
 Alexandra Paye
Alexandra Paye
- Laboratory of Tumor and Developmental Biology, Groupe Interdisciplinaire de Génoprotéomique Appliquée-Cancer, University of Liege, Liège, Belgium
The development of vascular system depends on the coordinated activity of a number of distinct families of molecules including growth factors and their receptors, cell adhesion molecules, extracellular matrix (ECM) molecules, and proteolytic enzymes. Matrix metalloproteases (MMPs) are a family of ECM degrading enzymes required for both physiological and pathological angiogenesis. Increasing evidence, point to a direct role of membrane type-MMPs (MT-MMPs) in vascular system stabilization, maturation, and leakage. Our understanding of the nature of MT-MMP interaction with extracellular and cell surface molecules and their multiple roles in vessel walls and perivascular stroma may provide new insights into mechanisms underlying vascular cell–ECM interactions and cell fate decisions in pathological conditions. Regulation of vascular leakage by MT-MMP interactions with the ECM could also lead to novel targeting opportunities for drug delivery in tumor. This review will shed lights on the emerging roles of MT1-MMP and MT4-MMP in vascular system alterations associated with cancer progression.
Introduction
In cancer, the extracellular matrix (ECM) within and surrounding a tumor undergoes continuous remodeling. It differs from that of normal tissue, and consequentially influences vascular cell behaviors (DeClerck et al., 2004). Tumor blood vessels are torturous, irregular in dimension and shape, and lack the hierarchical organization of arterioles, capillaries, and venules as found in normal vessels (Baluk et al., 2005). The endothelium of tumor vessels contains large number of fenestrations and the basement membrane surrounding vessels has altered morphology due to extensive vessel remodeling. Altered blood vessel structure causes disorganized blood flow within the tumor and increased leakage that lead to increased interstitial fluid pressure (IFP) (Baluk et al., 2005). Enhanced IFP in the tumor interstitium results not only, from leaky blood vessels but also from dysfunctional lymphatics. Such an increased IFP forms a barrier for transcapillary transport and represents a potential obstacle to the delivery of therapeutic molecules (Heldin et al., 2004; Wiig et al., 2010). Many current anti-cancer agents are not able to disseminate through tumors because of the high intratumoral IFP. Recently, the normalization of tumor vessels emerged as a new challenge for anti-angiogenic therapy and is now recognized as a requisite step to improve delivery of cytotoxic drugs inside tumors (Jain, 2005).
The ECM in the vascular wall contains a variety of molecules including collagens, elastic fibers, hyaluronan (HA), fibronectin, vitronectin, laminins, glycosaminoglycans (GAG), proteoglycans, and adhesive glycoproteins that provide structural and mechanical support to the cells. Vascular and perivascular cells are connected to major structural ECM components by cell surface receptors including integrin and non-integrin molecules (Kalluri, 2003; Davis and Senger, 2008). Interactions of these receptors with ECM molecules influence vascular cell shape, behavior, and the response to cytokines and growth factors (Boudreau and Jones, 1999). ECM remodeling in vascular wall or perivascular stroma involves several enzymes among which Matrix metalloproteases (MMPs) produced by endothelial, inflammatory, or malignant cells are key players. MMPs are a family of zinc endopeptidases composed of 24 currently known human enzymes that share several functional domains. MMPs are often referred to soluble (MMPs) or membrane type-MMPs (MT-MMPs) that are anchored to the cell surface through transmembrane domain or glycosylphosphatidylinositol (GPI) linker. For a description of the structure, function, and regulation of MT-MMPs, the reader is referred to previous reviews (Sounni and Noel, 2005; Sohail et al., 2008). MT1-MMP (membrane type 1-matrix metalloproteinase-1, also known as MMP-14) a MMP with transmembrane and cytoplasmic domains is a unique cell surface activator of pro-MMP2 through the formation of trimolecular complex with TIMP-2 (tissue inhibitor of metalloproteinases). In addition, MT1-MMP has a large number of substrates including ECM and non-ECM molecules (Figure 1). MT4-MMP however has a glycophosphatidylinositol anchor instead of the type-1 transmembrane and is a poor activator of pro-MMP2. Despite the high degree of similarity between its ectodomain structure and that of MT1-MMP, MT4-MMP has a restricted repertoire of substrates with no activity against collagens (Figure 1). Collagenases are the only known mammalian enzymes able of degrading triple-helical fibrillar collagen into distinctive 3/4 and 1/4 fragments. Collagenolytic MMPs including MMP1, MMP8, MMP13, MMP14/MT1-MMP play pivotal roles in multiple physiological and pathological processes involving extensive and aberrant collagenolysis. MMP-mediated collagen remodeling regulates tissue homeostasis and development through several mechanisms, such as the generation of space for cell migration, production of cryptic peptides with novel biological activity or activation of signaling molecules (Page-McCaw et al., 2007). Latest technological progresses have obviously advanced our consideration of MMPs as important modulators of the tumor microenvironment (Kessenbrock et al., 2010). Beside their capacity to remodel ECM, these enzymes mediate the release of ECM-associated growth factors such as VEGF, TGFβ as well as unmask cryptic sites within ECM molecules which can simulate malignant cells and endothelial cells to communicate and escape from homeostatic control (Rundhaug, 2005; Page-McCaw et al., 2007; Kessenbrock et al., 2010). However, the contribution of MMPs to vessel maturation and stabilization is an innovative emergent concept. Understanding the complex roles of MT-MMPs in vessel wall assembly and function in homeostasis or disease will open new avenues to maintain vessel stability and functionality. The present review will highlight the emerging roles of MT1-MMP and MT4-MMP in vascular cell fate and function during cancer progression.
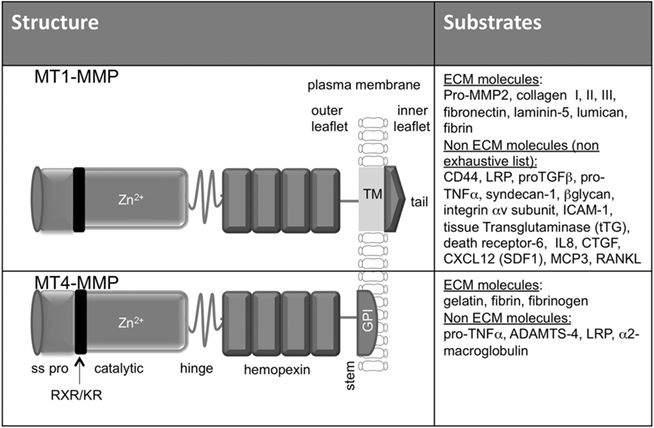
Figure 1. Structure and substrate specificity of MT1-MMP and MT4-MMP. The structure of MT1-MMP and MT4-MMP share the identical domain organization of most MMPs, including a signal sequence (SS), propeptide domain (pro), a zinc-containing catalytic domain (catalytic) with 37 identity (50% similarity), a hing region, and a hemopexin like domain (hemopexin). In addition, MT1-MMP and MT4-MMP are anchored to the cell surface through a hydrophobic a transmembrane domain (TM) and a GPI anchor, respectively. Both the transmembrane MT1-MMP and the GPI–MT4-MMP contain proprotein convertase recognition RXR/KR motif in the propeptide domain and a hydrophilic region also called stem region at the end of hemopexin like domain. Whereas MT1-MMP exhibits activity against many ECM and non-ECM components both in vitro and in vivo, MT4-MMP was found in vitro to have a minimal or no activity against ECM molecules. The substrates of MT4-MMP in vivo are not yet identified.
Collagen Proteolysis in Perivascular Stroma
Collagen is the most abundant molecule of the ECM. It includes fibrillar collagen types I, II, and III, with type I collagen representing the major type within ECM stroma, with 25% of total protein in the body (Shoulders and Raines, 2009). Initial studies examining the role of type I collagen in new blood vessel function started 28 years ago. Montesano et al. (1983) have demonstrated that, when cultured in vitro within 3D matrix of type I collagen, endothelial cells reorganize into capillary-like structures. Later, different studies revealed that the interaction between endothelial cells and type I collagen is mediated by several β1 integrins including α1β1, α2β1, and α3β1 (Xu et al., 2000). Moreover, inhibition of integrin α1β1 and α2β1 either by blocking antibody or silencing by small interferon RNA, disrupts VEGF signaling and inhibits endothelial cell migration and tumor angiogenesis (Senger et al., 2002).
Type I collagen is cleaved into characteristic 3/4 and 1/4 fragments by collagenolytic MMPs, including MMP1, MMP8, MMP13, and MT1-MMP (Ohuchi et al., 1997). Among the collagen remodeling enzymes, MT1-MMP-mediated cleavage of type I collagen stimulates migration, guidance, and organization of endothelial cells to form tubular structures (Chun et al., 2004). Type I collagen remodeling by MT1-MMP during tumor growth has been demonstrated by the study of Hotary et al. (2003) showing that MT1-MMP expression in cancer cells enables these cells to escape the mechanical barriers that confine them to collagen matrix and stimulates tumor growth in vivo. Moreover, the generation of Mt1-mmp−/− mice pinpointed the role of MT1-MMP in collagen remodeling during embryonic and post-natal development growth. Indeed, Mt1-mmp−/− mice exhibit skeletal defects with craniofacial abnormalities, osteopenia, and angiogenesis (Holmbeck et al., 1999; Zhou et al., 2000). MMP2 is activated primarily by MT1-MMP present at the cell surface. Although MMP2 exhibits mainly a gelatinolytic activity, it is able to cleave type I collagen by a mechanism distinct from the classic collagenases (Tam et al., 2004). MMP2 emerged as a new collagenase with higher activity against type I collagen harboring a mutation that renders it resistant to collagenase-mediated cleavage to 3/4 and 1/4 fragments (Col1a1r/r ) than the normal type I collagen (Egeblad et al., 2007). Interestingly, the generation of Mmp2−/− mice with similar mutation in colla1 gene (Mmp2−/−; Col1a1r/r mice) showed severe skeletal defects and developmental abnormalities resembling those observed in MMP2-null humans and those seen in Mt1-mmp−/− mice (Egeblad et al., 2007). While Mmp2−/− mice develop normally with no major skeletal abnormalities, the perturbation of collagen degradation in vivo increases the phenotypic alteration, suggesting that MMP2 is important for type I collagen remodeling during development.
Type I collagen remodeling in perivascular stroma represents an important step for endothelial cell reorganization into tubular structures during normal and pathological angiogenesis. In addition, a direct link between collagen metabolism and the regulation of vascular leakage has been demonstrated in transgenic Col1a1r/r mice with collagenase-resistant collagen (Liu et al., 1995). The steady-state vascular leakage is dramatically reduced in these mice. Furthermore, the evaluation of acute vascular leakage in response to tissue assault revealed a failure to develop normal vascular response to tissue injuries and damage due to reduction of vascular openings within vessel wall in these transgenic mice (Sounni et al., 2010a). In cancer, collagen cross-linking and stiffening is known to induce malignant progression through integrin clustering and signaling, activation of focal adhesion kinase (FAK), PI3 kinase, and Akt (Levental et al., 2009). However, the effect of collagen abundance and cross-linking on endothelial cell behaviors and its effect on signaling and leakage in tumor vessels remain to be explored.
Vascular leakage and ECM remodeling are considered as a critical and requisite step in angiogenesis initiation in response to tissue damage, wound repair, or tumor. It is recognized that collagen architecture controls the diffusion and transport of macromolecules in tissue. A genetic alteration of ECM structure in the vessel wall impacts blood vessel integrity and the extravasation of fluids and plasma proteins (Wiig et al., 2008). Collagen accumulation together with proteoglycans and myofibroblasts in the interstitial space contributes also to the higher IFP observed in solid tumors which regulates drug diffusion through the tumor (Heldin et al., 2004). Thus, perivascular ECM emerged recently as an important determinant of intratumoral IFP and a drug delivery modulator (Netti et al., 2000). In tumor stroma, increased collagen synthesis and accumulation are correlated to restrictive delivery of high molecular weight anti-cancer drugs (Netti et al., 2000; Brown et al., 2003). Furthermore, enzymatic digestion of collagen and decorin, an associated proteoglycan, improves macromolecule diffusion inside tumors (Magzoub et al., 2008). In addition, a major consequence of macrophage and other inflammatory cell infiltration into the tumor stroma is the release of cytokines and growth factors which, by acting on stromal fibroblasts and blood vessels, stimulate the production of ECM molecules, further reinforcing matrix accumulation and IFP enhancement within tumors (Heldin et al., 2004). One of the clinical impacts of the control of collagen remodeling in perivascular stroma would be to normalize tumor vasculature, lower IFP, and modulate hydraulic conductivity of the stroma and fluid balance. Consequently, this would improve tumor perfusion and delivery of cytotoxic drugs in solid tumors (Kessenbrock et al., 2010; Sounni et al., 2010a).
Soluble MMP in Vessel Maturation
Perturbations of tumor blood vessels have been attributed to their failure to mature and to restore quiescence as normal vessels. Among the vascular cells, pericytes are major players contributing to vessel maturation (Feron, 2004). Due to their contractile activity, pericytes control the blood flow in normal and tumor vessels and consequently, regulate drug delivery into tumor tissue (Feron, 2004; Raza et al., 2010). The pericyte recruitment around endothelial cells during embryonic development, atherosclerosis, and tumor angiogenesis relies on different pathways which involve PDGFB/PDGFR-β, sphingosine 1-phosphate (S1P)/endothelial differentiation gene-1 (EDG-1), Ang1/Tie-2, and TGF-β/activin-like kinase receptor (ALK5) axis (Jain, 2003; Gerhardt and Semb, 2008; Park et al., 2009). For instance, genetic ablation of PDGFB or PDGFR-β in mice is associated with a lack of pericyte coverage around vessels and lead to microvascular aneurysms and lethal microhemorrhages (Lindahl et al., 1997). Beside the role of growth factors in pericyte recruitment and vessel maturation, soluble MMPs are known to facilitate pericyte migration and vessel coverage (Sounni and Noel, 2005; Chantrain et al., 2006). MMP inhibition in different experimental tumor models of melanoma and neuroblastoma show reduced pericyte recruitment and decreased tumor vessel perfusion (Spurbeck et al., 2002; Chantrain et al., 2004; Noel et al., 2008). Among soluble MMPs, MMP9 is considered as the most important candidate in pericyte recruitment from bone marrow. Pericyte coverage along tumor microvessels is decreased in Mmp9−/− mice xenotransplanted with human neuroblastoma. Indeed, the formation of mature tumor vessels is restored when Mmp9−/− mice are transplanted with bone marrow from wild type mice (Chantrain et al., 2004). In addition, MMPs increase the bioavailability of factors that affect pericyte phenotype through the activation and release of growth factor from cell surface or ECM. Among these factors, HB–EGF shedding from cell membrane is mediated by several proteases of the MMP family membrane to activate receptor tyrosine kinase (RTK) that may promote pericyte recruitment and vessel maturation in vivo (Eguchi et al., 2001). For instance, MMP3 cleavage of proHB–EGF at specific juxtamembrane site enhances growth factor bioavailability in physiological and pathological processes (Suzuki et al., 1997), CD44 mediates MMP7/proHB–EGF complex formation and active HB–EGF release (Yu et al., 2002). MMP2 and MMP9 contribute to EGFR activation in vivo in a mouse model of pressure-induced myogenic tone of resistance arteries (Lucchesi et al., 2004). In several tumor models, MMPs have been shown to release soluble factors sequestered in the ECM and regulate vascular cell formation and maturation. For instance, inflammatory cell-derived MMP9 promotes tumor angiogenesis through the release of ECM-bound angiogenic factor such as VEGF (Bergers et al., 2000; Huang et al., 2002). Furthermore, in vitro, MMP2, MMP3, and MMP7 induce the release of TGFβ1 from decorin, a proteoglycan that sequester TGFβ in the matrix (Imai et al., 1997).
In addition to these multiple actions of soluble MMPs, these enzymes also contribute to the selective recruitment of bone marrow-derived stem cells occurring in post-natal organogenesis and tumorigenesis (Coussens et al., 2000; Lyden et al., 2001). Recruitment of hematopoietic stem cells (HSCs), but also other types of undifferentiated cells, such as endothelial progenitor cells (EPCs) seem to be dependent on proteolytic activation of their effector cytokines. MMP9 has been identified as a key player in mobilization of both HSCs and VEGFR2+ CEPs to the circulation from BM. In this context, MMP9 converts Kit-ligand from a membrane-bound molecule to a soluble survival/mitogenic factor sKit-ligand (Heissig et al., 2002). MMP2 and MMP9 can play a major role in EPCs migration and vessel maturation during several physiological processes. Yoon et al. (2005) have isolated and characterized early EPCs and late outgrowth endothelial cells (OECs) for their angiogenic potential and demonstrate that MMP9 and MMP2 secretion by both EPCs and OECs are essential for maturation and repair during ischemia. However, the contribution of EPCs to the angiogenic process during cancer development or their implication in vessel maturity is still not clear. Recently, Purhonen et al. (2008) have provided evidences that EPCs are not required for tumor angiogenesis. Another study has proven that hematopoietic cells infiltrating tumors are not EPCs but rather CD11b-positive myelomonocytic cells. Furthermore, these cells are likely responsible for tumor vasculogenesis but not angiogenesis, their recruitment to tumor is dependent on MMP9 expression (Ahn and Brown, 2008). Despite, their role in angiogenesis, soluble MMPs are also important in vascular homeostasis. For instance, MMP9 plays an important role in edema prevention. Experimental secondary lymphedema in the mouse tail of MMP-9 null mice were unable to sufficiently counteract the swelling due to the lack of matrix remodeling around lymphatic vessels (Rutkowski et al., 2006).
All together, soluble MMPs might participate in vessel maturation through the control of bioavailability of growth factors and cytokines that regulate pericyte recruitment and coverage of new formed vessels in solid tumors. However, it cannot be excluded that one or a group of these soluble MMPs acting in perivascular stroma or in the vessel wall, deregulates vessel permeability and contributes to pathological vascular leakage associated with cancer.
Role of MT1-MMP in Vessel Maturation
The contribution of MT1-MMP in angiogenesis is well documented and reported in the literature (Lafleur et al., 2003; Sounni et al., 2003; Zucker et al., 2003; Stratman et al., 2009). MT1-MMP is likely the most important MMP regulating endothelial cell functions. The generation of Mt1-mmp mice has shed light on the key role played by MT1-MMP during angiogenesis (Zhou et al., 2000; Sounni and Noel, 2005). MT1-MMP stimulates angiogenesis in fibrin gel more efficiently than serine, cysteine proteases, and other MMPs (Hiraoka et al., 1998; Hotary et al., 2002; Collen et al., 2003). The proangiogenic mechanisms of MT1-MMP include: (1) cleavage of ECM molecules, (2) the production of angiogenic factors, such as VEGF, (3) interaction with cells surface molecules, such as CD44 and S1P, (4) the degradation of anti-angiogenic factors such as decorin in cornea (Mimura et al., 2009). These regulatory mechanisms could enhance angiogenesis in both physiological and pathological conditions. Alternatively, additional signaling molecules, such as proTGFβ and endoglin (TGFβ receptor) are substrates of MT1-MMP, which can play a dual role on vessel maturation and angiogenesis.
We and others have previously demonstrated that MT1-MMP overexpression in cancer cells stimulates tumor angiogenesis in vivo through VEGF upregulation (Deryugina et al., 2002; Sounni et al., 2002, 2004; Deng et al., 2009). MT1-MMP regulation of VEGF expression is dependent on Src signaling and requires the proteolytic activity of MT1-MMP (Sounni et al., 2004). Recently, Eisenach et al. (2010) have provided a complete mechanism of VEGF regulation by MT1-MMP and Src signaling. This study demonstrates that MT1-MMP regulates VEGF through a complex with VEGFR-2 and Src, activation of Akt and mTOR signaling pathway. MT1-MMP activity and signaling through cytoplasmic domain are confirmed as required for transcriptional regulation of VEGF expression in cancer cells. MT1-MMP interaction with Src has also been investigated in several in vitro models of angiogenesis and cell migration (Labrecque et al., 2004; Ouyang et al., 2010; Sabbota et al., 2010). MT1-MMP-mediated Src activation has also been described during cell migration. Recently, Sabbota et al. (2010) reported that MT1-MMP induces receptor activator of NF-κB ligand (RANKL) shedding from prostate cancer cell surface and signaling through its receptor RANK and downstream Src activation and consequently stimulates cell migration. Src activation by MT1-MMP has also been linked with ERK1/2 activation in cell migration within 3D collagen gel (Takino et al., 2010). However, MT1-MMP can form a complex with TIMP2 at the cell surface that can stimulate cancer cell migration in a non-proteolytic manner. This non-proteolytic mechanism implicates the activation of MEK1/2-ERK1/2-p90RSK signaling cascade by the MT1-MMP/TIMP-2 complex (Sounni et al., 2010b). Beside its role in ECM remodeling, MT1-MMP promotes endothelial cell migration, lumen formation, and vascular guidance tunnels in collagen matrices (Stratman et al., 2009). The observation of Lehti et al. (2005) that Mt1-mmp−/− mice present a marked reduction in mural cell density and abnormal vessel morphology in brain, suggests that membrane-associated MMP acts as a cofactor in propagating signaling through vascular smooth muscle cells (VSMC). This study, demonstrates that MT1-MMP processing of PDGFR-β is required for PDBFB/PDGFR-β signaling in VSMC and migration in vitro. Moreover, MT1-MMP induces VSMC dedifferentiation and acquisition of migratory and invasive phenotype during vascular injury through low density lipoprotein (LDL) receptor-related protein (LRP) proteolysis that promotes signaling through PDGFB/PDGFR-β axis (Lehti et al., 2009). Indeed, MT1-MMP cooperates with platelet derived S1P to induce endothelial cell migration and morphogenic differentiation (Langlois et al., 2004). Furthermore, MT1-MMP regulates signaling of the advanced glycation end products (AGE)/a receptor for AGE (RAGE) axis in the vasculature, a target known to play a key role in diabetic vascular complications (Kamioka et al., 2011). Interestingly, expression of MT1-MMP correlates with the clinical mobilization of human CD43+ cell in patient with lymphoid malignancies (Vagima et al., 2009). Of interest, MT1-MMP expression and its association with lipid rafts facilitates G-CSF-induced hematopoietic stem/progenitor cell (HSPC) mobilization in human and murine (Shirvaikar et al., 2010). The molecular mechanisms of MT1-MMP relay on CD44 cleavage, activation of pro-MMP2 and inactivation of SDF-1 that facilitates HSPC egress from the bone marrow into circulation (Shirvaikar et al., 2011). MT1-MMP signaling in cancer cells, pericytes, and endothelial cells are schematically illustrated in Figure 2.
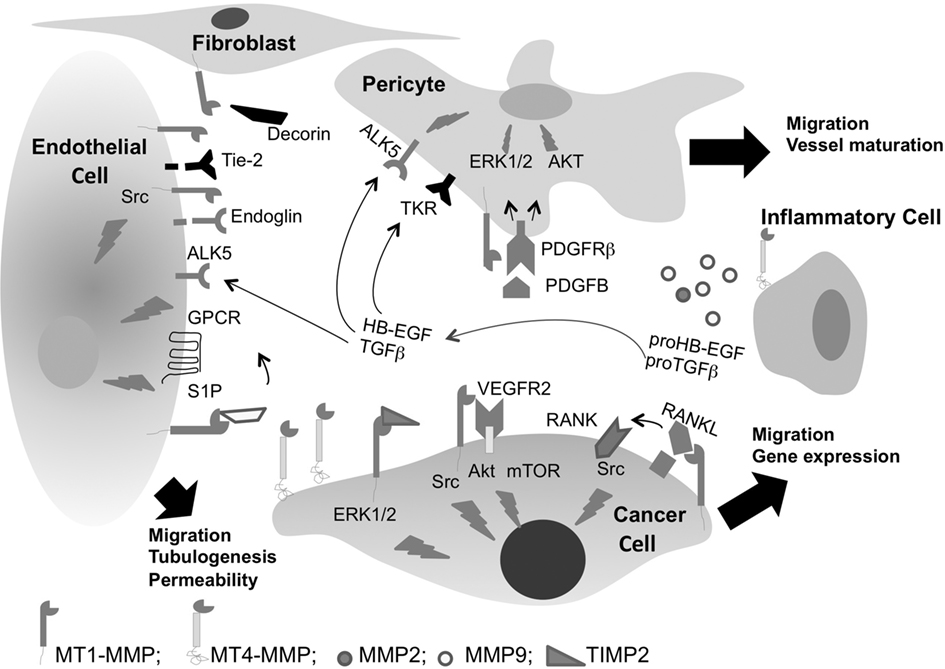
Figure 2. Membrane type-MMPs regulate vascular cell signaling in cancer. MT1-MMP regulates cell migration through both ECM proteolysis and non-proteolytic-dependent TIMP-2 activation of ERK1/2 pathway. MT1-MMP regulates VEGF gene expression through Src, Akt, and mTOR activation and stimulates tumor angiogenesis. In addition, it induces RANKL shedding and signaling through Src leading to cell migration. MT1-MMP cooperates with platelet derived S1P to induce signaling through G protein-coupled-receptors (GPCRs) and regulates endothelial cell migration and tubulogenesis. MT1-MMP regulates TGFβ bioavailability and its signaling through ALK5 leading to vessel maturation. Shedding of Tie-2 or degradation of decorin by MT1-MMP stimulates angiogenesis, but shedding of endoglin from endothelial cell surface inhibits angiogenesis. ProTGFβ can also be activated by MMP2 and MMP9. MT1-MMP activates PDGFRβ signaling and regulates pericyte migration and vessel maturation. MT4-MMP a metastatic MMP, is highly expressed by cancer cells and induces tumor vessel destabilization through pericyte detachment.
Recently, a spatial and temporal expression of MT1-MMP during angiogenesis has been monitored in the transgenic Mt1-mmp+/LacZ mice (Yana et al., 2007). This study shows that vascular maturation coordinated by crosstalk between endothelial cells and VSMC reorients MT1-MMP expression at leading edges of newly developed vessels. Implication of MT1-MMP in vessel stabilization/destabilization has been reinforced by the study of Onimaru et al. (2010) which reveals an interaction between MT1-MMP and angiopoietin (Ang)–Tie-2 axis. It is well known that VEGF regulates Ang–Tie-2 signaling by inducing proteolytic cleavage and shedding of Tie-2 (Findley et al., 2007). MT1-MMP has been recently shown to induce shedding of Tie-2 from cultured endothelial cells and modulates Ang1-dependent Tie-2-associated signaling pathways (Onimaru et al., 2010). MT1-MMP activity leads to a Tie-2 shedding-mediated decrease in endothelial Tie-2 level, resulting in the induction of vessel destabilization and subsequent endothelial migration. Although the role of Ang–Tie-2 signaling in vascular growth and remodeling is quite complex, it is generally accepted that Ang1-mediated activation of Tie-2 promotes vascular stabilization and quiescence, whereas Ang2 acts in opposition to Ang1 to facilitate VEGF mediated angiogenesis. The role of MT1-MMP in Ang2–Tie-2 signaling is still not investigated and, whether MT1-MMP activity is required for vessel maturation step Ang–Tie-2 dependent pathway in pericyte are required to define the precise role of this enzyme during all angiogenesis steps.
We recently investigated MT1-MMP activity in perivascular stroma and the regulation of vascular permeability in vivo (Sounni et al., 2010a). We directly assessed whether MMP inhibition in both healthy and mouse model of age-related dermal fibrosis (i.e., Col1a1r/r mice Liu et al., 1995) could impact vascular leakage in an in vivo vascular leakage assay (Miles and Miles, 1952). Surprisingly, prior treatment with broad spectrum MMP inhibitor GM6001 (Ilomastat, Galardin) significantly increases vascular leakage in both wild type and Col1a1r/r mice. This indicates that the inhibition of steady-state MMP activity renders vessels more susceptible to induced acute leakage, and implies a link between steady-state MMP activity and vascular leakage. In vivo vascular assays applied to the skin of different MMP-deficient mice such as Mmp2−/−, Mmp9−/−, Mmp8−/−, and Mmp13−/− lead to similar results except for Mt1-mmp−/− mice. Mice lacking MT1-MMP show a distinct higher steady-state vascular leakage and low levels of active TGFβ compared to their corresponding age-matched wild type littermates. Maintaining vascular homeostasis relies on molecular mechanisms coordinated by MT1-MMP activity which controls TGFβ bioavailability and signaling through ALK5 in vascular wall. In vivo inhibition of TGFβ signaling in mice treated with small amount of ALK5 inhibitor (ALK5I) for 1 week, strongly increases vascular leakage both in normal and fibrotic skin, and it also enhances macromolecules delivery in neoplastic tissues (Figure 3). The treatment of transgenic mice developing skin carcinoma (K14-HPV16 transgenic mice) and mammary adenocarcinomas (MMTV-PyMT mice) with ALK5I results in enhanced delivery and bio-distribution of macromolecules in tumors compared to mice treated with vehicle alone. Taken together, these data provide evidence for a central role for MT1-MMP-activated TGFβ in mediating vascular homeostasis and remodeling. They further indicate that TGFβ and/or MT1-MMP-selective antagonists may enhance vascular leakage and therapeutic delivery to tissues where hemodynamic limits efficient drug delivery.
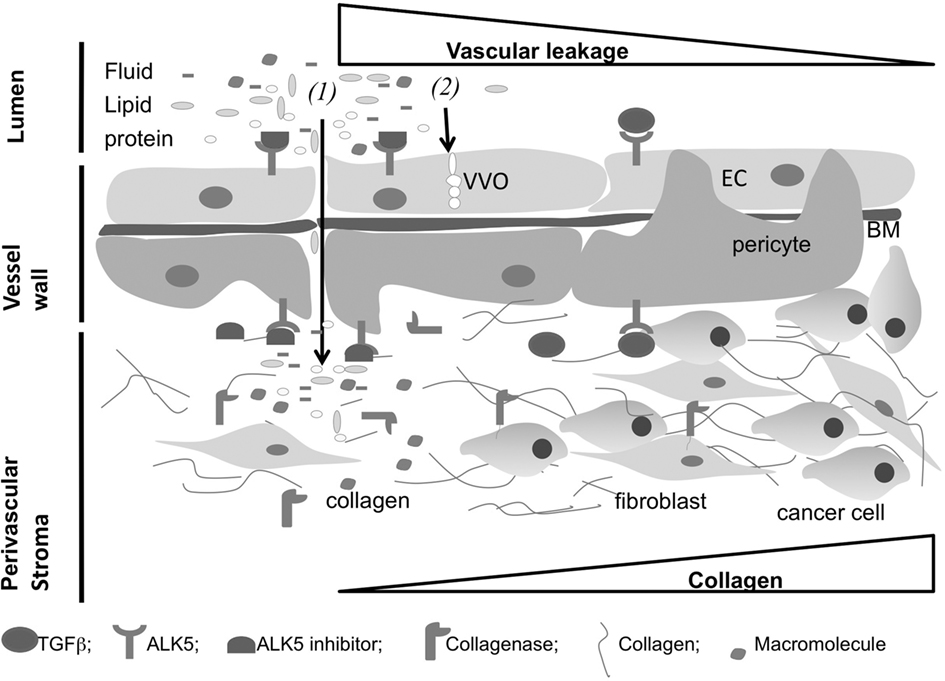
Figure 3. Extracellular matrix (ECM) components regulate vascular permeability in tumor. Transport of fluids and macromolecules across the vessel wall can be regulated by both (1) paracellular diffusion through the apparition of openings between vascular cells and (2) transendothelial transport through vesiculo-vacuolar organelle (VVO) across the endothelium. Inhibition of ALK5 results in increased vascular leakage and increased bio-distribution of macromolecules within tumor. Collagen and fibroblast accumulation within tumor opposes to macromolecule penetration and reduces drug delivery. Collagen metabolism by collagenases results in increased vascular leakage and accumulation of macromolecules within tumors. Short inhibition of TGFβ signaling in vascular wall and control of the ECM metabolism in perivascular stroma could enhance drug delivery in solid tumors (EC, endothelial cell; BM, basement membrane).
Recent report, by Hawinkels et al. (2010) shows that MT1-MMP dependent shedding of endoglin inhibits angiogenesis in vitro. MT1-MMP activity at endothelial cell surface increases soluble endoglin that acts as decoy for TGFβ and blocks downstream signaling in endothelial cells. Although the study shows that soluble form of circulating endoglin decreases in colorectal cancer patients, it did not correlate this with MT1-MMP levels and tumor angiogenesis in patients. The effect of MT1-MMP interaction with TGFβ signaling on angiogenesis in vivo, is a complex regulatory mechanism that depends on its spatial distribution, such as, perivascular stroma or endothelial cells compartment. A direct shedding of endoglin from endothelial cell surface inhibits angiogenesis (Hawinkels et al., 2010), but the cleavage of LTBP-1 (Dallas et al., 2002; Tatti et al., 2008) or LAP–TGFβ from the stroma in fibrotic tissue increases TGFβ availability which in turn activates TGFβ signaling in vascular cells (Sato, 1995; Sounni et al., 2010a).
MT1-MMP/TGFβ signaling axis is regulated by type I collagen, the most abundant ECM molecules. Type I collagen induces MT1-MMP expression which in turn regulates TGFβ signaling in stromal and cancer cells (Gilles et al., 1997; Ottaviano et al., 2006; Shields et al., 2011). Moreover, a recent report by Dangi-Garimella et al. (2011) shows that collagen induces MT1-MMP expression which in turn repress tumor suppressor let-7 family of micro RNA expression in pancreatic ductal adenocarcinoma tumor. Interestingly, the mechanism regulated by MT1-MMP in this study is dependent on TGFβ signaling and ERK1/2 activation, which is in accordance with our previous reports on vessel maturation and cell migration (Sounni et al., 2010a,b). In contrast, MT1-MMP may play an anti-tumor effect through the induction of cell aggregation and Rho-ROCK-Myosin signaling in squamous cell carcinoma or keratinocytes (Dangi-Garimella et al., 2010). The formation of cell aggregate by MT1-MMP is contradictory to it migration promoting effect widely attributed to this enzyme during the last decade. These controversial data require more investigation to determine whether MT1-MMP is a pro- or an anti-tumorigenic enzyme and, whether these effects are context dependent.
Role of MT4-MMP in Vessel Destabilization
The microvessel pericyte coverage in several human tumors and their colocalization with endothelial cell markers varied from 10 to 20% in human glioblastoma and renal carcinoma, to 70% in mammary and colon carcinoma (Eberhard et al., 2000). In tumor angiogenesis, pericytes contribute to the stabilization of nascent microvessels (Raza et al., 2010). They detach from the preexistent vessels and are recruited around the endothelial cell sprouts, leading to the reduction of endothelial cell proliferation and the formation of larger perfused microvessels (Gee et al., 2003). It is possible that tumor cells trigger metastasis through the alteration of pericyte-endothelial cell–cell interactions. The recent study by Xian et al. (2006) demonstrates that pericytes limit tumor cell metastasis, and the lack or dysfunction of pericytes within tumor vessels increases the metastatic potential of solid tumors. Indeed, pancreatic beta cell tumorigenesis induced in primary pericyte-deficient Pdgfb(ret/ret) mice results in increased tumor cell metastases in local lymph nodes and distant organs. In breast carcinoma, the correlation between the lack of pericyte coverage in tumor and the increase of metastatic spread has been attributed to MT4-MMP activity in vivo (Chabottaux et al., 2009). The overexpression of MT4-MMP in breast cancer MDA-MB-231 cells enhances subcutaneous tumor growth and stimulates the development of lung metastases in 100% of inoculated RAG-1 immunodeficient mice (Chabottaux et al., 2006). Intriguingly, functional studies failed to reveal an effect of MT4-MMP overexpression on in vitro cell migration and invasion, pro-MMP2 activation, VEGF production, and angiogenesis. The ultrastructural analysis of vessel pericyte coverage in tumor overexpressing MT4-MMP by electron microscopy revealed unique features of pericytes with irregular shape, extended cytoplasm, and poor association with endothelial cells (Chabottaux et al., 2009). A computer-assisted quantification confirmed a significant detachment of pericytes from vessels in MT4-MMP overexpressing tumors. The tumor leakiness and delivery of high molecular weight compounds were investigated by measuring the bio-distribution of fluorescent polymeric macromolecule of 250 kDa AngioSense 680 into tumor interstitium. MT4-MMP overexpression confers a significant increase in macromolecule leakage into tumors. Furthermore, while MT4-MMP has no effect on angiogenic factor expression such as VEGF, TIEs, or PDGFRs and their ligands, mRNA expression of the human anti-angiogenic factor thrombospondin-2 (TSP-2) is modulated in MT4-MMP overexpressing tumors. These data suggest that MT4-MMP expression at tumor cell surface affects vessel integrity and thereafter increases vascular leakage and metastatic cell intravasation. Taken together, MT4-MMP and MT1-MMP play a major role in pericyte migration and recruitment to the vessels (Figure 2). These distinct effects of MT-MMPs in vessel wall function and integrity is of great interest in delivery of therapeutics in vivo. These two molecules are able to modify the tumor physiology and to affect vessel permeability in vivo. At this end, understanding, the cellular and the molecular mechanisms of MT-MMPs and their interaction with ECM and non-ECM molecules, permeability, and IFP of tumor, will open new targeting opportunities to enhance vascular leakage and delivery of therapeutics to tissues where hemodynamics limit efficient drug delivery (Figure 3).
It is well known that vessel destabilization is one of series of events during the angiogenic process. In addition, immature blood vessels in human tumors have been reported to regress as a consequence of VEGF withdrawal (Benjamin et al., 1999). Kazlauskas works have highlighted a new concept of vascular regression and show that is not merely a default of apoptosis. Instead, it is a response that is intrinsic to the angiogenic program and is controlled by specific signaling enzymes (Im and Kazlauskas, 2006; Im et al., 2010). A subtle coordination change in tissue remodeling during angiogenesis and regression must occur during normal and pathological events. It is possible that vascular regression occurs as consequence of impaired vascular cell–ECM contact, and the role MT-MMPs may be important in this process. We cannot exclude that MT4-MMP induction of pericyte detachment from endothelial cells in tumor may drive the destabilization step of the angiogenic program (Chabottaux et al., 2009). More investigations are required to determine the exact role of MT4-MMP at all steps of the angiogenic process. MT1-MMP and MT4-MMP can be considered as angiomodulator which could control vessel maturation or regression. Therefore, vessel regression or maturation dependent-MT-MMPs would be potential diagnostic and/or therapeutic targets for vascular diseases and tumor angiogenesis.
Cryptic Sites in Vascular Basement Membrane
The vascular basement membrane is commonly considered as altered or incomplete in tumor associated blood vessels (Paku, 1998). Structural analysis and immunoreactivities for basement membrane components (collagen type IV, laminin, entactin/nidogen, and fibronectin) have been performed in various tumor mouse models including spontaneous RIP-Tag2 pancreatic islet tumors, MCa-IV mammary carcinomas, and Lewis lung carcinoma. These analyses revealed that basement membrane covers most tumor vessels, but it exhibits multiple abnormalities and incomplete association with endothelial cells and pericytes in these tumors (Baluk et al., 2003; Page-McCaw et al., 2007). Vessel basement membrane is considered as a reservoir for various growth factors, cytokines that are released by proteases during vascular response to physiological or pathological stimuli (Kessenbrock et al., 2010). In addition, some basement membrane cleavage products play a crucial role in vessel sprouting and vessel permeability.
Laminin is the major basement membrane ligand for epithelial, endothelial, and carcinoma cells. It supports cell attachment, migration, and invasion through its interaction with two major receptors, α1β1 and α6β4 integrins (Nishiuchi et al., 2006). Laminin also contains several functional domains that can regulate endothelial cell processes. For example, the RGD tripeptide containing domain within the α chain of laminin promotes cell adhesion through the binding to β1 integrin. Moreover, a second domain (YIGSR) within the β1 chain of laminin induces cell–cell interactions, regulates morphogenesis, and reorganizes endothelial cells into tube-like structures (Grant et al., 1989). This pentapeptide YIGSR sequence binds to endothelial cells via the non-integrin laminin receptor (LR; Basson et al., 1990). While many regulatory sequences of angiogenesis within laminin are readily available to cells, there are also relevant cryptic sequences that require proteolytic remodeling for exposure. For example, the proteolytic cleavage of the Laminin-5γ2 chain into several short γ2′ and γ2× chains by MT1-MMP and/or MMP2 promotes the formation of vasculogenic mimicry formed by tumor cells (Hendrix et al., 2003). In contrast, laminin α5 chain (component of laminin-10 and laminin-11) derived peptide (RLVSYNGIIFFLK) inhibits angiogenesis and metastasis through the disruption of FGF2 binding to the heparan sulfate side chain of CD44 (Hibino et al., 2004). Several peptides corresponding to regions of laminin-1 have been implicated in the regulation of angiogenesis and tumor metastasis (Malinda et al., 1999; Ponce and Kleinman, 2003). Akalu et al. (2007) have identified STQ peptide which recognize proteolysed or denatured laminin-1, but not intact laminin-1 or native and denatured laminin-5. STQ peptide inhibits endothelial and tumor cell adhesion, migration, proliferation in vitro and blocks angiogenesis, tumor growth, and metastasis in vivo. Abnormal synthesis of the endothelial cell basement membrane components could also contribute to vascular leakage as shown in diabetic retina. It remains to be explored whether the basement membrane laminin proteolysis and unmasking of cryptic sites, exert their effect on the maturation or destabilization of vascular walls in tumors (Oshitari et al., 2006).
During pathological angiogenesis such as in tumor angiogenesis and vascular diseases, specific cryptic type IV collagen epitopes are exposed. For instance, a cryptic site hidden within the 3D structure of type IV collagen is exposed by MMP2 and promotes tumor growth and angiogenesis in vivo (Xu et al., 2001). The recognition of a cryptic epitope (HU177) present exclusively in type IV collagen by a specific mouse antibody (HUIV26) inhibits endothelial cell proliferation and tubule-like structure formation in vitro and tumor angiogenesis in vivo (Cretu et al., 2007). The type IV collagen epitope (HU177) is present in ECM of tumor blood vessels but not in quiescent vessels. Its inhibition results in the expression of the cell cycle inhibitor the cyclin-dependent kinase (CDK) inhibitor p27kip1 in endothelial cells (Cretu et al., 2007). Interestingly, cryptic activities embedded within intact type IV collagen molecules are unmasked as a consequence of MT1-MMP, MT2-MMP, and MT3-MMP during morphogenesis (Rebustini et al., 2009). MT-MMPs activities release NC1 domains of type IV collagen and stimulate branching of cultured mouse submandibular gland (SMG) explants. In choroidal neovascularization (CNV), the angiogenic process associated with age-related macular degeneration, exposure of cryptic collagen type IV epitopes is associated with increased CNV incidence. The humanized antibody H8 directed against a cryptic collagen type IV epitope inhibits CNV progression (Gocheva et al., 2006). However, more investigations are needed for a better understanding of the role of MT-MMPs on unmasking cryptic sites within type IV collagen of vessel basement membrane and its implication in vascular permeability/leakage in cancer and vascular diseases.
Perlecan, an additional widely expressed vascular basement membrane component plays an important role in angiogenesis and promotes endothelial cell migration (Aviezer et al., 1994). However, endorepellin, a fragment generated by the processing of the perlecan C-terminal domain by MMPs (Gonzalez et al., 2005), inhibits angiogenesis through its interaction with α1β1 and triggers a signaling cascade that leads to the disruption of actin cytoskeleton in endothelial cells and ultimately to angiostasis (Iozzo, 2005). Endorepellin can promote platelet response to collagen, clot formation, and healing, through distinct interaction with α2β1 receptor integrin (Bix et al., 2007).
The vascular basement membrane in tumor or vascular pathologies is exposed to intensive remodeling by MMPs that can create a less restrictive microenvironment. MMPs release growth factors sequestered in the matrix or generate bioactive peptides that promote endothelial cell proliferation, motility, and new blood vessel growth. However, ECM proteolysis may release a number of endogenous angiogenesis inhibitors, including endostatin, tumstatin, and other fragments of ECM proteins that may have anti-angiogenic activity (Egeblad and Werb, 2002).
Conclusion
The ECM in vessel wall is no longer seen as a simple scaffold for vascular cells, but is now viewed as a dynamic actor in vascular cell signaling and physiology control. Deregulation of the balance in ECM synthesis and degradation contributes to vascular pathologies and tumor angiogenesis. Expression of MT-MMPs in tumor microenvironment results in blood vessel abnormalities and dysfunctions. These molecules could also affect vascular wall destabilization and dysfunctions in vascular disorders. MMPs and ECM like thrombospondin-1 modulate vessel integrity and progenitor cell recruitment in cardiovascular disease and ischemia (Silvestre et al., 2001; Favier et al., 2005; Smadja et al., 2011). Many similarities exist in the molecular and cellular alteration common to blood vessels in cancer and vascular diseases, such as arthrosclerosis, diabetic retinopathy, hypertension, and aneurism rupture. However, therapeutic benefits derived from ECM degrading enzyme inhibition may lead to vessel stabilization in cancer and hyperactive tissues. The complexity of targeting proteolytic enzymes due to their pro and anti-angiogenic function in cancer or vascular diseases (Lopez-Otin and Matrisian, 2007), renders targeting cryptic collagen sites a highly selective approach for regulating angiogenesis. Further investigations are required to understand how basement membrane molecules regulate vessel permeability and leakage in cancer and vascular diseases processes. Particularly, this may be beneficial if combined with other target pathway inhibitors or cytotoxic drugs for cancer treatment. An increased understanding of regulation and function of cryptic fragments within ECM and proteolytic peptides in the control of vascular cell physiology will likely lead to more effective strategies for therapy, which can be targeted specifically to tumor angiogenesis or vascular pathologies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the FP7-HEALTH-2007-A Proposal No. 201279 “MICROENVIMET,” the Fonds de la Recherche Scientifique – FNRS (F.R.S.-FNRS, Belgium), the Foundation against Cancer (foundation of public interest, Belgium), the Centre Anticancéreux près l’Université de Liège, the Fonds Léon Fredericq (University of Liège), the Direction Générale Opérationnelle de l’Economie, de l’Emploi et de la Recherche from the S.P.W. (Région Wallonne, Belgium), the Interuniversity Attraction Poles Programme – Belgian Science Policy (Brussels, Belgium).
References
Ahn, G. O., and Brown, J. M. (2008). Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell 13, 193–205.
Akalu, A., Roth, J. M., Caunt, M., Policarpio, D., Liebes, L., and Brooks, P. C. (2007). Inhibition of angiogenesis and tumor metastasis by targeting a matrix immobilized cryptic extracellular matrix epitope in laminin. Cancer Res. 67, 4353–4363.
Aviezer, D., Hecht, D., Safran, M., Eisinger, M., David, G., and Yayon, A. (1994). Perlecan, basal lamina proteoglycan, promotes basic fibroblast growth factor-receptor binding, mitogenesis, and angiogenesis. Cell 79, 1005–1013.
Baluk, P., Hashizume, H., and McDonald, D. M. (2005). Cellular abnormalities of blood vessels as targets in cancer. Curr. Opin. Genet. Dev. 15, 102–111.
Baluk, P., Morikawa, S., Haskell, A., Mancuso, M., and McDonald, D. M. (2003). Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 163, 1801–1815.
Basson, C. T., Knowles, W. J., Bell, L., Albelda, S. M., Castronovo, V., Liotta, L. A., and Madri, J. A. (1990). Spatiotemporal segregation of endothelial cell integrin and nonintegrin extracellular matrix-binding proteins during adhesion events. J. Cell Biol. 110, 789–801.
Benjamin, L. E., Golijanin, D., Itin, A., Pode, D., and Keshet, E. (1999). Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J. Clin. Invest. 103, 159–165.
Bergers, G., Brekken, R., McMahon, G., Vu, T. H., Itoh, T., Tamaki, K., Tanzawa, K., Thorpe, P., Itohara, S., Werb, Z., and Hanahan, D. (2000). Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2, 737–744.
Bix, G., Iozzo, R. A., Woodall, B., Burrows, M., McQuillan, A., Campbell, S., Fields, G. B., and Iozzo, R. V. (2007). Endorepellin, the C-terminal angiostatic module of perlecan, enhances collagen-platelet responses via the alpha2beta1-integrin receptor. Blood 109, 3745–3748.
Boudreau, N. J., and Jones, P. L. (1999). Extracellular matrix and integrin signalling: the shape of things to come. Biochem. J. 339(Pt 3), 481–488.
Brown, E., McKee, T., diTomaso, E., Pluen, A., Seed, B., Boucher, Y., and Jain, R. K. (2003). Dynamic imaging of collagen and its modulation in tumors in vivo using second-harmonic generation. Nat. Med. 9, 796–800.
Chabottaux, V., Ricaud, S., Host, L., Blacher, S., Paye, A., Thiry, M., Garofalakis, A., Pestourie, C., Gombert, K., Bruyere, F., Lewandowsky, D., Tavitian, B., Foidart, J. M., Duconge, F., and Noel, A. (2009). Membrane-type 4 matrix metalloproteinase (MT4-MMP) induces lung metastasis by alteration of primary breast tumour vascular architecture. J. Cell. Mol. Med. 13, 4002–4013.
Chabottaux, V., Sounni, N. E., Pennington, C. J., English, W. R., van den Brule, F., Blacher, S., Gilles, C., Munaut, C., Maquoi, E., Lopez-Otin, C., Murphy, G., Edwards, D. R., Foidart, J. M., and Noël, A. (2006). Membrane-type 4 matrix metalloproteinase promotes breast cancer growth and metastases. Cancer Res. 66, 5165–5172.
Chantrain, C., Shimada, H., Jodele, S., Groshen, S., Ye, W., Shalinsky, D., Werb, Z., Coussens, L. M., and DeClerck, Y. A. (2004). Stromal matrix metalloproteinase-9 regulates the vascular architecture in neuroblastoma by promoting pericyte recruitment. Cancer Res. 64, 1675–1686.
Chantrain, C. F., Henriet, P., Jodele, S., Emonard, H., Feron, O., Courtoy, P. J., DeClerck, Y. A., and Marbaix, E. (2006). Mechanisms of pericyte recruitment in tumour angiogenesis: a new role for metalloproteinases. Eur. J. Cancer 42, 310–318.
Chun, T. H., Sabeh, F., Ota, I., Murphy, H., McDonagh, K. T., Holmbeck, K., Birkedal-Hansen, H., Allen, E. D., and Weiss, S. J. (2004). MT1-MMP-dependent neovessel formation within the confines of the three-dimensional extracellular matrix. J. Cell Biol. 167, 757–767.
Collen, A., Hanemaaijer, R., Lupu, F., Quax, P. H., Van Lent, N., Grimbergen, J., Peters, E., Koolwijk, P., and Van Hinsbergh, V. W. (2003). Membrane-type matrix metalloproteinase-mediated angiogenesis in a fibrin-collagen matrix. Blood 101, 1810–1817.
Coussens, L. M., Tinkle, C. L., Hanahan, D., and Werb, Z. (2000). MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 103, 481–490.
Cretu, A., Roth, J. M., Caunt, M., Akalu, A., Policarpio, D., Formenti, S., Gagne, P., Liebes, L., and Brooks, P. C. (2007). Disruption of endothelial cell interactions with the novel HU177 cryptic collagen epitope inhibits angiogenesis. Clin. Cancer Res. 13, 3068–3078.
Dallas, S. L., Rosser, J. L., Mundy, G. R., and Bonewald, L. F. (2002). Proteolysis of latent transforming growth factor-beta (TGF-beta)- binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J. Biol. Chem. 277, 21352–21360.
Dangi-Garimella, S., Redig, A. J., Shields, M. A., Siddiqui, M. A., and Munshi, H. G. (2010). Rho-rock-myosin signaling mediates membrane type 1 matrix metalloproteinase-induced cellular aggregation of keratinocytes. J. Biol. Chem. 285, 28363–28372.
Dangi-Garimella, S., Strouch, M. J., Grippo, P. J., Bentrem, D. J., and Munshi, H. G. (2011). Collagen regulation of let-7 in pancreatic cancer involves TGF-beta1-mediated membrane type 1-matrix metalloproteinase expression. Oncogene 30, 1002–1008.
Davis, G. E., and Senger, D. R. (2008). Extracellular matrix mediates a molecular balance between vascular morphogenesis and regression. Curr. Opin. Hematol. 15, 197–203.
DeClerck, Y. A., Mercurio, A. M., Stack, M. S., Chapman, H. A., Zutter, M. M., Muschel, R. J., Raz, A., Matrisian, L. M., Sloane, B. F., Noel, A., Hendrix, M. J., Coussens, L., and Padarathsingh, M. (2004). Proteases, extracellular matrix, and cancer: a workshop of the path B study section. Am. J. Pathol. 164, 1131–1139.
Deng, Y. P., Li, W., Li, Y. L., Xu, H., Liang, S. S., and Zhang, L. H. (2009). MT1-MMP up-regulates VEGF expression in human breast carcinoma MCF-7 cells and induces tumor angiogenesis. Zhonghua Zhong Liu Za Zhi 31, 727–731.
Deryugina, E. I., Soroceanu, L., and Strongin, A. Y. (2002). Up-regulation of vascular endothelial growth factor by membrane-type 1 matrix metalloproteinase stimulates human glioma xenograft growth and angiogenesis. Cancer Res. 62, 580–588.
Eberhard, A., Kahlert, S., Goede, V., Hemmerlein, B., Plate, K. H., and Augustin, H. G. (2000). Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res. 60, 1388–1393.
Egeblad, M., Shen, H. C., Behonick, D. J., Wilmes, L., Eichten, A., Korets, L. V., Kheradmand, F., Werb, Z., and Coussens, L. M. (2007). Type I collagen is a genetic modifier of matrix metalloproteinase 2 in murine skeletal development. Dev. Dyn. 236, 1683–1693.
Egeblad, M., and Werb, Z. (2002). New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2, 161–174.
Eguchi, S., Dempsey, P. J., Frank, G. D., Motley, E. D., and Inagami, T. (2001). Activation of MAPKs by angiotensin II in vascular smooth muscle cells. Metalloprotease-dependent EGF receptor activation is required for activation of ERK and p38 MAPK but not for JNK. J. Biol. Chem. 276, 7957–7962.
Eisenach, P. A., Roghi, C., Fogarasi, M., Murphy, G., and English, W. R. (2010). MT1-MMP regulates VEGF-A expression through a complex with VEGFR-2 and Src. J. Cell Sci. 123, 4182–4193.
Favier, J., Germain, S., Emmerich, J., Corvol, P., and Gasc, J. M. (2005). Critical overexpression of thrombospondin 1 in chronic leg ischaemia. J. Pathol. 207, 358–366.
Feron, O. (2004). Targeting the tumor vascular compartment to improve conventional cancer therapy. Trends Pharmacol. Sci. 25, 536–542.
Findley, C. M., Cudmore, M. J., Ahmed, A., and Kontos, C. D. (2007). VEGF induces Tie2 shedding via a phosphoinositide 3-kinase/Akt dependent pathway to modulate Tie2 signaling. Arterioscler. Thromb. Vasc. Biol. 27, 2619–2626.
Gee, M. S., Procopio, W. N., Makonnen, S., Feldman, M. D., Yeilding, N. M., and Lee, W. M. (2003). Tumor vessel development and maturation impose limits on the effectiveness of anti-vascular therapy. Am. J. Pathol. 162, 183–193.
Gerhardt, H., and Semb, H. (2008). Pericytes: gatekeepers in tumour cell metastasis? J. Mol. Med. 86, 135–144.
Gilles, C., Polette, M., Seiki, M., Birembaut, P., and Thompson, E. W. (1997). Implication of collagen type I-induced membrane-type 1-matrix metalloproteinase expression and matrix metalloproteinase-2 activation in the metastatic progression of breast carcinoma. Lab. Invest. 76, 651–660.
Gocheva, V., Zeng, W., Ke, D., Klimstra, D., Reinheckel, T., Peters, C., Hanahan, D., and Joyce, J. A. (2006). Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 20, 543–556.
Gonzalez, E. M., Reed, C. C., Bix, G., Fu, J., Zhang, Y., Gopalakrishnan, B., Greenspan, D. S., and Iozzo, R. V. (2005). BMP-1/tolloid-like metalloproteases process endorepellin, the angiostatic C-terminal fragment of perlecan. J. Biol. Chem. 280, 7080–7087.
Grant, D. S., Tashiro, K., Segui-Real, B., Yamada, Y., Martin, G. R., and Kleinman, H. K. (1989). Two different laminin domains mediate the differentiation of human endothelial cells into capillary-like structures in vitro. Cell 58, 933–943.
Hawinkels, L. J., Kuiper, P., Wiercinska, E., Verspaget, H. W., Liu, Z., Pardali, E., Sier, C. F., and ten Dijke, P. (2010). Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 70, 4141–4150.
Heissig, B., Hattori, K., Dias, S., Friedrich, M., Ferris, B., Hackett, N. R., Crystal, R. G., Besmer, P., Lyden, D., Moore, M. A., Werb, Z., and Rafii, S. (2002). Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell 109, 625–637.
Heldin, C. H., Rubin, K., Pietras, K., and Ostman, A. (2004). High interstitial fluid pressure – an obstacle in cancer therapy. Nat. Rev. Cancer 4, 806–813.
Hendrix, M. J., Seftor, E. A., Hess, A., and Seftor, R. E. (2003). Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat. Rev. Cancer 3, 411–421.
Hibino, S., Shibuya, M., Engbring, J. A., Mochizuki, M., Nomizu, M., and Kleinman, H. K. (2004). Identification of an active site on the laminin alpha5 chain globular domain that binds to CD44 and inhibits malignancy. Cancer Res. 64, 4810–4816.
Hiraoka, N., Allen, E., Apel, I. J., Gyetko, M. R., and Weiss, S. J. (1998). Matrix metalloproteinases regulate neovascularization by acting as pericellular fibrinolysins. Cell 95, 365–377.
Holmbeck, K., Bianco, P., Caterina, J., Yamada, S., Kromer, M., Kuznetsov, S. A., Mankani, M., Robey, P. G., Poole, A. R., Pidoux, I., Ward, J. M., and Birkedal-Hansen, H. (1999). MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell 99, 81–92.
Hotary, K. B., Allen, E. D., Brooks, P. C., Datta, N. S., Long, M. W., and Weiss, S. J. (2003). Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell 114, 33–45.
Hotary, K. B., Yana, I., Sabeh, F., Li, X. Y., Holmbeck, K., Birkedal-Hansen, H., Allen, E. D., Hiraoka, N., and Weiss, S. J. (2002). Matrix metalloproteinases (MMPs) regulate fibrin-invasive activity via MT1-MMP-dependent and -independent processes. J. Exp. Med. 195, 295–308.
Huang, S., Van Arsdall, M., Tedjarati, S., McCarty, M., Wu, W., Langley, R., and Fidler, I. J. (2002). Contributions of stromal metalloproteinase-9 to angiogenesis and growth of human ovarian carcinoma in mice. J. Natl. Cancer Inst. 94, 1134–1142.
Im, E., and Kazlauskas, A. (2006). New insights regarding vessel regression. Cell Cycle 5, 2057–2059.
Im, E., Motiejunaite, R., Aranda, J., Park, E. Y., Federico, L., Kim, T. I., Clair, T., Stracke, M. L., Smyth, S., and Kazlauskas, A. (2010). Phospholipase Cgamma activation drives increased production of autotaxin in endothelial cells and lysophosphatidic acid-dependent regression. Mol. Cell. Biol. 30, 2401–2410.
Imai, K., Hiramatsu, A., Fukushima, D., Pierschbacher, M. D., and Okada, Y. (1997). Degradation of decorin by matrix metalloproteinases: identification of the cleavage sites, kinetic analyses and transforming growth factor-beta1 release. Biochem. J. 322, 809–814.
Iozzo, R. V. (2005). Basement membrane proteoglycans: from cellar to ceiling. Nat. Rev. Mol. Cell Biol. 6, 646–656.
Jain, R. K. (2005). Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307, 58–62.
Kalluri, R. (2003). Basement membranes: structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 3, 422–433.
Kamioka, M., Ishibashi, T., Ohkawara, H., Eguchi, R., Sugimoto, K., Uekita, H., Matsui, T., Yamagishi, S. I., Ando, K., Sakamoto, T., Sakamoto, N., Takuwa, Y., Wada, I., Shiomi, M., Maruyama, Y., and Takeishi, Y. (2011). Involvement of membrane type 1-matrix metalloproteinase (MT1-MMP) in rage activation signaling pathways. J. Cell. Physiol. 226, 1554–1563.
Kessenbrock, K., Plaks, V., and Werb, Z. (2010). Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67.
Labrecque, L., Nyalendo, C., Langlois, S., Durocher, Y., Roghi, C., Murphy, G., Gingras, D., and Beliveau, R. (2004). Src-mediated tyrosine phosphorylation of caveolin-1 induces its association with membrane type 1 matrix metalloproteinase. J. Biol. Chem. 279, 52132–52140.
Lafleur, M. A., Handsley, M. M., and Edwards, D. R. (2003). Metalloproteinases and their inhibitors in angiogenesis. Expert Rev. Mol. Med. 5, 1–39.
Langlois, S., Gingras, D., and Beliveau, R. (2004). Membrane type 1-matrix metalloproteinase (MT1-MMP) cooperates with sphingosine 1-phosphate to induce endothelial cell migration and morphogenic differentiation. Blood 103, 3020–3028.
Lehti, K., Allen, E., Birkedal-Hansen, H., Holmbeck, K., Miyake, Y., Chun, T. H., and Weiss, S. J. (2005). An MT1-MMP-PDGF receptor-beta axis regulates mural cell investment of the microvasculature. Genes Dev. 19, 979–991.
Lehti, K., Rose, N. F., Valavaara, S., Weiss, S. J., and Keski-Oja, J. (2009). MT1-MMP promotes vascular smooth muscle dedifferentiation through LRP1 processing. J. Cell Sci. 122, 126–135.
Levental, K. R., Yu, H., Kass, L., Lakins, J. N., Egeblad, M., Erler, J. T., Fong, S. F., Csiszar, K., Giaccia, A., Weninger, W., Yamauchi, M., Gasser, D. L., and Weaver, V. M. (2009). Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906.
Lindahl, P., Johansson, B. R., Leveen, P., and Betsholtz, C. (1997). Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 277, 242–245.
Liu, X., Wu, H., Byrne, M., Jeffrey, J., Krane, S., and Jaenisch, R. (1995). A targeted mutation at the known collagenase cleavage site in mouse type I collagen impairs tissue remodeling. J. Cell Biol. 130, 227–237.
Lopez-Otin, C., and Matrisian, L. M. (2007). Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 7, 800–808.
Lucchesi, P. A., Sabri, A., Belmadani, S., and Matrougui, K. (2004). Involvement of metalloproteinases 2/9 in epidermal growth factor receptor transactivation in pressure-induced myogenic tone in mouse mesenteric resistance arteries. Circulation 110, 3587–3593.
Lyden, D., Hattori, K., Dias, S., Costa, C., Blaikie, P., Butros, L., Chadburn, A., Heissig, B., Marks, W., Witte, L., Wu, Y., Hicklin, D., Zhu, Z., Hackett, N. R., Crystal, R. G., Moore, M. A., Hajjar, K. A., Manova, K., Benezra, R., and Rafii, S. (2001). Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat. Med. 7, 1194–1201.
Magzoub, M., Jin, S., and Verkman, A. S. (2008). Enhanced macromolecule diffusion deep in tumors after enzymatic digestion of extracellular matrix collagen and its associated proteoglycan decorin. FASEB J. 22, 276–284.
Malinda, K. M., Nomizu, M., Chung, M., Delgado, M., Kuratomi, Y., Yamada, Y., Kleinman, H. K., and Ponce, M. L. (1999). Identification of laminin alpha1 and beta1 chain peptides active for endothelial cell adhesion, tube formation, and aortic sprouting. FASEB J. 13, 53–62.
Miles, A. A., and Miles, E. M. (1952). Vascular reactions to histamine, histamine-liberator and leukotaxine in the skin of guinea pigs. J. Physiol. 118, 228–257.
Mimura, T., Han, K. Y., Onguchi, T., Chang, J. H., Kim, T. I., Kojima, T., Zhou, Z., and Azar, D. T. (2009). MT1-MMP-mediated cleavage of decorin in corneal angiogenesis. J. Vasc. Res. 46, 541–550.
Montesano, R., Mouron, P., Amherdt, M., and Orci, L. (1983). Collagen matrix promotes reorganization of pancreatic endocrine cell monolayers into islet-like organoids. J. Cell Biol. 97, 935–939.
Netti, P. A., Berk, D. A., Swartz, M. A., Grodzinsky, A. J., and Jain, R. K. (2000). Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 60, 2497–2503.
Nishiuchi, R., Takagi, J., Hayashi, M., Ido, H., Yagi, Y., Sanzen, N., Tsuji, T., Yamada, M., and Sekiguchi, K. (2006). Ligand-binding specificities of laminin-binding integrins: a comprehensive survey of laminin-integrin interactions using recombinant alpha3beta1, alpha6beta1, alpha7beta1 and alpha6beta4 integrins. Matrix Biol. 25, 189–197.
Noel, A., Jost, M., and Maquoi, E. (2008). Matrix metalloproteinases at cancer tumor-host interface. Semin. Cell Dev. Biol. 19, 52–60.
Ohuchi, E., Imai, K., Fujii, Y., Sato, H., Seiki, M., and Okada, Y. (1997). Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J. Biol. Chem. 272, 2446–2451.
Onimaru, M., Yonemitsu, Y., Suzuki, H., Fujii, T., and Sueishi, K. (2010). An autocrine linkage between matrix metalloproteinase-14 and Tie-2 via ectodomain shedding modulates angiopoietin-1-dependent function in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 30, 818–826.
Oshitari, T., Polewski, P., Chadda, M., Li, A. F., Sato, T., and Roy, S. (2006). Effect of combined antisense oligonucleotides against high-glucose- and diabetes-induced overexpression of extracellular matrix components and increased vascular permeability. Diabetes 55, 86–92.
Ottaviano, A. J., Sun, L., Ananthanarayanan, V., and Munshi, H. G. (2006). Extracellular matrix-mediated membrane-type 1 matrix metalloproteinase expression in pancreatic ductal cells is regulated by transforming growth factor-beta1. Cancer Res. 66, 7032–7040.
Ouyang, M., Huang, H., Shaner, N. C., Remacle, A. G., Shiryaev, S. A., Strongin, A. Y., Tsien, R. Y., and Wang, Y. (2010). Simultaneous visualization of protumorigenic Src and MT1-MMP activities with fluorescence resonance energy transfer. Cancer Res. 70, 2204–2212.
Page-McCaw, A., Ewald, A. J., and Werb, Z. (2007). Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 8, 221–233.
Park, J. S., Seo, J., Kim, Y. O., Lee, H. S., and Jo, I. (2009). Coordinated regulation of angiopoietin-1 and vascular endothelial growth factor by arsenite in human brain microvascular pericytes: implications of arsenite-induced vascular dysfunction. Toxicology 264, 26–31.
Ponce, M. L., and Kleinman, H. K. (2003). Identification of redundant angiogenic sites in laminin alpha1 and gamma1 chains. Exp. Cell Res. 285, 189–195.
Purhonen, S., Palm, J., Rossi, D., Kaskenpaa, N., Rajantie, I., Yla-Herttuala, S., Alitalo, K., Weissman, I. L., and Salven, P. (2008). Bone marrow-derived circulating endothelial precursors do not contribute to vascular endothelium and are not needed for tumor growth. Proc. Natl. Acad. Sci. U.S.A. 105, 6620–6625.
Raza, A., Franklin, M. J., and Dudek, A. Z. (2010). Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am. J. Hematol. 85, 593–598.
Rebustini, I. T., Myers, C., Lassiter, K. S., Surmak, A., Szabova, L., Holmbeck, K., Pedchenko, V., Hudson, B. G., and Hoffman, M. P. (2009). MT2-MMP-dependent release of collagen IV NC1 domains regulates submandibular gland branching morphogenesis. Dev. Cell 17, 482–493.
Rutkowski, J. M., Moya, M., Johannes, J., Goldman, J., and Swartz, M. A. (2006). Secondary lymphedema in the mouse tail: lymphatic hyperplasia, VEGF-C upregulation, and the protective role of MMP-9. Microvasc. Res. 72, 161–171.
Sabbota, A. L., Kim, H. R., Zhe, X., Fridman, R., Bonfil, R. D., and Cher, M. L. (2010). Shedding of RANKL by tumor-associated MT1-MMP activates Src-dependent prostate cancer cell migration. Cancer Res. 70, 5558–5566.
Sato, Y. (1995). Activation of latent TGF-beta at the vascular wall-roles of endothelial cells and mural pericytes or smooth muscle cells. J. Atheroscler. Thromb. 2, 24–29.
Senger, D. R., Perruzzi, C. A., Streit, M., Koteliansky, V. E., de Fougerolles, A. R., and Detmar, M. (2002). The alpha(1)beta(1) and alpha(2)beta(1) integrins provide critical support for vascular endothelial growth factor signaling, endothelial cell migration, and tumor angiogenesis. Am. J. Pathol. 160, 195–204.
Shields, M. A., Dangi-Garimella, S., Krantz, S. B., Bentrem, D. J., and Munshi, H. G. (2011). Pancreatic cancer cells respond to type i collagen by inducing snail expression to promote membrane type 1 matrix metalloproteinase-dependent collagen invasion. J. Biol. Chem. 286, 10495–10504.
Shirvaikar, N., Marquez-Curtis, L. A., Ratajczak, M. Z., and Janowska-Wieczorek, A. (2011). Hyaluronic acid and thrombin upregulate MT1-MMP through PI3K and Rac-1 signaling and prime the homing-related responses of cord blood hematopoietic stem/progenitor cells. Stem Cells Dev. 20, 19–30.
Shirvaikar, N., Marquez-Curtis, L. A., Shaw, A. R., Turner, A. R., and Janowska-Wieczorek, A. (2010). MT1-MMP association with membrane lipid rafts facilitates G-CSF-induced hematopoietic stem/progenitor cell mobilization. Exp. Hematol. 38, 823–835.
Shoulders, M. D., and Raines, R. T. (2009). Collagen structure and stability. Annu. Rev. Biochem. 78, 929–958.
Silvestre, J. S., Mallat, Z., Tamarat, R., Duriez, M., Tedgui, A., and Levy, B. I. (2001). Regulation of matrix metalloproteinase activity in ischemic tissue by interleukin-10: role in ischemia-induced angiogenesis. Circ. Res. 89, 259–264.
Smadja, D. M., d’Audigier, C., Bieche, I., Evrard, S., Mauge, L., Dias, J. V., Labreuche, J., Laurendeau, I., Marsac, B., Dizier, B., Wagner-Ballon, O., Boisson-Vidal, C., Morandi, V., Duong-Van-Huyen, J. P., Bruneval, P., Dignat-George, F., Emmerich, J., and Gaussem, P. (2011). Thrombospondin-1 is a plasmatic marker of peripheral arterial disease that modulates endothelial progenitor cell angiogenic properties. Arterioscler. Thromb. Vasc. Biol. 31, 551–559.
Sohail, A., Sun, Q., Zhao, H., Bernardo, M. M., Cho, J. A., and Fridman, R. (2008). MT4-(MMP17) and MT6-MMP (MMP25), a unique set of membrane-anchored matrix metalloproteinases: properties and expression in cancer. Cancer Metastasis Rev. 27, 289–302.
Sounni, N. E., Dehne, K., van Kempen, L., Egeblad, M., Affara, N. I., Cuevas, I., Wiesen, J., Junankar, S., Korets, L., Lee, J., Shen, J., Morrison, C. J., Overall, C. M., Krane, S. M., Werb, Z., Boudreau, N., and Coussens, L. M. (2010a). Stromal regulation of vessel stability by MMP14 and TGFbeta. Dis. Model Mech. 3, 317–332.
Sounni, N. E., Rozanov, D. V., Remacle, A. G., Golubkov, V. S., Noel, A., and Strongin, A. Y. (2010b). Timp-2 binding with cellular MT1-MMP stimulates invasion-promoting MEK/ERK signaling in cancer cells. Int. J. Cancer 126, 1067–1078.
Sounni, N. E., Devy, L., Hajitou, A., Frankenne, F., Munaut, C., Gilles, C., Deroanne, C., Thompson, E. W., Foidart, J. M., and Noel, A. (2002). MT1-MMP expression promotes tumor growth and angiogenesis through an up-regulation of vascular endothelial growth factor expression. FASEB J. 16, 555–564.
Sounni, N. E., Janssen, M., Foidart, J. M., and Noel, A. (2003). Membrane type-1 matrix metalloproteinase and TIMP-2 in tumor angiogenesis. Matrix Biol. 22, 55–61.
Sounni, N. E., and Noel, A. (2005). Membrane type-matrix metalloproteinases and tumor progression. Biochimie 87, 329–342.
Sounni, N. E., Roghi, C., Chabottaux, V., Janssen, M., Munaut, C., Maquoi, E., Galvez, B. G., Gilles, C., Frankenne, F., Murphy, G., Foidart, J. M., and Noel, A. (2004). Up-regulation of vascular endothelial growth factor-A by active membrane-type 1 matrix metalloproteinase through activation of Src-tyrosine kinases. J. Biol. Chem. 279, 13564–13574.
Spurbeck, W. W., Ng, C. Y., Strom, T. S., Vanin, E. F., and Davidoff, A. M. (2002). Enforced expression of tissue inhibitor of matrix metalloproteinase-3 affects functional capillary morphogenesis and inhibits tumor growth in a murine tumor model. Blood 100, 3361–3368.
Stratman, A. N., Saunders, W. B., Sacharidou, A., Koh, W., Fisher, K. E., Zawieja, D. C., Davis, M. J., and Davis, G. E. (2009). Endothelial cell lumen and vascular guidance tunnel formation requires MT1-MMP-dependent proteolysis in 3-dimensional collagen matrices. Blood 114, 237–247.
Suzuki, M., Raab, G., Moses, M. A., Fernandez, C. A., and Klagsbrun, M. (1997). Matrix metalloproteinase-3 releases active heparin-binding EGF-like growth factor by cleavage at a specific juxtamembrane site. J. Biol. Chem. 272, 31730–31737.
Takino, T., Tsuge, H., Ozawa, T., and Sato, H. (2010). MT1-MMP promotes cell growth and ERK activation through c-Src and paxillin in three-dimensional collagen matrix. Biochem. Biophys. Res. Commun. 396, 1042–1047.
Tam, E. M., Moore, T. R., Butler, G. S., and Overall, C. M. (2004). Characterization of the distinct collagen binding, helicase and cleavage mechanisms of matrix metalloproteinase 2 and 14 (gelatinase A and MT1-MMP): the differential roles of the MMP hemopexin c domains and the MMP-2 fibronectin type II modules in collagen triple helicase activities. J. Biol. Chem. 279, 43336–43344.
Tatti, O., Vehvilainen, P., Lehti, K., and Keski-Oja, J. (2008). MT1-MMP releases latent TGF-beta1 from endothelial cell extracellular matrix via proteolytic processing of LTBP-1. Exp. Cell Res. 314, 2501–2514.
Vagima, Y., Avigdor, A., Goichberg, P., Shivtiel, S., Tesio, M., Kalinkovich, A., Golan, K., Dar, A., Kollet, O., Petit, I., Perl, O., Rosenthal, E., Resnick, I., Hardan, I., Gellman, Y. N., Naor, D., Nagler, A., and Lapidot, T. (2009). MT1-MMP and RECK are involved in human CD34+ progenitor cell retention, egress, and mobilization. J. Clin. Invest. 119, 492–503.
Wiig, H., Gyenge, C., Iversen, P. O., Gullberg, D., and Tenstad, O. (2008). The role of the extracellular matrix in tissue distribution of macromolecules in normal and pathological tissues: potential therapeutic consequences. Microcirculation 15, 283–296.
Wiig, H., Tenstad, O., Iversen, P. O., Kalluri, R., and Bjerkvig, R. (2010). Interstitial fluid: the overlooked component of the tumor microenvironment? Fibrogenesis Tissue Repair 3, 12.
Xian, X., Hakansson, J., Stahlberg, A., Lindblom, P., Betsholtz, C., Gerhardt, H., and Semb, H. (2006). Pericytes limit tumor cell metastasis. J. Clin. Invest. 116, 642–651.
Xu, J., Rodriguez, D., Petitclerc, E., Kim, J. J., Hangai, M., Moon, Y. S., Davis, G. E., Brooks, P. C., and Yuen, S. M. (2001). Proteolytic exposure of a cryptic site within collagen type IV is required for angiogenesis and tumor growth in vivo. J. Cell Biol. 154, 1069–1079.
Xu, Y., Gurusiddappa, S., Rich, R. L., Owens, R. T., Keene, D. R., Mayne, R., Hook, A., and Hook, M. (2000). Multiple binding sites in collagen type I for the integrins alpha1beta1 and alpha2beta1. J. Biol. Chem. 275, 38981–38989.
Yana, I., Sagara, H., Takaki, S., Takatsu, K., Nakamura, K., Nakao, K., Katsuki, M., Taniguchi, S., Aoki, T., Sato, H., Weiss, S. J., and Seiki, M. (2007). Crosstalk between neovessels and mural cells directs the site-specific expression of MT1-MMP to endothelial tip cells. J. Cell. Sci. 120, 1607–1614.
Yoon, C. H., Hur, J., Park, K. W., Kim, J. H., Lee, C. S., Oh, I. Y., Kim, T. Y., Cho, H. J., Kang, H. J., Chae, I. H., Yang, H. K., Oh, B. H., Park, Y. B., and Kim, H. S. (2005). Synergistic neovascularization by mixed transplantation of early endothelial progenitor cells and late outgrowth endothelial cells: the role of angiogenic cytokines and matrix metalloproteinases. Circulation 112, 1618–1627.
Yu, W. H., Woessner, J. F. Jr., McNeish, J. D., and Stamenkovic, I. (2002). CD44 anchors the assembly of matrilysin/MMP-7 with heparin-binding epidermal growth factor precursor and ErbB4 and regulates female reproductive organ remodeling. Genes Dev. 16, 307–323.
Zhou, Z., Apte, S. S., Soininen, R., Cao, R., Baaklini, G. Y., Rauser, R. W., Wang, J., Cao, Y., and Tryggvason, K. (2000). Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc. Natl. Acad. Sci. U.S.A. 97, 4052–4057.
Keywords: MT-MMPs, MMPs, collagen, vessel permeability, interstitial fluid pressure
Citation: Sounni NE, Paye A, Host L and Noël A (2011) MT-MMPS as regulators of vessel stability associated with angiogenesis. Front. Pharmacol. 2:111. doi: 10.3389/fphar.2011.00111
Received: 17 February 2011; Paper pending published: 18 March 2011;
Accepted: 27 April 2011; Published online: 13 May 2011.
Edited by:
Annarosa Arcangeli, University of Florence, ItalyReviewed by:
David Smadja, Paris Descartes University, FranceJulien Verrax, Université Catholique de Louvain, Belgium
Copyright: © 2011 Sounni, Paye, Host and Noël. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Agnès Noël and Nor Eddine Sounni, Laboratory of Tumor and Developmental Biology, University of Liège, Tour de Pathologie (B23), Sart-Tilman, B-4000 Liège, Belgium. e-mail:YWduZXMubm9lbEB1bGcuYWMuYmU=;bmVzb3VubmlAdWxnLmFjLmJl