 Jingjing Fu1‡
Jingjing Fu1‡ Yangming Tang2‡
Yangming Tang2‡ Xueqin Ruan2Runfa Wang1Tong Chen1Li Jiang1Yanli He3Zhihong Xu1Balian Wang2Haiqin Zhang1Jing Zhou1Mei Lan2*†Hongrui Li1*†
Xueqin Ruan2Runfa Wang1Tong Chen1Li Jiang1Yanli He3Zhihong Xu1Balian Wang2Haiqin Zhang1Jing Zhou1Mei Lan2*†Hongrui Li1*†- 1The Department of Cytogenetics, Kindstar Globalgene Technology, Inc, Wuhan, China
- 2The Department of Hematology, Guangxi Academy of Medical Sciences & The People’s Hospital of Guangxi Zhuang Autonomous Region, Nanning, China
- 3Center for Stem Cell Research and Application, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
In chronic myeloid leukemia (CML), less than 2% of cases express atypical or rare BCR::ABL1 transcripts. The e8a2 BCR::ABL1 fusion transcript, a rare variant, has been reported in only 20 cases to date, primarily in case reports or case series. The direct junction between BCR exon 8 and ABL1 exon 2 generates a premature stop codon at position 7 after the fusion, while the insertion of certain sequences can result in the formation of an in-frame e8a2 transcript. To date, the insertion of SPECC1L gene sequences into e8a2 BCR::ABL1 fusion transcripts has been reported in two CML cases, and the V379I mutation (in ABL1) has been identified in two additional CML cases. We describe a case of accelerated-phase CML involving three key molecular abnormalities: the insertion of a 154 bp SPECC1L exon 4 sequence into the e8a2 BCR::ABL1 fusion transcript, a concomitant ABL1 V379I mutation, and deletions near the t(9;22) breakpoint on derivative chromosome 9 (der(9)). The patient’s clinical manifestations, cytogenetic features, and molecular genetic characteristics were summarized and discussed. Despite sequential therapy with full-dose dasatinib for 10 months and the third-generation tyrosine-kinase inhibitor (TKI) olverembatinib for 7 months, the patient experienced progressive disease. She ultimately achieved Major Molecular Response (MMR) after haploidentical hematopoietic stem-cell transplantation (haplo-HSCT). This case highlights the importance of comprehensive molecular profiling at diagnosis and the need to develop alternative therapeutic strategies for rare BCR::ABL1 variants.
Introduction
CML is a hematopoietic stem cell malignancy characterized by a reciprocal translocation between chromosomes 9 and 22 [t(9;22)(q34;q11.2)], which results in the formation of the Philadelphia (Ph) chromosome and the BCR::ABL1 fusion gene (1). This fusion gene encodes a protein with constitutively activated tyrosine kinase activity (2). Most CML cases harbor a BCR::ABL1 fusion gene encoding the 210-kDa BCR::ABL1 protein, which arises from e13a2 (b2a2) and/or e14a2 (b3a2) junctions depending on the breakpoint location within the BCR gene (3). However, in less than 2% of cases (4), the breakpoint occurs outside the major BCR (M-BCR) region or is fused with an exon other than ABL1 exon 2, leading to the generation of atypical transcripts such as e1a2, e19a2, e13a3, e14a3, e1a3, e6a2, e8a2, and others (2, 3). The translation of each different transcript results in distinct protein tyrosine kinases, which may potentially affect the biological characteristics of the disease and the response to treatment (2).
Although e8a2 BCR::ABL1 transcripts have been reported previously (5, 6), we describe a rare case of CML in which the e8a2 transcript contains an in-frame 154 bp insertion derived from SPECC1L exon 4 between BCR exon 8 and ABL1 exon 2. Additionally, an acquired ABL1 V379I mutation in exon 7 and deletions near the t(9;22) breakpoint on der(9) were identified. The patient became resistant to imatinib, and subsequent therapy with dasatinib and olverembatinib failed to achieve the expected outcomes. She therefore underwent haplo-HSCT in July 2025 and subsequently achieved major molecular response (MMR). To our knowledge, this represents the first reported case of CML harboring both a SPECC1L exon 4 insertion within the e8a2 transcript and a concurrent ABL1 V379I mutation. This unique genetic profile may confer a synergistic mechanism of TKI resistance and aggressive disease behavior.
Case presentation
A 22-year-old woman underwent treatment at an external hospital in October 2016. Lab tests showed a white blood cell count of 226.72×109/L, platelet count of 107×109/L, and hemoglobin level of 5.8 g/dL. Bone marrow morphological examination was consistent with CML. Cytogenetic analysis confirmed the presence of the Philadelphia chromosome, with a karyotype of 46,XX,t(9;22)(q34;q11.2). She received imatinib 400 mg daily for 3 years but discontinued it due to persistent nausea and vomiting. On 2 March 2024, she was admitted to our hospital with a ten-day history of fever, cough, and fatigue. C-reactive protein (73.90 mg/L; normal rang: 0–10 mg/L) and procalcitonin (11.39 ng/mL; normal rang: 0-0.05 ng/mL) were elevated. Abdominal ultrasound revealed hepatosplenomegaly: the left hepatic lobe measured 92 × 74mm, the right hepatic subcostal oblique diameter was 155mm, and the spleen was 208mm in length with an 84-mm hilar depth. Chest CT revealed bilateral pulmonary infiltrates indicative of pneumonia. The T-SPOT.TB assay was positive. Bronchoscopy revealed chronic inflammation in bilateral bronchi. Microbiological NGS analysis of bronchoalveolar lavage fluid identified Enterococcus faecalis (189 reads), Epstein-Barr virus (19,344 reads), and Mycobacterium tuberculosis (760 reads). The patient reported a prior tuberculosis (TB) exposure history, which further supports the diagnosis of secondary pulmonary tuberculosis.
The complete blood count revealed white blood cell count of 119.28×109/L, platelet count of 82×109/L, hemoglobin level of 3.5g/dL, granulocyte count of 85.11×109/L, and monocyte count of 22.47×109/L. Peripheral blood smear: blasts 14.5%, promyelocytes 1.5%, metamyelocytes 20.5%, myelocytes 13.5%, neutrophilic band forms 23.5%, segmented neutrophils 17.5%, eosinophils 2%, basophils 4%, lymphocytes 3%, intermediate normoblasts 4%, and late normoblasts 16%. Morphological analysis of bone marrow revealed markedly increased proliferation of nucleated cells, with granulocytes accounting for 87.5%, including 12.0% blast cells. Flow cytometry immunophenotyping revealed that immature cells constituted 8.68%, co-expressing CD117, CD34, CD13, HLA-DR, and MPO, with partial expression of CD33, CD38, and CD9. These results are consistent with a diagnosis of accelerated-phase CML.
The typical e13a2/e14a2 transcripts, as well as the atypical e19a2 and e1a2 transcripts, were not detected by RT-PCR. However, karyotype analysis showed 46,XX,t(9;22)(q34;q11.2) in all analyzed metaphase cells (Figure 1A). Interphase FISH with the BCR/ABL DF probe revealed a variant 1R1G1F pattern (Figure 1B). Therefore, metaphase FISH was performed. The results showed 46,XX,t(9;22)(q34;q11.2)[20].ish t(9;22)(ABL1-,BCR-;BCR+,ABL1+)[20], indicating that the fusion signal was located on chromosome 22, with no corresponding signal detected on der(9) (Figures 1C, D). Additionally, an abnormal 1R pattern was detected using the ASS probe (Figure 1E).
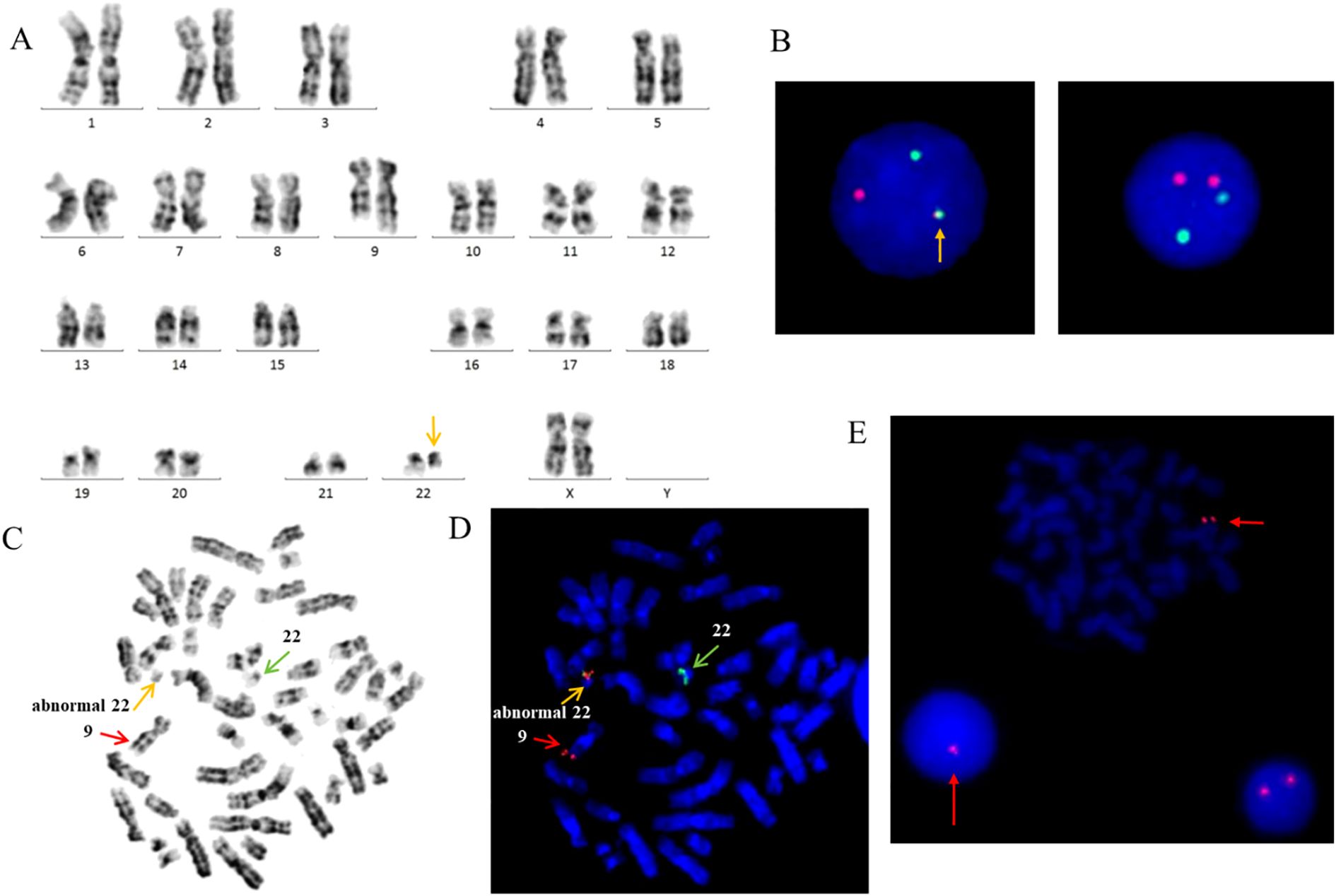
Figure 1. (A) Karyotype showing the typical translocation between chromosome 9 and 22. (B) FISH analysis using ABL(red) and BCR(green) dual-fusion probes. (C) Giemsa-banded metaphase preparation. (D) FISH analysis using BCR/ABL dual-fusion probe, demonstrating BCR::ABL1 fusion signals on chromosomes 22. (E) FISH analysis using a LSP probe for ASS at 9q34 reveals a single red signal, indicative of a deletion at the ASS locus.
Whole-transcriptome sequencing revealed the presence of fusion genes SPECC1L::ABL1 (SPECC1L exon 4 fused to ABL1 exon 2), BCR::SPECC1L (BCR exon 8 fused to SPECC1L exon 4), and LILRB1::LILRA2, along with a missense mutation (c.1135G > A, p.V379I) in the ABL1 gene. The rare e8a2 BCR::ABL1 variant was confirmed through the use of e8a2 RT-PCR primers (Supplementary Figure 1A). Sequence analysis of the amplified RT-PCR products demonstrated that a 154-bp sequence corresponding to exon 4 of the SPECC1L gene had been inserted at the e8a2 fusion junction (Supplementary Figure 1B).
Following six months of treatment, an Optical Genome Mapping (OGM) test was performed, the result was: ogm[GRCh38] 9q34.11q34.13(129791854_132041726)×1[0.078], t(9;22)(q34.11;q11.23)(129792636;24315572)[VAF0.18],22q11.23(23577634_24380537)×1[0.146],t(22;9)(q11.23;q34.12)(23261125;130777258)(BCR::ABL1)[VAF0.15]. The findings indicate an unbalanced translocation between chromosomes 9 and 22 involving the BCR::ABL1 fusion gene. The variant allele frequency of this BCR::ABL1 fusion allele was approximately 15%. Deletions of 2.250 Mb (including ASS1) and 0.803 Mb (including CABIN1) were detected at the breakpoint regions on chromosomes 22 and 9, respectively (Supplementary Figure 2). It is important to note that the accurate determination of these translocation breakpoints is challenging due to the inherent resolution limitations of OGM and the potential difficulty in precisely identifying unlabeled DLS regions.
After one year of treatment, whole exome sequencing analysis was performed to determine whether new clonal evolution had occurred in the patient. No additional mutations were identified in ABL1; however, a germline missense mutation, c.185A>G (p.His62Arg), was detected in G6PD. CNV-seq revealed the following copy number variations: seq[GRCh37] 9q34.11q34.12(132562879_133643956)×1,22q11.23(23597083_24677393)×1. These findings indicate a segmental deletion on chromosome 9 with the breakpoint located within the ABL1 gene, as well as a segmental deletion on chromosome 22 involving breakpoints within the BCR gene (22q11.23) and the SPECC1L gene (22q11.23), respectively.
According to the results of various molecular biological analyses, the patient was found to have two breaks in the long arms of chromosomes 9 and 22, with sequence deletions, leading to the formation of an unbalanced translocation [t(9;22)]. Additionally, an e8a2 BCR::ABL1 fusion transcript was detected; notably, the insertion of a 154 bp sequence from exon 4 of the SPECC1L gene at the fusion junction resulted in a novel in-frame variant of this transcript, designated as e8-[ins]-a2. Moreover, a p.V379I missense mutation was identified in exon 7 of the ABL1 gene.
The patient was diagnosed with CML in the accelerated phase (AP), complicated by concurrent pneumonia. Hydroxyurea (1g twice daily) was administered to control leukocytosis, along with antimicrobial therapy and supportive blood transfusions. The patient was also diagnosed with secondary pulmonary tuberculosis and commenced on anti-tuberculosis treatment, which was discontinued in October 2024. The patient started dasatinib 140 mg once daily on March 5, 2024. After six months of treatment, the SPECC1L::ABL1/ABL1 fusion gene ratio was 23.49% determined by Quantitative Real-time PCR, indicating resistance to dasatinib. A third-generation TKI was recommended; however, the patient declined the treatment due to financial constraints.
Following nine months of continued dasatinib treatment, the fusion gene ratio decreased to 9.64%. On January 23, 2025, the patient agreed to initiate treatment with olverembatinib. After six months of olverembatinib therapy, the SPECC1L::ABL1/ABL1 fusion gene ratios were recorded as 15.75%, indicating resistance to olverembatinib as well. The patient has undergone haplo-HSCT and has achieved a MMR. The regular monitoring results are presented in Table 1, and the treatment process and outcomes are summarized in Figure 2.
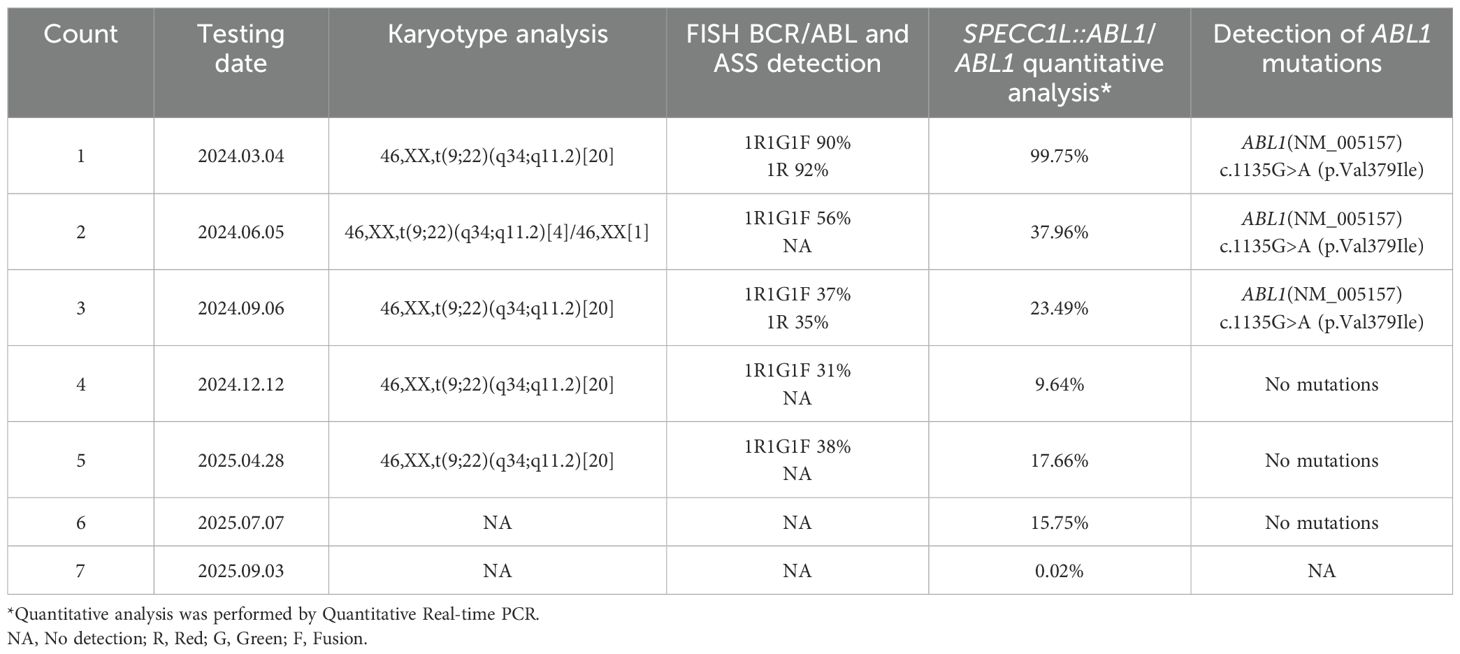
Table 1. Genetic and molecular characteristics of patient.
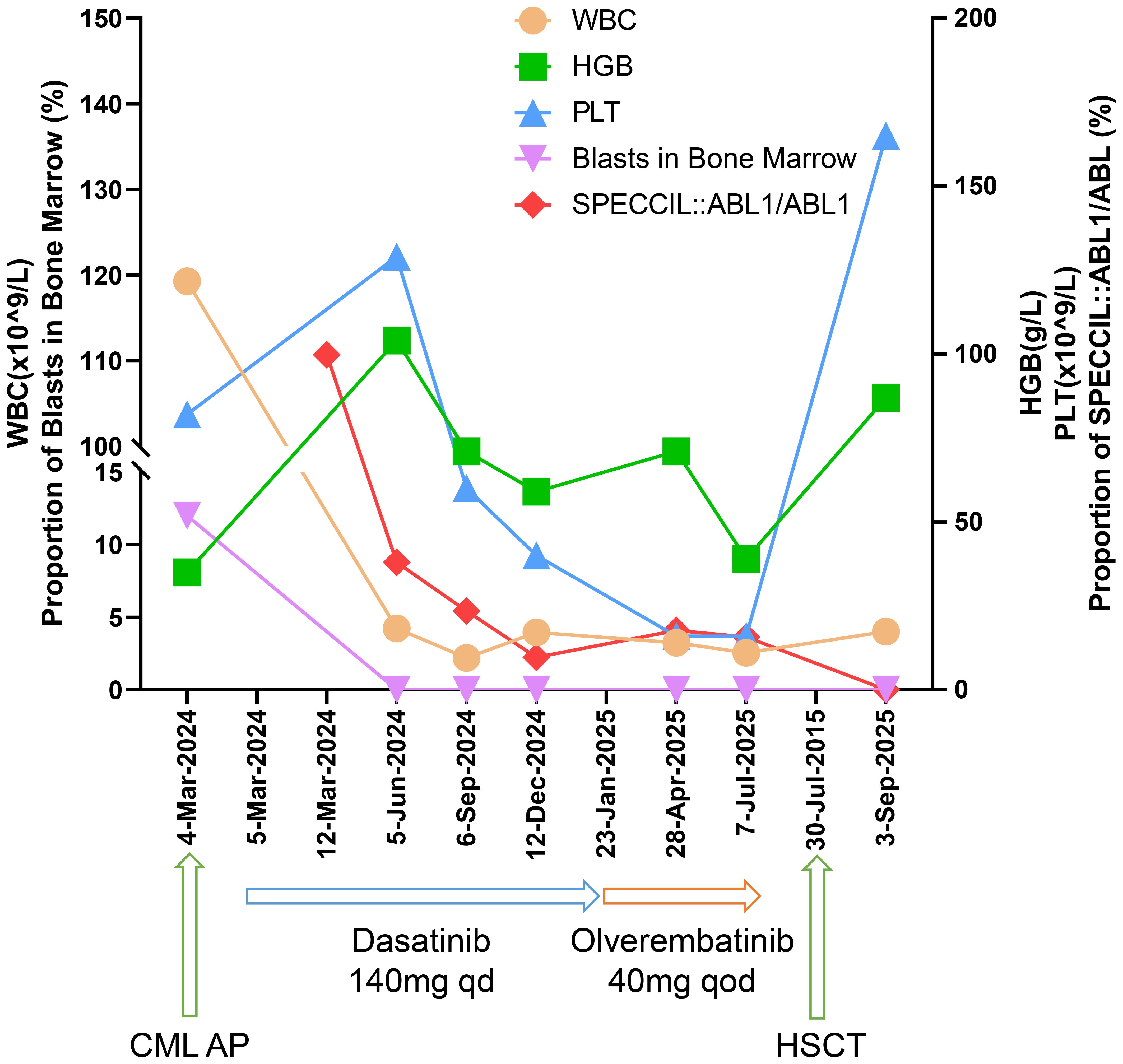
Figure 2. The graph illustrates the quantitative changes in leukocyte count, hemoglobin levels, platelet count, bone marrow blast percentage, and SPECCIL::ABL1/ABL1 fusion ratio following TKI treatment.
Discussion
This study reports an unusual case of CML identified via whole-transcriptome sequencing. The case was defined by two concurrent genetic aberrations: a rare e8a2 BCR::ABL1 fusion transcript harboring an in-frame insertion of SPECC1L exon 4, and a missense mutation (V379I) in the ABL1 kinase domain. Neither of these molecular features is common individually, and their co-occurrence has not been previously documented in TKI-treated CML. This finding provides critical insights into multilayered mechanisms underlying therapeutic resistance in CML.
CML with e8a2 BCR::ABL1 transcripts is relatively rare, with about 20 cases reported to date (Table 2). It is widely acknowledged that a direct junction between BCR exon 8 (e8) and ABL1 exon 2 (a2) generates a premature stop codon (UAG) at position 7 after the fusion (5, 7). However, insertion of certain sequences can result in the formation of an in-frame e8a2 transcript. These insertion sequences generally derived from ABL intron Ia/Ib, BCR intron 8, and other specific genes including PRDM12, SPECC1L, and MAST2. Although these inserted sequences do not participate in encoding functional protein domains, they can indirectly affect the expression level of the BCR::ABL1 fusion protein by adjusting the splice sites of the fusion gene or regulating mRNA stability, thereby leading to differences in the clinical phenotype of CML patients and their responses to drug treatment.
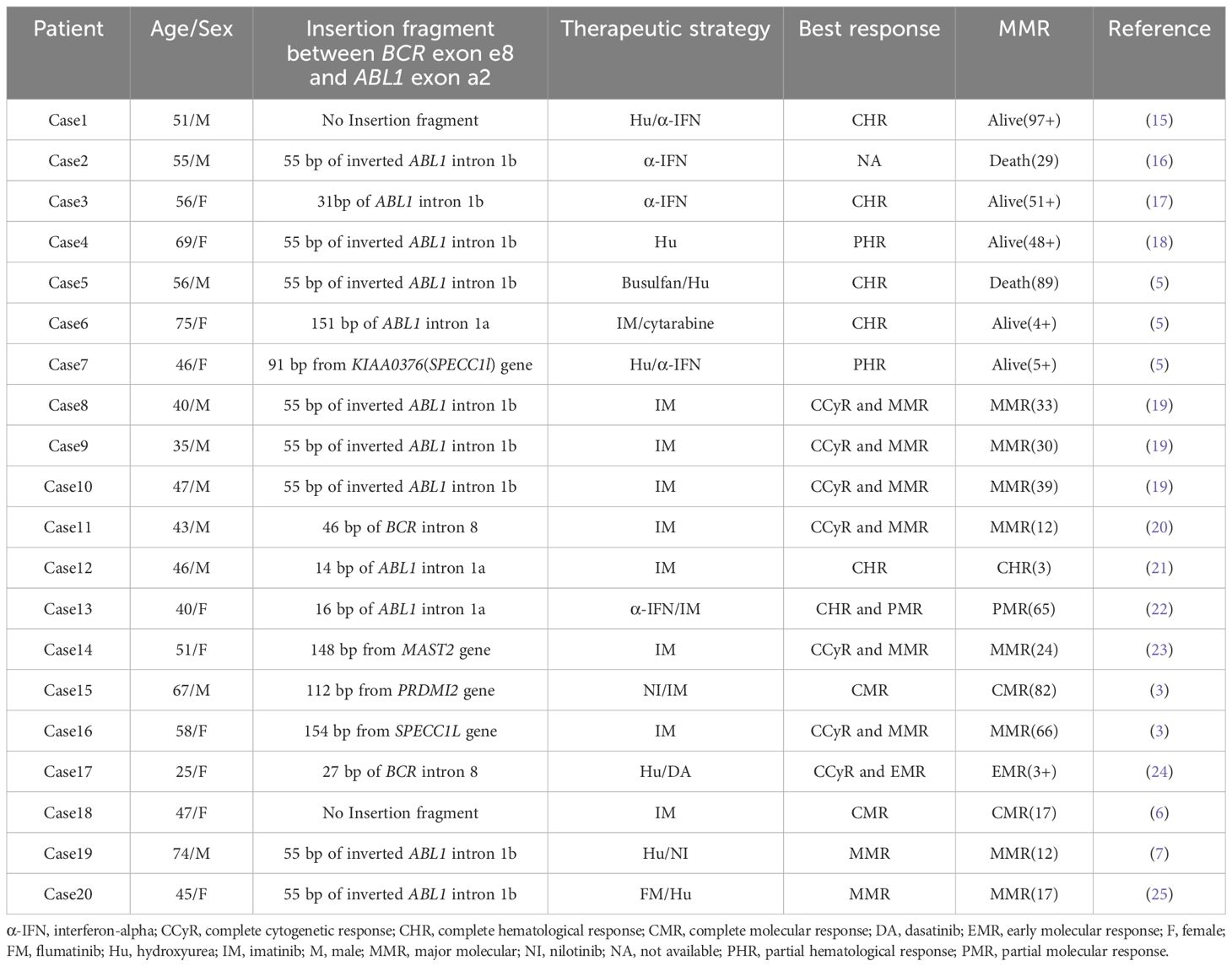
Table 2. Summary of CML cases with e8a2 BCR::ABL1 fusion transcript.
Among these, the insertion of SPECC1L sequences into the e8a2 transcript is rare, with only two documented cases reported prior to this study (3, 5). In these two earlier studies, the patients primarily received treatment with interferon-α or imatinib, achieving only partial hematological response and MMR, respectively. Our case represents the first documented instance of this specific genotype being sequentially treated with second- and third-generation TKIs (dasatinib and olverembatinib). Notably, the patient also harbored an ABL1 V379I mutation, allowing for a comparative assessment of therapeutic efficacy against this genetic background.
The poor treatment response observed in our patient is likely driven primarily by the genetic alterations. The V379I mutation is located between the catalytic domain and the activation loop of the ABL1 kinase, where it plays a critical role in abrogating binding to BCR::ABL1 (8). To date, this mutation has been reported in only two cases (8, 9). One patient was treated with imatinib for 17 years, achieving a complete hematological response but failing attain a MMR. The other patient progressed to the accelerated phase after 34 months of imatinib treatment, at which point the V379I mutation was identified. Treatment was switched to dasatinib; 10 months later, a compound mutation involving V379I and T315I emerged. Like the second reported case, our patient progressed to the accelerated phase following imatinib treatment and later harbored the V379I mutation. Subsequently, treatment with dasatinib and olverembatinib failed to achieve disease control. In conjunction with previously reported findings, this observation suggests that the V379I mutation may confer resistance not only to first- and second-generation TKIs but also to the third-generation agent olverembatinib. Furthermore, the insertion of SPECC1L may contribute to increased resistance to drug therapy. SPECC1L is critical for cytoskeletal organization, cytokinesis, and cell migration (3, 10). Aberrant integration of SPECC1L may contribute to genomic instability and could potentially impair TKI binding through an allosteric effect. Most critically, we propose that the co-occurrence of the V379I mutation and the SPECC1L insertion engenders a synergistic resistance phenotype. Neither alteration alone may be sufficient to confer broad TKI resistance, but together they likely destabilize the ABL1 kinase domain in a manner that precludes effective drug binding. This model is strongly corroborated by the failure of olverembatinib, despite its documented activity against most single ABL1 mutations (11), to control the disease in our patient.
Certain clinical factors may also influence the treatment outcomes of patients. The advanced phase of disease (AP) at the time of second-line treatment, the prior history of TKIs exposures, and the prolonged interval from diagnosis to the initiation of olverembatinib are all associated with a diminished therapeutic response. Furthermore, a drug-drug interaction with rifampicin may have initially reduced dasatinib exposure, but the persistence of resistance after the discontinuation of rifampicin confirms an inherent resistance mechanism (12).
It is important to emphasize that olverembatinib demonstrates notable efficacy and favorable safety in the treatment of CML. It exhibits anti-leukemic activity against nearly all ABL1 kinase domain mutations that cause TKI resistance (11). However, the therapeutic efficacy in patients in the accelerated phase is lower than that in patients in the chronic phase (13). The treatment failure in this case highlights the potential clinical severity of synergism between a SPECC1L insertion and the V379I mutation in the kinase domain, which may compromise the efficacy of even third-generation TKI.
Finally, among patients with CML, about 10-15% have been reported to have deletions adjacent to the t(9;22) breakpoint on der(9). CML-AP patients who carry this deletion may demonstrate a suboptimal response to imatinib. This may be attributed to the loss of critical genes, such as tumor suppressor genes, within the deleted region and is associated with increased genomic instability (14).
In conclusion, this case underscores the clinical imperative of utilizing molecular diagnostics, particularly whole-transcriptome sequencing, in instances where conventional BCR::ABL1 assays yield negative results despite cytogenetic evidence of Ph+ leukemia. For patients with atypical e8a2 transcript, particularly younger individuals, the earlier administration of second- or third-generation TKIs, in conjunction with ABL1 kinase domain mutation analysis, may yield more favorable clinical outcomes. Larger, multicenter studies are necessary to establish the most effective treatment strategy for patients with these rare transcripts.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
JF: Writing – original draft, Data curation, Formal Analysis, Writing – review & editing. YT: Writing – original draft, Data curation, Formal Analysis, Writing – review & editing. XR: Data curation, Formal Analysis, Writing – original draft. RW: Methodology, Validation, Writing – original draft. TC: Methodology, Validation, Writing – original draft. LJ: Project administration, Supervision, Writing – original draft. YH: Project administration, Resources, Writing – original draft. ZX: Project administration, Resources, Writing – original draft. BW: Methodology, Validation, Writing – original draft. HZ: Methodology, Validation, Writing – original draft. JZ: Methodology, Validation, Writing – original draft. ML: Writing – review & editing, Resources. HL: Writing – review & editing, Resources.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Conflict of interest
Author JF, RW, TC, LJ, ZX, HZ, JZ, and HL were employed by the company Kindstar Globalgene Technology, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2025.1711888/full#supplementary-material
References
1. Jabbour E and Kantarjian H. Chronic myeloid leukemia: 2025 update on diagnosis, therapy, and monitoring. Am J Hematol. (2024) 99:2191–212. doi: 10.1002/ajh.27443
2. Baccarani M, Castagnetti F, Gugliotta G, Rosti G, Soverini S, Albeer A, et al. The proportion of different BCR-ABL1 transcript types in chronic myeloid leukemia. Int overview Leukemia. (2019) 33:1173–83. doi: 10.1038/s41375-018-0341-4
3. Huet S, Dulucq S, Chauveau A, Ménard A, Chomel JC, Maisonneuve H, et al. Molecular characterization and follow-up of five CML patients with new BCR-ABL1 fusion transcripts. Genes Chromosomes Cancer. (2015) 54:595–605. doi: 10.1002/gcc.22263
4. Qin YZ, Jiang Q, Jiang H, Lai YY, Shi HX, Chen WM, et al. Prevalence and outcomes of uncommon BCR-ABL1 fusion transcripts in patients with chronic myeloid leukaemia: data from a single centre. Br J Haematol. (2018) 182:693–700. doi: 10.1111/bjh.15453
5. Demehri S, Paschka P, Schultheis B, Lange T, Koizumi T, Sugimoto T, et al. e8a2 BCR-ABL: more frequent than other atypical BCR-ABL variants? Leukemia. (2005) 19:681–4. doi: 10.1038/sj.leu.2403604
6. Jin C, Zhu X, Xiao M, Liu S, Liu X, Liu J, et al. A novel e8a2BCR-ABL1 fusion transcript without insertion sequence in a patient with chronic myeloid leukemia. Ann Lab Med. (2018) 38:169–71. doi: 10.3343/alm.2018.38.2.169
7. Burmeister T, Bullinger L, and le Coutre P. The recurrent atypical e8a2 BCR::ABL1 transcript with insertion of an inverted 55 base pair ABL1 intron 1b sequence: A detailed molecular analysis. Acta Haematologica. (2023) 146:413–8. doi: 10.1159/000531128
8. Rejali L, Poopak B, Hasanzad M, Sheikhsofla F, Varnoosfaderani AS, Safari N, et al. Characterizing of four common BCR-ABL kinase domain mutations (T315I, Y253H, M351T and E255K) in Iranian chronic myelogenous leukemia patients with imatinib resistance. Iranian J Cancer Prev. (2015) 8:e2334. doi: 10.17795/ijcp2334
9. Sorel N, Roy L, Martineau G, Guilhot F, Turhan AG, Chomel JC, et al. Sequential emergence of ABL-kinase mutations with loss of unmutated BCR-ABL allele during targeted therapies of CML. Blood. (2006) 108:1782–3. doi: 10.1182/blood-2006-03-011668
10. Saadi I, Goering JP, Hufft-Martinez BM, and Tran PV. SPECC1L: A cytoskeletal protein that regulates embryonic tissue dynamics. Biochem Soc Trans. (2023) 51:949–58. doi: 10.1042/BST20220461
11. Wu A, Liu X, Fruhstorfer C, and Jiang X. Clinical insights into structure, regulation, and targeting of ABL kinases in human leukemia. Int J Mol Sci. (2024) 25(6):3307. doi: 10.3390/ijms25063307
12. Haouala A, Widmer N, Duchosal MA, Montemurro M, Buclin T, Decosterd LA, et al. Drug interactions with the tyrosine kinase inhibitors imatinib, dasatinib, and nilotinib. Blood. (2011) 117:e75–87. doi: 10.1182/blood-2010-07-294330
13. Jiang Q, Li Z, Qin Y, Li W, Xu N, Liu B, et al. Olverembatinib (HQP1351), a well-tolerated and effective tyrosine kinase inhibitor for patients with T315I-mutated chronic myeloid leukemia: results of an open-label, multicenter phase 1/2 trial. J Hematol Oncol. (2022) 15:113. doi: 10.1186/s13045-022-01334-z
14. Chandran RK, Geetha N, Sakthivel KM, Aswathy CG, Gopinath P, Nair J, et al. Prognostic implications of derivative chromosome 9 deletions in patients with advanced-stage chronic myelogenous leukemia. J Environ Pathol Toxicol Oncol. (2018) 37:117–26. doi: 10.1615/JEnvironPatholToxicolOncol.2018026023
15. How GF, Lim LC, Kulkarni S, Tan LT, Tan P, Cross NC, et al. Two patients with novel BCR/ABL fusion transcripts (e8/a2 and e13/a2) resulting from translocation breakpoints within BCR exons. Br J Haematol. (1999) 105:434–6. doi: 10.1111/j.1365-2141.1999.01372.x
16. Branford S, Rudzki Z, and Hughes TP. A novel BCR-ABL transcript (e8a2) with the insertion of an inverted sequence of ABL intron 1b in a patient with Philadelphia-positive chronic myeloid leukaemia. Br J Haematol. (2000) 109:635–7. doi: 10.1046/j.1365-2141.2000.02042.x
17. Martinelli G, Terragna C, Amabile M, Montefusco V, Testoni N, Ottaviani E, et al. Alu and translisin recognition site sequences flanking translocation sites in a novel type of chimeric BCR-ABL transcript suggest a possible general mechanism for BCR-ABL breakpoints. Haematologica. (2000) 85:40–6.
18. Sugimoto T, Ijima K, Hisatomi H, Murayama T, Mizuno I, Hato A, et al. Second case of CML with aberrant BCR-ABL fusion transcript (e8/a2) with insertion of an inverted ABL intron 1b sequence. Am J Hematol. (2004) 77:164–6. doi: 10.1002/ajh.20138
19. Cayuela JM, Rousselot P, Nicolini F, Espinouse D, Ollagnier C, Bui-Thi MH, et al. Identification of a rare e8a2 BCR-ABL fusion gene in three novel chronic myeloid leukemia patients treated with imatinib. Leukemia. (2005) 19:2334–6. doi: 10.1038/sj.leu.2403986
20. Tchirkov A, Couderc JL, Périssel B, Goumy C, Regnier A, Uhrhammer N, et al. Major molecular response to imatinib in a patient with chronic myeloid leukemia expressing a novel form of e8a2 BCR-ABL transcript. Leukemia. (2006) 20:167–8. doi: 10.1038/sj.leu.2404012
21. Park IJ, Lim YA, Lee WG, Park JS, Kim HC, Lee HJ, et al. A case of chronic myelogenous leukemia with e8a2 fusion transcript. Cancer Genet Cytogenetics. (2008) 185:106–8. doi: 10.1016/j.cancergencyto.2008.06.001
22. Qin YZ, Jiang B, Jiang Q, Zhang Y, Jiang H, Li JL, et al. Imatinib mesylate resistance in a chronic myeloid leukemia patient with a novel e8a2 BCR-ABL transcript variant. Acta Haematologica. (2008) 120:146–9. doi: 10.1159/000178145
23. Riva E, Manrique Arechavaleta G, De Almeida C, Costa V, Fernandez Del Campo M, Ifran González S, et al. A novel e8a2 BCR-ABL1 fusion with insertion of MAST2 exon 2 in a four-way translocation t (1;17;9;22) (p35;q24;q44;q11) in a patient with chronic myeloid leukemia. Leukemia Lymphoma. (2016) 57:203–5. doi: 10.3109/10428194.2015.1043549
24. Chen L, Wu Y, You Y, Xiao M, Yao Y, Li W, et al. A novel e8a2 BCR-ABL1 intronic fusion through insertion of a chromosome 22 BCR gene fragment into chromosome 9 in an atypical Philadelphia (Ph) chromosome chronic myeloid leukemia patient. Leukemia Lymphoma. (2016) 57:2930–3. doi: 10.3109/10428194.2016.1173211
Keywords: BCR::ABL1, e8a2, SPECC1L, chronic myeloid leukemia (CML), accelerated phase, V379I mutation
Citation: Fu J, Tang Y, Ruan X, Wang R, Chen T, Jiang L, He Y, Xu Z, Wang B, Zhang H, Zhou J, Lan M and Li H (2025) Case Report: Dual resistance to dasatinib/olverembatinib in accelerated-phase cml: identification of a novel SPECC1L-inserted e8a2 BCR::ABL1 transcript and ABL1 V379I mutation. Front. Oncol. 15:1711888. doi: 10.3389/fonc.2025.1711888
Received: 24 September 2025; Accepted: 09 October 2025;
Published: 24 October 2025.
Edited by:
Shimin Hu, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Qianghua Zhou, North York General Hospital, CanadaWen Shuai, Duke University, United States
Copyright © 2025 Fu, Tang, Ruan, Wang, Chen, Jiang, He, Xu, Wang, Zhang, Zhou, Lan and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongrui Li, bGlob25ncnVpQGtpbmRzdGFyLmNvbS5jbg==; Mei Lan, Mjc1MDQ3ODI2N0BxcS5jb20=
†These authors have contributed equally to this work
‡These authors have contributed equally to this work and share first authorship