 Xingyu Chen
Xingyu Chen Michael Bründl
Michael Bründl Theres Friesacher
Theres Friesacher Anna Stary-Weinzinger
Anna Stary-Weinzinger- Department of Pharmacology and Toxicology, University of Vienna, Vienna, Austria
Inwardly rectifying potassium (KIR) channels play important roles in controlling cellular excitability and K+ ion homeostasis. Under physiological conditions, KIR channels allow large K+ influx at potentials negative to the equilibrium potential of K+ but permit little outward current at potentials positive to the equilibrium potential of K+, due to voltage dependent block of outward K+ flux by cytoplasmic polyamines. These polycationic molecules enter the KIR channel pore from the intracellular side. They block K+ ion movement through the channel at depolarized potentials, thereby ensuring, for instance, the long plateau phase of the cardiac action potential. Key questions concerning how deeply these charged molecules migrate into the pore and how the steep voltage dependence arises remain unclear. Recent MD simulations on GIRK2 (=Kir3.2) crystal structures have provided unprecedented details concerning the conduction mechanism of a KIR channel. Here, we use MD simulations with applied field to provide detailed insights into voltage dependent block of putrescine, using the conductive state of the strong inwardly rectifying K+ channel GIRK2 as starting point. Our µs long simulations elucidate details about binding sites of putrescine in the pore and suggest that voltage-dependent rectification arises from a dual mechanism.
Introduction
Inwardly rectifying potassium (KIR) channels play important roles in controlling cellular excitability and K+ ion homeostasis. Under physiological conditions, KIR channels pass inward currents more easily than outward currents due to voltage-dependent block by intracellular cations such as Mg2+ and polyamines (Hibino et al., 2010). These polycationic molecules enter the KIR channel pore from the intracellular side and block K+ ion movement through the channel at depolarized potentials, thereby ensuring, for instance, the long plateau phase of the cardiac action potential (Nichols and Lee, 2018). The degree of inward rectification varies among the KIR channel family. In strong inward rectifier channels, such as Kir2 and Kir3, very little outward current occurs at voltages positive to the K+ equilibrium (EK) potential. While in weak rectifier channels, e.g. Kir1 and Kir6 family members, conductance at voltages positive to EK is reduced relative to inward currents. Intermediate rectification properties are found in Kir4.x channels (Hibino et al., 2010).
Atomic and near-atomic resolution structures of different KIR channels, albeit without polyamines bound, have been solved mainly in the closed state, revealing a conserved architecture of the KIR channel family. Further, the transmembrane domain (TMD) is conserved throughout the whole K+ channel superfamily (Doyle, 1998). It contains two transmembrane helices (M1 and M2), an extracellular turret region, a short P-helix and a selectivity filter region. A unique structural feature of KIR channels is a long cytoplasmic domain (CTD), which contains another constriction, the so-called G-loop gate and extends the ion conduction pore to > 85 Å (Kuo et al., 2003).
Unlike voltage dependent gating of Kv channel proteins, voltage-dependent “inward rectification” in KIR channels is not an intrinsic property. Voltage dependent rectification arises from voltage dependent channel blockade by cytoplasmic polycations, particularly polyamines, such as putrescine, spermine, and spermidine (Ficker et al., 1994; Lopatin et al., 1994; Fakler et al., 1995). Residues influencing rectification properties by polyamines have been identified in the CTD as well as the TMD. Two negatively charged residues in Kir2.1 (carrying IK1 current), E224 in the cytoplasm and D172, located in the transmembrane domain, have been shown to be critical for strong rectification. Interestingly, swapping residue D172 between strong and weak rectifier channels resulted in substantial transfer of rectification properties (Lopatin et al., 1994; Lu and MacKinnon, 1994; Stanfield et al., 1994; Wible et al., 1994). Due to the critical importance of this residue, this site has been referred to as rectification controller. Nevertheless, this position does not exert an all-or-nothing effect and surprisingly, channels such as Kir3.2, which exhibit relatively strong rectification lack this acidic residue (for alignment, see Supplemental Figure 1). Further, mutating D172 to N (Kir2.1) does weaken but not abolish polyamine binding (Wible et al., 1994; Yi et al., 2001; Guo et al., 2003). Importantly it has been shown that introducing a negative charge in the transmembrane domain is not position dependent (Kurata et al., 2004). This is in line with a computational study by the Roux group (Robertson et al., 2008), revealing that many of the residues influencing rectification interact with ions over very long distances up to 40 Å, likely affecting blocker energetics over the entire pore.
The CTD contributes to rectification to a lesser extent than the TMD. For example, interchanging the cytoplasmic domains of Kir2.1 and Kir1.1 resulted in partial transfer of rectification properties (Taglialatela et al., 1995). Mutagenesis identified two negatively charged residues in Kir2.1 (E224 and E299), being important determinants of rectification (Taglialatela et al., 1995; Kubo and Murata, 2001; Guo et al., 2003). Importantly, neutralization of these charges slowed the kinetics and reduced the affinity of spermine block (Taglialatela et al., 1995; Guo et al., 2003; Fujiwara and Kubo, 2006). However, mutations also changed the biophysics of the channel, complicating interpretation of the results.
Despite the existence of more and more crystal and cryo-EM structures, none of the different eukaryotic structures could be solved in the presence of polyamines so far, leaving important questions concerning the location of the polyamine binding site(s) within the KIR channel pore open. Further, the nature of the charge movement associated with block and the origin of the steepness of the voltage dependence of these compounds, which roughly correlates with the number of charges of the polyamines remain unresolved.
Recent MD simulations on KIR crystal structures have provided unprecedented details concerning the K+ conduction mechanism of Kir3.2 and 2.2 channels (Bernsteiner et al., 2019; Zangerl-Plessl et al., 2020) and can serve as excellent starting point to address inward rectification. Here, we perform multi-microsecond-timescale MD simulations with applied field to provide first atomistic insights into the voltage dependent block of putrescine (PUT2+), using the conductive state of the strong inwardly rectifying Kir3.2 channel as a starting point.
Results
Putrescine Block of Kir3.2 Channels Under Applied Field
Starting from a conductive snapshot from a previous simulation (Kir3.2 also known as GIRK2), a bona-fide strong rectifier, we addressed the dynamics of the “shortest” polyamine putrescine (PUT2+), a 1,4- butanediamine, which has a net charge of +2 under physiological pH. Figure 1 illustrates K+ and PUT2+ movement along the z-axis, monitored in six one µs atomistic simulations with an applied field of 40 mV/nm, which amounts to 580 mV. In four out of six runs, the molecule rapidly permeated from the initial binding site in the CTD to the selectivity filter (position SC or S4). In the other two runs, PUT2+ remained close to the initial binding site in the CTD. Interestingly, in all except one run, at least a second, and in the case of run six, a third molecule entered from the cytoplasm to bind to the negatively charged residues in the CTD, mainly E236, revealing that the cytoplasmic pore can accommodate more than one blocker molecule. However, in none of the runs, more than one PUT2+ traversed up to the selectivity filter (SF). K+ permeation was effectively prevented only when the charged blocker reached its binding site at the SF but not when the polyamine was bound to the original binding region in the CTD. As illustrated in Figure 1 in runs three, four and six, ions were observed to pass by PUT2+ when bound to the CTD (ion slippage examples are indicated by black arrows). This finding is in agreement with experimental data, suggesting a shallow blocked state for polyamines in the CTD (Kurata et al., 2007). The timescales for PUT2+ permeation to the SF varied from ~ 250 ns to ~650 ns, with the molecule remaining at its binding site for the rest of the simulation time. Binding to the CTD was reasonably stable when analyzing second and/or third PUT2+ molecules entering the CTD, with retention times of up to 850 ns, after accessing the binding site from the intracellular site. To analyze binding at these different sites in more detail, PUT2+ and K+ occupancies along the z-axis were plotted as function of time.
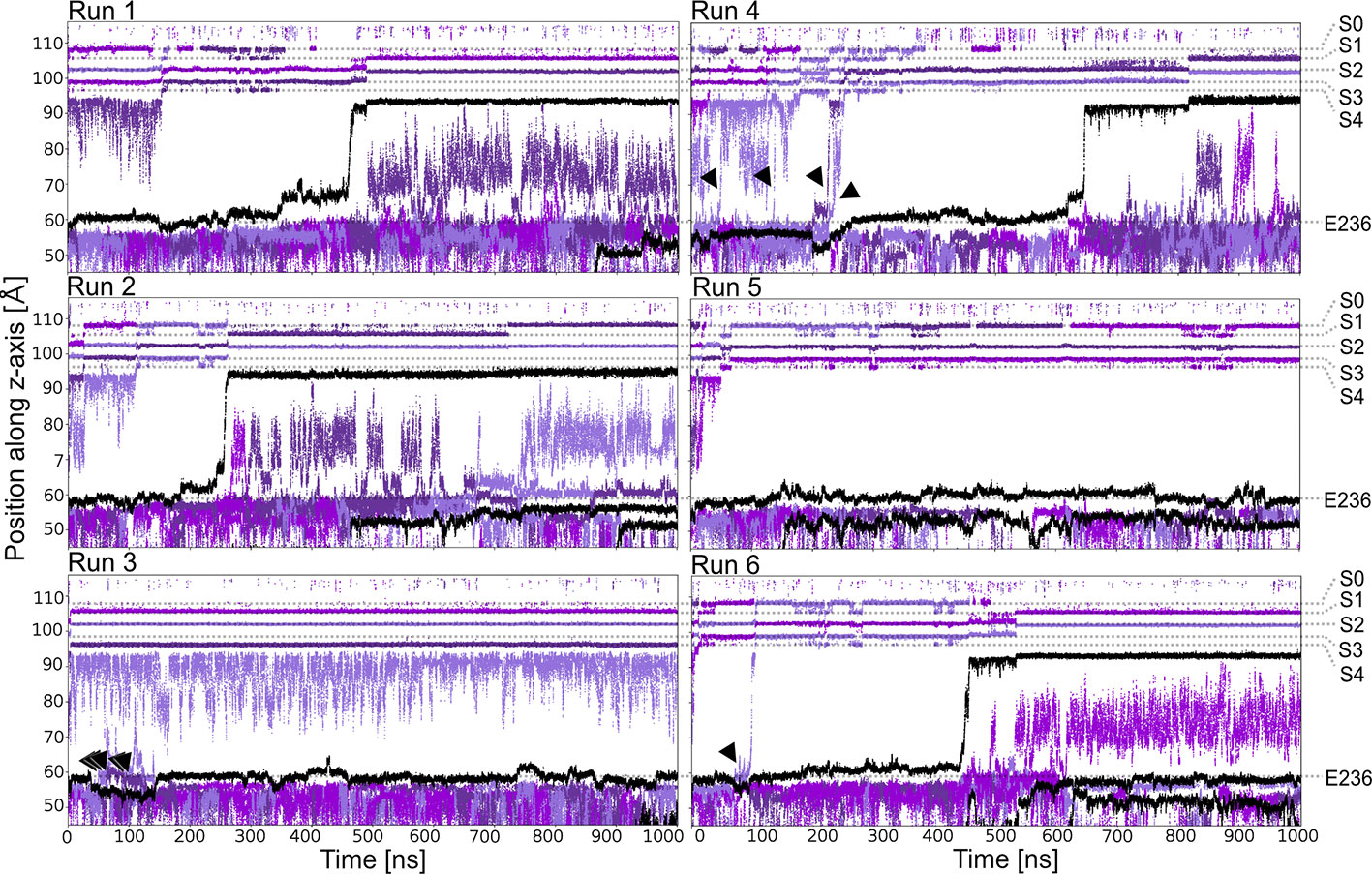
Figure 1 PUT2+ blockage of Kir3.2 channels under an applied field. Positions of K+ and PUT2+ molecules (center of mass) are plotted as function of time along the pore (z-axis). K+ ions are colored in different shades of purple and PUT2+ molecules are shown in black. Slippage of K+ ions is indicated with black arrows.
Binding Sites of Putrescine in Kir3.2
Based on analyzing PUT2+ and K+ ion occupancies extracted from 4µs MD simulations (only from runs, where PUT2+ reached the SF binding site, compare Figure 2B), two binding areas emerged, one located close to E236 in the CTD, and a second, higher occupancy site close to T154 from the SF (Figure 2A). At the binding site in the CTD, frequent hydrogen bonds between the nitrogen atoms of PUT2+ and side-chains of E236 (equal to the known rectification-influencing residue E224 in Kir2.1) were observed. Less frequent hydrogen bond interactions to G318 and T320, close to the G-loop gate, were seen as well (see Figure 2B, right panel). When the molecule left this “shallow” blocking site, it rapidly passed the helix bundle-crossing gate (lined by residues F192) and the transmembrane cavity, without further intermediate binding sites. PUT2+ reached its final binding site at the bottom of the SF, where it oriented mainly perpendicular to the filter axis, which in one run was preceded by a horizontal orientation below the SF at site SC (for representative snapshots, see Figure 2B, right panel). When PUT2+ was bound to the SF, it frequently formed hydrogen bonds with residue T154. Nitrogen atoms of the blocker interacted with the backbone carbonyl oxygen atoms as well as the side-chain of T154. Polyamine binding to the SF lead to displacement of a single K+ ion from the SF (Figure 1 and Supplemental Movies 1–4). Comparison of ion occupancies between apo (taken from Bernsteiner et al., 2019) and holo runs revealed important differences. While, binding sites S2 and S3 were predominantly occupied in the apo runs, with significant K+ occupancy also observed in site SC, PUT2+ binding to the SF or at site SC lead to displacement of ions from site SC, S4 or S3 upwards, expelling the uppermost K+ ion from the filter. Interestingly, a water molecule frequently occupied the site subsequent to PUT2+. K+ occupancy in site S2 was similar between apo and holo runs, while sites S1 and S2 revealed higher occupancy when PUT2+ was bound to the SF, as shown in Supplemental Figure 2.
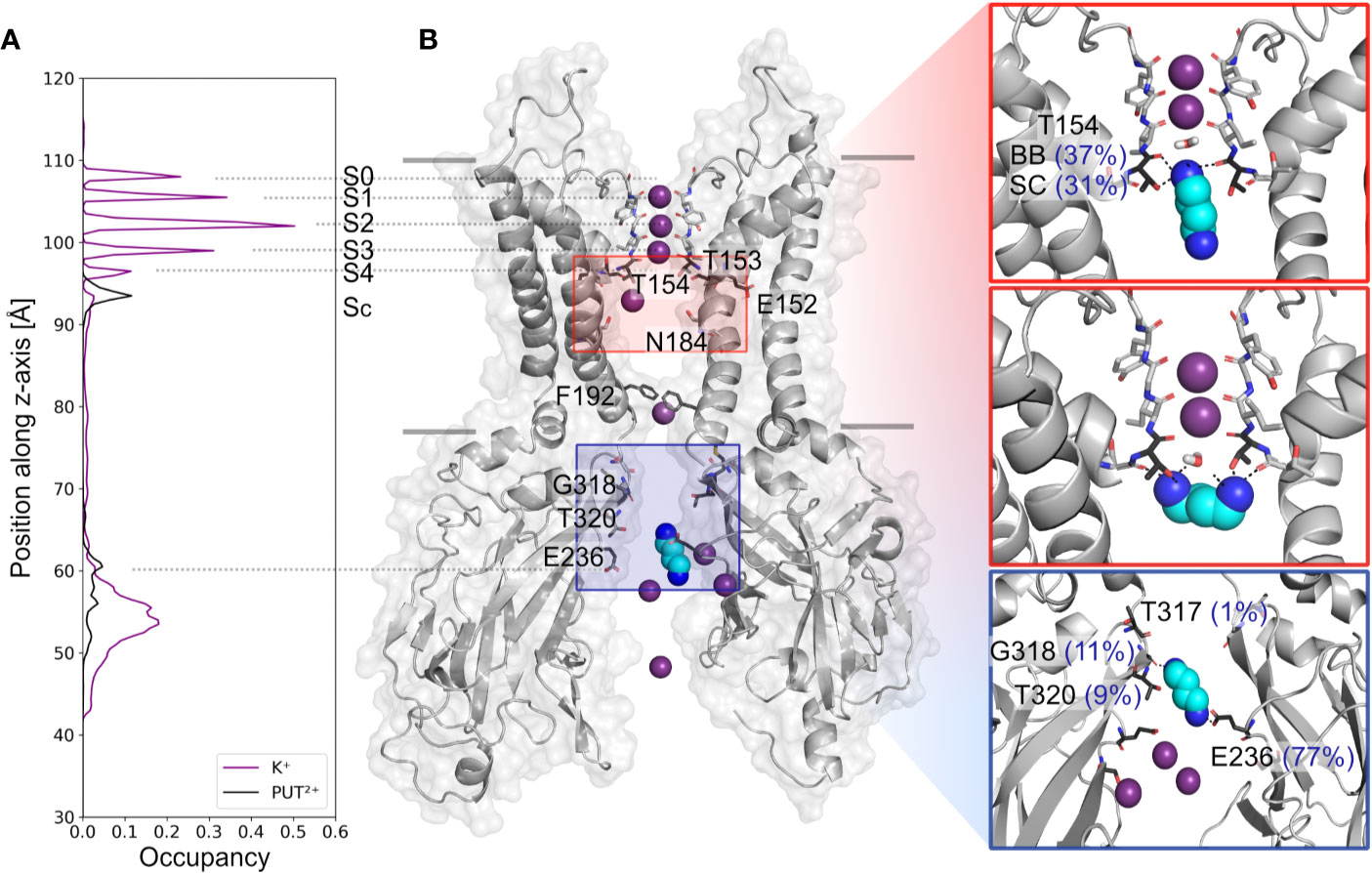
Figure 2 Putrescine occupancies and interactions with Kir3.2. (A) Histograms of K+ (purple line) and PUT2+ (black line) occupancies along the pore axis (z-axis) averaged from 4 µs MD simulations (runs 1, 2, 4, and 6). (B) Two opposing subunits of the equilibrated starting conformation of Kir3.2 (pdb: 3SYA), with K+ ions shown as purple spheres and PUT2+ shown as cyan spheres colored by atom name (nitrogen atoms colored in blue). Key residues for PUT2+ binding are labeled and shown in stick representation with oxygen atoms colored red and nitrogen atoms colored blue. Right panels: representative snapshots of PUT2+ binding sites at the SF (top and middle box) and the CTD (lower box) are shown, with hydrogen bond frequencies indicated in brackets (averaged over runs 1, 2, 4, and 6). BB, backbone; SC, sidechain.
Simulations of Spermine Block (SPM4+)
Next, we performed µs MD simulations with spermine, a polyamine with a net charge of +4 under physiological pH. In contrast to simulations with PUT2+, SPM4+ did not traverse up towards the SF, but remained bound at the CTD site. As illustrated in Figure 3C, the compound mainly interacts with residues E236, T320, G318, and T317. Interestingly, in contrast to simulations with PUT2+ no slippage of K+ ions was observed in two times 1 µs runs. This suggests that SPM4+ completely blocks the channel already at this binding site. To probe if SPM4+ might be able to bind deep within the pore as observed for the much smaller PUT2+, we performed pulling simulations as reported previously for polymine block in a Kainate receptor (Brown et al., 2016). SPM4+ was pulled into the transmembrane cavity up to the position at the SF, observed for PUT2+ binding by applying a harmonic force along the z-axis, as detailed in the Methods section. Subsequently, the molecule was pulled in the reverse direction, towards the intracellular solvent and the work involved in this process calculated as described previously (Brown et al., 2016). Similar pulling simulations were also performed with PUT2+. As illustrated in Figure 3, the work needed for PUT2+ to traverse up to the SF binding site is much smaller compared to the larger SPM4+ molecule. Overall, similar trends have been reported for the Kainate receptor, even though we did not attempt to pull the polyamines through the selectivity filter. These data suggest that SPM4+ might be able to reach a binding site deep in the pore, below the SF, as observed for PUT2+. In the future, more extensive simulations will be required to assess SPM4+ block in Kir3.2 channels in more detail.
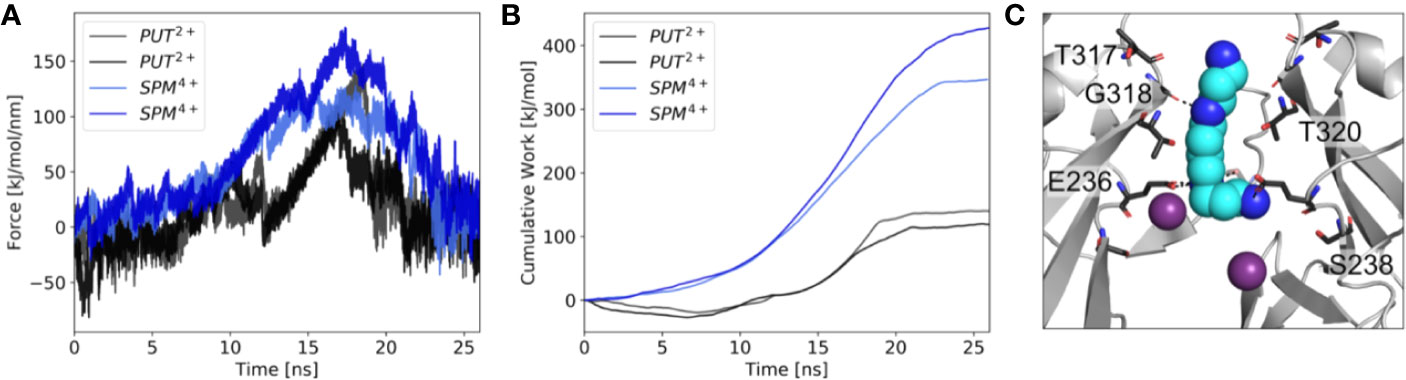
Figure 3 Force probe simulations of PUT2+ and SPM4+ pulling. (A) Force profiles of the polyamines, when pulled along the pore axis of Kir3.2 (B) work profiles for pulling PUT2+ (black) and SPM4+ (blue). (C) Representative snapshot of SPM4+ binding site at the CTD. K+ ions are shown as purple spheres, with interacting residues shown in stick representation.
Where Does the Steep Voltage-Dependence of Polyamine Block Come From?
A long standing question in the field concerns the steepness of the voltage dependence of polyamine block. It roughly correlates with the charge of the polyamine molecules itself, ranging from +2 (in the case of the diamine PUT2+) to +4 for SPM4+, which contains four nitrogen atoms (Nichols and Lee, 2018). To assess the contribution that blocker charge migration through the electric field might have on the steepness of the voltage dependence, we calculated the electrostatic profile along the Kir3.2 channel pore, extracting 21 frames from a 1µs trajectory, using the PME method as implemented in VMD (Aksimentiev and Schulten, 2005), as detailed in the Methods section. The electrostatic contour plots shown in Figure 4, reveal a previously unappreciated feature of the open state of Kir3.2. In accordance to the much wider pore of the MthK channel (27.3 Å, measured at position L95) (Jiang et al., 2002), the membrane electrostatic profile of the narrower Kir3.2 channel pore (Cα-Cα distances of 17.7 Å measured at F192, which is equivalent to position L95 in MthK) is predominantly focused on the SF region. Remarkably, however, there is also a significant fraction of the electric field focused on the narrow G-loop region (Figure 4). As previously reported, the region around residue G318, which forms the narrowest part of the G-loop gate, fluctuates between 5.9 to 6.8 Å, with oxygen atoms of G318 participating in K+ coordination (Bernsteiner et al., 2019). This suggests that the access resistance for a K+ ion diffusing between the cytoplasm and the SF might be higher in Kir3.2 compared to MthK. This agrees well with the much lower conductance rates in KIR channels [~30 pS for Kir3.2 (Kofuji et al., 1996) vs. 170 pS for MthK channels (Li et al., 2007)]. Due to the narrower pore radius of the open KIR channel, ions and other charged molecules such as PUT2+ experience a significant fraction of the membrane potential, when traversing from the CTD towards the SF. The electrostatic maps reveal that polyamine movement from the CTD towards a deep binding site below the SF might substantially contribute to the steepness of the voltage dependence of block (Figure 3). Further, partial binding to the SF, as observed for PUT2+ (Figure 2, left panel), contributes to voltage-dependence as well. This data also suggests that the electrical field distribution renders K+ conduction through the channel voltage-dependent. Indeed, such behavior has been previously reported by Spassova and Lu (Spassova and Lu, 1998).
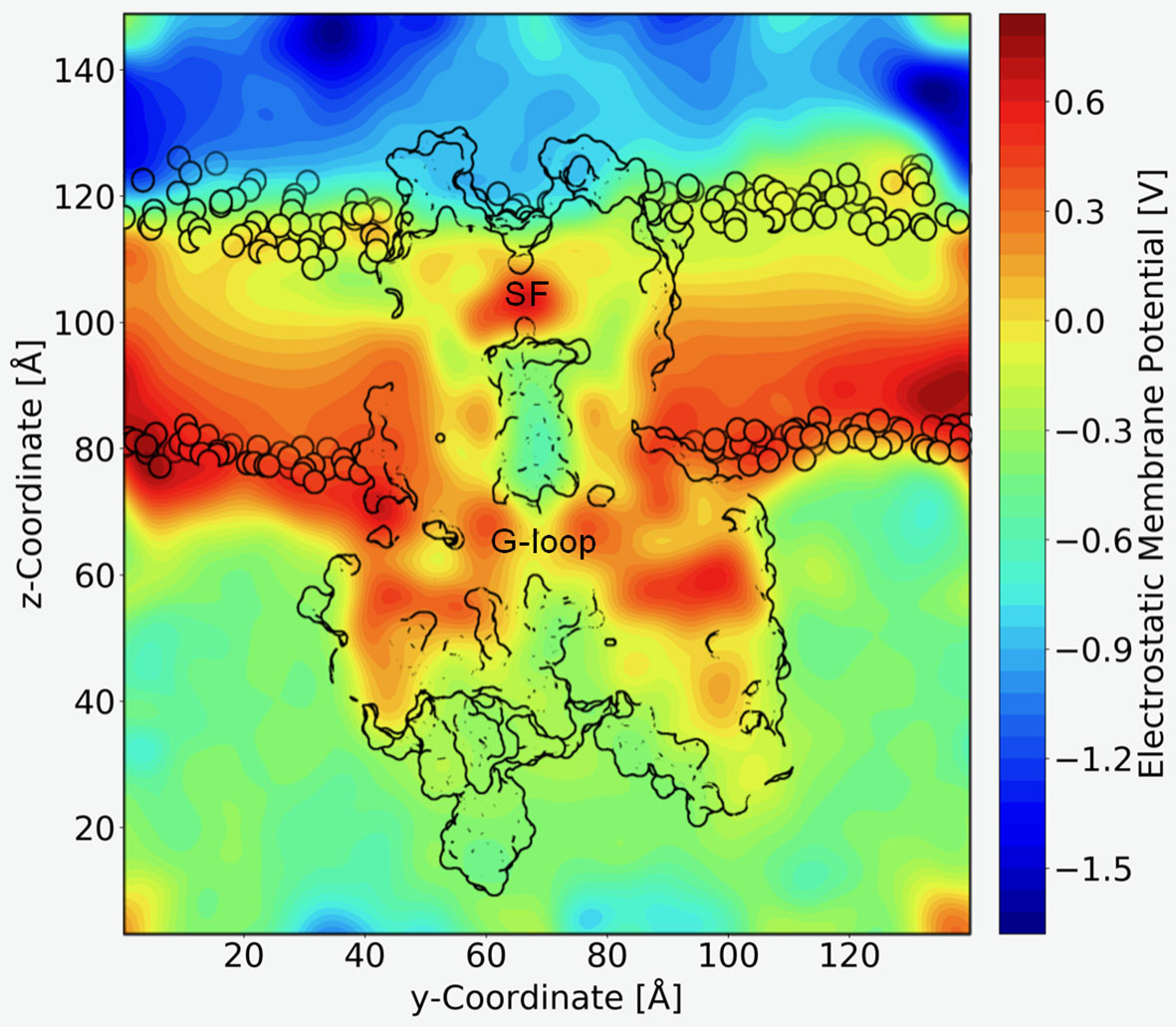
Figure 4 Electrostatic profile of Kir3.2. An external electric field of 0.5 V was introduced, as applied in free MD simulations. 21 snapshots from the trajectory were extracted and smoothed electrostatic potential grids with a resolution of 1 Å per grid point are shown. The overall shape of the Kir3.2 channels (side view), as well has phospholipid head groups are outlined in black. The selectivity filter (SF) and G-loop area are indicated.
Discussion
In this work, we aimed to obtain first structural insights into the mechanism of PUT2+ and SPM4+ interactions, two naturally occurring polyamines, with the strong inward rectifier K+ channel Kir3.2. Polyamines are essential components in cells and bear ionisable amine moieties in their aliphatic chains. They play critical roles in protein synthesis, cell division and cell growth (Pegg, 2016; Handa et al., 2018). Their role in KIR channel inhibition, due to pore block from the intracellular side was extensively studied (for detailed reviews see Nichols and Lopatin, 1997; Lu, 2004; Baronas and Kurata, 2014; Nichols and Lee, 2018), however, critical questions including the exact binding sites and the nature of the strong voltage-dependence are still unresolved.
We analysed blockade of the channel by performing multiple µs atomistic MD simulations on the GIRK2 crystal structure with an applied field to mimic a positive membrane potential, and performed pulling simulations to probe the work required for polyamines binding below the SF.
Two distinct, sequential polyamine binding sites were observed in our simulations with PUT2+, in agreement with kinetic modelling studies (Lopatin et al., 1995). A “shallow” binding site in the CTD was predicted, where the drug mainly interacted with a negatively charged ring, formed by E236, which is equivalent to E224 in Kir2.1 (Figure 2B, right panel, Supplemental Figure 1, for alignment), the best studied strong inward rectifier channel. This residue has previously been implicated in polyamine block. Neutralization has been shown to slow the rate of block, and reduce the apparent affinity of a second deep site block (Taglialatela et al., 1995; Kubo and Murata, 2001). In contrast, in simulations with SPM4+, only the CTD binding site was observed in the 1 µs runs, due to limited sampling. Thus, by applying enhanced sampling (pulling simulations) on the polyamines, we were able to sample binding of SPM4+ at the “deep” binding site, below the SF. Binding of this larger polyamine required three times more work, compared to the much smaller PUT2+, suggesting that it is much easier for PUT2+ to move up to the SF site, compared to SPM4+. A similar trend has previously been reported for Kainate receptor polyamine interactions (Brown et al., 2016).
Remarkably, our simulations revealed that PUT2+ could not completely inhibit ion conduction when interacting with the negatively charged residues in the CTD, since “slippage” of K+ ions past the bound polyamine upwards into the transmembrane region was observed in several runs (Figure 1). This observation agrees with previous suggestions of incomplete block at the CTD, based on functional studies on Kir2.1 (Xu et al., 2009). Interestingly, no slippage was observed, when SPM4+ was bound to the same site in the CTD. However, longer sampling will be required to address, if this difference indeed holds true on longer simulation time scales.
Further, MD simulations revealed that PUT2+ could move through the electric field up to a steep binding site, at the SF where it stably interacted with T154, located at the base of the filter. This residue has not been tested experimentally for polyamine block, but it has been implicated in high-affinity block of Ba2+ in Kir2.1 channels (Hsieh et al., 2015). Support for blocker migration deep into the pore comes from blocker protection experiments with MTS reagents, suggesting that polyamines might bind above the rectification controller position D172 (Kurata et al., 2008).
Another important observation of our study is that PUT2+ migration to the SF binding site is coupled with displacement of a permeant K+ ion. Such a scenario has recently been observed in CP-AMPAR channels, where polyamines with different tail length have been analyzed, using cryo-EM. Structures with short polyamines reveal that these molecules partially protrude into the SF, while longer molecules bind deep within the SF. The cryo-EM structure demonstrates that a combination of blocker entry into the filter and ion displacement can lead to steep voltage dependence (Twomey et al., 2018). Our simulations suggest a similar mechanism for inward rectifier channels. Further, such a mechanism has been previously envisioned, based on experimental observation, showing that the blocker dependence on the membrane voltage varied as a function of extracellular K+ concentration (Spassova and Lu, 1998; Whorton and MacKinnon, 2011).
An important, previously largely unappreciated finding of our study concerns the nature of the electric field. In contrast to previous expectations, based on calculations of K+ channel pores with wider gate diameters (e.g. Jiang et al., 2002), the membrane potential does not only drop over the selectivity filter, but there is a significant voltage drop over the G-loop area, as illustrated in Figure 4. Based on these qualitative calculations, polyamine binding to the CTD site is likely to depend on the prevailing voltage (without ion displacement from the SF), due to molecules traversing a fraction of the membrane potential, when passing through the G-loop gate area.
In addition, our simulations consistently predict that binding of PUT2+ to the SF site leads to outward displacement of a K+ ion (Figure 1 and Supplemental Movies). These predictions agree very well with functional studies, supporting sequential binding with weak and strong voltage dependence (Lopatin et al., 1995; Shin and Lu, 2005; Kurata et al., 2007).
The predicted differences in electrostatic potential along the pore further suggest that the access resistance for K+ ions diffusing between the cytoplasm and the SF is higher in Kir3 channels, compared to ion channels with wider open pores such as MthK. This is in line with the different conductance rates reported for these channels, which amount to ~ 30 pS in Kir3.2 channels (Kofuji et al., 1996), but are much higher in MthK channels, where values of ~ 200 pS have been published (Li et al., 2007). The electrostatic data further suggests that the electrical field distribution should render K+ conduction voltage dependent, which is in agreement with previous reports by Spassova and Lu (1998).
A clear limitation of our study is the fact that so far we tested only two polyamines PUT2+ and SPM4+ on an “atypical” strong inward rectifier channel that has been poorly studied in the homotetrameric form. Studies investigating polyamine block in Kir3 channels have almost exclusively focused on the heterotetrameric versions composed of Kir3.1 and Kir3.4 (carrying IKACh currents) (Yamada and Kurachi, 1995; Salazar-Fajardo et al., 2018). However, strong inward rectification was confirmed for the crystal structure construct of Kir3.2 (Whorton and MacKinnon, 2011), justifying the use of this construct to study the mechanisms of inward rectification. Further, the open state structure of the extremely well characterized Kir2.1 channel is still not available.
Given the very recent success to crystallize Kir2.2 channels in an open, conductive conformation (Zangerl-Plessl et al., 2020) future studies will need to be performed to address polyamine block in the classic strong rectifier Kir2 family. Comparison of conductance studies on Kir3.2 and Kir2.2 channels reveal very interesting differences, which might affect polyamine block. While the inner (transmembrane) cavity of Kir3.2 contains on average only one K+ ion (Bernsteiner et al., 2019), simulations on Kir2.2 channels revealed that the inner cavity in these channels can harbour up to five K+ ions, located close to the acidic charges of the rectification controller, which is lacking in Kir3.2 channels. Thus, it might be envisioned that perhaps polyamine entry into the inner cavity would lead to different ion displacement between these different channels. Further, the diameter of the G-loop region is quite different in the two Kir families. While no Kir2.2 structure showing a narrow G-loop region has been reported so far, various constricted conformations have been reported for Kir3 structures. Our recent simulations on WT Kir3.2 channels (Bernsteiner et al., 2019) vs. Kir2.2 mutant channels (Zangerl-Plessl et al., 2020) support a different behaviour of this region in the two channels. Future studies will be necessary to address similarities and differences inward rectification mechanisms in other Kir channels in detail.
Summarizing our simulations predict that blocker amines can indeed interact with the SF, and that voltage dependence of inward rectification arises in part from the movement of the blocker through the electric field and energetic coupling between blocker binding and permeating K+ ions.
Material and Methods
Molecular Dynamics Simulations
Gromacs (version 5.1.2) (Abraham et al., 2015) was used to perform molecular dynamics simulations, with the conductive state of the Kir3.2 channel, embedded in a lipid bilayer consisting of 588 POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) lipids, taken from Bernsteiner et al., (Bernsteiner et al., 2019). PIP2 parameters were taken from our previous work (Lee et al., 2016). Berger lipid parameters (Berger et al, 1997) were used for the POPC lipids, the amber99sb forcefield (Hornak et al., 2006) for the protein, and corrected monovalent Lennard-Jones parameters (Joung and Cheatham, 2008) were used for K+ ions. After neutralization with K+ ions, 150 mM KCl was added to the simulation system. For Lennard-Jones and electrostatic interactions, a cut-off of 1.0 nm was employed, with long-range electrostatic interactions calculated with the Particle-Mesh Ewald algorithm (Essmann et al., 1995). Bonds were constrained using the LINCS algorithm (Hess et al., 1997), allowing for an integration timestep of 2 fs. Temperature was coupled to 310 K using the v-rescale thermostat (Bussi et al., 2007), with a coupling constant of 0.1 ps. The pressure was kept constant semi-isotropically at 1 bar by the Parrinello-Rahman (Parrinello and Rahman, 1981) barostat (τ = 2 ps). Putrescine was parameterized using the Hartree-Fock geometry optimization with the 6–31G* basis set in Gaussian (Frisch et al, 2016) and the force field parameters are obtained from AmberTools (Case et al., 2018). All amine nitrogen atoms were treated as charged. Six times one µs runs with an applied electric field of 40 mV nm−1 along the z-axis of the simulation box were performed. With a z-axis box length of ∼14.5 nm, this resulted in transmembrane potentials of 580 mV (Treptow et al., 2004; Roux, 2008). 20 putrescine molecules were added to the solvent, with one molecule initially placed at the entrance of the CTD, as shown in Figure 2B (left panel), using gromacs tools. PUT2+ and K+ occupancies were calculated as described previously (Bernsteiner et al., 2019).
Force Probe (Steered) MD Simulations
The work needed to bind and unbind PUT2+ and SPM4+ from the Kir3.2 channel was assessed using force probe MD simulations. Polyamines were placed perpendicular to the z-axis and pulled into the transmembrane cavity towards position T154 (bottom of SF) and in the reverse direction towards the cytoplasm with a harmonic force of 100 kJ mol-1 nm-2 and a velocity of 0.25 nm ns-1. The work was calculated from the force profiles as previously described (Brown et al., 2016) using the following equation:
where v is the constant velocity, f(t) is the force at a specific time step given in the force probe output and dt is the time step between data points (100 fs).
Electrostatic Profile Calculations
The electrostatic profile along the Kir3.2 channel was calculated based on a 1 µs trajectory of a conductive WT Kir3.2 run taken from Bernsteiner et al., 2019. In total, 21 frames were extracted (one frame each 50 ns) and aligned at the selectivity filter backbone. We subsequently applied the VMD Plugin PME Electrostatics to each of the frames, which generates smoothed electrostatic potential grids by evaluating the reciprocal sum of the smooth particle-mesh Ewald method (PME) (Aksimentiev and Schulten, 2005; Treptow et al., 2009). By setting the size of the three dimensional grid to 144x144x160 points, we established a resolution of 1 Å per grid point. The Ewald factor was set to 0.25. Charge information was included by converting the PDBs to PSF files with Amber18 (Case et al., 2018) based on the Amber99sb force field with Berger lipids (see MD methods). Following the protocol for grid manipulation of the Aksimentiev group, an external electric field of 0.5 V was introduced to the system. The electrostatic profile was averaged over the 21 frames and plotted by extracting the y-z plane of the grid, which slices through the selectivity filter. A python script was kindly provided by Kumar Sarthak from the Aksimentiev group.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository and accession number(s) are: Zenodo, http://doi.org/10.5281/zenodo.3760965.
Author Contributions
AS-W designed the study. XC and MB performed simulations. XC, MB, TF, and AS-W analyzed the data. AS-W wrote the paper, which was edited by XC, MB, and TF.
Funding
This work was supported by the doctoral program “Molecular drug targets” W1232, from the Austrian Science Fund (FWF; http://www.fwf.ac.at).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The computational results presented have been achieved in part using the Vienna Scientific Cluster (VSC).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2020.00721/full#supplementary-material
References
Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., et al. (2015). GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25. doi: 10.1016/j.softx.2015.06.001
Aksimentiev, A., Schulten, K. (2005). Imaging Alpha-Hemolysin with Molecular Dynamics: Ionic Conductance, Osmotic Permeability, and the Electrostatic Potential Map. Biophys. J. 88 (6), 3745–3761. doi: 10.1529/biophysj.104.058727
Baronas, V. A., Kurata, H. T. (2014). Inward Rectifiers and Their Regulation by Endogenous Polyamines. Front. Physiol. 5. doi: 10.3389/fphys.2014.00325
Berger, O., Edholm, O., Jähnig, F. (1997). Molecular Dynamics Simulations of a Fluid Bilayer of Dipalmitoylphosphatidylcholine at Full Hydration, Constant Pressure, and Constant Temperature. Biophys. J. 72 (5), 2002–2013. doi: 10.1016/S0006-3495(97)78845-3
Bernsteiner, H., Zangerl-Plessl, E.-M., Chen, X., Stary-Weinzinger, A. (2019). Conduction through a Narrow Inward-Rectifier K+ Channel Pore. J. Gen. Physiol. 151 (10), 1231–1246. doi: 10.1085/jgp.201912359
Brown, P. M. G. E., Aurousseau, M. R. P., Musgaard, M., Biggin, P. C., Bowie, D. (2016). Kainate Receptor Pore-Forming and Auxiliary Subunits Regulate Channel Block by a Novel Mechanism. J. Physiol. 594 (7), 1821–1840. doi: 10.1113/JP271690
Bussi, G., Donadio, D., Parrinello, M.(2007). “Canonical sampling through velocity rescaling”. J. Chem. Phys. 126 (1), 014101. doi: 10.1063/1.2408420
Case, D. A., Ben-Shalom, I. Y., Brozell, S. R., Cerutti, D. S., Cheatham, T. E., Cruzeiro, V. W. D., et al. (2018). “AMBER 2018” (San Francisco: University of California).
Doyle, D. A., Morais, C. J., Pfuetzner, R. A., Kuo, A., Gulbis, J. M., Cohen, S. L., et al. (1998). The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280 (5360), 69–77. doi: 10.1126/science.280.5360.69
Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H., Pedersen, L. G. (1995). A smooth particle mesh Ewald method. J. Chem. Phys. 103 (19), 8577–8593. doi: 10.1063/1.470117
Fakler, B., Brändle, U., Glowatzki, E., Weidemann, S., Zenner, H. P., Ruppersberg, J. P. (1995). Strong Voltage-Dependent Inward Rectification of Inward Rectifier K+ Channels Is Caused by Intracellular Spermine. Cell 80 (1), 149–154. doi: 10.1016/0092-8674(95)90459-x
Ficker, E., Taglialatela, M., Wible, B. A., Henley, C. M., Brown, A. M. (1994). Spermine and Spermidine as Gating Molecules for Inward Rectifier K+ Channels. Science 266 (5187), 1068–1072. doi: 10.1126/science.7973666
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). “Gaussian 09” (Wallingford CT: Gaussian, Inc.).
Fujiwara, Y., Kubo, Y. (2006). Functional Roles of Charged Amino Acid Residues on the Wall of the Cytoplasmic Pore of Kir2.1. J. Gen. Physiol. 127 (4), 401–419. doi: 10.1085/jgp.200509434
Guo, D., Ramu, Y., Klem, A. M., Lu, Z. (2003). Mechanism of Rectification in Inward-Rectifier K+ Channels. J. Gen. Physiol. 121 (4), 261–275. doi: 10.1085/jgp.200208771
Handa, A. K., Fatima, T., Mattoo, A. K. (2018). Polyamines: Bio-Molecules with Diverse Functions in Plant and Human Health and Disease. Front. Chem. 6, 10. doi: 10.3389/fchem.2018.00010
Hess, B., Bekker, H., Berendsen, H. J. C., Fraaije, J. G. E. M. (1997). LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 18 (12), 1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H
Hibino, H., Inanobe, A., Furutani, K., Murakami, S., Findlay, I., Kurachi, Y. (2010). Inwardly Rectifying Potassium Channels: Their Structure, Function, and Physiological Roles. Physiol. Rev. 90 (1), 291–366. doi: 10.1152/physrev.00021.2009
Hornak, V., Abel, R., Okur, A., Strockbine, B., Roitberg, A., Simmerling, C. (2006). Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins 65 (3), 712–725. doi: 10.1002/prot.21123
Hsieh, C.-P., Kuo, C.-C., Huang, C.-W. (2015). Driving Force-Dependent Block by Internal Ba(2+) on the Kir2.1 Channel: Mechanistic Insight into Inward Rectification. Biophys. Chem. 202, 40–57. doi: 10.1016/j.bpc.2015.04.003
Jiang, Y., Lee, A., Chen, J., Cadene, M., Chait, B. T., MacKinnon, R. (2002). The open pore conformation of potassium channels. Nature 417 (6888), 523–526. doi: 10.1038/417523a
Joung, I. S., Cheatham, T. E. (2008). Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 112 (30), 9020–9041. doi: 10.1021/jp8001614
Kofuji, P., Hofer, M., Millen, K. J., Millonig, J. H., Davidson, N., Lester, H. A., et al. (1996). Functional Analysis of the Weaver Mutant GIRK2 K+ Channel and Rescue of Weaver Granule Cells. Neuron 16 (5), 941–952. doi: 10.1016/S0896-6273(00)80117-8
Kubo, Y., Murata, Y. (2001). Control of Rectification and Permeation by Two Distinct Sites after the Second Transmembrane Region in Kir2.1 K+ Channel. J. Physiol. 531 (Pt 3), 645–660. doi: 10.1111/j.1469-7793.2001.0645h.x
Kuo, A., Gulbis, J. M., Antcliff, J. F., Rahman, T., Lowe, E. D., Zimmer, J., et al. (2003). Crystal Structure of the Potassium Channel KirBac1.1 in the Closed State. Science 300 (5627), 1922–1926. doi: 10.1126/science.1085028
Kurata, H. T., Phillips, L. R., Rose, T., Loussouarn, G., Herlitze, S., Fritzenschaft, H., et al. (2004). Molecular Basis of Inward Rectification: Polyamine Interaction Sites Located by Combined Channel and Ligand Mutagenesis. J. Gen. Physiol. 124 (5), 541–554. doi: 10.1085/jgp.200409159
Kurata, H. T., Cheng, W. W., Arrabit, C., Slesinger, P. A., Nichols, C. G. (2007). The Role of the Cytoplasmic Pore in Inward Rectification of Kir2.1 Channels. J. Gen. Physiol. 130 (2), 145–155. doi: 10.1085/jgp.200709742
Kurata, H. T., Diraviyam, K., Marton, L. J., Nichols, C. G. (2008). Blocker Protection by Short Spermine Analogs: Refined Mapping of the Spermine Binding Site in a Kir Channel. Biophys. J. 95 (8), 3827–3839. doi: 10.1529/biophysj.108.133256
Lee, S.-J., Ren, F., Zangerl-Plessl, E.-M., Heyman, S., Stary-Weinzinger, A., Yuan, P., et al. (2016). Structural Basis of Control of Inward Rectifier Kir2 Channel Gating by Bulk Anionic Phospholipids. J. Gen. Physiol. 148 (3), 227–237. doi: 10.1085/jgp.201611616
Li, Y., Berke, I., Chen, L., Jiang, Y. (2007). Gating and Inward Rectifying Properties of the MthK K+ Channel with and without the Gating Ring. J. Gen. Physiol. 129 (2), 109–120. doi: 10.1085/jgp.200609655
Lopatin, A. N., Makhina, E. N., Nichols, C. G. (1994). Potassium Channel Block by Cytoplasmic Polyamines as the Mechanism of Intrinsic Rectification. Nature 372 (6504), 366–369. doi: 10.1038/372366a0
Lopatin, A. N., Makhina, E. N., Nichols, C. G. (1995). The mechanism of inward rectification of potassium channels: "long-pore plugging" by cytoplasmic polyamines. J Gen Physiol. 106 (5), 923–955. doi: 10.1085/jgp.106.5.923
Lu, Z., MacKinnon, R. (1994). Electrostatic Tuning of Mg2+ Affinity in an Inward-Rectifier K+ Channel. Nature 371 (6494), 243–246. doi: 10.1038/371243a0
Lu, Z. (2004). Mechanism of Rectification in Inward-Rectifier K+ Channels. Annu. Rev. Physiol. 66 (1), 103–129. doi: 10.1146/annurev.physiol.66.032102.150822
Nichols, C. G., Lee, S.-J. (2018). Polyamines and Potassium Channels: A 25-Year Romance. J. Biol. Chem. 293 (48), 18779–18788. doi: 10.1074/jbc.TM118.003344
Nichols, C. G., Lopatin, A. N. (1997). Inward rectifier potassium channels. Annu. Rev. Physiol. 59 (1), 171–191. doi: 10.1146/annurev.physiol.59.1.171
Parrinello, M., Rahman, A. (1981). “Polymorphic transitions in single crystals: A new molecular dynamics method”. J. Appl. Phys. 52 (12), 7182–7190. doi: 10.1063/1.328693
Pegg, A. E. (2016). “Functions of Polyamines in Mammals”. J. Biol. Chem. 291 (29), 14904–14912. doi: 10.1074/jbc.R116.731661
Robertson, J. L., Palmer, L. G., Roux, B. (2008). “Long-Pore Electrostatics in Inward-Rectifier Potassium Channels”. J. Gen. Physiol. 132 (6), 613–632. doi: 10.1085/jgp.200810068
Roux, B. (2008). “The Membrane Potential and Its Representation by a Constant Electric Field in Computer Simulations”. Biophys. J. 95 (9), 4205–4216. doi: 10.1529/biophysj.108.136499
Salazar-Fajardo, P. D., Aréchiga-Figueroa, I. A., López-Serrano, A. L., Rodriguez-Elias, J. C., Alamilla, J., Sánchez-Chapula, J. A., et al. (2018). The Voltage-Sensitive Cardiac M2 Muscarinic Receptor Modulates the Inward Rectification of the G Protein-Coupled, ACh-Gated K+ Current. Pflugers Archiv: Eur. J. Physiol. 470 (12), 1765–1776. doi: 10.1007/s00424-018-2196-y
Shin, H. G., Lu, Z. (2005). Mechanism of the voltage sensitivity of IRK1 inward-rectifier K+ channel block by the polyamine spermine. J. Gen. Physiol. 125 (4), 413–426. doi: 10.1085/jgp.200409242
Spassova, M., Lu, Z. (1998). Coupled Ion Movement Underlies Rectification in an Inward-Rectifier K+ Channel. J. Gen. Physiol. 112 (2), 211–221. doi: 10.1085/jgp.112.2.211
Stanfield, P. R., Davies, N. W., Shelton, P. A., Sutcliffe, M. J., Khan, I. A., Brammar, W. J., et al. (1994). A Single Aspartate Residue Is Involved in Both Intrinsic Gating and Blockage by Mg2+ of the Inward Rectifier, IRK1. J. Physiol. 478 ( Pt 1), 1–6. doi: 10.1113/jphysiol.1994.sp020225
Taglialatela, M., Ficker, E., Wible, B. A., Brown, A. M. (1995). C-Terminus Determinants for Mg2+ and Polyamine Block of the Inward Rectifier K+ Channel IRK1. EMBO J. 14 (22), 5532–5541. doi: 10.1002/j.1460-2075.1995.tb00240.x
Treptow, W., Maigret, B., Chipot, C., Tarek, M. (2004). Coupled Motions between Pore and Voltage-Sensor Domains: A Model for Shaker B, a Voltage-Gated Potassium Channel. Biophys. J. 87 (4), 2365–2379. doi: 10.1529/biophysj.104.039628
Treptow, W., Tarek, M., Klein, M. L. (2009). Initial Response of the Potassium Channel Voltage Sensor to a Transmembrane Potential. J. Am. Chem. Soc. 131 (6), 2107–2109. doi: 10.1021/ja807330g
Twomey, E. C., Yelshanskaya, M. V., Vassilevski, A. A., Sobolevsky, A. I. (2018). Mechanisms of Channel Block in Calcium-Permeable AMPA Receptors. Neuron 99 (5), 956–968.e4. doi: 10.1016/j.neuron.2018.07.027
Whorton, M. R., MacKinnon, R. (2011). Crystal Structure of the Mammalian GIRK2 K+ Channel and Gating Regulation by G Proteins, PIP2, and Sodium. Cell 147 (1), 199–208. doi: 10.1016/j.cell.2011.07.046
Wible, B. A., Taglialatela, M., Ficker, E., Brown, A. M. (1994). “Gating of Inwardly Rectifying K+ Channels Localized to a Single Negatively Charged Residue”. Nature 371 (6494), 246–249. doi: 10.1038/371246a0
Xu, Y., Shin, H. G., Szép, S., Lu, Z. (2009). Physical determinants of strong voltage sensitivity of K(+) channel block. Nat. Struct. Mol. Biol. 16 (12), 1252–1258. doi: 10.1038/nsmb.1717
Yamada, M., Kurachi, Y. (1995). Spermine Gates Inward-Rectifying Muscarinic but Not ATP-Sensitive K+ Channels in Rabbit Atrial Myocytes. Intracellular Substance-Mediated Mechanism of Inward Rectification. J. Biol. Chem. 270 (16), 9289–9294. doi: 10.1074/jbc.270.16.9289
Yi, B. A., Lin, Y. F., Jan, Y. N., Jan, L. Y. (2001). Yeast Screen for Constitutively Active Mutant G Protein-Activated Potassium Channels. Neuron 29 (3), 657–667. doi: 10.1016/s0896-6273(01)00241-0
Keywords: molecular dynamics simulation, charge movement, ion displacement, inward rectification, Kir3.2, putrescine
Citation: Chen X, Bründl M, Friesacher T and Stary-Weinzinger A (2020) Computational Insights Into Voltage Dependence of Polyamine Block in a Strong Inwardly Rectifying K+ Channel. Front. Pharmacol. 11:721. doi: 10.3389/fphar.2020.00721
Received: 30 November 2019; Accepted: 30 April 2020;
Published: 15 May 2020.
Edited by:
Gildas Loussouarn, Université de Nantes, FranceReviewed by:
Show-Ling Shyng, Oregon Health and Science University, United StatesMounir Tarek, Centre National de la Recherche Scientifique, France
Copyright © 2020 Chen, Bründl, Friesacher and Stary-Weinzinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Stary-Weinzinger, YW5uYS5zdGFyeUB1bml2aWUuYWMuYXQ=
†These authors have contributed equally to this work