 Marius Pollet
Marius Pollet Jean Krutmann1,2
Jean Krutmann1,2 Thomas Haarmann-Stemmann
Thomas Haarmann-Stemmann- 1IUF-Leibniz Research Institute for Environmental Medicine, Düsseldorf, Germany
- 2Medical Faculty, Heinrich-Heine University, Düsseldorf, Germany
A Commentary on
Usage of Mitogen-Activated Protein Kinase Small Molecule Inhibitors: More Than Just Inhibition!
by Meurer, S. K., and Weiskirchen, R. (2018). Front. Pharmacol. 9:98. doi: 10.3389/fphar.2018.00098
Steffen Meurer and Ralf Weiskirchen recently published an interesting and important study concerning the off-target effects of so-called “specific” protein kinase inhibitors (PKI) that are frequently applied in both, basic research and clinical applications (Meurer and Weiskirchen, 2018). A PKI-dependent inhibition of non-targeted protein kinases may occur due to the usage of concentrations that exceed the respective IC50 value by multiple factors. Meurer and Weiskirchen instead describe a PKI-mediated activation of non-targeted protein kinases. Specifically, the authors observed that a treatment of hepatic stellate cells, hepatocytes and portal myofibroblasts with a chemical inhibitor for a certain mitogen-activated protein kinase (MAPK) led to an activation of other members of the MAPK network. The authors called this phenomenon “activation by inhibition” and “cross-activation” (Meurer and Weiskirchen, 2018), terms which well describe the experimental observations but not the underlying molecular mechanism.
Regarding the latter one, we realized that four of the five MAPK inhibitors tested in the respective study are known to interfere with the activity of the aryl hydrocarbon receptor (AHR), a ligand-activated transcription factor and key regulator of xenobiotic metabolism (Murray et al., 2014). In its inactive form, the AHR is trapped in a cytosolic multiprotein complex. Upon binding of small molecular weight compounds, this complex dissociates and the AHR shuttles in the nucleus, dimerizes with its partner molecule ARNT and induces gene expression (Murray et al., 2014). The probably best-examined AHR target genes encode for the xenobiotic-metabolizing monooxygenases cytochrome P450 (CYP) 1A1, CYP1A2, and CYP1B1, which, in most cases, oxidize the invading chemicals to enhance their polarity and facilitate their excretion (Mescher and Haarmann-Stemmann, 2018). Importantly, the ligand-driven activation of AHR is frequently accompanied by a stimulation of other cellular signaling pathways, including NF-κB, epidermal growth factor receptor (EGFR) and MAPK signal transduction (Haarmann-Stemmann et al., 2009; Puga et al., 2009; Tian, 2009). The list of AHR ligands encompasses infamous environmental pollutants, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and benzo[a]pyrene, plant polyphenols, microbiota-derived indoles and phenazines as well as several pharmaceuticals (Murray et al., 2014). Interestingly, more than a dozen PKI, including the four MAPK inhibitors SB203580, U0126, PD98059, and SP600125 tested by Meurer and Weiskirchen, have been identified to date to modulate AHR activity and downstream gene expression (e.g., of CYP1A1) in either a positive or a negative manner (Table 1). Several PKI, such as the phosphoinositide 3-kinase inhibitor LY294002, bind to the AHR protein and antagonize its activation by the prototype ligand TCDD (Guo et al., 2000), whereas others, including the aforementioned U0126 and SB203580, were shown to interact with AHR and increase its transcriptional activity (Andrieux et al., 2004; Korashy et al., 2011). In this context, it is interesting to know that an activation of AHR by different ligands has been reported to stimulate the phosphorylation of ERK1/2, p38 MAPK, JNK, and upstream receptor tyrosine kinases in various human and rodent cells (Haarmann-Stemmann et al., 2009; Puga et al., 2009). In fact, Fumio Matsumura and his team have been among the first reporting a direct impact of AHR activation on protein kinase activity. Specifically, they observed an increased activity of protein kinase C and EGFR in hepatic tissue of rodents treated with TCDD (Madhukar et al., 1984; Bombick et al., 1985). Further examples for an impact of AHR activation on the signal transduction network, are the TCDD-induced phosphorylation of p38 MAPK observed in hepatoma cells (Weiss et al., 2005) and macrophages (Park et al., 2005), as well as the activation of EGFR and downstream ERK1/2 signaling by the AHR agonists 6-formylindolo[3,2-b]carbazole and TCDD in keratinocytes and colon cancer cells, respectively (Fritsche et al., 2007; Xie et al., 2012). It is thus tempting to speculate that at least some of the PKI-induced off-target effects observed by Meurer and Weiskirchen, such as the phosphorylation of ERK1/2 and JNK by the p38 MAPK inhibitor SB203580 or the activation of JNK and p38 MAPK by the MEK1/2 blocker U0126, were due to a stimulation of AHR activity. One may describe this phenomenon as an effect of a certain PKI on a non-kinase target (Munoz, 2017) or simply as the recognition of a foreign compound by the cellular defense system against xenobiotics. This notion is supported by the fact that some PKI have been found to interact with other xenobiotic receptors as well. The MEK1/2 inhibitor U0126, for instance, was shown to induce the expression of CYP3A4 in human hepatoma cells by binding to the pregnane X receptor (PXR) (Smutny et al., 2014). In addition, five out of nine tested clinically relevant PKI (erlotinib, gefitinib, nilotinib, sorafenib, and vandetanib) induced the expression of the ATP-binding cassette transporter P-glycoprotein in a PXR-dependent manner in human colon cancer cells (Harmsen et al., 2013).
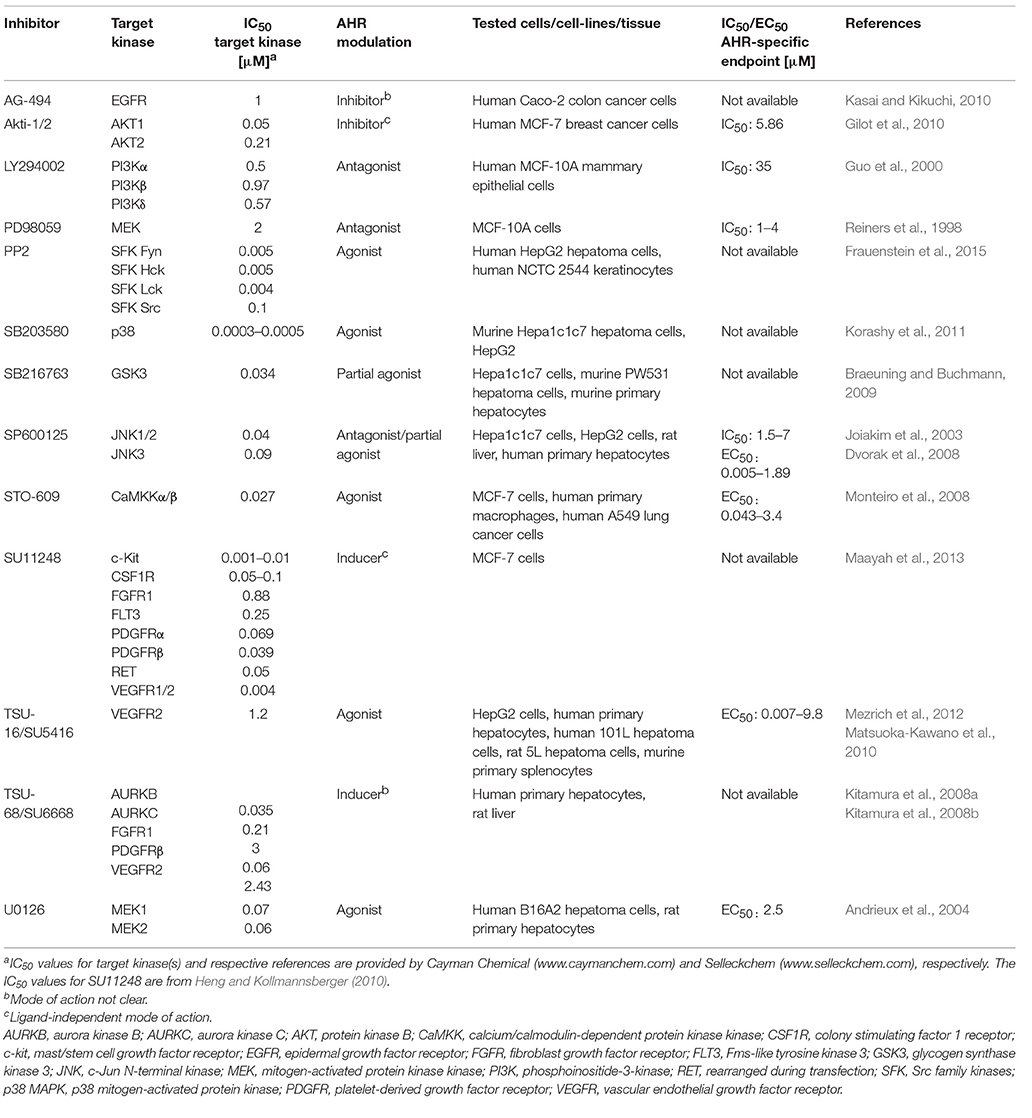
Table 1. Overview on protein kinase inhibitors known to manipulate AHR signaling.
The majority of PKI interact with the ATP-binding cleft of the target enzyme (Bain et al., 2007; Wu et al., 2015). Given that the 3D structure of the ATP-binding cleft is highly conserved amongst eukaryotic protein kinases, these inhibitors are limited in both their size and structural diversity (Bain et al., 2007; Wu et al., 2015). As implied by the growing list of PKI identified to interact with AHR and PXR, the structural prerequisites to bind to the ATP-binding cleft of protein kinases seem to resemble those required to interact with the ligand-binding domain of the xenobiotic receptors. Indeed, AHR and PXR contain a relatively large ligand-binding domain with a cavity volume of ~840 Å3 (Denison et al., 2002) and ~1,150 Å3 (Watkins et al., 2001), respectively, and share an extreme structural diversity of ligands (Denison and Faber, 2017). Notably, the known IC50/EC50 values of some PKI to modulate AHR activity are indeed in the range of the IC50 for their target kinases (Table 1). However, apart from ligand-binding, certain PKI may indirectly interfere with AHR signaling, for instance by inhibiting protein kinases contributing to AHR's nuclear translocation (Haarmann-Stemmann et al., 2009).
We agree with the authors that a detailed knowledge of the off-target effects induced by widely used PKI is urgently required, not only for the proper interpretation of experimental data, but in particular, to better forecast potential drug-drug interactions during therapy. When working with so-called “specific” PKI, one should be aware that these compounds will be at first recognized by the chemical defense system, i.e. by xenobiotic receptors, drug-metabolizing enzymes and drug transporters, of the exposed cells. The interaction with one or more xenobiotic receptor(s) is determined by the structural and physiochemical properties of a given PKI and may not only affect its own metabolism, but also the metabolism of eventually co-administered drugs, and the activity of other signaling pathways that might be tightly interconnected with the addressed chemosensory receptor.
Author Contributions
MP performed literature research and created the table. JK revised the manuscript critically for important intellectual content. TH-S performed literature research and wrote the manuscript. All authors approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Melina Mescher for critically reading the manuscript.
References
Andrieux, L., Langouët, S., Fautrel, A., Ezan, F., Krauser, J. A., Savouret, J. F., et al. (2004). Aryl hydrocarbon receptor activation and cytochrome P450 1A induction by the mitogen-activated protein kinase inhibitor U0126 in hepatocytes. Mol. Pharmacol. 65, 934–943. doi: 10.1124/mol.65.4.934
Bain, J., Plater, L., Elliott, M., Shpiro, N., Hastie, C. J., Mclauchlan, H., et al. (2007). The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315. doi: 10.1042/BJ20070797
Bombick, D. W., Madhukar, B. V., Brewster, D. W., and Matsumura, F. (1985). TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin) causes increases in protein kinases particularly protein kinase C in the hepatic plasma membrane of the rat and the guinea pig. Biochem. Biophys. Res. Commun. 127, 296–302. doi: 10.1016/S0006-291X(85)80158-3
Braeuning, A., and Buchmann, A. (2009). The glycogen synthase kinase inhibitor 3-(2,4-dichlorophenyl)-4-(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione (SB216763) is a partial agonist of the aryl hydrocarbon receptor. Drug Metab. Dispos. 37, 1576–1580. doi: 10.1124/dmd.109.027821
Denison, M. S., and Faber, S. C. (2017). And now for something completely different: diversity in ligand-dependent activation of Ah receptor responses. Curr Opin Toxicol 2, 124–131. doi: 10.1016/j.cotox.2017.01.006
Denison, M. S., Pandini, A., Nagy, S. R., Baldwin, E. P., and Bonati, L. (2002). Ligand binding and activation of the Ah receptor. Chem. Biol. Interact. 141, 3–24. doi: 10.1016/S0009-2797(02)00063-7
Dvorak, Z., Vrzal, R., Henklova, P., Jancova, P., Anzenbacherova, E., Maurel, P., et al. (2008). JNK inhibitor SP600125 is a partial agonist of human aryl hydrocarbon receptor and induces CYP1A1 and CYP1A2 genes in primary human hepatocytes. Biochem. Pharmacol. 75, 580–588. doi: 10.1016/j.bcp.2007.09.013
Frauenstein, K., Tigges, J., Soshilov, A. A., Kado, S., Raab, N., Fritsche, E., et al. (2015). Activation of the aryl hydrocarbon receptor by the widely used Src family kinase inhibitor 4-amino-5-(4-chlorophenyl)-7-(dimethylethyl)pyrazolo[3,4-d]pyrimidine (PP2). Arch. Toxicol. 89, 1329–1336. doi: 10.1007/s00204-014-1321-8
Fritsche, E., Schäfer, C., Calles, C., Bernsmann, T., Bernshausen, T., Wurm, M., et al. (2007). Lightening up the UV response by identification of the arylhydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc. Natl. Acad. Sci. U.S.A. 104, 8851–8856. doi: 10.1073/pnas.0701764104
Gilot, D., Giudicelli, F., Lagadic-Gossmann, D., and Fardel, O. (2010). Akti-1/2, an allosteric inhibitor of Akt 1 and 2, efficiently inhibits CaMKIalpha activity and aryl hydrocarbon receptor pathway. Chem. Biol. Interact. 188, 546–552. doi: 10.1016/j.cbi.2010.08.011
Guo, M., Joiakim, A., and Reiners, J. J., Jr. (2000). Suppression of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-mediated aryl hydrocarbon receptor transformation and CYP1A1 induction by the phosphatidylinositol 3-kinase inhibitor 2-(4-morpholinyl)-8-phenyl-4H-1- benzopyran-4-one (LY294002). Biochem. Pharmacol. 60, 635–642. doi: 10.1016/S0006-2952(00)00379-8
Haarmann-Stemmann, T., Bothe, H., and Abel, J. (2009). Growth factors, cytokines and their receptors as downstream targets of arylhydrocarbon receptor (AhR) signaling pathways. Biochem. Pharmacol. 77, 508–520. doi: 10.1016/j.bcp.2008.09.013
Harmsen, S., Meijerman, I., Maas-Bakker, R. F., Beijnen, J. H., and Schellens, J. H. (2013). PXR-mediated P-glycoprotein induction by small molecule tyrosine kinase inhibitors. Eur. J. Pharm. Sci. 48, 644–649. doi: 10.1016/j.ejps.2012.12.019
Heng, D. Y. C., and Kollmannsberger, C. (2010). “Sunitinib” in Small Molecules in Oncology ed U. M. Martens (Berlin: Springer), 71–82.
Joiakim, A., Mathieu, P. A., Palermo, C., Gasiewicz, T. A., and Reiners, J. J. Jr. (2003). The Jun N-terminal kinase inhibitor SP600125 is a ligand and antagonist of the aryl hydrocarbon receptor. Drug Metab. Dispos. 31, 1279–1282. doi: 10.1124/dmd.31.11.1279
Kasai, S., and Kikuchi, H. (2010). The inhibitory mechanisms of the tyrosine kinase inhibitors herbimycin a, genistein, and tyrphostin B48 with regard to the function of the aryl hydrocarbon receptor in Caco-2 cells. Biosci. Biotechnol. Biochem. 74, 36–43. doi: 10.1271/bbb.90438
Kitamura, R., Asanoma, H., Nagayama, S., and Otagiri, M. (2008a). Identification of human liver cytochrome P450 isoforms involved in autoinduced metabolism of the antiangiogenic agent (Z)-5-[(1,2-dihydro-2-oxo-3H-indol-3-ylidene)methyl]-2,4-dimethyl-1H-pyrrole-3-pr opanoic acid (TSU-68). Drug Metab. Dispos. 36, 1003–1009. doi: 10.1124/dmd.107.019877
Kitamura, R., Matsuoka, K., Nagayama, S., and Otagiri, M. (2008b). Time-dependent induction of rat hepatic CYP1A1 and CYP1A2 expression after single-dose administration of the anti-angiogenic agent TSU-68. Drug Metab. Pharmacokinet. 23, 421–427. doi: 10.2133/dmpk.23.421
Korashy, H. M., Anwar-Mohamed, A., Soshilov, A. A., Denison, M. S., and El-Kadi, A. O. (2011). The p38 MAPK inhibitor SB203580 induces cytochrome P450 1A1 gene expression in murine and human hepatoma cell lines through ligand-dependent aryl hydrocarbon receptor activation. Chem. Res. Toxicol. 24, 1540–1548. doi: 10.1021/tx200141p
Maayah, Z. H., El Gendy, M. A., El-Kadi, A. O., and Korashy, H. M. (2013). Sunitinib, a tyrosine kinase inhibitor, induces cytochrome P450 1A1 gene in human breast cancer MCF7 cells through ligand-independent aryl hydrocarbon receptor activation. Arch. Toxicol. 87, 847–856. doi: 10.1007/s00204-012-0996-y
Madhukar, B. V., Brewster, D. W., and Matsumura, F. (1984). Effects of in vivo-administered 2,3,7,8-tetrachlorodibenzo-p-dioxin on receptor binding of epidermal growth factor in the hepatic plasma membrane of rat, guinea pig, mouse, and hamster. Proc. Natl. Acad. Sci. U.S.A. 81, 7407–7411. doi: 10.1073/pnas.81.23.7407
Matsuoka-Kawano, K., Yoshinari, K., Nagayama, S., and Yamazoe, Y. (2010). TSU-16, (Z)-3-[(2,4-dimethylpyrrol-5-yl)methylidenyl]-2-indolinone, is a potent activator of aryl hydrocarbon receptor and increases CYP1A1 and CYP1A2 expression in human hepatocytes. Chem. Biol. Interact. 185, 33–41. doi: 10.1016/j.cbi.2010.02.014
Mescher, M., and Haarmann-Stemmann, T. (2018). Modulation of CYP1A1 metabolism: from adverse health effects to chemoprevention and therapeutic options. Pharmacol. Ther. 187, 71–87. doi: 10.1016/j.pharmthera.2018.02.012
Meurer, S. K., and Weiskirchen, R. (2018). Usage of mitogen-activated protein kinase small molecule inhibitors: more than just inhibition! Front. Pharmacol. 9:98. doi: 10.3389/fphar.2018.00098
Mezrich, J. D., Nguyen, L. P., Kennedy, G., Nukaya, M., Fechner, J. H., Zhang, X., et al. (2012). SU5416, a VEGF receptor inhibitor and ligand of the AHR, represents a new alternative for immunomodulation. PLoS ONE 7:e44547. doi: 10.1371/journal.pone.0044547
Monteiro, P., Gilot, D., Langouet, S., and Fardel, O. (2008). Activation of the aryl hydrocarbon receptor by the calcium/calmodulin-dependent protein kinase kinase inhibitor 7-oxo-7H-benzimidazo[2,1-a]benz[de]isoquinoline-3-carboxylic acid (STO-609). Drug Metab. Dispos. 36, 2556–2563. doi: 10.1124/dmd.108.023333
Munoz, L. (2017). Non-kinase targets of protein kinase inhibitors. Nat. Rev. Drug Discov. 16, 424–440. doi: 10.1038/nrd.2016.266
Murray, I. A., Patterson, A. D., and Perdew, G. H. (2014). Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat. Rev. Cancer 14, 801–814. doi: 10.1038/nrc3846
Park, S. J., Yoon, W. K., Kim, H. J., Son, H. Y., Cho, S. W., Jeong, K. S., et al. (2005). 2,3,7,8-Tetrachlorodibenzo-p-dioxin activates ERK and p38 mitogen-activated protein kinases in RAW 264.7 cells. Anticancer Res. 25, 2831–2836.
Puga, A., Ma, C., and Marlowe, J. L. (2009). The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem. Pharmacol. 77, 713–722. doi: 10.1016/j.bcp.2008.08.031
Reiners, J. J. Jr., Lee, J. Y., Clift, R. E., Dudley, D. T., and Myrand, S. P. (1998). PD98059 is an equipotent antagonist of the aryl hydrocarbon receptor and inhibitor of mitogen-activated protein kinase kinase. Mol. Pharmacol. 53, 438–445. doi: 10.1124/mol.53.3.438
Smutny, T., Bitman, M., Urban, M., Dubecka, M., Vrzal, R., Dvorak, Z., et al. (2014). U0126, a mitogen-activated protein kinase kinase 1 and 2 (MEK1 and 2) inhibitor, selectively up-regulates main isoforms of CYP3A subfamily via a pregnane X receptor (PXR) in HepG2 cells. Arch. Toxicol. 88, 2243–2259. doi: 10.1007/s00204-014-1254-2
Tian, Y. (2009). Ah receptor and NF-kappaB interplay on the stage of epigenome. Biochem. Pharmacol. 77, 670–680. doi: 10.1016/j.bcp.2008.10.023
Watkins, R. E., Wisely, G. B., Moore, L. B., Collins, J. L., Lambert, M. H., Williams, S. P., et al. (2001). The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science 292, 2329–2333. doi: 10.1126/science.1060762
Weiss, C., Faust, D., Dürk, H., Kolluri, S. K., Pelzer, A., Schneider, S., et al. (2005). TCDD induces c-jun expression via a novel Ah (dioxin) receptor-mediated p38-MAPK-dependent pathway. Oncogene 24, 4975–4983. doi: 10.1038/sj.onc.1208679
Wu, P., Nielsen, T. E., and Clausen, M. H. (2015). FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 36, 422–439. doi: 10.1016/j.tips.2015.04.005
Keywords: aryl hydrocarbon receptor, kinase inhibitor, off-target effects, signal transduction, pregnane X receptor
Citation: Pollet M, Krutmann J and Haarmann-Stemmann T (2018) Commentary: Usage of Mitogen-Activated Protein Kinase Small Molecule Inhibitors: More Than Just Inhibition! Front. Pharmacol. 9:935. doi: 10.3389/fphar.2018.00935
Received: 08 June 2018; Accepted: 30 July 2018;
Published: 20 August 2018.
Edited by:
Dagmar Meyer zu Heringdorf, Goethe-Universität Frankfurt am Main, GermanyReviewed by:
Krister Wennerberg, University of Helsinki, FinlandCopyright © 2018 Pollet, Krutmann and Haarmann-Stemmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Haarmann-Stemmann, dGhvbWFzLmhhYXJtYW5uLXN0ZW1tYW5uQGl1Zi1kdWVzc2VsZG9yZi5kZQ==