three main components: 1. Antibody: A chimeric or humanized monoclonal antibody that specifically binds to a tumor-associated antigen. 2. Cytotoxic payload: A highly potent agent (e.g., tubulin inhibitor, DNA-damaging agent, or topoisomerase inhibitor). 3. Linker: A cleavable or non-cleavable chemical bridge that connects the antibody to the payload. Abbreviations: ADC, antibody–drug conjugate.
ADCs selectively kill cancer cells that express the target antigen. Upon binding to the antigen on the tumor cell surface, the ADC–antigen complex is internalized via endocytosis. It then traffics through endosomal compartments and ultimately fuses with lysosomes (8). Within the lysosome, enzymatic or chemical cleavage of the linker releases the cytotoxic payload (9, 10). The freed payload can then disrupt critical cellular processes—such as DNA replication or microtubule assembly—resulting in tumor cell death (Figure 2).
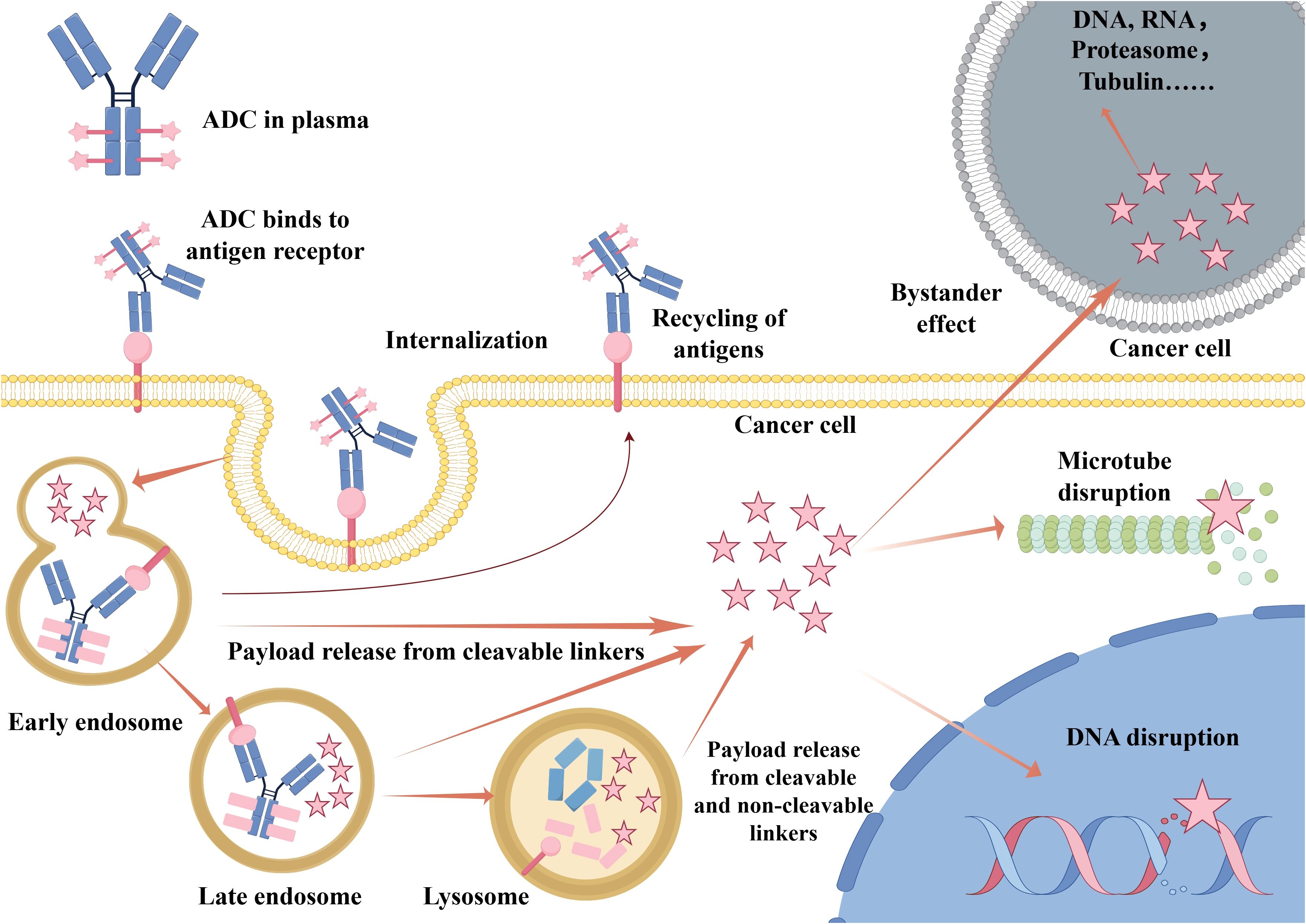
Figure 2. Mechanism of action of an antibody–drug conjugate (ADC). Upon administration, the ADC circulates in the plasma and specifically binds to target antigens on the tumor cell surface. The ADC–antigen complex is internalized via endocytosis and traffics through early endosomes, late endosomes, and finally lysosomes. The linker is cleaved and then the cytotoxic payload is released within the lysosome. The payload can disrupt critical cellular processes such as microtubule assembly or DNA integrity, leading to apoptosis. In some cases, the released payload may diffuse across the membrane to kill adjacent antigen-negative cells, which is known as the “bystander effect”. Some antigens may also undergo recycling back to the cell surface. Abbreviations: ADC, antibody–drug conjugate.
Furthermore, some released payloads are membrane-permeable and can diffuse into the tumor microenvironment. This ability to kill neighboring cancer cells that do not express the target antigen is referred to as the “bystander effect” (11–13). This phenomenon enhances ADC efficacy against heterogeneous tumors, which contain mixed populations of antigen-positive and antigen-negative cells. However, the bystander effect also raises the possibility of off-target toxicity. Achieving an optimal balance between therapeutic benefit and potential risk remains a crucial consideration in ADC development.
Since the U.S. Food and Drug Administration (FDA) approved Mylotarg® (gemtuzumab ozogamicin) in 2000 for the treatment of acute myeloid leukemia (AML) (14), a total of 18 ADCs had received approval worldwide as of mid-2025 (Table 1). Furthermore, more than 100 ADC candidates are currently in various stages of clinical development, over 80% of which are being evaluated in solid tumors (15–17). Approved ADCs are already being used in the treatment of lung cancer, breast cancer, and other malignancies (18, 19). With expanding molecular targets and clinical indications, ADCs are ushering in a new era of targeted anticancer therapy. Notably, ADCs can remain effective even in cases where resistance to other targeted agents has developed, and they also provide opportunities to target novel antigens (20). For example, sacituzumab govitecan (Trodelvy®), which targets trophoblast cell-surface antigen 2 (TROP-2), has emerged as an important therapeutic option for advanced triple-negative breast cancer (21).
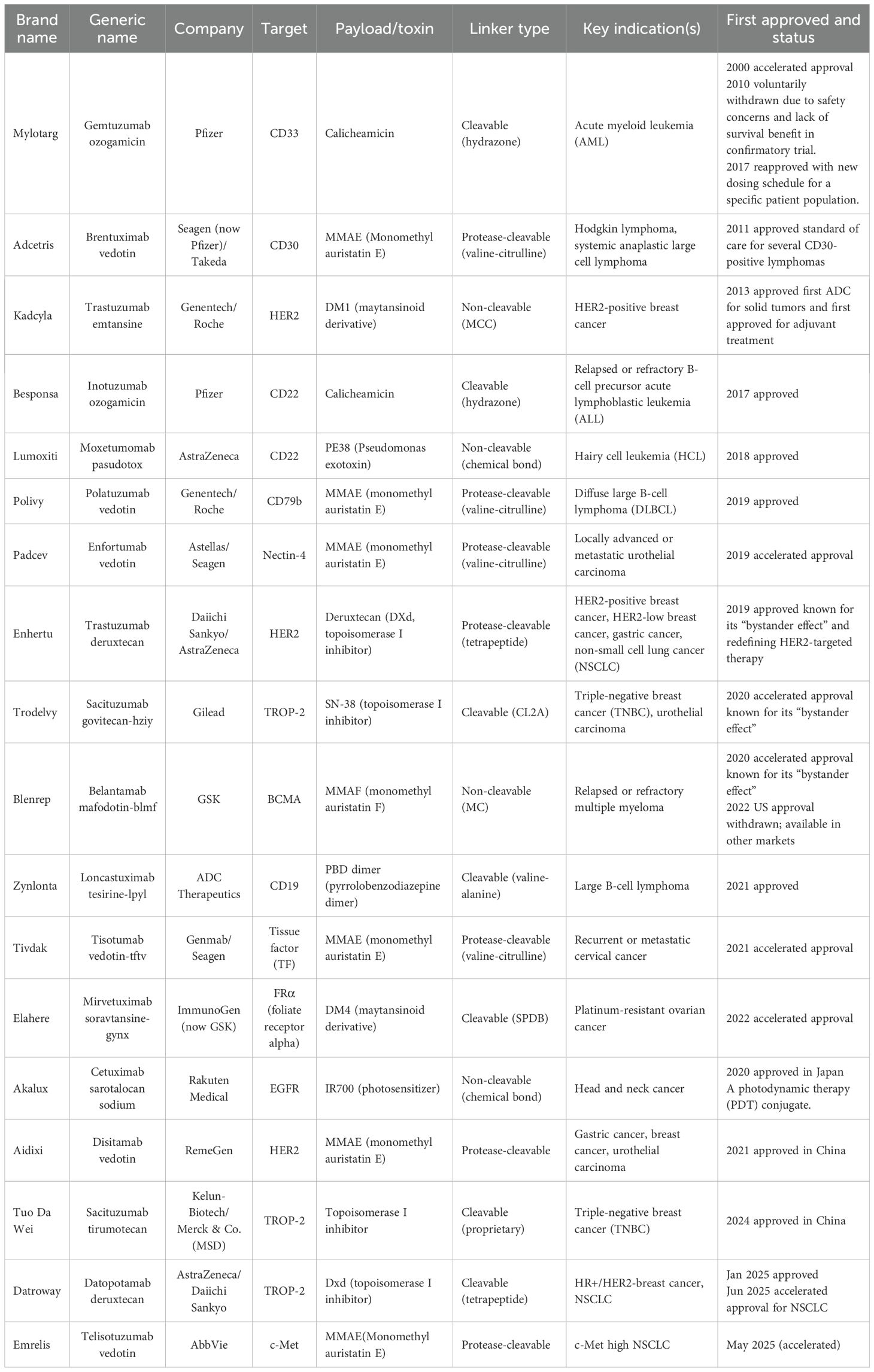
Table 1. Selected ADCs approved for marketing worldwide (as of mid-2025).
ADCs have begun to transform cancer treatment in recent years. However, their clinical translation requires addressing multifaceted challenges, including pharmacokinetic complexity, target antigen heterogeneity, controlled payload release, linker stability, toxicity, structural optimization, and resistance mechanisms (22, 23). Numerous studies are underway to tackle these issues. ADCs are expected to become a viable alternative to conventional chemotherapy in the future. In this review, we review the structure of ADCs and their molecular mechanisms of action, briefly summarize the advantages and limitations of approved ADC therapeutics, discuss existing challenges and strategies for achieving optimal efficacy, outline the prospects of novel ADCs under development, and examine the current landscape of combination therapies between ADCs and immune checkpoint inhibitors (ICIs).
Components and mechanisms of ADCs
Antibodies in ADCs
The ideal antibody should exhibit a high affinity for the target antigen, facilitate efficient internalization, demonstrate low immunogenicity, and have a long plasma half-life. High immunogenicity can cause the antibody to be recognized and destroyed by the body’s immune system before it can reach the target, reducing its efficacy (24). In the early days of antibody therapy, murine antibodies were commonly used. However, they were cleared rapidly from the bloodstream due to their high immunogenicity in humans, which led to serious side effects (25). With the emergence of recombinant DNA technology, murine antibodies have largely been replaced by less immunogenic chimeric and humanized antibodies (26).
For antibodies targeting tumor cells, efficacy largely depends on their binding affinity for the surface antigen. Generally, a higher affinity promotes more rapid internalization (27). However, excessively high affinity can hinder penetration into solid tumors. Treating solid tumors is more complex than treating hematological malignancies due to the “binding site barrier (BSB)” effect (26). Therefore, an optimal level of affinity must be determined to balance efficient tumor cell internalization with adequate tissue penetration and anticancer potency.
Antibody size is another critical factor influencing tumor penetration. The large molecular weight of immunoglobulin G (IgG) antibodies (approximately 150 kDa) often impedes their diffusion through capillary walls and the dense extracellular matrix of tumor tissues (28). In contrast, smaller antibody formats (e.g., scFv, Fab) not only retain high affinity and specificity but also penetrate tumors more effectively, thereby significantly enhancing antitumor efficacy (29). A trade-off of this size reduction, however, is a shorter in vivo half-life. Consequently, multiple factors must be carefully balanced when designing ADCs incorporating these miniaturized antibodies.
The antibodies currently used in ADCs are mostly IgG, which includes four subtypes: IgG1, IgG2, IgG3, and IgG4. The choice of IgG subclass primarily determines the effector functions and immunogenicity risk of the antibody component (30). Among the IgG subtypes, IgG1 is the dominant choice (found in 85% of clinical-stage ADCs) due to its superior Fcγ receptor (FcγR) binding capacity and extended serum half-life (14–21 days) (31, 32). IgG1 possesses strong effector functions, mediated through antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC), due to its high binding affinity for FcγRs (Figure 3) (33). When a target is highly and specifically overexpressed on tumor cells with minimal expression in normal tissues (e.g., HER2), selecting the IgG1 isotype leverages its potent effector functions (34). This activates the immune system to attack the tumor, complementing the payload’s cytotoxic effect and resulting in a multi-mechanistic killing strategy. However, IgG1 may also induce ADCC/ADCP effects against normal cells with low target expression, leading to on-target, off-tumor toxicity (35). Furthermore, by binding to FcγRs on immune cells, IgG1 may be internalized, causing unintended payload release and resulting in off-target toxicities such as myelosuppression (e.g., neutropenia) (36).
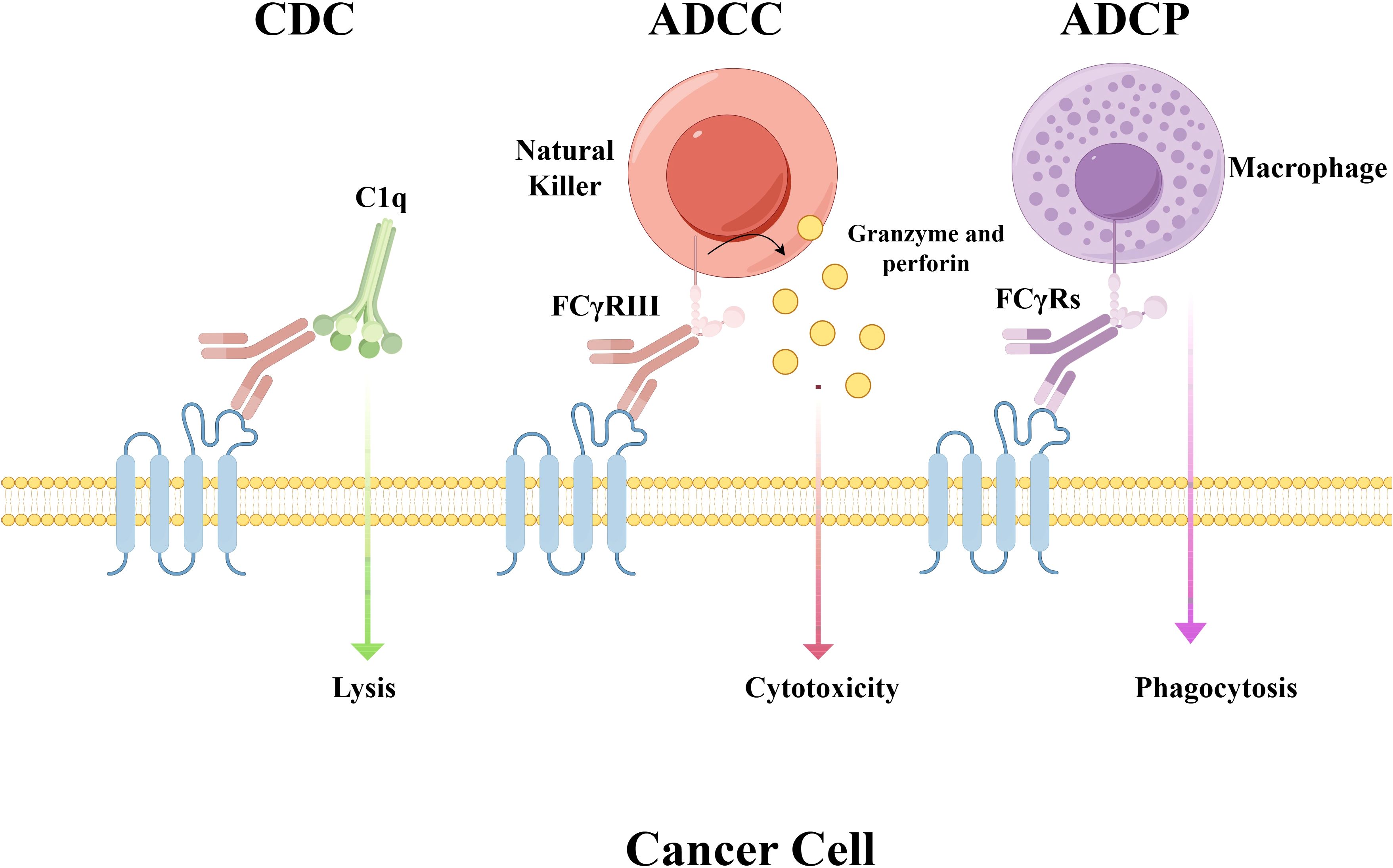
Figure 3. Mechanisms of Fc-mediated effector functions. This schematic illustrates antibody-dependent immune responses mediated through Fc gamma receptors (FcγRs). CDC: complement cascade activation and consequent cell lysis initiated by antibody-antigen binding ADCC: engagement of FcγRs (e.g., FcγRIII) on natural killer (NK) cells induces the release of cytolytic molecules such as granzyme and perforin, resulting in target cell death. ADCP: FcγRs on macrophages mediate this process, which enables the engulfment and damage of target cells. Abbreviations: FcγRs, Fc gamma receptors; CDC, complement-dependent cytotoxicity; ADCC, antibody-dependent cellular cytotoxicity; ADCP, antibody-dependent cellular phagocytosis.
IgG2 has minimal effector function due to its low affinity for most FcγRs and is a poor inducer of ADCC, ADCP, and CDC (37). However, a disadvantage of IgG2 is its structural complexity. The hinge region forms a unique, rigid structure stabilized by multiple disulfide bonds, a feature that can lead to product heterogeneity through the formation of multiple isoforms (38). IgG2 is selected for ADCs when the desired activity must rely entirely on the payload’s cytotoxicity to avoid risks associated with Fc effector functions. This is particularly relevant for targets that have essential physiological functions or are expressed at low levels in normal tissues, as this choice minimizes the risk of “on-target, off-tumor” toxicity mediated by effector functions (39, 40).
Among all human IgG subclasses, IgG3 has the most potent effector functions. It exhibits the highest affinity for FcγRIIIa and the greatest capacity to activate CDC (30). However, its core drawback is an extremely short half-life (approximately 7 days) resulting from its low affinity for FcRn. The key amino acid sequence in IgG3’s FcRn binding interface (e.g., H435) differs from that of other subtypes, resulting in significantly weaker binding at acidic pH. Consequently, IgG3 is more efficiently sorted to lysosomes for degradation rather than being recycled, leading to its rapid clearance. A key structural feature of IgG3 is its exceptionally long hinge region (approximately 62 amino acids), which is rich in cysteine and proline residues that form a complex structure (41). While this unique architecture confers exceptional flexibility and potent effector functions, it also renders the antibody highly susceptible to proteolytic cleavage by enzymes such as neutrophil elastase and matrix metalloproteinases (MMPs). This cleavage results in fragmentation into Fab and Fc segments in the bloodstream, further accelerating its clearance (37). This short half-life is a critical weakness for ADCs, which require prolonged circulation for optimal tumor distribution. Coupled with its inherent instability, these pharmacokinetic and physicochemical defects explain why IgG3 is rarely used in ADC development.
IgG4 has low affinity for FcγRs, resulting in negligible effector functions and an inability to activate CDC. Native IgG4 undergoes “Fab-arm exchange” in vivo. In this process, a half-molecule (one heavy chain and one light chain) from one IgG4 exchanges with a half-molecule from another IgG4 that has a different specificity, resulting in a bispecific antibody (42). This dynamic exchange is particularly problematic for ADCs because it results in a loss of targeting specificity—producing ineffective antibodies that fail to bind the target—or, worse, generates bispecific antibodies (BsAbs) that engage both tumor and healthy tissues, potentially causing severe off-target toxicity (43). The introduction of a point mutation (S228P; serine to proline) in the hinge region significantly stabilizes the IgG4 structure and prevents Fab-arm exchange (44). This stabilization is critical, and as such, all therapeutic antibodies based on the IgG4 isotype must incorporate this modification. When minimal effector function is desired, the engineered, stabilized IgG4 (IgG4-S228P) serves as a commonly used inert scaffold (45).
Modern ADC design is no longer limited to natural subclasses but employs genetic engineering techniques to modify the Fc region for precise control of effector functions, offering greater flexibility in IgG subclass selection (46). Fc silencing is the most common strategy, accomplished by introducing point mutations into an IgG1 backbone—chosen for its superior stability and long half-life (47, 48). Mutations such as L234A/L235A and P329G (termed the “LALA-PG” mutation) effectively abrogate binding to FcγRs and C1q (49). This approach retains the favorable stability, long half-life, and low immunogenicity risk of IgG1 while eliminating effector functions. It has become the platform of choice for many new-generation ADCs (e.g., T-DXd) (50), ensuring that efficacy stems solely from targeted payload delivery, thereby preventing Fc-mediated, off-target toxicity. Conversely, Fc enhancement involves introducing mutations (e.g., S239D/I332E) to increase affinity for FcγRIIIa, potently boosting ADCC (51). This strategy is employed when potent immune activation is desired to complement the ADC’s payload-mediated killing. However, it requires careful evaluation of on-target, off-tumor toxicity risks.
In summary, engineered Fc-silenced IgG1 is becoming the platform of choice in ADC development, as it merges the favorable pharmacokinetics (PK) of IgG1 with a superior safety profile devoid of effector function. This contrasts with the selection of native IgG1 or IgG4, which necessitates a careful, case-specific trade-off based on the target biology and mechanism of action.
Payloads in ADCs
First-generation payloads: conventional chemotherapy agents
The early development of ADC technology took place from the 1990s to the early 2000s. First-generation payloads were composed of conventional chemotherapeutic agents like methotrexate, doxorubicin, and mitomycin (52). While their mechanisms of action (e.g., inhibiting DNA synthesis) were well-established, these drugs exhibited relatively low potency, with cytotoxicity typically in the micromolar (μM) IC50 range (53). Their high hydrophilicity hindered cell membrane penetration, preventing a bystander killing effect on adjacent antigen-negative cells and significantly limiting efficacy against heterogeneous tumors (54). Furthermore, as substrates for efflux pumps like P-glycoprotein (P-gp), they were prone to multidrug resistance (MDR) (55). Due to insufficient potency, poor stability, and a narrow therapeutic window, most first-generation ADCs failed in clinical trials. Nonetheless, these pioneering efforts validated the core ADC concept and underscored the urgent need for more potent warheads.
Second-generation payloads: highly potent tubulin inhibitors
The maturation and regulatory approval of ADC technology spanned from the 2000s to the mid-2010s. This era was characterized by two potent payload classes: auristatins (e.g., MMAE, MMAF) and maytansinoids (e.g., DM1, DM4) (56, 57). These agents were 100 to 1,000 times more potent than their first-generation predecessors, with activity in the nM to pM range, ensuring tumor cell killing even with low ADC internalization (58). Their development was enabled by advanced linker technologies. For instance, MMAE-based ADCs used protease-cleavable linkers (Val-Cit) for stable circulation and targeted payload release in the lysosome. The released MMAE, due to its membrane permeability, mediates a potent bystander effect by diffusing into and killing adjacent antigen-negative cells—a key advantage for solid tumors (59). Conversely, MMAF is charged and hydrophilic, trapping it inside the original cell and eliminating this effect (60, 61). Second-generation ADCs yielded breakthrough drugs like Adcetris® and Kadcyla® (which features the maytansinoid DM1). A key limitation of these tubulin-inhibiting payloads was their primary efficacy against dividing cells, often sparing quiescent populations. Some also remained susceptible to efflux-mediated MDR (Table 2).
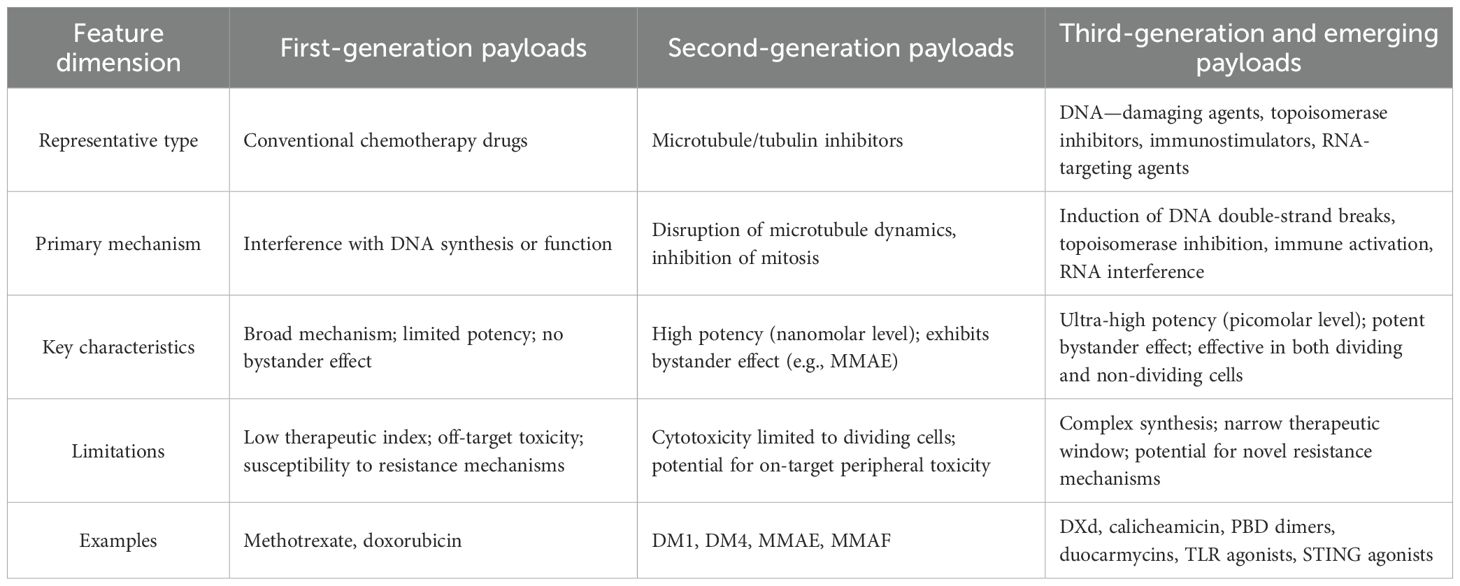
Table 2. Comparison of ADC payloads.
Third-generation payloads: DNA-damaging agents and emerging mechanisms
The breakthrough and innovation phase of ADC technology began in the late 2010s and continues to the present. Third-generation payloads include DNA topoisomerase I inhibitors, such as DXd (deruxtecan) and SN-38, and pyrrolobenzodiazepine (PBD) dimers like talirine. DXd is a revolutionary representative, serving as the payload for the third-generation ADC drug Enhertu® (DS-8201) (62, 63). PBD dimers exhibit extremely potent DNA cross-linking capabilities. Although calicheamicin was discovered earlier (used in Mylotarg®), its powerful DNA-cleaving mechanism aligns closely with the principles of third-generation payloads (64). These payloads demonstrate even higher potency than second-generation agents, particularly DNA-damaging agents, with IC50 values reaching the picomolar (pM) level. Their mechanism of action shifted from targeting microtubules to directly damaging DNA, enabling them to kill both actively dividing and quiescent cells, thereby broadening their therapeutic scope (65). Their strong membrane permeability contributes to a pronounced bystander effect, which is critical for overcoming tumor heterogeneity and acquired resistance. Many DNA-damaging agents are not substrates of P-gp efflux pumps, allowing them to remain effective against multidrug-resistant tumor cells (66). The third-generation concept also extends beyond cytotoxic payloads to include immune-stimulating agents (e.g., TLR agonists), designed to activate local immune responses rather than directly kill cells (67).
Characteristics of an ideal payload
High potency
The payload must possess extremely high cytotoxicity, typically in the pM to nM range. The antibody’s payload capacity is limited, typically with a drug-to-antibody ratio (DAR) of 2–4. Furthermore, only a small fraction of the administered ADC molecules are ultimately internalized into tumor cells (68). Consequently, each internalized payload molecule must be potent enough to kill a tumor cell, or even multiple cells via the bystander effect. The IC50 values of auristatins (e.g., MMAE) and exatecan derivatives (e.g., DXd) are in the pM–nM range, far exceeding the potency (by orders of magnitude) of traditional chemotherapy drugs, which are typically in the micromolar (μM) range (62).
Well-defined mechanism of action
The payload must have a well-defined and highly efficient mechanism of action capable of rapidly inducing cell death. A clear mechanism of action helps predict its efficacy, potential drug resistance, and toxic side effects. Inhibitors of tubulin polymerization (e.g., MMAE, DM1) prevent mitosis, leading to cell-cycle arrest and apoptosis (63). DNA-damaging agents (e.g., DXd, PBDs, and calicheamicin) cause DNA double-strand breaks—the most lethal type of DNA damage—which are effective against both dividing and quiescent cells (64). The payload DXd in DS-8201 (Enhertu) is a topoisomerase I inhibitor that exerts cytotoxicity by preventing DNA relegation (63).
Optimal hydrophilic–lipophilic balance
The payload must exhibit sufficient hydrophilicity to prevent excessive hydrophobicity. Excessive hydrophobicity (a high Log P value) promotes ADC aggregation in vivo, rapid clearance by the immune system, increased hepatic toxicity, and unfavorable PK (69). Conversely, for the treatment of solid tumors, the payload requires a degree of hydrophobicity to enable cell membrane penetration and to mediate the bystander effect, thereby killing adjacent tumor cells with low or no target expression (70). Therefore, a critical balance must be struck between hydrophilicity (which ensures ADC solubility and stability) and moderate hydrophobicity (which ensures membrane permeability and bystander killing). The comparison between auristatins MMAE and MMAF exemplifies this trade-off: MMAF, which features a terminal phenylalanine carboxylate group, is more hydrophilic than MMAE but consequently exhibits a minimal bystander effect due to its impaired membrane permeability (71).
Stability in circulation and efficient release in target cells
The payload must remain highly stable in the bloodstream after conjugation but be efficiently released upon internalization into target cells. Premature dissociation of the payload from the antibody in circulation can lead to severe off-target toxicity in healthy tissues (e.g., bone marrow, gastrointestinal tract) and is a primary driver of dose-limiting toxicities for ADCs (72). The payload is only active upon its specific release inside tumor cells. This efficacy is critically dependent on the linker design. Cleavable linkers (e.g., peptide linkers) are engineered to remain inert in plasma but undergo hydrolysis in the acidic environment of lysosomes or via enzymatic cleavage by specific proteases (e.g., cathepsin B, β-glucuronidase) (73). A prominent example is the tetrapeptide-based linker (GGFG) used in trastuzumab deruxtecan (T-DXd, Enhertu). This ADC demonstrates high stability in plasma due to its robust linker, minimizing off-target release. The active payload (DXd) is liberated specifically within tumor cells following cleavage by lysosomal proteases, ensuring potent and targeted cytotoxicity while sparing healthy tissues (23).
Low susceptibility to MDR
An ideal payload should avoid being a substrate for efflux pump proteins such as P-gp. Tumor cells often overexpress these pumps, which mediate the efflux of traditional chemotherapeutic agents (e.g., paclitaxel) from the cell, conferring drug resistance (74). If the ADC payload is also a substrate, it can be effluxed, leading to reduced intracellular concentration and treatment failure. Many tubulin inhibitors (e.g., MMAE and DM1) are vulnerable to MDR mechanisms. In contrast, certain DNA-damaging agents such PBD dimers are poor substrates for these pumps and can effectively circumvent this form of resistance (75). Therefore, developing payloads that evade efflux pumps is a critical design goal. For example, Mabwell Bioscience’s novel ADC, 7MW4911, utilizes a proprietary topoisomerase I inhibitor payload (MF-6). In preclinical studies, 7MW4911 demonstrated superior antitumor activity compared with MMAE- or DXd-based ADCs in models with ABC transporter-mediated MDR, highlighting its potential to overcome this major clinical challenge (76).
Linkers in ADCs
Linkers can be categorized into two main classes: cleavable (degradable) and non-cleavable (stable). Cleavable linkers take advantage of the environmental differences between the systemic circulation and tumor cells to accurately release the free cytotoxic drug. Cleavable linkers can be further categorized into chemically cleavable linkers, such as hydrazone bonds and disulfide bonds, and enzymatically cleavable linkers, such as glucuronide bonds and peptide bonds (14).
Hydrazone is a typical acid-sensitive linker. Hydrazone-linked ADCs are usually stable in the bloodstream but are hydrolyzed to release the cytotoxic payload in lysosomes and endosomes after internalization into target tumor cells (77). However, hydrolysis of the hydrazone bond is not completely confined to lysosomes, and occasional hydrolysis can also occur in the plasma, leading to reduced targeting efficiency and off-target effects (78). So far, ADCs containing hydrazone linkers have been used primarily in hematologic malignancies. For example, both gemtuzumab ozogamicin and inotuzumab ozogamicin use a hydrazone linker to conjugate calicheamicin to antibodies for the treatment of AML and acute lymphoblastic leukemia (ALL), respectively. The disulfide bond linker is another type of chemically cleavable linker, sensitive to reductive glutathione (GSH) (79). The concentration of GSH in the blood is considerably lower than the intracellular concentration in tumor cells (80). Therefore, disulfide bond linkers remain stable in the circulatory system while selectively releasing active payloads in tumor cells with elevated GSH levels.
As far as enzyme-sensitive linkers are concerned, peptide linkers are sensitive to lysosomal proteases and have been employed in many ADCs (81). Lysosomal proteases, such as cathepsin B, are usually overexpressed in tumor cells, enabling accurate drug release in the vicinity of the tumor. However, due to the presence of protease inhibitors in the blood, these linkers are generally stable in the bloodstream and help reduce the risk of side effects (82).
Among the approved ADC drugs, most utilize peptide linkers. For example, brentuximab vedotin uses a valine–citrulline linker. Beta-glucuronide linkers represent another type of enzyme-sensitive linker commonly used in ADCs. They can be cleaved by beta-glucuronidase in tumor cells to release the payload (83). Non-cleavable linkers are inert to common chemical and enzymatic conditions in vivo, such as thioether or maleimidocaproyl groups. These linkers exhibit high stability under various physiological conditions, including the tumor microenvironment’s pH, enzymatic activity, or reducing agents (84). After an ADC with a non-cleavable linker enters the tumor cell via target-mediated endocytosis, it is internalized into an endosome, which later fuses with a lysosome. Within the lysosome, the antibody backbone is extensively degraded by abundant proteases (e.g., cathepsins) and peptidases into small amino acids or short peptides. At this stage, the payload—which remains covalently attached to an amino acid residue (typically lysine or cysteine) via the non-cleavable linker—is released as a consequence of antibody degradation (85). However, the released entity is a payload derivative still connected to the linker and an amino acid remnant. The payload is considered “released” only after the hydrolysis of the specific antibody amino acid residue to which it is bound.
In the development of ADCs, the selection of an appropriate linker remains a major challenge. The ideal linker must be stable in the bloodstream while enabling rapid and effective release of the payload within tumor cells. As ADC technology continues to advance, the design and optimization of linkers will play a central role in driving the development of novel ADC therapeutics, ultimately improving treatment efficacy and reducing adverse effects.
Conjugation methods
In addition to the selection of the antibody, linker, and payload, the method used to conjugate the small-molecule moiety (i.e., the linker–payload complex) to the antibody is also critical for the successful construction of ADCs. Random conjugation remains a technology employed in some approved drugs. In lysine conjugation, the ϵ-amino groups of the abundant lysine residues on the antibody surface (typically 80–100 lysines, with 10–20 being reactive) react with activated esters (e.g., NHS esters) on the linker–payload (86). Because the conjugation occurs randomly, this process generates a heterogeneous mixture of ADC molecules with a wide range of DARs, such as DAR 0, 2, 4, 6, 8, or even higher (87). Although the resulting amide bonds are chemically stable, conjugation at sites within hydrophobic regions can promote ADC aggregation, whereas modification in the antigen-binding domain may impair antigen binding (88).
In cysteine conjugation, the four interchain disulfide bonds in the antibody hinge region are partially reduced to generate eight free cysteine thiol groups. These thiols then undergo alkylation by electrophilic moieties (e.g., maleimide) on the linker–payload (89). A major limitation of this approach is the instability of the maleimide–thioether bond. In plasma, this bond is prone to thiol exchange reactions with sulfhydryl-bearing molecules such as albumin, leading to premature payload release and potential off-target toxicity (90). The inherent limitations of random conjugation have driven the advancement of ADC technologies toward site-specific conjugation strategies (Table 3).
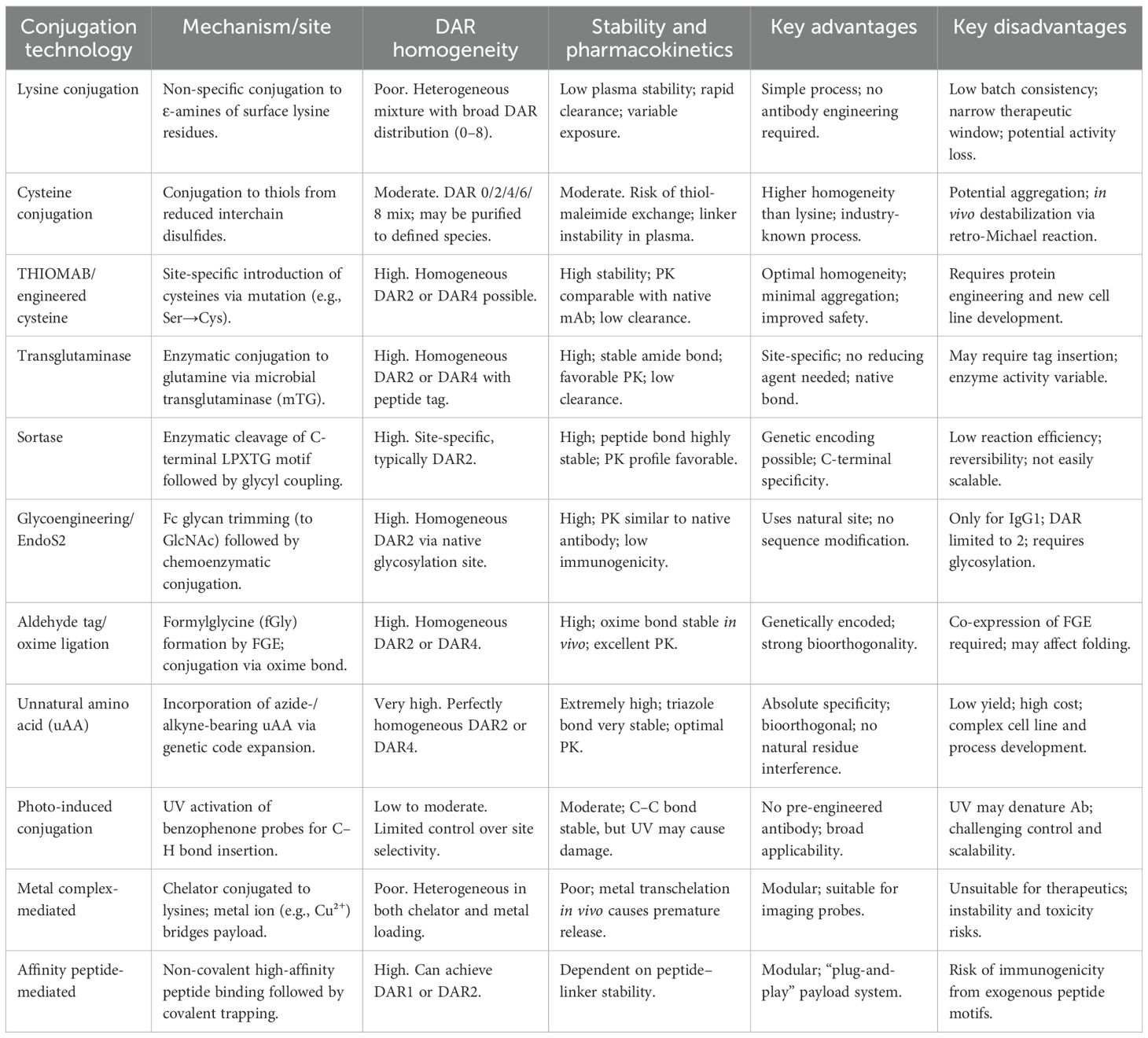
Table 3. Comparison of ADC conjugation technologies.
The engineered cysteine technology utilizes genetic engineering to replace specific amino acid residues (typically serine) at defined sites on the antibody (such as the native cysteine pairs on the heavy chain) with cysteine, thereby introducing new reactive thiol groups (–SH). These thiol groups can specifically react with linker–payload constructs containing functional moieties such as maleimide (91). Since the number of introduced thiol groups is precisely controlled (usually 2 or 4), all ADC molecules carry the same drug load, resulting in a highly homogeneous DAR (87). Furthermore, because the conjugation sites are engineered to avoid labile regions (e.g., the hinge region) and hydrophobic patches, the conjugate exhibits improved stability (92). The highly uniform DAR and enhanced stability significantly reduce clearance in systemic circulation, prolong the half-life, and ultimately contribute to prolonged efficacy and reduced systemic toxicity.
The microbial transglutaminase (mTG) technology utilizes mGT to specifically recognize the acyl donor glutamine (Q295) on the Fc region of antibodies and catalyze a cross-linking reaction between its amide group and a linker–payload construct containing a primary amine group (e.g., derived from a lysine side chain) (93). Alternatively, a “glutamine tag” (Q-tag) can be genetically introduced into specific antibody sequences to provide an enzymatic recognition site. The high specificity of the enzyme ensures that conjugation occurs exclusively at predetermined sites, resulting in nearly all ADC molecules carrying two payloads (due to the two symmetric Fc regions in IgG1) and achieving a highly uniform DAR 2 (94). This technology forms a stable amide bond that is resistant to chemical and enzymatic degradation in plasma, thereby minimizing the risk of cleavage and reducing the propensity for aggregation. The homogeneous molecular population and stable linkage promote consistent in vivo behavior, leading to more predictable PK and reduced interindividual variability (95).
The non-canonical amino acid (nnAA) technology involves the site-specific introduction of a nnnAA (such as para-acetylphenylalanine, pAcF) into the antibody sequence. This nnAA contains unique reactive functional groups (e.g., a ketone moiety) absent in natural amino acids, enabling highly efficient and specific conjugation to the linker–payload via bioorthogonal reactions such as oxime ligation (96). By controlling the number of incorporated nnAA residues, DAR can be precisely adjusted, allowing for the generation of ADCs with defined DAR values (e.g., DAR 2, 4, 6, etc.) (87). The stability of the resulting conjugate bond depends on the specific conjugation chemistry employed. For instance, linkages formed via oxime chemistry (analogous to those used in aldehyde tag approaches) may display reduced stability under acidic conditions relative to amide bonds (97).
The glycan remodeling/EndoS2 technology leverages the fact that antibodies are glycosylated proteins bearing a conserved N-glycan on their Fc region. The process begins by treating the antibody with an endoglycosidase (such as EndoS2) to remove the native glycan structures, leaving only a core N-acetylglucosamine (GlcNAc) residue on the Fc segment (98). An engineered glycosyltransferase (e.g., GalT/Y289L) then specifically attaches a sugar derivative (such as a UDP-sugar analog) carrying a linker–payload to this GlcNAc residue. Since each antibody Fc region contains a single conserved N-glycan site, the resulting ADC primarily exhibits a DAR of 2 (99). The newly formed glycosidic bond is natural in origin and demonstrates high stability in plasma, with strong resistance to hydrolysis. Moreover, because glycosylation is native to antibodies and the conjugation occurs within the Fc region, the antigen-binding function remains unaltered, enabling the ADC to retain a long circulation half-life comparable with that of unconjugated antibodies.
The aldehyde tag/oxime chemistry approach involves the genetic introduction of a short peptide sequence (“aldehyde tag”, LCXPXR) at the terminus of the antibody (e.g., the C-terminus of the heavy chain). This sequence is recognized by the formylglycine-generating enzyme (FGE), which catalytically oxidizes the cysteine residue within the tag to generate a unique formylglycine (fGly) residue. fGly contains a reactive aldehyde group (–CHO), which subsequently undergoes specific oxime ligation with a linker–payload construct containing an aminooxy group (–ONH2) (100, 101). Each introduced tag yields one aldehyde group, enabling precise conjugation with a DAR of 2. However, while the oxime bond is relatively stable under acidic conditions, it may undergo reversible reactions at neutral to alkaline pH. In vivo, it can also be susceptible to chemical and enzymatic degradation, posing a potential risk of cleavage in plasma and premature payload release (102). This relative instability may adversely affect PK, leading to accelerated clearance. Nevertheless, the highly uniform DAR achieved using this strategy remains superior to that obtained with random conjugation methods.
In summary, the conjugation technology field is rapidly moving from random conjugation toward site-specific strategies to produce homogeneous ADCs with excellent stability and PK. Future efforts are exploring ultra-precise techniques like unnatural amino acid incorporation and modular platforms, although each emerging approach still faces many challenges.
The ADCs approved for the market
ADCs have achieved remarkable clinical and commercial success, as demonstrated by the recent approval and strong market performance of agents such as trastuzumab deruxtecan (Enhertu®). These advancements have significantly influenced the pharmaceutical industry. According to industry reports, a total of 18 ADC drugs had received regulatory approval worldwide between 2000 and mid-2025. From the perspective of drug composition and technological features, the evolution of ADCs is commonly divided into three distinct generations (Table 4).
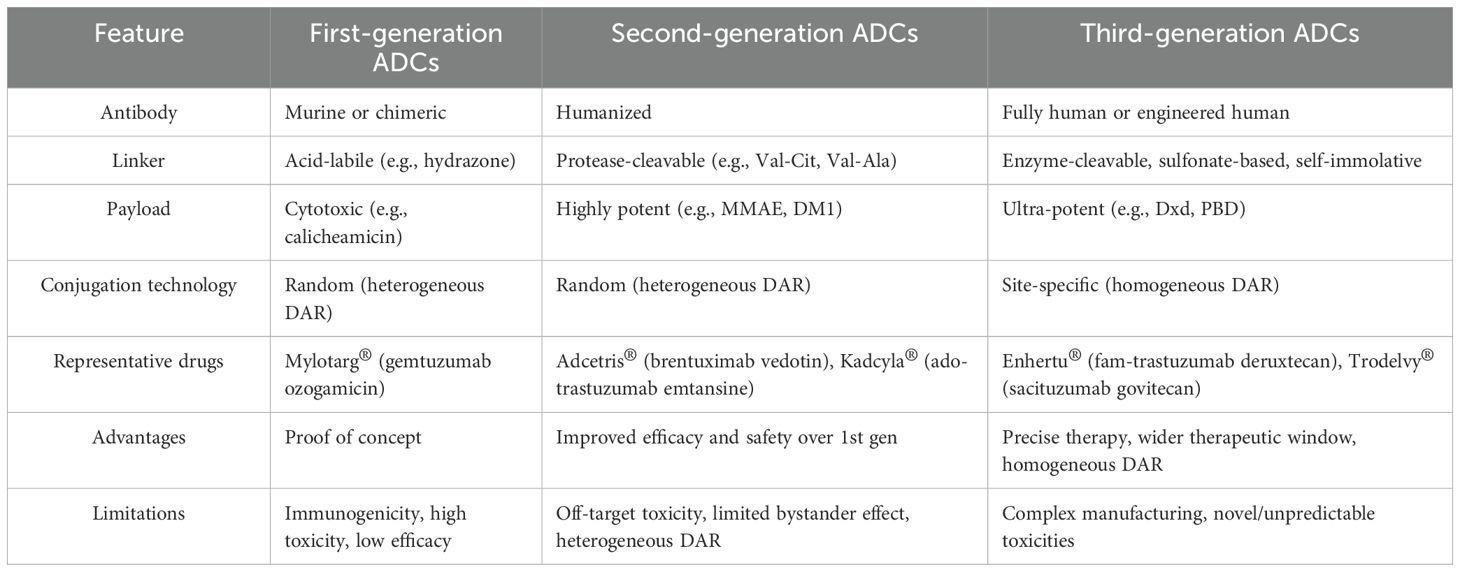
Table 4. Comparison of three generations of ADC technologies.
The first-generation ADCs
The first-generation ADCs, such as BR96-doxorubicin, gemtuzumab ozogamicin, and inotuzumab ozogamicin, employed non-cleavable linkers, traditional chemical cytotoxic payloads, and murine-derived antibodies (IgG4). Gemtuzumab ozogamicin (Mylotarg®) was the first ADC therapeutic approved for clinical use worldwide. It consists of a humanized monoclonal IgG4 antibody targeting CD33 and the cytotoxic agent N-acetyl-γ-calicheamicin, connected via a cleavable hydrazone linker (31). Mylotarg received initial approval in 2000 for the treatment of patients aged 60 years or older with CD33-positive relapsed acute AML who were not candidates for conventional chemotherapy (14).
In 2010, the Phase III SWOG S0106 trial revealed that patients treated with Mylotarg showed an increased incidence of hepatic veno-occlusive disease (VOD), with an early mortality rate of 5.7% compared with 1.4% in the control group, and failed to demonstrate a survival benefit compared with chemotherapy alone. As a result, Pfizer Inc. voluntarily withdrew the product from the market in October 2010 (103). In 2017, Mylotarg® was reevaluated and received renewed regulatory approval for the treatment of CD33-positive newly diagnosed AML in adult patients, with the single administration dose reduced from the original 9 mg/m² (used in 2000) to 3 mg/m² (104).
The antibodies used in first-generation ADCs were primarily murine or chimeric, exhibiting significant immunogenicity (15). The linkers, such as the hydrazone bond in Mylotarg, were chemically labile and exhibited poor stability, often leading to premature payload release in the bloodstream and resulting in off-target toxicity (73). The payloads had relatively low potency (e.g., calicheamicin with IC50 ≈ 10-8 M), contributing to a narrow therapeutic window. Random conjugation techniques resulted in heterogeneous DARs, typically ranging from 0 to 8, yielding highly heterogeneous products (87).
In summary, first-generation ADCs had several limitations, such as an extremely narrow therapeutic window, significant off-target toxicity, strong immunogenicity, and limited efficacy, emphasizing the need for further optimization and development.
The second-generation ADCs
Second-generation ADCs, represented by brentuximab vedotin and ado-trastuzumab emtansine, were developed through optimizations in mAb design, cytotoxic payloads, and linker technology. These ADCs exhibit improved targeting ability, more potent payloads, and reduced immunogenicity compared with their predecessors.
Brentuximab vedotin selectively binds to the CD30 antigen and is internalized via a clathrin-dependent mechanism (105). It is then transported into endosomes and lysosomes, where the linker is cleaved by cysteine proteases such as cathepsin B. The released free MMAE functions as an ultrapotent antimitotic agent that induces cell-cycle arrest and apoptosis by inhibiting tubulin polymerization.
In the ECHELON-2 trial, which evaluated therapies for CD30-positive peripheral T-cell lymphoma (PTCL), brentuximab vedotin combined with cyclophosphamide, doxorubicin, and prednisone (CHP) was compared with standard CHOP chemotherapy (cyclophosphamide, doxorubicin, vincristine, and prednisone) in CD30-positive PTCL patients. The brentuximab vedotin plus CHP regimen demonstrated a lower risk of death compared with CHOP (106). Based on these results, brentuximab vedotin received approval in 2018 for two additional clinical indications: certain types of peripheral T-cell lymphoma and previously untreated stage III or IV classical Hodgkin lymphoma (cHL) (107).
Second-generation ADCs took advantage of humanized antibodies, substantially reducing immunogenicity. The incorporation of cleavable linkers (e.g., valine-citrulline or VC linkers) enabled tumor-specific payload release with enhanced stability and diminished off-target toxicity. Additionally, more potent payloads improved antitumor efficacy significantly (15).
However, several limitations remained: random conjugation through cysteine or lysine residues continued to produce heterogeneous DAR. This resulted in ADC mixtures containing species with excessively high DAR (associated with increased toxicity) or suboptimal DAR (leading to reduced efficacy), thereby narrowing the therapeutic window. Additionally, some payloads exhibited limited membrane permeability, reducing bystander effects and impairing the elimination of adjacent antigen-negative tumor cells.
.
The third-generation ADCs
The third-generation ADCs are represented by polatuzumab vedotin, enfortumab vedotin, and fam-trastuzumab deruxtecan. Polatuzumab vedotin targets the CD79b antigen and is conjugated to the microtubule-disrupting agent MMAE via a protease-cleavable dipeptide linker (108). In July 2019, Polivy® (polatuzumab vedotin) received FDA approval for use in combination with bendamustine and rituximab for the treatment of relapsed/refractory diffuse large B-cell lymphoma (DLBCL) in patients who have received at least two prior therapies (109). It was the first ADC approved for DLBCL, the most common type of non-Hodgkin lymphoma (NHL).
Site-specific conjugation enables third-generation ADCs to achieve homogeneous composition, defined DAR (typically 2 or 4), and predictable cytotoxicity (87). These ADCs exhibit reduced off-target toxicity and improved PK profiles for their consistent DAR values. Furthermore, this generation takes advantage of fully humanized antibodies, minimizing immunogenicity compared with earlier chimeric types (15). Meanwhile, more potent payloads have been incorporated, such as PBDs, tubulysins, and novel immunomodulatory agents (52). Certain payloads (e.g., deruxtecan/DXd) possess cell membrane-penetrating capabilities, enabling potent bystander effects that eliminate heterogeneous tumor populations.
However, third-generation ADC technology is characterized by high complexity, significant manufacturing challenges, and extensive production costs. These ADCs also introduce novel toxicity risks. These include unique adverse events such as interstitial lung disease (ILD), which is a concern associated with ultrapotent payloads. A classic example is Enhertu® (fam-trastuzumab deruxtecan-nxki), which carries a black box warning specifically for ILD (110).
Novel ADCs
Biepitopic ADCs
Researchers are exploring novel strategies to overcome resistance to a single-target ADC. Biepitopic ADCs represent an innovative advancement in ADC technology by simultaneously binding to two distinct epitopes on a target antigen (Figure 4). This approach is designed to enhance tumor targeting, promote efficient internalization, and overcome drug resistance mechanisms (111). Biepitopic ADCs represent a novel therapeutic strategy in cancer treatment by enabling a single antibody to engage two different epitopes on a tumor-associated antigen. This design enhances binding affinity (particularly in tumors with low antigen expression such as HER2) and improves drug delivery efficiency (112). Although still in early-stage development, preliminary clinical results have shown promising efficacy. For example, TQB2102 has demonstrated a total pathological complete response (tpCR) rate of up to 73.1% in the neoadjuvant treatment of HER2-positive breast cancer (113). Nevertheless, the complex molecular design and manufacturing requirements of biepitopic ADCs present significant challenges. As more research data become available, biepitopic ADCs are expected to provide new therapeutic options for cancer patients.
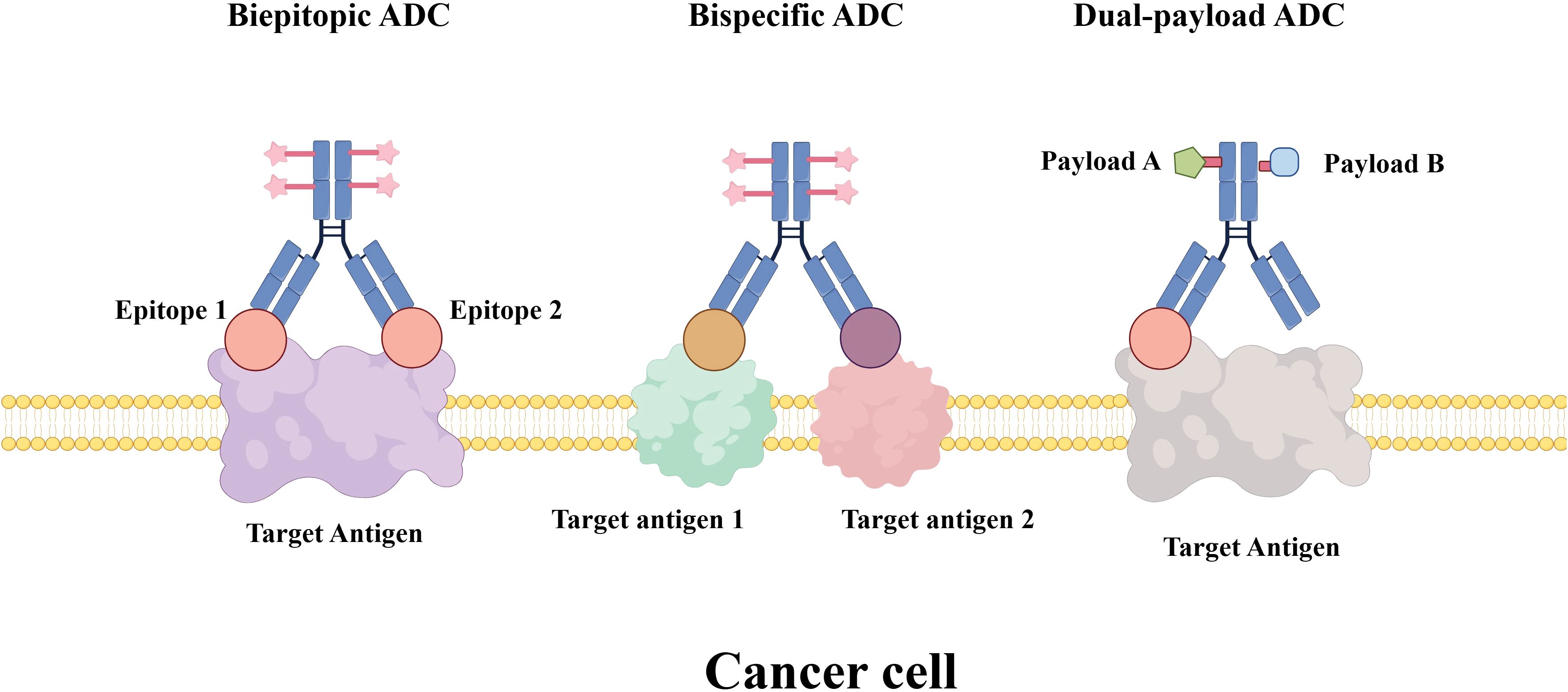
Figure 4. Novel ADC formats for cancer therapy. The illustration compares three advanced ADC design strategies: Biepitopic ADC: an ADC featuring a single antibody engineered to bind two distinct epitopes on the same target antigen, a strategy aimed at enhancing binding avidity and internalization efficiency. Bispecific ADC: an ADC incorporating a bispecific antibody that recognizes two different target antigens, improving tumor selectivity and potential to overcome antigen heterogeneity. Dual-payload ADC: an ADC conjugated with two different cytotoxic payloads (e.g., Payload A and Payload B) via a shared or separate linkers, enabling synergistic mechanisms of action and reduced risk of resistance.
Bispecific ADCs
Bispecific antibodies (BsAbs) serve as the carriers for bispecific ADCs (BsADCs). These antibodies possess two distinct antigen-binding domains, enabling them to simultaneously bind to two different antigens and thereby promote the delivery of cytotoxic payloads (114). The simultaneous binding to two different tumor cell surface targets enables bispecific ADCs to achieve greater specificity and more efficient internalization (115). This dual-targeting strategy can promote receptor internalization through cross-linking or enable simultaneous targeting of both tumor cells and components of the tumor microenvironment, leading to synergistic antitumor effects. Even if one target is mutated or downregulated, the other target can still mediate drug delivery. This strategy also helps to overcome resistance (116).
Novel tetravalent BsAbs have been developed to target c-Met/EGFR and c-Met/HER2. These BsAbs combine antibodies that induce rapid internalization and degradation of c-Met with single-chain variable fragments (scFvs) targeting EGFR or HER2 (117, 118). Furthermore, targeting HER2 in combination with other antigens such as B7-H3 and B7-H4 shows promise for broader therapeutic applications (119). For example, an HER2×CD63 bispecific ADC has demonstrated enhanced cytotoxicity in HER2-positive tumor models, underscoring its potential for more precise targeting (120). This HER2×CD63 bispecific ADC is currently under preclinical investigation. SI-B001, a bispecific antibody targeting EGFR and HER3, has been conjugated via a novel acid-cleavable (AC) linker to the topoisomerase I inhibitor ED04, forming the BsADC BL-B01D1 with a DAR of approximately 8. This conjugate exhibits improved targeting precision and safety. SI-B001 has shown promising efficacy in both Phase I and Phase II clinical trials, supporting its advancement to Phase III clinical studies (121).
Bispecific ADCs offer several advantages, including improved tumor selectivity, enhanced internalization efficiency, a potentially wider therapeutic window, and the ability to address tumor heterogeneity and drug resistance. However, the in vivo behavior of BsADCs is complex. Although dual targeting may theoretically improve tumor selectivity, it could also introduce novel and unpredictable toxicities, necessitating careful clinical monitoring and management.
Dual-payload ADCs
Dual-payload ADCs refer to ADCs armed with two distinct payloads that have different mechanisms of action, or the same payload conjugated via different linkers, on a single antibody (122). These ADCs have the potential to function as single therapeutic agents capable of eliciting synergistic effects and overcoming drug resistance in patients with treatment-refractory tumors (123).
For example, one study successfully achieved homogeneous co-conjugation of both MMAE and MMAF to anti-CD30 antibodies, attaining a DAR of 16 (124). These dual-payload ADCs demonstrated potent antitumor activity in a CD30-positive multidrug-resistant naplastic large cell lymphoma mouse xenograft model (125).
Dual-payload ADCs represent a significant advancement beyond conventional ADCs by integrating two distinct classes of payloads, rather than relying solely on cytotoxic agents such as MMAE and MMAF. For instance, a strategic approach co-conjugating a sterlin-like agent with TLR agonists to anti-folate receptor alpha (FolRα) antibodies has shown synergistic antitumor activity and induction of immune memory in mouse models (126).
However, not all dual-payload ADC configurations have demonstrated clear synergistic effects. In some studies combining mechanistically distinct payloads, enhanced efficacy was not observed. Researchers developed an anti-HER2 ADC co-loaded with MMAE and SG3457—an ultrapotent PBD dimer that induces DNA crosslinking damage (127). Similarly, another HER2-targeted ADC was designed to deliver both MMAF and the highly potent topoisomerase II inhibitor PNU-15968. Although both ADCs engaged dual mechanisms of action, neither exhibited superior potency compared with their single-payload counterparts in vitro (128).
Multiple challenges must be addressed in the development of dual-payload ADCs. The development of dual-payload ADCs requires complex linker chemistry to ensure both payloads are efficiently released at the correct time and site. Additive or synergistic toxicity from two potent agents may lead to unexpected adverse effects. Furthermore, differences in release kinetics, distribution, metabolism, and clearance profiles between the two payloads complicate PK and pharmacodynamic (PD) relationships, bringing significant challenges for clinical dosing regimen design.
Immune-stimulating ADCs
Immune-stimulating ADCs (ISACs) represent a novel class of therapeutics that share structural similarities with traditional ADCs, comprising three core components: a tumor-targeting antibody, a linker, and an immunostimulatory payload (67). Unlike conventional ADCs that employ cytotoxic agents, ISACs utilize immune agonists or modulators—such as Toll-like receptor (TLR) agonists or STING agonists—as their effector molecules. Upon release from tumor cells into the tumor microenvironment, immunostimulatory payloads are captured by surrounding antigen-presenting cells, leading to their potent activation. Alternatively, these payloads can directly activate innate immune signaling pathways within tumor cells, stimulating the production of chemokines or interferons (129). This response promotes the recruitment and activation of additional immune cells in the tumor microenvironment (Figure 5).
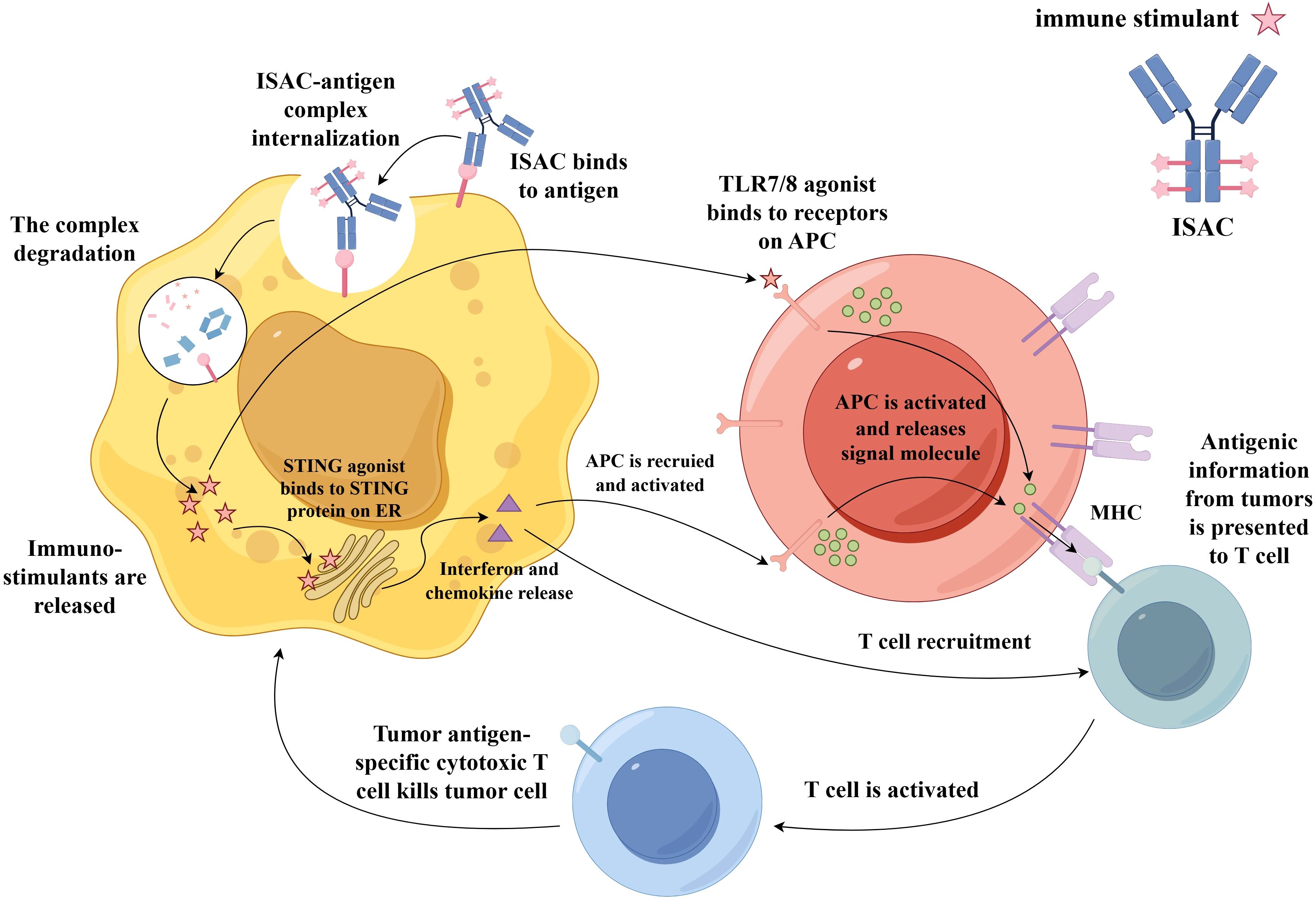
Figure 5. Mechanism of action of an ISAC. After binding specifically to a tumor-associated antigen on the cancer cell surface, the ISAC–antigen complex is internalized. Following degradation of the complex in the lysosome, immuno-stimulatory payloads (such as TLR7/8 or STING agonists) are released. The TLR7/8 binds to receptors on APCs, triggering the recruitment and activation of APCs toward tumor cells. These activated APCs then present tumor-associated antigens via MHC molecules to T cells, activating them into tumor antigen-specific cytotoxic T cell that kills the tumor cell. Additionally, STING agonists bind to the STING protein on the ER, inducing the production of interferons and chemokines. This promotes the recruitment and activation of APCs at the tumor site, which in turn can also activate T cells, further killing tumor cells. Abbreviations: TLR7/8, Toll-like receptor 7/8; APCs, antigen-presenting cells; ER, endoplasmic reticulum.
Antigen selection is critically important for ISACs and must satisfy criteria including high tumor-specific expression and minimal expression in healthy tissues. IgG1 is the preferred antibody subclass due to its high affinity for Fcγ receptors and extended serum half-life (130). ISACs primarily employ innate immune agonists such as TLR7/8, TLR9, or STING agonists, which effectively promote the priming and recruitment of tumor-specific immune cells. TLR7/8 agonists activate dendritic cells and macrophages, thereby bridging innate and adaptive immunity. TLR9 agonists enhance antigen presentation and cytokine production, whereas STING agonists activate type I interferon pathways to bolster antitumor immunity (131). ISACs are designed to combat tumors through targeted delivery and localized immune activation, theoretically mitigating the systemic toxicities associated with free immune agonists.
Recent preclinical studies have demonstrated that ISACs can effectively promote immune cell infiltration across various tumor models. Despite promising preclinical results, clinical outcomes have been modest. In a Phase 2 trial of BDC-1001 (a HER2-targeted ISAC conjugated to a TLR7/8 agonist), only 1 partial response and 12 cases of stable disease were observed among 57 participants, resulting in an overall response rate of 1.75% (132). Similarly, in a Phase 1 trial of SBT6050 (a HER2-targeted ISAC linked to a TLR8 agonist), among 14 evaluable patients, only one achieved a partial response and three had a stable disease, yielding an overall response rate of 7.1% (133).
The development of ISACs faces significant challenges, such as a narrow therapeutic window. While immune agonists with insufficient potency may be ineffective, excessive activation can lead to severe local or systemic inflammatory reactions, which makes it difficult to identify an optimal balance (131). The relationships among ISAC biodistribution, metabolism, payload release kinetics, and ultimate immunostimulatory effects and toxicity profiles remain highly complex, leading to substantial obstacles for predictive modeling and clinical evaluation (134). Furthermore, ISACs may induce anti-drug antibodies (ADAs), which could compromise efficacy and introduce additional safety concerns (130).
As an emerging strategy in cancer immunotherapy, ISAC development remains at an early stage. Although initial clinical data have fallen short of expectations, ISACs continue to represent a promising direction for novel approaches in tumor immunotherapy.
Challenges facing ADCs
Target antigen heterogeneity
Target antigen heterogeneity refers to the significant variation in the expression of target antigens on the surface of tumor cells, which can occur within a single tumor mass or across different metastatic lesions in the same patient (24). In other words, not all tumor cells uniformly express the target antigen for which an ADC is designed. Spatial heterogeneity describes the phenomenon wherein some regions within a tumor exhibit high antigen expression (antigen-positive), whereas other regions show low or no expression (antigen-negative). Temporal heterogeneity refers to the dynamic changes in antigen expression levels over time, which may occur as the disease progresses or in response to treatment (9). ADCs can effectively kill tumor cells with high antigen expression but are often ineffective against those with low or negative expression. These surviving cells may continue to proliferate, ultimately leading to disease relapse. Several strategies have been developed to address target antigen heterogeneity:
1. Utilizing the bystander effect: Employing a membrane-permeable payload allows the cytotoxic agent—once released inside an antigen-positive cell—to diffuse across the cell membrane into the surrounding tumor microenvironment. There, it can enter and kill adjacent antigen-negative tumor cells (77). For example, the DXd payload in trastuzumab deruxtecan (T-DXd/Enhertu®) exhibits a potent bystander effect. This property is a key reason for its notable efficacy in HER2-low breast cancer, as it can eliminate tumor cells with heterogeneous HER2 expression (130).
2. Developing payloads with novel mechanisms of action: Arming ADCs with immunostimulatory payloads creates ISACs. Such ADCs target antigen-positive cells to release immune agonists (e.g., TLR or STING agonists), which activate immune cells such as dendritic cells and macrophages in the tumor microenvironment (129, 134). These activated immune cells can attack tumor cells in an antigen-agnostic manner, thereby overcoming heterogeneity.
3. Developing bispecific ADCs: Designing antibodies capable of simultaneously binding two different tumor antigens increases the likelihood of ADC binding even if a tumor cell expresses only one of the target antigens (114, 116). This approach broadens the targetable tumor cell population and reduces the risk of resistance due to loss of a single antigen.
4. Optimizing target antigen: Choosing targets that are more uniformly expressed within tumor tissue can mitigate heterogeneity. Currently, the target antigens of approved ADC drugs are primarily specific proteins overexpressed on typical tumor cells, such as HER2, TROP-2, and nectin-4 in solid tumors, and CD19, CD22, CD33, and CD30 in hematologic malignancies (19, 111). A promising strategy involves targeting mutant proteins uniquely expressed in tumors, as these often exhibit higher levels of ubiquitination and are more readily internalized and degraded compared with their wild-type counterparts. Delivering ADCs specifically to cancer cells expressing such oncogenic mutant proteins may maximize treatment specificity (7, 78). Furthermore, with advances in basic research on tumor immunology, ADC target development has gradually expanded in recent years from classical tumor antigens to include antigens expressed on cells within the tumor microenvironment and cancer stem cells (9, 77).
Resistance mechanisms
Resistance to ADCs is a major challenge to their clinical efficacy. Gaining a deeper understanding of its mechanisms and developing corresponding strategies are crucial for improving patient outcomes. The primary mechanisms of ADC resistance include the following aspects (1). Target-Related resistance: Due to target antigen downregulation or loss, ADCs cannot recognize and bind to tumor cells. Epitope mutations in the target antigen result in reduced antibody-binding affinity22 (2). Internalization and processing resistance: Decreased internalization efficiency prevents ADC–antigen complexes from entering tumor cells. Lysosomal dysfunction (e.g., impaired acidification) hinders linker cleavage, preventing payload release. Alterations in the expression of lysosomal enzymes (e.g., cathepsins) reduce linker cleavage efficiency87 (3). Payload-related resistance: Upregulation of drug efflux pumps (e.g., P-gp, BCRP) enables cells to pump the payload out, reducing intracellular concentrations77. Mutations in the payload’s target prevent it from exerting its cytotoxic effect. Inactivation of apoptosis pathways or upregulation of antiapoptotic proteins allows cells to evade payload-induced cell death78.
Potential strategies to overcome the aforementioned ADC resistance mechanisms include the development of ADCs targeting novel antigens to expand the target repertoire (e.g., TROP-2, c-Met)117; the utilization of bispecific ADCs capable of simultaneously engaging two tumor antigens to mitigate resistance caused by single-antigen loss (114); and combination therapies with targeted agents, such as proteasome inhibitors, to prevent degradation of the target protein (24). Additional approaches involve the optimization of antibody selection for enhanced internalization efficiency through screening of antibodies with superior cellular uptake capabilities (135); the advancement of novel linker technologies designed for cleavage by a broader spectrum of enzymatic systems; and the substitution of conventional payloads (e.g., microtubule inhibitors like MMAE) with novel agents (e.g., topoisomerase I inhibitors like Dxd) (136). Further strategies include the design of payloads engineered to evade efflux pumps (e.g., P-gp) (137); the exploration of combination regimens with efflux pump inhibitors, although still under clinical investigation due to potential toxicity concerns; and the development of ADCs with potent bystander effects (e.g., DS-8201) to eliminate adjacent antigen-negative tumor cells and address tumor heterogeneity (138).
Pharmacokinetic complexity
The PK of ADCs represent one of the most complex and challenging aspects of their development. This complexity stems from the fact that ADCs are inherently heterogeneous mixtures that undergo multiple biotransformations in vivo. As a result, their PK cannot be adequately described using traditional models developed for conventional mAbs or small-molecule drugs. Following administration, three primary analyte forms can be present in systemic circulation: the intact ADC, the unconjugated (naked) antibody, and the free cytotoxic payload (22).
Intact ADC refers to the complete conjugate structure with the payload attached. It represents the active drug form responsible for target engagement and overall drug exposure. Intact ADCs are primarily eliminated via proteolytic degradation and exhibit a long half-life, typically ranging from days to weeks (87). Unconjugated antibody refers to the bare mAb after the payload has been cleaved and released. It may compete with the ADC for target binding but lacks cytotoxic activity (139). Free payload refers to the small-molecule cytotoxin released into circulation upon linker cleavage. Its PK follows patterns typical of small molecules, with a short half-life (minutes to hours) and very low plasma concentrations that are often challenging to quantify (140). Free payload is primarily metabolized in the liver and eliminated via renal or fecal excretion. This process may be influenced by drug–drug interactions and impaired hepatic or renal function (71).
The release kinetics, distribution, metabolism, and clearance pathways of these three components differ significantly yet are interrelated (141). Together, they shape the overall and safety profile of the ADC. Thus, the PK of ADCs constitute a dynamic, MTI-analyte, and multi-pathway system. A deep understanding of this intricate PK behavior is essential for optimizing therapeutic efficacy and guiding the development of next-generation ADCs.
Unavoidable side effect
One of the most common adverse effects of ADCs is hematologic toxicity, which manifests as neutropenia (the most frequent), anemia, and thrombocytopenia. These effects are likely due to the released payload—often an antimitotic agent—affecting rapidly dividing bone marrow hematopoietic cells (142). Regular monitoring of complete blood counts is essential during treatment. Granulocyte colony-stimulating factor (G-CSF) may be used for the prevention or treatment of neutropenia. Dose delays or adjustments may be necessary based on clinical indications (53).
Ocular toxicity is linked to specific payloads (e.g., MMAE, MMAF) and may include dry eye syndrome, keratopathy, and blurred vision (72). This may result from payload distribution to ocular tissues via tear secretion or systemic circulation, affecting proliferating corneal epithelial cells. Prophylactic use of artificial tears is recommended. In severe cases, ophthalmologic consultation is necessary, and ADC therapy may need to be interrupted or discontinued (143).
Hepatotoxicity, which is related to the hepatic metabolism of the payload or Fc-mediated uptake of the ADC (144), may manifest as elevated transaminases and bilirubin levels. Therefore, liver function should be monitored regularly during treatment, with dose adjustments or treatment interruptions implemented based on severity. Gastrointestinal adverse reactions are also common, including nausea, vomiting, diarrhea, and decreased appetite (53). Patients should be managed with prophylactic antiemetics and active supportive measures, including antidiarrheal agents and fluid replacement.
ILD/pneumonitis is one of the most serious and potentially life-threatening adverse reactions and requires heightened vigilance. It is strongly associated with certain ADCs, as highlighted by the boxed warning for Enhertu (T-DXd)-related ILD (110). Payloads with high membrane permeability (such as DXd and SN-38) can be released from target cells and diffuse into surrounding normal lung tissue, causing DNA damage and cell death, which leads to intense inflammatory and fibrotic responses. This is the primary supposed mechanism for Enhertu (T-DXd)-related ILD. Microtubule inhibitors (such as MMAE and DM1) may also result in lung injury by injuring normal alveolar epithelial cell function or inducing vascular leakage (145). Early recognition is critical. Upon symptom onset, treatment should be interrupted immediately, followed by radiographic evaluation and corticosteroid therapy (146).
Dermatologic toxicity manifests associated with ADCs present as rash, pruritus, and dryness; in severe cases, it may lead to extensive skin detachment and mucosal erosion (e.g., enfortumab vedotin) (53). This may be related to the expression of Nectin-4 in skin keratinocytes, non-specific killing caused by the ADC “bystander effect”, and synergistic exacerbation of inflammation due to immune activation (147). Before treatment, it is essential to assess the patient’s baseline skin condition and risk factors. During treatment, regular skin examinations should be conducted, with particular attention to new rashes or changes in existing rashes (148).
Peripheral neuropathy presents as tingling or numbness in the fingers or toes, as well as muscle weakness (149). This may be associated with MMAE payload disrupting microtubule function, affecting neuronal axonal transport and nerve fiber integrity (150). Before treatment, it is necessary to evaluate the patient’s existing neuropathy symptoms and risk factors. During each follow-up, actively inquire whether the patient has symptoms such as numbness, tingling, burning pain, or weakness in the hands and feet, and perform simple neurological examinations (e.g., pinprick sensation, vibration sense, tendon reflexes) (148).
Although not all ADCs induce hyperglycemia, those ADCs incorporating MMAE carry a well-established risk. MMAE, an antimicrotubule agent that inhibits cell division, can disrupt microtubule function. Pancreatic beta cells, which secrete insulin, are notably sensitive to such disruption despite their low rate of division (151). Patients with preexisting risk factors, such as a history of diabetes, obesity, or insulin resistance, are generally at increased risk for hyperglycemic events. ADC therapy may exacerbate these underlying metabolic disorders (53). Before the treatment with such ADCs, patients should receive a baseline assessment that includes blood glucose and glycated hemoglobin measurements, in addition to an evaluation of their diabetes history and risk profile. During therapy, blood glucose levels should be monitored regularly, particularly proximate to each treatment cycle (148).
Combination therapy of ADCs with immune checkpoint inhibitors
With the precise delivery of potent cytotoxic drugs into tumor cells, ADCs have revolutionized cancer treatment greatly. However, the complexity and adaptability of tumors often lead to the failure of single-agent therapies (152). Therefore, combining ADCs with other therapies—such as ICIs that enhance immune response, targeted drugs that block different signaling pathways, or traditional chemotherapy—has become an essential strategy to overcome drug resistance and enhance the depth and persistence of response (153).
ICIs have a completely different mechanism from that of ADCs. ICIs are designed to enhance the inherent antitumor activity of the immune system by removing inhibitory signals of T-cell activation to restore cytotoxic immune effector function against cancer cells. ADCs can reduce immunosuppressive cells, increase CD8+ T-cell infiltration, enhance the response of tumors to immunotherapy, and remodel the tumor microenvironment, thereby enhancing the efficacy of ICIs (154).
PD-1/PD-L1 inhibitors are a type of immune checkpoint inhibitor that can block the interaction between PD-1 and PD-L1 to prevent the immune escape of tumor cells. The combination therapy of ADCs with PD-1/PD-L1 inhibitors represents a cutting-edge direction in current cancer immunotherapy, demonstrating significantly enhanced antitumor efficacy through synergistic mechanisms (155). The combination regimen of enfortumab vedotin (EV), an ADC directed to Nectin-4, and pembrolizumab, a PD-1 inhibitor, was evaluated in EV-302, a Phase 3 study in patients with locally advanced or metastatic urothelial cancer. The combination showed a statistically significant and clinically meaningful improvement in overall survival (OS), progression-free survival (PFS), and the key secondary endpoint of objective response rate (ORR) compared with chemotherapy. The ORR was higher in the combination group than in the chemotherapy group (68% vs. 44%). The median PFS was 12.5 months in the combination group versus 6.3 months in the chemotherapy group (HR = 0.45), whereas the median OS was 31.5 versus 16.1 months (HR = 0.47) (156).
Sacituzumab govitecan, a Trop-2-directed ADC with a topoisomerase I inhibitor payload, improves PFS and OS compared with chemotherapy in patients with pretreated metastatic triple-negative breast cancer (TNBC) (157). The open-label, international, multicenter, randomized Phase III trial (AFT-65/ASCENT-05/OptimICE-RD) will determine whether the combination of sacituzumab govitecan and pembrolizumab (a PD-1 inhibitor) can improve interval disease-free survival compared with pembrolizumab alone or in combination with capecitabine in patients with stage II-III TNBC who have residual invasive disease after neoadjuvant therapy. This clinical trial is currently underway (158). In a study of patients treated with RC-48-ADC and toripalimab, HER2 was positive in 59% of patients, and the objective response rate was 73.2% (159). These results demonstrate promising efficacy for this combination regimen. Other combination trials are ongoing, and their results are eagerly awaited.
The combination of enfortumab vedotin (EV) and pembrolizumab brings new hope to patients with advanced urothelial carcinoma. However, its associated safety issues—particularly adverse events such as cutaneous toxicity (147), neuropathy (150), and hyperglycemia (151)—require closer attention and proper management by clinicians, as previously discussed in the section on unavoidable side effects of ADC-based therapeutics.
Overall, the combination of ADCs and ICIs is a highly promising direction in the field of cancer therapy. Its core advantage lies in a strong synergistic effect at the mechanism level, which has been proven in multiple clinical trials of specific tumor types to significantly improve efficacy. Despite challenges in safety, biomarker development, and protocol optimization, this strategy shows great promise for improving cancer therapy and becomes a key direction for future research and clinical practice.
Conclusion
ADCs represent a pioneering class of targeted anticancer therapeutics. ADCs have been designed to combine the specificity of monoclonal antibodies with the potent cytotoxicity of chemotherapeutic agents. With the selective delivery of highly toxic payloads to tumor cells expressing specific antigens, ADCs significantly improve the therapeutic index and minimize damage to healthy tissues. ADCs have achieved remarkable clinical and commercial success and significantly influenced the pharmaceutical industry. However, ADCs still face numerous challenges. Target antigen heterogeneity can limit efficacy and drive resistance. Mechanisms of resistance include antigen downregulation, impaired internalization, payload efflux, and altered apoptosis pathways. ADC pharmacokinetics are complex due to the coexistence of intact conjugates, naked antibodies, and free payloads, each with distinct behaviors. Toxicity is a major concern. Common adverse effects include hematologic toxicity, neuropathy, ocular damage, hepatotoxicity, and pneumonitis.
To overcome these limitations, innovative strategies are being explored. The conjugation technology field is rapidly moving from random conjugation toward site-specific strategies to produce homogeneous ADCs with excellent stability and PK. New ADCs are under exploration, such as bsADCs, dual-payload ADCs, and ISACs. The combination of ADCs with ICIs such as PD-1/PD-L1 inhibitors has demonstrated synergistic efficacy by enhancing antitumor immune responses. Trials of enfortumab vedotin plus pembrolizumab have shown significantly improved response and survival rates in urothelial carcinoma and other cancers. In conclusion, ADCs have ushered in a new era of targeted cancer therapy, offering substantial benefits over traditional treatments. Ongoing research focuses on optimizing antibody engineering, linker stability, payload potency, and conjugation methods to enhance efficacy and reduce toxicity. The integration of ADCs with other therapeutic modalities holds great potential to address resistance, improve outcomes, and expand treatment options for cancer patients.
Author contributions
ZT: Writing – review & editing, Writing – original draft, Conceptualization. YX: Writing – review & editing. YZ: Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AC, acid-cleavable;AML, acute myeloid leukemia; ADC, Antibody–drug conjugate; ADCC, antibody-dependent cell-mediated cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; ADCs, antibody–drug conjugates; ADAs, anti-drug antibodies; BSB, binding site barrier;BsAbs, bispecific antibodies; BsADCs, bispecific antibody-drug conjugates; cHL, classical Hodgkin lymphoma; CDC, complement-dependent cytotoxicity; DLBCL, diffuse large B-cell lymphoma; DAR, drug-to-antibody ratio; DARs, drug-to-antibody ratios; DXd, deruxtecan; EV, enfortumab vedotin; FcγR, Fcγ receptor; FDA, Food and Drug Administration; FGE, formylglycine-generating enzyme; G-CSF, granulocyte colony-stimulating factor; ICIs, immune checkpoint inhibitors; ISACs, immune-stimulating antibody-drug conjugates; IgG, immunoglobulin G; ILD, interstitial lung disease; MMPs, matrix metalloproteinases; mTG, microbial transglutaminase; mAb, monoclonal antibody; MDR, multidrug resistance; nM, nanomolar; nnAA, non-canonical amino acid; NHL, non-Hodgkin lymphoma; ORR, objective response rate; OS, overall survival; PTCL, peripheral T-cell lymphoma; P-gp, P-glycoprotein; PK, pharmacokinetics; pM, picomolar; PFS, progression-free survival; PBD, pyrrolobenzodiazepine; scFvs, single-chain variable fragments; TLR, Toll-like receptor; tpCR, total pathological complete response; TNBC, triple-negative breast cancer; TROP-2, trophoblast cell-surface antigen 2; VOD, veno-occlusive disease.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
2. Wang Y, Yan Q, Fan C, Mo Y, Wang Y, Li X, et al. Overview and countermeasures of cancer burden in China. Sci China Life Sci. (2023) 66:2515–26. doi: 10.1007/s11427-022-2240-6
3. Chen PC, Liu X, and Lin Y. Drug repurposing in anticancer reagent development. Comb Chem High Throughput Screen. (2017) 20:395–402. doi: 10.2174/1386207319666161226143424
5. Lindley C, McCune JS, Thomason TE, Lauder D, Sauls A, Adkins S, et al. Perception of chemotherapy side effects cancer versus noncancer patients. Cancer Pract. (1999) 7:59–65. doi: 10.1046/j.1523-5394.1999.07205.x
6. Ferrara N, Hillan KJ, and Novotny W. Bevacizumab (Avastin), a humanized antiVEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun. (2005) 333:328–35. doi: 10.1016/j.bbrc.2005.05.132
7. Hafeez U, Parakh S, Gan HK, and Scott AM. Antibody-drug conjugates for cancer therapy. Molecules. (2020) 25:4764. doi: 10.3390/molecules25204764
8. Coleman N, Yap TA, Heymach JV, Meric-Bernstam F, and Le X. Antibody-drug conjugates in lung cancer: dawn of a new era? NPJ Precis Oncol. (2023) 7:5. doi: 10.1038/s41698-022-00338-9
9. Marks S and Naidoo J. Antibody drug conjugates in non-small cell lung cancer: An emerging therapeutic approach. Lung Cancer. (2022) 163:59–68. doi: 10.1016/j.lungcan.2021.11.016
10. Nakada T, Masuda T, Naito H, Yoshida M, Ashida S, Morita K, et al. Novel antibody drug conjugates containing exatecan derivative-based cytotoxic payloads. Bioorg Med Chem Lett. (2016) 26:1542–5. doi: 10.1016/j.bmcl.2016.02.020
11. Guo Y, Shen Z, Zhao W, Lu J, Song Y, Shen L, et al. Rational identification of novel antibody-drug conjugate with high bystander killing effect against heterogeneous tumors. Adv Sci (Weinh). (2024) 11:e2306309. doi: 10.1002/advs.202306309
12. Wang Y, Xia B, Cao L, Yang J, Feng C, Jiang F, et al. Preclinical studies of BB-1701, a HER2-targeting eribulin-containing ADC with potent bystander effect and ICD activity. Antib Ther. (2024) 7:221–32. doi: 10.1093/abt/tbae019
13. Singh AP and Shah DK. A “Dual” Cell-level systems PK-PD model to characterize the bystander effect of ADC. J Pharm Sci. (2019) 108:2465–75. doi: 10.1016/j.xphs
14. Sorokin P. Mylotarg approved for patients with CD33+ acute myeloid leukemia. Clin J Oncol Nurs. (2000) 4:279–80.
15. Ruan DY, Wu HX, Meng Q, and Xu RH. Development of antibody-drug conjugates in cancer: Overview and prospects. Cancer Commun (Lond). (2024) 44:3–22. doi: 10.1002/cac2.12517
16. Long R, Zuo H, Tang G, Zhang C, Yue X, Yang J, et al. Antibody-drug conjugates in cancer therapy: applications and future advances. Front Immunol. (2025) 16:1516419. doi: 10.3389/fimmu.2025.1516419
17. Abuhelwa Z, Alloghbi A, and Nagasaka M. A comprehensive review on antibody-drug conjugates (ADCs) in the treatment landscape of non-small cell lung cancer (NSCLC). Cancer Treat Rev. (2022) 106:102393. doi: 10.1016/j.ctrv.2022.102393
18. Diamantis N and Udai B. Antibody-drug conjugates–an emerging class of cancer treatment. Br J Cancer. (2016) 114:362–7. doi: 10.1038/bjc.2015.435
19. Sievers EL and Senter PD. Antibody-drug conjugates in cancer therapy. Annu Rev Med. (2013) 64:15–29. doi: 10.1146/annurev-med-050311-201823
20. Jaracz S, Chen J, Kuznetsova LV, and Ojima I. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg MedChem. (2005) 13:5043–54. doi: 10.1016/j.bmc
21. Rugo HS, Bardia A, Marmé F, Cortés J, Schmid P, Loirat D, et al. Overall survival with sacituzumab govitecan in hormone receptor-positive and human epidermal growth factor receptor 2-negative metastatic breast cancer (TROPiCS-02): a randomised, open-label, multicentre, phase 3 trial. Lancet. (2023) 402:1423–33. doi: 10.1016/S0140-6736(23)01245-X
22. Shefet-Carasso L and Benhar I. Antibody-targeted drugs and drug resistance— challenges and solutions. Drug Resist Update. (2015) 18:36–46. doi: 10.1016/j.drup.2014.11.001
23. Hejmady S, Pradhan R, Kumari S, Pandey M, Dubey SK, and Taliyan R. Pharmacokinetics and toxicity considerations for antibodydrug conjugates: an overview. Bioanalysis. (2023) 15:1193–202. doi: 10.4155/bio-2023-0104
24. Hock MB, Thudium KE, Carrasco-Triguero M, and Schwabe NF. Immunogenicity of antibody drug conjugates: bioanalytical methods and monitoring strategy for a novel therapeutic modality. AAPS J. (2015) 17:35–43. doi: 10.1208/s12248-014-9684-6
25. De Cecco M and Galbraith DN. McDermott LL What makes a good antibody-drug conjugate? Expert Opin Biol Ther. (2021) 21:841–7. doi: 10.1080/14712598.2021.1880562
26. Singh AP, Guo L, Verma A, Wong GG, Thurber GM, and Shah DK. Antibody coadministration as a strategy to overcome binding-site barrier for ADCs: a quantitative investigation. AAPS J. (2020) 22:28. doi: 10.1208/s12248-019-0387-x
27. Xu S. Internalization, trafficking, intracellular processing and actions of antibody-drug conjugates. Pharm Res. (2015) 32:3577–83. doi: 10.1007/s11095-015-1729-8
28. Tsumura R, Manabe S, Takashima H, Koga Y, Yasunaga M, and Matsumura Y. Influence of the dissociation rate constant on the intra-tumor distribution of antibody-drug conjugate against tissue factor. J Control. Release. (2018) 284:49–56. doi: 10.1016/j.jconrel.2018.06.016
29. Saunders KO. Conceptual approaches to modulating antibody effector functions and circulation half-life. Front Immunol. (2019) 10:1296. doi: 10.3389/fimmu.2019.01296
30. Dekkers G, Bentlage AEH, Stegmann TC, Howie HL, Lissenberg-Thunnissen S, Zimring J, et al. Vidarsson G.Affinity of human IgG subclasses to mouse Fc gamma receptors. MAbs. (2017) 9:767–73. doi: 10.1080/19420862.2017.1323159
31. Bahou C, Love EA, Leonard S, Spears RJ, Maruani A, Armour K, et al. Disulfide modified igG1: an investigation of biophysical profile and clinically relevant fc interactions. Bioconjug Chem. (2019) 30:1048–54. doi: 10.1021/acs.bioconjchem.9b00174
32. Golay J, Andrea AE, and Cattaneo I. Role of fc core fucosylation in the effector function of igG1 antibodies. Front Immunol. (2022) 13:929895. doi: 10.3389/fimmu.2022.929895
33. Yu J, Song Y, and Tian WJ. How to select IgG subclasses in developing anti-tumor therapeutic antibodies. Hematol Oncol. (2020) 13:45. doi: 10.1186/s13045-020-00876-4
34. Wu HH, Crames M, Wei Y, Liu D, Gueneva-Boucheva K, Son I, et al. Effect of the ADCC-modulating mutations and the selection of human igG isotypes on physicochemical properties of fc. J Pharm Sci. (2022) 111:2411–21. doi: 10.1016/j.xphs.2022.06.014
35. Lee CH and Delidakis G. Engineering igG1 fc domains that activate the complement system. Methods Mol Biol. (2022) 2421:187–200. doi: 10.1007/978-1-0716-1944-5_13
36. Weitzman SA and Stossel TP. Drug-induced immunological neutropenia. Lancet. (1978) 1:1068–72. doi: 10.1016/s0140-6736(78)90915-7
37. de Taeye SW, Bentlage AEH, Mebius MM, Meesters JI, Lissenberg-Thunnissen S, Falck D, et al. FcγR binding and ADCC activity of human igG allotypes. Front Immunol. (2020) 11:740. doi: 10.3389/fimmu.2020.00740
38. Ayalew L, D’Souza AD, Chen Y, and Kim MT. Identification and quantitation of hinge cysteinylation on endogenous IgG2 antibodies from human serum. MAbs. (2020) 12:1854923. doi: 10.1080/19420862.2020.1854923
39. Teplyakov A, Zhao Y, Malia TJ, Obmolova G, and Gilliland GL. IgG2 Fc structure and the dynamic features of the IgG CH2-CH3 interface. Mol Immunol. (2013) 56:131–9. doi: 10.1016/j.molimm.2013.03.018
40. Liu-Shin L, Fung A, Malhotra A, and Ratnaswamy G. Influence of disulfide bond isoforms on drug conjugation sites in cysteine-linked IgG2 antibody-drug conjugates. MAbs. (2018) 10:583–95. doi: 10.1080/19420862.2018.1440165
41. Stapleton NM, Andersen JT, Stemerding AM, Bjarnarson SP, Verheul RC, Gerritsen J, et al. Competition for FcRn-mediated transport gives rise to short half-life of human IgG3 and offers therapeutic potential. Nat Commun. (2011) 2:599. doi: 10.1038/ncomms1608
42. Handlogten MW, Peng L, Christian EA, Xu W, Lin S, Venkat R, et al. Prevention of Fab-arm exchange and antibody reduction via stabilization of the IgG4 hinge region. MAbs. (2020) 12:1779974. doi: 10.1080/19420862.2020.1779974
43. Labrijn AF, Buijsse AO, van den Bremer ET, Verwilligen AY, Bleeker WK, Thorpe SJ, et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol. (2009) 27:767–71. doi: 10.1038/nbt.1553
44. Lai PK, Ghag G, Yu Y, Juan V, Fayadat-Dilman L, and Trout BL. Differences in human IgG1 and IgG4 S228P monoclonal antibodies viscosity and self-interactions: Experimental assessment and computational predictions of domain interactions. MAbs. (2021) 13:1991256. doi: 10.1080/19420862.2021.1991256
45. Gromisch CM, Tan GLA, Pasion KA, Moran AM, Gromisch MS, Grinstaff MW, et al. Humanized anti-DEspR IgG4(S228P) antibody increases overall survival in a pancreatic cancer stem cell-xenograft peritoneal carcinomatosis rat(nu/nu) model. BMC Cancer. (2021) 21:407. doi: 10.1186/s12885-021-08107-w
46. Saxena A and Wu D. Advances in therapeutic fc engineering - modulation of igG-associated effector functions and serum half-life. Front Immunol. (2016) 7:580. doi: 10.3389/fimmu.2016.00580
47. Pejchal R, Cooper AB, Brown ME, Vásquez M, and Krauland EM. Profiling the biophysical developability properties of common igG1 fc effector silencing. Variants.Antibodies (Basel). (2023) 12:54. doi: 10.3390/antib12030054
48. Wang L, Hoseini SS, Xu H, Ponomarev V, and Cheung NK. Silencing fc domains in T cell-engaging bispecific antibodies improves T-cell trafficking and antitumor potency. Cancer Immunol Res. (2019) 7:2013–24. doi: 10.1158/2326-6066.CIR-19-0121
49. Lo M, Kim HS, Tong RK, Bainbridge TW, Vernes JM, Zhang Y, et al. Effector-attenuating substitutions that maintain antibody stability and reduce toxicity in mice. J Biol Chem. (2017) 292:3900–8. doi: 10.1074/jbc.M116.767749
50. Okamoto H, Oitate M, Hagihara K, Shiozawa H, Furuta Y, Ogitani Y, et al. Pharmacokinetics of trastuzumab deruxtecan (T-DXd), a novel anti-HER2 antibody-drug conjugate, in HER2-positive tumour-bearing mice. Xenobiotica. (2020) 50:1242–50. doi: 10.1080/00498254.2020.1755909
51. Jebamani P, Sriramulu DK, and Lee SG. Residue interaction network and molecular dynamics simulation study on the binding of S239D/I332E Fc variant with enhanced affinity to FcgammaRIIIa receptor. J Mol Graph Model. (2023) 118:108327. doi: 10.1016/j.jmgm.2022.108327
52. Wang Z, Li H, Gou L, Li W, and Wang Y. Antibody-drug conjugates: Recent advances in payloads. Acta Pharm Sin B. (2023) 13:4025–59. doi: 10.1016/j.apsb.2023.06.015
53. Masters JC, Nickens DJ, Xuan D, Shazer RL, and Amantea M. Clinical toxicity of antibody drug conjugates: a meta-analysis of payloads. Invest New Drugs. (2018) 36:121–35. doi: 10.1007/s10637-017-0520-6
54. Fleming R. ADC analysis by hydrophobic interaction chromatography. Methods Mol Biol. (2020) 2078:147–61. doi: 10.1007/978-1-4939-9929-3_10
55. Abelman RO, Wu B, Spring LM, Ellisen LW, and Bardia A. Mechanisms of resistance to antibody-drug conjugates. Cancers (Basel). (2023) 15:1278. doi: 10.3390/cancers15041278
56. Bordeau BM, Nguyen TD, Polli JR, Chen P, and Balthasar JP. Payload-binding fab fragments increase the therapeutic index of MMAE antibody-drug conjugates. Mol Cancer Ther. (2023) 22:459–70. doi: 10.1158/1535-7163.MCT-22-0440
57. Scribner JA, Hicks SW, Sinkevicius KW, Yoder NC, Diedrich G, Brown JG, et al. Preclinical evaluation of IMGC936, a next-generation maytansinoid-based antibody-drug conjugate targeting ADAM9-expressing tumors. Mol Cancer Ther. (2022) 21:1047–59. doi: 10.1158/1535-7163.MCT-21-0915
58. Yip V, Saad OM, Leipold D, Li C, Kamath A, Shen BQ, et al. (MMAE), a payload for multiple antibody drug conjugates (ADCs), demonstrates differential red blood cell partitioning across human and animal species. Xenobiotica. (2024) 54:511–20. doi: 10.1080/00498254.2024.2345849
59. Nittoli T, Delfino F, Kelly M, Carosso S, Markotan T, Kunz A, et al. Antibody drug conjugates of cleavable amino-benzoyl-maytansinoids. Bioorg Med Chem. (2020) 28:115785. doi: 10.1016/j.bmc.2020.115785
60. Shi B, Wu M, Li Z, Xie Z, Wei X, Fan J, et al. Antitumor activity of a 5T4 targeting antibody drug conjugate with a novel payload derived from MMAF via C-Lock linker. Cancer Med. (2019) 8:1793–805. doi: 10.1002/cam4.2066
61. D’Agostino D, Gentile R, Ponziani S, Di Vittorio G, Dituri F, Giannelli G, et al. EV20-sss-vc/MMAF, an HER-3 targeting antibody-drug conjugate displays antitumor activity in liver cancer. Oncol Rep. (2021) 45:776–85. doi: 10.3892/or.2020.7893
62. Hurvitz SA, Hegg R, Chung WP, Im SA, Jacot W, Ganju V, et al. Trastuzumab deruxtecan versus trastuzumab emtansine in patients with HER2-positive metastatic breast cancer: updated results from DESTINY-Breast03, a randomised, open-label, phase 3 trial. Lancet. (2023) 401:105–17. doi: 10.1016/S0140-6736(22)02420-5
63. Ogitani Y, Aida T, Hagihara K, Yamaguchi J, Ishii C, Harada N, et al. DS-8201a, A novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin Cancer Res. (2016) 22:5097–108. doi: 10.1158/1078-0432.CCR-15-2822
64. Hartley JA. Antibody-drug conjugates (ADCs) delivering pyrrolobenzodiazepine (PBD) dimers for cancer therapy. Expert Opin Biol Ther. (2021) 21:931–43. doi: 10.1080/14712598.2020.1776255
65. Cardillo TM, Govindan SV, Sharkey RM, Trisal P, and Goldenberg DM. Humanized anti-Trop-2 IgG-SN-38 conjugate for effective treatment of diverse epithelial cancers: preclinical studies in human cancer xenograft models and monkeys. Clin Cancer Res. (2011) 17:3157–69. doi: 10.1158/1078-0432.CCR-10-2939
66. Vollmar BS, Frantz C, Schutten MM, Zhong F, Del Rosario G, Go MAT, et al. Calicheamicin antibody-drug conjugates with improved properties. Mol Cancer Ther. (2021) 20:1112–20. doi: 10.1158/1535-7163.MCT-20-0035
67. Ackerman E, Pearson CI, Gregorio JD, JC G, JA K, FJ H, et al. Immune-stimulating antibody conjugates elicit robust myeloid activation and durable antitumor immunity. Nat Cancer. (,2021) 2:18–33. doi: 10.1038/s43018-020-00136-x
68. Li M, Zhao X, Yu C, and Wang L. Antibody-drug conjugate overview: A state-of-theart manufacturing process and control strategy. Pharmaceut Res. (2024) 41:419–40. doi: 10.1007/s11095-023-03649-z
69. Mahmood I. Clinical pharmacology of antibody-drug conjugates. Antibod (Basel Switzerland). (2021) 10:20. doi: 10.3390/antib10020020
70. Samantasinghar A, Sunildutt NP, Ahmed F, Soomro AM, Salih ARC, Parihar P, et al. A comprehensive review of key factors affecting the efficacy of antibody drug conjugate. BioMed Pharmacother. (2023) 161:114408. doi: 10.1016/j.biopha.2023.114408
71. Boghaert ER, Cox MC, and Vaidya KS. Pathophysiologic and pharmacologic considerations to improve the design and application of antibody-drug conjugates. Cancer Res. (2022) 82:1858–69. doi: 10.1158/0008-5472.CAN-21-3236
72. Mahalingaiah PK, Ciurlionis R, Durbin KR, Yeager RL, Philip BK, Bawa B, et al. Potential mechanisms of target-independent uptake and toxicity of antibody-drug. conjugates. Pharmacol.Ther. (.2019) 200:110–25. doi: 10.1016/j.pharmthera.2019.04.008
73. Sun X, Ponte JF, Yoder NC, Laleau R, Coccia J, Lanieri L, et al. Effects of drug antibody ratio on pharmacokinetics, biodistribution, efficacy, and tolerability of antibody-maytansinoid conjugates. Bioconjugate Chem. (2017) 28:1371–81. doi: 10.1021/acs.bioconjchem.7b00062
74. Weng W, Meng T, Zhao Q, Shen Y, Fu G, Shi J, et al. Antibody-exatecan conjugates with a novel self-immolative moiety overcome resistance in colon and lung cancer. Cancer Discov. (2023) 13:950–73. doi: 10.1158/2159-8290.CD-22-1368
75. Khoury R, Saleh K, Khalife N, Saleh M, Chahine C, Ibrahim R, et al. Mechanisms of resistance to antibody-drug conjugates. Int J Mol Sci. (2023) 24:9674. doi: 10.3390/ijms24119674
76. Wang R, Fang P, Chen X, Ji J, Yu D, Mei F, et al. Overcoming multidrug resistance in gastrointestinal cancers with a CDH17-targeted ADC conjugated to a DNA topoisomerase inhibitor. Cell Rep Med. (2025) 6:102213. doi: 10.1016/j.xcrm.2025.102213
77. suchikama K and An Z. Antibody-drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell. (2018) 9:33–46. doi: 10.1007/s13238-016-0323-0
78. Flygare JA, Pillow TH, and Aristoff P. Antibody-drug conjugates for the treatment of cancer. Chem Biol Drug Des. (2013) 81:113–21. doi: 10.1111/cbdd.12085
79. Zhang D, Fourie-O’Donohue A, Dragovich PS, Pillow TH, Sadowsky JD, Kozak KR, et al. Catalytic cleavage of disulfide bonds in small molecules and linkers of antibody–drug conjugates. Drug Metab Dispos. (2019) 47:1156–63. doi: 10.1124/dmd.118.086132
80. Pallardó FV, Markovic J, García JL, and Viña J. Role of nuclear glutathione as a key regulator of cell proliferation. Mol Asp. Med. (2009) 30:77–85. doi: 10.1016/j.mam.2009.01.001
81. Doronina SO, Bovee TD, Meyer DW, Miyamoto JB, Anderson ME, Morris-Tilden CA, et al. Novel peptide linkers for highly potent antibody– auristatin conjugate. Bioconjug. Chem. (2008) 19:1960–3. doi: 10.1021/bc800289a
82. Dubowchik GM, RA F, Padilla L, Willner D, SJ H, Mosure K, et al. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: model studies of enzymatic drug release and antigen-specific. Vitro Anticancer activity. Bioconjug. Chem. (2002) 13:855–69. doi: 10.1021/bc025536j
83. Jeffrey SC, Andreyka JB, Bernhardt SX, Kissler KM, Kline T, Lenox JS, et al. Development and properties of beta-glucuronide linkers for monoclonal antibody-drug conjugates. Bioconjug Chem. (2006) 17:831–40. doi: 10.1021/bc0600214
84. Walles M, Connor A, and Hainzl D. ADME and safety aspects of non-cleavable linkers in drug discovery and development. Curr Top Med Chem. (2017) 17:3463–75. doi: 10.2174/1568026618666180118153502
85. Sheyi R, de la Torre BG, and Albericio F. Linkers: an assurance for controlled delivery of antibody-drug conjugate. Pharmaceutics. (2022) 14:396. doi: 10.3390/pharmaceutics14020396
86. Sang H, Wan N, Lu G, Tian Y, Wang G, and Ye H. Conjugation site analysis of lysine-conjugated ADCs. Methods Mol Biol. (2020) 2078:235–50. doi: 10.1007/978-1-4939-9929-3_16
87. Tang SC, Wynn C, Le T, McCandless M, Zhang Y, Patel R, et al. Influence of antibody-drug conjugate cleavability, drug-to-antibody ratio, and free payload concentration on systemic toxicities: A systematic review and meta-analysis. Cancer Metastasis Rev. (2024) 44:18. doi: 10.1007/s10555-024-10231-5
88. Zeini D, Glover JC, Knudsen KD, and Nyström B. Influence of lysine and TRITC conjugation on the size and structure of dextran nanoconjugates with potential for biomolecule delivery to neurons. ACS Appl Bio Mater. (2021) 4:6832–42. doi: 10.1021/acsabm.1c00544
89. Zhang C, Dai P, Vinogradov AA, Gates ZP, and Pentelute BL. Site-selective cysteine-cyclooctyne conjugation. Angew Chem Int Ed Engl. (2018) 57:6459–63. doi: 10.1002/anie.201800860
90. Cal PM, Bernardes GJ, and Gois PM. Cysteine-selective reactions for antibody conjugation. Angew Chem Int Ed Engl. (2014) 53:10585–7. doi: 10.1002/anie.201405702
91. Zhang C, Welborn M, Zhu T, Yang NJ, Santos MS, Van Voorhis T, et al. pi-Clamp-mediated cysteine conjugation. Nat Chem. (2016) 8:120–8. doi: 10.1038/nchem.2413
92. Adhikari P, Zacharias N, Ohri R, and Sadowsky J. Site-specific conjugation to cys-engineered THIOMAB antibodies. Methods Mol Biol. (2020) 2078:51–69. doi: 10.1007/978-1-4939-9929-3_4
93. Dickgiesser S, Deweid L, Kellner R, Kolmar H, and Rasche N. Site-specific antibody-drug conjugation using microbial transglutaminase. Methods Mol Biol. (2019) 2012:135–49. doi: 10.1007/978-1-4939-9546-2_8
94. Zhao Y, Kim S, Zheng X, Kim SH, Han A, Chen TH, et al. Investigation of high molecular weight size variant formation in antibody-drug conjugates: microbial transglutaminase-mediated crosslinking. J Pharm Sci. (2023) 112:2629–36. doi: 10.1016/j.xphs.2023.08.006
95. Hadjabdelhafid-Parisien A, Bitsch S, Macarrón Palacios A, Deweid L, Kolmar H, and Pelletier JN. Tag-free, specific conjugation of glycosylated IgG1 antibodies using microbial transglutaminase. RSC Adv. (2022) 12:33510–5. doi: 10.1039/d2ra05630e
96. Pagar AD, Jeon H, Khobragade TP, Sarak S, Giri P, Lim S, et al. Non-canonical amino acid-based engineering of (R)-amine transaminase. Front Chem. (2022) 10:839636. doi: 10.3389/fchem.2022.839636
97. Oller-Salvia B. Genetic encoding of a non-canonical amino acid for the generation of antibody-drug conjugates through a fast bioorthogonal reaction. J Vis Exp. (2018) 139):58066. doi: 10.3791/58066
98. Zhang X, Ou C, Liu H, Prabhu SK, Li C, Yang Q, et al. General and robust chemoenzymatic method for glycan-mediated site-specific labeling and conjugation of antibodies: facile synthesis of homogeneous antibody-drug conjugates. ACS Chem Biol. (2021) 16:2502–14. doi: 10.1021/acschembio.1c00597
99. Ou C, Li C, Zhang R, Yang Q, Zong G, Dai Y, et al. One-pot conversion of free sialoglycans to functionalized glycan oxazolines and efficient synthesis of homogeneous antibody-drug conjugates through site-specific chemoenzymatic glycan remodeling. Bioconjug Chem. (2021) 32:1888–97. doi: 10.1021/acs.bioconjchem.1c00314
100. Hudak JE, Yu HH, and Bertozzi CR. Protein glycoengineering enabled by the versatile synthesis of aminooxy glycans and the genetically encoded aldehyde tag. J Am Chem Soc. (. 2011) 133:16127–35. doi: 10.1021/ja206023e
101. Smith EL, Giddens JP, Iavarone AT, Godula K, Wang LX, and Bertozzi CR. Chemoenzymatic Fc glycosylation via engineered aldehyde tags. Bioconjug Chem. (2014) 25:788–95. doi: 10.1021/bc500061s
102. Xia F, Liu Z, Hang J, Xu H, Xiao Y, Niu S, et al. Harnessing acylhydrazone-oxime exchange reaction to achieve diverse synthesis of glycosite-specific antibody-drug conjugates. Org Biomol Chem. (2025) 23:1448–56. doi: 10.1039/d4ob01826e
103. Petersdorf SH, Kopecky KJ, Slovak M, Willman C, Nevill T, Brandwein J, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. blood. (2013) 121:4854–60. doi: 10.1182/blood-2013-01-466706
104. Norsworthy KJ, Ko CW, Lee JE, Liu J, John CS, Przepiorka D, et al. FDA approval summary: mylotarg for treatment of patients with relapsed or refractory CD33-positive acute myeloid leukemia. Oncologist. (2018) 23:1103–8. doi: 10.1634/theoncologist.2017-0604
105. Herrera AF, Zain J, Savage KJ, Feldman T, Brammer JE, Chen L, et al. Iyer SP.Brentuximab vedotin plus cyclophosphamide, doxorubicin, etoposide, and prednisone followed by brentuximab vedotin consolidation in CD30-positive peripheral T-cell lymphomas: a multicentre, single-arm, phase 2 study. Lancet Haematol. (2024) 11:e671–81. doi: 10.1016/S2352-3026(24)00171-6
106. Horwitz S, O’Connor OA, Pro B, Trümper L, Iyer S, Advani R, et al. The ECHELON-2 Trial: 5-year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann Oncol. (2022) 33:288–98. doi: 10.1016/j.annonc.2021.12.002
107. Horwitz SM, Scarisbrick JJ, Dummer R, Whittaker S, Duvic M, Kim YH, et al. Randomized phase 3 ALCANZA study of brentuximab vedotin vs physician’s choice in cutaneous T-cell lymphoma: final data. Blood Adv. (2021) 5:5098–106. doi: 10.1182/bloodadvances.2021004710
108. Fuh FK, Looney C, Li D, Poon KA, Dere RC, Danilenko DM, et al. Anti-CD22 and anti-CD79b antibody-drug conjugates preferentially target proliferating B cells. Br J Pharmacol. (2017) 174:628–40. doi: 10.1111/bph.13697
109. Lugtenburg PJ, de Nully Brown P, van der Holt B, D’Amore FA, Koene HR, de Jongh E, et al. Rituximab-CHOP with early rituximab intensification for diffuse large B-cell lymphoma: A randomized phase III trial of the HOVON and the nordic lymphoma group (HOVON-84). J Clin Oncol. (2020) 38:3377–87. doi: 10.1200/JCO.19.03418
110. Wekking D, Porcu M, Pellegrino B, Lai E, Mura G, Denaro N, et al. Multidisciplinary clinical guidelines in proactive monitoring, early diagnosis, and effective management of trastuzumab deruxtecan (T-DXd)-induced interstitial lung disease (ILD) in breast cancer patients. ESMO Open. (2023) 8:102043. doi: 10.1016/j.esmoop.2023.102043
111. Alexander S, Aleem U, Jacobs T, Frizziero M, Foy V, Hubner RA, et al. Antibody-drug conjugates and their potential in the treatment of patients with biliary tract cancer. Cancers(Basel). (2024) 16:3345. doi: 10.3390/cancers16193345
112. Akkapeddi P, Azizi SA, Freedy AM, Cal P, Gois PMP, and Bernardes GJL. Construction of homogeneous antibody-drug conjugates using site-selective protein chemistry. Chem Sci. (2016) 7:2954–63. doi: 10.1039/c6sc00170j
113. Jiang Z. TQB2102 in combination with pertuzumab and docetaxel for neoadjuvant treatment of patients with HER2-positive early or locally advanced breast cancer: A multi-center, single-arm, phase II trial. *Presented at. ESMO Asia Congress. (2023), 2023295.
114. Gu Y, Wang Z, and Wang Y. Bispecific antibody drug conjugates: Making 1 + 1>2. Acta Pharm Sin B. (2024) 14:1965–86. doi: 10.1016/j.apsb.2024.01.009
115. Thieblemont C, Phillips T, Ghesquieres H, Cheah CY, Clausen MR, Cunningham D, et al. Epcoritamab, a novel, subcutaneous CD3xCD20 bispecific T-cell-engaging antibody, in relapsed or refractory large B-cell lymphoma: dose expansion in a phase I/II trial. J Clin Oncol. (2023) 41:2238–47. doi: 10.1200/JCO.22.01725
116. Panowski SH, Kuo TC, Zhang Y, Chen A, Geng T, Aschenbrenner L, et al. Preclinical efficacy and safety comparison of CD3 bispecific and ADC modalities targeting BCMA for the treatment of multiple myeloma. Mol Cancer Ther. (2019) 18:2008–20. doi: 10.1158/1535-7163.MCT-19-0007
117. Rinnerthaler G, Gampenrieder SP, and Greil R. Her2 directed antibody-drugconjugates beyond T-dm1 in breast cancer. Int J Mol Sci. (2019) 20:1115. doi: 10.3390/ijms20051115
118. Pegram MD, Hamilton EP, Tan AR, Storniolo AM, Balic K, Rosenbaum AI, et al. First-in-human, phase 1 dose-escalation study of biparatopic anti-her2 antibodydrug conjugate medi4276 in patients with her2-positive advanced breast or gastric cancer. Mol Cancer Ther. (2021) 20:1442–53. doi: 10.1158/1535-7163.MCT-20-0014
119. Faria M, Peay M, Lam B, Ma E, Yuan M, WRM WMC.OMMAJ.R.X.X.X, et al. Multiplex lc-ms/ms assays for clinical bioanalysis of medi4276, an antibody-drug conjugate of tubulysin analogue attached via cleavable linker to a biparatopic humanized antibody against her-2. Antibodies (Basel). (2019) 8:11. doi: 10.3390/antib8010011
120. Xue J, Kong D, Yao Y, Yang L, Yao Q, Zhu Y, et al. Prediction of human pharmacokinetics and clinical effective dose of SI-B001, an EGFR/HER3 bi-specific monoclonal antibody. J Pharm Sci. (2020) 109:3172–80. doi: 10.1016/j.xphs.2020.06.015
121. Xue J, Ma Y, Zhao Y, Wang Y, Hong W, Huang Y, et al. Izalontamab (SI-B001), a novel EGFRxHER3 bispecific antibody in patients with Locally Advanced or Metastatic Epithelial Tumor: Results from first-in-human phase I/Ib study. Clin Cancer Res. (2025) 21. doi: 10.1158/1078-0432.CCR-25-0206
122. Mckertish CM and Kayser V. A novel dual-payload ADC for the treatment of HER2+ Breast and colon cancer. Pharmaceutics. (2023) 15:2020. doi: 10.3390/pharmaceutics15082020
123. Watanabe T, Iwai Y, Stofleth JT, Fujii T, Seki T, and Matsuda Y. Homogeneous dual-payload antibody-drug conjugates produced by combined distinct conjugation strategies. ACS Med Chem Lett. (2025) 16:1334–9. doi: 10.1021/acsmedchemlett.5c00209
124. Li F, Emmerton KK, Jonas M, Zhang X, Miyamoto JB, Setter JR, et al. Intracellular released payload influences potency and bystander-killing effects of antibody-drug conjugates in preclinical models. Cancer Res. (2016) 76:2710–9. doi: 10.1158/0008-5472.CAN-15-1795
125. Levengood MR, Zhang X, Hunter JH, Emmerton KK, Miyamoto JB, Lewis TS, et al. Orthogonal cysteine protection enables homogeneous multi-drug antibody-drugconjugates. Angew Chem Int Ed Engl. (2017) 56:733–7. doi: 10.1002/anie.201608292
126. Yamazaki CM, Yamaguchi A, Anami Y, Xiong W, Otani Y, Lee J, et al. Antibodydrug conjugates with dual payloads for combating breast tumor heterogeneity and drug resistance. Nat Commun. (2021) 12:3528. doi: 10.1038/s41467-021-23793-7
127. Kumar A, Kinneer K, Masterson L, Ezeadi E, Howard P, Wu H, et al. Synthesis of a heterotrifunctional linker for the site-specific preparation of antibody-drug conjugates with two distinct warheads. Bioorg Med Chem Lett. (2018) 28:3617–21. doi: 10.1016/j.bmcl.2018.10.043
128. Nilchan N, Li X, Pedzisa L, Nanna AR, Roush WR, and Rader C. Dual-mechanistic antibody-drug conjugate via site-specific selenocysteine/cysteine conjugation. Antib Ther. (2019) 2:71–8. doi: 10.1093/abt/tbz009
129. Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, et al. Tlr7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat BioMed Eng. (2018) 2:578–88. doi: 10.1038/s41551-018-0236-8
130. Fuentes-Antras J, Genta S, Vijenthira A, and Siu LL. Antibody-drug conjugates: in search of partners of choice. Trends Cancer. (2023) 9:339–54. doi: 10.1016/j.trecan.2023.01.003
131. Zhang BD, Wu JJ, Li WH, Hu HG, Zhao L, He PY, et al. Sting and tlr7/8 agonists-based nanovaccines for synergistic antitumor immune activation. Nano Res. (2022) 15:6328–39. doi: 10.1007/s12274-022-4282-x
132. Hamilton E, Specht JM, Puhalla SA, Ward AM, Stout AJ, et al. Phase 2 study of BDC-1001 in patients with advanced HER2-expressing solid tumors: Efficacy and safety results. In: Presented at: european society for medical oncology (ESMO) congress 2023; october 20-24, 2023. (2023) p. 508.
133. Hamilton E, LoRusso P, Wang ES, Bauman JR, Bhatia SS, Specht JM, et al. Preliminary results from a phase 1 study of SBT6050, a first-in-class HER2-targeted TLR8 agonist antibody-conjugate, in patients with advanced HER2-expressing solid tumors. In: Presented at: american association for cancer research (AACR) annual meeting 2022; april 8-13, 2022. (2022) p. CT014.
134. Huang L, Ge X, Liu Y, Li H, and Zhang Z. The role of toll-like receptor agonists and their nanomedicines for tumor immunotherapy. Pharmaceutics. (2022) 14:1228. doi: 10.3390/pharmaceutics14061228
135. Wu S-G and Shih J-Y. Management of acquired resistance to EGFR TKI–targeted therapy in advanced non-small cell lung cancer. Mol Cancer. (2018) 17:38. doi: 10.1186/s12943-018-0777-1
136. Irie H, Kawabata R, Fujioka Y, Nakagawa F, Itadani H, Nagase H, et al. Acquired resistance to trastuzumab/pertuzumab or to T-DM1 in vivo can be overcome by HER2 kinase inhibition with TAS0728. Cancer Sci. (2020) 111:2123–31. doi: 10.1111/cas.14407
137. Sipos G and Kuchler K. Fungal ATP-binding cassette (ABC) transporters in drug resistance & detoxification. Curr Drug Targets. (2006) 7:471–81. doi: 10.2174/138945006776359403
138. Buongervino S, Lane MV, Garrigan E, Zhelev DV, Dimitrov DS, and Bosse KR. Antibody-drug conjugate efficacy in neuroblastomarole of payload, resistance mechanisms, target density, and antibody internalization. Mol Cancer Ther. (2021) 20:2228–39. doi: 10.1158/1535-7163.MCT-20-1034
139. Guo J, Kumar S, Chipley M, Marcq O, Gupta D, Jin Z, et al. Characterization and higher-order structure assessment of an interchain cysteine-based ADC: impact of drug loading and distribution on the mechanism of aggregation. Bioconjug Chem. (2016) 27:604–15. doi: 10.1021/acs.bioconjchem.5b00603
140. Malik P, Phipps C, Edginton A, and Blay J. Pharmacokinetic considerations for antibody-drug conjugates against cancer. Pharm Res. (2017) 34:2579–95. doi: 10.1007/s11095-017-2259-3
141. Khera E and Thurber GM. Pharmacokinetic and immunological considerations for expanding the therapeutic window of next-generation antibody–drug conjugates. BioDrugs. (2018) 32:465–80. doi: 10.1007/s40259-018-0302-5
142. Ruan J, Moskowitz A, Mehta-Shah N, Sokol L, Chen Z, Kotlov N, et al. Martin P.Multicenter phase 2 study of oral azacitidine (CC-486) plus CHOP as initial treatment for PTCL. Blood. (2023) 141:2194–205. doi: 10.1182/blood.2022018254
143. Zhao H, Atkinson J, Gulesserian S, Zeng Z, Nater J, Ou J, et al. Doñate F.Modulation of macropinocytosis-mediated internalization decreases ocular toxicity of antibody-drug conjugates. Cancer Res. (2018) 78:2115–26. doi: 10.1158/0008-5472.CAN-17-3202
144. Yang P, Zhang H, Li J, Li Z, Liu Z, Wang M, et al. Incidence of antibody-drug conjugate-related hepatotoxicity in breast cancer: a systematic review and meta-analysis. Ther Adv Drug Saf. (2024) 15:20420986241304680. doi: 10.1177/20420986241304680
145. Desai A, Subbiah V, Roy-Chowdhuri S, Sheshadri A, Deshmukh S, and Peters S. Association of antibody-drug conjugate (ADC) target expression and interstitial lung disease (ILD) in non-small-cell lung cancer (NSCLC): association or causation or neither? Cancers (Basel). (2024) 16:3753. doi: 10.3390/cancers16223753
146. Powell CA, Modi S, Iwata H, Takahashi S, Smit EF, Siena S, et al. Camidge DR.Pooled analysis of drug-related interstitial lung disease and/or pneumonitis in nine trastuzumab deruxtecan monotherapy studies. ESMO Open. (2022) 7:100554. doi: 10.1016/j.esmoop.2022.100554
147. Lacouture ME, Patel AB, Rosenberg JE, and O’Donnell PH. Management of dermatologic events associated with the nectin-4-directed antibody-drug conjugate enfortumab vedotin. Oncologist. (2022) 27:e223–32. doi: 10.1093/oncolo/oyac001
148. Yu M, Zhou L, Cao M, Ji C, and Zheng Y. Post-marketing drug safety surveillance of enfortumab vedotin: an observational pharmacovigilance study based on a real-world database. Front Immunol. (2024) 15:1397692. doi: 10.3389/fimmu.2024.1397692
149. Hoimes CJ, Flaig TW, Milowsky MI, Friedlander TW, Bilen MA, Gupta S, et al. Enfortumab vedotin plus pembrolizumab in previously untreated advanced urothelial cancer. J Clin Oncol. (2023) 41:22–31. doi: 10.1200/JCO.22.01643
150. Taoka R, Kamada M, Izumi K, Tanimoto R, Daizumoto K, Hayashida Y, et al. Sugimoto M.Peripheral neuropathy and nerve electrophysiological changes with enfortumab vedotin in patients with advanced urothelial carcinoma: a prospective multicenter cohort study. Int J Clin Oncol. (2024) 29:602–11. doi: 10.1007/s10147-024-02481-8
151. Weekes CD, LE L, MJ B, Voortman J, RR M, JR D, et al. Phase I study of DMOT4039A, an antibody-drug conjugate targeting mesothelin, in patients with unresectab le pancreatic or platinum-resistant ovarian cancer. Mol Cancer Ther. (2016) 15:439–47. doi: 10.1158/1535-7163.MCT-15-0693
152. Colombo R, Tarantino P, Rich JR, LoRusso PM, and de Vries EGE. The journey of antibody-drug conjugates: lessons learned from 40 years of development. Cancer Discov. (2024) 14:2089–108. doi: 10.1158/2159-8290
153. Wei Q, Li P, Yang T, Zhu J, Sun L, Zhang Z, et al. The promise and challenges of combination therapies with antibody-drug conjugates in solid tumors. J Hematol Oncol. (2024) 17:1. doi: 10.1186/s13045-023-01509-2
154. Wabitsch S, McCallen JD, Kamenyeva O, Ruf B, McVey JC, Kabat J, et al. Metformin treatment rescues CD8(+) T-cell response to immune checkpoint inhibitor therapy in mice with NAFLD. J Hepatol. (2022) 77:748–60. doi: 10.1016/j.jhep.2022.03.010
155. Powles TB, van der Heijden MS, Loriot Y, Bedke J, Valderrama BP, Iyer G, et al. Enfortumab vedotin plus pembrolizumab in untreated locally advanced or metastatic urothelial carcinoma: 2.5-year median follow-up of the phase III EV-302/KEYNOTE-A39 trial. Ann Oncol. (2025) 36:1212–9. doi: 10.1016/j.annonc.2025.05.536
156. Powles T, Valderrama BP, Gupta S, Bedke J, Kikuchi E, Hoffman-Censits J, et al. EV-302 trial investigators. Enfortumab vedotin and pembrolizumab in untreated advanced urothelial cancer. N Engl J Med. (2024) 390:875–88. doi: 10.1056/NEJMoa2312117
157. Bardia A, Hurvitz SA, Tolaney SM, Loirat D, Punie K, Oliveira M, et al. ASCENT clinical trial investigators.Sacituzumab govitecan in metastatic triple-negative breast cancer. N Engl J Med. (2021) 384:1529–41. doi: 10.1056/NEJMoa2028485
158. A study of sacituzumab govitecan and pembrolizumab versus pembrolizumab alone in participants with previously untreated, locally advanced, inoperable, or metastatic triple-negative breast cancer (ASCENT-05). ClinicalTrials.gov (2024). Bethesda (MD): National Library of Medicine (US). p. NCT05633654. Available online at: https://clinicaltrials.gov/study/NCT05633654. (Accessed April 20, 2025)
Keywords: antibody drug conjugate, monoclonal antibody, linker, cytotoxic payload, resistance, toxicity, immune checkpoint inhibitor
Citation: Tang Z, Xie Y and Zeng Y (2025) Antibody–drug conjugate: a newly developed biological missile for tumor treatment. Front. Oncol. 15:1688057. doi: 10.3389/fonc.2025.1688057
Received: 18 August 2025; Accepted: 30 September 2025;
Published: 29 October 2025.
Edited by:
Zheng Zhong, University of Michigan, United StatesReviewed by:
Zhennan Wu, Guangzhou University of Chinese Medicine, ChinaDevesh Aggarwal, AstraZeneca, United States
Copyright © 2025 Tang, Xie and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhuoran Tang, MjAyMDIzMjE4OEBzdHUuY3B1LmVkdS5jbg==