 Jatin Sharma
Jatin Sharma Joseph Larkin III
Joseph Larkin III- Department of Microbiology and Cell Science, Institute of Food and Agricultural Sciences, University of Florida, Gainesville, FL, United States
The suppressor of cytokine signaling (SOCS) family of intracellular proteins has a vital role in the regulation of the immune system and resolution of inflammatory cascades. SOCS1, also called STAT-induced STAT inhibitor (SSI) or JAK-binding protein (JAB), is a member of the SOCS family with actions ranging from immune modulation to cell cycle regulation. Knockout of SOCS1 leads to perinatal lethality in mice and increased vulnerability to cancer, while several SNPs associated with the SOCS1 gene have been implicated in human inflammation-mediated diseases. In this review, we describe the mechanism of action of SOCS1 and its potential therapeutic role in the prevention and treatment of autoimmunity and cancer. We also provide a brief outline of the other JAK inhibitors, both FDA-approved and under investigation.
Introduction
Cytokines are small glycoproteins secreted by a variety of immune and non-immune cells. These molecules govern a range of processes including, but not limited to, hematopoiesis, inflammation, cell proliferation, survival, apoptosis, and chemotaxis. As such, intricate modulation of cytokine signaling is required for maintaining immune system homeostasis and regulation of inflammatory responses. Cytokine receptors typically belong to one of the following families: IL-1 receptor superfamily, TNF-receptor family, IL-17 receptor superfamily, G-protein coupled receptor (GPCR) superfamily, transforming growth factor superfamily, receptor tyrosine kinase superfamily (RTK), and type I and II cytokine receptor superfamily. Among these, only type I and II cytokine receptors are physically associated with JAKs (Gadina et al., 2001). JAKs are tyrosine kinases whose primary targets are Signal transduction and activator of transcription (STAT) proteins. JAK-STAT signaling is the canonical pathway induced by cytokines binding to type I or II cytokine receptors, though other major pathways such as PI3K/AKT and MAPK (p38, JNK, and ERK1/2) are also involved either directly or indirectly depending on the cytokine and the target cell type. Cytokine signaling can be regulated in the following ways - (1) by modulation of cytokine gene expression, (2) regulation at the receptor level or (3) at the stage of signal transduction. Cytokine receptor modulation can occur through changes in expression of the cytokine receptor, blockade of receptors via natural antagonists/decoy ligands (e.g., IL-1Ra blocks IL-1R) (Seckinger et al., 1987), or blockade of ligand via decoy receptors (e.g., sgp130 blocks sIL-6R signaling) (Jostock et al., 2001). In terms of signal transduction, changes in the expression of signal transducing elements, or their respective regulators, may serve to modulate the signal. Signal transduction regulation may occur as either post-transcriptional or post-translational regulation. While regulation at the ligand or the receptor level is more specific, modulation at the signal transduction stage allows control over multiple cytokine signals at once. The JAK family of non-receptor tyrosine kinases comprises of four members: JAK1, JAK2, JAK3, and TYK2. Their canonical targets are the STAT family proteins which includes STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. The STAT proteins are transiently active when phosphorylated by JAKs. Phosphorylated STATs form homo/heterodimers to act as transcription factors, though unphosphorylated STAT dimers may be present and exert biological activity (Braunstein et al., 2003; Yang and Stark, 2008; Sgrignani et al., 2015; Butturini et al., 2016). JAK/STAT signaling cascades, under normal conditions, are regulated by Protein inhibitor of activated STAT (PIAS), phosphatases such as SHP-1 and SHP-2, and the members of the SOCS family of proteins (Chung et al., 1997; Naka et al., 1997; Liu et al., 1998; Barber, 2001; Kirito, 2002; Kelley, 2008). Recent reports have elucidated that JAK1/2 may also be able to activate the PI3K-AKT pathway via phosphorylation of p85 (the regulatory subunit of the PI3K enzyme) (Yamada et al., 2012). Hence, the regulators of the JAK family indirectly modulate PI3K signaling as well.
The SOCS family consists of a group of 8 intracellular proteins: SOCS 1–7 and CIS, the first member to be discovered, (Barber, 2001; Krebs and Hilton, 2001) all possessing an SH2 domain, C-terminal SOCS box, N-terminal extended SH2 subdomain (ESS), and a variable N-terminal region (see Figure 1). Additionally, SOCS1 and SOCS3 also possess a KIR. The SH2 domain imparts specificity to the protein by binding to specific phosphotyrosine residues on the target (Koch et al., 1991; Liau et al., 2017), allowing the KIR domain to inhibit kinase activity by acting as a pseudosubstrate, in case of SOCS1 and SOCS3. The SOCS box can recruit factors to form E3 ligase complex that tags the target protein for ubiquitination, leading to its proteasomal degradation (Zhang et al., 1999; Bullock et al., 2007; Liau et al., 2018; Figure 2 demonstrates the mechanism briefly). Notably, only SOCS1 has been reported to have a nuclear localization signal (NLS) (Baetz et al., 2008). The SOCS1 NLS is known to enable p65 destabilization in the nucleus and it was shown in mouse CD11c+ cells that SOCS1ΔNLS has impaired ability to inhibit NF-κB-induced inflammation as compared to the complete SOCS1 protein (Nakagawa et al., 2002; Ryo et al., 2003; Strebovsky et al., 2011; Zimmer et al., 2016). SOCS1 mRNA is naturally regulated through microRNA-155 at the post-transcriptional level (Yao et al., 2012), while post-translational regulation of SOCS1 includes phosphorylation by kinases like v-abl, pim1, and pim2. These kinases prevent the SOCS Box from binding to Elongin C, an important intermediate for E3 complex recruitment (Chen et al., 2002; Limnander et al., 2004). SOCS1 was discovered by three different groups simultaneously in the year 1997 led by Tadamitsu Kishimoto at the Osaka University Medical School, (Naka et al., 1997) Akihiko Yoshimura at the Institute of Life Sciences in Karume, (Endo et al., 1997) and by Douglas Hilton at the Walter and Elisa Hall Institute in Melbourne (Starr et al., 1997). SOCS1 expression can be induced by a number of signaling molecules including IL-2, 4, 7, 10, 15, type I and II IFNs, TNFα, and Colony stimulating factors (CSFs) (Sakamoto et al., 1998; Sporri, 2001; Federici et al., 2002; Cornish et al., 2003; Ding et al., 2003; van de Geijn et al., 2004). The SOCS1 gene is located on Chromosome 16.
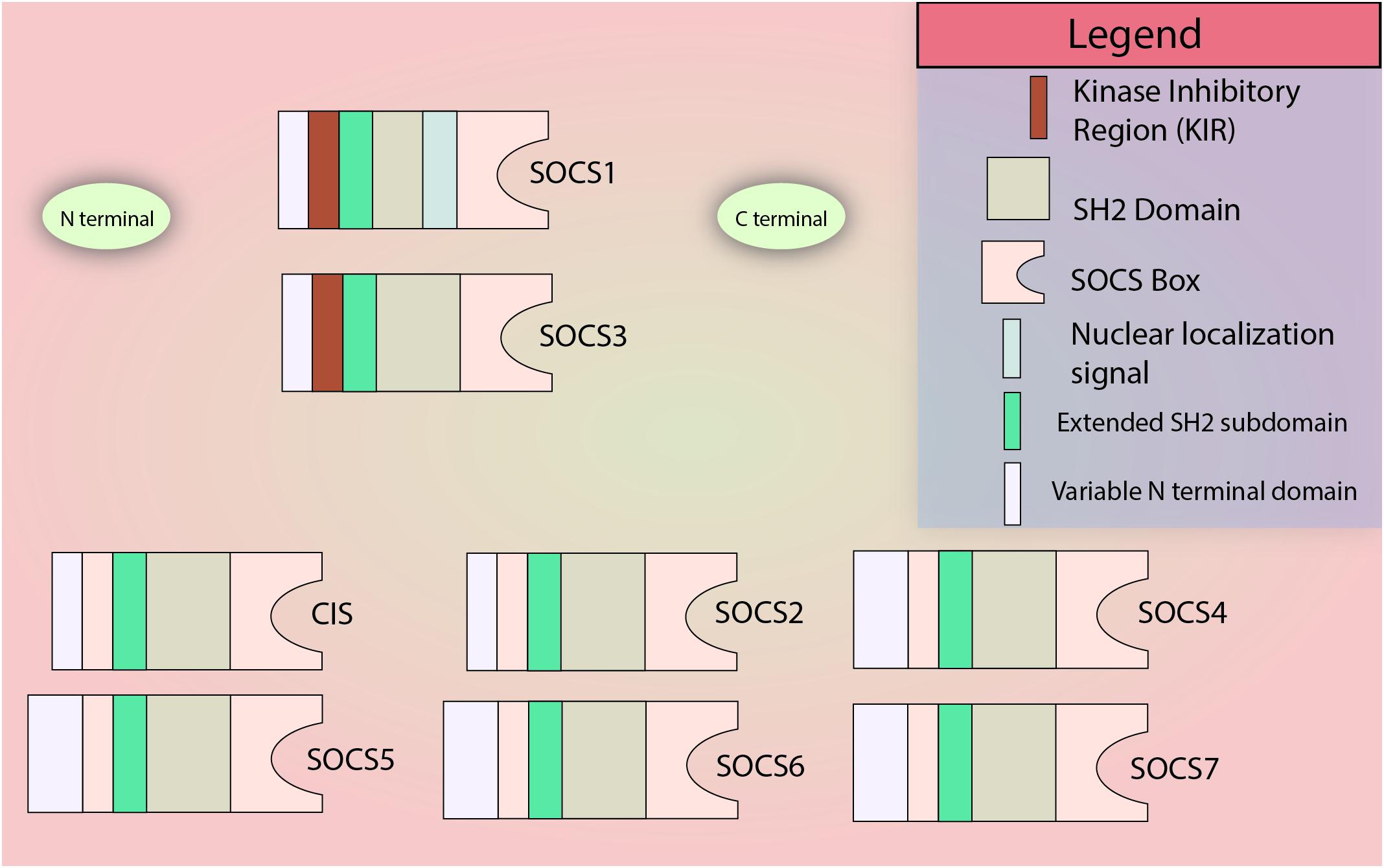
Figure 1. SOCS family members. All members of the SOCS family contain a variable N terminal domain, an SH2 domain, an extended SH2 subdomain (ESS), and the C-terminal SOCS Box domain. The N-terminal KIR domain is restricted to SOCS1 and SOCS3. Only SOCS1 is known to contain a nuclear localization signal. Please note: In most SOCS proteins, there is a little C-terminal sequence left after the SOCS Box which has not been illustrated in the figure for simplicity.
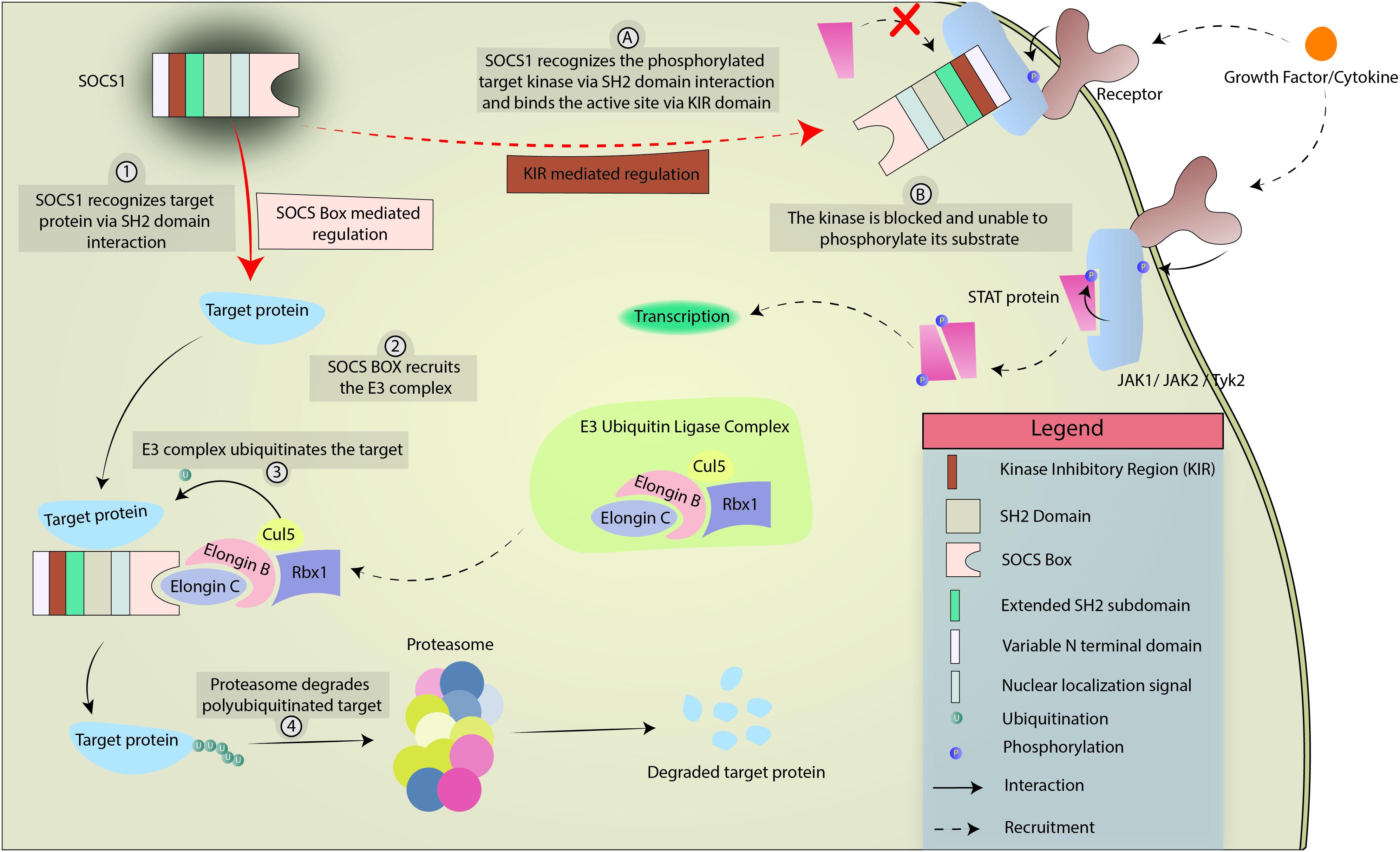
Figure 2. Mechanism of SOCS1-mediated regulation of cytokine and growth factor signaling. SOCS1 regulates intracellular processes in 2 ways, limned as either numerical (SOCS box-mediated) or alphabetical (KIR-mediated). In SOCS Box mediated regulation, SOCS1 interacts with target protein via SH2 domain interaction and uses the SOCS Box to recruit the E3 ubiquitin ligase complex. The E3 complex polyubiquitinates the target which is eventually degraded by the proteasome. In KIR-mediated regulation, SOCS1 interacts with a target kinase (JAK1, JAK2, or TYK2) via SH2 domain interaction. The KIR acts as a pseudosubstrate and blocks the phosphorylation site of the kinase, preventing the kinase from phosphorylating its target.
Suppressor of cytokine signaling 1 not only modulates JAK/STAT pathways, but it can also regulate TLR signaling. TLRs are pattern recognition receptors that can identify conserved microbial molecules and upregulate immune response against them (Mogensen, 2009). SOCS1 regulates these responses by targeting intracellular signal transduction elements MAL (MyD88-adaptor-like protein / TIRAP), IRAK1 (IL-1 receptor-associated kinase), TRAF6 (TNF receptor-associated factor 6), and p65 (a subunit of NF-κB) for ubiquitin-mediated proteasomal degradation and can bind IRAK1 to modulate TLR4 responses. SOCS1 is also induced in a feedback mechanism followed by TLR activation and STAT1 signaling (Nakagawa et al., 2002; Mansell et al., 2006; Jager et al., 2011; Strebovsky et al., 2011; Zhou et al., 2015). A recent report has elucidated that the mechanism of SOCS1-mediated inhibition of kinase activity of JAK1, JAK2, and TYK2 is through binding to the GQM motif on the αG helix of the three above-mentioned kinases (Liau et al., 2018).
Suppressor of cytokine signaling 1 can regulate responses of type I IFN, which function through IFNAR1/2 and TYK2/JAK1; and type II IFN (IFN γ), which functions through IFNGR1/IFNGR2 and JAK1/JAK2 (Federici et al., 2002; Platanias, 2005). Additionally, SOCS1 modulates IL-12 signaling, gp130 (CD130) utilizing cytokines such as IL-6 and LIF, and common γ chain (CD132) utilizing cytokines such as IL-2 and IL-21 (Losman et al., 1999; Sporri, 2001; Eyles et al., 2002). Since SOCS1 has a profound role in T cell homeostasis, it is a prominent player in both autoimmunity and cancer. SOCS1-/- mice die of perinatal autoinflammatory disease or lymphoid deficiencies, develop polycystic kidneys, and inflammatory lesions. While these mice can be partially saved by IFNγ deletion, these mice still develop fatal inflammatory diseases later (Starr et al., 1998; Alexander et al., 1999; Metcalf et al., 2002; Collins et al., 2011). SOCS1 deficiency or dysregulated JAK/STAT signaling has been correlated with a number of immune disorders in humans, including SLE, scleritis, and asthma (Lee et al., 2009; Wang et al., 2010; Yu et al., 2011; Sukka-Ganesh and Larkin, 2016). SOCS1-/- Dendritic cells have an increased sensitivity to LPS and can often result in system autoimmune diseases (Hanada et al., 2003). Moreover, SOCS1-/- peripheral T cells show increased responsiveness to IL-2 and tend to have a skewed ratio of CD4/CD8 population (Cornish et al., 2003; Ilangumaran et al., 2003a,b).
A novel approach to combat SOCS1 deficiency is the use of SOCS1 mimetics. A SOCS1 mimetic peptide containing only the n-terminal kinase inhibitory region (KIR 53- DTHFRTFRSHSDYRRI-68) domain has gained attention due to its effectiveness in JAK1/2 and TYK2 inhibition activity (Waiboci et al., 2007). The KIR domain binds to the activation loop of JAK1/2 and TYK2 to prevent them from phosphorylating their targets. It is an intrinsically disordered protein (IDP), lacking a tertiary structure prior to substrate engagement (Jirgensons, 1966; Uversky et al., 2000, 2005). It has been shown using circular dichroism that SOCS1 mimetic peptide can take up an α-helical structure upon addition of trifluoroethanol which highlights the peptide’s propensity to form stable secondary structure, allowing it to carry out its function (Recio et al., 2014). Of note, SOCS1-/- mice have prolonged survival after treatment with this SOCS1 mimetic peptide (Collins et al., 2011). This approach has also been proven to have a beneficial effect in animal models of inflammatory diseases like EAE and Uveitis (Jager et al., 2011; He et al., 2016).
In this review, we highlight the importance of SOCS1 as a regulator of immune responses contributing to autoimmunity/autoinflammation and cancer and the potential use of SOCS1 mimetic peptide or gene therapy as treatment tactic.
Psoriasis
Psoriasis Vulgaris (PsO) is a dermatological disease marked by plaques and erythematosus on the skin. Histological analyses reveal excess keratinocyte proliferation (acanthosis) and lymphocyte infiltration into the epidermis (Griffiths and Barker, 2007; Menter et al., 2008; Chiricozzi et al., 2011; Guttman-Yassky et al., 2011; Lin et al., 2011). Five types of PsO exist – (1) Plaque-type: most common form of psoriasis and manifests as marked erythematous plaques and squamous lesions localized on elbows, scalp, knees, and sacral zone; (2) Inverse type: plaques localized to skin folds but squamous lesions do not form; (3) Glutate type: frequent in children and adolescents, lesions appear like small droplets and often manifest after a streptococcal infection; (4) Pustular type: rarely occurs and is marked by the presence of pustules on the skin; (5) Erythrodermic: lesions affect about 80% of the body surface accompanied by widespread vasodilation (Koca, 2016). The national psoriasis foundation (NPF) defines three levels of severity – mild psoriatic skin (<3% of body surface covered), moderate psoriatic skin (3–10% body surface covered, and severe psoriatic skin (>10% body surface affected) (Pariser et al., 2007). PASI (Psoriasis area severity index; a measure of average redness, thickness, and scaliness of the lesions) and PGA (Physician global assessment; based on a single estimate to represent the patient’s disease severity as assigned by the physician) are other classifications used to define the disease severity (Langley and Ellis, 2004; Feldman, 2005). There are multiple genetic susceptibility loci known including – AIM2 [dsDNA cytosolic receptor aim2], IL1RL1 [Interleukin 1 receptor-like 1], IFNGR2 [Interferon γ receptor 2], IL12B [interleukin 12β], TNIP1 [TNFAIP3-interacting protein 1], TNFAIP3 [TNFα induced protein 3], and NFKBIA (NF-κB inhibitor A) (Loft et al., 2018; Tejasvi et al., 2012; Zuo et al., 2015). Even though the etiology is not clearly known, PsO can begin with bacterial infection followed by the release of anti-microbial peptides (Weisenseel, 2002). In a study by Munz et al. (2010), 16S rRNA sequencing of blood samples from 20 patients of psoriasis shed light on the presence of Staphylococci and/or Streptococci, depending on the type of psoriasis, suggesting an association between psoriasis and bacteremia (Munz et al., 2010). Certain anti-microbial peptides – LL37 and β-Defensins have been vastly implicated in the pathogenesis of psoriasis. LL37 complexes with host self-nucleic acids and ligate dendritic cells via TLR7 and TLR9, leading to loss of self-nucleic acid tolerance (Lande et al., 2007; Hollox et al., 2008; Ganguly et al., 2009). LL37 can protect keratinocytes from apoptosis, further aiding to psoriasis phenotype. LL37 and keratin-7 are some of the autoantigens targeted by T lymphocytes in psoriasis. Previously thought to be Th1-dominant disease, it is now known to be both Th1 and Th17 driven disease (Lee et al., 2004; Blauvelt, 2007). Since SOCS1 is a classical regulator of IFN-γ and IL-6 signaling, SOCS1 can skew T cells responses away from Th1 and Th17 (Starr et al., 1997; Alexander et al., 1999; Diehl et al., 2000). Interestingly, Foxp3+ regulatory T cells tend to show increased plasticity and lean toward Th1/Th17 phenotype when deficient in SOCS1 (Collins et al., 2011; Takahashi et al., 2011, 2017).
Psoriasis-like lesions can be induced on mouse skin by topical application of imiquimod, a TLR7 agonist (van der Fits et al., 2009; Lande et al., 2014). The imiquimod-induced mouse model for psoriasis shows similar histological and phenotypical characteristics to human plaque psoriasis and is believed to be a reliable induced model for studying the human disease (Palamara et al., 2004; van der Fits et al., 2009). TLR7/8 signaling in pDCs (Plasmacytoid dendritic cells) and MoDCs, in synergy with TLR4 signaling, can stimulate expression of IL12p35, IL23p19, and IL-6 which mediate Th1/Th17 polarization resulting in secretion of IFN-γ or IL-17 by Th1/NK and Th17 cells, respectively (Mosmann and Sad, 1996; Hamalainen et al., 2001; Acosta-Rodriguez et al., 2007; Nakae et al., 2007; Xu et al., 2007). IFN-γ is a potent activator of macrophages and inducer of CXCL9 (MIG) and CXCL10 (IP-10) in the epidermis, which then aids to recruit CXCR3+ Th1 cells, NK cells, and neutrophils to aggravate inflammation during early stages of the disease (Klunker et al., 2003; Ottaviani et al., 2006; Ferrari et al., 2015). It should be noted that CXCL10 is a strong biomarker of psoriasis, found in psoriatic plaques, and an active therapeutic target (Gottlieb, 1988; Ferrari et al., 2015). In a similar manner, TLR7 agonists can induce CCL2 (MCP-1) secretion by macrophages which then recruits CCR2+ Th17 cells and monocytes to the epidermis (Lembo et al., 2014). SOCS1 also maintains the expression of CCR7 on naïve T lymphocytes and aids in retaining them in the secondary lymphoid organs, highlighting a key role of SOCS1 is preventing unwarranted infiltration of naïve T cells into peripheral tissues like the skin (Yu et al., 2008). IL-17 can signal in both hematopoietic and non-hematopoietic cells. Apart from its regular housekeeping functions and synergizing with TNF-α, IL-17 can act as an amplifier of inflammation as it can stabilize other cytokines’ mRNA by inducing expression of RNA stabilizing intracellular protein HuR (Amatya et al., 2017). TLR7 signaling is particularly amplified in pDCs due to their relatively high expression of TLR7 (Jarrossay et al., 2001; Kadowaki et al., 2001; Hänsel et al., 2011). Furthermore, TLR7 and 8 signaling have been known to upregulate their own gene expression, in an autocrine fashion (Lombardi et al., 2009). Of note, a recent study by Yu et al. (2018) has demonstrated TLR7 signaling also induces SOCS1 gene expression and that SOCS1 can suppress TLR7-mediated type-1 IFN secretion by pDCs, another vital element in psoriasis pathogenesis. The mechanism is both through IRF7 degradation, required for TLR7 signaling, and the inhibition of tyk2, required for type-1 IFN signaling (Gilliet et al., 2008; Piganis et al., 2011; Baldwin et al., 2013; Gui et al., 2016; Yu et al., 2018). UV-B narrow band can also reduce type-1 IFN signaling via facilitating phosphorylation-dependent ubiquitination of the IFN receptor chain – IFNAR1 (Gui et al., 2016). Grine et al. (2015) reported that IFNAR1-deficient mice were partially protected from Imiquimod-induced inflammation.
Th17 cells are major players in imiquimod-induced psoriasis as well, and pDCs have been reported to secrete pro-Th17 cytokines (e.g., IL-6) in response to TLR7 ligation (Yu et al., 2010). Kim et al. (2016) showed that upon imiquimod application, the CD27-Vγ1- γδ T cell population was significantly increased. Moreover, anti-p40 (a subunit shared by IL-12 and IL-23) and PD-L1-F fusion protein therapy resulted in assuagement of the disease (Krueger et al., 2007; Kim et al., 2016). Moreover, IL-22, a Th17 cytokine, has been implicated in promoting acanthosis and immune cell recruitment in the epidermis and high serum levels of IL-22 are correlated with disease severity (Boniface et al., 2005; Wolk et al., 2006). IL-17A, the first member of the IL-17 family, has a distinct pattern of gene regulation in differentiated and undifferentiated keratinocytes. Spleen tyrosine kinase (Syk) can mediate IL-17 induced gene expression in keratinocytes and is also involved in TLR7 signaling, making it an interesting candidate to study in the context of psoriasis (Chiricozzi et al., 2014; Wu et al., 2015; Aouar et al., 2016). In a Syk-independent branch of the pathway, IL-17 signaling has a unique ability to synergize with other cytokine signaling pathways by stabilizing their downstream gene transcripts and preventing their degradation, leading to an amplified inflammatory response (Amatya et al., 2017). Anti-IL-17A MAb therapy has been approved for treatment of moderate to severe psoriasis plaques, though some patients with moderate to severe psoriasis plaques have been known to suffer from unexpected side effects like nasopharyngitis, arthralgia, and upper respiratory tract infections (Papp et al., 2013; Rich et al., 2013; Langley et al., 2014; Gordon et al., 2016). Moreover, the application of anti-IL-17A therapy in patients with mild psoriasis is limited in the context of risk versus benefit as IL-17 plays a protective and reparative role in the gut and barrier tissues (Song et al., 2015). As such, therapeutic alternatives to anti-IL-17 therapies remain an unmet need in individuals with mild to moderate disease.
TLR4 expression is upregulated in PBMCs in human patients with psoriasis, and variants of TLR4 are implicated in both plaque-type psoriasis and psoriatic arthritis (Garcia-Rodriguez et al., 2013; Panzer et al., 2014; Smith et al., 2016). TLR4 can interact with bacteria endotoxin LPS and initiate an inflammatory signaling cascade (Janssens and Beyaert, 2003). SOCS1 can regulate TLR4 mediated inflammation by inducing degradation of TRAF6, IRAK1, and Mal protein, which subsequently prevents p65 phosphorylation and activation (Nakagawa et al., 2002; Mansell et al., 2006; Jager et al., 2011; Zhou et al., 2015). The importance of SOCS1 is highlighted in that SOCS1 knockout macrophages have increased sensitivity to LPS (Sachithanandan et al., 2011). TLR4 signaling may also mediate crosstalk with STAT3 signaling via MyD88-induced IL-6 (Yamawaki et al., 2010). STAT3 is a widely accepted oncogene and inflammatory mediator that will be discussed later in this article. STAT3 is an interesting target in the context of psoriasis as it is required for IL-6, IL-22, and IL-23 signaling and has been linked with the development of psoriasis in a transgenic mouse model (Sano et al., 2005; Figure 3 touches upon SOCS1-mediated TLR regulation briefly).
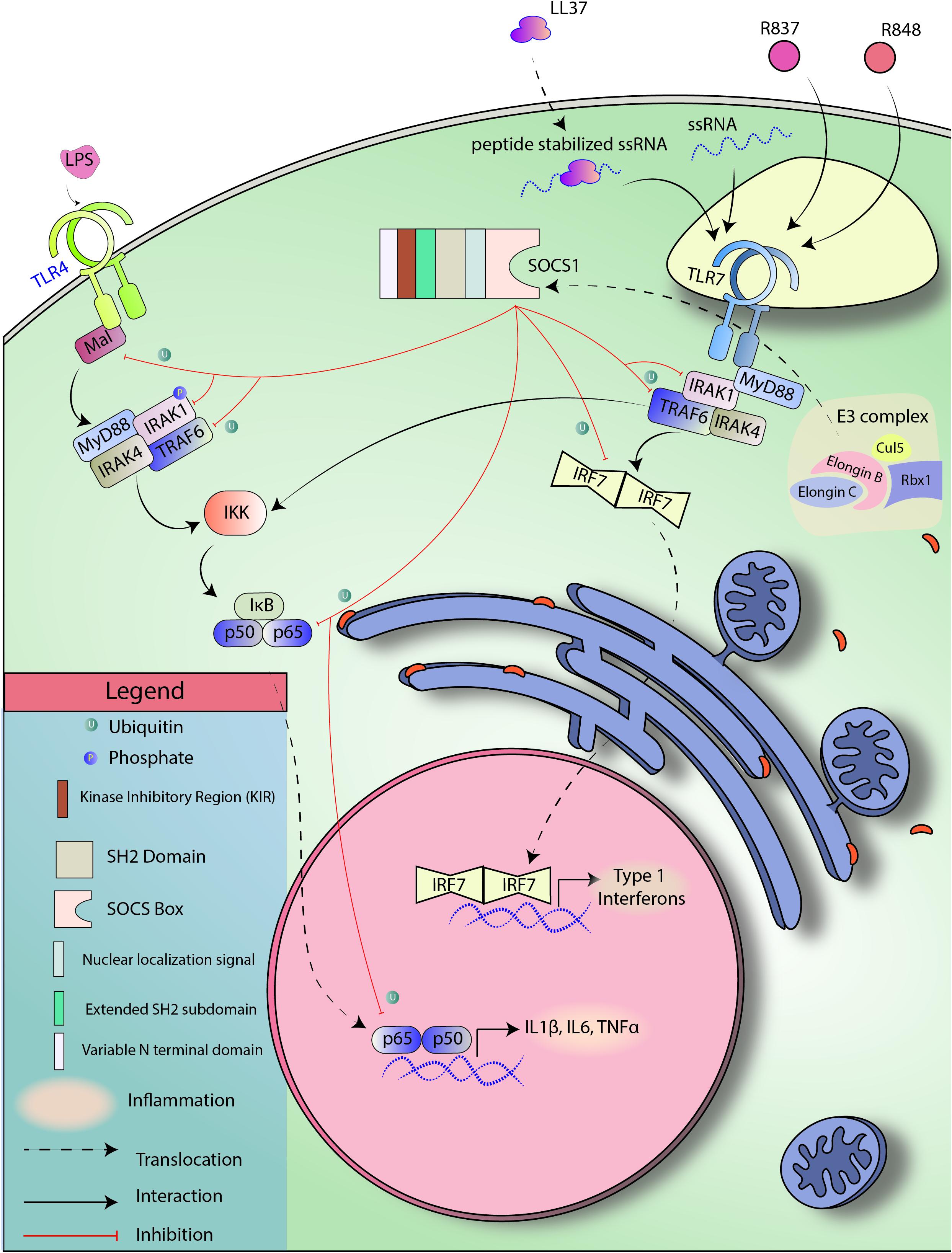
Figure 3. SOCS1 regulates TLR signaling. TLR4 and TLR7 are pattern recognition receptors known to be responsive toward LPS and ssRNA, respectively. TLR4 and TLR7 signal through NF-kB and IRF7, respectively to induce the expression of target genes. SOCS1 can induce SOCS Box-mediated ubiquitination of Mal, TRAF6, p65, and IRF7 to block both the signaling cascades.
Cancer
Anomalous gene expression by cancer cells can lead to cell transformations. Such transformed cells can sustain unimpeded growth, evade contact inhibition, ignore apoptotic signals, undergo metastasis and angiogenesis, and evade the immune response (Hanahan and Weinberg, 2011). While the JAK/STAT pathway is required for cytokine signaling and alerting the immune system for tumor clearance, it can also facilitate tissue survival and neoplasia (Classen et al., 2009; Bunker et al., 2015; La Fortezza et al., 2016). SOCS-1, being a classical regulator of JAK/STAT signaling, is a potent tumor suppressor as aberrant SOCS1 gene methylation and allelic mutations have been linked to different types of malignant cancers (Fujitake et al., 2004; Melzner, 2005, Melzner et al., 2006). Epigenetic inactivation due to CpG methylation of SOCS1 is frequently linked to Hepatocellular carcinoma, human gastric carcinoma, melanoma, multiple myeloma, pancreatic ductal neoplasm, and acute myeloid leukemia (Franke, 2001; Yoshikawa et al., 2001; Chen et al., 2003; Fukushima et al., 2003; Galm, 2003; Oshimo et al., 2004; Mottok et al., 2007; Liu S. et al., 2008). SOCS1 mediated negative feedback signaling is paramount for not only reducing inflammation, but also to curb unchecked cell growth.
Suppressor of cytokine signaling 1 has been shown to regulate, directly or indirectly, a number of molecules and pathways that have been implicated in cancer – CDK2, CDK4, Cyclin D1, MAPK/p38, PDL1, STAT1, STAT3, STAT6, p53, p21, FAK, E-cadherins, Met tyrosine kinase, type I and II IFN, and numerous proinflammatory cytokines (Liu, 2003; Ritz et al., 2008; Neuwirt et al., 2009; Souma et al., 2012; David et al., 2014; Gui et al., 2015; Yeganeh et al., 2016; Liau et al., 2018; Figure 4 briefly explains the mode of regulation by SOCS1).
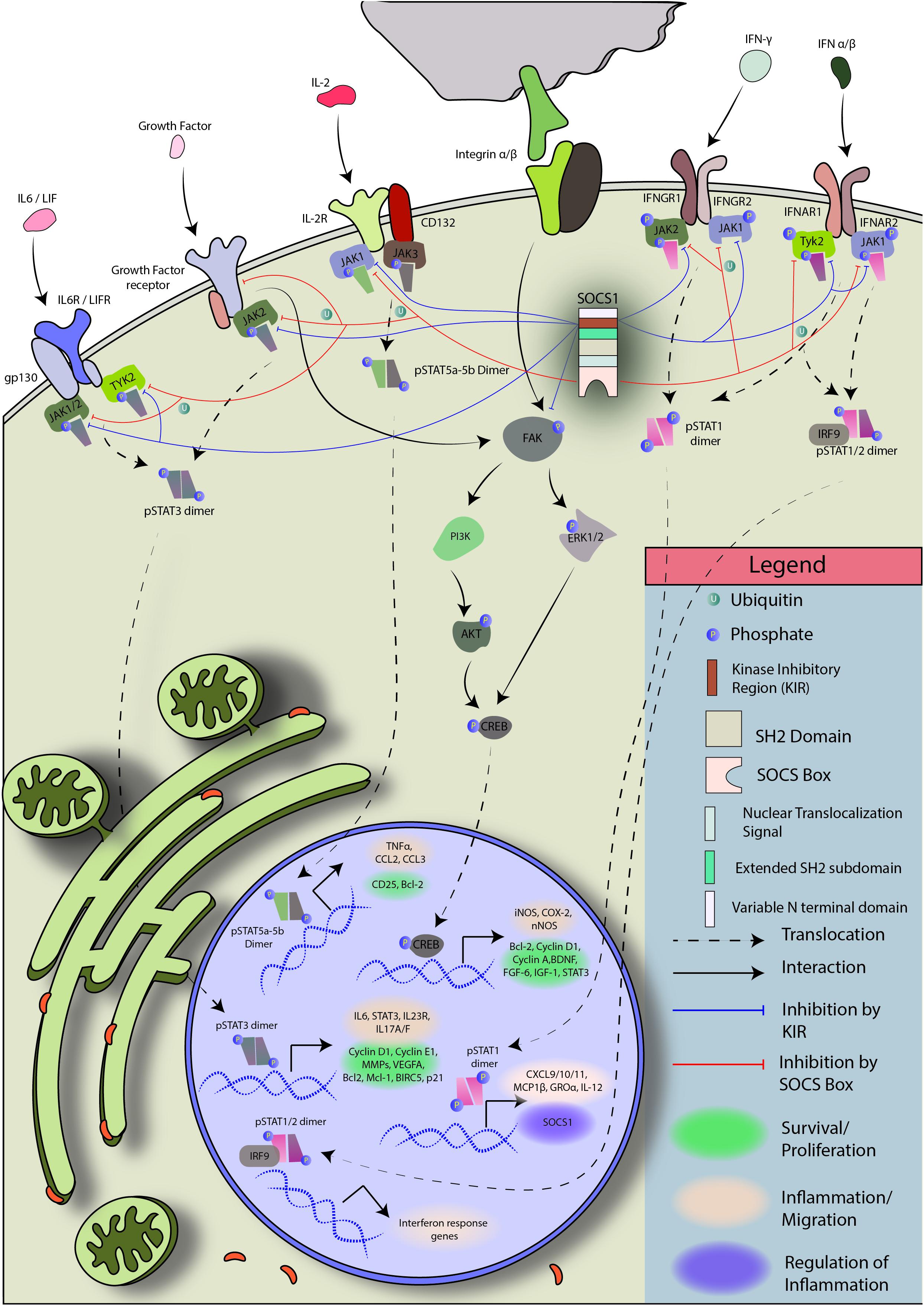
Figure 4. Regulation of JAK/STAT and FAK signaling by SOCS1: Janus kinases (JAKs) are physically close to cytokine receptors. When a cytokine binds its cognate receptor, the respective JAK phosphorylates itself and the cytokine receptor. Receptor phosphorylation creates docking sites for STAT protein binding and brings the associated JAKs in propinquity. JAK autophosphorylation is required for enzymatic activation. The activated JAKs then phosphorylate their target pre-formed STAT dimers or STAT monomers, which form homo/heterodimers and enter the nucleus to initiate transcription. SOCS1 is a regulator of JAK1/2 and TYK2. It can block phosphorylated JAK1, JAK2, and TYK2 to prevent STAT activation and dimer formation, putting a halt to JAK/STAT signal transduction (Braunstein et al., 2003; McNally and Eck, 2014).
One of the ways non-immune cells, including cancer cells, promote immune-suppression and induction of tolerance is by expressing PDL1 (also known as B7.H1 or CD274), a ligand for PD1 expressed by T cells. The interaction between PD1 and PDL1 can cause anergy or even programmed cell death in T cells (Dong et al., 2002). Anti-PDL1 is a common anti-cancer therapy facilitating immune cells recognition and obliteration of tumor cells. IFN β and γ receptor signaling contribute to significantly increase the expression of PDL1 through the JAK1/2-STAT1/2/3-IRF1 axis (Garcia-Diaz et al., 2017). Since SOCS1 is a natural regulator of both type-I and II IFN signaling and JAK1/2/STAT1 signaling, it also indirectly regulates PDL1 expression. Nonetheless, there are also contradicting reports that suggest increased SOCS1 expression as an accomplice in melanoma, colorectal cancer, breast cancer, and neuroendocrine cancer (Raccurt et al., 2003; Li et al., 2004; Laner-Plamberger et al., 2013; Tobelaim et al., 2015; Berzaghi et al., 2017). Hence, further studies are required to elucidate alternative pathways modulated by SOCS1 and cell-type specific functions. It should also be noted that SOCS1 overexpression has been reported to subvert IFN-α therapy in chronic myeloid leukemia, therefore, a balance, rather than an excess, of SOCS1 is required for normal cell functioning.
STAT3 (also known as acute phase response factor) is DNA-binding, an intracellular signaling protein that has pleiotropic effects on embryogenesis, oncogenesis, tumor suppression, cell differentiation, growth, and both innate and adaptive immunity (Akira, 2000; de la Iglesia et al., 2008, 2009). IL-6 signaling is known to induce STAT3 gene expression and its phosphorylation-mediated activation resulting in the transcription of STAT3 target genes (Ichiba et al., 1998). The activation of STAT3 can be induced by a variety of cytokines including IL-6 and IFN-α (Puthier et al., 1999). Intriguingly, unphosphorylated STAT3, in response to IL-6, has also been reported to aid in inflammation by interacting with NF-κB and upregulating CCL5, IL8, IFNβ, and ICAM1 (Matikainen et al., 1999; Yang et al., 2007). In addition to non-receptor kinases such as JAKs, Bcr-abl1, and Src., STAT3 can also be phosphorylated by growth factor associated kinases, like Trk, (Ng et al., 2006; Al Zaid Siddiquee and Turkson, 2008). Under physiological conditions, STAT3 signaling is highly regulated. However, under pathological conditions activated STAT3 has been implicated in hematological and non-hematological tumors, largely through promotion of autocrine IL-6 signaling and secretion that drives cancer progression and multidrug resistance (Koudstaal et al., 1967; Buettner et al., 2002; Yu and Jove, 2004; Yeh et al., 2006; Al Zaid Siddiquee and Turkson, 2008; Huang et al., 2010; Mace et al., 2013; Cheng et al., 2016). Mouse fibroblasts artificially induced to have constitutive expression of dimerized pSTAT3 were autonomously capable of causing tumors when transferred to nude mice (Bromberg et al., 1999). Moreover, aberrant IL6/JAK/STAT3 signaling has been observed in human patients of cervical, breast, ovarian, head and neck, colorectal, prostate, renal, oesophageal, non-small cancers, brain cancers, sezary syndrome, retinoblastoma, and lymphoma (Eriksen et al., 2001; Rahaman et al., 2002; Zhang et al., 2002; Chung and Chang, 2003; Chang C.H. et al., 2013; Konnikova et al., 2003; Riedel et al., 2005; Lane et al., 2011; Lesina et al., 2011; Culig and Puhr, 2012; Chen et al., 2013; Jo et al., 2014; Jinno et al., 2015; Kotowicz et al., 2016; Kitamura et al., 2017; Johnson et al., 2018). STAT3 phosphorylation followed by irradiation therapy and chemotherapy presents a challenge for cancer treatment since pSTAT3 contributes to the transcription of anti-apoptotic genes including Mcl1, Bcl2, Bcl-xL, and BIRC5 (Bromberg et al., 1999; Catlett-Falcone et al., 1999; Alas and Bonavida, 2001; Real et al., 2002; Diaz, 2006; Kujawski et al., 2008; Yu et al., 2013). Irradiated breast cancer cells have been reported to secrete SASP factors, including IL-6, which aides in tumor progression, angiogenesis, and metastasis (Kujawski et al., 2008; Barbieri et al., 2010; Yu et al., 2013). STAT3 has been known to induce HIF-1α gene expression, required for tumors to survive in hypoxia, and can also regulate p53, Cyclin D1, E1, and p21 (Kortylewski et al., 2005; Niu et al., 2005, 2008; Chang Q. et al., 2013). Furthermore, aberrant STAT3 expression may play a role in maintaining survival and plasticity of cancer stem cells, as STAT3 is known to support pluripotency by upregulating sox2 [SRY-box 2], Nanog [Homeobox protein nanog], and c-myc [MYC proto-onco gene] (Kiuchi et al., 1999; Foshay and Gallicano, 2008; Gregory et al., 2008; Kamiya et al., 2011). Constitutive pSTAT3 signaling is also known to provide resistance to chemotherapy in breast cancer cells via a similar downstream process as mentioned previously (Real et al., 2002). It is worth noting that STAT3 integrates with the PI3K pathway, another major signaling pathway governing cell survival and apoptosis, by regulating the expression of the regulatory subunits of the Class IA PI3K enzyme during lactation and involution (Abell and Watson, 2005). Various STAT3 inhibitors have been studied to inhibit cell proliferation in cancer cell lines (Swiatek-Machado et al., 2012). Moreover, STAT6, a typical Th2 signaling molecule, has been reported to play role in glioma progression as well, both alone, and as an accomplice to STAT3 (Merk et al., 2011; Yan et al., 2016). SOCS1 is one of the natural regulators of STAT3 and STAT6 signaling and has tremendous potential as therapeutic. SOCS1 can localize to the nucleus via NLS and aid in p53 phosphorylation, hence, it is also a part of DNA damage response (Mallette et al., 2010). This partially explains why SOCS1-/- mice are relatively more susceptible to cancer. SOCS1 can also regulate many cell cycle components directly. Natatsuka et al. (2015) demonstrated that SOCS1 can bind ATR through p53 and cause a G2/M arrest of gastric cancer cell lines. Of note, SOCS1 adenoviral gene therapy has been shown to impede cell growth in gastric cancer cells by reducing levels of pSTAT3 (Natatsuka et al., 2015; Sugase et al., 2018). Similarly, SOCS1 gene therapy was also shown to augment irradiation mediated DNA damage in Esophageal Squamous Cell Carcinoma (ESCC) (Souma et al., 2012; Sugase et al., 2017). The twist in the story comes with STAT1, which is primarily associated as a target of SOCS1-meditated regulation. STAT1 is known to have both tumor suppressive and oncogenic effects (Rock et al., 2018). Aberrantly low STAT1 expression is reported to have a poor clinical outcome in several cancers, including melanoma and breast cancer. STAT1-/-mice are more prone to experimentally induced tumors, as well as develop cancer spontaneously (Lesinski et al., 2003; Chan et al., 2012; Hosui et al., 2012; Hix et al., 2013). Activated STAT1 is also known to cooperate with p53 to induce apoptosis in malignant cancer cells (Forys et al., 2014; Youlyouz-Marfak et al., 2008). Part of the tumor suppressive effects can be ascribed to heterodimer formation with STAT3. The STAT1-STAT3 heterodimer governs transcription of a different set of genes, often resulting in apoptosis instead of survival (Thyrell et al., 2007; Regis et al., 2008). Moreover, STAT1 and STAT3 reciprocally regulate each other’s expression and activity and even compete for JAK docking sites for phosphorylation (You et al., 2013; Friedrich et al., 2017). On the other hand, several studies have portrayed STAT1 masquerading as an oncogene, more of which is described in the following review (Rock et al., 2018). This scenario highlights the complexity of the effects of SOCS-mediated regulation.
Focal Adhesion Kinases (or protein tyrosine kinase 2) reside within focal adhesions where the cell cytoskeleton contacts the extracellular matrix. FAK activation can result from receptor signaling via receptor tyrosine kinases (RTK), cytokine receptors, growth factor receptors, G-protein coupled receptors, and integrins through FERM domain interactions (Frame et al., 2010). There is increasing evidence that FAK autophosphorylation at Y397 is associated with oncogenesis by aiding in cell migration, FAK activity can lead to turnover of focal adhesion points and upregulation of MMPs, and the inhibition of the autophosphorylation has been shown to reduce tumor growth (Sieg et al., 2000; Hauck, 2002; Dunty et al., 2004; Cui et al., 2006; Hochwald et al., 2009; Heffler et al., 2013). One of the reported mechanisms for focal adhesion turnover is through phosphorylation of actin-binding protein cortactin (Tomar et al., 2012). The blockade of FAK-Cortactin signaling pathway has been shown to make cells susceptible to radiation therapy in head and neck cancer (Eke et al., 2012). Furthermore, MMP9 expression by FAK signaling is implicated in orthotopic breast cancer metastasis (Mitra et al., 2006). FAK governs not only tumors, but also stromal cell biology (Sulzmaier et al., 2014). Besides the traditional roles of FAK, it is also involved in epithelial-to-mesenchymal transition (EMT), bypassing apoptosis, and angiogenesis (Xu et al., 2000; Kurenova et al., 2004; McLean et al., 2005; Weis et al., 2008; Zouq et al., 2009; Canel et al., 2013; Fan et al., 2013). Activated FAK protein can interact with src-kinase to form a dual-kinase complex and then upregulate MAPK-ERK kinase cascade to induce migration (Schlaepfer and Hunter, 1997). αvβ5 integrin–FAK–AKT signaling pathway blockade has been reported to prevent attachment-dependent apoptosis in murine ovarian carcinoma cells (Lane et al., 2010). FAK pharmacological inhibitors are being investigated as cancer chemotherapeutics and FAK inhibition has been shown to ameliorate tumor growth, metastasis, and angiogenesis in mouse models of adenocarcinoma, ovarian carcinoma, pancreatic cancer, and non-small lung cancer (Halder et al., 2007; Slack-Davis et al., 2009; Lane et al., 2010; Stokes et al., 2011; Chen et al., 2012; Jean et al., 2014). SOCS1 is known to be induced by PDGF and integrin signaling. SOCS1 binds directly with Y397-phosphorylated FAK through the SH2 and KIR domains and induce ubiquitination followed by proteasomal degradation (Liu, 2003). SOCS1 also modulates JAK/STAT signaling of other growth factors which lie upstream of FAK in mice therefore indirectly regulating FAK signaling. Ergo, SOCS1 holds great potential in the treatment of FAK-driven cancers. Of note, a quite recent study showed that SOCS1 gene therapy can prohibit proliferation of gastrointestinal stromal tumors by interfering with FAK and PI3K pathway (Sugase et al., 2018). One of the challenges with FAK inhibition therapy would be the fact that PYK2, a homolog with similar function, can substitute for FAK activity in its absence, and hence, the inhibitors need to be carefully designed or a combinatorial therapy should be preferred. While there is some evidence that SOCS1 may become associated with PYK2 for activity modulation, there is a need to elucidate the mechanism and confirm the finding before drawing a strong conclusion (Masuhara et al., 1997). Another challenge would be to consider the pleiotropic functions of FAK in housekeeping tasks.
Lupus
Like most autoimmune diseases, Lupus’ etiology is not clearly known. A well-established model for studying SLE is MRL/LPR mice, which are known to develop lupus-like pathology and clinical manifestation closely resembling the human condition, spontaneously (Perry et al., 2011). Splenomegaly and lymphadenopathy due to hyperproliferation of CD3+ CD4- CD8- T cells is an immunological feature of these mice (Zhang et al., 2009). Lupus-like pathology can also be created by topical treatment of mice with TLR7 agonists like R837 (imiquimod) and R848 (resiquimod) (Yokogawa et al., 2014). While TLR7 signaling exacerbates the disease condition, a recent study has found that TLR9 may have a protective role in SLE since TLR9-/- mice had an accelerated disease phenotype (Liu et al., 2018). Polymorphism in SOCS1 gene has been correlated with the occurrence of SLE (Chan et al., 2010).
Malignant NETosis by neutrophils can lead to ANA production, one of the hallmarks of SLE (Yu and Su, 2013). These circulating ANA and nucleic acid can cause type I IFN secretion via TLR7/8 activation. Multiple reports have hinted toward the contribution of type I and II IFNs in disease priming and progression (Vallin et al., 1999; Baechler et al., 2003; Han et al., 2003; Hua et al., 2006; Lit, 2006; Elkon and Santer, 2012; Munroe et al., 2014). The ANA can induce more NETosis and perpetuate the cycle of inflammation (Murphy et al., 1998; Marzocchi-Machado et al., 2002; Lande et al., 2007, 2011; Garcia-Romo et al., 2011). The condition can be exacerbated by LL37-mediated stabilization of DNA (Lande et al., 2011). This anti-microbial peptide has also been known to induce M1-phenotype in macrophages and activating inflammasomes resulting in increased IL-18, another biomarker for SLE (Kahlenberg et al., 2011). TLR7-mediated self RNA ligation and duplication in the TLR7 gene have been known to upregulate autoreactive B cell responses. Similar to psoriasis, LL37 can also complex with self-RNA, stabilize, and internalize it for TLR7 activation and promote inflammation and ANA production (Blanco et al., 1991; Pisitkun, 2006; Ganguly et al., 2009). Since the pro-inflammatory cytokines secreted in the abovementioned processes signal through the JAK/STAT pathway, the pathway becomes a particularly effective therapeutic target, especially in Lupus. It has also been reported that monocytes of SLE patients have hyperactive JAK/STAT signaling (Li et al., 2011).
Reduced expression of SOCS1 and/or increased IFN-γ/IL-6 signaling are rampant in SLE rodent models and human patients (Baechler et al., 2003; Fujimoto, 2004; Harigai et al., 2008; Sharabi et al., 2009; Sukka-Ganesh and Larkin, 2016). Since SOCS1 is known to regulate JAK/STAT pathway and multiple TLR responses including TLR4 and TLR7, directly or indirectly, SOCS1 mimetics have a remarkable therapeutic potential which should be explored (Kinjyo et al., 2002; Nakagawa et al., 2002; Strebovsky et al., 2011; Yu et al., 2018). Recently, a small-molecule inhibitor of JAK1 and 3, tofacitinib, has been shown to assuage lupus progression in MRL/LPR mice (Clark et al., 2014; Furumoto et al., 2017).
The ANA, in complex with their target epitopes, make their way to the kidneys for clearance where they are phagocytosed by mesangial cells, leading to a condition called lupus nephritis (LN) which is a major cause of morbidity in SLE patients (Mak et al., 2007; Almaani et al., 2017). These phagocytosed antibodies can further create a nuisance by causing T cell infiltration into the mesangial membrane and induce upregulation of MHC II of the mesangial cells, leading to incessant inflammation and mesangial cell hypertrophy. Therefore, successful therapy for SLE should also clear these infiltrating leucocytes and reduce nephritis intensity. Our group has previously shown the importance of SOCS1 pathway in SLE and reduced SOCS1 expression in patients (Sukka-Ganesh and Larkin, 2016).
Recurrent Uveitis
Uveitis is a severe disease of the eye that accounts for more than 10% of the visually handicapped population in the United States (Acharya et al., 2013; González et al., 2018). It can manifest as anterior uveitis, in the front of the eye, posterior uveitis, in the back of the eye, or pan uveitis, throughout the eye (Nussenblatt, 1990). The disease can be induced by immunizing with retinal antigens, using appropriate adjuvants (Wiechert et al., 2001; Deeg et al., 2002). The pathogenesis is mostly due to immune cells infiltrating the eye and causing inflammation, with pathologic T cells being the prominent drivers of the disease. Studies are carried out in mouse or rat model of EAU, however, the equine disease resembles the most to the human condition (Gilger et al., 1999; Wiechert et al., 2001; Deeg et al., 2004; Malalana et al., 2015). In equine recurrent uveitis, S-antigen and IRBP are the primary antigens but the polyclonal T cell expansion causes epitope spreading and hence brings out the recurrent nature of the disease. A similar pattern may be expected in human disease (Deeg et al., 2006). Migration of immune cells to the eye is an important event for the disease to progress. Chemokines containing the CC motif, including MIP-1α (CCL3), MCP-1 (CCL2), and RANTES (CCL5), have a prominent role in recruiting T cells and monocytes to the inflamed eye (Crane et al., 2001). CCL2 can be upregulated by macrophages in response to IFN γ (Bauermeister, 1998), and RANTES expression is known to be modulated by STAT3 signaling (Yang et al., 2007). In addition, STAT3-/- T cells were unable to mount a spontaneous autoimmune response. This highlights the prime role of JAKs in disease progression as both STAT3 and IFN-γ signaling depend on it for signal propagation. The importance of JAKs in the disease underscores the basis for using SOCS1 as a therapeutic approach (Liu X. et al., 2008). Current therapies include corticosteroids and NSAIDs, however, they have been known to cause severe side effects like glaucoma and cataract (Nussenblatt, 2002). Constitutive expression of SOCS1 in the retina was reported to reduce recruitment of lymphocytes, resulting in reduced inflammation (Yu et al., 2011). Moreover, ocular topical treatment with a SOCS1 mimetic, containing the KIR region called SOCS1-KIR, has been shown to provide alleviation in the disease condition in Lewis rats and B10.RIII mice (He et al., 2015, 2016; Ahmed et al., 2018). As previously described, SOCS1 prevents naïve T cells from infiltrating ocular tissues by maintaining the expression of CCR7, required for retention of these cells in the secondary lymphoid organs where CCL19 and CCL21 are constitutively expressed (Yu et al., 2008). SOCS1 is a therapeutic candidate worth exploring in the context of uveitis (see Figure 5 for summary).
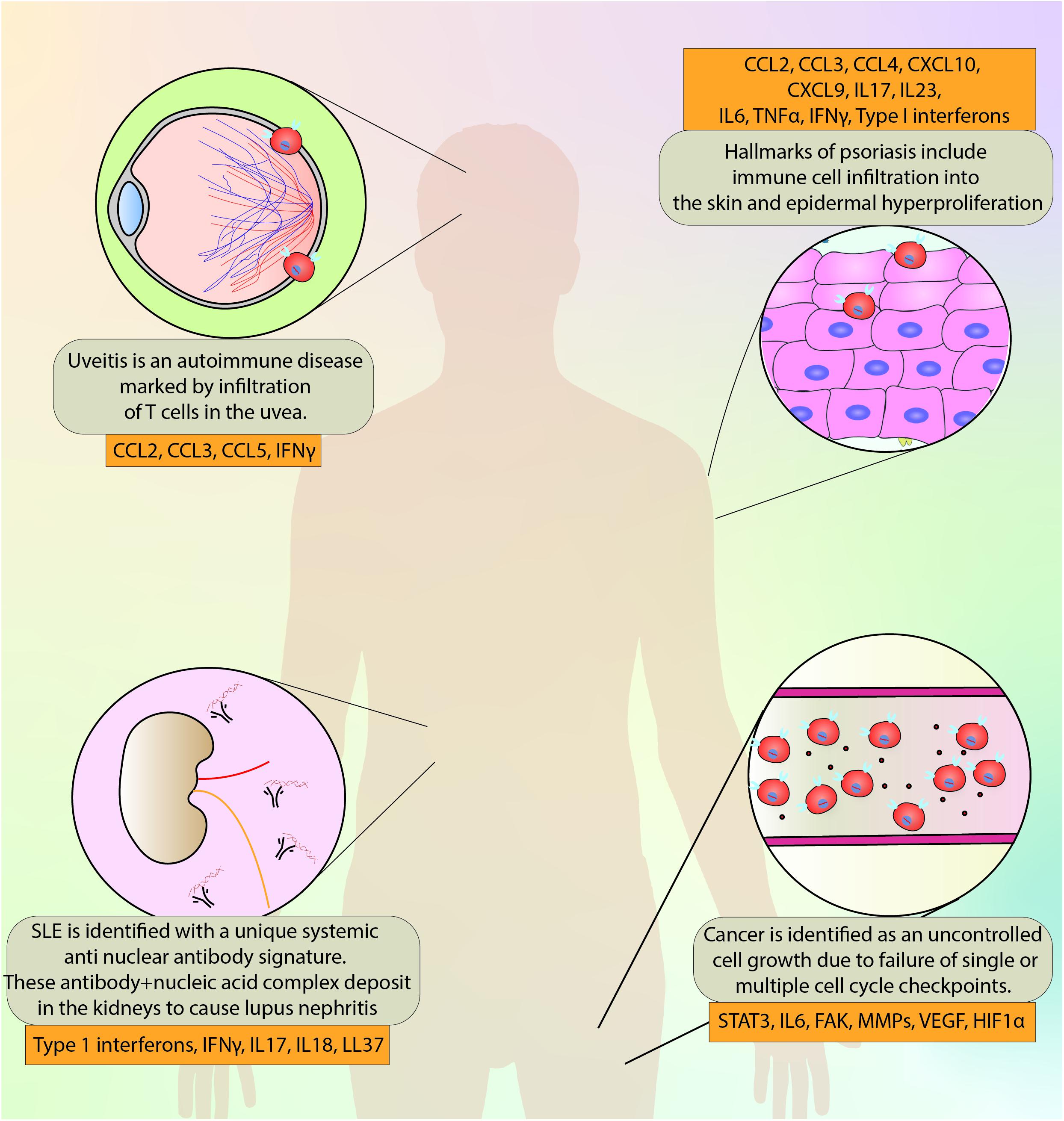
Figure 5. A brief overview of the disorders discussed in this review and the major molecules implicated in the pathogenesis and progression.
Other JAK Inhibitors–Approved and Under Investigation
Initial tests for the feasibility of tyrosine kinase inhibitors were done in the context of cancers in early 2000. Imatinib, a BCR-ABL tyrosine kinase inhibitor, was the first such inhibitor shown to be effective in chronic myelogenous leukemia (CML) and since then a number of kinase inhibitors have been approved by FDA for cancers (Druker et al., 2001). The success of these tyrosine kinase inhibitors undoubtedly paved a way for JAK inhibitors to undergo clinical trials. Ruxolitinib was the first FDA approved JAK inhibitor targeting tumors with hyperactive JAK/STAT signaling pathways (Harrison et al., 2012; Verstovsek et al., 2012). The approval of Ruxolitinib not only confirmed that JAK inhibitors were feasible but also safe for use as therapeutics. JAK inhibitors or jakinibs can regulate multiple cytokine and growth factor signals, while still allowing non-JAK signaling cytokines like TNFα, IL-1β, IL-17, and IL-8 to function normally, precluding an immunocompromised condition for the patient. Although, the same fact also underscores the limitation of this therapy, hence, jakinibs must not be confused for an autoimmunity panacea.
The first generation of jakinibs were pan-inhibitors, i.e., they blocked multiple JAKs. The shortcoming of the first generation of jakinibs is (1) they would also block JAKs required for anti-inflammatory cytokines, like IL-10, to signal (2) they could expose the patient to infections (3) they can block hematopoietic cytokines that typically signal through JAK1/JAK2 from functioning and cause cytopenia, which may not be desirable. Tofacitinib, a first generation JAK1/3 inhibitor, was the first jakinib to be approved for autoimmunity in humans (Meyer et al., 2010). It is a reversible competitive inhibitor for the ATP binding site of JAK1 and 3, and to a much less extent, of JAK2 and TYK2 (Philip et al., 1987). Initially, it was particularly advised for RA patients where methotrexate could not be administered or did not work, and later, it was proven to be non-inferior to the standard care – adalimumab, a TNF blocker (Fleischmann et al., 2017; Kotyla, 2018). Tofacitinib is also the most studied jakinib. There are other first-generation jakinibs in clinical trials for autoimmune diseases: Ruxolitinib (JAK1 and JAK2 inhibitor) for GVHD (NCT02913261), Psoriasis (NCT00617994), and Vitiligo (NCT02809976); Baricitinib (JAK1 and JAK2 inhibitor) for GVHD (NCT02759731) and Diabetic nephropathy (NCT01683409). In contrast, the second generation jakinibs are specific to a certain JAKs. This allows for a better targeting tactic with relatively reduced side effects than first gen jakinibs. Some of the second generation jakinibs being investigated are Upadacitinib (JAK1 inhibitor) for Atopic dermatitis and PF-06651600 (JAK3 inhibitor) for RA (NCT02969044) and ulcerative colitis (NCT02958865). Jakinibs have become a promising treatment for a range of immunity-related disorders including psoriasis, vitiligo, GVHD, lymphoma, solid tumors, SLE, ulcerative colitis, and atopic dermatitis (Buchert et al., 2016; Schwartz et al., 2017; Hosking et al., 2018).
Suppressor of cytokine signaling 1 KIR, a SOCS1 mimetic containing only the KIR domain, acts as a pseudosubstrate for JAK1, JAK2, and TYK2, with no known interaction with JAK3 (Liau et al., 2018). However, SOCS1 KIR can also interact with FAK, setting it apart from every other jakinib. What makes SOCS1 KIR more attractive as a therapeutic is its similarity to the naturally occurring protein SOCS1. Nonetheless, mimetic peptide drugs have disadvantages to their small molecule counterparts in terms of high cost, low permeability, proteolytic instability, and poor oral bioavailability, though a number of strategies are being used to improve these features in peptide drugs (Otvos and Wade, 2014; Di, 2015). For example, modifications like N-acetylation and c-amidation can improve peptide stability (Volonterio et al., 2003; Sato et al., 2006), use of lipid membrane and/or transporter systems for better bioavailability (Mahato et al., 2003; Griffin and O’Driscoll, 2011), and increasing serum protein binding of the peptide to reduce renal clearance (Pollaro and Heinis, 2010). Even though peptide drugs share an extremely small market share compared to small molecules (Vlieghe et al., 2010), their better specificity, low toxicity profile, and low drug-drug interaction potential makes them viable choice for the future once the challenges around their ADME (absorption, distribution, metabolism, and excretion) are overcome with progress in computational biology, metabolomics, and proteomics.
Discussion
Suppressor of cytokine signaling 1 is an essential molecule for maintaining immune homeostasis and subverting inflammation. Disorders arising from excess inflammation or SOCS1 deficiency can be potentially treated with SOCS1 mimetics (Ahmed et al., 2015). While SOCS1 has promising potential in many disorders, it should be noted that new targets and actions of SOCS1 are still being discovered and not all the effects of this protein are beneficial in autoimmune diseases and cancer. For instance, SOCS1 degrades IRS1 and IRS2, required for insulin signaling, via the SOCS Box domain, thus, limiting its potential in type-2 diabetes (Rui et al., 2002). However, such challenges can be met by using SOCS1 mimetic peptide lacking the SOCS Box domain. As of now, SOCS1 gene therapy and mimetic-peptide biologics are active areas of research around the globe. Jakinibs have gained a great deal of attention in the last two decades for their efficacy in cancer and autoimmune diseases and we believe SOCS1 mimetics would be a great addition to the arsenal of jakinibs. Nonetheless, detailed safety and efficacy studies need to be carried before directly comparing SOCS1 mimetics to other jakinibs.
Author Contributions
JS and JL wrote the manuscript. JS designed the figures. JL reviewed the figures.
Funding
Research in JL lab was supported by the Lupus Research Institute, National Psoriasis Foundation Awards to the University of Florida.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ANA, Antinuclear antibodies; ATR, Ataxia telangiectasia and Rad3 related; dsDNA, Double stranded deoxyribonucleic acid; EAE, Experimental autoimmune/allergic encephalomyelitis; EAU, Experimental autoimmune uveitis; ERU, Equine recurrent uveitis; FAK, Focal adhesion kinases; GVHD, Graft vs. Host Disease; IFN, Interferons; IL, Interleukin; IRBP, Interphotoreceptor retinoid-binding protein; IRF, Interferon regulatory factor; JAK, Janus Kinase; KIR, Kinase inhibitory region; MAb, Monoclonal antibody; MCP-1, Monocyte chemoattractant protein; MIP-1ββ, Macrophage inflammatory protein; MMP, Matrix Metalloproteinases; MoDCs, Monocyte-derived Dendritic Cells.; MS, Multiple sclerosis; NET, Neutrophil extracellular trap; NF κκB, Nuclear Factor κκB; NSAID, Non-steroidal anti-inflammatory drugs; PD1, Programmed cell death-1; PDL1, Programmed cell death ligand-1; pSTAT, phosphorylated Signal Transducer and Activator of Transcription; RANTES, Regulated on activation of normal T-cell-expressed and secreted; RNA, Ribonucleic acid; SASP, Senescence-associated secretory phenotype; SLE, Systemic lupus erythematosus; SOCS1, Suppressor of cytokine signaling 1; STAT, Signal transducer and activator of transcription; Th, T helper; TLR, Toll-like receptor.
References
Abell, K., and Watson, C. J. (2005). The Jak/Stat pathway: a novel way to regulate PI3K activity. Cell Cycle 4, 897–900. doi: 10.4161/cc.4.7.1837
Acharya, N. R., Tham, V. M., Esterberg, E., Borkar, D. S., Parker, J. V., Vinoya, A. C., et al. (2013). Incidence and prevalence of uveitis: results from the Pacific Ocular Inflammation Study. JAMA Ophthalmol. 131, 1405–1412. doi: 10.1001/jamaophthalmol.2013.4237
Acosta-Rodriguez, E. V., Napolitani, G., Lanzavecchia, A., and Sallusto, F. (2007). Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17–producing human T helper cells. Nat. Immunol. 8, 942–949. doi: 10.1038/ni1496
Ahmed, C. M., Massengill, M. T., Brown, E. E., Ildefonso, C. J., Johnson, H. M., and Lewin, A. S. (2018). A cell penetrating peptide from SOCS-1 prevents ocular damage in experimental autoimmune uveitis. Exp. Eye Res. 177, 12–22. doi: 10.1016/j.exer.2018.07.020
Ahmed, C. M. I., Larkin, J., and Johnson, H. M. (2015). SOCS1 mimetics and antagonists: a complementary approach to positive and negative regulation of immune function. Front. Immunol. 6:183. doi: 10.3389/fimmu.2015.00183
Akira, S. (2000). Roles of STAT3 defined by tissue-specific gene targeting. Oncogene 19, 2607–2611. doi: 10.1038/sj.onc.1203478
Al Zaid Siddiquee, K., and Turkson, J. (2008). STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 18, 254–267. doi: 10.1038/cr.2008.18
Alas, S., and Bonavida, B. (2001). Rituximab inactivates signal transducer and activation of transcription 3 (STAT3) activity in B-non-Hodgkin’s lymphoma through inhibition of the interleukin 10 autocrine/paracrine loop and results in down-regulation of Bcl-2 and sensitization to cytotoxic. Cancer Res. 61, 5137–5144.
Alexander, W. S., Starr, R., Fenner, J. E., Scott, C. L., Handman, E., Sprigg, N. S., et al. (1999). SOCS1 is a critical inhibitor of interferon γ signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell 98, 597–608. doi: 10.1016/S0092-8674(00)80047-1
Almaani, S., Meara, A., and Rovin, B. H. (2017). Update on lupus nephritis. Clin. J. Am. Soc. Nephrol. 12, 825–835. doi: 10.2215/CJN.05780616
Amatya, N., Garg, A. V., and Gaffen, S. L. (2017). IL-17 signaling: the Yin and the Yang. Trends Immunol. 38, 310–322. doi: 10.1016/j.it.2017.01.006
Aouar, B., Kovarova, D., Letard, S., Font-Haro, A., Florentin, J., Weber, J., et al. (2016). Dual role of the tyrosine kinase Syk in regulation of toll-like receptor signaling in plasmacytoid dendritic cells. PLoS One 11:e0156063. doi: 10.1371/journal.pone.0156063
Baechler, E. C., Batliwalla, F. M., Karypis, G., Gaffney, P. M., Ortmann, W. A., Espe, K. J., et al. (2003). Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. U.S.A. 100, 2610–2615. doi: 10.1073/pnas.0337679100
Baetz, A., Koelsche, C., Strebovsky, J., Heeg, K., and Dalpke, A. H. (2008). Identification of a nuclear localization signal in suppressor of cytokine signaling 1. FASEB J. 22, 4296–4305. doi: 10.1096/fj.08-116079
Baldwin, H. M., Pallas, K., King, V., Jamieson, T., McKimmie, C. S., Nibbs, R. J. B., et al. (2013). Microarray analyses demonstrate the involvement of type I interferons in psoriasiform pathology development in D6-deficient mice. J. Biol. Chem. 288, 36473–36483. doi: 10.1074/jbc.M113.491563
Barber, D. L. (2001). A common epitope is shared by activated signal transducer and activator of transcription-5 (STAT5) and the phosphorylated erythropoietin receptor: implications for the docking model of STAT activation. Blood 97, 2230–2237. doi: 10.1182/blood.V97.8.2230
Barbieri, I., Pensa, S., Pannellini, T., Quaglino, E., Maritano, D., Demaria, M., et al. (2010). Constitutively active Stat3 enhances Neu-mediated migration and metastasis in mammary tumors via upregulation of cten. Cancer Res. 70, 2558–2567. doi: 10.1158/0008-5472.CAN-09-2840
Bauermeister, K. (1998). Distinct regulation of IL-8 and MCP-1 by LPS and interferon- gamma-treated human peritoneal macrophages. Nephrol. Dial. Transplant. 13, 1412–1419. doi: 10.1093/ndt/13.6.1412
Berzaghi, R., Maia, V. S. C., Pereira, F. V., Melo, F. M., Guedes, M. S., Origassa, C. S. T., et al. (2017). SOCS1 favors the epithelial-mesenchymal transition in melanoma, promotes tumor progression and prevents antitumor immunity by PD-L1 expression. Sci. Rep. 7:40585. doi: 10.1038/srep40585
Blanco, F., Kalsi, J., and Isenberg, D. A. (1991). Analysis of antibodies to RNA in patients with systemic lupus erythematosus and other autoimmune rheumatic diseases. Clin. Exp. Immunol. 86, 66–70. doi: 10.1111/j.1365-2249.1991.tb05775.x
Blauvelt, A. (2007). New concepts in the pathogenesis and treatment of psoriasis: key roles for IL-23, IL-17A and TGF-β 1. Expert Rev. Dermatol. 2, 69–78. doi: 10.1586/17469872.2.1.69
Boniface, K., Bernard, F.-X., Garcia, M., Gurney, A. L., Lecron, J.-C., and Morel, F. (2005). IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J. Immunol. 174, 3695–3702. doi: 10.4049/jimmunol.174.6.3695
Braunstein, J., Brutsaert, S., Olson, R., and Schindler, C. (2003). STATs Dimerize in the absence of phosphorylation. J. Biol. Chem. 278, 34133–34140. doi: 10.1074/jbc.M304531200
Bromberg, J. F., Wrzeszczynska, M. H., Devgan, G., Zhao, Y., Pestell, R. G., Albanese, C., et al. (1999). Stat3 as an oncogene. Cell 98, 295–303. doi: 10.1016/S0092-8674(00)81959-5
Buchert, M., Burns, C. J., and Ernst, M. (2016). Targeting JAK kinase in solid tumors: emerging opportunities and challenges. Oncogene 35, 939–951. doi: 10.1038/onc.2015.150
Buettner, R., Mora, L. B., and Jove, R. (2002). Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention 1. Clin. Cancer Res. 8, 945–954.
Bullock, A. N., Rodriguez, M. C., Debreczeni, J. É, Songyang, Z., and Knapp, S. (2007). Structure of the SOCS4-ElonginB/C complex reveals a distinct SOCS box interface and the molecular basis for SOCS-dependent EGFR degradation. Structure 15, 1493–1504. doi: 10.1016/j.str.2007.09.016
Bunker, B. D., Nellimoottil, T. T., Boileau, R. M., Classen, A. K., and Bilder, D. (2015). The transcriptional response to tumorigenic polarity loss in Drosophila. eLife 4:e03189. doi: 10.7554/eLife.03189
Butturini, E., Gotte, G., Dell’Orco, D., Chiavegato, G., Marino, V., Canetti, D., et al. (2016). Intermolecular disulfide bond influences unphosphorylated STAT3 dimerization and function. Biochem. J. 473, 3205–3219. doi: 10.1042/BCJ20160294
Canel, M., Serrels, A., Frame, M. C., and Brunton, V. G. (2013). E-cadherin-integrin crosstalk in cancer invasion and metastasis. J. Cell Sci. 126, 393–401. doi: 10.1242/jcs.100115
Catlett-Falcone, R., Landowski, T. H., Oshiro, M. M., Turkson, J., Levitzki, A., Savino, R., et al. (1999). Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 10, 105–115. doi: 10.1016/S1074-7613(00)80011-4
Chan, H., Ke, L., Chang, L., Liu, C., Hung, Y., Lin, C., et al. (2010). Suppressor of cytokine signaling 1 gene expression and polymorphisms in systemic lupus erythematosus. Lupus 19, 696–702. doi: 10.1177/0961203309357437
Chan, S. R., Vermi, W., Luo, J., Lucini, L., Rickert, C., Fowler, A. M., et al. (2012). STAT1-deficient mice spontaneously develop estrogen receptor α-positive luminal mammary carcinomas. Breast Cancer Res. 14:R16. doi: 10.1186/bcr3100
Chang, C. H., Hsiao, C. F., Yeh, Y. M., Chang, G. C., Tsai, Y. H., Chen, Y. M., et al. (2013). Circulating interleukin-6 level is a prognostic marker for survival in advanced nonsmall cell lung cancer patients treated with chemotherapy. Int. J. Cancer 132, 1977–1985. doi: 10.1002/ijc.27892
Chen, C.-Y., Tsay, W., Tang, J.-L., Shen, H.-L., Lin, S.-W., Huang, S.-Y., et al. (2003). SOCS1 methylation in patients with newly diagnosed acute myeloid leukemia. Genes Chromosomes Cancer 37, 300–305. doi: 10.1002/gcc.10222
Chang, Q., Bournazou, E., Sansone, P., Berishaj, M., Gao, S. P., Daly, L., et al. (2013). The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 15, 848–IN45. doi: 10.1593/neo.13706
Chen, M.-F., Chen, P.-T., Lu, M. S., Lin, P. Y., Chen, W.-C., and Lee, K.-D. (2013). IL-6 expression predicts treatment response and outcome in squamous cell carcinoma of the esophagus. Mol. Cancer 12:26. doi: 10.1186/1476-4598-12-26
Chen, X. L., Nam, J.-O., Jean, C., Lawson, C., Walsh, C. T., Goka, E., et al. (2012). VEGF-induced vascular permeability is mediated by FAK. Dev. Cell 22, 146–157. doi: 10.1016/j.devcel.2011.11.002
Chen, X. P., Losman, J. A., Cowan, S., Donahue, E., Fay, S., Vuong, B. Q., et al. (2002). Pim serine/threonine kinases regulate the stability of Socs-1 protein. Proc. Natl. Acad. Sci. U.S.A. 99, 2175–2180. doi: 10.1073/pnas.042035699
Cheng, J., Deng, Y., Yi, H., Wang, G., Fu, B., Chen, W., et al. (2016). Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis 5:e198. doi: 10.1038/oncsis.2016.7
Chiricozzi, A., Guttman-Yassky, E., Suárez-Fariñas, M., Nograles, K. E., Tian, S., Cardinale, I., et al. (2011). Integrative responses to IL-17 and TNF-α in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J. Invest. Dermatol. 131, 677–687. doi: 10.1038/jid.2010.340
Chiricozzi, A., Nograles, K. E., Johnson-Huang, L. M., Fuentes-Duculan, J., Cardinale, I., Bonifacio, K. M., et al. (2014). IL-17 induces an expanded range of downstream genes in reconstituted human epidermis model. PLoS One 9:e90284. doi: 10.1371/journal.pone.0090284
Chung, C. D., Liao, J., Liu, B., Rao, X., Jay, P., Berta, P., et al. (1997). Specific inhibition of Stat3 signal transduction by PIAS3. Science 278, 1803–1805. doi: 10.1126/science.278.5344.1803
Chung, Y.-C., and Chang, Y.-F. (2003). Serum interleukin-6 levels reflect the disease status of colorectal cancer. J. Surg. Oncol. 83, 222–226. doi: 10.1002/jso.10269
Clark, J. D., Flanagan, M. E., and Telliez, J.-B. (2014). Discovery and development of Janus Kinase (JAK) inhibitors for inflammatory diseases. J. Med. Chem. 57, 5023–5038. doi: 10.1021/jm401490p
Classen, A.-K., Bunker, B. D., Harvey, K. F., Vaccari, T., and Bilder, D. (2009). A tumor suppressor activity of Drosophila Polycomb genes mediated by JAK-STAT signaling. Nat. Genet. 41, 1150–1155. doi: 10.1038/ng.445
Collins, E. L., Jager, L. D., Dabelic, R., Benitez, P., Holdstein, K., Lau, K., et al. (2011). Inhibition of SOCS1-/- lethal autoinflammatory disease correlated to enhanced peripheral Foxp3+ regulatory T cell homeostasis. J. Immunol. 187, 2666–2676. doi: 10.4049/jimmunol.1003819
Cornish, A. L., Chong, M. M., Davey, G. M., Darwiche, R., Nicola, N. A., Hilton, D. J., et al. (2003). Suppressor of cytokine signaling-1 regulates signaling in response to interleukin-2 and other γc-dependent cytokines in peripheral T cells. J. Biol. Chem. 278, 22755–22761. doi: 10.1074/jbc.M303021200
Crane, I. J., McKillop-Smith, S., Wallace, C. A., Lamont, G. R., and Forrester, J. V. (2001). Expression of the Chemokines MIP-1α, MCP-1, and RANTES in experimental autoimmune uveitis. Invest. Ophthalmol. Vis. Sci. 42, 1547–1552.
Cui, X., Kim, H.-J., Kuiatse, I., Kim, H., Brown, P. H., and Lee, A. V. (2006). Epidermal growth factor induces insulin receptor substrate-2 in breast cancer cells via c-Jun NH 2 -terminal kinase/activator protein-1 signaling to regulate cell migration. Cancer Res. 66, 5304–5313. doi: 10.1158/0008-5472.CAN-05-2858
Culig, Z., and Puhr, M. (2012). Interleukin-6: a multifunctional targetable cytokine in human prostate cancer. Mol. Cell. Endocrinol. 360, 52–58. doi: 10.1016/j.mce.2011.05.033
David, M., Naudin, C., Letourneur, M., Polrot, M., Renoir, J.-M., Lazar, V., et al. (2014). Suppressor of cytokine signaling 1 modulates invasion and metastatic potential of colorectal cancer cells. Mol. Oncol. 8, 942–955. doi: 10.1016/j.molonc.2014.03.014
de la Iglesia, N., Konopka, G., Lim, K.-L., Nutt, C. L., Bromberg, J. F., Frank, D. A., et al. (2008). Deregulation of a STAT3-interleukin 8 signaling pathway promotes human glioblastoma cell proliferation and invasiveness. J. Neurosci. 28, 5870–5878. doi: 10.1523/JNEUROSCI.5385-07.2008
de la Iglesia, N., Puram, S., and Bonni, A. (2009). STAT3 regulation of glioblastoma pathogenesis. Curr. Mol. Med. 9, 580–590. doi: 10.2174/156652409788488739
Deeg, C. A., Amann, B., Raith, A. J., and Kaspers, B. (2006). Inter- and intramolecular epitope spreading in equine recurrent uveitis. Investig. Opthalmol. Vis. Sci. 47, 652–656. doi: 10.1167/iovs.05-0789
Deeg, C. A., Reese, S., Gerhards, H., Wildner, G., and Kaspers, B. (2004). The uveitogenic potential of retinal S-antigen in horses. Investig. Opthalmol. Vis. Sci. 45, 2286–2292. doi: 10.1167/iovs.03-1226
Deeg, C. A., Thurau, S. R., Gerhards, H., Ehrenhofer, M., Wildner, G., and Kaspers, B. (2002). Uveitis in horses induced by interphotoreceptor retinoid-binding protein is similar to the spontaneous disease. Eur. J. Immunol. 32, 2598–2606. doi: 10.1002/1521-4141(200209)32:9<2598::AID-IMMU2598>3.0.CO;2-
Di, L. (2015). Strategic approaches to optimizing peptide ADME properties. AAPS J. 17, 134–143. doi: 10.1208/s12248-014-9687-3
Diaz, N. (2006). Activation of Stat3 in primary tumors from high-risk breast cancer patients is associated with elevated levels of activated Src and survivin expression. Clin. Cancer Res. 12, 20–28. doi: 10.1158/1078-0432.CCR-04-1749
Diehl, S., Anguita, J., Hoffmeyer, A., Zapton, T., Ihle, J. N., Fikrig, E., et al. (2000). Inhibition of Th1 differentiation by IL-6 Is mediated by SOCS1. Immunity 13, 805–815. doi: 10.1016/S1074-7613(00)00078-9
Ding, Y., Chen, D., Tarcsafalvi, A., Su, R., Qin, L., and Bromberg, J. S. (2003). Suppressor of cytokine signaling 1 Inhibits IL-10-mediated immune responses. J. Immunol. 170, 1383–1391. doi: 10.4049/jimmunol.170.3.1383
Dong, H., Strome, S. E., Salomao, D. R., Tamura, H., Hirano, F., Flies, D. B., et al. (2002). Erratum: tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 8, 793–800. doi: 10.1038/nm730
Druker, B. J., Talpaz, M., Resta, D. J., Peng, B., Buchdunger, E., Ford, J. M., et al. (2001). Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 344, 1031–1037. doi: 10.1056/NEJM200104053441401
Dunty, J. M., Gabarra-Niecko, V., King, M. L., Ceccarelli, D. F. J., Eck, M. J., and Schaller, M. D. (2004). FERM domain interaction promotes FAK signaling. Mol. Cell. Biol. 24, 5353–5368. doi: 10.1128/MCB.24.12.5353-5368.2004
Eke, I., Deuse, Y., Hehlgans, S., Gurtner, K., Krause, M., Baumann, M., et al. (2012). β1 Integrin/FAK/cortactin signaling is essential for human head and neck cancer resistance to radiotherapy. J. Clin. Invest. 122, 1529–1540. doi: 10.1172/JCI61350
Elkon, K. B., and Santer, D. M. (2012). Complement, interferon and lupus. Curr. Opin. Immunol. 24, 665–670. doi: 10.1016/j.coi.2012.08.004
Endo, T. A., Masuhara, M., Yokouchi, M., Suzuki, R., Sakamoto, H., Mitsui, K., et al. (1997). A new protein containing an SH2 domain that inhibits JAK kinases. Nature 387, 921–924. doi: 10.1038/43213
Eriksen, K., Kaltoft, K., Mikkelsen, G., Nielsen, M., Zhang, Q., Geisler, C., et al. (2001). Constitutive STAT3-activation in Sezary syndrome: tyrphostin AG490 inhibits STAT3-activation, interleukin-2 receptor expression and growth of leukemic Sezary cells. Leukemia 15, 787–793. doi: 10.1038/sj.leu.2402093
Eyles, J. L., Metcalf, D., Grusby, M. J., Hilton, D. J., and Starr, R. (2002). Negative regulation of interleukin-12 signaling by suppressor of cytokine signaling-1. J. Biol. Chem. 277, 43735–43740. doi: 10.1074/jbc.M208586200
Fan, H., Zhao, X., Sun, S., Luo, M., and Guan, J.-L. (2013). Function of focal adhesion kinase scaffolding to mediate endophilin A2 phosphorylation promotes epithelial-mesenchymal transition and mammary cancer stem cell activities in vivo. J. Biol. Chem. 288, 3322–3333. doi: 10.1074/jbc.M112.420497
Federici, M., Giustizieri, M. L., Scarponi, C., Girolomoni, G., and Albanesi, C. (2002). Impaired IFN- -dependent inflammatory responses in human keratinocytes overexpressing the suppressor of cytokine signaling 1. J. Immunol. 169, 434–442. doi: 10.4049/jimmunol.169.1.434
Feldman, S. R. (2005). Psoriasis assessment tools in clinical trials. Ann. Rheum. Dis. 64, ii65–ii68. doi: 10.1136/ard.2004.031237
Ferrari, S. M., Ruffilli, I., Colaci, M., Antonelli, A., Ferri, C., and Fallahi, P. (2015). CXCL10 in psoriasis. Adv. Med. Sci. 60, 349–354. doi: 10.1016/j.advms.2015.07.011
Fleischmann, R., Mysler, E., Hall, S., Kivitz, A. J., Moots, R. J., Luo, Z., et al. (2017). Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): a phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet 390, 457–468. doi: 10.1016/S0140-6736(17)31618-5
Forys, J. T., Kuzmicki, C. E., Saporita, A. J., Winkeler, C. L., Maggi, L. B., and Weber, J. D. (2014). ARF and p53 coordinate tumor suppression of an oncogenic IFN-β-STAT1-ISG15 signaling axis. Cell Rep. 7, 514–526. doi: 10.1016/j.celrep.2014.03.026
Foshay, K. M., and Gallicano, G. I. (2008). Regulation of Sox2 by STAT3 initiates commitment to the neural precursor cell fate. Stem Cells Dev. 17, 269–278. doi: 10.1089/scd.2007.0098
Frame, M. C., Patel, H., Serrels, B., Lietha, D., and Eck, M. J. (2010). The FERM domain: organizing the structure and function of FAK. Nat. Rev. Mol. Cell Biol. 11, 802–814. doi: 10.1038/nrm2996
Franke, S. (2001). Lymphocyte predominance Hodgkin disease is characterized by recurrent genomic imbalances. Blood 97, 1845–1853. doi: 10.1182/blood.V97.6.1845
Friedrich, K., Dolznig, H., Han, X., and Moriggl, R. (2017). Steering of carcinoma progression by the YIN/YANG interaction of STAT1/STAT3. Biosci. Trends 11, 1–8. doi: 10.5582/bst.2016.01250
Fujimoto, M. (2004). Inadequate induction of suppressor of cytokine signaling-1 causes systemic autoimmune diseases. Int. Immunol. 16, 303–314. doi: 10.1093/intimm/dxh030
Fujitake, S., Hibi, K., Okochi, O., Kodera, Y., Ito, K., Akiyama, S., et al. (2004). Aberrant methylation of SOCS-1 was observed in younger colorectal cancer patients. J. Gastroenterol. 39, 120–124. doi: 10.1007/s00535-003-1262-0
Fukushima, N., Sato, N., Sahin, F., Su, G. H., Hruban, R. H., and Goggins, M. (2003). Aberrant methylation of suppressor of cytokine signalling-1 (SOCS-1) gene in pancreatic ductal neoplasms. Br. J. Cancer 89, 338–343. doi: 10.1038/sj.bjc.6601039
Furumoto, Y., Smith, C. K., Blanco, L., Zhao, W., Brooks, S. R., Thacker, S. G., et al. (2017). Tofacitinib ameliorates murine lupus and its associated vascular dysfunction. Arthritis Rheumatol. 69, 148–160. doi: 10.1002/art.39818
Gadina, M., Hilton, D., Johnston, J. A., Morinobu, A., Lighvani, A., Zhou, Y.-J., et al. (2001). Signaling by Type I and II cytokine receptors: ten years after. Curr. Opin. Immunol 13, 363–373. doi: 10.1016/S0952-7915(00)00228-4
Galm, O. (2003). SOCS-1, a negative regulator of cytokine signaling, is frequently silenced by methylation in multiple myeloma. Blood 101, 2784–2788. doi: 10.1182/blood-2002-06-1735
Ganguly, D., Chamilos, G., Lande, R., Gregorio, J., Meller, S., Facchinetti, V., et al. (2009). Self-RNA–antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J. Exp. Med. 206, 1983–1994. doi: 10.1084/jem.20090480
Garcia-Diaz, A., Shin, D. S., Moreno, B. H., Saco, J., Escuin-Ordinas, H., Rodriguez, G. A., et al. (2017). Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 19, 1189–1201. doi: 10.1016/j.celrep.2017.04.031
Garcia-Rodriguez, S., Arias-Santiago, S., Perandrés-López, R., Castellote, L., Zumaquero, E., Navarro, P., et al. (2013). Increased gene expression of Toll-like receptor 4 on peripheral blood mononuclear cells in patients with psoriasis. J. Eur. Acad. Dermatol. Venereol. 27, 242–250. doi: 10.1111/j.1468-3083.2011.04372.x
Garcia-Romo, G. S., Caielli, S., Vega, B., Connolly, J., Allantaz, F., Xu, Z., et al. (2011). Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 3:73ra20. doi: 10.1126/scitranslmed.3001201
Gilger, B. C., Malok, E., Cutter, K. V., Stewart, T., Horohov, D. W., and Allen, J. B. (1999). Characterization of T-lymphocytes in the anterior uvea of eyes with chronic equine recurrent uveitis. Vet. Immunol. Immunopathol. 71, 17–28. doi: 10.1016/S0165-2427(99)00082-3
Gilliet, M., Cao, W., and Liu, Y.-J. (2008). Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 8, 594–606. doi: 10.1038/nri2358
González, M. M., Solano, M. M., Porco, T. C., Oldenburg, C. E., Acharya, N. R., Lin, S. C., et al. (2018). Epidemiology of uveitis in a US population-based study. J. Ophthalmic Inflamm. Infect 8:6. doi: 10.1186/s12348-018-0148-5
Gordon, K. B., Blauvelt, A., Papp, K. A., Langley, R. G., Luger, T., Ohtsuki, M., et al. (2016). Phase 3 Trials of Ixekizumab in moderate-to-severe plaque psoriasis. N. Engl. J. Med. 375, 345–356. doi: 10.1056/NEJMoa1512711
Gottlieb, A. B. (1988). Detection of a gamma interferon-induced protein IP-10 in psoriatic plaques. J. Exp. Med. 168, 941–948. doi: 10.1084/jem.168.3.941
Gregory, L., Came, P. J., and Brown, S. (2008). Stem cell regulation by JAK/STAT signaling in Drosophila. Semin. Cell Dev. Biol. 19, 407–413. doi: 10.1016/j.semcdb.2008.06.003
Griffin, B., and O’Driscoll, C. (2011). Opportunities and challenges for oral delivery of hydrophobic versus hydrophilic peptide and protein-like drugs using lipid-based technologies. Ther. Deliv. 2, 1633–1653. doi: 10.4155/tde.11.128
Griffiths, C. E., and Barker, J. N. (2007). Pathogenesis and clinical features of psoriasis. Lancet 370, 263–271. doi: 10.1016/S0140-6736(07)61128-3
Grine, L., Dejager, L., Libert, C., and Vandenbroucke, R. E. (2015). Dual Inhibition of TNFR1 and IFNAR1 in imiquimod-induced psoriasiform skin inflammation in mice. J. Immunol. 194, 5094–5102. doi: 10.4049/jimmunol.1403015
Gui, J., Gober, M., Yang, X., Katlinski, K. V., Marshall, C. M., Sharma, M., et al. (2016). Therapeutic elimination of the type 1 interferon receptor for treating psoriatic skin inflammation. J. Invest. Dermatol. 136, 1990–2002. doi: 10.1016/j.jid.2016.06.608
Gui, Y., Yeganeh, M., Donates, Y.-C., Tobelaim, W.-S., Chababi, W., Mayhue, M., et al. (2015). Regulation of MET receptor tyrosine kinase signaling by suppressor of cytokine signaling 1 in hepatocellular carcinoma. Oncogene 34, 5718–5728. doi: 10.1038/onc.2015.20
Guttman-Yassky, E., Nograles, K. E., and Krueger, J. G. (2011). Contrasting pathogenesis of atopic dermatitis and psoriasis—Part I: clinical and pathologic concepts. J. Allergy Clin. Immunol. 127, 1110–1118. doi: 10.1016/j.jaci.2011.01.053
Halder, J., Lin, Y. G., Merritt, W. M., Spannuth, W. A., Nick, A. M., Honda, T., et al. (2007). Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 67, 10976–10983. doi: 10.1158/0008-5472.CAN-07-2667
Hamalainen, H., Zhou, H., Chou, W., Hashizume, H., Heller, R., and Lahesmaa, R. (2001). Distinct gene expression profiles of human type 1 and type 2 T helper cells. Genome Biol. 2:RESEARCH0022. doi: 10.1186/gb-2001-2-7-research0022
Han, G.-M., Chen, S.-L., Shen, N., Ye, S., Bao, C.-D., and Gu, Y.-Y. (2003). Analysis of gene expression profiles in human systemic lupus erythematosus using oligonucleotide microarray. Genes Immun. 4, 177–186. doi: 10.1038/sj.gene.6363966
Hanada, T., Yoshida, H., Kato, S., Tanaka, K., Masutani, K., Tsukada, J., et al. (2003). Suppressor of cytokine signaling-1 is essential for suppressing dendritic cell activation and systemic autoimmunity. Immunity 19, 437–450. doi: 10.1016/S1074-7613(03)00240-1
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Hänsel, A., Günther, C., Ingwersen, J., Starke, J., Schmitz, M., Bachmann, M., et al. (2011). Human slan (6-sulfo LacNAc) dendritic cells are inflammatory dermal dendritic cells in psoriasis and drive strong Th17/Th1 T-cell responses. J. Allergy Clin. Immunol. 127, 787.e–794.e. doi: 10.1016/j.jaci.2010.12.009
Harigai, M., Kawamoto, M., Hara, M., Kubota, T., Kamatani, N., and Miyasaka, N. (2008). Excessive production of IFN- in patients with systemic lupus erythematosus and its contribution to induction of B lymphocyte stimulator/B cell-activating factor/TNF ligand superfamily-13B. J. Immunol. 181, 2211–2219. doi: 10.4049/jimmunol.181.3.2211
Harrison, C., Kiladjian, J.-J., Al-Ali, H. K., Gisslinger, H., Waltzman, R., Stalbovskaya, V., et al. (2012). JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 366, 787–798. doi: 10.1056/NEJMoa1110556
Hauck, C. R. (2002). FRNK blocks v-Src-stimulated invasion and experimental metastases without effects on cell motility or growth. EMBO J. 21, 6289–6302. doi: 10.1093/emboj/cdf631
He, C., Yu, C.-R., Mattapallil, M. J., Sun, L., Larkin, J. III, and Egwuagu, C. E. (2016). SOCS1 mimetic peptide suppresses chronic intraocular inflammatory disease (Uveitis). Mediators Inflamm. 2016, 1–15. doi: 10.1155/2016/2939370
He, C., Yu, C.-R., Sun, L., Mahdi, R. M., Larkin, J. III, and Egwuagu C. E. (2015). Topical administration of a suppressor of cytokine signaling-1 (SOCS1) mimetic peptide inhibits ocular inflammation and mitigates ocular pathology during mouse uveitis. J. Autoimmun. 62, 31–38. doi: 10.1016/j.jaut.2015.05.011
Heffler, M., Golubovskaya, V. M., Dunn, K. M. B., and Cance, W. (2013). Focal adhesion kinase autophosphorylation inhibition decreases colon cancer cell growth and enhances the efficacy of chemotherapy. Cancer Biol. Ther. 14, 761–772. doi: 10.4161/cbt.25185
Hix, L. M., Karavitis, J., Khan, M. W., Shi, Y. H., Khazaie, K., and Zhang, M. (2013). Tumor STAT1 transcription factor activity enhances breast tumor growth and immune suppression mediated by myeloid-derived suppressor cells. J. Biol. Chem. 288, 11676–11688. doi: 10.1074/jbc.M112.441402
Hochwald, S. N., Nyberg, C., Zheng, M., Zheng, D., Wood, C., Massoll, N. A., et al. (2009). A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell Cycle 8, 2435–2443. doi: 10.4161/cc.8.15.9145
Hollox, E. J., Huffmeier, U., Zeeuwen, P. L. J. M., Palla, R., Lascorz, J., Rodijk-Olthuis, D., et al. (2008). Psoriasis is associated with increased β-defensin genomic copy number. Nat. Genet. 40, 23–25. doi: 10.1038/ng.2007.48
Hosking, A.-M., Juhasz, M., and Mesinkovska, N. A. (2018). Topical Janus kinase inhibitors: a review of applications in dermatology. J. Am. Acad. Dermatol. 79, 535–544. doi: 10.1016/j.jaad.2018.04.018
Hosui, A., Klover, P., Tatsumi, T., Uemura, A., Nagano, H., Doki, Y., et al. (2012). Suppression of signal transducers and activators of transcription 1 in hepatocellular carcinoma is associated with tumor progression. Int. J. Cancer 131, 2774–2784. doi: 10.1002/ijc.27580
Hua, J., Kirou, K., Lee, C., and Crow, M. K. (2006). Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti–RNA binding protein autoantibodies. Arthritis Rheum. 54, 1906–1916. doi: 10.1002/art.21890
Huang, W.-L., Yeh, H.-H., Lin, C.-C., Lai, W.-W., Chang, J.-Y., Chang, W.-T., et al. (2010). Signal transducer and activator of transcription 3 activation up-regulates interleukin-6 autocrine production: a biochemical and genetic study of established cancer cell lines and clinical isolated human cancer cells. Mol. Cancer 9:309. doi: 10.1186/1476-4598-9-309
Ichiba, M., Nakajima, K., Yamanaka, Y., Kiuchi, N., and Hirano, T. (1998). Autoregulation of the Stat3 gene through cooperation with a cAMP-responsive element-binding protein. J. Biol. Chem. 273, 6132–6138. doi: 10.1074/jbc.273.11.6132
Ilangumaran, S., Ramanathan, S., La Rose, J., Poussier, P., and Rottapel, R. (2003a). Suppressor of cytokine signaling 1 regulates IL-15 receptor signaling in CD8+CD44high memory T lymphocytes. J. Immunol. 171, 2435–2445. doi: 10.4049/jimmunol.171.5.2435
Ilangumaran, S., Ramanathan, S., Ning, T., La Rose, J., Reinhart, B., Poussier, P., et al. (2003b). Suppressor of cytokine signaling 1 attenuates IL-15 receptor signaling in CD8+thymocytes. Blood 102, 4115–4122.
Jager, L. D., Dabelic, R., Waiboci, L. W., Lau, K., Haider, M. S., Ahmed, C. M. I., et al. (2011). The kinase inhibitory region of SOCS-1 is sufficient to inhibit T-helper 17 and other immune functions in experimental allergic encephalomyelitis. J. Neuroimmunol. 232, 108–118. doi: 10.1016/j.jneuroim.2010.10.018
Janssens, S., and Beyaert, R. (2003). Role of toll-like receptors in pathogen recognition. Clin. Microbiol. Rev. 16, 637–646. doi: 10.1128/CMR.16.4.637-646.2003
Jarrossay, D., Napolitani, G., Colonna, M., Sallusto, F., and Lanzavecchia, A. (2001). Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur. J. Immunol. 31, 3388–3393. doi: 10.1002/1521-4141(200111)31:11<3388::AID-IMMU3388>3.0.CO;2-Q
Jean, C., Chen, X. L., Nam, J.-O., Tancioni, I., Uryu, S., Lawson, C., et al. (2014). Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function. J. Cell Biol. 204, 247–263. doi: 10.1083/jcb.201307067
Jinno, T., Kawano, S., Maruse, Y., Matsubara, R., Goto, Y., Sakamoto, T., et al. (2015). Increased expression of interleukin-6 predicts poor response to chemoradiotherapy and unfavorable prognosis in oral squamous cell carcinoma. Oncol. Rep. 33, 2161–2168. doi: 10.3892/or.2015.3838
Jirgensons, B. (1966). Classification of proteins according to conformation. Makromol. Chem. 91, 74–86. doi: 10.1002/macp.1966.020910105
Jo, D. H., Kim, J. H., Cho, C. S., Cho, Y.-L., Jun, H. O., Yu, Y. S., et al. (2014). STAT3 inhibition suppresses proliferation of retinoblastoma through down-regulation of positive feedback loop of STAT3/miR-17-92 clusters. Oncotarget 5, 11513–11525. doi: 10.18632/oncotarget.2546
Johnson, D. E., O’Keefe, R. A., and Grandis, J. R. (2018). Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 15, 234–248. doi: 10.1038/nrclinonc.2018.8
Jostock, T., Müllberg, J., Özbek, S., Atreya, R., Blinn, G., Voltz, N., et al. (2001). Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 268, 160–167. doi: 10.1046/j.1432-1327.2001.01867.x
Kadowaki, N., Ho, S., Antonenko, S., de Waal Malefyt, R., Kastelein, R. A., Bazan, F., et al. (2001). Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J. Exp. Med. 194, 863–870. doi: 10.1084/jem.194.6.863
Kahlenberg, J. M., Thacker, S. G., Berthier, C. C., Cohen, C. D., Kretzler, M., and Kaplan, M. J. (2011). Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus. J. Immunol. 187, 6143–6156. doi: 10.4049/jimmunol.1101284
Kamiya, S., Okumura, M., Chiba, Y., Fukawa, T., Nakamura, C., Nimura, N., et al. (2011). IL-27 suppresses RANKL expression in CD4+ T cells in part through STAT3. Immunol. Lett. 138, 47–53. doi: 10.1016/j.imlet.2011.02.022
Kelley, K. W. (2008). NIH public access policy. Brain Behav. Immun. 22:629. doi: 10.1016/j.bbi.2008.05.010
Kim, J. H., Choi, Y. J., Lee, B. H., Song, M.-Y., Ban, C. Y., Kim, J., et al. (2016). Programmed cell death ligand 1 alleviates psoriatic inflammation by suppressing IL-17A production from programmed cell death 1–high T cells. J. Allergy Clin. Immunol 137, 1466.e–1476.e. doi: 10.1016/j.jaci.2015.11.021
Kinjyo, I., Hanada, T., Inagaki-Ohara, K., Mori, H., Aki, D., Ohishi, M., et al. (2002). SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity 17, 583–591. doi: 10.1016/S1074-7613(02)00446-6
Kirito, K. (2002). Identification of the human erythropoietin receptor region required for Stat1 and Stat3 activation. Blood 99, 102–110. doi: 10.1182/blood.V99.1.102
Kitamura, H., Ohno, Y., Toyoshima, Y., Ohtake, J., Homma, S., Kawamura, H., et al. (2017). Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 108, 1947–1952. doi: 10.1111/cas.13332
Kiuchi, N., Nakajima, K., Ichiba, M., Fukada, T., Narimatsu, M., Mizuno, K., et al. (1999). STAT3 is required for the gp130-mediated full activation of the c- myc gene. J. Exp. Med. 189, 63–73. doi: 10.1084/jem.189.1.63
Klunker, S., Trautmann, A., Akdis, M., Verhagen, J., Schmid-Grendelmeier, P., Blaser, K., et al. (2003). A second step of chemotaxis after transendothelial migration: keratinocytes undergoing apoptosis release IFN-γ-inducible protein 10, monokine induced by IFN-γ, and IFN-γ-inducible α-chemoattractant for T cell chemotaxis toward epidermis in atopic dermatit. J. Immunol. 171, 1078L–1084. doi: 10.4049/jimmunol.171.2.1078
Koca, T. T. (2016). A short summary of clinical types of psoriasis. North. Clin. Istanbul. 3, 79–82. doi: 10.14744/nci.2016.16023
Koch, C., Anderson, D., Moran, M., Ellis, C., and Pawson, T. (1991). SH2 and SH3 domains: elements that control interactions of cytoplasmic signaling proteins. Science 252, 668–674. doi: 10.1126/science.1708916
Konnikova, L., Kotecki, M., Kruger, M. M., and Cochran, B. H. (2003). Knockdown of STAT3 expression by RNAi induces apoptosis in astrocytoma cells. BMC Cancer 3:23. doi: 10.1186/1471-2407-3-23
Kortylewski, M., Kujawski, M., Wang, T., Wei, S., Zhang, S., Pilon-Thomas, S., et al. (2005). Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 11, 1314–1321. doi: 10.1038/nm1325
Kotowicz, B., Fuksiewicz, M., Jonska-Gmyrek, J., Bidzinski, M., and Kowalska, M. (2016). The assessment of the prognostic value of tumor markers and cytokines as SCCAg, CYFRA 21.1, IL-6, VEGF and sTNF receptors in patients with squamous cell cervical cancer, particularly with early stage of the disease. Tumor Biol. 37, 1271–1278. doi: 10.1007/s13277-015-3914-0
Kotyla, P. J. (2018). Are Janus kinase inhibitors superior over classic biologic agents in RA patients? Biomed Res. Int. 2018:7492904. doi: 10.1155/2018/7492904
Koudstaal, J., Frensdorf, E. L., Kremer, J., Mudde, J. M., and Hardonk, M. J. (1967). THE HISTOCHEMICAL PATTERN OF THE HUMAN ADULT TESTES. Acta Endocrinol. 55, 415–426. doi: 10.1530/acta.0.0550415
Krebs, D. L., and Hilton, D. J. (2001). SOCS proteins: negative regulators of cytokine signaling. Stem Cells 19, 378–387. doi: 10.1634/stemcells.19-5-378
Krueger, G. G., Langley, R. G., Leonardi, C., Yeilding, N., Guzzo, C., Wang, Y., et al. (2007). A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. N. Engl. J. Med. 356, 580–592. doi: 10.1056/NEJMoa062382
Kujawski, M., Kortylewski, M., Lee, H., Herrmann, A., Kay, H., and Yu, H. (2008). Stat3 mediates myeloid cell–dependent tumor angiogenesis in mice. J. Clin. Invest. 118, 3367–3377. doi: 10.1172/JCI35213
Kurenova, E., Xu, L.-H., Yang, X., Baldwin, A. S., Craven, R. J., Hanks, S. K., et al. (2004). Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol. Cell. Biol. 24, 4361–4371. doi: 10.1128/MCB.24.10.4361-4371.2004
La Fortezza, M., Schenk, M., Cosolo, A., Kolybaba, A., Grass, I., and Classen, A.-K. (2016). JAK/STAT signalling mediates cell survival in response to tissue stress. Development 143, 2907–2919. doi: 10.1242/dev.132340
Lande, R., Botti, E., Jandus, C., Dojcinovic, D., Fanelli, G., Conrad, C., et al. (2014). The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat. Commun. 5:5621. doi: 10.1038/ncomms6621
Lande, R., Ganguly, D., Facchinetti, V., Frasca, L., Conrad, C., Gregorio, J., et al. (2011). Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 3, ra19–ra73. doi: 10.1126/scitranslmed.3001180
Lande, R., Gregorio, J., Facchinetti, V., Chatterjee, B., Wang, Y.-H., Homey, B., et al. (2007). Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449, 564–569. doi: 10.1038/nature06116
Lane, D., Goncharenko-Khaider, N., Rancourt, C., and Piché, A. (2010). Ovarian cancer ascites protects from TRAIL-induced cell death through αvβ5 integrin-mediated focal adhesion kinase and Akt activation. Oncogene 29, 3519–3531. doi: 10.1038/onc.2010.107
Lane, D., Matte, I., Rancourt, C., and Piché, A. (2011). Prognostic significance of IL-6 and IL-8 ascites levels in ovarian cancer patients. BMC Cancer 11:210. doi: 10.1186/1471-2407-11
Laner-Plamberger, S., Wolff, F., Kaser-Eichberger, A., Swierczynski, S., Hauser-Kronberger, C., Frischauf, A.-M., et al. (2013). Hedgehog/GLI signaling activates suppressor of cytokine signaling 1 (SOCS1) in epidermal and neural tumor cells. PLoS One 8:e75317. doi: 10.1371/journal.pone.0075317
Langley, R. G., Elewski, B. E., Lebwohl, M., Reich, K., Griffiths, C. E. M., Papp, K., et al. (2014). Secukinumab in plaque psoriasis — results of two phase 3 trials. N. Engl. J. Med. 371, 326–338. doi: 10.1056/NEJMoa1314258
Langley, R. G., and Ellis, C. N. (2004). Evaluating psoriasis with psoriasis area and severity index, psoriasis global assessment, and lattice system physician’s global assessment. J. Am. Acad. Dermatol. 51, 563–569. doi: 10.1016/j.jaad.2004.04.012
Lee, C., Kolesnik, T. B., Caminschi, I., Chakravorty, A., Carter, W., Alexander, W. S., et al. (2009). Suppressor of cytokine signalling 1 (SOCS1) is a physiological regulator of the asthma response. Clin. Exp. Allergy 39, 897–907. doi: 10.1111/j.1365-2222.2009.03217.x
Lee, E., Trepicchio, W. L., Oestreicher, J. L., Pittman, D., Wang, F., Chamian, F., et al. (2004). Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J. Exp. Med. 199, 125–130. doi: 10.1084/jem.20030451
Lembo, S., Capasso, R., Balato, A., Cirillo, T., Flora, F., Zappia, V., et al. (2014). MCP-1 in psoriatic patients: effect of biological therapy. J. Dermatolog. Treat. 25, 83–86. doi: 10.3109/09546634.2013.782091
Lesina, M., Kurkowski, M. U., Ludes, K., Rose-John, S., Treiber, M., Klöppel, G., et al. (2011). Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 19, 456–469. doi: 10.1016/j.ccr.2011.03.009
Lesinski, G. B., Anghelina, M., Zimmerer, J., Bakalakos, T., Badgwell, B., Parihar, R., et al. (2003). The antitumor effects of IFN-α are abrogated in a STAT1-deficient mouse. J. Clin. Invest. 112, 170–180. doi: 10.1172/JCI16603
Li, J., Zhao, S., Yi, M., Hu, X., Le, J., Xie, H., et al. (2011). Activation of JAK-STAT1 signal transduction pathway in lesional skin and monocytes from patients with systemic lupus erythematosus. Zhong Nan Da Xue Xue Bao Yi Xue Ban 36, 109–115. doi: 10.3969/j.issn.1672-7347.2011.02.003
Li, Z., Metze, D., Nashan, D., Müller-Tidow, C., Serve, H. L., Poremba, C., et al. (2004). Expression of SOCS-1, suppressor of cytokine signalling-1, in human melanoma. J. Invest. Dermatol. 123, 737–745. doi: 10.1111/j.0022-202X.2004.23408.x
Liau, N. P. D., Laktyushin, A., and Babon, J. J. (2017). Purification of SOCS (suppressor of cytokine signaling) SH2 domains for structural and functional studies. Methods Mol. Biol. 1555, 173–182. doi: 10.1007/978-1-4939-6762-9_10
Liau, N. P. D., Laktyushin, A., Lucet, I. S., Murphy, J. M., Yao, S., Whitlock, E., et al. (2018). The molecular basis of JAK/STAT inhibition by SOCS1. Nat. Commun. 9:1558. doi: 10.1038/s41467-018-04013-1
Limnander, A., Danial, N. N., and Rothman, P. B. (2004). v-Abl signaling disrupts SOCS-1 function in transformed Pre-B cells. Mol. Cell 15, 329–341. doi: 10.1016/j.molcel.2004.06.041
Lin, A. M., Rubin, C. J., Khandpur, R., Wang, J. Y., Riblett, M., Yalavarthi, S., et al. (2011). Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J. Immunol. 187, 490–500. doi: 10.4049/jimmunol.1100123
Lit, L. C. W. (2006). Raised plasma concentration and ex vivo production of inflammatory chemokines in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 65, 209–215. doi: 10.1136/ard.2005.038315
Liu, B., Liao, J., Rao, X., Kushner, S. A., Chung, C. D., Chang, D. D., et al. (1998). Inhibition of Stat1-mediated gene activation by PIAS1. Proc. Natl. Acad. Sci. U.S.A. 95, 10626–10631. doi: 10.1073/pnas.95.18.10626
Liu, E. (2003). Negative regulation of FAK signaling by SOCS proteins. EMBO J. 22, 5036–5046. doi: 10.1093/emboj/cdg503
Liu, S., Ren, S., Howell, P., Fodstad, O., and Riker, A. I. (2008). Identification of novel epigenetically modified genes in human melanoma via promoter methylation gene profiling. Pigment Cell Melanoma Res. 21, 545–558. doi: 10.1111/j.1755-148X.2008.00484.x
Liu, X., Lee, Y. S., Yu, C.-R., and Egwuagu, C. E. (2008). Loss of STAT3 in CD4 + T cells prevents development of experimental autoimmune diseases. J. Immunol. 180, 6070–6076. doi: 10.4049/jimmunol.180.9.6070
Liu, Y., Seto, N. L., Carmona-Rivera, C., and Kaplan, M. J. (2018). Accelerated model of lupus autoimmunity and vasculopathy driven by toll-like receptor 7/9 imbalance. Lupus Sci. Med. 5:e000259. doi: 10.1136/lupus-2018-000259
Loft, N. D., Skov, L., Rasmussen, M. K., Gniadecki, R., Dam, T. N., Brandslund, I., et al. (2018). Genetic polymorphisms associated with psoriasis and development of psoriatic arthritis in patients with psoriasis. PLoS One 13:e0192010. doi: 10.1371/journal.pone.0192010
Lombardi, V., Van Overtvelt, L., Horiot, S., and Moingeon, P. (2009). Human dendritic cells stimulated via TLR7 and/or TLR8 induce the sequential production of Il-10, IFN-, and IL-17A by Naive CD4+ T cells. J. Immunol. 182, 3372–3379. doi: 10.4049/jimmunol.0801969
Losman, J. A., Chen, X. P., Hilton, D., and Rothman, P. (1999). Cutting edge: SOCS-1 is a potent inhibitor of IL-4 signal transduction. J. Immunol. 162, 3770–3774.
Mace, T. A., Ameen, Z., Collins, A., Wojcik, S., Mair, M., Young, G. S., et al. (2013). Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 73, 3007–3018. doi: 10.1158/0008-5472.CAN-12-4601
Mahato, R. I., Narang, A. S., Thoma, L., and Miller, D. D. (2003). Emerging trends in oral delivery of peptide and protein drugs. Crit. Rev. Ther. Drug Carrier Syst. 20, 153–214. doi: 10.1615/CritRevTherDrugCarrierSyst.v20.i23.30
Mak, A., Mok, C. C., Chu, W. P., To, C. H., Wong, S. N., and Au, T. C. (2007). Renal damage in systemic lupus erythematosus: a comparative analysis of different age groups. Lupus 16, 28–34. doi: 10.1177/0961203306074469
Malalana, F., Stylianides, A., and McGowan, C. (2015). Equine recurrent uveitis: human and equine perspectives. Vet. J. 206, 22–29. doi: 10.1016/j.tvjl.2015.06.017
Mallette, F. A., Calabrese, V., Ilangumaran, S., and Ferbeyre, G. (2010). SOCS1, a novel interaction partner of p53 controlling oncogene-induced senescence. Aging 2, 445–452. doi: 10.18632/aging.100163
Mansell, A., Smith, R., Doyle, S. L., Gray, P., Fenner, J. E., Crack, P. J., et al. (2006). Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat. Immunol. 7, 148–155. doi: 10.1038/ni1299
Marzocchi-Machado, C. M., Alves, C. M. O. S., Azzolini, A. E. C. S., Polizello, A. M. N., Carvalho, I. F., and Lucisano-Valim, Y. M. (2002). Fcgamma and complement receptors: expression, role and co-operation in mediating the oxidative burst and degranulation of neutrophils of Brazilian systemic lupus erythematosus patients. Lupus 11, 240–248. doi: 10.1191/0961203302lu172oa
Masuhara, M., Sakamoto, H., Matsumoto, A., Suzuki, R., Yasukawa, H., Mitsui, K., et al. (1997). Cloning and characterization of novel CIS family genes. Biochem. Biophys. Res. Commun. 239, 439–446. doi: 10.1006/bbrc.1997.7484
Matikainen, S., Sareneva, T., Ronni, T., Lehtonen, A., Koskinen, P. J., and Julkunen, I. (1999). Interferon-alpha activates multiple STAT proteins and upregulates proliferation-associated IL-2Ralpha, c-myc, and pim-1 genes in human T cells. Blood 93, 1980–1991.
McLean, G. W., Carragher, N. O., Avizienyte, E., Evans, J., Brunton, V. G., and Frame, M. C. (2005). The role of focal-adhesion kinase in cancer — a new therapeutic opportunity. Nat. Rev. Cancer 5, 505–515. doi: 10.1038/nrc1647
McNally, R., and Eck, M. J. (2014). JAK–cytokine receptor recognition, unboxed. Nat. Struct. Mol. Biol. 21, 431–433. doi: 10.1038/nsmb.2824
Melzner, I. (2005). Biallelic mutation of SOCS-1 impairs JAK2 degradation and sustains phospho-JAK2 action in the MedB-1 mediastinal lymphoma line. Blood 105, 2535–2542. doi: 10.1182/blood-2004-09-3701
Melzner, I., Weniger, M. A., Bucur, A. J., Brüderlein, S., Dorsch, K., Hasel, C., et al. (2006). Biallelic deletion within 16p13.13 including SOCS-1 in Karpas1106P mediastinal B-cell lymphoma line is associated with delayed degradation of JAK2 protein. Int. J. Cancer 118, 1941–1944. doi: 10.1002/ijc.21485
Menter, A., Gottlieb, A., Feldman, S. R., Van Voorhees, A. S., Leonardi, C. L., Gordon, K. B., et al. (2008). Guidelines of care for the management of psoriasis and psoriatic arthritis. J. Am. Acad. Dermatol. 58, 826–850. doi: 10.1016/j.jaad.2008.02.039
Merk, B. C., Owens, J. L., Lopes, M.-B. S., Silva, C. M., and Hussaini, I. M. (2011). STAT6 expression in glioblastoma promotes invasive growth. BMC Cancer 11:184. doi: 10.1186/1471-2407-11-184
Metcalf, D., Mifsud, S., Di Rago, L., Nicola, N. A., Hilton, D. J., and Alexander, W. S. (2002). Polycystic kidneys and chronic inflammatory lesions are the delayed consequences of loss of the suppressor of cytokine signaling-1 (SOCS-1). Proc. Natl. Acad. Sci. U.S.A. 99, 943–948. doi: 10.1073/pnas.022628499
Meyer, D. M., Jesson, M. I., Li, X., Elrick, M. M., Funckes-Shippy, C. L., Warner, J. D., et al. (2010). Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J. Inflamm. 7:41. doi: 10.1186/1476-9255-7-41
Mitra, S. K., Lim, S.-T., Chi, A., and Schlaepfer, D. D. (2006). Intrinsic focal adhesion kinase activity controls orthotopic breast carcinoma metastasis via the regulation of urokinase plasminogen activator expression in a syngeneic tumor model. Oncogene 25, 4429–4440. doi: 10.1038/sj.onc.1209482
Mogensen, T. H. (2009). Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 22, 240–273. doi: 10.1128/CMR.00046-08
Mosmann, T. R., and Sad, S. (1996). The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol. Today 17, 138–146. doi: 10.1016/0167-5699(96)80606-2
Mottok, A., Renné, C., Willenbrock, K., Hansmann, M. L., and Bräuninger, A. (2007). Somatic hypermutation of SOCS1 in lymphocyte-predominant Hodgkin lymphoma is accompanied by high JAK2 expression and activation of STAT6. Blood 110, 3387–3390. doi: 10.1182/blood-2007-03-082511
Munroe, M. E., Vista, E. S., Guthridge, J. M., Thompson, L. F., Merrill, J. T., and James, J. A. (2014). Proinflammatory adaptive cytokine and shed tumor necrosis factor receptor levels are elevated preceding systemic lupus erythematosus disease flare. Arthritis Rheumatol. 66, 1888–1899. doi: 10.1002/art.38573
Munz, O. H., Sela, S., Baker, B. S., Griffiths, C. E. M., Powles, A. V., and Fry, L. (2010). Evidence for the presence of bacteria in the blood of psoriasis patients. Arch. Dermatol. Res. 302, 495–498. doi: 10.1007/s00403-010-1065-0
Murphy, H. S., Bakopoulos, N., Dame, M. K., Varani, J., and Ward, P. A. (1998). Heterogeneity of vascular endothelial cells: differences in susceptibility to neutrophil-mediated injury. Microvasc. Res. 56, 203–211. doi: 10.1006/mvre.1998.2110
Naka, T., Narazaki, M., Hirata, M., Matsumoto, T., Minamoto, S., Aono, A., et al. (1997). Structure and function of a new STAT-induced STAT inhibitor. Nature 387, 924–929. doi: 10.1038/43219
Nakae, S., Iwakura, Y., Suto, H., and Galli, S. J. (2007). Phenotypic differences between Th1 and Th17 cells and negative regulation of Th1 cell differentiation by IL-17. J. Leukoc. Biol. 81, 1258–1268. doi: 10.1189/jlb.1006610
Nakagawa, R., Naka, T., Tsutsui, H., Fujimoto, M., Kimura, A., Abe, T., et al. (2002). SOCS-1 participates in negative regulation of LPS responses. Immunity 17, 677–687. doi: 10.1016/S1074-7613(02)00449-1
Natatsuka, R., Takahashi, T., Serada, S., Fujimoto, M., Ookawara, T., Nishida, T., et al. (2015). Gene therapy with SOCS1 for gastric cancer induces G2/M arrest and has an antitumour effect on peritoneal carcinomatosis. Br. J. Cancer 113, 433–442. doi: 10.1038/bjc.2015.229
Neuwirt, H., Puhr, M., Santer, F. R., Susani, M., Doppler, W., Marcias, G., et al. (2009). Suppressor of cytokine signaling (SOCS)-1 Is expressed in human prostate cancer and exerts growth-inhibitory function through down-regulation of cyclins and cyclin-dependent kinases. Am. J. Pathol. 174, 1921–1930. doi: 10.2353/ajpath.2009.080751
Ng, Y. P., Cheung, Z. H., and Ip, N. Y. (2006). STAT3 as a downstream mediator of Trk signaling and functions. J. Biol. Chem. 281, 15636–15644. doi: 10.1074/jbc.M601863200
Niu, G., Briggs, J., Deng, J., Ma, Y., Lee, H., Kortylewski, M., et al. (2008). Signal transducer and activator of transcription 3 is required for hypoxia-inducible factor-1 RNA expression in both tumor cells and tumor-associated myeloid cells. Mol. Cancer Res. 6, 1099–1105. doi: 10.1158/1541-7786.MCR-07-2177
Niu, G., Wright, K. L., Ma, Y., Wright, G. M., Huang, M., Irby, R., et al. (2005). Role of Stat3 in regulating p53 expression and function. Mol. Cell. Biol. 25, 7432–7440. doi: 10.1128/MCB.25.17.7432-7440.2005
Nussenblatt, R. B. (1990). The natural history of uveitis. Int. Ophthalmol. 14, 303–308. doi: 10.1007/BF00163549
Nussenblatt, R. B. (2002). BENCH TO BEDSIDE: NEW APPROACHES TO THE IMMUNOTHERAPY OF UVEITIC DISEASE. Int. Rev. Immunol. 21, 273–289. doi: 10.1080/08830180212067
Oshimo, Y., Kuraoka, K., Nakayama, H., Kitadai, Y., Yoshida, K., Chayama, K., et al. (2004). Epigenetic inactivation ofSOCS-1 by CpG island hypermethylation in human gastric carcinoma. Int. J. Cancer 112, 1003–1009. doi: 10.1002/ijc.20521
Ottaviani, C., Nasorri, F., Bedini, C., de Pità, O., Girolomoni, G., and Cavani, A. (2006). CD56brightCD16- NK cells accumulate in psoriatic skin in response to CXCL10 and CCL5 and exacerbate skin inflammation. Eur. J. Immunol. 36, 118–128. doi: 10.1002/eji.200535243
Otvos, L., and Wade, J. D. (2014). Current challenges in peptide-based drug discovery. Front. Chem. 2:62. doi: 10.3389/fchem.2014.00062
Palamara, F., Meindl, S., Holcmann, M., Lührs, P., Stingl, G., and Sibilia, M. (2004). Identification and characterization of pDC-Like cells in normal mouse skin and melanomas treated with imiquimod. J. Immunol. 173, 3051–3061. doi: 10.4049/jimmunol.173.5.3051
Panzer, R., Blobel, C., Fölster-Holst, R., and Proksch, E. (2014). TLR2 and TLR4 expression in atopic dermatitis, contact dermatitis and psoriasis. Exp. Dermatol. 23, 364–366. doi: 10.1111/exd.12383
Papp, K. A., Langley, R. G., Sigurgeirsson, B., Abe, M., Baker, D. R., Konno, P., et al. (2013). Efficacy and safety of secukinumab in the treatment of moderate-to-severe plaque psoriasis: a randomized, double-blind, placebo-controlled phase II dose-ranging study. Br. J. Dermatol. 168, 412–421. doi: 10.1111/bjd.12110
Pariser, D. M., Bagel, J., Gelfand, J. M., Korman, N. J., Ritchlin, C. T., Strober, B. E., et al. (2007). National psoriasis foundation clinical consensus on disease severity. Arch. Dermatol. 143, 239–242. doi: 10.1001/archderm.143.2.239
Perry, D., Sang, A., Yin, Y., Zheng, Y.-Y., and Morel, L. (2011). Murine models of systemic lupus erythematosus. J. Biomed. Biotechnol. 2011, 1–19. doi: 10.1155/2011/271694
Philip, T., Bernard, J. L., Zucker, J. M., Pinkerton, R., Lutz, P., Bordigoni, P., et al. (1987). High-dose chemoradiotherapy with bone marrow transplantation as consolidation treatment in neuroblastoma: an unselected group of stage IV patients over 1 year of age. J. Clin. Oncol. 5, 266–271. doi: 10.1200/JCO.1987.5.2.266
Piganis, R. A. R., De Weerd, N. A., Gould, J. A., Schindler, C. W., Mansell, A., Nicholson, S. E., et al. (2011). Suppressor of Cytokine signaling (SOCS) 1 inhibits type I interferon (IFN) signaling via the interferon α receptor (IFNAR1)-associated Tyrosine Kinase Tyk2. J. Biol. Chem. 286, 33811–33818. doi: 10.1074/jbc.M111.270207
Pisitkun, P. (2006). Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science 312, 1669–1672. doi: 10.1126/science.1124978
Platanias, L. C. (2005). Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386. doi: 10.1038/nri1604
Pollaro, L., and Heinis, C. (2010). Strategies to prolong the plasma residence time of peptide drugs. Med. Chem. Commun. 1, 319–324. doi: 10.1039/c0md00111b
Puthier, D., Bataille, R., and Amiot, M. (1999). IL-6 up-regulates Mcl-1 in human myeloma cells through JAK / STAT rather than Ras / MAP kinase pathway. Eur. J. Immunol. 29, 3945–3950. doi: 10.1002/(SICI)1521-4141(199912)29:12<3945::AID-IMMU3945>3.0.CO;2-O
Raccurt, M., Tam, S. P., Lau, P., Mertani, H. C., Lambert, A., Garcia-Caballero, T., et al. (2003). Suppressor of cytokine signalling gene expression is elevated in breast carcinoma. Br. J. Cancer 89, 524–532. doi: 10.1038/sj.bjc.6601115
Rahaman, S. O., Harbor, P. C., Chernova, O., Barnett, G. H., Vogelbaum, M. A., and Haque, S. J. (2002). Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene 21, 8404–8413. doi: 10.1038/sj.onc.1206047
Real, P. J., Sierra, A., de Juan, A., Segovia, J. C., Lopez-Vega, J. M., and Fernandez-Luna, J. L. (2002). Resistance to chemotherapy via Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer cells. Oncogene 21, 7611–7618. doi: 10.1038/sj.onc.1206004
Recio, C., Oguiza, A., Lazaro, I., Mallavia, B., Egido, J., and Gomez-Guerrero, C. (2014). Suppressor of cytokine signaling 1–derived peptide inhibits Janus Kinase/signal transducers and activators of transcription pathway and improves inflammation and atherosclerosis in diabetic mice. Arterioscler. Thromb. Vasc. Biol. 34, 1953–1960. doi: 10.1161/ATVBAHA.114.304144
Regis, G., Pensa, S., Boselli, D., Novelli, F., and Poli, V. (2008). Ups and downs: The STAT1:STAT3 seesaw of Interferon and gp130 receptor signalling. Semin. Cell Dev. Biol. 19, 351–359. doi: 10.1016/j.semcdb.2008.06.004
Rich, P., Sigurgeirsson, B., Thaci, D., Ortonne, J.-P., Paul, C., Schopf, R. E., et al. (2013). Secukinumab induction and maintenance therapy in moderate-to-severe plaque psoriasis: a randomized, double-blind, placebo-controlled, phase II regimen-finding study. Br. J. Dermatol. 168, 402–411. doi: 10.1111/bjd.12112
Riedel, F., Zaiss, I., Herzog, D., Götte, K., Naim, R., and Hörmann, K. (2005). Serum levels of interleukin-6 in patients with primary head and neck squamous cell carcinoma. Anticancer Res. 25, 2761–2765.
Ritz, O., Guiter, C., Dorsch, K., Dusanter-Fourt, I., Wegener, S., Jouault, H., et al. (2008). STAT6 activity is regulated by SOCS-1 and modulates BCL-XL expression in primary mediastinal B-Cell lymphoma. Leukemia 22, 2106–2110. doi: 10.1038/leu.2008.85
Rock, K., Huang, S. H., Tiong, A., Lu, L., Xu, W., Ringash, J., et al. (2018). Partial laryngeal IMRT for T2N0 Glottic cancer: impact of image guidance and radiation therapy intensification. Int. J. Radiat. Oncol. 102, 941–949. doi: 10.1016/j.ijrobp.2018.03.034
Rui, L., Yuan, M., Frantz, D., Shoelson, S., and White, M. F. (2002). SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J. Biol. Chem. 277, 42394–42398. doi: 10.1074/jbc.C200444200
Ryo, A., Suizu, F., Yoshida, Y., Perrem, K., Liou, Y.-C., Wulf, G., et al. (2003). Regulation of NF-κB signaling by Pin1-dependent Prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell 12, 1413–1426. doi: 10.1016/S1097-2765(03)00490-8
Sachithanandan, N., Graham, K. L., Galic, S., Honeyman, J. E., Fynch, S. L., Hewitt, K. A., et al. (2011). Macrophage deletion of SOCS1 increases sensitivity to LPS and palmitic acid and results in systemic inflammation and hepatic insulin resistance. Diabetes 60, 2023–2031. doi: 10.2337/db11-0259
Sakamoto, H., Yasukawa, H., Masuhara, M., Tanimura, S., Sasaki, A., Yuge, K., et al. (1998). “Summary for Policymakers,” in Climate Change 2013 - The Physical Science Basis, Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, eds T. F. Stocker, D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen, J. Boschung, et al. (Cambridge: Cambridge University Press), 1–30.
Sano, S., Chan, K. S., Carbajal, S., Clifford, J., Peavey, M., Kiguchi, K., et al. (2005). Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat. Med. 11, 43–49. doi: 10.1038/nm1162
Sato, A. K., Viswanathan, M., Kent, R. B., and Wood, C. R. (2006). Therapeutic peptides: technological advances driving peptides into development. Curr. Opin. Biotechnol. 17, 638–642. doi: 10.1016/j.copbio.2006.10.002
Schlaepfer, D. D., and Hunter, T. (1997). Focal adhesion kinase overexpression enhances Ras-dependent integrin signaling to ERK2/mitogen-activated protein kinase through interactions with and activation of c-Src. J. Biol. Chem. 272, 13189–13195. doi: 10.1074/jbc.272.20.13189
Schwartz, D. M., Kanno, Y., Villarino, A., Ward, M., Gadina, M., and O’Shea, J. J. (2017). JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 16, 843–862. doi: 10.1038/nrd.2017.201
Seckinger, P., Lowenthal, J. W., Williamson, K., and Macdonald, H. R. (1987). A urine inhibitor of interleukin 1 activity that blocks ligand binding. J. Immunol. 139, 1546–1549.
Sgrignani, J., Olsson, S., Ekonomiuk, D., Genini, D., Krause, R., Catapano, C. V., et al. (2015). Molecular determinants for unphosphorylated STAT3 dimerization determined by integrative modeling. Biochemistry 54, 5489–5501. doi: 10.1021/bi501529x
Sharabi, A., Sthoeger, Z. M., Mahlab, K., Lapter, S., Zinger, H., and Mozes, E. (2009). A tolerogenic peptide that induces suppressor of cytokine signaling (SOCS)-1 restores the aberrant control of IFN-γ signaling in lupus-affected (NZB × NZW)F1 mice. Clin. Immunol. 133, 61–68. doi: 10.1016/j.clim.2009.06.010
Sieg, D. J., Hauck, C. R., Ilic, D., Klingbeil, C. K., Schaefer, E., Damsky, C. H., et al. (2000). FAK integrates growth-factor and integrin signals to promote cell migration. Nat. Cell Biol. 2, 249–256. doi: 10.1038/35010517
Slack-Davis, J. K., Hershey, E. D., Theodorescu, D., Frierson, H. F., and Parsons, J. T. (2009). Differential requirement for focal adhesion kinase signaling in cancer progression in the transgenic adenocarcinoma of mouse prostate model. Mol. Cancer Ther. 8, 2470–2477. doi: 10.1158/1535-7163.MCT-09-0262
Smith, R. L., Hébert, H. L., Massey, J., Bowes, J., Marzo-Ortega, H., Ho, P., et al. (2016). Association of Toll-like receptor 4 (TLR4) with chronic plaque type psoriasis and psoriatic arthritis. Arch. Dermatol. Res. 308, 201–205. doi: 10.1007/s00403-016-1620-4
Song, X., Dai, D., He, X., Zhu, S., Yao, Y., Gao, H., et al. (2015). Growth factor FGF2 cooperates with interleukin-17 to repair intestinal epithelial damage. Immunity 43, 488–501. doi: 10.1016/j.immuni.2015.06.024
Souma, Y., Nishida, T., Serada, S., Iwahori, K., Takahashi, T., Fujimoto, M., et al. (2012). Antiproliferative effect of SOCS-1 through the suppression of STAT3 and p38 MAPK activation in gastric cancer cells. Int. J. Cancer 131, 1287–1296. doi: 10.1002/ijc.27350
Sporri, B. (2001). JAB/SOCS1/SSI-1 is an interleukin-2-induced inhibitor of IL-2 signaling. Blood 97, 221–226. doi: 10.1182/blood.V97.1.221
Starr, R., Metcalf, D., Elefanty, A. G., Brysha, M., Willson, T. A., Nicola, N. A., et al. (1998). Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc. Natl. Acad. Sci. U.S.A. 95, 14395–14399. doi: 10.1073/pnas.95.24.14395
Starr, R., Willson, T. A., Viney, E. M., Murray, L. J. L., Rayner, J. R., Jenkins, B. J., et al. (1997). A family of cytokine-inducible inhibitors of signalling. Nature 387, 917–921. doi: 10.1038/43206
Stokes, J. B., Adair, S. J., Slack-Davis, J. K., Walters, D. M., Tilghman, R. W., Hershey, E. D., et al. (2011). Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol. Cancer Ther. 10, 2135–2145. doi: 10.1158/1535-7163.MCT-11-0261
Strebovsky, J., Walker, P., Lang, R., and Dalpke, A. H. (2011). Suppressor of cytokine signaling 1 (SOCS1) limits NFκB signaling by decreasing p65 stability within the cell nucleus. FASEB J. 25, 863–874. doi: 10.1096/fj.10-170597
Sugase, T., Takahashi, T., Serada, S., Fujimoto, M., Hiramatsu, K., Ohkawara, T., et al. (2017). SOCS1 gene therapy improves radiosensitivity and enhances irradiation-induced DNA damage in esophageal squamous cell carcinoma. Cancer Res. 77, 6975–6986. doi: 10.1158/0008-5472.CAN-17-1525
Sugase, T., Takahashi, T., Serada, S., Fujimoto, M., Ohkawara, T., Hiramatsu, K., et al. (2018). SOCS1 gene therapy has antitumor effects in imatinib-resistant gastrointestinal stromal tumor cells through FAK/PI3 K signaling. Gastric Cancer 21, 968–976. doi: 10.1007/s10120-018-0822-1
Sukka-Ganesh, B., and Larkin, J. (2016). Therapeutic potential for targeting the suppressor of cytokine signalling-1 pathway for the treatment of SLE. Scand. J. Immunol. 84, 299–309. doi: 10.1111/sji.12475
Sulzmaier, F. J., Jean, C., and Schlaepfer, D. D. (2014). FAK in cancer: mechanistic findings and clinical applications. Nat. Rev. Cancer 14, 598–610. doi: 10.1038/nrc3792
Swiatek-Machado, K., Mieczkowski, J., Ellert-Miklaszewska, A., Swierk, P., Fokt, I., Szymanski, S., et al. (2012). Novel small molecular inhibitors disrupt the JAK/STAT3 and FAK signaling pathways and exhibit a potent antitumor activity in glioma cells. Cancer Biol. Ther. 13, 657–670. doi: 10.4161/cbt.20083
Takahashi, R., Nakatsukasa, H., Shiozawa, S., and Yoshimura, A. (2017). SOCS1 is a key molecule that prevents regulatory T cell plasticity under inflammatory conditions. J. Immunol. 199, 149–158. doi: 10.4049/jimmunol.1600441
Takahashi, R., Nishimoto, S., Muto, G., Sekiya, T., Tamiya, T., Kimura, A., et al. (2011). SOCS1 is essential for regulatory T cell functions by preventing loss of Foxp3 expression as well as IFN-γ and IL-17A production. J. Exp. Med. 208, 2055–2067. doi: 10.1084/jem.20110428
Tejasvi, T., Stuart, P. E., Chandran, V., Voorhees, J. J., Gladman, D. D., Rahman, P., et al. (2012). TNFAIP3 gene polymorphisms are associated with response to TNF blockade in psoriasis. J. Invest. Dermatol. 132(3 Pt 1), 593–600. doi: 10.1038/jid.2011.376
Thyrell, L., Arulampalam, V., Hjortsberg, L., Farnebo, M., Grandér, D., and Pokrovskaja Tamm, K. (2007). Interferon alpha induces cell death through interference with interleukin 6 signaling and inhibition of STAT3 activity. Exp. Cell Res. 313, 4015–4024. doi: 10.1016/j.yexcr.2007.08.007
Tobelaim, W. S., Beaurivage, C., Champagne, A., Pomerleau, V., Simoneau, A., Chababi, W., et al. (2015). Tumour-promoting role of SOCS1 in colorectal cancer cells. Sci. Rep. 5:14301. doi: 10.1038/srep14301
Tomar, A., Lawson, C., Ghassemian, M., and Schlaepfer, D. D. (2012). Cortactin as a Target for FAK in the regulation of focal adhesion dynamics. PLoS One 7:e44041. doi: 10.1371/journal.pone.0044041
Uversky, V. N., Gillespie, J. R., and Fink, A. L. (2000). Why are?natively unfolded? proteins unstructured under physiologic conditions? Proteins Struct. Funct. Genet. 41, 415–427. doi: 10.1002/1097-0134(20001115)41:3<415::AID-PROT130>3.0.CO;2-7
Uversky, V. N., Oldfield, C. J., and Dunker, A. K. (2005). Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 18, 343–384. doi: 10.1002/jmr.747
Vallin, H., Blomberg, S., Alm, G. V., Cederblad, B., and Rönnblom, L. (1999). Patients with systemic lupus erythematosus (SLE) have a circulating inducer of interferon-alpha (IFN-alpha) production acting on leucocytes resembling immature dendritic cells. Clin. Exp. Immunol. 115, 196–202. doi: 10.1046/j.1365-2249.1999.00772.x
van de Geijn, G.-J. M., Gits, J., and Touw, I. P. (2004). Distinct activities of suppressor of cytokine signaling (SOCS) proteins and involvement of the SOCS box in controlling G-CSF signaling. J. Leukoc. Biol. 76, 237–244. doi: 10.1189/jlb.0104041
van der Fits, L., Mourits, S., Voerman, J. S. A., Kant, M., Boon, L., Laman, J. D., et al. (2009). Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 Axis. J. Immunol. 182, 5836–5845. doi: 10.4049/jimmunol.0802999
Verstovsek, S., Mesa, R. A., Gotlib, J., Levy, R. S., Gupta, V., DiPersio, J. F., et al. (2012). A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 366, 799–807. doi: 10.1056/NEJMoa1110557
Vlieghe, P., Lisowski, V., Martinez, J., and Khrestchatisky, M. (2010). Synthetic therapeutic peptides: science and market. Drug Discov. Today 15, 40–56. doi: 10.1016/j.drudis.2009.10.009
Volonterio, A., Bellosta, S., Bravin, F., Bellucci, M. C., Bruché, L., Colombo, G., et al. (2003). Synthesis, structure and conformation of partially-modified retro- and retro-Inversoψ[NHCH(CF3)]Gly Peptides. Chemistry 9, 4510–4522. doi: 10.1002/chem.200304881
Waiboci, L. W., Ahmed, C. M., Mujtaba, M. G., Flowers, L. O., Martin, J. P., Haider, M. I., et al. (2007). Both the suppressor of cytokine signaling 1 (SOCS-1) kinase inhibitory region and SOCS-1 mimetic bind to JAK2 autophosphorylation site: implications for the development of a SOCS-1 antagonist. J. Immunol. 178, 5058–5068. doi: 10.4049/jimmunol.178.8.5058
Wang, S., Yang, N., Zhang, L., Huang, B., Tan, H., Liang, Y., et al. (2010). Jak/STAT signaling is involved in the inflammatory infiltration of the kidneys in MRL/lpr mice. Lupus 19, 1171–1180. doi: 10.1177/0961203310367660
Weis, S. M., Lim, S.-T., Lutu-Fuga, K. M., Barnes, L. A., Chen, X. L., Göthert, J. R., et al. (2008). Compensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell FAK. J. Cell Biol. 181, 43–50. doi: 10.1083/jcb.200710038
Weisenseel, P. (2002). Streptococcal infection distinguishes different types of psoriasis. J. Med. Genet. 39, 767–768. doi: 10.1136/jmg.39.10.767
Wiechert, W., Möllney, M., Petersen, S., and de Graaf, A. A. (2001). A Universal framework for 13C metabolic flux analysis. Metab. Eng. 3, 265–283. doi: 10.1006/mben.2001.0188
Wolk, K., Witte, E., Wallace, E., Döcke, W.-D., Kunz, S., Asadullah, K., et al. (2006). IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur. J. Immunol. 36, 1309–1323. doi: 10.1002/eji.200535503
Wu, N.-L., Huang, D.-Y., Tsou, H.-N., Lin, Y.-C., and Lin, W.-W. (2015). Syk Mediates IL-17-Induced CCL20 Expression by Targeting Act1-Dependent K63-Linked Ubiquitination of TRAF6. J. Invest. Dermatol. 135, 490–498. doi: 10.1038/jid.2014.383
Xu, L., Kitani, A., Fuss, I., and Strober, W. (2007). Cutting Edge: Regulatory T Cells Induce CD4+CD25-Foxp3- T Cells or Are Self-Induced to Become Th17 Cells in the Absence of Exogenous TGF-. J. Immunol. 178, 6725–6729. doi: 10.4049/jimmunol.178.11.6725
Xu, L.-H., Yang, X., Bradham, C. A., Brenner, D. A., Baldwin, A. S., Craven, R. J., et al. (2000). The focal adhesion kinase suppresses transformation-associated, anchorage-independent apoptosis in human breast cancer cells. J. Biol. Chem. 275, 30597–30604. doi: 10.1074/jbc.M910027199
Yamada, O., Ozaki, K., Akiyama, M., and Kawauchi, K. (2012). JAK-STAT and JAK-PI3K-mTORC1 pathways regulate telomerase transcriptionally and posttranslationally in ATL cells. Mol. Cancer Ther. 11, 1112–1121. doi: 10.1158/1535-7163.MCT-11-0850
Yamawaki, Y., Kimura, H., Hosoi, T., and Ozawa, K. (2010). MyD88 plays a key role in LPS-induced Stat3 activation in the hypothalamus. Am. J. Physiol. Integr. Comp. Physiol. 298, R403–R410. doi: 10.1152/ajpregu.00395.2009
Yan, D., Wang, H.-W., Bowman, R. L., and Joyce, J. A. (2016). STAT3 and STAT6 signaling pathways synergize to promote cathepsin secretion from macrophages via IRE1α activation. Cell Rep. 16, 2914–2927. doi: 10.1016/j.celrep.2016.08.035
Yang, J., Liao, X., Agarwal, M. K., Barnes, L., Auron, P. E., and Stark, G. R. (2007). Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NF B. Genes Dev. 21, 1396–1408. doi: 10.1101/gad.1553707
Yang, J., and Stark, G. R. (2008). Roles of unphosphorylated STATs in signaling. Cell Res. 18, 443–451. doi: 10.1038/cr.2008.41
Yao, R., Ma, Y.-L., Liang, W., Li, H.-H., Ma, Z.-J., Yu, X., et al. (2012). MicroRNA-155 modulates Treg and Th17 cells differentiation and Th17 cell function by targeting SOCS1. PLoS One 7:e46082. doi: 10.1371/journal.pone.0046082
Yeganeh, M., Gui, Y., Kandhi, R., Bobbala, D., Tobelaim, W.-S., Saucier, C., et al. (2016). Suppressor of cytokine signaling 1-dependent regulation of the expression and oncogenic functions of p21CIP1/WAF1 in the liver. Oncogene 35, 4200–4211. doi: 10.1038/onc.2015.485
Yeh, H.-H., Lai, W.-W., Chen, H. H. W., Liu, H.-S., and Su, W.-C. (2006). Autocrine IL-6-induced Stat3 activation contributes to the pathogenesis of lung adenocarcinoma and malignant pleural effusion. Oncogene 25, 4300–4309. doi: 10.1038/sj.onc.1209464
Yokogawa, M., Takaishi, M., Nakajima, K., Kamijima, R., Fujimoto, C., Kataoka, S., et al. (2014). Epicutaneous application of toll-like receptor 7 agonists leads to systemic autoimmunity in wild-type mice: a new model of systemic lupus erythematosus. Arthritis Rheumatol. 66, 694–706. doi: 10.1002/art.38298
Yoshikawa, H., Matsubara, K., Qian, G. S., Jackson, P., Groopman, J. D., Manning, J. E., et al. (2001). SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat. Genet. 28, 29–35. doi: 10.1038/88225
You, W., Tang, Q., Zhang, C., Wu, J., Gu, C., Wu, Z., et al. (2013). IL-26 promotes the proliferation and survival of human gastric cancer cells by regulating the balance of STAT1 and STAT3 activation. PLoS One 8:e63588. doi: 10.1371/journal.pone.0063588
Youlyouz-Marfak, I., Gachard, N., Le Clorennec, C., Najjar, I., Baran-Marszak, F., Reminieras, L., et al. (2008). Identification of a novel p53-dependent activation pathway of STAT1 by antitumour genotoxic agents. Cell Death Differ. 15, 376–385. doi: 10.1038/sj.cdd.4402270
Yu, C., Mahdi, R. M., Liu, X., Zhang, A., Naka, T., Kishimoto, T., et al. (2008). “Summary for policymakers,” in Climate Change 2013 - The Physical Science Basis, eds T. F. Stocker, D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen, J. Boschung et al. (Cambridge: Cambridge University Press), 1–30.
Yu, C.-F., Peng, W.-M., Oldenburg, J., Hoch, J., Bieber, T., Limmer, A., et al. (2010). Human plasmacytoid dendritic cells support Th17 Cell effector function in response to TLR7 Ligation. J. Immunol. 184, 1159–1167. doi: 10.4049/jimmunol.0901706
Yu, C.-F., Peng, W.-M., Schlee, M., Barchet, W., Eis-Hübinger, A. M., Kolanus, W., et al. (2018). SOCS1 and SOCS3 target IRF7 Degradation to suppress TLR7-mediated type I IFN production of human plasmacytoid dendritic cells. J. Immunol. 200, 4024–4035. doi: 10.4049/jimmunol.1700510
Yu, C.-R., Mahdi, R. R., Oh, H.-M., Amadi-Obi, A., Levy-Clarke, G., Burton, J., et al. (2011). Suppressor of cytokine signaling-1 (SOCS1) inhibits lymphocyte recruitment into the retina and protects SOCS1 transgenic rats and mice from ocular inflammation. Investig. Opthalmol. Vis. Sci. 52, 6978–6986. doi: 10.1167/iovs.11-7688
Yu, H., and Jove, R. (2004). The STATs of cancer — new molecular targets come of age. Nat. Rev. Cancer 4, 97–105. doi: 10.1038/nrc1275
Yu, Y., and Su, K. (2013). Neutrophil extracellular traps and systemic lupus erythematosus. J. Clin. Cell. Immunol. 4:139. doi: 10.4172/2155-9899.1000139
Yu, Y.-C., Yang, P.-M., Chuah, Q.-Y., Huang, Y.-H., Peng, C.-W., Lee, Y.-J., et al. (2013). Radiation-induced senescence in securin-deficient cancer cells promotes cell invasion involving the IL-6/STAT3 and PDGF-BB/PDGFR pathways. Sci. Rep. 3:1675. doi: 10.1038/srep01675
Zhang, J.-G., Farley, A., Nicholson, S. E., Willson, T. A., Zugaro, L. M., Simpson, R. J., et al. (1999). The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc. Natl. Acad. Sci. U.S.A. 96, 2071–2076. doi: 10.1073/pnas.96.5.2071
Zhang, Q., Raghunath, P. N., Xue, L., Majewski, M., Carpentieri, D. F., Odum, N., et al. (2002). Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/Null-cell lymphoma. J. Immunol. 168, 466–474. doi: 10.4049/jimmunol.168.1.466
Zhang, Z., Kyttaris, V. C., and Tsokos, G. C. (2009). The role of IL-23/IL-17 axis in lupus nephritis. J. Immunol. 183, 3160–3169. doi: 10.4049/jimmunol.0900385
Zhou, X., Liu, Z., Cheng, X., Zheng, Y., Zeng, F., and He, Y. (2015). Socs1 and Socs3 degrades Traf6 via polyubiquitination in LPS-induced acute necrotizing pancreatitis. Cell Death Dis. 6:e2012. doi: 10.1038/cddis.2015.342
Zimmer, J., Weitnauer, M., Boutin, S., Küblbeck, G., Thiele, S., Walker, P., et al. (2016). Nuclear localization of suppressor of cytokine signaling-1 regulates local immunity in the lung. Front. Immunol. 7:514. doi: 10.3389/fimmu.2016.00514
Zouq, N. K., Keeble, J. A., Lindsay, J., Valentijn, A. J., Zhang, L., Mills, D., et al. (2009). FAK engages multiple pathways to maintain survival of fibroblasts and epithelia - differential roles for paxillin and p130Cas. J. Cell Sci. 122, 357–367. doi: 10.1242/jcs.030478
Keywords: autoimmunity, SOCS1, JAK/STAT, jakinibs, cancer, cytokines
Citation: Sharma J and Larkin J III (2019) Therapeutic Implication of SOCS1 Modulation in the Treatment of Autoimmunity and Cancer. Front. Pharmacol. 10:324. doi: 10.3389/fphar.2019.00324
Received: 05 December 2018; Accepted: 18 March 2019;
Published: 11 April 2019.
Edited by:
Bernhard Brüne, Goethe-Universität Frankfurt am Main, GermanyReviewed by:
Subburaj Ilangumaran, Université de Sherbrooke, CanadaFred Schaper, Otto-von-Guericke Universität Magdeburg, Germany
Vida A. Dennis, Alabama State University, United States
Copyright © 2019 Sharma and Larkin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph Larkin III, amxhcmtpbjNAdWZsLmVkdQ==