 Ruiying Wang
Ruiying Wang Min Wang
Min Wang Shuaibing He
Shuaibing He Guibo Sun
Guibo Sun Xiaobo Sun
Xiaobo Sun- 1Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
- 2Beijing Key Laboratory of Innovative Drug Discovery of Traditional Chinese Medicine (Natural Medicine) and Translational Medicine, Institute of Medicinal Plant Development, Peking Union Medical College & Chinese Academy of Medical Sciences, Beijing, China
- 3Key Laboratory of Bioactive Substances and Resources Utilization of Chinese Herbal Medicine, Ministry of Education, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
- 4Key Laboratory of Efficacy Evaluation of Chinese Medicine Against Glycolipid Metabolic Disorders, State Administration of Traditional Chinese Medicine, Institute of Medicinal Plant Development, Peking Union Medical College & Chinese Academy of Medical Sciences, Beijing, China
- 5Key Laboratory of New Drug Discovery Based on Classic Chinese Medicine Prescription, Chinese Academy of Medical Sciences, Beijing, China
Calcium homeostasis plays an essential role in maintaining excitation–contraction coupling (ECC) in cardiomyocytes, including calcium release, recapture, and storage. Disruption of calcium homeostasis may affect heart function, leading to the development of various heart diseases. Myocardial ischemia/reperfusion (MI/R) injury may occur after revascularization, which is a treatment used in coronary heart disease. MI/R injury is a complex pathological process, and the main cause of increased mortality and disability after treatment of coronary heart disease. However, current methods and drugs for treating MI/R injury are very scarce, not ideal, and have limitations. Studies have shown that MI/R injury can cause calcium overload that can further aggravate MI/R injury. Therefore, we reviewed the effects of critical calcium pathway regulators on MI/R injury and drew an intuitive diagram of the calcium homeostasis pathway. We also summarized and analyzed calcium pathway-related or MI/R drugs under research or marketing by searching Therapeutic Target and PubMed Databases. The data analysis showed that six drugs and corresponding targets are used to treat MI/R injury and involved in calcium signaling pathways. We emphasize the relevance of further detailed investigation of MI/R injury and calcium homeostasis and the therapeutic role of calcium homeostasis in MI/R injury, which bridges basic research and clinical applications of MI/R injury.
Introduction
Myocardial ischemia/reperfusion (MI/R) is an inevitable process in the treatment of cardiovascular diseases such as acute myocardial infarction, thrombolysis, coronary angioplasty, and cardiac arrest (Zhu et al., 2017). MI/R injury refers to a more serious damage caused by ischemic myocardial tissues after blood perfusion is restored. MI/R can aggravate reversible damage to cardiac tissue; it may also promote reversible damage into irreversible damage of cardiac tissue, eventually worsening the patient's condition and even causing death (Fiolet and Baartscheer, 2000). It can be clinically manifested as arrhythmia, decreased cardiac function, and enlarged myocardial necrotic area (Murphy and Steenbergen, 2008). Recently, research has mainly focused on various theories such as calcium overload, energy metabolism disorders, free radical effects, oxidative stress, inflammatory response, and mitochondrial dysfunction (Thind et al., 2015). Although vast studies have reported on the mechanism underlying MI/R injury, the clinical transformation is limited and the therapeutic effects are not ideal. At present, there are few clinical treatments for MI/R injury, mainly through ischemic post-conditioning and drugs of anti-free radicals, dilating blood vessels, and reducing calcium overload. Therefore, further exploring the mechanism of MI/R injury and developing more effective anti-MI/R drugs are necessary (Turer and Hill, 2010; Ibanez et al., 2015). Calcium homeostasis is particularly important for myocardial cell structure and function. So, it is of significance to focus on some cardiac protective reagents related to calcium overload to treat MI/R injury (Kleinbongard et al., 2012).
Ca2+ is an important cytoplasmic signaling molecule in most cellular responses and is mainly distributed in extracellular and intracellular organelles, such as sarcoplasmic reticulum (SR) and mitochondria. Calcium plays a vital role in excitation–contraction coupling (ECC) of heart tissue. When cardiac action potential occurs, extracellular Ca2+ enters the cell through L-type Ca2+ channels (LTCC), and intracellular Ca2+ activates ryanodine receptor 2 (RyR2) and more Ca2+ is released from the sarcoplasmic reticulum releases (Marks, 2013). When the intracellular Ca2+ is present at a certain level, it binds to the myofilament protein troponin C causing myocardial contraction. On the one hand, sarco (endo) plasmic reticulum Ca2+-ATPase (SERCA) recaptures intracellular Ca2+ back to SR. On the other hand, sodium/calcium exchanger (NCX) expels Ca2+ from the cells and the dissociation of Ca2+ from myofilament protein relaxes the myocardial cells (Hadri and Hajjar, 2011). The ECC in myocardial cells can also be attributed to multiple organelles together to maintain intracellular calcium homeostasis. Calcium levels in myocardial cells are regulated by LTCC, NCX, SERCA, RyR2, and mitochondria, which participate in calcium overload during MI/R injury (Landstrom et al., 2017). The study of cardiac calcium homeostasis has always been a hot topic. Common experimental subjects include human induced pluripotent stem cells (hiPSCs), isolated hearts, primary cardiomyocytes and whole animals(Pall et al., 2003; Lee et al., 2011). Experimental methods of calcium homeostasis are also constantly evolving usually through electrophysiological methods, such as field potential and patch clamp; the direct capture technology of intracellular calcium sparks, calcium leakage, and calcium transients; traditional gene and protein qualitative or quantitative techniques for in-depth study (Huffaker et al., 2004; Eden et al., 2016). Continuously updated research methods are conducive to clarifying the role of calcium homeostasis in MI/R injury, thus accelerating drug development for the treatment of MI/R injury to alleviate limitations of clinical drugs.
Calcium homeostasis disorder is regarded as an important treatment target for clinical intervention of MI/R injury. However, there is currently no article that summarizes and introduces the interaction between calcium homeostasis and MI/R injury. Therefore, we review the role of calcium homeostasis in MI/R injury and the contribution of calcium signal pathway-related drugs currently under research or in the market to MI/R injury. Our work aimed to provide strong evidence for further development of MI/R candidate drugs treatment for MI/R injury.
Calcium Homeostasis
Calcium Release
During cardiac action potential, Ca2+ enters through LTCC, which triggers Ca2+ release from SR through RyR2. And FKBP12.6 stabilizes RyR2 and prevents abnormal contraction of cardiomyocytes (Bers, 2008).
LTCC
Voltage-gated calcium channels include Cav1 (LTCC), and Cav2 and Cav3, which are non-LTCCs. Cav1.2 and Cav1.3 are co-expressed in multiple tissues (sinus node, atrium, and neurons) and play similar regulatory roles. Cav1.1 and Cav1.4 are expressed only in skeletal muscle and retina, respectively (Striessnig et al., 2010). LTCC channel consists of α1, β, α2δ, γ subunits, and calmodulin. The α1 subunit is the main pore-forming structure with the most significant effect on the LTCC current intensity. As a dual regulator, calmodulin is capable of Ca2+-dependent inactivation and facilitation. The T-tubule on the plasma membrane of ventricular myocytes is a typical recessed structure with a large amount of LTCC distribution (Shaw and Colecraft, 2013). At myocardial cell action potential, LTCC is activated by depolarization and opened for extracellular Ca2+ into the cell. This enriched T-tubule spatial advantage is helpful for RyR2 of nearby SR to complete calcium-induced calcium release. β2 adrenergic receptor (β2AR) interacts directly with LTCC (Cav1.2); LTCC is regulated by G protein, adenylate cyclase, PKA, PP1, and PP2A, which affects channel activity. Some studies have shown that loss or deficiency of LTCC function can lead to short QT intervals and sudden cardiac death (Shaw and Colecraft, 2013). Cav1.2 subtype blockers, such as nifedipine, verapamil, diltiazem, are clinically called calcium channel blockers and are used for the treatment of hypertension, angina pectoris, and arrhythmia (Kleinbongard et al., 2012). Therefore, LTCC plays a pivotal role in many cardiovascular diseases, and more research is needed to elaborate its specific mechanisms.
RyR2
RyR is a homotetrameric structure located on the SR membrane. There are three RyR subtypes distributed in mammals: RyR1 is mainly distributed in skeletal muscle, RyR2 primarily in cardiac muscle tissue (Laver, 2018), and RyR3 mainly in brain tissue. This article mainly introduces RyR2 function of in myocardial tissue. RyR2 is a calcium channel that releases calcium from SR to the cytoplasm after LTCC activation (Dulhunty et al., 2012). The amount of calcium released by SR during a cardiac cycle is related to LTCC current, RyR2 Ca2+ sensitivity, and SR Ca2+ content (Cully et al., 2018). RyR2 and inositol triphosphate receptor (IP3R) have the same conservative structure, and therefore, may have evolved from a common precursor structure; the common allosteric activation mechanism of both is N-terminus displacement of two adjacent domains (Dulhunty et al., 2012). PKA hyperphosphorylated RyR2 increases calcium-dependent calcium release, leading to diastolic calcium leakage in cardiomyocytes and decreased myocardial contractility. Importantly, FKBP12.6 can stabilize RyR2, thereby preventing excessive calcium release by RyR2 and intracellular calcium homeostasis imbalance (Oda et al., 2015). The role of RyR2 in M/R remains debatable; however, reducing RyR2 activity has a protective effect on cardiomyocytes. Specifically, reducing RyR2 opening before ischemia can reduce the calcium overload (Yang et al., 2015). Studies have shown that during MI/R, RyR2 structure gets damaged, which also accounts for large amount of calcium leaks in SR and calcium overload. Therefore, reducing calcium leakage may be a novel strategy for treating MI/R (Zucchi et al., 1994; Fauconnier et al., 2013).
FKBP12.6
The FK506 binding proteins (FKBP) are expressed in cardiomyocytes and act as essential RyR2 regulators. FKBP12 and FKBP12.6 subtypes correspond to low and high RyR2 affinity, respectively. Reportedly, FKBP12.6 and not FKBP12 can inhibit RyR2 activity. FKBP12.6 and RyR2 association keeps RyR2 closed, preventing calcium release from the SR, thereby, ensuring heart relaxation (Xiao et al., 2018). PKA, Ca2+-calmodulin–dependent protein kinase II (CaMKII), and phosphatases are involved in RyR2 phosphorylation. PKA-induced RyR2 phosphorylation (Ser-2808) promotes FKBP12.6 and RyR2 dissociation, thereby opening RyR2 and promoting calcium-activated calcium release (Zhao et al., 2017). This process may be caused by the reduction of PP1 and PP2A levels.
RyR2 phosphorylation caused by CaMKII (Ser-2814) is more critical than that by PKA. Studies have shown that atrial fibrillation induced in FKBP12.6 KO mice can be achieved by inhibiting CaMKII phosphorylation RyR2 instead of PKA, thereby inhibiting SR calcium leakage and delayed depolarization (Bito et al., 2013). Decreasing RyR2 open frequency has been shown to reduce heart failure. The overexpression of cardiac FKBP12.6 in the mouse model has shown to reduce the probability of RyR2 opening, but this method is not sufficient to prevent and mitigate remodeling after myocardial infarction. Besides, TGF-β has a protective effect on MI/R injury, and FKBP12 is a ligand of TGF-β. FKBP12 can also promote the production of interleukin-2, which is used as a transcription factor against MI/R injury (Åström-Olsson et al., 2009). Therefore, FKBP12.6 reportedly has potential in treatment of cardiovascular diseases.
Calcium Recapture
After myocardial contraction, intracellular Ca2+ is returned to SR through SERCA; excessive Ca2+ can also be expelled from cells through the NCX, resulting in myocardial cell relaxation (Zhu et al., 2017).
NCX
Mammals have three NCX gene types, namely, SLC8A1 (NCX1), SLC8A2 (NCX2), SLC8A3 (NCX3) (Giladi et al., 2016). In mammals, NCX1 is widely distributed in a variety of cells, NCX2 primarily in brain tissue, and NCX3 mainly in brain and skeletal muscle. Besides, SLC8A4 (NCX4) is expressed in amphibians, reptiles, and fish. NCX regulates myocardial ECC, long-term enhancement, neuron development, immune response, and mitochondrial function. NCX structure mainly consists of 10 transmembrane helices and a large cytosolic regulatory loop (f-loop) (Khananshvili, 2013). Unlike SERCA (high affinity-low volume), NCX (low affinity-high volume) responds quickly to changes in intracellular Ca2+ based on [Ca2+]i. NCX performs intracellular and external ion-exchange, according to 3Na+:Ca2+ (Chu et al., 2016; Giladi et al., 2016). The direction of calcium movement depends on [Na+]i, [Ca2+]i, and membrane potential inside and outside the cell. NCX cannot be phosphorylated by kinases but can be reversibly regulated by cellular effectors, including Ca2+, Na+, H+, NO, PIP2, phosphoarginine, phosphocreatine, ATP, endogenous NCX inhibitor, and f-loop (Khananshvili, 2013).
NCX participates intracellular calcium homeostasis regulation and is implicated in the occurrence and development of various diseases, such as heart failure, arrhythmia, diabetes, hypertension, and cerebral ischemia. The role of NCX in cardiovascular diseases is debatable. Some studies have shown that heart-specific NCX knockout mice can resist MI/R injury, which may be associated with calcium overload inhibition (Chu et al., 2016). Other studies have shown that NCX can alleviate cardiomyocyte function in patients with heart failure and overexpression reduces the development of systolic and diastolic dysfunction (Khananshvili, 2013). In conclusion, inhibiting reverse transport of NCX is an ideal method for reducing calcium overload and a promising treatment strategy for MI/R.
SERCA
There are three SERCA genotypes: ATP2A1 (SERCA1a, SERCA1b), ATP2A2 (SERCA2a, SERCA2b, SERCA2c), and ATP2A3 (SERCA3a-f), each of containing multiple subunits. SERCA2a is the most important, mainly expressed in the heart and slow-twitch skeletal muscle, and mainly involved in cardiomyocyte ECC. SERCA1 is mainly expressed in fast-twitch skeletal muscle, SERCA2b mainly in smooth muscle and non-muscle tissue, SERCA2c mainly in epithelial and hematopoietic cells and SERCA3 primarily in hematopoietic cells (Lipskaia et al., 2014). SERCA has a molecular weight of 110 kDa and hydrolyzes one ATP to transport two Ca2+ back to the SR, causing myocardial relaxation (Balcazar et al., 2018). SERCA consists of three distinct domains, including the cytoplasmic head, the transmembrane region of ten helical segments that contain two Ca2+ binding sites, and luminal loops (Stammers et al., 2015).
A variety of molecules regulate SERCA; SERCA also affects intracellular calcium homeostasis. Phospholamban (PLB) phosphorylation can release the inhibitory effect on SERCA; PKA affects SERCA sensitivity to calcium (Hayward et al., 2015). SERCA overexpression increases calcium reuptake and calcium transients, but has no effect on calcium leakage caused by RyR. Hence, SERCA agonists are used to treat heart failure, diabetes, and metabolic diseases (Bassani and Bassani, 2005). Studies have shown that soluble guanylyl cyclase activation can stimulate cGMP signals, increase Ca2+ uptake by increasing SERCA activity, and reduce reperfusion-induced calcium overload. SERCA is an essential calcium homeostasis and is associated with the pathological mechanisms of various diseases; SERCA is expected to become an important target for cardiovascular disease.
PLB
PLB located in SR contains 52 amino acids (6.1 kD), easily forms a polymer (22 kD), which interacts with SERCA and inhibits its activity (Ablorh and Thomas, 2015). PLB consists of three parts: cytoplasmic domain a (including phosphorylation sites serine16 and threonine17), cytoplasmic domain b (rich in amidated amino acids), and domain c (transmembrane structure). PLB can be phosphorylated by PKA (serine16) and CaMKII (threonine17), thereby releasing its inhibitory effect on SERCA (Haghighi et al., 2014). When cytosolic calcium levels are low, PLB combines with SERCA2a to reduce calcium reuptake; when cytosolic calcium levels iare high, CaMKII or PKA can phosphorylate PLB, thereby promoting PLB and SERCA2a depolymerization and allowing SERCA2a to uptake calcium again (Frank and Kranias, 2000). Sarcolipin is a functional PLB homolog with a serine/threonine kinase 16 phosphorylation site (Bhupathy et al., 2007). SERCA activity is super-inhibited by the synergistic action of PLB and SLN (Morita et al., 2008). Disrupting PLB–SERCA2a interactions can reduce cardiac systolic dysfunction, but eliminating PLB is not useful in treating all forms of heart failure (MacLennan and Kranias, 2003). Further research on the structure and function of PLB–SERCA2a will provide new insights into the role of PLB in cardiovascular disease.
CaMKII
CaMKII has four genotypes α, β, γ, and δ; α and β are mainly expressed in the nervous system, and γ and δ generally in various cells (Grueter et al., 2006). Among them, δ is the most prevalent subtype in cardiac tissue. CaMKII structure includes amino-terminal catalytic, the central regulatory, and the carboxy-terminal associated domains that oligomerize (Grueter et al., 2006). CaMKII holoenzyme is a wheel-like structure composed of 6-12 homologous or heterologous kinase subunit. CaMKII can act on Ca2+-releasing proteins such as RyR2, LTCC, IP3R, and regulate calcium recalculation by PLB and SERCA phosphorylation (Maier and Bers, 2002). CaMKII phosphorylation promotes maintenance of high active LTCC, resulting in excessive extracellular influx into myocardial cells (Mattiazzi and Kranias, 2014). The effect of CaMKII on RyR2 is controversial, as RyR2-mediated increased and decreased calcium release has been reported. CaMKII phosphorylates PLB, which in turn releases its inhibitory effect on SERCA and enhances calcium reuptake capacity.
A study of an animal model of structural heart disease proved that CaMKII overexpression can lead to myocardial dilatation, dysfunction, and calcium homeostasis imbalance in cardiomyocytes (Grueter et al., 2006). βAR increases CaMKII activity and expression. Therefore, CaMKII inhibition can combat cardiac hypertrophy, cardiac dilatation, and myocardial infarction-induced dysfunction, and βAR overstimulation (Mattiazzi and Kranias, 2014). In MI/R injury, CaMKII could activate NF-κb, promote inflammatory response leading to apoptosis, and aggravate MI/R injury (Ling et al., 2013). In simple terms, CaMKII has a significant impact on calcium cycling and various diseases, and it is worth carrying out an in-depth study.
Calcium Storage
The main cardiomyocyte Ca2+ storing organelles are SR and mitochondria. SR and mitochondria are essential during heart contraction and relaxation and provide enough oxygen and ATP to body tissues via blood circulation. Although the functional characteristics of SR and mitochondria are different, their cooperation also ensures normal heart operation. Therefore, both mitochondrial and SR reductions and dysfunction can cause heart-related diseases (Chernorudskiy and Zito, 2017).
Sarcoplasmic Reticulum
Myocardial SR is composed of longitudinal tubes and terminal cisternae. The longitudinal tube membrane mainly contains SERCA and PLB for calcium reuptake. The terminal cisternae (RyR2) stores and releases Ca2+. There are also several soluble proteins in the SR cavity, including calsequestrin and calreticulin. They have low Ca2+ affinity, but a large binding capacity. Their primary function is to bind Ca2+ in the SR cavity. These proteins can reduce [Ca2+]i in the SR cavity, facilitate transport of calcium pumps, and store Ca2+ for release at the subsequent myocardial contraction. Therefore, SR regulates cytosolic [Ca2+]i mainly in three steps such as calcium release, reuptake, and storage, and different proteins perform their functions (Bers, 2008).
Studies have found that SR calcium balance is very vital for normal physiological function and information transmission of cardiomyocytes. SR calcium homeostasis is closely related to the development of cardiovascular diseases, such as ischemic heart disease, cardiac hypertrophy, hypertension, and heart failure (Fiolet and Baartscheer, 2000). Ca2+ release other than that in the normal calcium cycle is called SR calcium leakage, which is mainly mediated by RyR2. Calcium leakage can cause cardiac systolic dysfunction by reducing effective SR Ca2+ release, cause diastolic dysfunction by increasing diastolic cytosolic Ca2+, and induce arrhythmias (Hulot et al., 2012). Therefore, further study on SR calcium regulatory channels will help reveal the role of Ca2+ regulation in cardiovascular disease and provide new insights and strategies for cardiovascular disease treatment and prognosis.
Mitochondria
A double membrane structure characterizes the mitochondria; there is a cavity between both membranes due to the folding of inner membrane to form ridges. Mitochondria are the main sites for intracellular oxidative phosphorylation and adenosine triphosphate (ATP) formation. Additionally, mitochondria are also involved in processes such as apoptosis, signal transduction, cell proliferation, and metabolism (Williams et al., 2015). The cardiomyocyte activity is energy consuming. The mitochondria of each ventricular muscle cell account for about 33% of cell volume, which also reflects their importance for cardiomyocytes. Ca2+ enters the mitochondrial matrix through a highly selective and low-conductivity mitochondrial calcium uniporter channel. It is pumped into the membrane space through the mitochondrial sodium-calcium exchanger, which maintains calcium dynamic balance in the mitochondria (Williams et al., 2015). Calcium directly or indirectly regulates many components in ATP production, such as crucial enzymes and proteins of the tricarboxylic acid cycle.
Mitochondria are not involved in myocardial ECC regulation under physiological conditions. However, due to the large calcium capacity of mitochondria, which accounts for about 30% of the calcium capacity in plasma and tissue cells, they compensate by alleviating intracellular calcium accumulation during calcium overload in myocardial cells (Dorn and Maack, 2013). Studies have shown that mitochondrial reactive oxygen species production is an early trigger for MI/R injury and causes respiratory chain dysfunction. In I/R heart, mitochondrial channel dysfunction of myocardial cells causes changes in mitochondria membrane potentials (Chouchani et al., 2014). This extends to the entire cell, causing structural and functional disorders. Therefore, mitochondrial damage causes ATP depletion, which accelerates myocardial cell death and promotes the reversible damage of myocardial tissue to irreversible damage (Paradies et al., 2018)
Interaction of Calcium Overload and Myocardial Ischemia-Reperfusion
MI/R Protection Signaling Pathway
Ca2+ is driven by a large electrochemical gradient across the plasma membrane into the cells. Cells use this external source of signal Ca2+ by activating various entry channels, such as voltage-operated channels (Shaw and Colecraft, 2013). The other principal source of Ca2+ for signaling is the internal stores that are located primarily in the SR, and IP3R, RyR, and SERCA regulate the release and recycle of Ca2+ (Chernorudskiy and Zito, 2017). The principal activator of these channels is Ca2+ itself, and this process of Ca2+-induced Ca2+ release is central to the mechanism of calcium signaling (Bers, 2008). The signaling pathway of MI/R protection is very complicated and involves many factors (Fiolet and Baartscheer, 2000). Obviously, the intervention of the above proteins is potential to promote MI/R protection. And there are some other signaling pathways, which start with G protein-coupled receptors or cytokine receptors activated by adenosine or opioids (Murphy and Steenbergen, 2008). These processes enable protection signals into cardiac cell. Intracellular signals then activate the PI3K-AKT, JAK-STAT, and cGMP-protein kinase G (PKG) pathways. Signaling further activates downstream endothelial nitric oxide synthase (eNOS), glycogen synthase kinase (GSK)-3β, hexokinase II (HK II), and protein kinase Cϵ (PKCϵ). The above process further opens the mitochondrial ATP-dependent potassium channel and inhibits opening of the mitochondrial permeability transition pore (MPTP), which prevents apoptosis factor cytochrome C and apoptosis-inducing factor (AIF) from entering the cytoplasm to induce apoptosis (Hausenloy and Yellon, 2016). In fact, intervention in one of the above pathways may achieve cardiac protection. For example, meprobamate and KAI-9803 (Table 1) are two drugs in clinical research, which protect MI/R injury via PKC-ϵ activation and PKC-δ inhibition, respectively (Mochly-Rosen et al., 2012). There are insufficient drugs for treating MI/R injury, so it is necessary to conduct further research on MI/R signaling pathways to find targets with cardioprotective effects.
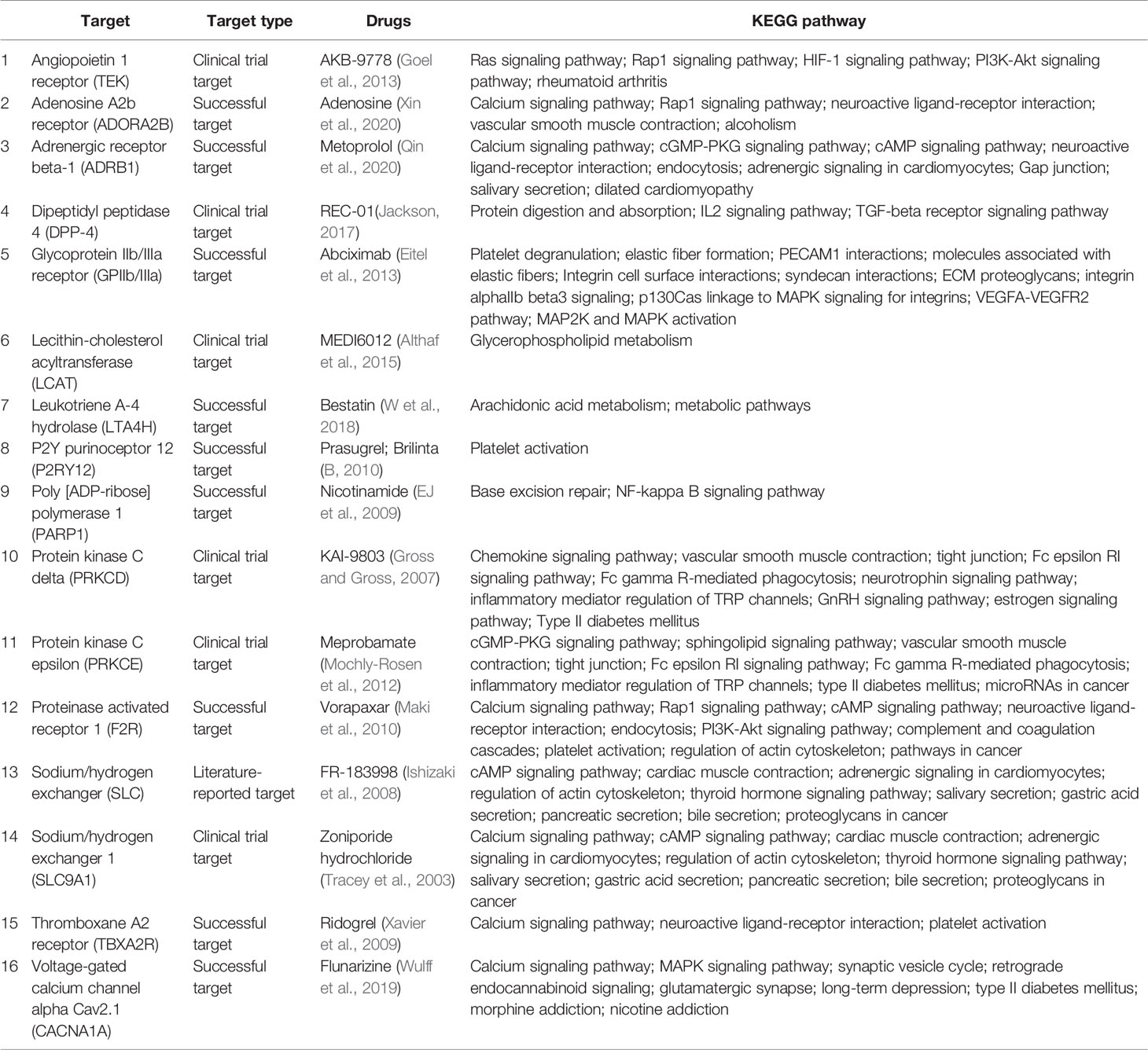
Table 1 Summary of MI/R injury drugs.
Mechanism of Calcium Overload Generation During MI/R
After a period of ischemia and hypoxia, myocardial cells have metabolic abnormalities, including increased anaerobic fermentation. Furthermore, intracellular H+ aggregation causes a low intracellular pH, and intracellular Na+ increases through H+/Na+ exchange (HNX). Excessive intracellular Na+ will promote Na+ excretion and Ca2+ intake by NCX, which significantly increases intracellular calcium levels, hence, leading to calcium overload. When blood flow and oxygen supply to the cardiac tissue returns to normal, extracellular pH level is further increased, HNX and NCX activities are enhanced, and intracellular calcium overload is further aggravated (Murphy and Steenbergen, 2008). Direct H+/Ca2+ exchange can also directly cause calcium overload. Elevated intracellular calcium levels also activate intracellular calcium-activated calcium release. Existing studies have shown that NCX and HNX inhibition has a protective effect on MI/R injury (Mozaffari et al., 2013). Two candidate drugs, FR-183998 and zoniporide hydrochloride which target HNX, are currently under development for MI/R injury treatment(Tracey et al., 2003; Ishizaki et al., 2008). Opening RyR2 channel effluxes SR Ca2+ into the cytoplasm, exacerbating calcium overload. Calcium overload can cause a series of irreversible cell injury responses, such as cardiac contractile dysfunction and apoptosis.
Additionally, MI/R produces some toxic substances, such as oxygen-free radicals (OFR), in the myocardial cells. OFR can decompose cell membrane phospholipid components and damage membrane structure, which leads to increased membrane permeability and excessive extracellular Ca2+ influx (Fiolet and Baartscheer, 2000). OFR also damages to the SR membrane, eventually increasing intracellular calcium levels and further exacerbating calcium overload (Mozaffari et al., 2013).
Mechanism of Calcium Overload Aggravating MI/R Injury
Ca2+ plays a vital role in cardiac ECC, so calcium overload will further exacerbate the degree of MI/R injury in cardiac muscle cells. Excessive intracellular calcium will enter the mitochondria, resulting in mitochondrial calcium overload; this inhibits ATP production, exacerbates energy metabolism disorders, and eventually leads to myocardial cell apoptosis (Dorn and Maack, 2013). Mitochondrial damage is regarded as a sign of myocardial cell transformation from reversible to irreversible damage (Williams et al., 2015).
Intracellular calcium can also activate some phospholipases, mainly protein kinase C and phospholipase A, which destroy the cell membrane skeleton. Furthermore, the reaction produces some toxic substances such as free fatty acids, leukotrienes, prostaglandins, and oxygen-free radicals (Hausenloy and Yellon, 2016). These cause mitochondrial dysfunction, increase membrane permeability, impede signal transmission in cells, and promote vast myocardial cell apoptosis. Excessive calcium can also activate caspase, calpain, endonuclease, and phospholipase, which induce intracellular digestion of proteins and fats (Chouchani et al., 2014; Lesnefsky et al., 2017).
Myocardial calcium overload can also cause changes in the structure and function of coronary blood vessels and microvascular endothelial cells. It causes adhesion, accumulation, and infiltration of neutrophils; a series of inflammatory factors release; and further the heart vascular tissue damage (Kleinbongard et al., 2012). Energy metabolism disorder caused by calcium overload can also cause myocardial spasm, and cause pathological changes, such as arrhythmia (Zile and Gaasch, 2011). Therefore, calcium overload plays a vital role in MI/R injury occurrence and development and will also play a key role in preventing and treating MI/R injury.
Comprehensive Analysis of MI/R Injury and Calcium Signaling Pathway
MI/R Injury Drugs
Tissue ischemia is due to insufficient hemoperfusion under various pathological conditions. Moreover, MI/R injury is characterized by increased tissue and cell dysfunction, metabolic damage, and structural damage after blood supply is restored and reperfusion occurs (Murphy and Steenbergen, 2008; Lesnefsky et al., 2017). Studies have shown that MI/R causes cardiac dysfunction as well as peripheral vascular injury. Recently, research on the mechanism of MI/R injury and the search for effective measures to reduce reperfusion injury have become hot spots in the cardiovascular field. To more fully understand and summarize research findings and clinical use of MI/R drugs, we obtained relevant information (including target, disease, drug, and KEGG pathway) via “Therapeutic Target Database” and verified it with “PubMed” database. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway is a database that integrates genomic, chemical, and system function information, linking gene catalogs from genomes that have been fully sequenced to higher-level system functions at cell, species, and ecosystem levels (Kanehisa et al., 2017).
In Table 1, we have summarized drugs and candidates for MI/R injury treatment. Over half of the drugs are marketed, three eighth are drug under investigation, and the remaining are potential candidates. Nearly half of the drugs target calcium regulators that are mainly involved in calcium signaling pathway regulation. Except for calcium channels, MI/R targets include angiopoietin 1 receptor, P2Y purinoceptor 12, protein kinase C, lecithin-cholesterol acyltransferase, and dipeptidyl peptidase 4, indicating complexity of MI/R injury mechanisms and the diversity of targets. In fact, the drugs used to treat MI/R injury are very limited in clinical practice, and most of the drugs are selected according to the symptoms (Ibanez et al., 2015). Some drugs, such as prasugrel, brilinta, and AKB-9778, prevent blood vessels from being clogged due to platelet activation or vasodilation (B, 2010; Goel et al., 2013), and some drugs, such as flunarizine, meprobamate, and KAI-9803, reduce cardiomyocyte damage by calcium overload and PKC pathway inhibition (Mochly-Rosen et al., 2012). Furthermore, according to the KEGG pathway, most drugs are not involved in a single signal pathway, but in an interconnected signal network, which also reflects the intricate connection between various reactions in the body. This also helps us to understand the role of drugs in signaling networks and links between disease and various signaling pathways. Some of the drugs in Table 1 are still at different stages of research, so basic and clinical researchers need to work rigorously to develop more safe and effective drugs to alleviate the pain and distress of MIR patients.
Calcium Signaling Pathway Drugs
We also searched for the calcium signaling pathway-related drugs using the “Therapeutic Target Database” to better understand the role of calcium homeostasis in various diseases. As shown in Table 2, calcium signaling pathway drugs are mainly used to treat cardiovascular and cerebrovascular diseases, such as myocardial (cerebral) I/R injury, hypertension, cardiac failure, and myocardial infarction. About half of the drugs are marketed, about two fifths are undergoing clinical trials, and the remaining are potential active candidates.
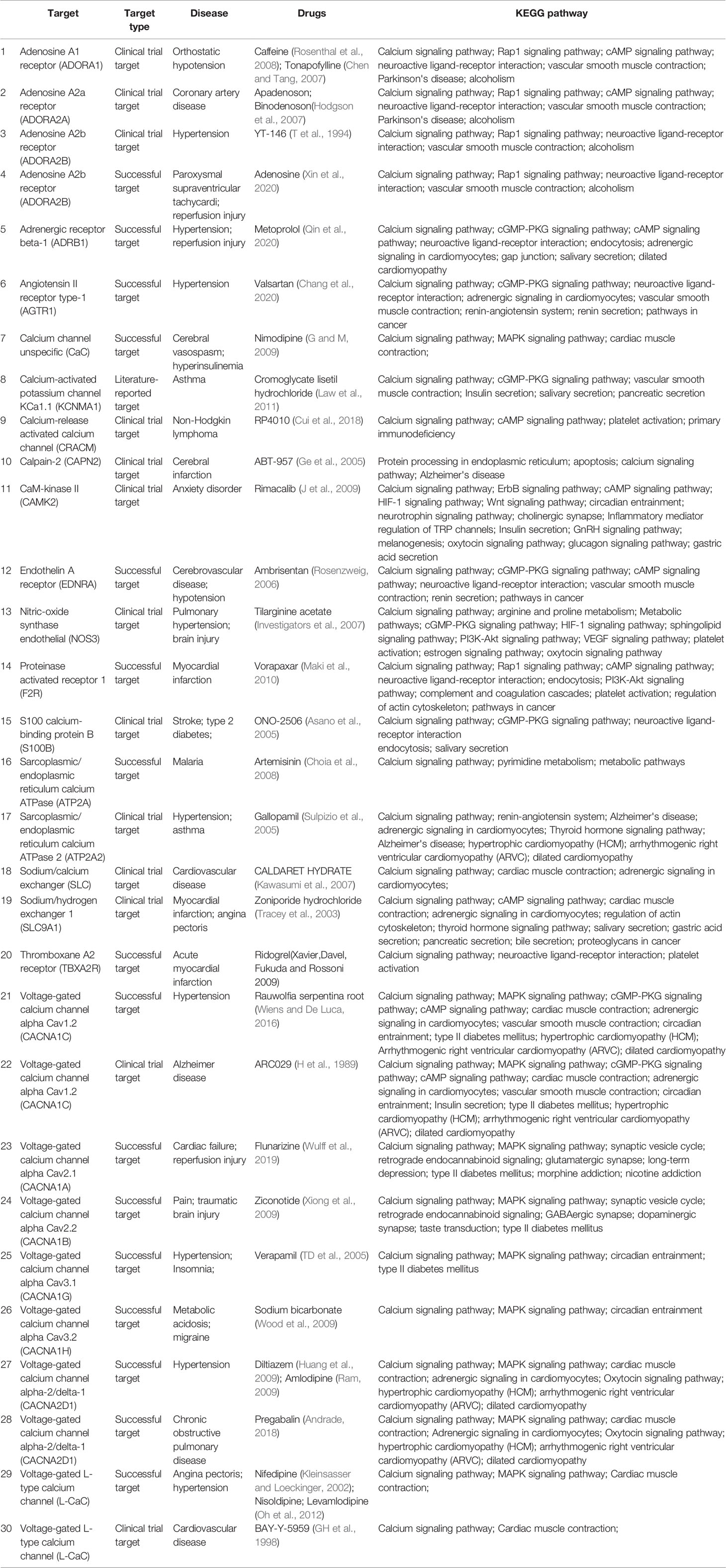
Table 2 Summary of calcium signaling pathway drugs.
Calcium channel blockers (CCBs), such as nifedipine, nisoldipine, diltiazem, verapamil, nimodipine, and amlodipine (Table 2), can be widely used to treat cardiovascular diseases including myocardial infarction, hypertension, arrhythmia, and angina pectoris. CCBs reduce calcium levels in myocardial cells, which triggers a series of physiological effects against multiple diseases. Most CCBs have significant effects and reasonable prices, but there are still many adverse reactions. CCBs only target angina pectoris, caused by coronary spasm, but are not effective against other types of angina pectoris (Capiod, 2011; Kleinbongard et al., 2012). Therefore, for, appropriate drugs should be selected for treating different patients, according to the disease type, to reduce adverse reactions as much as possible. In addition, calcium overload aggravates cerebral I/R injury, and drugs targeting calcium channels and calcium pathway-related factors have good clinical effects on cerebral I/R injury (Ge et al., 2005; Zhao et al., 2013). The drug targets are mainly voltage-dependent calcium channels, calcium-activated ion channels, and adenosine receptors. According to KEGG analysis, the drugs listed in Table 2 also involve multiple signaling pathways. In addition to the calcium pathway, other pathways, such as the cAMP signaling pathway, Adrenergic signaling pathway, cardiac muscle contraction, platelet activation, also occur more frequently, and there is an upstream and downstream connection between these pathways and the calcium signaling pathways. Cardiac muscle contraction is a complex process initiated by the electrical excitation of cardiac myocytes, which is the processes of ECC (Bers, 2008). In other signaling pathways, calcium serves as an important second messenger. Adrenergic receptor as the pre-dominate receptor, induces positive inotropic and chronotropic effects, the most effective mechanism to acutely increase output of the heart, by coupling to Gs, formation of cAMP by adenylyl cyclase, and PKA-dependent phosphorylation of various target proteins (such as RyR2, PLB, and LTCC) (Qin et al., 2020). During platelet activation, different receptors are stimulated by various agonists, almost converging in increasing intracellular Ca2+ concentration that stimulate platelet shape change and granule secretion (Maki et al., 2010). In a word, some important proteins, such as RyR2, PLB, FKBP12.6, and NCX, participate in the calcium cycling process and maintain calcium homeostasis, thereby ensuring that intracellular Ca2+ transfers different signals to downstream signaling pathways. Therefore, the importance of calcium pathways is self-evident, and future research should focus on calcium pathway drugs for treating cardiovascular and cerebrovascular diseases.
Comprehensive Analysis
As a second messenger in cells, Ca2+ plays an essential role in maintaining cell proliferation, division, and energy metabolism. The calcium signal pathway-related protein expression and ion channel regulation mechanism connect arrhythmia, myocardial hypertrophy, and heart failure in cardiovascular diseases, making the development of these diseases a cause and effect (Bers, 2008; Hidalgo, 2017). As MI/R injury-mediated calcium overload is multifactorial, it is vital to regulate intracellular calcium homeostasis to maintain normal physiological function and information transmission in cells (Fiolet and Baartscheer, 2000). Based on data of tables, about two fifths of diseases (Table 1) involve calcium signaling pathways, three fifth of the drugs (Table 2) are used to treat cardiovascular disease, and more than one-fifth of the drugs (Table 2) to treat MI/R injury. The above results also prove that calcium homeostasis plays an irreplaceable role in normal cell operation and cardiovascular disease development, especially MI/R injury and hypertension.
As shown in Figure 1, the six drugs including adenosine, ridogrel, vorapaxar, metoprolol, flunarizine, and zoniporide hydrochloride have the potential to treat MIR, but their treatment mechanisms are different. Our analysis of the data in Tables 1 and 2 shows that voltage-dependent calcium channels, adrenergic receptor β1, proteinase-activated receptor 1 (PAR1), HNX, adenosine A2b receptor, and thromboxane A2 (TXA2) receptor are important targets for MI/R injury treatment and calcium pathways. As mentioned in Mechanism of Calcium Overload Generation During MI/R, when MI/R, a huge difference in intracellular and extracellular pH of the myocardial cells occurs, and HNX and NCX change the movement of H+, Na+, and Ca2+, which aggravates calcium overload (Chu et al., 2016). The role of LTCC as ECC activating signal in the calcium pathway is self-evident. Besides, adrenergic receptors can effectively induce downstream signaling pathways, including calcium cycling pathways by cAMP formation and PKA phosphorylation, thereby increasing cardiac output (Qin et al., 2020). Studies also have shown that angiotensin-induced increase in TXA2 has a protective effect on MI/R injury, and this regulatory effect is related to maintaining calcium homeostasis (Dogan et al., 1997). Therefore, these targets directly or indirectly play a role in relieving MI/R injury by regulating calcium signaling pathways.
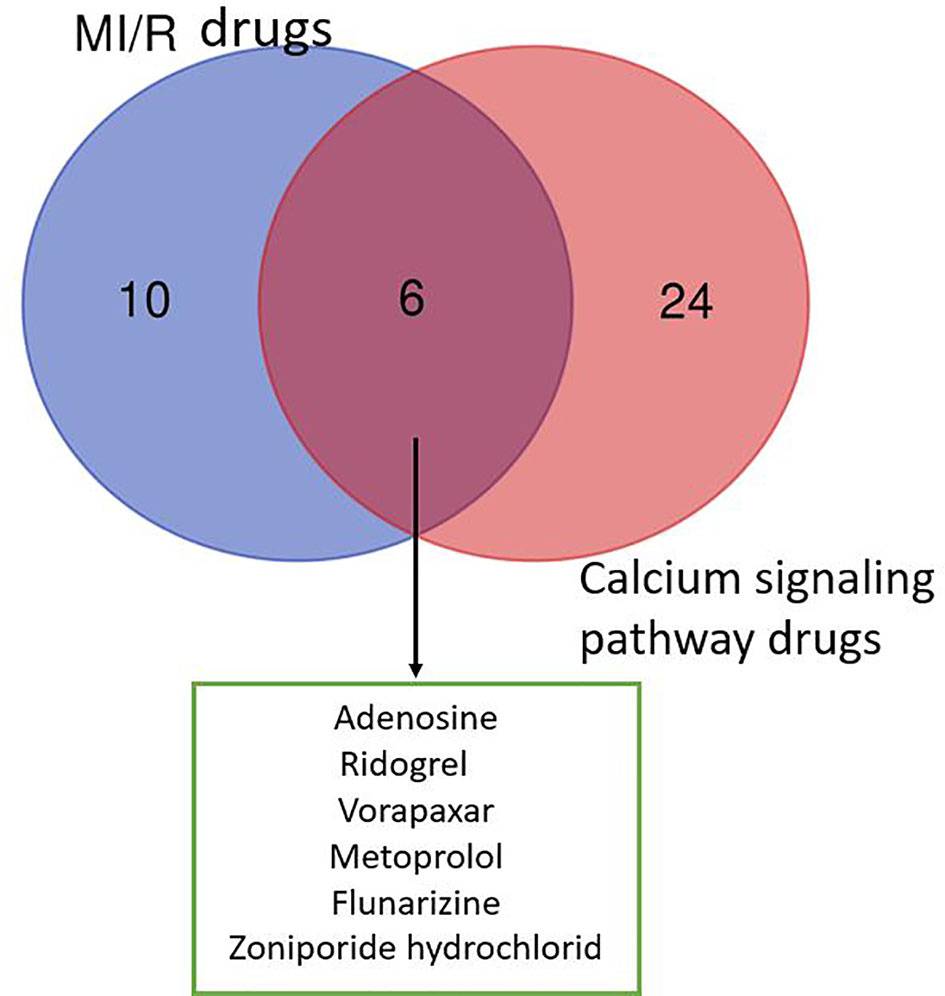
Figure 1 Venn diagram of drugs from MI/R injury and calcium signaling pathway. We collected 16 drugs for the treatment of MIR injury and 30 drugs involved in calcium signaling pathway. The MI/R drugs related to calcium signaling pathway include: adenosine, ridogrel, vorapaxar, metoprolol, flunarizine, and zoniporide hydrochloride.
Besides, the drugs we listed are basically chemical drugs, which generally have specific targets. But, the composition of traditional Chinese medicine (TCM) is complex, and the targets are not single. For cardiovascular diseases, which involve multiple signaling pathways, TCM has its unique advantages. For example, Rauwolfia serpentina root was clinically proven to treat hypertension, fever, liver disease, and other diseases in the past (Wiens and De Luca, 2016; Dong et al., 2018). Besides, current research also proved its good effect on MI/R injury. Therefore, investing TCM for cardiovascular disease treatment should be increased.
In fact, nearly half of the drugs listed in Tables 1 and 2 are in preclinical or clinical research, such as FR-183998 and zoniporide. In order to ensure the safety and effectiveness of drugs, drug development is a long and costly process. If there is a problem in any part of the research and development process, the drugs will be forced to stop. Therefore, not all of the drugs we listed can be successfully marketed in the end and solve problems for patients. The disease characteristics of MI/R injury determine that more effective targets and even drugs related to calcium pathways can be discovered in the future. These still need persevering research by scientific researchers.
Conclusions
Normal heart function depends on coordinated Ca2+ movement into and out of the plasma membrane and sarcoplasmic reticulum; calcium circulation disorders can cause various heart diseases. Changes in the Ca2+ steady-state, including decreased SR Ca2+ reuptake, abnormal calcium channels, SR Ca2+ leakage, or significantly decreased SERCA2a expression can cause various heart diseases, including ischemic heart disease, reperfusion injury, hypertrophic cardiomyopathy, diastolic cardiomyopathy, and heart failure (Bers, 2008). With continuous improvement of electrophysiology and pharmacology technology, especially combination and application of patch-clamp technology and molecular cloning technology, researcher can gain an in-depth understanding of drugs and the ion channel effects of cardiovascular active ingredients from cell to the molecular levels. These methods are significant in understanding the impact of drugs on ion channel activity and mechanism of action against MI/R injury. By studying MI/R injury mechanism and better understanding the underlying signaling pathways, we will have better opportunities to develop treatments to protect against MI/R injury. We look forward to the development of new therapies to reduce MI/R injury and incorporate them into clinical trials as soon as possible.
In summary, understanding the role of calcium pathway-related proteins in MI/R injury is of great benefit in elucidating the pathogenesis of MI/R injury. Copious literature summarize that MI/R injury can cause calcium overload, which can further aggravate MI/R injury. Therefore, these regulatory proteins, such as LTCC, NCX, SERCA, RyR2, PLB, and FKBP12.6, provide potential targets for the prevention and treatment of clinical MI/R injury, and multi-target therapy may also have potential application. After analyzing the data in the Tables, we found that six drugs including adenosine, ridogrel, vorapaxar, metoprolol, flunarizine, and zoniporide hydrochloride were used to treat MI/R injury and the treatment mechanisms were related to the calcium signaling pathway (Figure 2). The specific targets of these six drugs, such as voltage-dependent calcium channels, adrenergic receptors β1 and HNX are different and involved in calcium signaling pathway, which also reflects the complexity of the MI/R mechanism and the important role of maintaining calcium homeostasis on MI/R injury. In our statistics, some drugs are still in the research stage, and the clinical demand has also pushed researchers to make greater efforts to find more effective and safe drugs to treat MI/R injury. Therefore, this article reviews the role of calcium overload in the development of MI/R injury and the current research progress of marketed and candidate drugs, aiming to provide some help for further studying the protection mechanism and therapeutic reagents of MI/R injury.
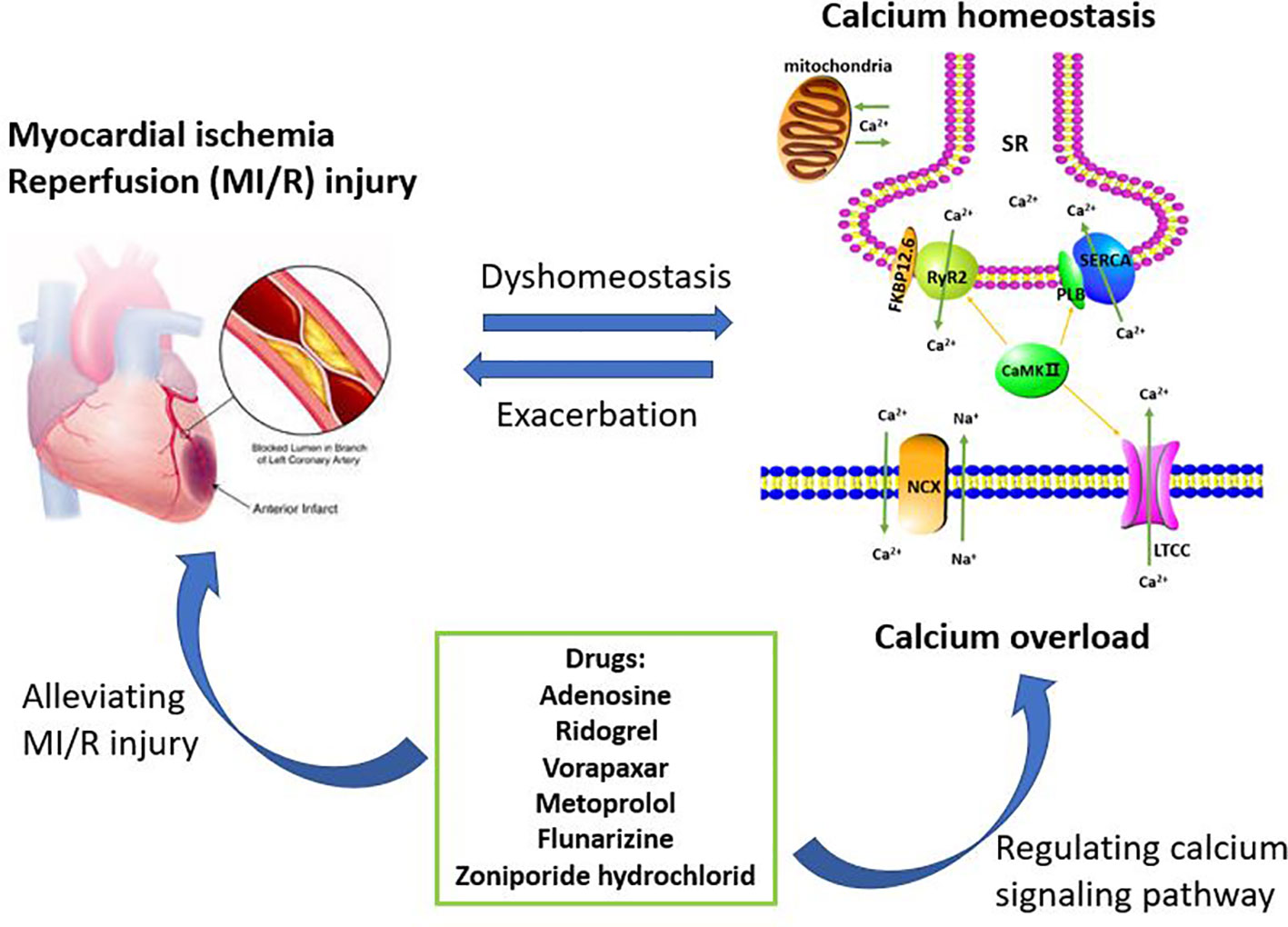
Figure 2 Calcium overload is the important target of treating myocardial ischemia/reperfusion (MI/R) injury. The maintenance of calcium homeostasis requires the participation of multiple regulatory proteins including LTCC, RyR2, SERCA, NCX, PLB, FKBP12.6, and CaMK II. When MI/R occurs, calcium homeostasis will be broken and further developed into calcium overload. Calcium overload further exacerbates MI/R injury. Therefore, inhibiting calcium overload is an effective way to reduce MI/R injury. These drugs currently on the market or under investigation including adenosine, ridogrel, vorapaxar, metoprolol, flunarizine, and zoniporide hydrochloride, are for the treatment of MI/R injury or have the potential to treat MI/R injury. These drugs can play a cardioprotective role by regulating the calcium signaling pathway.
However, the current study is still in its initial stage, and there are many problems, mainly with regard to two aspects. Firstly, MI/R injury involves multiple signaling pathways, and the cross-talk between multiple MI/R-related mechanism and calcium regulation can be further studied. Additionally, different calcium pathway drug combinations or multi-target chemical synthetic drugs can be tested in MI/R injury treatment to cope with the complex pathological mechanism underlying MI/R. Second, to study calcium homeostasis in MI/R injury, application and development of new technologies can promote research breakthroughs, such as real-time calcium channel current and calcium concentration detection, and real-time capture detection for calcium channel protein modification, which broaden the scope of basic research and accelerate the search for MI/R candidate drugs.
Author Contributions
Conceptualization: RW and MW. Writing—original draft preparation: RW. Writing—review and editing: MW, GS, and XS. Visualization: SH. Funding acquisition: GS and XS. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the Drug Innovation Major Project (grant number 2018ZX09711001-009), National Key Research and Development Project (grant number 2017YFC1702504 and 2018YFC1707408), and the National Natural Sciences Foundation of China (grant number 81891012).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Åström-Olsson, K., Karlsson, L., Mattsson Hultén, L., Davidsson, P., Mantovani, V., Månsson, C., et al. (2009). Myocardial release of FKBP12 and increased production of FKBP12.6 in ischemia and reperfusion experimental models. Biochem. Biophys. Res. Commun. 390, 1299–1304. doi: 10.1016/j.bbrc.2009.10.140
Ablorh, N. D., Thomas, D. D. (2015). Phospholamban phosphorylation, mutation, and structural dynamics: a biophysical approach to understanding and treating cardiomyopathy. Biophys. Rev. 7, 63–76. doi: 10.1007/s12551-014-0157-z
Althaf, M. M., Almana, H., Abdelfadiel, A., Amer, S. M., Al-Hussain, T. O. (2015). Familial lecithin-cholesterol acyltransferase (LCAT) deficiency; a differential of proteinuria. J. Nephropathol. 4, 25–28. doi: 10.12860/jnp.2015.05
Andrade, C. (2018). Safety of Pregabalin in Pregnancy. J. Clin. Psychiatry 79, 18f12568. doi: 10.4088/JCP.18f12568
Asano, T., Mori, T., Shimoda, T., Shinagawa, R., Satoh, S., Yada, N., et al. (2005). Arundic Acid (ONO-2506) Ameliorates Delayed Ischemic Brain Damage by Preventing Astrocytic Overproduction of S100B. Curr. Drug Targets - CNS Neurol. Disord. 4, 127–142. doi: 10.2174/1568007053544084
Balcazar, D., Regge, V., Santalla, M., Meyer, H., Paululat, A., Mattiazzi, A., et al. (2018). SERCA is critical to control the Bowditch effect in the heart. Sci. Rep. 8, 12447. doi: 10.1038/s41598-018-30638-9
Bassani, J. W., Bassani, R. A. (2005). SERCA upregulation: breaking the positive feedback in heart failure? Cardiovasc. Res. 67, 581–582. doi: 10.1016/j.cardiores.2005.06.019
Bers, D. M. (2008). Calcium Cycling and Signaling in Cardiac Myocytes. Annu. Rev. Physiol. 70, 23–49. doi: 10.1146/annurev.physiol.70.113006.100455
Bhupathy, P., Babu, G. J., Periasamy, M. (2007). Sarcolipin and phospholamban as regulators of cardiac sarcoplasmic reticulum Ca2+ ATPase. J. Mol. Cell Cardiol. 42, 903–911. doi: 10.1016/j.yjmcc.2007.03.738
Bito, V., Biesmans, L., Gellen, B., Antoons, G., Macquaide, N., Rouet-Benzineb, P., et al. (2013). FKBP12.6 overexpression does not protect against remodelling after myocardial infarction. Exp. Physiol. 98, 134–148. doi: 10.1093/cvr/cvw247
Capiod, T. (2011). Cell proliferation, calcium influx and calcium channels. Biochimie 93, 2075–2079. doi: 10.1016/j.biochi.2011.07.015
Chang, P.-C., Wo, H.-T., Lee, H.-L., Lin, S.-F., Chu, Y., Wen, M.-S., et al. (2020). Sacubitril/Valsartan Therapy Ameliorates Ventricular Tachyarrhythmia Inducibility in a Rabbit Myocardial Infarction Model. J. Cardiac. Failure 19, 31581–31587. doi: 10.1016/j.cardfail.2020.03.007
Chen, S. Y., Tang, W. H. W. (2007). Emerging drugs for acute and chronic heart failure: current and future developments. Expert Opin. Emerging Drugs 12, 75–95. doi: 10.1517/14728214.12.1.75
Chernorudskiy, A. L., Zito, E. (2017). Regulation of Calcium Homeostasis by ER Redox: A Close-Up of the ER/Mitochondria Connection. J. Mol. Biol. 429, 620–632. doi: 10.1016/j.jmb.2017.01.017
Choia, S.-R., Mukherjeea, P., Avery, M. A. (2008). The Fight Against Drug-Resistant Malaria Novel Plasmodial Targets and Antimalarial Drugs. Curr. Med. Chem. 15, 161–171. doi: 10.2174/092986708783330575
Chouchani, E. T., Pell, V. R., Gaude, E., Aksentijević, D., Sundier, S. Y., Robb, E. L., et al. (2014). Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. nature 515, 431–435. doi: 10.1038/nature13909
Chu, L., L, J., Greenstein andL.Winslow, R. (2016). Modeling Na+-Ca2 + exchange in the heart Allosteric activation, spatial localization, sparks and excitation-contraction coupling. J. Mol. Cell. Cardiol. 99, 174–187. doi: 10.1016/j.yjmcc.2016.06.068
Cui, C., Chang, Y., Zhang, X., Choi, S., Tran, H., Penmetsa, K. V., et al. (2018). Targeting Orai1-mediated store-operated calcium entry by RP4010 for anti-tumor activity in esophagus squamous cell carcinoma. Cancer Lett. 432, 169–179. doi: 10.1016/j.canlet.2018.06.006
Cully, T. R., Choi, R. H., Bjorksten, A. R., Stephenson, D. G., Murphy, R. M., Launikonis, B. S. (2018). Junctional membrane Ca(2+) dynamics in human muscle fibers are altered by malignant hyperthermia causative RyR mutation. Proc. Natl. Acad. Sci. U. S. A 115, 8215–8220. doi: 10.1073/pnas.1800490115
Dogan, S., Turnbaugh, D., Zhang, M., Cofie, D. Q., Fugate, R. D., Kern, D. C. (1997). Thromboxane A2 receptor mediation of calcium and calcium transients in rat cardiomyocytes. Life Sci. 60, 943–952. doi: 10.1016/S0024-3205(97)00024-6
Dong, R., Chen, P., Chen, Q. (2018). Inhibition of pancreatic cancer stem cells by Rauwolfia vomitoria extract. Oncol. Rep. 40, 3144–3154. doi: 10.3892/or.2018.6713
Dorn, G. W., Maack, C. (2013). SR and mitochondria: Calcium cross-talk between kissing cousins. J. Mol. Cell. Cardiol. 55, 42–49. doi: 10.1016/j.yjmcc.2012.07.015
Dulhunty, A. F., Wium, E., Li, L., Hanna, A. D., Mirza, S., Talukder, S., et al. (2012). Proteins within the intracellular calcium store determine cardiac RyR channel activity and cardiac output. Clin. Exp. Pharmacol. Physiol. 39, 477–484. doi: 10.1111/j.1440-1681.2012.05704.x
Eden, M., Meder, B., Völkers, M., Poomvanicha, M., Domes, K., Branchereau, M., et al. (2016). Myoscape controls cardiac calcium cycling and contractility via regulation of L-type calcium channel surface expression. Nat. Commun. 7, 11317. doi: 10.1038/ncomms11317
Eitel, I., Wöhrle, J., Suenkel, H., Meissner, J., Kerber, S., Lauer, B., et al. (2013). Intracoronary Compared With Intravenous Bolus Abciximab Application During Primary Percutaneous Coronary Intervention in ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 61, 1447–1454. doi: 10.1016/j.jacc.2013.01.048
EJ, A., E, A., M, D., ME, M., M, H. (2009). beta-1,2,3-Triazolyl-nucleosides as nicotinamide riboside mimics. Nucleosides Nucleotides Nucleic Acids 28, 238–259. doi: 10.1080/15257770902865415
Fauconnier, J., Roberge, S., Saint, N., Lacampagne, A. (2013). Type 2 ryanodine receptor: A novel therapeutic target in myocardial ischemia/reperfusion. Pharmacol. Ther. 138, 323–332. doi: 10.1016/j.pharmthera.2013.01.015
Fiolet, J. W. T., Baartscheer, A. (2000). Cellular calcium homeostasis during ischemia; a thermodynamic approach. Cardiovasc. Res. 45, 100–106. doi: 10.1016/S0008-6363(99)00294-1
Frank, K., Kranias, E. G. (2000). Phospholamban and cardiac contractility. Ann. Med. 32, 572–578. doi: 10.3109/07853890008998837
G, B., M, K. (2009). Influence of bupropion and calcium channel antagonists on the nicotine-induced memory-related response of mice in the elevated plus maze. Pharmacol. Rep. 61, 236–244. doi: 10.1016/s1734-1140(09)70027-1
Ge, X., Yamamoto, S., Tsutsumi, S., Midorikawa, Y., Ihara, S., Wang, S. M., et al. (2005). Interpreting expression profiles of cancers by genome-wide survey of breadth of expression in normal tissues. Genomics 86, 127–141. doi: 10.1016/j.ygeno.2005.04.008
GH, Y., D, B., H, Z., SM, Z., D, Z., J, W. (1998). Hemodynamic effects of a calcium channel promoter, BAY y 5959, are preserved after chronic administration in ischemic heart failure in conscious dogs. J. Pharmacol. Exp. Ther. 286, 760–766.
Giladi, M., Tal, I., Khananshvili, D. (2016). Structural Features of Ion Transport and Allosteric Regulation in Sodium-Calcium Exchanger (NCX) Proteins. Front. Physiol. 7, 30. doi: 10.3389/fphys.2016.00030
Goel, S., Gupta, N., Walcott, B. P., Snuderl, M., Kesler, C. T., Kirkpatrick, N. D., et al. (2013). Effects of vascular-endothelial protein tyrosine phosphatase inhibition on breast cancer vasculature and metastatic progression. J. Natl. Cancer Inst. 105, 1188–1201. doi: 10.1093/jnci/djt164
Gross, E. R., Gross, G. J. (2007). Pharmacologic therapeutics for cardiac reperfusion injury. Expert Opin. Emerg. Drugs 12, 367–388. doi: 10.1517/14728214.12.3.367
Grueter, C. E., Colbran, R. J., Anderson, M. E. (2006). CaMKII, an emerging molecular driver for calcium homeostasis, arrhythmias, and cardiac dysfunction. J. Mol. Med. 85, 5–14. doi: 10.1007/s00109-006-0125-6
H, C., M, U., K, S., A, M., M, H., Y, O., et al. (1989). Dihydropyrimidines: novel calcium antagonists with potent and long-lasting vasodilative and antihypertensive activity. J. Med. Chem. 32, 2399–2406. doi: 10.1021/jm00130a029
Hadri, L., Hajjar, R. J. (2011). Calcium Cycling Proteins and Their Association With Heart Failure. Clin. Pharmacol. Ther. 90, 620–624. doi: 10.1038/clpt.2011.161
Haghighi, K., Bidwell, P., Kranias, E. G. (2014). Phospholamban interactome in cardiac contractility and survival: A new vision of an old friend. J. Mol. Cell. Cardiol. 77, 160–167. doi: 10.1016/j.yjmcc.2014.10.005
Hausenloy, D. J., Yellon, D. M. (2016). Ischaemic conditioning and reperfusion injury. Nat. Rev. Cardiol. 13, 193–209. doi: 10.1038/nrcardio.2016.5
Hayward, C., Banner, N. R., Morley-Smith, A., Lyon, A. R., Harding, S. E. (2015). The Current and Future Landscape of SERCA Gene Therapy for Heart Failure: A Clinical Perspective. Hum. Gene Ther. 26, 293–304. doi: 10.1089/hum.2015.018
Hidalgo, C. (2017). Calcium Rules. Circulation. 135, 1379–1381. doi: 10.1161/CIRCULATIONAHA.117.024244
Hodgson, J. M., Dib, N., Kern, M. J., Bach, R. G., Barrett, R. J. (2007). Coronary Circulation Responses to Binodenoson, a Selective Adenosine A2A Receptor Agonist. Am. J. Cardiol. 99, 1507–1512. doi: 10.1016/j.amjcard.2006.12.086
Huang, Z., Li, H., Guo, F., Jia, Q., Zhang, Y., Liu, X., et al. (2009). Egr-1, the Potential Target of Calcium Channel Blockers in Cardioprotection with Ischemia Reperfusion Injury in Rats. Cell. Physiol. Biochem. 24, 17–24. doi: 10.1159/000227809
Huffaker, R., Lamp, S. T., Weiss, J. N., Kogan, B. (2004). Intracellular calcium cycling, early afterdepolarizations, and reentry in simulated long QT syndrome. Heart Rhythm. 1, 441–448. doi: 10.1016/j.hrthm.2004.06.005
Hulot, J. S., Senyei, G., Hajjar, R. J. (2012). Sarcoplasmic reticulum and calcium cycling targeting by gene therapy. Gene Ther. 19, 596–599. doi: 10.1038/gt.2012.34
Ibanez, B., Heusch, G., Ovize, M., Van de Werf, F. (2015). Evolving therapies for myocardial ischemia/reperfusion injury. J. Am. Coll. Cardiol. 65, 1454–1471. doi: 10.1016/j.jacc.2015.02.032
Investigators, T., JH, A., HR, R., AL, S., V, D., RA, H., et al. (2007). Effect of Tilarginine Acetate in Patients With Acute Myocardial Infarction and Cardiogenic Shock. JAMA 297, 1657–1666. doi: 10.1001/jama.297.15.joc70035
Ishizaki, M., Kaibori, M., Uchida, Y., Hijikawa, T., Tanaka, H., Ozaki, T., et al. (2008). Protective effect of FR183998, a Na+ H+ exchanger inhibitor, and its inhibition of iNOS induction in hepatic ischemia-reperfusion injury in rats. SHOCK 30, 311–317. doi: 10.1097/SHK.0b013e318164ef14
J, W., E, B., E, B., B, D.-V.D.M., MD, P., MA, V. L., et al. (2009). Role for CaMKII inhibition in rheumatoid arthritis: effects on HIF-1-induced VEGF production by rheumatoid synovial fibroblasts. Ann. N. Y. Acad. Sci. 1173, 706–711. doi: 10.1111/j.1749-6632.2009.04736.x
Jackson, E. K. (2017). Context-dependent effects of dipeptidyl peptidase 4 inhibitors. Curr. Opin. Nephrol. Hypertens. 26, 83–90. doi: 10.1097/MNH.0000000000000303
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., Morishima, K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. doi: 10.1093/nar/gkw1092
Kawasumi, H., Satoh, N., Kitada, Y. (2007). Caldaret, an Intracellular Ca2+ Handling Modulator, Limits Infarct Size of Reperfused Canine Heart. J. Pharmacol. Sci. 103, 222–233. doi: 10.1254/jphs.FP0060765
Khananshvili, D. (2013). The SLC8 gene family of sodium-calcium exchangers (NCX) - structure, function, and regulation in health and disease. Mol. Aspects Med. 34, 220–235. doi: 10.1016/j.mam.2012.07.003
Kleinbongard, P., Baars, T., Heusch, G. (2012). Calcium antagonists in myocardial ischemia reperfusion—update 2012. Wiener Medizinische Wochenschrift 162, 302–310. doi: 10.1089/dna.2016.3391
Kleinsasser, A., Loeckinger, A. (2002). Are sildenafil and theophylline effective in the prevention of high-altitude pulmonary edema. Med. Hypotheses 59, 223–225. doi: 10.1016/S0306-9877(02)00198-6
Landstrom, A. P., Dobrev, D., Wehrens, X. H. T. (2017). Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 120, 1969–1993. doi: 10.1161/circresaha.117.310083
Laver, D. R. (2018). Regulation of the RyR channel gating by Ca(2+) and Mg(2). Biophys. Rev. 10, 1087–1095. doi: 10.1007/s12551-018-0433-4
Law, J. K. Y., Yeung, C. K., Wan, S. P., Ingebrandt, S., Lau, H. Y. A., Rudd, J. A., et al. (2011). The significance of chloride in the inhibitory action of disodium cromoglycate on immunologically-stimulated rat peritoneal mast cells. Biochim. Biophys. Acta (BBA) - Gen. Subj. 1810, 867–874. doi: 10.1016/j.bbagen.2011.05.007
Lee, Y. K., Ng, K. M., Lai, W. H., Chan, Y. C., Lau, Y. M., Lian, Q., et al. (2011). Calcium homeostasis in human induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Rev. Rep. 7, 976–986. doi: 10.1007/s12015-011-9273-3
Lesnefsky, E. J., Chen, Q., Tandler, B., Hoppel, C. L. (2017). Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion Implications for Novel Therapies. Annu. Rev. Pharmacol. Toxicol. 57, 535–565. doi: 10.1146/annurev-pharmtox-010715-103335
Ling, H., Gray, C. B., Zambon, A. C., Grimm, M., Gu, Y., Dalton, N., et al. (2013). Ca2+/Calmodulin-dependent protein kinase II delta mediates myocardial ischemia/reperfusion injury through nuclear factor-kappaB. Circ. Res. 112, 935–944. doi: 10.1161/CIRCRESAHA.112.276915
Lipskaia, L., Keuylian, Z., Blirando, K., Mougenot, N., Jacquet, A., Rouxel, C., et al. (2014). Expression of sarco (endo) plasmic reticulum calcium ATPase (SERCA) system in normal mouse cardiovascular tissues, heart failure and atherosclerosis. Biochim. Biophys. Acta 1843, 2705–2718. doi: 10.1016/j.bbamcr.2014.08.002
MacLennan, D. H., Kranias, E. G. (2003). Phospholamban: a crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 4, 566–577. doi: 10.1038/nrm1151
Maier, L. S., Bers, D. M. (2002). Calcium, Calmodulin, and Calcium-Calmodulin Kinase II Heartbeat to Heartbeat and Beyond. J. Mol. Cell Cardiol. 34, 919–939. doi: 10.1006/jmcc.2002.2038
Maki, J., Hirano, M., Hoka, S., Kanaide, H., Hirano, K. (2010). Thrombin activation of proteinase-activated receptor 1 potentiates the myofilament Ca2+ sensitivity and induces vasoconstriction in porcine pulmonary arteriesbph. Br. J. Pharmacol. 159, 919–927. doi: 10.1111/j.1476-5381.2009.00591.x
Marks, A. R. (2013). Calcium cycling proteins and heart failure: mechanisms and therapeutics. J. Clin. Invest. 123, 46–52. doi: 10.1172/jci62834
Mattiazzi, A., Kranias, E. G. (2014). The role of CaMKII regulation of phospholamban activity in heart disease. Front. Pharmacol. 5, 5. doi: 10.3389/fphar.2014.00005
Mochly-Rosen, D., Das, K., Grimes, K. V. (2012). Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discovery 11, 937–957. doi: 10.1038/nrd3871
Morita, T., Hussain, D., Asahi, M., Tsuda, T., Kurzydlowski, K., Toyoshima, C., et al. (2008). Interaction sites among phospholamban, sarcolipin, and the sarco(endo)plasmic reticulum Ca(2+)-ATPase. Biochem. Biophys. Res. Commun. 369, 188–194. doi: 10.1016/j.bbrc.2007.11.098
Mozaffari, M. S., Liu, J. Y., Abebe, W., Baban, B. (2013). Mechanisms of load dependency of myocardial ischemia reperfusion injury. Am. J. Cardiovasc. Dis. 3, 180–196. doi: 2160-200X/AJCD1307005
Murphy, E., Steenbergen, C. (2008). Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 88, 581–609. doi: 10.1152/physrev.00024.2007
Oda, T., Yang, Y., Uchinoumi, H., Thomas, D. D., Chen-Izu, Y., Kato, T., et al. (2015). Oxidation of ryanodine receptor (RyR) and calmodulin enhance Ca release and pathologically alter, RyR structure and calmodulin affinity. J. Mol. Cell. Cardiol. 85, 240–248. doi: 10.1016/j.yjmcc.2015.06.009
Oh, G.-C., Lee, H.-Y., Kang, H.-J., Zo, J.-H., Choi, D.-J., Oh, B.-H. (2012). Quantification of Pedal Edema During Treatment With S(-)-Amlodipine Nicotinate Versus Amlodipine Besylate in Female Korean Patients With Mild to Moderate Hypertension: A 12-Week, Multicenter, Randomized, Double-Blind, Active-Controlled, Phase IV Clinical Trial. Clin. Ther. 34, 1940–1947. doi: 10.1016/j.clinthera.2012.08.003
Pall, G. S., Johnson, K. J., Smith, G. L. (2003). Abnormal contractile activity and calcium cycling in cardiac myocytes isolated from dmpk knockout mice. Physiol. Genomics 13, 139–146. doi: 10.1152/physiolgenomics.00107.2002
Paradies, G., Paradies, V., Ruggiero, F. M., Petrosillo, G. (2018). Mitochondrial bioenergetics and cardiolipin alterations in myocardial ischemia-reperfusion injury: implications for pharmacological cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 315, H1341–H1352. doi: 10.1152/ajpheart.00028.2018
Qin, W., Zhang, L., Li, Z., Xiao, D., Zhang, Y., Yang, H., et al. (2020). Metoprolol protects against myocardial infarction by inhibiting miR-1 expression in rats. J. Pharm. Pharmacol. 72, 76–83. doi: 10.1111/jphp.13192
Ram, C. V. S. (2009). Antihypertensive efficacy of olmesartan medoxomil or valsartan in combination with amlodipine: A review of factorial-design studies. Curr. Med. Res. Opin. 25, 177–185. doi: 10.1185/03007990802597456
Rosenthal, T. C., Majeroni, B. A. A., Pretorius, R., Malik, K. (2008). Fatigue An Overview. Am. Family Physician 78, 1173–1179.
Rosenzweig, E. B. (2006). Emerging treatments for pulmonary arterial hypertension. Expert Opin. Emerg. Drugs 11, 609–619. doi: 10.1517/14728214.11.4.609
Shaw, R. M., Colecraft, H. M. (2013). L-type calcium channel targeting and local signalling in cardiac myocytes. Cardiovasc. Res. 98, 177–186. doi: 10.1093/cvr/cvt021
Stammers, A. N., Susser, S. E., Hamm, N. C., Hlynsky, M. W., Kimber, D. E., Kehler, D. S., et al. (2015). The regulation of sarco(endo)plasmic reticulum calcium-ATPases (SERCA). Can. J. Physiol. Pharmacol. 93, 843–854. doi: 10.1139/cjpp-2014-0463
Striessnig, J., Bolz, H. J., Koschak, A. (2010). Channelopathies in Cav1.1, Cav1.3, and Cav1.4 voltage-gated L-type Ca2+ channels. Pflügers Archiv. - Eur. J. Physiol. 460, 361–374. doi: 10.1007/s00424-010-0800-x
Sulpizio, A. C., Pullen, M. A., Edwards, R. M., Louttit, J. B., West, R., Brooks, D. P. (2005). Mechanism of vasopeptidase inhibitor-induced plasma extravasation: comparison of omapatrilat and the novel neutral endopeptidase 24.11/angiotensin-converting enzyme inhibitor GW796406. J. Pharmacol. Exp. Ther. 315, 1306–1313. doi: 10.1124/jpet.105.084749
T, I., S, U., Y, T., T, U., K, K., N, T., et al. (1994). Identification of adenosine A2 receptor-cAMP system in human aortic endothelial cells. Biochem. Biophys. Res. Commun. 199, 905–910. doi: 10.1006/bbrc.1994.1314
TD, B., DJ, T., MJ, W., DH, M. (2005). Mechanism of tissue-selective drug action in the cardiovascular system. Mol. Interventions 5, 84–93. doi: 10.1124/mi.5.2.6
Thind, G. S., Agrawal, P. R., Hirsh, B., Saravolatz, L., Chen-Scarabelli, C., Narula, J., et al. (2015). Mechanisms of myocardial ischemia– reperfusion injury and the cytoprotective role of minocycline scope and limitations. Future Cardiol. 11, 61–76. doi: 10.2217/fca.14.76
Tracey, W. R., Allen, M. C., Frazier, D. E., Fossa, A. A., Johnson, C. G., Marala, R. B., et al. (2003). Zoniporde a potent and selective inhibitor of the human sodium-hydrogen exchanger isoform 1. Cardiovasc. Drug Rev. 21, 17–32. doi: 10.1111/j.1527-3466.2003.tb00103.x
Turer, A. T., Hill, J. A. (2010). Pathogenesis of Myocardial Ischemia-Reperfusion Injury and Rationale for Therapy. Am. J. Cardiol. 106, 360–368. doi: 10.1016/j.amjcard.2010.03.032
W, Y., BT, R., X, L., BT, P., I, K., AM, B., et al. (2018). Mapping the Pathway and Dynamics of Bestatin Inhibition of the Plasmodium falciparum M1 Aminopeptidase PfA-M1. ChemMedChem 13, 2504–0513. doi: 10.1002/cmdc.201800563
Wiens, B., De Luca, V. (2016). Molecular and biochemical characterization of a benzenoid/phenylpropanoid meta/para-O-methyltransferase from Rauwolfia serpentina roots. Phytochemistry 132, 5–15. doi: 10.1016/j.phytochem.2016.10.004
Williams, G. S. B., Boyman, L., Lederer, W. J. (2015). Mitochondrial calcium and the regulation of metabolism in the heart. J. Mol. Cell. Cardiol. 78, 35–45. doi: 10.1016/j.yjmcc.2014.10.019
Wood, D. M., Dargan, P. I., Hoffman, R. S. (2009). Management of cocaine-induced cardiac arrhythmias due to cardiac ion channel dysfunction. Clin. Toxicol. 47, 14–23. doi: 10.1080/15563650802339373
Wulff, H., Christophersen, P., Colussi, P., Chandy, K. G., Yarov-Yarovoy, V. (2019). Antibodies and venom peptides: new modalities for ion channels. Nat. Rev. Drug Discovery 18, 339–357. doi: 10.1038/s41573-019-0013-8
Xavier, F. E., Davel, A. P. C., Fukuda, L. V. E., Rossoni, L. V. (2009). Chronic ouabain treatment exacerbates blood pressure elevation in spontaneously hypertensive rats the role of vascular mechanisms. J. Hypertension 27, 1233–1242. doi: 10.1097/HJH.0b013e32832a391f
Xiao, Y. F., Zeng, Z. X., Guan, X. H., Wang, L. F., Wang, C. J., Shi, H., et al. (2018). FKBP12.6 protects heart from AngII-induced hypertrophy through inhibiting Ca(2+) /calmodulin-mediated signalling pathways in vivo and in vitro. J. Cell Mol. Med. 22, 3638–3651. doi: 10.1111/jcmm.13645
Xin, M., Feng, J., Hao, Y., You, J., Wang, X., Yin, X., et al. (2020). Cyclic adenosine monophosphate in acute ischemic stroke: some to update, more to explore. J. Neurol. Sci. 413, 116775. doi: 10.1016/j.jns.2020.116775
Xiong, Y., Mahmood, A., Chopp, M. (2009). Emerging treatments for traumatic brain injury. Expert Opin. Emerg. Drugs 14, 67–84. doi: 10.1517/14728210902769601
Yang, Y., Lu, X., Rong, X., Jiang, W., Lai, D., Ma, Y., et al. (2015). Inhibition of the mevalonate pathway ameliorates anoxia-induced down-regulation of FKBP12.6 and intracellular calcium handling dysfunction in H9c2 cells. J. Mol. Cell Cardiol. 80, 166–174. doi: 10.1016/j.yjmcc.2015.01.010
Zhao, Q. I. N., Wang, S., Li, Y. U., Wang, P., Li, S., Guo, Y., et al. (2013). The role of the mitochondrial calcium uniporter in cerebral ischemia/reperfusion injury in rats involves regulation of mitochondrial energy metabolism. Mol. Med. Rep. 7, 1073–1080. doi: 10.3892/mmr.2013.1321
Zhao, Y.-T., Guo, Y.-B., Gu, L., Fan, X.-X., Yang, H.-Q., Chen, Z., et al. (2017). Sensitized signalling between L-type Ca21 channels and ryanodine receptors in the absence or inhibition of FKBP12.6 in cardiomyocytes. Cardiovasc. Res. 113, 332–342. doi: 10.1093/cvr/cvw247
Zhu, S., Xu, T., Luo, Y., Zhang, Y., Xuan, H., Ma, Y., et al. (2017). Luteolin Enhances Sarcoplasmic Reticulum Ca2+-ATPase Activity through p38 MAPK Signaling thus Improving Rat Cardiac Function after Ischemia/Reperfusion. Cell. Physiol. Biochem. 41, 999–1010. doi: 10.1159/000460837
Zile, M. R., Gaasch, W. H. (2011). Abnormal Calcium Homeostasis One Mechanism in Diastolic Heart Failure. J. Am. Coll. Cardiol. 58, 156–157. doi: 10.1016/j.jacc.2010.10.068
Keywords: calcium homeostasis, myocardial ischemia/reperfusion injury, calcium signaling pathway, therapeutic reagents, Therapeutic Target Database
Citation: Wang R, Wang M, He S, Sun G and Sun X (2020) Targeting Calcium Homeostasis in Myocardial Ischemia/Reperfusion Injury: An Overview of Regulatory Mechanisms and Therapeutic Reagents. Front. Pharmacol. 11:872. doi: 10.3389/fphar.2020.00872
Received: 06 April 2020; Accepted: 27 May 2020;
Published: 09 June 2020.
Edited by:
Chrishan S. Samuel, Monash University, AustraliaReviewed by:
Ágota Apáti, Hungarian Academy of Sciences (MTA), HungaryTamer M. A. Mohamed, University of Louisville, United States
Copyright © 2020 Wang, Wang, He, Sun and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guibo Sun, c3VuZ3VpYm9AMTI2LmNvbQ==; Xiaobo Sun, c3VuX3hpYW9ibzE2M0AxNjMuY29t