 Milena De Nicola
Milena De Nicola Lina Ghibelli
Lina Ghibelli- 1Dipartimento di Biologia, Universita' di Roma Tor Vergata, Roma, Italy
- 2Dipartimento di Scienze e Tecnologie Chimiche, Universita' di Roma Tor Vergata, Roma, Italy
GSH Extrusion in Apoptosis
Damaged cells opting for apoptosis actively extrude glutathione in the reduced form as an apical event in the apoptotic signaling (Ghibelli et al., 1995; Van den Dobbelsteen et al., 1996). Apoptotic GSH efflux is very rapid and, as a consequence, cells undergo redox imbalance and become susceptible to oxidative stress due to loss of ROS scavenging power (D'Alessio et al., 2003); moreover, the sudden redox disequilibrium causes the formation of inter-protein disulfides among reactive exposed cysteines, altering cell functions and signaling in a pro-apoptotic fashion (Circu and Aw, 2012).
GSH Efflux in the Life and Death of a Cell
Glutathione is synthesized in the cell cytosol, and acts essentially as an intracellular antioxidant. However, viable cells (mainly hepatocytes and macrophages) extrude GSH as part of their physiological functions; this provides antioxidant protection to the extracellular environment (Bachhawat et al., 2013). The passage of GSH through plasma membrane is regulated by a switch in the open/closed configuration of the transporters, and occurs according to gradient, being in fact uni-directional (i.e., export rather than import) because intracellular levels are much higher than those of body fluids. GSH transporters were originally functionally identified as belonging to the sinusoidal or canalicular type according to their position in the hepatic anatomy, and to their responsiveness to specific inhibitors (Yi et al., 1995). However, their molecular nature is still elusive (Bachhawat et al., 2013). Based on several experimental evidences, the prevalent current opinion is that GSH transporters coincide with the multi-drug resistance associated proteins (MRP) (Ballatori et al., 2009; Franco and Cidlowski, 2012), which constitute the complexes responsible for cell detoxification from xenobiotics through the export of glutathione-S conjugates produced by glutathione-S-transferase (Pompella et al., 2003). The regulation of the activity of GSH transporters is still unexplored, possibly being controlled by the differential concentration of GSH in the vicinity of the carriers in the internal vs. external face of the cell membrane, thereby relying in the zonal control of GSH levels by intracellular trafficking.
A different scenario applies to the export of the oxidized glutathione dimers (GSSG) formed during oxidative stress (and not reduced back to GSH by glutathione reductase). Even though GSSG was shown to be target of MRP (Ballatori et al., 2009), in conditions of oxidative stress GSSG exits from cells passively crossing the plasma membrane. This mechanism helps avoiding a dangerous drop in the GSH/GSSG ratio due to the accumulation of GSSG, and the consequent redox imbalance.
Glutathione loss in apoptosis has been subject of intense investigation. Most apoptotic cells become devoid of glutathione even before rupture of the plasma membrane (Ghibelli et al., 1995), which indicates an active phenomenon of extrusion. In instances of apoptosis induced by oxidative insults, a passive efflux of GSSG was observed (Esteve et al., 1999); in this case, GSH loss is a consequence of oxidative stress. In instances of apoptosis induced by non-oxidative insults, glutathione is extruded outside cells in the reduced form immediately prior to the execution of apoptosis (Ghibelli et al., 1995; Van den Dobbelsteen et al., 1996), followed by ROS production as a consequence (D'Alessio et al., 2003); in this case, GSH loss is the cause, rather than the consequence of oxidative stress.
Apoptotic GSH extrusion is prevented by inhibitors of the sinusoidal GSH transporters (Ghibelli et al., 1998), indicating that apoptotic GSH efflux follows the same route as physiological GSH export. However, while healthy cells shed only a limited fraction of their GSH, in apoptosis the efflux leads to complete GSH depletion, suggesting that the transporters remain stably in an open configuration. The events that cause this phenomenon are still unknown; we have preliminary evidence that the activation of the apoptotic protease caspase-2, but not the other caspases, is required for apoptotic GSH efflux, suggesting that a proteolytic event (the digestion of an inhibitor of the transporters?), might cause an irreversible opening of the GSH transporters (De Nicola et al., in preparation).
Role of Apoptotic GSH Extrusion
GSH extrusion is a necessary step of the apoptotic signal transduction (Coppola and Ghibelli, 2000), occurring as an apical and causative event, as shown by studies demonstrating that inhibitors of GSH efflux also inhibit apoptosis (Ghibelli et al., 1998). The block of the apoptotic signaling occurs as a very early step, because even apical events such as Bax translocation are prevented. Antioxidants do not hamper apoptotic GSH efflux, but protect GSH-depleted cells from apoptosis (Liuzzi et al., 2003), indicating that the sudden GSH drop activates a redox-sensitive target among the apical pro-apoptotic proteins. A likely target is Bax, a pro-apoptotic member of the Bcl-2 family that in apoptosis moves to mitochondria, where it is responsible for the release of cytochrome c and the consequent caspase activation and cell demise (Ghibelli and Diederich, 2010). Bax has two exposed cysteines (Suzuki et al., 2000) able to form inter-protein disulfide bridges upon oxidation (D'Alessio et al., 2005), and therefore it can be at the same time a sensor of oxidation and an effector of apoptosis. Indeed, Bax was experimentally shown to assemble in an oxidative dimer configuration, putatively forming a disulfide between cysteine 62 and cysteine 126, thereby acquiring the ability to translocate to mitochondria (D'Alessio et al., 2005) and promote cytochrome c release (Ghibelli et al., 1999).
Damage-induced apoptosis was shown to follow either redox-sensitive or -insensitive routes, characterized by GSH depletion or retention, respectively, through alternative biochemical routes that can be morphologically identified (De Nicola et al., 2006). Leukemic cells display multiple nuclear apoptotic morphologies; in particular, two types of nuclear vesiculation (budding and cleavage) are the result of two independent morphological routes, since they never interconvert and are differently modulated by damage vs. physiological apoptogenic agents (Dini et al., 1996). In situ analyses showed that intracellular GSH is completely extruded in cells undergoing apoptosis by cleavage, whereas it is fully maintained in apoptotic budding cells (De Nicola et al., 2006). Accordingly, in situ measurement of ROS levels showed that only cells in cleavage develop oxidative stress (Celardo et al., 2011). GSH depletion, and the consequent oxidation, is not only concomitant to, but is the determinant of the cleavage route, since inhibition of the sinusoidal GSH transporters completely prevents the appearance of the apoptotic cells displaying the budding morphology (De Nicola et al., 2006).
Pro-Survival Pathways Induced by GSH Depletion
GSH, as a co-factor of the glutathione-S-transferase set of enzymes, plays a major role in the detoxification of cells from exogenous molecules, including pharmaceutical drugs. The depletion of GSH, achieved in clinics with the specific GSH synthesis inhibitor buthionine sulphoxymine (BSO), is an important strategy to improve drug efficacy in anticancer therapies, since many aggressive tumor cells are very efficient in drug extrusion (multi-drug resistant, MDR), thereby nullifying the effects of chemotherapies. In these cases BSO acts a chemosensitizer, since GSH depletion allows bypassing the MDR obstacle, achieving efficacious levels of antitumor drug within MDR cells (Fojo and Bates, 2003).
BSO is a specific inhibitor of glutathione synthase, thereby in BSO-treated cells the GSH pool is consumed but not replaced, and its level declines according to the consumption rate, with a kinetics that depends on the oxidative status of the cells. In any case, the depletion is much slower compared with what occurs during apoptosis (hours vs. minutes). This slow rate gives time to the BSO-treated cells to adapt to the progressively oxidizing environment, setting up stress response pathways aimed at increasing cell survival (Rahman et al., 2005; Ishii and Mann, 2014). Such responses can be very robust, and in non-MDR tumor cells BSO, rather than acting as a chemo-sensitizer, paradoxically potentiates cell survival, thereby decreasing the pro-apoptotic efficiency of anti-tumor drugs (D'Alessio et al., 2004).
The survival responses to GSH depletion include the transcriptional activation of genes encoding for cell protective proteins, via the nuclear factor kappaB (NF-kappaB) (Filomeni et al., 2005) or the NF-E2-related factor 2 (Nrf-2) (Lee et al., 2008) transcriptional activators.
NF-kappaB is a transcription complex that in response to many insults, including oxidative stress, mediates the transactivation of key cell protective proteins (Morgan and Liu, 2011). BSO-mediated activation of NF-kappaB leads to multiple cell-protective responses, such as the trans-activation of heat shock protein 27, which stops the apoptotic signal after Bax translocation and cytochrome c release (Filomeni et al., 2005), and the main anti-apoptotic protein Bcl-2. Bcl-2 is over-expressed in BSO-treated cells via transcriptional activation, both in tumor cells (D'Alessio et al., 2004) and primary lymphocytes (Cristofanon et al., 2006) and monocytes (Cristofanon et al., 2008). The increased Bcl-2 synthesis compensates for, and overcome, the faster catabolism of the Bcl-2 protein occurring in cells with low GSH levels (Meredith et al., 1998; D'Alessio et al., 2004), increasing the intracellular Bcl-2 levels up to three-four folds (D'Alessio et al., 2004). This is achieved through the activation of the so-called non-canonical NF-kappaB pathway by a two-step process. First, as a fast response to GSH depletion, a ternary disulfide complex forms between two subunits of the NF-kappaB component p50 and its tutoring protein Bcl-3 (Cristofanon et al., 2009). Second, the complex is then activated via a mechanism dependent on the mitogen-activated protein p38, a kinase that requires high ROS levels (Filomeni et al., 2003); this occurs as a consequence of GSH depletion only several hours later (Limón-Pacheco et al., 2007; Cristofanon et al., 2009).
Nrf-2 is a transcription factor activated by redox imbalance via the removal of its redox-sensitive anchor Keap1 (Kundu and Surh, 2010), and is devoted to the transcription of genes encoding for glutathione metabolism and antioxidant enzymes (Li and Kong, 2009; Ramprasath and Selvam, 2013), thus providing cells with extra antioxidant defense, and reducing the apoptotic outcome. It was found that murine embryonic fibroblasts survive to BSO treatment due to a strong Nrf-2 response, whereas Nrf-2 deficient cells, or cells treated with Nrf2 siRNA became sensitive to BSO-induced apoptosis (Lee et al., 2008).
Therefore, the straightforward use of BSO as coadjuvant in anticancer therapies should be limited to tumors bearing MDR cells. Importantly, non-MDR cells could develop chemo-resistance as a result of BSO treatment, unless they are targeted with specific RNA interfering agents to prevent the over-expression of survival proteins.
Conclusions
Summarizing, GSH depletion may promote either pro-apoptotic or pro-survival pathways depending on the kinetic of depletion; swift GSH extrusion activates pro-apoptotic pathways, whereas slower GSH depletion activates pro-survival pathways (Figure 1). Intriguingly, both types of response require increased ROS levels and formation of inter-protein disulfides to be activated. The reactivity of cysteines depends on the surrounding aminoacids determining protein conformation, and each inter-protein cystine forms at fixed levels of oxidation of the cell microenvironment. It is conceivable that the reactive cysteines of the proteins involved in the opposite responses (e.g., the cell-protective NF-kappaB component p50 vs. the pro-apoptotic Bax) may be differently modulated according to the swiftness of the redox imbalance, possibly differently relying in the tutoring effect of thiol-modifying enzymes such as thioredoxin or protein disulfide isomerase.
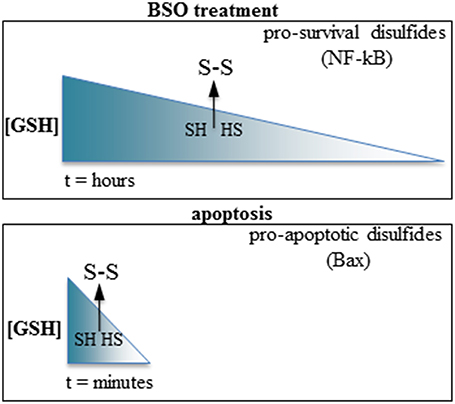
Figure 1. Slow vs. fast GSH depletion produce opposite effects on cell survival. Top: BSO induce slow GSH depletion (hours); the disulfides formed after reaching a threshold level of redox imbalance possess a cell-survival activity (e.g., the p50 subunit of NF-kappaB). Bottom: the very rapid GSH extrusion occurring in apoptosis promote the formation of pro-apoptotic disulfides (e.g., Bax). The arrows indicate a hypothetical threshold level of GSH depletion after which sulfhydryls (SH, HS) are oxidized to disulfides (S-S) in each condition.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Bachhawat, A. K., Thakur, A., Kaur, J., and Zulkifli, M. (2013). Glutathione transporters. Biochim. Biophys. Acta 1830, 3154–3164. doi: 10.1016/j.bbagen.2012.11.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ballatori, N., Krance, S. M., Marchan, R., and Hammond, C. L. (2009). Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol. Aspects Med. 30, 13–28. doi: 10.1016/j.mam.2008.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Celardo, I., De Nicola, M., Mandoli, C., Pedersen, J. Z., Traversa, E., and Ghibelli, L. (2011). Ce3+ ions determine redox-dependent anti-apoptotic effect of cerium oxide nanoparticles. ACS Nano 5, 4537–4549. doi: 10.1021/nn200126a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Circu, M. L., and Aw, T. Y. (2012). Glutathione and modulation of cell apoptosis. Biochim. Biophys. Acta 1823, 1767–1777. doi: 10.1016/j.bbamcr.2012.06.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Coppola, S., and Ghibelli, L. (2000). GSH extrusion and the mitochondrial pathway of apoptotic signalling. Biochem. Soc. Trans. 28, 56–61.
Cristofanon, S., Morceau, F., Scovassi, A. I., Dicato, M., Ghibelli, L., and Diederich, M. (2009). Oxidative, multistep activation of the noncanonical NF-kappaB pathway via disulfide Bcl-3/p50 complex. FASEB J. 23, 45–57. doi: 10.1096/fj.07-104109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cristofanon, S., Nuccitelli, S., D'Alessio, M., Dicato, M., Diederich, M., and Ghibelli, L. (2008). Oxidation-dependent maturation and survival of explanted blood monocytes via Bcl-2 up-regulation. Biochem. Pharmacol. 76, 1533–1543. doi: 10.1016/j.bcp.2008.07.042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cristofanon, S., Nuccitelli, S., D'Alessio, M., Radogna, F., De Nicola, M., Bergamaschi, A., et al. (2006). Oxidative upregulation of Bcl-2 in healthy lymphocytes. Ann. N.Y. Acad. Sci. 1091, 1–9. doi: 10.1196/annals.1378.049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D'Alessio, M., Cerella, C., Amici, C., Pesce, C., Coppola, S., Fanelli, C., et al. (2004). Glutathione depletion up-regulates Bcl-2 in BSO-resistant cells. FASEB J. 18, 1609–1611. doi: 10.1096/fj.04-1813fje
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D'Alessio, M., Cerella, C., De Nicola, M., Bergamaschi, A., Magrini, A., Gualandi, G., et al. (2003). Apoptotic GSH extrusion is associated with free radical generation. Ann. N.Y. Acad. Sci. 1010, 449–452. doi: 10.1196/annals.1299.082
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D'Alessio, M., De Nicola, M., Coppola, S., Gualandi, G., Pugliese, L., Cerella, C., et al. (2005). Oxidative Bax dimerization promotes its translocation to mitochondria independently of apoptosis. FASEB J. 19, 1504–1506. doi: 10.1096/fj.04-3329fje
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Nicola, M., Gualandi, G., Alfonsi, A., Cerella, C., D'Alessio, M., Bergamaschi, A., et al. (2006). Different fates of intracellular glutathione determine different modalities of apoptotic nuclear vesiculation. Biochem. Pharmacol. 72, 1405–1416. doi: 10.1016/j.bcp.2006.06.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dini, L., Coppola, S., Ruzittu, M. T., and Ghibelli, L. (1996). Multiple pathways for apoptotic nuclear fragmentation. Exp. Cell Res. 223, 340–347.
Esteve, J. M., Mompo, J., Garcia de la Asuncion, J., Sastre, J., Asensi, M., Boix, J., et al. (1999). Oxidative damage to mitochondrial DNA and glutathione oxidation in apoptosis: studies in vivo and in vitro. FASEB J. 13, 1055–1064.
Filomeni, G., Aquilano, K., Rotilio, G., and Ciriolo, M. R. (2005). Antiapoptotic response to induced GSH depletion: involvement of heat shock proteins and NF-kappaB activation. Antioxid. Redox Signal. 7, 446–455. doi: 10.1089/ars.2005.7.446
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Filomeni, G., Rotilio, G., and Ciriolo, M. R. (2003). Glutathione disulfide induces apoptosis in U937 cells by a redox-mediated p38 MAP kinase pathway. FASEB J. 17, 64–66. doi: 10.1096/fj.02-0105fje
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fojo, T., and Bates, S. (2003). Strategies for reversing drug resistance. Oncogene 22, 7512–7523. doi: 10.1038/sj.onc.1206951
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Franco, R., and Cidlowski, J. A. (2012). Glutathione efflux and cell death. Antioxid. Redox Signal. 17, 1694–1713. doi: 10.1089/ars.2012.4553
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ghibelli, L., Coppola, S., Fanelli, C., Rotilio, G., Civitareale, P., Scovassi, A. I., et al. (1999). Glutathione depletion causes cytochrome c release even in the absence of cell commitment to apoptosis. FASEB J. 13, 2031–2036.
Ghibelli, L., Coppola, S., Rotilio, G., Lafavia, E., Maresca, V., and Ciriolo, M. R. (1995). Non-oxidative loss of glutathione in apoptosis via GSH extrusion. Biochem. Biophys. Res. Commun. 216, 313–320. doi: 10.1006/bbrc.1995.2626
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ghibelli, L., and Diederich, M. (2010). Multistep and multitask Bax activation. Mitochondrion 10, 604–613. doi: 10.1016/j.mito.2010.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ghibelli, L., Fanelli, C., Rotilio, G., Lafavia, E., Coppola, S., Colussi, C., et al. (1998). Rescue of cells from apoptosis by inhibition of active GSH extrusion. FASEB J. 12, 479–486.
Ishii, T., and Mann, G. E. (2014). Redox status in mammalian cells and stem cells during culture in vitro: critical roles of Nrf2 and cystine transporter activity in the maintenance of redox balance. Redox. Biol. 2, 786–794. doi: 10.1016/j.redox.2014.04.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kundu, J. K., and Surh, Y. J. (2010). Nrf2-Keap1 signaling as a potential target for chemoprevention of inflammation-associated carcinogenesis. Pharm. Res. 27, 999–1013. doi: 10.1007/s11095-010-0096-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, H. R., Cho, J. M., Shin, D. H., Yong, C. S., Choi, H. G., Wakabayashi, N., et al. (2008). Adaptive response to GSH depletion and resistance to L: -buthionine-(S,R)-sulfoximine: involvement of Nrf2 activation. Mol. Cell. Biochem. 318, 23–31. doi: 10.1007/s11010-008-9853-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, W., and Kong, A. N. (2009). Molecular mechanisms of Nrf2-mediated antioxidant response. Mol. Carcinog. 48, 91–104. doi: 10.1002/mc.20465
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Limón-Pacheco, J. H., Hernández, N. A., Fanjul-Moles, M. L., and Gonsebatt, M. E. (2007). Glutathione depletion activates mitogen-activated protein kinase (MAPK) pathways that display organ-specific responses and brain protection in mice. Free Radic. Biol. Med. 43, 1335–1347. doi: 10.1016/j.freeradbiomed.2007.06.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liuzzi, F., Fanelli, C., Ciriolo, M. R., Cerella, C., D'Alessio, M., Denicola, M., et al. (2003). Rescue of cells from apoptosis by antioxidants occurs downstream from GSH extrusion. Ann. N.Y. Acad. Sci. 1010, 441–445. doi: 10.1196/annals.1299.080
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meredith, M. J., Cusick, C. L., Soltaninassab, S., Sekhar, K. S., Lu, S., and Freeman, M. L. (1998). Expression of Bcl-2 increases intracellular glutathione by inhibiting methionine-dependent GSH efflux. Biochem. Biophys. Res. Commun. 248, 458–463. doi: 10.1006/bbrc.1998.8998
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Morgan, M. J., and Liu, Z. G. (2011). Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 21, 103–115. doi: 10.1038/cr.2010.178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pompella, A., Visvikis, A., Paolicchi, A., De Tata, V., and Casini, A. F. (2003). The changing faces of glutathione, a cellular protagonist. Biochem. Pharmacol. 66, 1499–1503. doi: 10.1016/S0006-2952(03)00504-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rahman, I., Biswas, S. K., Jimenez, L. A., Torres, M., and Forman, H. J. (2005). Glutathione, stress responses, and redox signaling in lung inflammation. Antioxid. Redox Signal. 7, 42–59. doi: 10.1089/ars.2005.7.42
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ramprasath, T., and Selvam, G. S. (2013). Potential impact of genetic variants in Nrf2 regulated antioxidant genes and risk prediction of diabetes and associated cardiac complications. Curr. Med. Chem. 20, 4680–4693. doi: 10.2174/09298673113209990154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suzuki, M., Youle, R. J., and Tjandra, N. (2000). Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 103, 645–654. doi: 10.1016/S0092-8674(00)00167-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van den Dobbelsteen, D., Nobel, S., Schlegel, J., Cotgreave, I. A., Orrenius, S., and Slater, A. (1996). GSH effflux in FAS/APO-1-induced apoptosis. J. Biol. Chem. 271, 15420–15427.
Yi, J. R., Lu, S., Fernández-Checa, J., and Kaplowitz, N. (1995). Expression cloning of the cDNA for a polypeptide associated with rat hepatic sinusoidal reduced glutathione transport: characteristics and comparison with the canalicular transporter. Proc. Natl. Acad. Sci. U.S.A. 92, 1495–1499. doi: 10.1073/pnas.92.5.1495
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: apoptosis, GSH, GSH transporters, BSO, NF-KappaB
Citation: De Nicola M and Ghibelli L (2014) Glutathione depletion in survival and apoptotic pathways. Front. Pharmacol. 5:267. doi: 10.3389/fphar.2014.00267
Received: 26 September 2014; Paper pending published: 07 November 2014;
Accepted: 15 November 2014; Published online: 04 December 2014.
Edited by:
Alfonso Pompella, Università di Pisa, ItalyReviewed by:
Muzamil Ahmad, Indian Institute of Integrative Medicine, IndiaCopyright © 2014 De Nicola and Ghibelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:Z2hpYmVsbGlAdW5pcm9tYTIuaXQ=