 Tomohiko Aoe1,2*
Tomohiko Aoe1,2*- 1Pain Center, Teikyo University Chiba Medical Center, Ichihara, Japan
- 2Department of Medicine, Teikyo University, Tokyo, Japan
Coronavirus disease 2019 (COVID-19), the seventh human coronavirus infectious disease, was first reported in Wuhan, China, in December 2019, followed by its rapid spread globally (251,059 deaths, on May 5, 2020, by Johns Hopkins University). An early clinical report showed that fever, cough, fatigue, sputum production, and myalgia were initial symptoms, with the development of pneumonia as the disease progressed. Increases in the level of serum liver enzymes, D-dimer, cardiac troponin I, and creatinine have been observed in severely ill patients, indicating that multiple organ failure had occurred in these cases. Lymphopenia and an increase in interleukin-6 (IL-6) were also observed. Although COVID-19 patients are administered glucocorticoid therapy to treat the excessive immune response to severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) infection, the efficacy of this form of therapy is unclear. Viremia is observed in severe cases, suggesting that in addition to type II alveolar epithelial cells, many cell types, such as vascular endothelial cells, cardiomyocytes, renal tubular cells, neuronal cells, and lymphocytes, may be damaged. The improvement of survival rates requires elucidation of the mechanism by which cellular damage occurs during viral infection. Cellular therapy, along with organ support systems such as oxygen therapy, artificial ventilation, extra corporeal membrane oxygenation and dialysis, as well as antiviral therapy, are required. Viral replication in infected host cells may perturb protein folding in the endoplasmic reticulum (ER), causing ER stress. Although an adaptive cellular response, i.e. the unfolded protein response, can compensate for the misfolded protein burden to some extent, continued viral proliferation may induce inflammation and cell death. Therefore, we propose that proteostasis dysfunction may cause conformational disorders in COVID-19. The application of pharmacological chaperone therapy to treat COVID-19 patients is additionally discussed.
Introduction
The severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), which causes coronavirus disease 2019 (COVID-19), belongs to the genus Betacoronavirus of human coronaviruses (Wu et al., 2020; Zhou et al., 2020b); SARS-CoV-2 and the closely related SARS-CoV belong to lineage B, while the Middle East respiratory syndrome coronavirus (MERS-CoV) belongs to lineage C. SARS-CoV and MERS-CoV have caused severe respiratory disorders with higher mortality rates (Fung and Liu, 2019b).
SARS-CoV-2 is an enveloped virus whose genome comprises a single-stranded RNA molecule of 29,903 bases (Wu et al., 2020). The virus is composed of the RNA genome, a lipid membrane, and four constituent proteins, namely the spike glycoprotein, nucleocapsid protein, membrane protein, and envelope protein (Wu et al., 2020). As in SARS-CoV, the spike glycoprotein, membrane protein, and envelope protein are synthesized by the ribosome and inserted into the endoplasmic reticulum (ER) membrane during the replication of SARS-CoV-2. The newly synthesized polypeptides interact with ER molecular chaperones such as calnexin (Fukushi et al., 2012) to ensure correct protein folding. Mature viral proteins are transported from the ER to the ER-Golgi intermediate compartment (Masters, 2006), where viral particles are assembled from these proteins and the nucleocapsid protein, RNA, and lipid membrane (Fung and Liu, 2019b). Viral particles are transported along the secretory pathway to the plasma membrane, and secreted from the cell. Among the proteins that form SARS-CoV-2 viral particles, the spike glycoprotein, with a large molecular weight of 1,273 amino acids, exhibits N-linked glycosylation and plays an important role in mediating SARS-CoV-2 entry into host cells and the ensuing cellular damage (Walls et al., 2020; Zhou et al., 2020b).
SARS-CoV-2 enters epithelial cells in the mucosal membrane of the eyes, nose, and mouth, or the respiratory tract. As in SARS-CoV infection, during viral entry, the spike glycoprotein of SARS-CoV-2 attaches to the angiotensin-converting enzyme 2 (ACE2) protein, which is expressed on the cell surface of host cells, and uses it as a receptor (Walls et al., 2020; Zhou et al., 2020b). The SARS-CoV-2 spike glycoprotein is primed by the host serine protease, TMPRSS2, which confers the ability to bind to ACE2 (Hoffmann et al., 2020). ACE2 is expressed in various organs, such as the oral and nasal mucosa, nasopharynx, lung, stomach, small intestine, colon, skin, lymph nodes, thymus, bone marrow, spleen, liver, kidney, and brain (Hamming et al., 2004). It has been suggested that the spike glycoprotein recognizes molecules other than ACE2 expressed on the cell surface. A structural analysis of the spike glycoprotein of SARS-CoV-2 suggests that this protein can additionally bind to glucose-regulated protein 78 (GRP78) on the cell surface (Ibrahim et al., 2020). GRP78 is also known as binding immunoglobulin protein (BiP), an ER chaperone.
ER Stress and the UPR
Secretory proteins and membrane proteins are synthesized by ribosomes on the ER membrane and inserted into the ER. These nascent polypeptides interact with ER molecular chaperones such as BiP, protein disulfide isomerase (PDI), calreticulin, and calnexin to undergo correct folding, sugar chain modification, and complex formation (Wang and Kaufman, 2016; Rapoport et al., 2017). Properly folded proteins are secreted from the ER by coat protein complex II (COPII) vesicles to the Golgi apparatus, and then transported to the plasma membrane. Proteins such as hormones are secreted from the cell, while others, such as G protein coupled receptors (GPCRs), are expressed on the plasma membrane as cell-surface receptors (Peotter et al., 2019) (Figure 1).
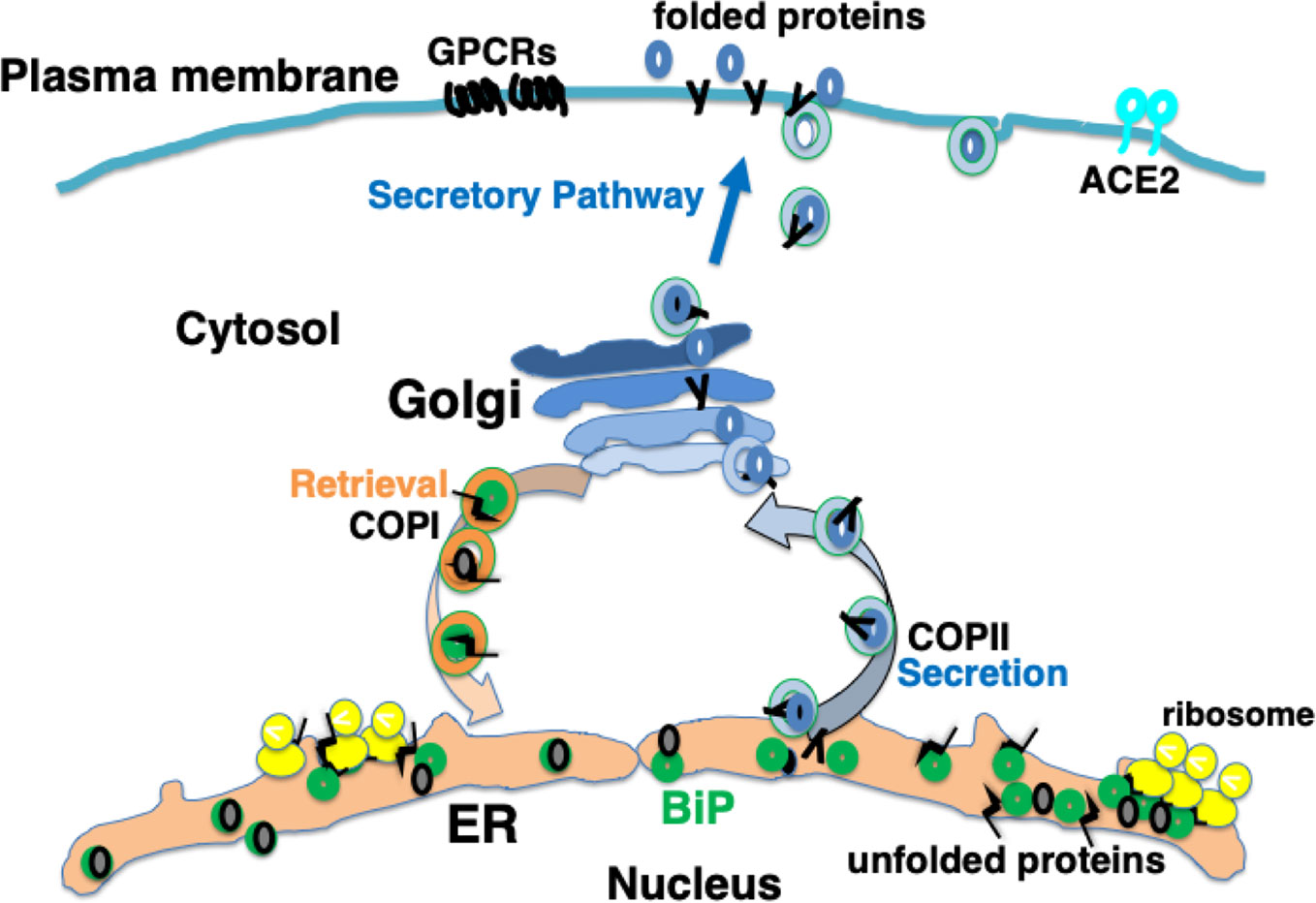
Figure 1 Newly synthesized polypeptides are inserted into the endoplasmic reticulum (ER). Properly folded proteins are transported to the secretory pathway. Interaction with ER chaperones such as BiP facilitates proper folding of nascent proteins. Mature proteins are released into the secretory pathway via coat protein II (COPII) vesicular transport. A certain fraction of KDEL proteins, such as BiP, associating with unfolded proteins are secreted; these are retrieved via KDEL receptors from the cis-Golgi to the ER through COPI vesicular transport.
BiP is a major ER molecular chaperone; most of them are distributed to the ER lumen by binding to ER membrane proteins such as activated transcription factor 6 (ATF6), inositol requiring kinase-1 (IRE1), and PKR-like ER related kinase (PERK). BiP additionally binds unfolded nascent polypeptides to ensure proper folding without the formation of aggregates (Hendershot, 2004). External factors such as ischemia, oxidative stress, malnutrition, and exposure to toxic substances, as well as internal factors such as genetic abnormalities resulting in the production of mutant proteins, disrupt protein folding in the ER. The accumulation of misfolded proteins that are unable to form proper three-dimensional structures or complexes in the ER causes ER stress, leading to an adaptive cellular response known as the unfolded protein response (UPR) (Hetz and Papa, 2018). BiP binds to misfolded proteins and dissociates from ATF6, IRE1, and PERK, resulting in the activation of these membrane proteins. ATF6 is a type 2 transmembrane protein that is transported from the ER to the Golgi complex, where it undergoes cleavage of its cytoplasmic amino-terminal side to become a transcription factor. ATF6 translocates to the nucleus and induces the transcription of genes involved in the UPR such as those that encode ER molecular chaperones (Shen et al., 2002). IRE1 and PERK form a homo-multimer and are activated by autophosphorylation. IRE1, which exhibits RNase activity, splices X-box binding protein-1 (XBP-1) mRNA. The spliced form is translated into XBP-1 protein, which acts as a transcription factor and mediates progression of the UPR (Lee et al., 2002). IRE1 also activates mitogen-activated protein (MAP) kinases such as c-jun N-terminal kinase (JNK) (Urano et al., 2000), triggering inflammation (Hasnain et al., 2012), promoting autophagy (Fung and Liu, 2019a; Sozen et al., 2020). PERK phosphorylates eukaryotic initiation factor 2α (eIF2α), leading to the suppression of general protein translation, which reduces the load of newly synthesized unfolded polypeptides on the ER (Hughes and Mallucci, 2019). The UPR promotes increased production of cytoprotective molecules such as ER molecular chaperones, suppression of the protein translation of most proteins, accelerated degradation of misfolded proteins (ER-associated degradation; ERAD) (Figure 2).
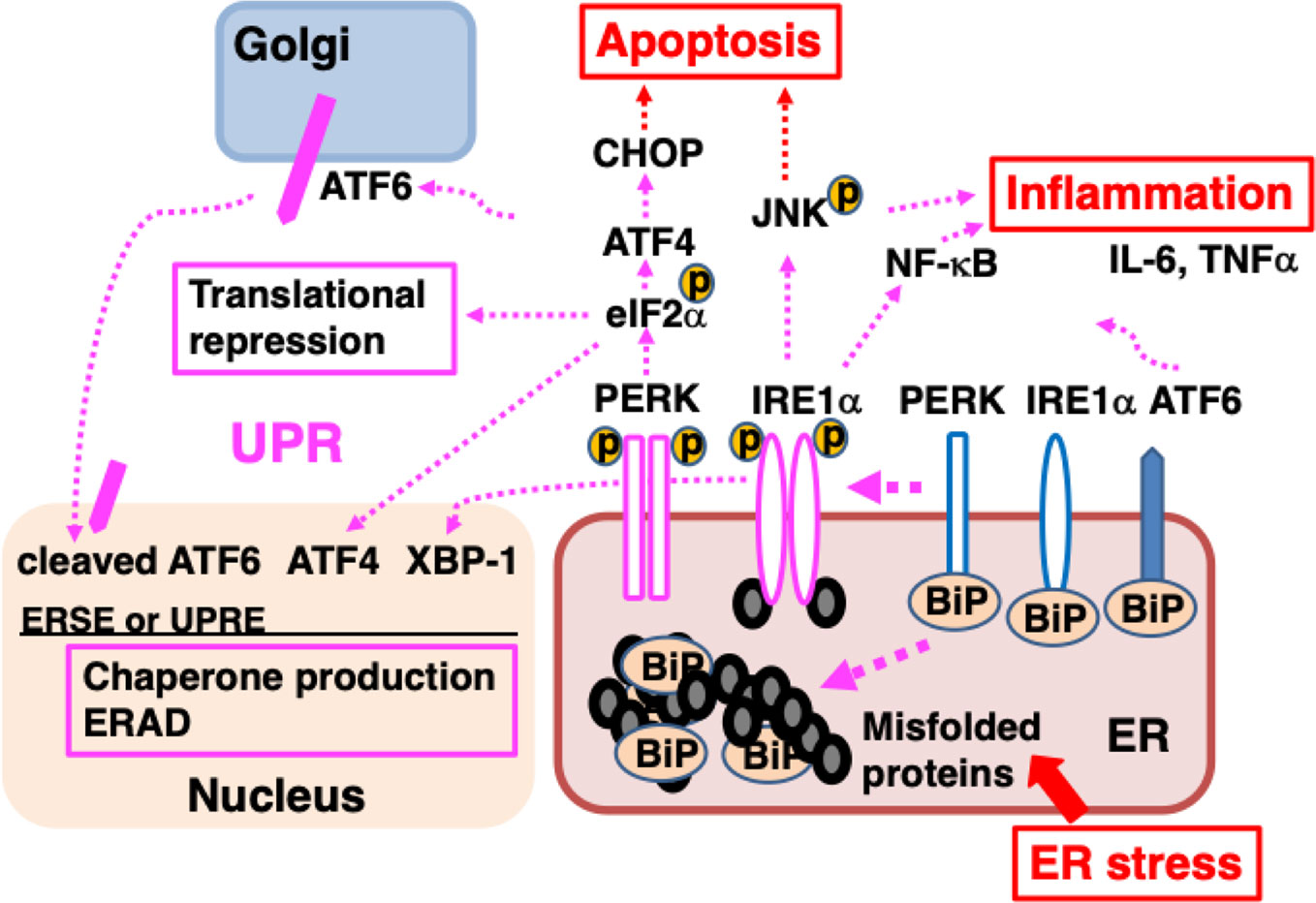
Figure 2 ER stress causes the accumulation of unfolded proteins in the ER, inducing the unfolded protein response (UPR). Misfolded proteins accumulate in the ER when protein folding is disturbed due to ER stress such as intrinsic defects (e.g., mutated sequences, missing subunits) or extrinsic injuries (e.g., ischemia, malnutrition, hypoxia, and toxicity). BiP dissociates from ATF6, IRE1α, and PERK, and associates with misfolded proteins; dissociation from BiP initiates the UPR. ATF6 is transported to the Golgi, where it is cleaved and the cytoplasmic portion of ATF6 is transported to the nucleus; there, it functions as a transcription factor, binding to ER stress response element (ERSE) and promoting the transcription of genes important for the UPR. After dissociating from BiP, IRE1α, and PERK multimerize and are activated by autophosphorylation. IRE1α is a type I membrane protein containing a serine/threonine kinase domain and an endoribonuclease domain at its cytoplasmic carboxyl terminus. Activated IRE1α splices XBP1 mRNA. The XBP1 protein binds to UPR elements (UPREs) in the transcriptional region of various genes required for the UPR and promotes their transcription. PERK is a serine/threonine kinase that phosphorylates and inactivates eIF2α, thereby halting the translation of most proteins. However, the translation of ATF4 is induced. ATF4 acts as a transcription factor. The UPR enhances the ability of cells to deal with increased levels of misfolded proteins through chaperone production, translational repression, and ER-associated protein degradation (ERAD). Activation of IRE1α leads to inflammation through the activation of MAP kinases (JNK) and NF-κB, inducing elevated expression of pro-inflammatory cytokines, such as TNFα and IL-6. Persistent ER stress induces the expression of CHOP, leading to apoptosis.
Thus, the capacity of cells to handle misfolded proteins in the ER is expanded, and cells can tolerate ER stress to a certain degree (Hetz and Papa, 2018). However, in response to excessive load of misfolded proteins due to a greater invasion like a higher viral load, the UPR is prolonged. The activation of PERK induces the expression of ATF4, leading to the production of the transcription factor C/EBP homologous protein (CHOP), which induces the downregulation of anti-apoptotic Bcl-2 and results in apoptotic cell death (Oyadomari and Mori, 2004; Rozpedek et al., 2016). Persistent ER stress alleviates Ca2+ homeostasis. The ER comprises a major pool of Ca2+, and ER molecular chaperones such as BiP are capable of binding Ca2+. ER stress induces the outflow of Ca2+ from the ER and the influx of Ca2+ into mitochondria, leading to the release of cytochrome C and the activation of caspases 9 and 3, resulting in apoptosis, cell death (Ye et al., 2008; Bahar et al., 2016).
Proteostasis and Conformational Diseases
The proteostasis network, which is mediated by the interaction with ER molecular chaperones and cytosolic molecular chaperones such as heat shock protein 70 (HSP70), serves as a quality control process for ensuring proper protein folding and complex assembly. Failure of proteostasis leads to the accumulation of misfolded proteins, which triggers a persistent integrated stress response (ISR), including the UPR in the ER and heat shock response in the cytosol (Labbadia and Morimoto, 2015).
The accumulation of misfolded proteins and ER stress are associated with neurodegenerative diseases such as Alzheimer's disease (Ferreira and Klein, 2011), Parkinson's disease (Gibb and Lees, 1988), amyotrophic lateral sclerosis (ALS) (Rosen et al., 1993), and prion disease (Park et al., 2017), as well as other diseases such as cardiomyopathy (Hamada et al., 2004), pulmonary fibrosis (Peca et al., 2015), chronic renal tubular disease (Kimura et al., 2008), diabetes (Ozcan et al., 2006), obesity (Shan et al., 2017), and aging in human and animal models (Labbadia and Morimoto, 2015). Acute diseases such as respiratory failure and ischemia-reperfusion injury additionally involve the induction of ER stress and the UPR (Tajiri et al., 2004; Mimura et al., 2007; Klymenko et al., 2019).
Owing to genetic mutations, some proteins may evade the proteostasis network, causing protein aggregation both within and outside of cells. Alpha 1-antitrypsin deficiency (AATD) is a genetic disease caused by a mutation in the SERPINA1 gene (serine proteinase inhibitor, group A, member 1), which encodes a secretory protein, alpha 1-antitrypsin (AAT). The Z mutant of AAT, which is the most common variant of this protein in AATD, accumulates in the ER of hepatocytes without undergoing proper folding; this leads to ER stress and cellular dysfunction. In addition, AAT inhibits the activity of neutrophil serine proteases in blood. Thus, the deficiency of properly folded AAT causes the activation of neutrophil serine proteases, resulting in lung injury (Karatas and Bouchecareilh, 2020). Cystic fibrosis, another example of a genetic disorder, is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) protein, which is a chloride ion channel expressed on the epithelial cells of the lung, pancreas, and other organs. The most common mutation of the CFTR gene, F508del results in the translation of a mutant CFTR, which exhibits impaired trafficking (Cheng et al., 1990). The loss of cell-surface expression of CFTR results in the production of thick mucus and clog formation. Accumulation of the F508del mutant CFTR in the ER induces ER stress, causing cellular dysfunction and inflammation in airway epithelia (Ribeiro and Boucher, 2010) and production of interleukin-6 (IL-6) and tumor necrosis factor alpha (TNFα) by innate immune cells (Lara-Reyna et al., 2019).
Pulmonary surfactant, which is composed of phospholipids and the pulmonary surfactant proteins (SP)-A, -B, -C, and -D, reduces alveolar surface tension to facilitate spontaneous physiological respiration. The SP-C precursor contains the BRICHOS domain, which prevents amyloid formation (Dolfe et al., 2016). Several mutations in the BRICHOS domain of the SP-C gene have been reported to induce protein aggregation and ER stress, resulting in cell death and chronic interstitial lung diseases (Beers and Mulugeta, 2005; Peca et al., 2015). Owing to their native structure, some proteins, such as olfactory GPCR, are difficult to fold properly. GPCR is a membrane protein with a highly complex structure that penetrates the membrane seven times; because of this complex structure, nascent polypeptides, despite having normal gene sequences, may not all undergo proper folding. Many nascent polypeptides may be misfolded, retained in the ER, and degraded by ERAD and autophagy (Lu et al., 2003; Lu et al., 2004).
Diseases related to the dysfunction of proteostasis, which are known as conformational diseases (Carrell and Lomas, 1997; Kopito and Ron, 2000), occur in various cell types and are characterized by organ-specific symptoms corresponding to them. Although such conformational diseases are sometimes caused by a mutation in the gene encoding a specific protein, resulting in aggregation of the misfolded mutant protein, they can also be caused by dysfunction of the proteostasis system itself. BiP is a luminal chaperone present in the ER; a fraction of BiP proteins associating with unfolded proteins, are secreted from the ER by COPII vesicles (Raykhel et al., 2007). At the Golgi apparatus, the Lys-Asp-Glu-Leu (KDEL) carboxyl terminal of BiP is recognized by the KDEL receptor, which facilitates the return of BiP to the ER via COPI vesicles. The KDEL-retrieval system is part of the proteostasis network (Yamamoto et al., 2001). Homozygous knock-in mice expressing mutant BiP with the KDEL amino acid sequence deleted, instead of normal BiP, are born according to Mendelian law. As a result, all homozygous mutant BiP mice reportedly died from neonatal acute respiratory distress syndrome (ARDS) on the first day after birth (Mimura et al., 2007). In the type II alveolar epithelial cells of these mice, folding of surfactant proteins was impaired, leading to the loss of the secretion of pulmonary surfactant. In addition, the accumulation of misfolded surfactant proteins in the ER causes ER stress, inducing the expression of CHOP and triggering apoptosis. The proper assembly of correctly folded individual surfactant proteins is required for the production and secretion of pulmonary surfactant, which enables breathing by keeping the alveolar space open. Proteostasis therefore plays an important role in the production and secretion of pulmonary surfactant.
Chronic Comorbidities and Proteostasis
Sporadic neurodegenerative diseases often accompany aging (Labbadia and Morimoto, 2015). Heterozygous mutant BiP mice are capable of survival, and their lifespan is not significantly different from that of wild-type mice. However, in addition to wild-type BiP, these mice also express mutant BiP lacking the KDEL sequence. Misfolded proteins bound to the mutant BiP may escape from the proteostasis network without being recognized by the KDEL receptor at the Golgi complex, even if secreted from the ER (Jin et al., 2017). In aged mutated BiP mice, ubiquitinated protein aggregation was observed in the spinal cord and brain (Jin et al., 2014). Motor and cognitive dysfunction were additionally observed (Jin et al., 2018). With aging, organ symptoms such as renal tubular injury were also found (Kimura et al., 2008). Attenuation of proteostasis can lead to acute cellular dysfunction and cell death, as well as chronic cellular dysfunction and organ failure. The function of ER chaperones that play an important role in the proteostasis network declines with aging (Brown and Naidoo, 2012).
Preclinical studies suggest that vascular endothelial cells, cardiomyocytes, alveolar epithelial cells, neuronal cells, and lymphocytes may already be subjected to ER stress in individuals with comorbidities such as hypertension (Hasty and Harrison, 2012; Young, 2017; Ochoa et al., 2018), diabetes (Back and Kaufman, 2012), obesity (Chen et al., 2016), chronic obstructive pulmonary diseases (Liu et al., 2018), and neurodegenerative diseases (Labbadia and Morimoto, 2015). Obesity causes chronic inflammation in adipocytes as well as macrophages that induces elevated expression of pro-inflammatory cytokines, TNFα, and IL-6 (Solinas et al., 2007). Alleviation of ER stress by pharmacological chaperones suppresses NF-κB activity and inflammation in diet-induced obese mice (Chen et al., 2016).
It is considered that among patients with complications and elderly people, protein folding in the ER is impaired to a certain extent; therefore, the ability of the UPR to accommodate further ER stress may be limited. As a consequence, proteostasis in such individuals may be sensitive to further stress in the ER, for example, in conditions such as viral protein overload (Hrincius et al., 2015; Guan et al., 2020; Zhou et al., 2020a) (Figure 3).
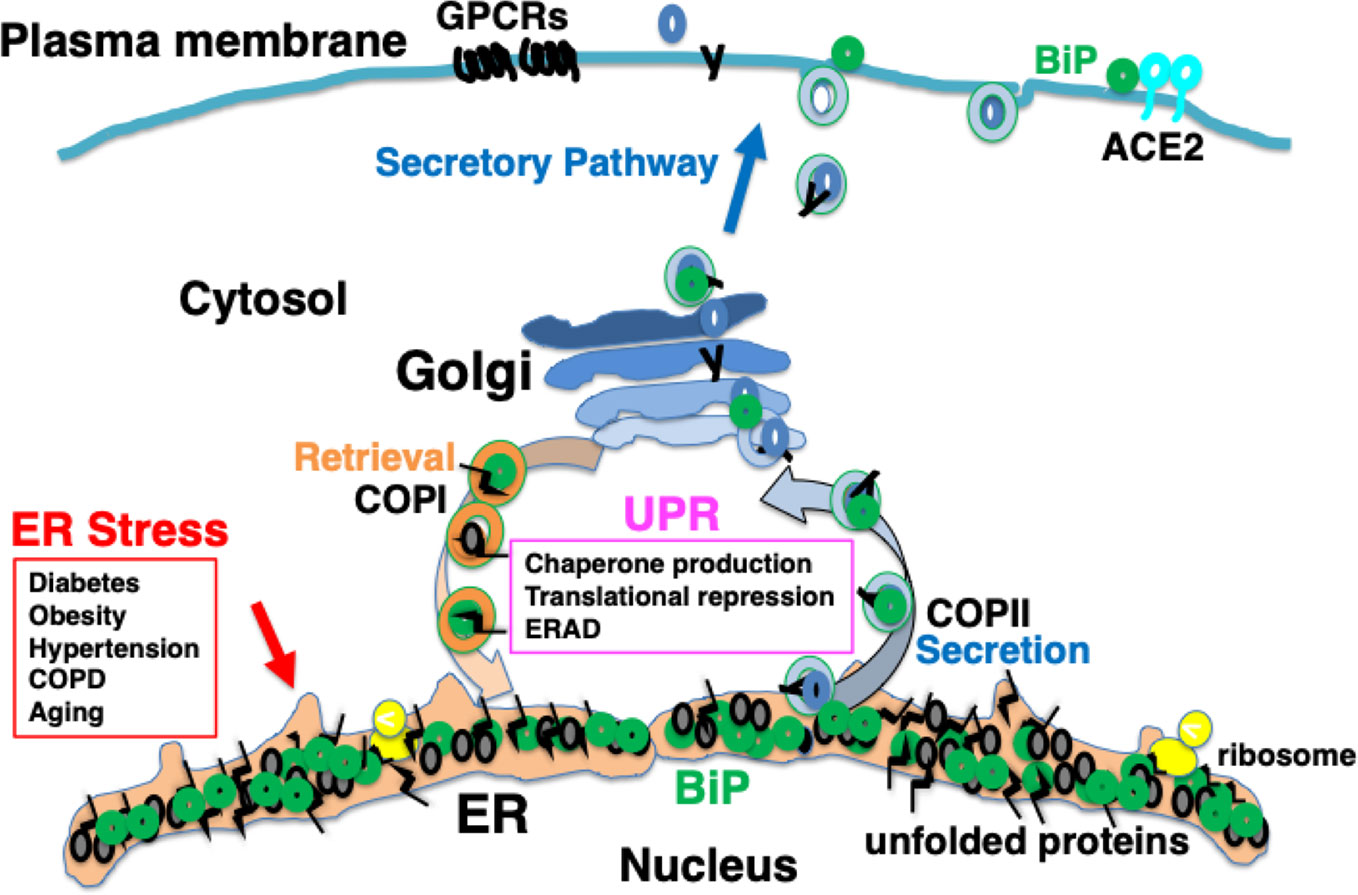
Figure 3 Chronic comorbidity is associated with ER stress. Among patients with complications and elderly people, protein folding in the ER is impaired to a certain extent; therefore, the ability of the UPR to accommodate further ER stress may be limited.
SARS-COV, SARS-CoV-2, and ER Stress
The organization of the genome of SARS-CoV-2 is closely related to that of SARS-CoV, and the structures of the proteins of both viruses are similar (Wu et al., 2020). Furthermore, both SARS-CoV and SARS-CoV-2 recognize the same ACE2 receptor expressed on host cells, and viral entry is mediated via their spike glycoproteins (Walls et al., 2020; Zhou et al., 2020b). Both viruses utilize the protein translation machinery of host cells for their replication. The spike glycoprotein, membrane protein, and envelope protein of SARS-CoV and SARS-CoV-2 are inserted into the ER after translation by ribosomes, where they undergo protein folding and glycosylation. Preclinical studies on SARS-CoV and other viruses have revealed that a result of rapid viral replication, nascent unfolded viral polypeptides accumulate in the ER, possibly causing ER stress (Huang et al., 2016).
In SARS-CoV infected patients, apoptosis in pneumocytes, T and B lymphocytes, and monocytes has been found to occur in the lung, spleen, and lymph nodes (Zhang et al., 2003). Lymphopenia and reduced counts of CD4-positive and CD8-positive T cells have been observed early in the disease course in SARS patients (Wong et al., 2003). When SARS-CoV was expressed in cultured cells, the spike glycoprotein accumulated in the ER and induced the UPR (Chan et al., 2006). In addition to the transcription of molecular chaperones such as BiP and GRP94, the expression of CHOP was also induced. This study suggested that when SARS-CoV induced the UPR, it did not activate ATF6, instead preferentially triggering the activation of PERK, leading to CHOP induction through the phosphorylation of eIF2α and the activation of transcription factor ATF4. Accumulation of the accessory 3α protein in the ER was found in SARS-CoV-infected cultured cells, where the activation of PERK, leading to the induction of ATF4 and CHOP were additionally observed (Minakshi et al., 2009). The SARS-CoV open reading frame (ORF) 8b protein has also been found to undergo aggregation in infected cells, and induce ER stress (Shi et al., 2019). These clinical observations in SARS patients and preclinical studies of SARS-CoV suggests that SARS-CoV infection induces ER stress and subsequent apoptosis in infected cells.
Several studies suggest that ACE2-mediated signaling reduces the effects of ER stress (Zhang et al., 2018). Stimulation of ACE2 by the ligands angiotensin 1–7 suppresses apoptosis mediated by ER stress in alveolar epithelial cells with accumulation of mutant surfactant protein C (Uhal et al., 2013). Shedding of ACE2 by spike glycoproteins may exacerbate ER stress. Individuals with diseases such as asthma and chronic obstructive pulmonary disease have similar levels of ACE2 expression to healthy people, but cigarette smokers have the elevated expression (Li et al., 2020a). It is unclear whether people with these comorbidities are at increased risk of SARS-CoV-2 infection based on their ACE2 expression levels.
The spike glycoprotein of MERS-CoV recognizes dipeptidyl peptidase 4 (DPP4) as a receptor; DPP4 bound to BiP is recognized more strongly than DPP4 alone, resulting in enhanced infectivity (Chu et al., 2018). Under ER stress, the UPR induces the production of BiP, which saturates reverse transport by the KDEL receptor. This is considered to result in missorting of BiP from the Golgi to the cell surface (Kokubun et al., 2019). Some BiP molecules are secreted extracellularly, whereas others are attached to the cell membrane via the proline-rich region near the carboxyl terminus (Tseng et al., 2019). BiP expressed on the plasma membrane may be involved in signal transduction as a cell-surface receptor (Tsai et al., 2018). The spike glycoprotein of SARS-CoV-2 may recognize BiP on the cell surface (Ibrahim et al., 2020); therefore, ER stress may increase infectivity in patients with various comorbidities and elderly individuals (Figure 4).
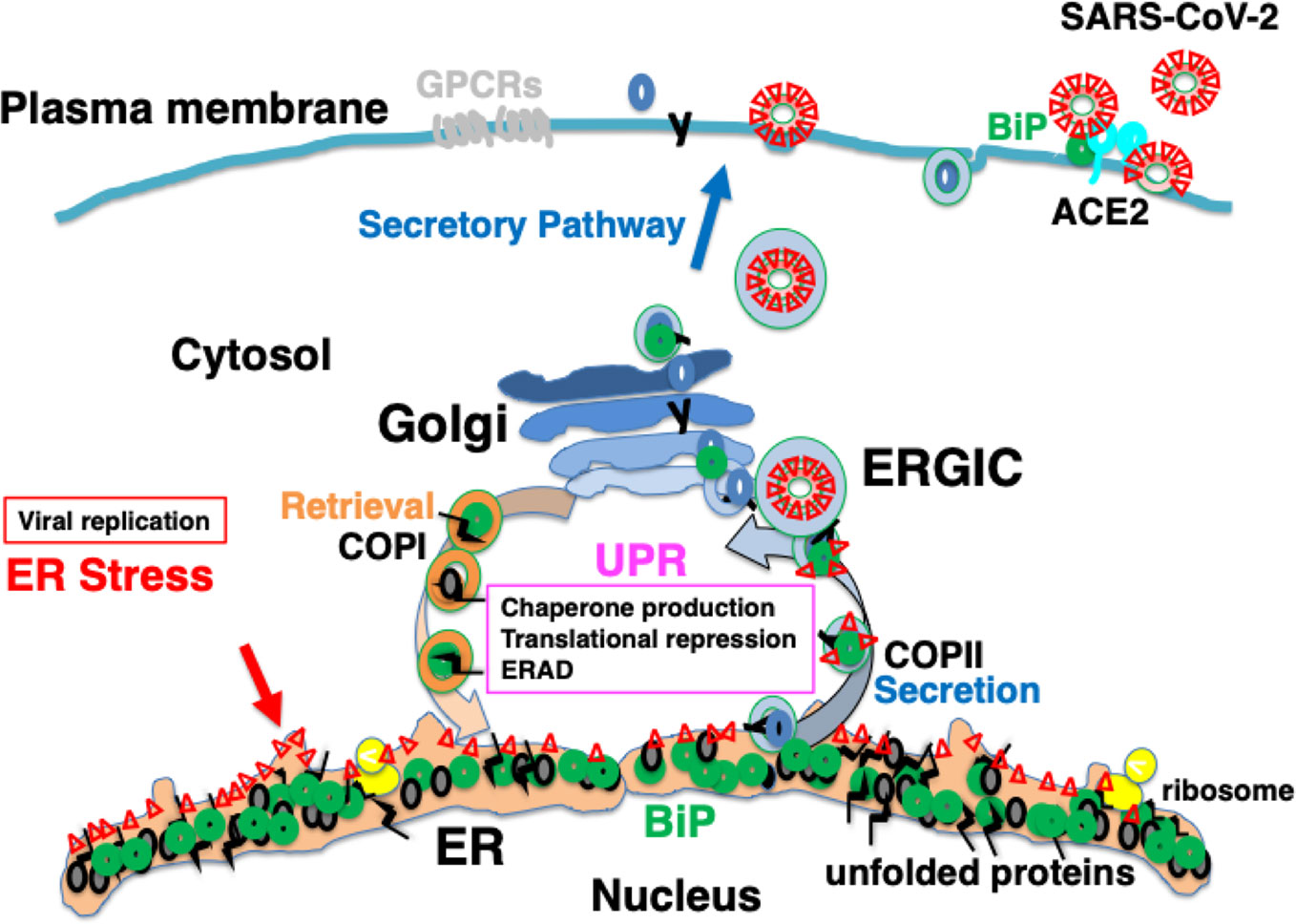
Figure 4 Infection with severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) may cause ER stress, inducing an adaptive response known as the UPR. SARS-CoV-2 infects ACE2-expressing cells. BiP on the cell surface may increase infectability as observed in Middle East respiratory syndrome coronavirus (MERS-CoV) infection. Mature viral proteins are transported from the ER to the ER-Golgi intermediate compartment (ERGIC), where viral particles are assembled from these proteins as well as nucleocapsid proteins, RNA, and lipid membranes. The replication of viral proteins causes ER stress, which is tolerated by the cell because of the UPR. Unstable proteins such as GPCRs, like olfactory receptors and taste receptors, may be susceptible to ERAD.
COVID-19 as a Conformational Disease
SARS-CoV-2 entry occurs through the mucous membranes of the eyes, nose, and mouth. This is followed by the appearance of symptoms such as fever, cough, fatigue, sputum production, shortness of breath, and myalgia (Guan et al., 2020; Zhou et al., 2020a), although many infected people do not show clear symptoms (Arons et al., 2020). Some people complain of abnormal taste and smell as initial symptoms (Xydakis et al., 2020). Smell is sensed by various olfactory receptors expressed on the surface of olfactory cells. Olfactory receptors are GPCRs, which have a complex structure that penetrates the membrane seven times. As a result, nascent peptides of olfactory receptors show high rates of misfolding, and misfolded peptides are retained in the ER and degraded by ERAD and autophagy (Lu et al., 2003; Lu et al., 2004). Taste is sensed by taste receptor type 1 (T1R) and taste receptor type 2 (T2R) receptors, which are also seven-transmembrane GPCR proteins expressed in taste cells distributed in the taste buds of the tongue, soft palate, and epiglottis (Nuemket et al., 2017). The folding of these receptor proteins is considered susceptible to impaired proteostasis by SARS-CoV-2 infection.
The most common fatal complication in COVID-19 patients is pneumonia, followed by ARDS (Guan et al., 2020; Huang et al., 2020; Yang et al., 2020; Zhou et al., 2020a). Some patients are not aware of hypoxemia (Ottestad et al., 2020). Among COVID-19 patients with CT findings of pneumonia, those without other symptoms such as fever and cough have been reported (Zhang et al., 2020). This asymptomatic silent pneumonia may be caused by impaired synthesis of pulmonary surfactant in type II alveolar epithelial cells infected with SARS-CoV-2 rather than inflammation.
In critically ill patients, serum levels of cytokines such as IL-6 (Zhou et al., 2020a), IL-2, IL-7, IL-10, GCSF, IP10, MCP1, MIP1A, and TNFα (Huang et al., 2020) are elevated. Pneumonia and ARDS in COVID-19 are considered to result from excessive systemic inflammation, a severe cytokine storm; therefore, corticosteroids are administered to these patients (Guan et al., 2020; Huang et al., 2020; Zhou et al., 2020a). However, the clinical data for SARS, MERS, and COVID-19 do not support the clinical efficacy of corticosteroids (Stockman et al., 2006; Russell et al., 2020). Complications such as avascular necrosis, delayed viral clearance, psychosis, and diabetes have been observed in SARS patients (Stockman et al., 2006; Russell et al., 2020). The World Health Organization does not recommend the use of corticosteroids for COVID-19 (World Health Organization, 2020). In COVID-19 patients with severe disease, low lymphocyte counts have been observed (Guan et al., 2020; Huang et al., 2020; Li et al., 2020b; Zhou et al., 2020a). Further, it remains unclear to what extent excessive systemic inflammation contributes to the cellular damage observed in patients with COVID-19.
An additional explanation for the etiology of COVID-19 is that the intracellular replication of SARS-CoV-2 itself is cytotoxic to host cells. SARS-CoV-2 infects ACE2-expressing cells such as type II alveolar epithelial cells, and proliferates by hijacking the protein translational machinery of host cells. The spike glycoprotein, membrane protein, and envelope protein, which are the main constituent proteins of the novel coronavirus, are co-translationally inserted into the ER. The explosive production of nascent viral polypeptides may cause ER stress and induce the UPR. In most individuals, cells have the ability to tolerate ER stress caused by viral infection because of the UPR, which expands the capacity of proteostasis, enabling infected cells to survive (Figure 4). However, when the burden of viral protein replication cannot be accommodated by the UPR, the production of functional proteins in the host cells is suppressed and cellular function is impaired, eventually leading to apoptosis (Figure 5). The assembly and secretion of pulmonary surfactant in type II alveolar epithelial cells is sensitive to impaired proteostasis, and the loss of pulmonary surfactant secretion in addition to apoptosis of type II alveolar epithelial cells may cause ARDS. Ultrastructural study of lung tissues of COVID-19 patients revealed viral particles in cytoplasm of type II cells with markedly swollen mitochondria and dilated ER (Li et al., 2020b). Pyroptosis and apoptosis were observed in the lung cells. Massive infiltration of CD68+ macrophages and neutrophils in lung tissues accompanied with elevated serum cytokines including IL6, IL8, TNFα, IP10, MCP1, and RANTES were also observed (Li et al., 2020b). In SARS patients, the expression of pro-inflammatory cytokines such as MCP-1, TGF-β, TNF-α, IL-1β, and IL-6 was upregulated in SARS-CoV infected, ACE2-expressing cells such as alveolar epithelial cells and macrophages in the lung (He et al., 2006). Innate immunity activated by viral ER stress may contribute to the disease (Hrincius et al., 2015).
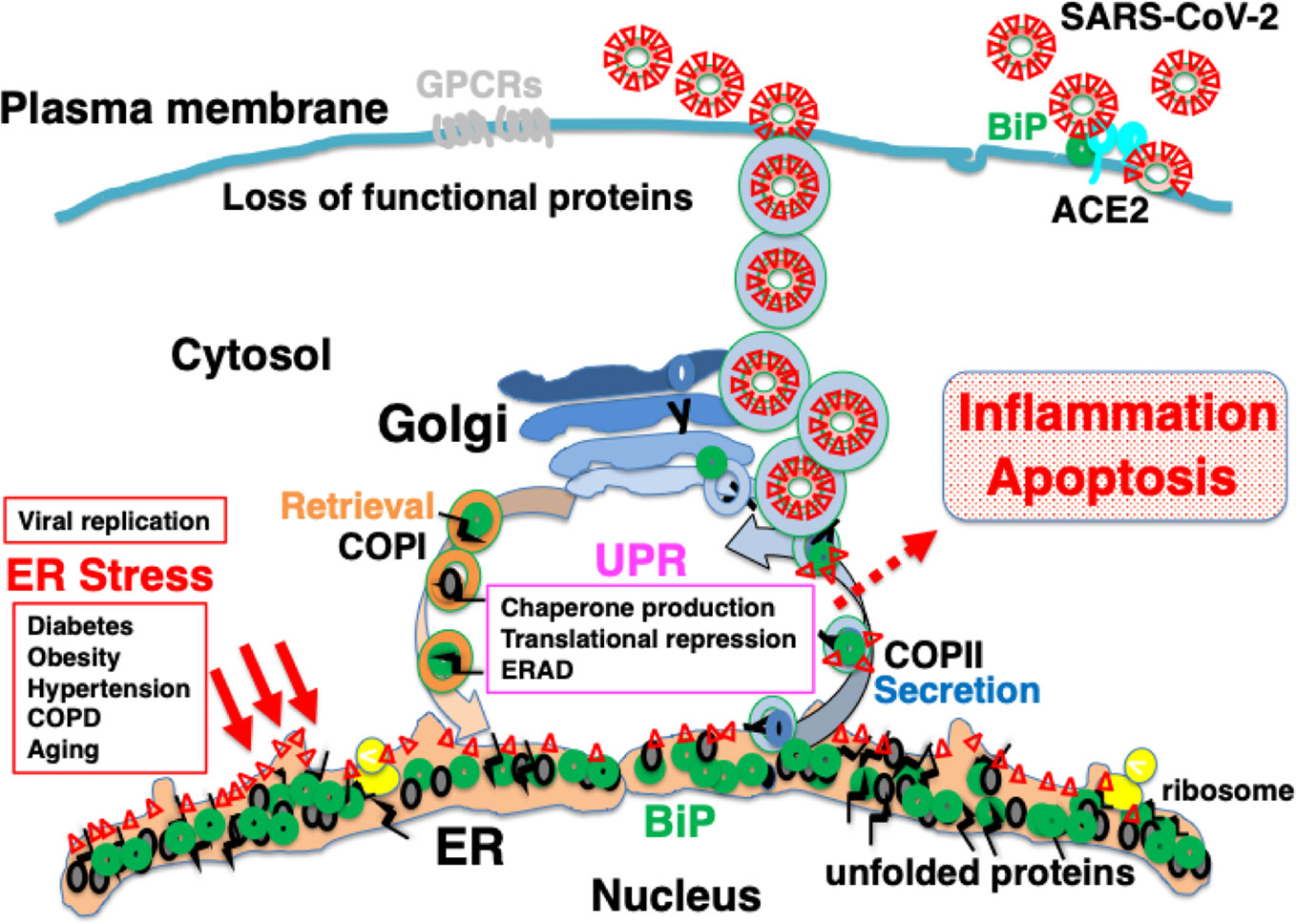
Figure 5 Explosive SARS-CoV-2 replication may induce persistent ER stress, leading to inflammation and apoptosis. The extremely rapid rate of replication of SARS-CoV-2 increases ER stress to levels that cannot be accommodated by the UPR, leading to inflammation and apoptosis. Patients with comorbidities may have limited capacity for proteostasis, and be sensitive to ER stress caused by viral protein overload. Pharmacological chaperones may reduce ER stress.
SARS-CoV-2 initially invades epithelial cells in the upper and lower airway, and viremia has been observed in patients with severe COVID-19 (Huang et al., 2020). ACE2 is expressed in multiple organs such as the kidney, testis, intestinal tract, and heart, as well as in vascular endothelial cells (Hamming et al., 2004). The explosive production of viral proteins in infected cells and the resulting elevated ER stress may impair proteostasis in host cells, inducing suppression of the translation of functional proteins, and triggering apoptosis. Increases in the levels of serum liver enzymes, D-dimer, cardiac troponin I, creatinine, and lymphopenia have been observed in severely ill patients (Zhou et al., 2020a). The Sequential Organ Failure Assessment (SOFA) score of patients who died from COVID-19 is reported to be high, indicating that these individuals had suffered multiple organ failure, including renal dysfunction, liver dysfunction, cardiovascular dysfunction, gastrointestinal disorders, neuronal disorders, and immune suppression (Guan et al., 2020; Mao et al., 2020; Zhou et al., 2020a). Pathological analysis of patients with COVID-19 indicated the presence of viral particles in vascular endothelial cells. Direct viral infection caused diffuse vascular endothelial inflammation in multiple organs including lung, heart, kidney, liver, and small intestine (Varga et al., 2020). The induction of caspase 3 and apoptotic bodies in endothelial cells and mononuclear cells in small bowel and lung specimens has been observed (Varga et al., 2020).
Although the removal of SARS-CoV-2 or infected cells would represent a “cure” for COVID-19, SARS-CoV-2 may continue to proliferate in infected cells in a state where cellular dysfunction is tolerable by the host's UPR. The duration of the resulting subclinical infection is unclear. It remains unknown whether chronic infection with SARS-CoV-2 may cause chronic conformational diseases in infected cells and organs such as interstitial pneumonia, tubulointerstitial renal fibrosis, and neurodegenerative disease.
The Use of Pharmacological Chaperones as a Potential Therapeutic Strategy for COVID-19
Several drugs like Remdesivir, a nucleotide analogue prodrug that inhibits viral RNA polymerases seem to be promising for anti-SARS-CoV-2 therapy (Grein et al., 2020; Lythgoe and Middleton, 2020). If antiviral therapy is administered at the later stages of the infection, i.e. when viral propagation occurs and cell death is initiated, the effect may be limited, and cell death and mortality may be inevitable. Antiviral therapy should be administered prior to the onset of rapid viral replication. Surfactant replacement therapy may be effective in improving respiratory function (Hentschel et al., 2020). In addition, the therapeutic effect of pharmacological chaperones that promote protein folding in the ER, is considered to involve alleviation of ER stress, and suppression of cellular dysfunction, inflammation and apoptosis, and therefore represents a promising treatment strategy in COVID-19. Pharmacological chaperone therapy may additionally “buy more time” to effectively treat patients through antiviral therapy with infected cells alive.
Several candidate pharmacological chaperones, such as tauroursodeoxycholic acid (TUDCA) and 4-phenyl butyric acid (PBA), may offer promising therapeutic avenues in this context (Chen et al., 2016). TUDCA has been most well studied, preclinically and clinically, to date (Kusaczuk, 2019). TUDCA is a taurine conjugate of ursodeoxycholic acid (UDCA), which promotes bile acid secretion and exerts a hepatocyte-protective effect. Clinical use of UDCA for the treatment of primary biliary cholangitis has been approved by the U.S. Food and Drug Administration. Human bile acids contain UDCA; TUDCA/UDCA is also expected to be clinically applicable in the treatment of various diseases as a pharmacological chaperone that promotes protein folding in the ER (Vang et al., 2014). TUDCA is water-soluble, and can be administered orally or intravenously. It suppresses the dissociation of BiP from PERK, and inhibits the phosphorylation of eIF2α and induction of ATF4-CHOP. As a result, the expression of anti-apoptotic BCL-2 expression is increased, while that of pro-apoptotic Bax is decreased, and caspase-3 activation is suppressed (Rodrigues et al., 1999). The activity of pharmacological chaperones and the anti-apoptotic effect of TUDCA against acute conditions such as ischemia, hypoxia, and pressure overload on the cardiovascular system have been demonstrated in various preclinical studies using cultured cells and mouse models (Kusaczuk, 2019). Chronic ER stress due to obesity induces adipose inflammation and suppresses signaling from the insulin receptor to insulin receptor substrate-1 (IRS-1), leading to insulin resistance. TUDCA ameliorates these signaling abnormalities (Ozcan et al., 2006), and reduces adipose tissue inflammation (Chen et al., 2016). In neurodegenerative disease models such as prion disease and ALS, ER stress due to aggregated proteins in neuronal cells is relieved and neuronal cell death is reduced (Kusaczuk, 2019). TUDCA and PBA relieve influenza A virus-induced inflammation, and suppress the production of IL-6 in lung epithelial cells (Hrincius et al., 2015).
Several clinical trials of TUDCA for conformational diseases induced by ER stress have been reported (Table 1). ALS is a neurodegenerative disease in which unstable proteins, such as mutant Cu/Zn superoxide dismutase 1 (SOD1), aggregate in motor neurons, inducing cellular dysfunction and cell death, and resulting in motor disability. In one trial, TUDCA was orally administered to 14 patients at 2,000 mg daily for 54 weeks; TUDCA-treated patients had significantly suppressed progression of ALS symptoms compared with 15 placebo controls. Mild diarrhea occurred in two individuals in each group, but no other side effects were observed. During the study period, three subjects in the placebo control group and one subject in the TUDCA-treated group died (Elia et al., 2016). Obesity causes ER stress, inducing insulin resistance. Ten obese patients, whose BMI was above 35, were treated with 1,750 mg of TUDCA daily for 4 weeks in another clinical trial. In TUDCA-treated patients, the insulin sensitivity in liver and muscle was significantly increased compared with that before treatment; however, no significant change was observed in the 10 patients who received a placebo control. In muscle cells of TUDCA-treated patients, phosphorylation of IRS-1 and Akt (protein kinase B) was enhanced, and insulin receptor signaling was improved. No side effects were reported (Kars et al., 2010).
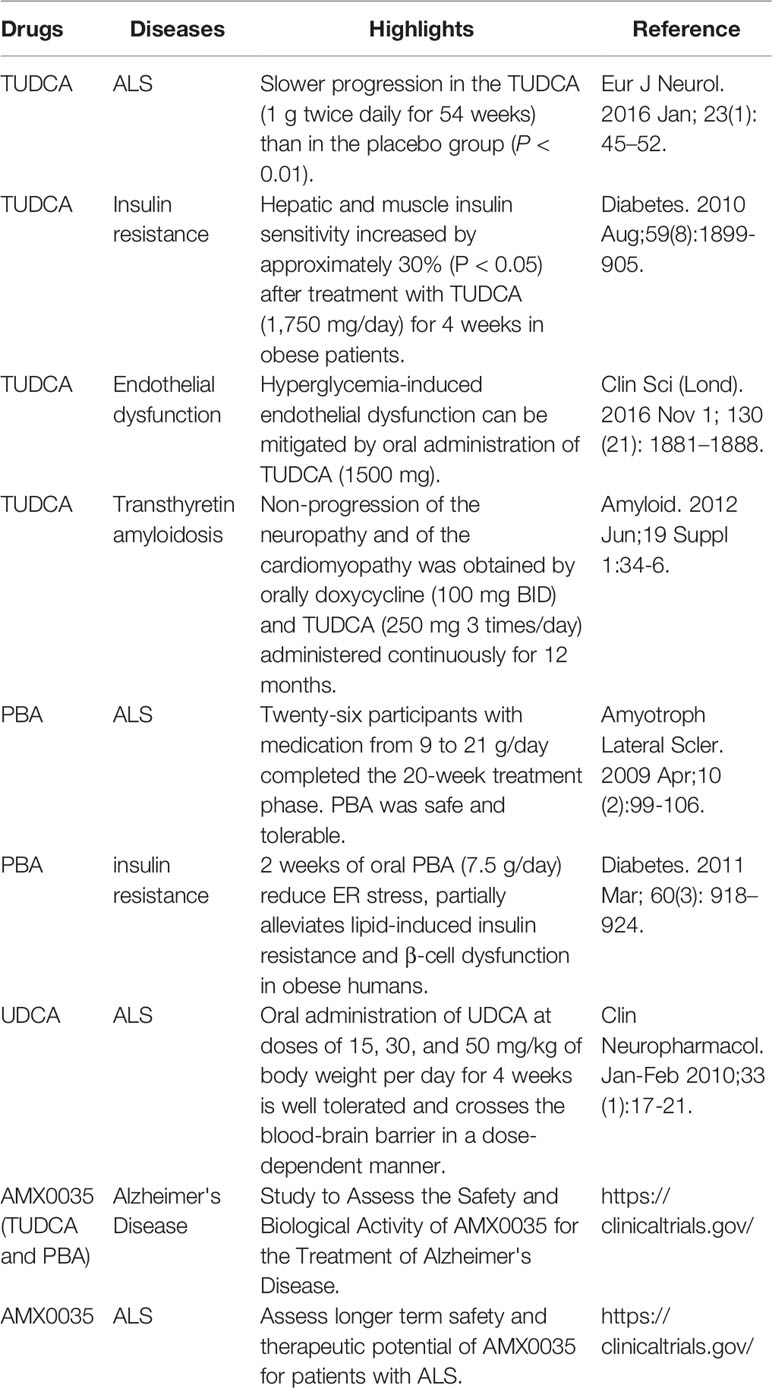
Table 1 Overview of clinical trials using pharmacological chaperones.
Orally administered TUDCA is mostly metabolized in the liver because of entero-hepatic circulation (Kusaczuk, 2019). This may limit its distribution to the systemic circulation in sufficient amounts; therefore, intravenous or subcutaneous administration may be more effective systemically.
Conclusion
Pharmacological chaperones such as TUDCA are considered to promote proteostasis in infected cells, maintain proper folding of functional proteins, and suppress the progression of inflammation and apoptosis. This may result in the survival of SARS-CoV-2-infected cells, and in turn, the virus. Therefore, by the simultaneous administration of antiviral therapy and pharmacological chaperone therapy in combination, the two modalities may constitute a promising therapeutic strategy for COVID-19.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was funded by Grants-in-Aid from the Japan Society for the Promotion of Science (KAKENHI) to TA (17K11114).
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is the product of a discussion with Dr. Teruhiko Ishikawa. I would like to thank Dr. Hisayo Jin, Dr. Hiroshi Kokubun, and Ms. Kayoko Yamaguchi for their contributions to our research. The authors hope that a solution to the global COVID-19 public health crisis will emerge soon.
References
Arons, M. M., Hatfield, K. M., Reddy, S. C., Kimball, A., James, A., Jacobs, J. R., et al. (2020). Presymptomatic SARS-CoV-2 Infections and Transmission in a Skilled Nursing Facility. N. Engl. J. Med. 382 (22), 2081–2090. doi: 10.1056/NEJMoa2008457
Back, S. H., Kaufman, R. J. (2012). Endoplasmic reticulum stress and type 2 diabetes. Annu. Rev. Biochem. 81, 767–793. doi: 10.1146/annurev-biochem-072909-095555
Bahar, E., Kim, H., Yoon, H. (2016). ER Stress-Mediated Signaling: Action Potential and Ca(2+) as Key Players. Int. J. Mol. Sci. 17 (9), 1558. doi: 10.3390/ijms17091558
Beers, M. F., Mulugeta, S. (2005). Surfactant protein C biosynthesis and its emerging role in conformational lung disease. Annu. Rev. Physiol. 67, 663–696. doi: 10.1146/annurev.physiol.67.040403.101937
Brown, M. K., Naidoo, N. (2012). The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol. 3, 263. doi: 10.3389/fphys.2012.00263
Carrell, R. W., Lomas, D. A. (1997). Conformational disease. Lancet 350 (9071), 134–138. doi: 10.1016/S0140-6736(97)02073-4
Chan, C. P., Siu, K. L., Chin, K. T., Yuen, K. Y., Zheng, B., Jin, D. Y. (2006). Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 80 (18), 9279–9287. doi: 10.1128/JVI.00659-06
Chen, Y., Wu, Z., Zhao, S., Xiang, R. (2016). Chemical chaperones reduce ER stress and adipose tissue inflammation in high fat diet-induced mouse model of obesity. Sci. Rep. 6, 27486. doi: 10.1038/srep27486
Cheng, S. H., Gregory, R. J., Marshall, J., Paul, S., Souza, D. W., White, G. A., et al. (1990). Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63 (4), 827–834. doi: 10.1016/0092-8674(90)90148-8
Chu, H., Chan, C. M., Zhang, X., Wang, Y., Yuan, S., Zhou, J., et al. (2018). Middle East respiratory syndrome coronavirus and bat coronavirus HKU9 both can utilize GRP78 for attachment onto host cells. J. Biol. Chem. 293 (30), 11709–11726. doi: 10.1074/jbc.RA118.001897
Dolfe, L., Winblad, B., Johansson, J., Presto, J. (2016). BRICHOS binds to a designed amyloid-forming beta-protein and reduces proteasomal inhibition and aggresome formation. Biochem. J. 473 (2), 167–178. doi: 10.1042/BJ20150920
Elia, A. E., Lalli, S., Monsurro, M. R., Sagnelli, A., Taiello, A. C., Reggiori, B., et al. (2016). Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 23 (1), 45–52. doi: 10.1111/ene.12664
Ferreira, S. T., Klein, W. L. (2011). The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer's disease. Neurobiol. Learn. Mem. 96 (4), 529–543. doi: 10.1016/j.nlm.2011.08.003
Fukushi, M., Yoshinaka, Y., Matsuoka, Y., Hatakeyama, S., Ishizaka, Y., Kirikae, T., et al. (2012). Monitoring of S protein maturation in the endoplasmic reticulum by calnexin is important for the infectivity of severe acute respiratory syndrome coronavirus. J. Virol. 86 (21), 11745–11753. doi: 10.1128/JVI.01250-12
Fung, T. S., Liu, D. X. (2019a). The ER stress sensor IRE1 and MAP kinase ERK modulate autophagy induction in cells infected with coronavirus infectious bronchitis virus. Virology 533, 34–44. doi: 10.1016/j.virol.2019.05.002
Fung, T. S., Liu, D. X. (2019b). Human Coronavirus: Host-Pathogen Interaction. Annu. Rev. Microbiol. 73, 529–557. doi: 10.1146/annurev-micro-020518-115759
Gibb, W. R., Lees, A. J. (1988). The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. J. Neurol. Neurosurg. Psychiatry 51 (6), 745–752. doi: 10.1136/jnnp.51.6.745
Grein, J., Ohmagari, N., Shin, D., Diaz, G., Asperges, E., Castagna, A., et al. (2020). Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 382 (24), 2327–2336. doi: 10.1056/NEJMoa2007016
Guan, W. J., Ni, Z. Y., Hu, Y., Liang, W. H., Ou, C. Q., He, J. X., et al. (2020). Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 382 (18), 1708–1720. doi: 10.1056/NEJMoa2002032
Hamada, H., Suzuki, M., Yuasa, S., Mimura, N., Shinozuka, N., Takada, Y., et al. (2004). Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol. Cell Biol. 24 (18), 8007–8017. doi: 10.1128/MCB.24.18.8007-8017.2004
Hamming, I., Timens, W., Bulthuis, M. L., Lely, A. T., Navis, G., van Goor, H. (2004). Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 203 (2), 631–637. doi: 10.1002/path.1570
Hasnain, S. Z., Lourie, R., Das, I., Chen, A. C., McGuckin, M. A. (2012). The interplay between endoplasmic reticulum stress and inflammation. Immunol. Cell Biol. 90 (3), 260–270. doi: 10.1038/icb.2011.112
Hasty, A. H., Harrison, D. G. (2012). Endoplasmic reticulum stress and hypertension - a new paradigm? J. Clin. Invest. 122 (11), 3859–3861. doi: 10.1172/JCI65173
He, L., Ding, Y., Zhang, Q., Che, X., He, Y., Shen, H., et al. (2006). Expression of elevated levels of pro-inflammatory cytokines in SARS-CoV-infected ACE2+ cells in SARS patients: relation to the acute lung injury and pathogenesis of SARS. J. Pathol. 210 (3), 288–297. doi: 10.1002/path.2067
Hendershot, L. M. (2004). The ER function BiP is a master regulator of ER function. Mt. Sinai J. Med. 71 (5), 289–297.
Hentschel, R., Bohlin, K., van Kaam, A., Fuchs, H., Danhaive, O. (2020). Surfactant replacement therapy: from biological basis to current clinical practice. Pediatr. Res. doi: 10.1038/s41390-020-0750-8
Hetz, C., Papa, F. R. (2018). The Unfolded Protein Response and Cell Fate Control. Mol. Cell 69 (2), 169–181. doi: 10.1016/j.molcel.2017.06.017
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Kruger, N., Herrler, T., Erichsen, S., et al. (2020). SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 181 (2), 271–280 e278. doi: 10.1016/j.cell.2020.02.052
Hrincius, E. R., Liedmann, S., Finkelstein, D., Vogel, P., Gansebom, S., Samarasinghe, A. E., et al. (2015). Acute Lung Injury Results from Innate Sensing of Viruses by an ER Stress Pathway. Cell Rep. 11 (10), 1591–1603. doi: 10.1016/j.celrep.2015.05.012
Huang, M., Xu, A., Wu, X., Zhang, Y., Guo, Y., Guo, F., et al. (2016). Japanese encephalitis virus induces apoptosis by the IRE1/JNK pathway of ER stress response in BHK-21 cells. Arch. Virol. 161 (3), 699–703. doi: 10.1007/s00705-015-2715-5
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395 (10223), 497–506. doi: 10.1016/S0140-6736(20)30183-5
Hughes, D., Mallucci, G. R. (2019). The unfolded protein response in neurodegenerative disorders - therapeutic modulation of the PERK pathway. FEBS J. 286 (2), 342–355. doi: 10.1111/febs.14422
Ibrahim, I. M., Abdelmalek, D. H., Elshahat, M. E., Elfiky, A. A. (2020). COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 80 (5), 554–562. doi: 10.1016/j.jinf.2020.02.026
Jin, H., Mimura, N., Kashio, M., Koseki, H., Aoe, T. (2014). Late-onset of spinal neurodegeneration in knock-in mice expressing a mutant BiP. PloS One 9 (11), e112837. doi: 10.1371/journal.pone.0112837
Jin, H., Komita, M., Aoe, T. (2017). The Role of BiP Retrieval by the KDEL Receptor in the Early Secretory Pathway and its Effect on Protein Quality Control and Neurodegeneration. Front. Mol. Neurosci. 10, 222. doi: 10.3389/fnmol.2017.00222
Jin, H., Komita, M., Aoe, T. (2018). Decreased Protein Quality Control Promotes the Cognitive Dysfunction Associated With Aging and Environmental Insults. Front. Neurosci. 12, 753. doi: 10.3389/fnins.2018.00753
Karatas, E., Bouchecareilh, M. (2020). Alpha 1-Antitrypsin Deficiency: A Disorder of Proteostasis-Mediated Protein Folding and Trafficking Pathways. Int. J. Mol. Sci. 21 (4), 1493. doi: 10.3390/ijms21041493
Kars, M., Yang, L., Gregor, M. F., Mohammed, B. S., Pietka, T. A., Finck, B. N., et al. (2010). Tauroursodeoxycholic Acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 59 (8), 1899–1905. doi: 10.2337/db10-0308
Kimura, K., Jin, H., Ogawa, M., Aoe, T. (2008). Dysfunction of the ER chaperone BiP accelerates the renal tubular injury. Biochem. Biophys. Res. Commun. 366 (4), 1048–1053. doi: 10.1016/j.bbrc.2007.12.098
Klymenko, O., Huehn, M., Wilhelm, J., Wasnick, R., Shalashova, I., Ruppert, C., et al. (2019). Regulation and role of the ER stress transcription factor CHOP in alveolar epithelial type-II cells. J. Mol. Med. (Berl) 97 (7), 973–990. doi: 10.1007/s00109-019-01787-9
Kokubun, H., Jin, H., Aoe, T. (2019). Pathogenic Effects of Impaired Retrieval between the Endoplasmic Reticulum and Golgi Complex. Int. J. Mol. Sci. 20 (22), 5614. doi: 10.3390/ijms20225614
Kopito, R. R., Ron, D. (2000). Conformational disease. Nat. Cell Biol. 2 (11), E207–E209. doi: 10.1038/35041139
Kusaczuk, M. (2019). Tauroursodeoxycholate-Bile Acid with Chaperoning Activity: Molecular and Cellular Effects and Therapeutic Perspectives. Cells 8 (12), 1471. doi: 10.3390/cells8121471
Labbadia, J., Morimoto, R. I. (2015). The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 84, 435–464. doi: 10.1146/annurev-biochem-060614-033955
Lara-Reyna, S., Scambler, T., Holbrook, J., Wong, C., Jarosz-Griffiths, H. H., Martinon, F., et al. (2019). Metabolic Reprograming of Cystic Fibrosis Macrophages via the IRE1alpha Arm of the Unfolded Protein Response Results in Exacerbated Inflammation. Front. Immunol. 10, 1789. doi: 10.3389/fimmu.2019.01789
Lee, K., Tirasophon, W., Shen, X., Michalak, M., Prywes, R., Okada, T., et al. (2002). IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 16 (4), 452–466. doi: 10.1101/gad.964702
Li, G., He, X., Zhang, L., Ran, Q., Wang, J., Xiong, A., et al. (2020a). Assessing ACE2 expression patterns in lung tissues in the pathogenesis of COVID-19. J. Autoimmun. 102463. doi: 10.1016/j.jaut.2020.102463
Li, S., Jiang, L., Li, X., Lin, F., Wang, Y., Li, B., et al. (2020b). Clinical and pathological investigation of severe COVID-19 patients. JCI Insight. 5 (12), e138070doi: 10.1172/jci.insight.138070
Liu, J. Q., Zhang, L., Yao, J., Yao, S., Yuan, T. (2018). AMPK alleviates endoplasmic reticulum stress by inducing the ER-chaperone ORP150 via FOXO1 to protect human bronchial cells from apoptosis. Biochem. Biophys. Res. Commun. 497 (2), 564–570. doi: 10.1016/j.bbrc.2018.02.095
Lu, M., Echeverri, F., Moyer, B. D. (2003). Endoplasmic reticulum retention, degradation, and aggregation of olfactory G-protein coupled receptors. Traffic 4 (6), 416–433. doi: 10.1034/j.1600-0854.2003.00097.x
Lu, M., Staszewski, L., Echeverri, F., Xu, H., Moyer, B. D. (2004). Endoplasmic reticulum degradation impedes olfactory G-protein coupled receptor functional expression. BMC Cell Biol. 5, 34. doi: 10.1186/1471-2121-5-34
Lythgoe, M. P., Middleton, P. (2020). Ongoing Clinical Trials for the Management of the COVID-19 Pandemic. Trends Pharmacol. Sci. 41 (6), 363–382. doi: 10.1016/j.tips.2020.03.006
Mao, L., Jin, H., Wang, M., Hu, Y., Chen, S., He, Q., et al. (2020). Neurologic Manifestations of Hospitalized Patients With Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 77 (6), 683–690. doi: 10.1001/jamaneurol.2020.1127
Masters, P. S. (2006). The molecular biology of coronaviruses. Adv. Virus Res. 66, 193–292. doi: 10.1016/S0065-3527(06)66005-3
Mimura, N., Hamada, H., Kashio, M., Jin, H., Toyama, Y., Kimura, K., et al. (2007). Aberrant quality control in the endoplasmic reticulum impairs the biosynthesis of pulmonary surfactant in mice expressing mutant BiP. Cell Death Differ. 14 (8), 1475–1485. doi: 10.1038/sj.cdd.4402151
Minakshi, R., Padhan, K., Rani, M., Khan, N., Ahmad, F., Jameel, S. (2009). The SARS Coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PloS One 4 (12), e8342. doi: 10.1371/journal.pone.0008342
Nuemket, N., Yasui, N., Kusakabe, Y., Nomura, Y., Atsumi, N., Akiyama, S., et al. (2017). Structural basis for perception of diverse chemical substances by T1r taste receptors. Nat. Commun. 8, 15530. doi: 10.1038/ncomms15530
Ochoa, C. D., Wu, R. F., Terada, L. S. (2018). ROS signaling and ER stress in cardiovascular disease. Mol. Aspects Med. 63, 18–29. doi: 10.1016/j.mam.2018.03.002
Ottestad, W., Seim, M., Maehlen, J. O. (2020). COVID-19 with silent hypoxemia. Tidsskr Nor. Laegeforen 140 (7). doi: 10.4045/tidsskr.20.0299
Oyadomari, S., Mori, M. (2004). Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11 (4), 381–389. doi: 10.1038/sj.cdd.4401373
Ozcan, U., Yilmaz, E., Ozcan, L., Furuhashi, M., Vaillancourt, E., Smith, R. O., et al. (2006). Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313 (5790), 1137–1140. doi: 10.1126/science.1128294
Park, K. W., Eun Kim, G., Morales, R., Moda, F., Moreno-Gonzalez, I., Concha-Marambio, L., et al. (2017). The Endoplasmic Reticulum Chaperone GRP78/BiP Modulates Prion Propagation in vitro and in vivo. Sci. Rep. 7, 44723. doi: 10.1038/srep44723
Peca, D., Boldrini, R., Johannson, J., Shieh, J. T., Citti, A., Petrini, S., et al. (2015). Clinical and ultrastructural spectrum of diffuse lung disease associated with surfactant protein C mutations. Eur. J. Hum. Genet. 23 (8), 1033–1041. doi: 10.1038/ejhg.2015.45
Peotter, J., Kasberg, W., Pustova, I., Audhya, A. (2019). COPII-mediated trafficking at the ER/ERGIC interface. Traffic 20 (7), 491–503. doi: 10.1111/tra.12654
Rapoport, T. A., Li, L., Park, E. (2017). Structural and Mechanistic Insights into Protein Translocation. Annu. Rev. Cell Dev. Biol. 33, 369–390. doi: 10.1146/annurev-cellbio-100616-060439
Raykhel, I., Alanen, H., Salo, K., Jurvansuu, J., Nguyen, V. D., Latva-Ranta, M., et al. (2007). A molecular specificity code for the three mammalian KDEL receptors. J. Cell Biol. 179 (6), 1193–1204. doi: 10.1083/jcb.200705180
Ribeiro, C. M., Boucher, R. C. (2010). Role of endoplasmic reticulum stress in cystic fibrosis-related airway inflammatory responses. Proc. Am. Thorac. Soc. 7 (6), 387–394. doi: 10.1513/pats.201001-017AW
Rodrigues, C. M., Ma, X., Linehan-Stieers, C., Fan, G., Kren, B. T., Steer, C. J. (1999). Ursodeoxycholic acid prevents cytochrome c release in apoptosis by inhibiting mitochondrial membrane depolarization and channel formation. Cell Death Differ. 6 (9), 842–854. doi: 10.1038/sj.cdd.4400560
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362 (6415), 59–62. doi: 10.1038/362059a0
Rozpedek, W., Pytel, D., Mucha, B., Leszczynska, H., Diehl, J. A., Majsterek, I. (2016). The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 16 (6), 533–544. doi: 10.2174/1566524016666160523143937
Russell, C. D., Millar, J. E., Baillie, J. K. (2020). Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet 395 (10223), 473–475. doi: 10.1016/S0140-6736(20)30317-2
Shan, B., Wang, X., Wu, Y., Xu, C., Xia, Z., Dai, J., et al. (2017). The metabolic ER stress sensor IRE1alpha suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat. Immunol. 18 (5), 519–529. doi: 10.1038/ni.3709
Shen, J., Chen, X., Hendershot, L., Prywes, R. (2002). ER Stress Regulation of ATF6 Localization by Dissociation of BiP/GRP78 Binding and Unmasking of Golgi Localization Signals. Dev. Cell 3 (1), 99–111. doi: 10.1016/S1534-5807(02)00203-4
Shi, C. S., Nabar, N. R., Huang, N. N., Kehrl, J. H. (2019). SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discovery 5, 101. doi: 10.1038/s41420-019-0181-7
Solinas, G., Vilcu, C., Neels, J. G., Bandyopadhyay, G. K., Luo, J. L., Naugler, W., et al. (2007). JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. 6 (5), 386–397. doi: 10.1016/j.cmet.2007.09.011
Sozen, E., Yazgan, B., Tok, O. E., Demirel, T., Ercan, F., Proto, J. D., et al. (2020). Cholesterol induced autophagy via IRE1/JNK pathway promotes autophagic cell death in heart tissue. Metabolism 106, 154205. doi: 10.1016/j.metabol.2020.154205
Stockman, L. J., Bellamy, R., Garner, P. (2006). SARS: systematic review of treatment effects. PloS Med. 3 (9), e343. doi: 10.1371/journal.pmed.0030343
Tajiri, S., Oyadomari, S., Yano, S., Morioka, M., Gotoh, T., Hamada, J. I., et al. (2004). Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 11 (4), 403–415. doi: 10.1038/sj.cdd.4401365
Tsai, Y. L., Ha, D. P., Zhao, H., Carlos, A. J., Wei, S., Pun, T. K., et al. (2018). Endoplasmic reticulum stress activates SRC, relocating chaperones to the cell surface where GRP78/CD109 blocks TGF-beta signaling. Proc. Natl. Acad. Sci. U. S. A. 115 (18), E4245–E4254. doi: 10.1073/pnas.1714866115
Tseng, C. C., Zhang, P., Lee, A. S. (2019). The COOH-Terminal Proline-Rich Region of GRP78 Is a Key Regulator of Its Cell Surface Expression and Viability of Tamoxifen-Resistant Breast Cancer Cells. Neoplasia 21 (8), 837–848. doi: 10.1016/j.neo.2019.05.008
Uhal, B. D., Nguyen, H., Dang, M., Gopallawa, I., Jiang, J., Dang, V., et al. (2013). Abrogation of ER stress-induced apoptosis of alveolar epithelial cells by angiotensin 1-7. Am. J. Physiol. Lung Cell Mol. Physiol. 305 (1), L33–L41. doi: 10.1152/ajplung.00001.2013
Urano, F., Wang, X., Bertolotti, A., Zhang, Y., Chung, P., Harding, H. P., et al. (2000). Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287 (5453), 664–666. doi: 10.1126/science.287.5453.664
Vang, S., Longley, K., Steer, C. J., Low, W. C. (2014). The Unexpected Uses of Urso- and Tauroursodeoxycholic Acid in the Treatment of Non-liver Diseases. Glob. Adv. Health Med. 3 (3), 58–69. doi: 10.7453/gahmj.2014.017
Varga, Z., Flammer, A. J., Steiger, P., Haberecker, M., Andermatt, R., Zinkernagel, A. S., et al. (2020). Endothelial cell infection and endotheliitis in COVID-19. Lancet. 395 (10234), 1417–1418. doi: 10.1016/S0140-6736(20)30937-5
Walls, A. C., Park, Y. J., Tortorici, M. A., Wall, A., McGuire, A. T., Veesler, D. (2020). Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 181 (2), 281–292 e286. doi: 10.1016/j.cell.2020.02.058
Wang, M., Kaufman, R. J. (2016). Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529 (7586), 326–335. doi: 10.1038/nature17041
Wong, R. S., Wu, A., To, K. F., Lee, N., Lam, C. W., Wong, C. K., et al. (2003). Haematological manifestations in patients with severe acute respiratory syndrome: retrospective analysis. BMJ 326 (7403), 1358–1362. doi: 10.1136/bmj.326.7403.1358
Wu, F., Zhao, S., Yu, B., Chen, Y. M., Wang, W., Song, Z. G., et al. (2020). A new coronavirus associated with human respiratory disease in China. Nature 579 (7798), 265–269. doi: 10.1038/s41586-020-2008-3
Xydakis, M. S., Dehgani-Mobaraki, P., Holbrook, E. H., Geisthoff, U. W., Bauer, C., Hautefort, C., et al. (2020). Smell and taste dysfunction in patients with COVID-19. Lancet Infect. Dis. doi: 10.1016/S1473-3099(20)30293-0
Yamamoto, K., Fujii, R., Toyofuku, Y., Saito, T., Koseki, H., Hsu, V. W., et al. (2001). The KDEL receptor mediates a retrieval mechanism that contributes to quality control at the endoplasmic reticulum. EMBO J. 20 (12), 3082–3091. doi: 10.1093/emboj/20.12.3082
Yang, X., Yu, Y., Xu, J., Shu, H., Xia, J., Liu, H., et al. (2020). Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir. Med. 8 (5), 475–481. doi: 10.1016/S2213-2600(20)30079-5
Ye, Z., Wong, C. K., Li, P., Xie, Y. (2008). A SARS-CoV protein, ORF-6, induces caspase-3 mediated, ER stress and JNK-dependent apoptosis. Biochim. Biophys. Acta 1780 (12), 1383–1387. doi: 10.1016/j.bbagen.2008.07.009
Young, C. N. (2017). Endoplasmic reticulum stress in the pathogenesis of hypertension. Exp. Physiol. 102 (8), 869–884. doi: 10.1113/EP086274
Zhang, Q. L., Ding, Y. Q., He, L., Wang, W., Zhang, J. H., Wang, H. J., et al. (2003). [Detection of cell apoptosis in the pathological tissues of patients with SARS and its significance]. Di Yi Jun Yi Da Xue Xue Bao 23 (8), 770–773.
Zhang, M., Gao, Y., Zhao, W., Yu, G., Jin, F. (2018). ACE-2/ANG1-7 ameliorates ER stress-induced apoptosis in seawater aspiration-induced acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 315 (6), L1015–L1027. doi: 10.1152/ajplung.00163.2018
Zhang, R., Ouyang, H., Fu, L., Wang, S., Han, J., Huang, K., et al. (2020). CT features of SARS-CoV-2 pneumonia according to clinical presentation: a retrospective analysis of 120 consecutive patients from Wuhan city. Eur. Radiol. 30 (8), 4417–4426. doi: 10.1007/s00330-020-06854-1
Zhou, F., Yu, T., Du, R., Fan, G., Liu, Y., Liu, Z., et al. (2020a). Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395 (10229), 1054–1062. doi: 10.1016/S0140-6736(20)30566-3
Keywords: SARS-CoV-2, COVID-19, ER stress, unfolded protein response (UPR), apoptosis, proteostasis, pharmacological chaperone, BiP
Citation: Aoe T (2020) Pathological Aspects of COVID-19 as a Conformational Disease and the Use of Pharmacological Chaperones as a Potential Therapeutic Strategy. Front. Pharmacol. 11:1095. doi: 10.3389/fphar.2020.01095
Received: 05 May 2020; Accepted: 09 June 2020;
Published: 10 July 2020.
Edited by:
Paola Patrignani, University of Studies G. d'Annunzio Chieti and Pescara, ItalyReviewed by:
Stefania Tacconelli, University of Studies G. d'Annunzio Chieti and Pescara, ItalyPatrizia Ballerini, University of Studies G. d'Annunzio Chieti and Pescara, Italy
Copyright © 2020 Aoe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomohiko Aoe, dG9tb2hpa29BQG1lZC50ZWlreW8tdS5hYy5qcA==