 Xin Zhi1†
Xin Zhi1† Qian Chen2†
Qian Chen2† Shaojun Song3†Zhengrong Gu4Wenqiang Wei4Huiwen Chen5
Shaojun Song3†Zhengrong Gu4Wenqiang Wei4Huiwen Chen5 Xiao Chen5
Xiao Chen5 Weizong Weng5
Weizong Weng5 Qirong Zhou5
Qirong Zhou5 Jin Cui5*
Jin Cui5* Liehu Cao4*
Liehu Cao4*- 1Department of Orthopedics, PLA General Hospital, Beijing, China
- 2Basic Medical School, Naval Military Medical University, Shanghai, China
- 3Department of Emergency, General Hospital of Central Theather Command, Wuhan, China
- 4Department of Orthopedics, Shanghai Baoshan Luodian Hospital, Shanghai, China
- 5Department of Orthopedics Trauma, Shanghai Changhai Hospital, Naval Military Medical University, Shanghai, China
Myostatin is a crucial cytokine that is widely present in skeletal muscle and that negatively regulates the growth and development of muscle cells. Recent research has shown that myostatin might play an essential role in bone metabolism. In RAW264.7 cells and bone marrow monocytes (BMMCs), myostatin activates the expression of the II type receptor ActR II B. Here, we report that myostatin significantly promoted RANKL/M-CSF-induced osteoclastogenesis and activated NF-κB and MAPK pathways in vitro via the Ccdc50 gene. Overexpression of myostatin promoted osteoclastogenesis and osteoclastogenesis-related markers including c-Src, MMP9, CTR, CK, and NFATc1. Specifically, myostatin increased the phosphorylation of Smad2, which led to the activation of NF-κB and MAPK pathways to activate osteoclastogenesis. Ccdc50 was identified as a gene whose expression was highly decreased in osteoclastogenesis upon myostatin treatment, and it could inhibit the function of myostatin in osteoclastogenesis by blocking NF-κB and MAPKs pathways. Our study indicates that myostatin is a promising candidate target for inhibiting RANKL-mediated osteoclastogenesis and might participate in therapy for osteoporosis, and that the Ccdc50 gene plays a significant role in the regulatory process.
Introduction
Normal bone tissue is in a constant dynamic equilibrium state of bone formation and bone resorption. Bone formation is mediated by osteoblasts, while bone resorption is mainly mediated by osteoclasts (Kular et al., 2012). Differentiating from the monocyte-macrophage lineage, osteoclasts have essential physiological functions in bone resorption (Boyce et al., 2009). Receptor activator of NF-κB ligand (RANKL) stimulates osteoclast precursors and activates RANK by combining it with the co-effector M-CSF (Mizukami et al., 2002), which can promote bone resorption in the process of normal or pathological bone remodeling.
Nuclear factor-kappa B (NF-κB) belongs to a family including p65, p52, p50, RelB, and c-Rel, and positively regulates the expression of many genes related to inflammation and other reactions (Courtois and Gilmore, 2006). Osteoclast precursor cells rapidly initiate the classical NF-κB pathway in response to RANKL (Asagiri et al., 2005). After the activation of the classical NF-κB pathway, the non-classical NF-κB pathway is activated and lasts for several hours (Boyce et al., 2015). The NF-κB pathway is activated as TRAF I is recruited, and then the nuclear factor of activated T-cells cytoplasmic 2 (NFATc2) binds to NF-κB to induce activation and expansion of nuclear factor of activated T-cells cytoplasmic 1 (NFATc1) (Takayanagi et al., 2002). NFATc1 promotes the expression of genes related to the proliferation and differentiation of osteoclasts. The induction of NFATc1/NFAT2 and the classical and non-classical NF-κB pathways are necessary for transcription and translation of osteoclastogenesis-critical genes under the induction of RANKL/RANK-mediated activation. In this way, monocyte-derived macrophages fuse and differentiate into osteoclasts with mature osteolytic function (Walsh and Choi, 2014). The mitogen-activated protein kinase (MAPK) pathway represents a series of serine-threonine kinases in eukaryotes, and functions by phosphorylating other cytoplasmic proteins and directly regulating transcription after translocating from the cytoplasm to the nucleus (Hagemann and Blank, 2001). The MAPK pathway is organized into three-tiered cascades comprising three types of molecules: MAPKs, MAPK kinases (MAPKK or MEK), and MAPKK kinases (MAPKKK or MEKK) (Lee et al., 2018). Generic MAPK signaling includes the c-Jun N-terminal kinase (JNK), extracellular signal-related kinase (ERK), and p38 (Sun et al., 2015). JNK and p38 mediate osteoclast precursor proliferation and osteoclast differentiation when induced by RANKL and M-CSF, while ERK accelerates the proliferation of osteoclast precursors via the M-CSF/c-Fms axis (Lee et al., 2018). Moreover, p38 phosphorylates its transactivation domain with the co-stimulation of c-fos both in vivo and in vitro (Tanos et al., 2005).
Myostatin, which is also called GDF-8, is a member of the TGF-β family. Previous research has shown that myostatin is synthesized by muscle cells and is a critical autocrine/paracrine inhibitor of skeletal muscle growth (Nissinen et al., 2016). The binding of myostatin and the II type receptor ActR II B occurs on the cytomembrane, then Smad2/3-mediated transcription is activated and inhibits muscle protein synthesis (Han et al., 2013). A recent study showed that bone formation is enhanced when the expression of myostatin is deficient (Hamrick, 2003; Bialek et al., 2014). RANKL-mediated osteoclastogenesis is accelerated by myostatin under transcription factor SMAD2-dependent regulation of NFATc1 (Dankbar et al., 2015). In the present study, we investigated the mechanism by which myostatin regulates the RANKL-induced downstream pathway.
Method
Reagents and Inhibitor
Myostatin was provided by Phoenix pep (San Francisco, CA, United States). RANKL was provided by Raybiotech (Guangzhou, China). The Smad2 inhibitor LY209761 was supplied by Selleck (Houston, TX, United States).
Osteoclastogenesis Assay In Vitro
RAW264.7 cells were provided by the Chinese Academy of Sciences (Shanghai, China). BMMCs were separated by flushing the bone marrow of femurs from 4-week-old C57BL/6 mice. Cells were seeded into 24-well plates at a density of 8 × 103 cells/well. Cells of the third generation were induced with RANKL (50 ng/ml) and M-CSF (30 ng/ml) with or without myostatin treatment (30 ng/ml). DMEM medium with low-glucose used to culture RAW264.7 cells and BMMCs was mixed with 10% fetal bovine serum and 1% penicillin-streptomycin as supplied by the Experiment Center of Changhai Hospital (Shanghai, China). Cells were cultured at 37°C and 5% CO2.
TRAP Staining
Osteoclasts were stained by a TRAP staining kit from Sigma-Aldrich (St. Louis, MO, United States), and the procedure was strictly according to the protocol of the manufacturer. The number and size of osteoclasts were observed under an upright metallurgical microscope. Huge fused multinucleated (greater than or equal to three) cells were considered as osteoclasts.
Immunofluorescence Staining
The localization of ActR II B in RAW264.7 cells and BMMCs was observed by immunofluorescence. Cell medium was removed and replaced with 4% paraformaldehyde to fix cell morphology. Then paraformaldehyde was washed out by Triton X-100, and the fixed cells were incubated with myostatin conjugated with fluorescein. The cellular localization of ActR II B was observed by using protocols as previously described (Wen et al., 2019).
Western Blotting
Cells were collected and lyzed for extracting protein by the M-PER mammalian protein extraction reagent supplied by Pierce (Rockford, IL, United States). The concentration of protein was evaluated by a bicinchoninic acid (BCA) protein assay kit supplied by Pierce. Equal amounts of protein were added into SDS-PAGE gels with 10 μg protein per lane. The blots were probed with a monoclonal antibody against human anti-ActR II B (1:100), anti-c-Src (1:200), anti-MMP-9 (1:200), anti-CTR (1:200), anti-CK (1:200), anti-P-Smad2 (1:200), anti-Smad2 (1:200), anti-P-p65, anti-p65 (1:300) (1:500), anti-P-p50 (1:400), anti-p50 (1:250), anti-P-ERK (1:400), anti-ERK (1:200), anti-P-JNK (1:400), anti-JNK (1:500), anti-P-p38 (1:300), anti-p38 (1:300), anti-P-c-fos (1:400), anti-c-fos (1:400), and anti-β-actin (1:1000) from Santa Cruz (TX, United States), and the secondary HRP-conjugated anti-mouse/rabbit antibody was purchased from Santa Cruz. After washing with TBST, chemiluminescence was performed to detect the bands. β-actin was added as an internal control for eliminating errors in protein quantification and loading. Proteins were transferred onto nitrocellulose membranes after electrophoresis. After transfer, primary antibodies were used for incubation, and then secondary antibodies were used for exposure and development. Finally, film exposure was performed, and images were analyzed by Image-Pro Plus software. All the experiments were detected three times, and the average was calculated.
RT-PCR
Total RNA was extracted from RAW264.7 cells to measure the expressions of ActR II B, c-Src, MMP-9, CTR, NFATc1, and Ccdc50, using Trizol reagent from Invitrogen (Carlsbad, CA, United States) according to the manufacturer’s protocol. Reverse transcription was performed on extracted RNA to synthesize cDNAs by using a SYBR1 Premix Dimmer Eraser kit (TaKaRa Biotechnology, Otsu, Japan). Realtime PCR was then executed using SYBR® Premix Ex TaqTM II (Tli RNaseH Plus) purchased from TaKaRa Biotechnology (Dalian, China) and detected by an ABI 7500 Sequencing Detection System from Applied Biosystems (Foster City, CA, United States). The primer sequences used were as follows: MMP-9: (F) GGACCCGAAGCGGACATTG; (R) CGTCGTCGAAATGGGCATCT, C-Src: (F) CAACTTCGGCACAGCAAC; (R) TCAGACACCAGCACATTCC, ActRIIB: (F) GCTCCCTCACGGATTACC; (R) CACGACACCACGGCACAT, CTR: (F) CCTGGTTGAGGTTGTGCC; (R) GCGTTGCTCGTCGGTAAA, NFATc1: (F) ACCACTCCACCCACTTCTG; (R) GCTGCCTTCCGTCTCATAG, Ccdc50: (R) AAAGAGGGTGATGAAGCA; (F) ATGGAAGCCTTTCTGTGA.
Pit-formation Assays
RAW264.7 cells and BMMCs for detecting differentiation were seeded on hydroxyapatite-coated surfaces of biomimetic synthetic bone from Corning (Bedford, MA, United States). Culture medium was changed on day 3. After incubation for 7 days, the medium was washed out with PBS and dried in the air at room temperature for 5 h. A representative region of osteoclast-resorbing pits was photographed using a light microscope (BX53) provided by Olympus (Tokyo, Japan). Further research and analysis were carried out by Image-Pro Plus software.
CHIP Assay
RAW264.7 cells treated with myostatin were subjected to chromatin immunoprecipitation assays by a CHIP kit (EpiGentek, NY, United States). In brief, cells cultured in 24-well plates were fixed by formaldehyde. Lysates digested by cell lysis buffer were collected, followed by mixing with Smad2-specific IgG. Then the mixture was incubated at room temperature overnight for 12 h. Protein Agarose was added for combining antibody-target protein-DNA complexes. The supernatant was discarded after centrifugation at 4°C for 30 min at 3,000 g, followed by washing to obtain the target protein-DNA complex. Then the crosslinked purified DNA was removed. Further analysis was conducted by qRT-PCR. Nonprobe-based analyses were performed using SYBR® green as an intercalating dye for target detection. Based on the quantification of housekeeping genes, the procedures and conditions of the genes of interest had internal controls to prevent influence by the experimental conditions. Enrichment analysis for differentially expressed genes that were up- or down-regulated was performed by Gene Ontology (GO) functional and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis.
Lentivirus Infection of RAW264.7 Cells
An siRNA sequence with a target sequence homologous to Ccdc50 was chemically synthesized to inhibit the transcription of mouse Ccdc50. The artificially synthesized oligonucleotide templates were cloned into the linear pSIH-H1-copGFP siRNA vector (System Biosciences, Palo Alto, CA, United States) to form pSIH-shRNA-Ccdc50. A negative control (NC) was originated from an invalid siRNA sequence, and the negative control expression vector pSIH-NC was constructed. DNA of Ccdc50 was obtained from total mouse RNA extracted from different tissues with treatment by Trizol (Invitrogen) followed by reverse transcription and PCR. Then, the Ccdc50 expression vector pcDH-Ccdc50 was constructed after the artificial DNA was cloned into pcDH-GFP Lentivector (CD511A-1, System Biosciences).
The plasmids (pSIH-NC, pSIH-shRNA-Ccdc50, and pcDH-Ccdc50) were transfected into 293T cells together with the packaging vector (System Biosciences) to produce lentiviruses Lv-NC, Lv-shRNA-Ccdc50, and Lv-Ccdc50. The harvested virus suspensions were concentrated by ultrafiltration and purification (Benskey and Manfredsson, 2016). RAW264.7 cell suspensions digested by trypsinization were divided into four groups and seeded on six-well plates, then cultured at 37°C and 5% CO2. The groups were 1) cells infected with Lv-NC; 2) cells infected with Lv-shRNA Ccdc50; 3) cells infected with Lv-Ccdc50; and 4) control group (not infected). After culturing for 12 h, the pre-configured lentiviral solution at MOI = 20 was added into the corresponding wells and mixed with medium. After 3 days, the cells were digested and extracted for total protein, and transfection efficiency was determined after the measurement of the amount of Ccdc50 by western blotting (Sakuma et al., 2012).
Statistical Analysis
All results were tested by a homogeneity of variance test and two samples independent t test by SPSS Statistics supplied by IBM (New York, United States). p < 0.05 was considered to be statistically significant.
Results
The Expression of ActR II B Increases During Osteoclastogenesis
To research the function of myostatin in osteoclastogenesis in vitro, we first detected the expression of ActR II B, which is a receptor of myostatin. We chose RAW264.7 and BMMCs, which can be induced to form osteoclasts (Battaglino et al., 2002; Takayanagi, 2007) in vitro (Figure 1A). Western blotting indicated that ActR II B had low expression in RAW264.7 cells and BMMCs before the induction of osteoclast differentiation (Figure 1B). Immunofluorescence staining and RT-PCR also indicated that little ActR II B was expressed in RAW264.7 cells and BMMCs (Figures 1C,D). Then, BMMCs were treated with RANKL and M-CSF, while RAW264.7 cells were treated with RANKL for induction (Bharti et al., 2004; Lee et al., 2006). ActR II B significantly increased after induction by RANKL (Figures 1E,F). Western blotting and RT-PCR showed similar results (Figures 1G–I).
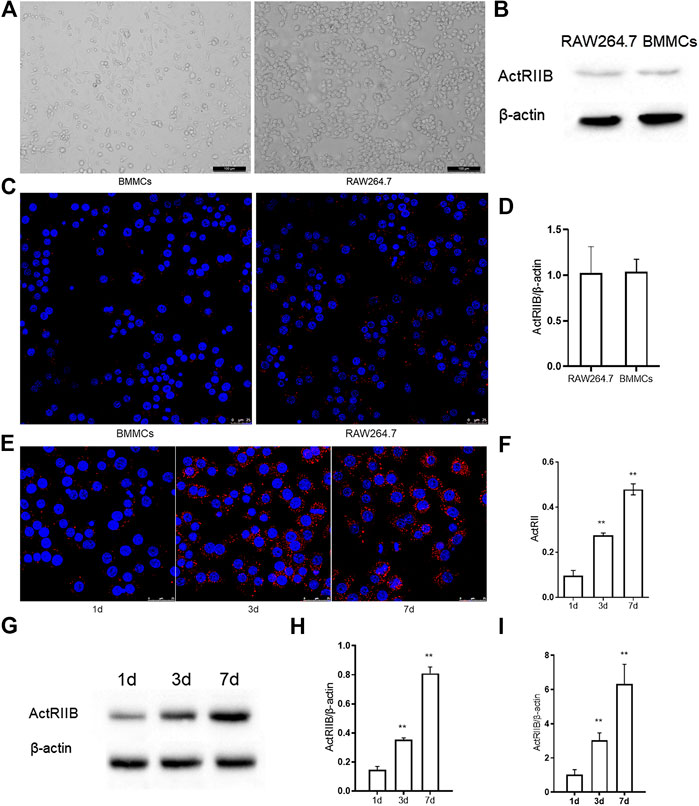
FIGURE 1. The expression of ActR II B increases during osteoclastogenesis. (A) Cells were visualized by an inverted optical microscope. (B) ActR II B protein was extracted for analysis by western blot. (C) Immunofluorescence staining of ActR II B in RAW264.7 cells and BMMCs. (D) RT-PCR was used to determine the expression of ActR II B. (E,F) Immunofluorescence staining of ActR II B in RAW264.7 cells and BMMCs on days 1, 3, and 7. (G,H) Lyzed protein from induced RAW264.7 cells was analyzed by western blotting. (I) Total RNA was analyzed by RT-PCR to examine the expression of mRNA. **p < 0.01.
Myostatin Promotes Osteoclastogenesis and Osteoclast Function
During the differentiation of osteoclasts, the expression of ActR II B was upregulated. We next examined whether myostatin promoted osteoclast precursors to differentiate into mature osteoclasts. We found that the size of osteoclasts was larger in the presence of myostatin than in its absence in the process of RANKL-induced osteoclastogenesis (Figure 2A). To further verify the mechanism by which myostatin influences the function of osteoclasts, we performed bone resorption experiments. When BMMCs and RAW264.7 were induced by RANKL, the surface of the biomimetic synthetic bone was resorbed, and the resorbed area was further expanded with the addition of myostatin (Figure 2B). Meanwhile, treatment with myostatin significantly increased the expression of osteoclast-related markers, including C-Src, MMP-9, CTR, and cathepsin K (Figure 2C; Supplementary Figure S1). We next assessed the effect of myostatin on the expression of NFATc1. The results showed an increase of NFATc1 transcription via RANKL induction, and this was further promoted by myostatin treatment (Figure 2D).
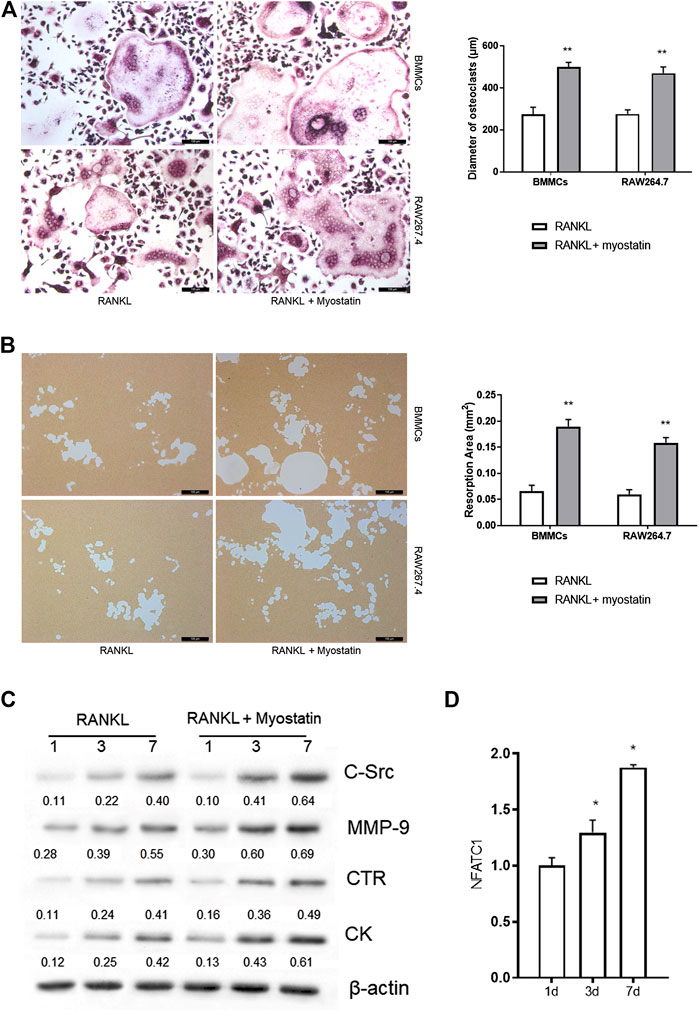
FIGURE 2. Myostatin promotes osteoclastogenesis and osteoclast function. (A) TRAP staining of osteoclasts (Scalebar, 100 μm). (B) Representative images of resorption pits localized on biomimetic synthetic bone; the resorptive area was analyzed in the histogram (Scale bar, 100 μm). (C) Expressions of C-Src, MMP-9, CTR, cathepsin K, and β-actin as analyzed by western blotting. (D) The total RNA of NFATc1 was analyzed by RT-PCR. **p < 0.01.
Myostatin Promotes the Differentiation of Osteoclasts by Activating Smad2 and Activates the NF-κB and MAPK Pathways
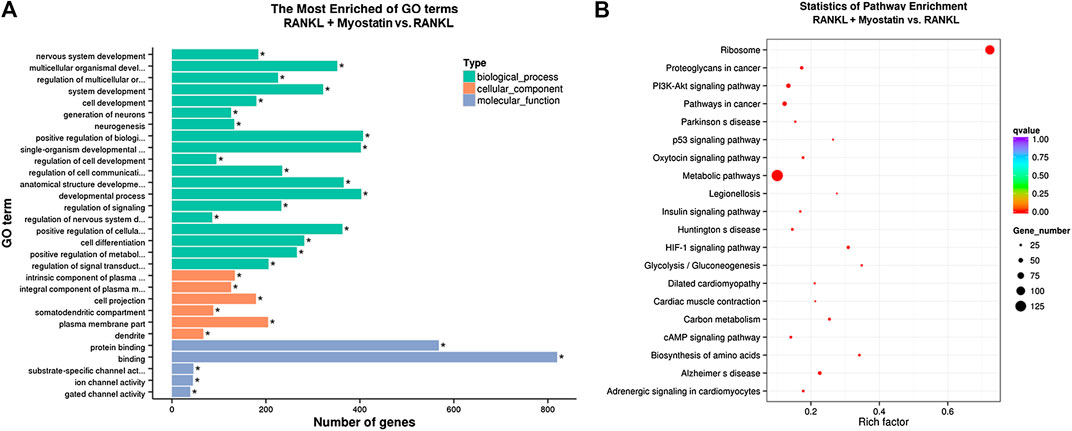
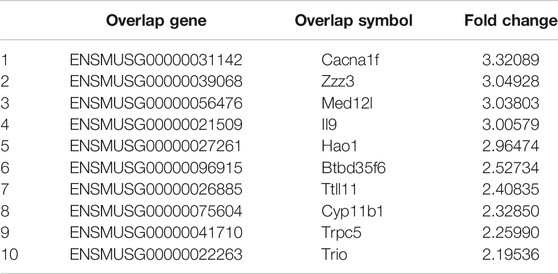
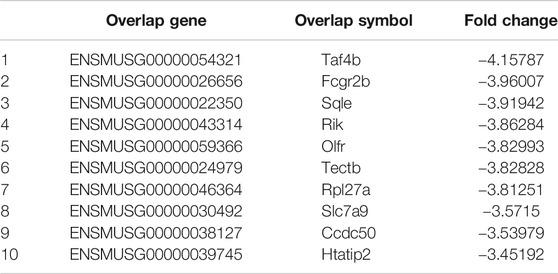
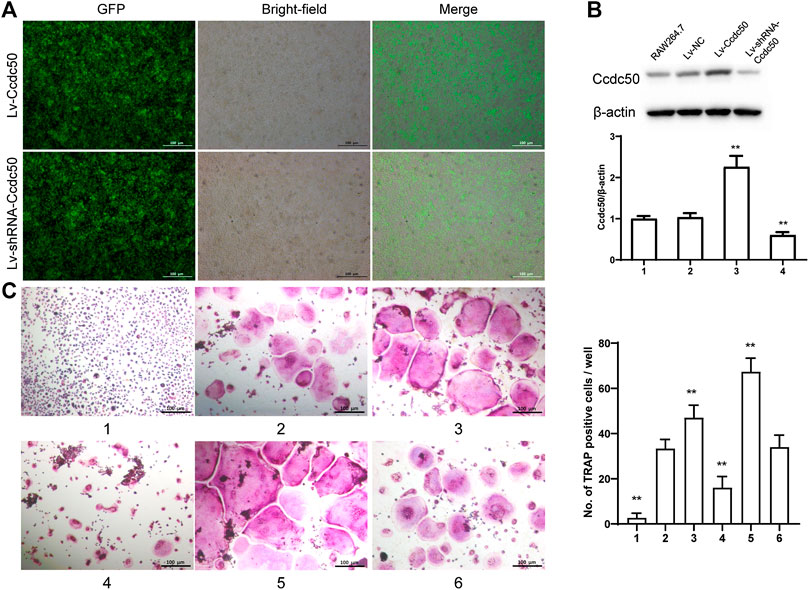
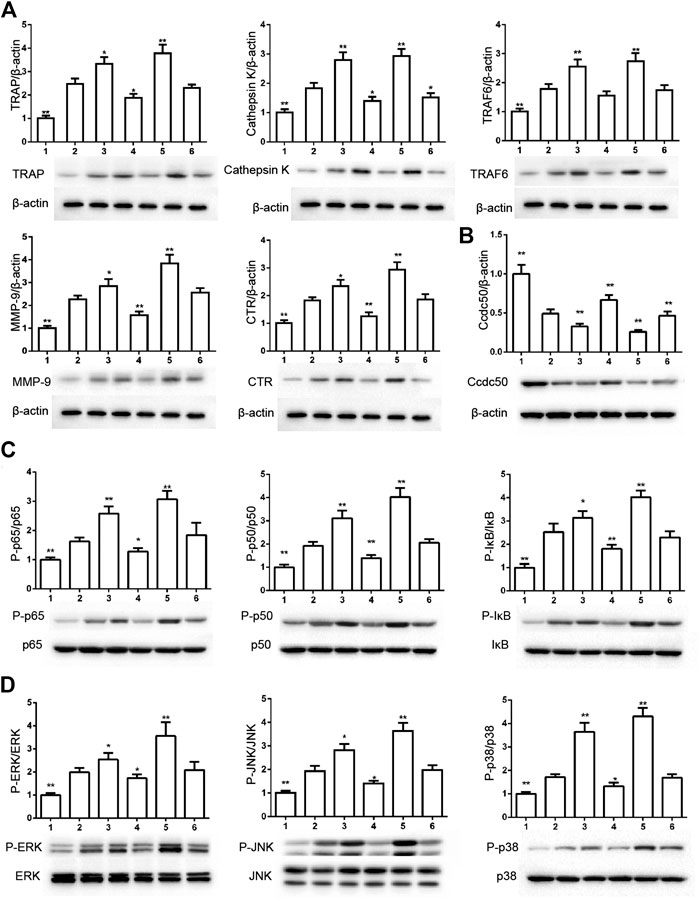
The phosphorylation of Smad2 increased in RAW264.7 cells with myostatin treatment (Figure 3A). In addition, treatment with myostatin significantly elevated the expressions of CTR, C-Src, MMP-9, and NFATc1, which are related to osteoclastogenesis (Figure 3B). NF-κB and MAPK pathways are crucial for osteoclast differentiation. The phosphorylation of p65 and p50, which is related to the activation of NF-κB, was enhanced by myostatin within 60 min during the differentiation process of RAW264.7 cells. In addition, the phosphorylation of JNK, ERK, p38, and c-fos were enhanced by myostatin, indicating that the MAPK pathway was activated by myostatin (Figure 3C; Supplementary Figure S2). However, the phosphorylation of p65, p50, JNK, ERK, p38, and c-fos were suppressed with the addition of LY209761, which is an inhibitor of Smad2 (Figure 3D; Supplementary Figure S3), indicating that NF-κB and MAPK pathways were regulated by Smad2. FIGURE 3. Myostatin promotes the differentiation of osteoclasts by activating Smad2 and activates NF-κB and MAPK pathways. (A) P-Smad2 and Smad2 were detected by western blot. (B) Total RNA from RAW264.7 cells treated with or without myostatin for 24 h was analyzed by RT-PCR to detect the expressions of CTR, C-SRC, MMP-9, and NFATc1. (C) The expressions of P-p65/p65, P-p50/p50, P-JNK/JNK, P-ERK/ERK, P-p38/p38, and P-c-fos/c-fos were analyzed by western blotting. (D) RAW264.7 cells were cultured with RANKL and M-CSF in the presence or absence of LY209761 and collected at 0, 15, 30, 45, and 60 min. The expressions of P-p65/p65, P-p50/p50, P-JNK/JNK, P-ERK/ERK, P-p38/p38, and P-c-fos/c-fos were analyzed by western blot. *p < 0.05, **p < 0.01. To illuminate the biological functions and signaling pathways of osteoclastogenesis after treatment with myostatin, GO functional enrichment and KEGG enrichment analyses were carried out. The differentially expressed genes (DEGs) were mainly involved in 30 GO terms (Figure 4A) and 20 pathways (Figure 4B). GO enrichment was markedly associated with biological processes, cellular components, and molecular functions. Notably, the number of pathways involved in biological processes was the most, including positive regulation of biological processes, single-organism developmental processes, and developing process. KEGG pathway enrichment revealed remarkable involvement of myostatin in pathways including metabolic pathways, ribosomes, pathways in cancer, and the PI3K-Akt signaling pathway. To investigate specific genes whose expression changed greatly in osteoclastogenesis under the effect of myostatin, we listed 10 relatively high expression genes (Table 1) and relatively 10 low expression genes (Table 2) after treatment with myostatin in osteoclastogenesis. FIGURE 4. Functional enrichment analysis of differentially expressed genes. (A) Differentially expressed genes in osteoclastogenesis were researched by GO analysis. (B) Differentially expressed genes in osteoclastogenesis were researched by KEGG analysis. TABLE 1. The high expression gene in osteoclastogenesis after treated with myostatin. TABLE 2. The low expression gene in osteoclastogenesis after treated with myostatin. We selected the Ccdc50 gene as a target gene that regulates osteoclastogenesis under the effect of myostatin. We first investigated whether osteoclastogenesis was regulated by Ccdc50. Ccdc50 levels were measured in RAW264.7 cells infected with Lv-Ccdc50, Lv-shRNA-Ccdc50, and Lv-NC. Ccdc50 was successfully overexpressed and silenced (Figures 5A,B). To further investigate whether myostatin could enhance osteoclastogenesis together with Ccdc50, RAW264.7 cells were treated with Lv-NC, Lv-shRNA-Ccdc50, or Lv-Ccdc50 in the presence of RANKL, then treated with 30 ng/ml myostatin. TRAP staining indicated that overexpressed Ccdc50 suppressed osteoclastogenesis in the presence of myostatin (Figure 5C). The effects of osteoclastogenesis were weakened when Ccdc50 was overexpressed, while when Ccdc50 was not expressed, the process of osteoclastogenesis was enhanced. FIGURE 5. Myostatin regulates osteoclastogenesis under the regulation of Ccdc50. (A) The localization of Ccdc50 expressed by lentivirus after 72 h was ascertained by fluorescence staining. (B) The expression of Ccdc50 of four groups were detected by western blot. (C) Representative images and quantitative analysis of RAW264.7 cells cultured in the six groups and stained for TRAP. 1) RAW264.7 cells. 2) RAW264.7 cells infected with Lv-NC, induced with M-CSF, RANKL. 3) RAW264.7 cells infected with Lv-NC, induced with M-CSF, RANKL, and 30 ng/ml myostatin. 4) RAW264.7 cells infected with Lv-Ccdc50, induced with M-CSF, RANKL. 5) RAW264.7 cells infected with Lv-shRNA-Ccdc50, induced with M-CSF, RANKL, and 30 ng/ml myostatin; 6) RAW264.7 cells infected with Lv-Ccdc50, induced with M-CSF, RANKL, and 30 ng/ml myostatin. The decreased expressions of TRAF6, CTR, TRAP, MMP-9, and cathepsin K indicated that overexpressed Ccdc50 inhibited osteoclast differentiation (Figure 6A). The expression levels of Ccdc50 (Figure 6B), p65, p50, and IκBa in the above-mentioned six groups indicated that myostatin suppressed the phosphorylation of p65, p50, and IκBa, which are crucial markers of the NF-κB pathway. The effects of myostatin were inhibited when Ccdc50 was overexpressed, and the silencing of Ccdc50 enhanced the effects of myostatin on the NF-κB pathway (Figure 6C). The MAPK pathway was also promoted by myostatin, as the phosphorylation of JNK, ERK, and p38 was promoted in the process. All these results showed that Ccdc50 inhibited the relative effects of myostatin (Figure 6D). FIGURE 6. Ccdc50 regulates NF-κB, and MAPK pathways. (A) The expressions of TRAP, cathepsin K, TRAF6, MMP9, and CTR were analyzed by western blot. (B) Changes in Ccdc50 in osteoclastogenesis. (C) Phosphorylation of p65, p50, and IκB as analyzed by western blot. (D) Phosphorylation of JNK, ERK, and p38 as analyzed by western blot. 1) RAW264.7 cells. 2) RAW264.7 cells infected with Lv-NC, induced with M-CSF, RANKL. 3) RAW264.7 cells infected with Lv-NC, induced with M-CSF, RANKL, and 30 ng/ml myostatin. 4) RAW264.7 cells infected with Lv-Ccdc50, induced with M-CSF, RANKL. 5) RAW264.7 cells infected with Lv-shRNA-Ccdc50, induced with M-CSF, RANKL, and 30 ng/ml myostatin; 6) RAW264.7 cells infected with Lv-Ccdc50, induced with M-CSF, RANKL, and 30 ng/ml myostatin.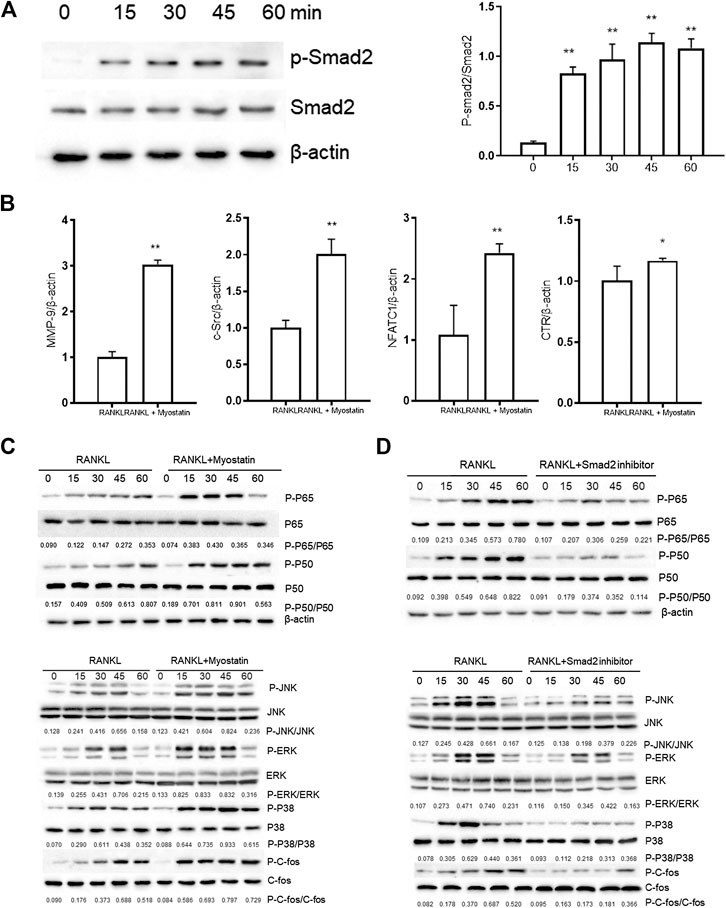
Functional Enrichment Analysis of Differentially Expressed Genes
Ccdc50 Regulates Osteoclastogenesis
Ccdc50 Regulates PI3K/Akt, MAPK, and NF-κB Pathways
Discussion
In this study, we demonstrated that myostatin promotes osteoclastogenesis under the regulation of Ccdc50 in vitro. By its connection with ActR II B, myostatin promotes activation of NF-κB and MAPK pathways. This process requires Smad2 to mediate the downstream signals, including NF-κB and MAPKs, as the inhibition of Smad2 suppressed the activation of the two osteoclastogenesis-related pathways. Our experiments indicated that myostatin regulates osteoclastogenesis via expression of Ccdc50. Overexpression of Ccdc50 inhibited osteoclastogenesis, while silencing Ccdc50 enhanced osteoclastogenesis. These results indicated that Ccdc50 is essential for myostatin’s promotion of osteoclastogenesis and for NF-κB and MAPK pathways. Myostatin can potentially serve as a target for the regulation of osteoclastogenesis-related physiological and pathological processes, since it significantly promotes osteoclastogenesis and related pathways.
The process of bone remodeling is highly regulated and maintained by the dynamic equilibrium of bone formation and bone resorption. The core of this process is composed of two parts including the resorption of inorganic minerals in bone by osteoclasts and the formation of matrix and collagenous fiber by osteoblasts (Xiao et al., 2016b). In osteoporosis, the balance of osteoclasts and osteoblasts is broken, and the rate of bone resorption is increased in comparison with bone formation (Xiao et al., 2016a). Recent studies have demonstrated that myostatin may play an essential role in bone metabolism, even though its main function is negatively regulating muscle fiber growth (Bray, 2015; Tagliaferri et al., 2015). The bone density of myostatin-knockout mice is increased significantly, and bone volume is greater than that of wild-type mice by as much as 60% (Hamrick et al., 2002; Hamrick et al., 2006; Kellum et al., 2009). Likewise, after injection with myostatin inhibitors, bone mass is increased significantly in rats (Chiu et al., 2013). This indicates that myostatin is an important regulator of bone resorption. Furthermore, the inhibition of myostatin reduces bone destruction caused by osteoarthritis, so myostatin could be a target for treating osteoarthritis (Bray, 2015; Dankbar et al., 2015; Onuora, 2015). All these studies show that myostatin may participate in the activation of osteoclasts and could be a potential target for curing orthopedic diseases.
We previously found that ActR II B exists on BMMC and RAW264.7 cell membranes at low levels. However, ActR II B increases sharply in the process of osteoclastogenesis, which means that interaction of myostatin and its receptor would be enhanced during the process, and that myostatin could play an important role in osteoclastogenesis.
It has been reported that RANKL/RANK/OPG participates in the differentiation of osteoclasts (Pepene et al., 2011; Walsh and Choi, 2014). RANKL expression is stimulated in postmenopausal osteoporosis and glucocorticoid osteoporosis patients (Liu and Zhang, 2015). The current study demonstrates that myostatin promotes osteoclastogenesis and osteoclast function. In the early stage of the RANKL pathway, RANKL combines with RANK, and then tumor necrosis factor receptor-associated factor 6 (TRAF6) is recruited and activates the MAPK and NF-κB pathways (Jimi et al., 1998; Tan et al., 2017). The main function of NF-κB is to bind RANKL and transfer signals to regulate osteoclast formation (Miyazaki et al., 2000). Our results showed that phosphorylation of p65 and p50 was promoted after treatment with myostatin, which demonstrated that myostatin can promote the activation of the NF-κB pathway within a short time. Furthermore, we demonstrated the activation of MAPKs in the presence of myostatin by detecting the phosphorylation of c-JNK, ERK, and p38. Similar to the results for NF-κB, we found higher expression when induced by RANKL and myostatin than when induced by RANKL only, which indicated that myostatin promoted the activation of MAPKs.
A previous study suggested that target genes are transcribed by the phosphorylation of Smad2 after myostatin binds to ActR II B (Walsh and Celeste, 2005). Once TGF-β receptors are activated and Smad2 is phosphorylated in turn, a complex containing Smad2, Smad4, and FAST assembles in the nucleus and activates the transcription of multiple genes (Wrana and Attisano, 2000). One result of this transcriptional activation is an increase in osteoclast production and alveolar bone loss in mice (Alotaibi et al., 2016). Our study suggests that phosphorylation of Smad2 increases with the addition of myostatin in osteoclastogenesis, and a series of osteoclast-related markers increases at the same time. However, the phosphorylation of markers of NF-κB and MAPKs clearly decreased after the addition of LY209761, which means that the activation of NF-κB and MAPKs was regulated by Smad2 protein. This process connects the downstream channel of myostatin and classical osteoclast activation pathways.
Despite these results, the upstream signals that regulate myostatin in osteoclastogenesis have remained unclear. To find candidate genes that participate in osteoclastogenesis after treatment with myostatin, a CHIP assay was performed, and 20 genes that were clearly upregulated or downregulated were selected. These differentially expressed genes play important roles in inflammation (Slominski et al., 2020), calcium channels (Fenninger et al., 2019), transcription (Nizon et al., 2019; Gura et al., 2020), cell differentiation (Gobbi et al., 2019), and cell apoptosis (Morris et al., 2020). II9, for example, is critical for mast cell-driven diseases (Abdul Qayum et al., 2019), while Htatip2 can regulate the cell cycle and promote cell apoptosis (Zhao et al., 2020). Among them, we selected Ccdc50 as a gene for study. Previous studies have demonstrated that Ccdc50, which belonged to the downregulated group, is related to the NF-κB pathway (Farfsing et al., 2009; Kameda et al., 2009) and might regulate osteoclastogenesis specifically. However, the function of Ccdc50 remains largely unknown, as it also participates in other pathological and physiological processes. Our study, for the first time, proved that the enhancement of osteoclastogenesis by myostatin was associated with low expression of Ccdc50, as overexpression of Ccdc50 inhibited osteoclastogenesis and osteoclast function, while the silencing of Ccdc50 played the opposite role. The inhibition of NF-κB and MAPK pathways was also verified in osteoclastogenesis when Ccdc50 was overexpressed and vice versa. This indicated that Ccdc50 is significant for myostatin’s induction of osteoclastogenesis by acting on the NF-κB and MAPK pathways.
In summary, we found that myostatin promoted osteoclastogenesis induced by RANKL in vitro by activating Smad2 and further activated the NF-κB and MAPK pathways. Furthermore, the process of osteoclastogenesis through the enhancement of myostatin was inhibited by Ccdc50. These findings imply that myostatin can serve as a potential target for overactivation-related pathological processes and provide a basis for understanding the mechanism of bone remodeling in future research.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
LC and JC designed the study. XZ, QC, SS, HC, ZG, WW, and XC performed the experiments. WW and QZ analyzed the data. LC interpreted the data. XZ and QC wrote the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (81701364, 81901426), Shanghai Sailing Program (19YF1447400), Changhai Hospital Initial Foundation (2018QNA012).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Acknowledgments
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2020.565163/full#supplementary-material
References
Abdul Qayum, A., Koh, B., Martin, R. K., Kenworthy, B. T., Kharwadkar, R., Fu, Y., et al. (2019). The Il9 CNS-25 regulatory element controls mast cell and basophil IL-9 production. J. Immunol. 203, 1111–1121. doi:10.4049/jimmunol.1900272.
Alotaibi, M. K., Kitase, Y., and Shuler, C. F. (2016). Smad2 overexpression induces alveolar bone loss and up regulates TNF-α, and RANKL. Arch. Oral Biol. 71, 38–45. doi:10.1016/j.archoralbio.2016.06.023.
Asagiri, M., Sato, K., Usami, T., Ochi, S., Nishina, H., Yoshida, H., et al. (2005). Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 202, 1261–1269. doi:10.1084/jem.20051150.
Battaglino, R., Kim, D., Fu, J., Vaage, B., Fu, X.-Y., and Stashenko, P. (2002). c-myc is required for osteoclast differentiation. J. Bone Miner. Res. 17, 763–773. doi:10.1359/jbmr.2002.17.5.763.
Benskey, M. J., and Manfredsson, F. P. (2016). Lentivirus production and purification. Methods Mol. Biol. 1382, 107–114. doi:10.1007/978-1-4939-3271-9_8.
Bharti, A. C., Takada, Y., and Aggarwal, B. B. (2004). Curcumin (diferuloylmethane) inhibits receptor activator of NF-κB ligand-induced NF-κB activation in osteoclast precursors and suppresses osteoclastogenesis. J. Immunol. 172, 5940–5947. doi:10.4049/jimmunol.172.10.5940.
Bialek, P., Parkington, J., Li, X., Gavin, D., Wallace, C., Zhang, J., et al. (2014). A myostatin and activin decoy receptor enhances bone formation in mice. Bone 60, 162–171. doi:10.1016/j.bone.2013.12.002.
Boyce, B. F., Xiu, Y., Li, J., Xing, L., and Yao, Z. (2015). NF-κB-Mediated regulation of osteoclastogenesis. Endocrinol. Metab. 30, 35–44. doi:10.3803/enm.2015.30.1.35.
Boyce, B., Yao, Z., and Xing, L. (2009). Osteoclasts have multiple roles in bone in addition to bone resorption. Crit. Rev. Eukaryot. Gene Expr. 19, 171–180. doi:10.1615/critreveukargeneexpr.v19.i3.10.
Bray, N. (2015). Targeting myostatin for direct joint defence. Nat. Rev. Drug Discov. 14, 677. doi:10.1038/nrd4745.
Chiu, C.-S., Peekhaus, N., Weber, H., Adamski, S., Murray, E. M., Zhang, H. Z., et al. (2013). Increased muscle force production and bone mineral density in ActRIIB-Fc-treated mature rodents. J. Gerontol. A Biol. Sci. Med. Sci. 68, 1181–1192. doi:10.1093/gerona/glt030.
Courtois, G., and Gilmore, T. D. (2006). Mutations in the NF-κB signaling pathway: implications for human disease. Oncogene 25, 6831–6843. doi:10.1038/sj.onc.1209939.
Dankbar, B., Fennen, M., Brunert, D., Hayer, S., Frank, S., Wehmeyer, C., et al. (2015). Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat. Med. 21, 1085–1090. doi:10.1038/nm.3917.
Farfsing, A., Engel, F., Seiffert, M., Hartmann, E., Ott, G., Rosenwald, A., et al. (2009). Gene knockdown studies revealed CCDC50 as a candidate gene in mantle cell lymphoma and chronic lymphocytic leukemia. Leukemia 23, 2018–2026. doi:10.1038/leu.2009.144.
Fenninger, F., Han, J., Stanwood, S. R., Nohara, L. L., Arora, H., Choi, K. B., et al. (2019). Mutation of an L-type calcium channel gene leads to T lymphocyte dysfunction. Front. Immunol. 10, 2473. doi:10.3389/fimmu.2019.02473.
Gobbi, S., Hu, Q., Foschi, G., Catanzaro, E., Belluti, F., Rampa, A., et al. (2019). Benzophenones as xanthone-open model CYP11B1 inhibitors potentially useful for promoting wound healing. Bioorg. Chem. 86, 401–409. doi:10.1016/j.bioorg.2019.01.066.
Gura, M. A., Mikedis, M. M., Seymour, K. A., De Rooij, D. G., Page, D. C., and Freiman, R. N. (2020). Dynamic and regulated TAF gene expression during mouse embryonic germ cell development. PLoS Genet. 16, e1008515. doi:10.1371/journal.pgen.1008515.
Hagemann, C., and Blank, J. L. (2001). The ups and downs of MEK kinase interactions. Cell. Signal. 13, 863–875. doi:10.1016/s0898-6568(01)00220-0.
Hamrick, M. W. (2003). Increased bone mineral density in the femora of GDF8 knockout mice. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 272, 388–391. doi:10.1002/ar.a.10044.
Hamrick, M. W., Mcpherron, A. C., and Lovejoy, C. O. (2002). Bone mineral content and density in the humerus of adult myostatin-deficient mice. Calcif. Tissue Int. 71, 63–68. doi:10.1007/s00223-001-1109-8.
Hamrick, M. W., Samaddar, T., Pennington, C., and Mccormick, J. (2006). Increased muscle mass with myostatin deficiency improves gains in bone strength with exercise. J. Bone Miner. Res. 21, 477–483. doi:10.1016/s0736-0266(03)00105-0.
Han, H. Q., Zhou, X., Mitch, W. E., and Goldberg, A. L. (2013). Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. Int. J. Biochem. Cell Biol. 45, 2333–2347. doi:10.1016/j.biocel.2013.05.019.
Jimi, E., Nakamura, I., Ikebe, T., Akiyama, S., Takahashi, N., and Suda, T. (1998). Activation of NF-κB is involved in the survival of osteoclasts promoted by interleukin-1. J. Biol. Chem. 273, 8799–8805. doi:10.1074/jbc.273.15.8799.
Kameda, H., Watanabe, M., Bohgaki, M., Tsukiyama, T., and Hatakeyama, S. (2009). Inhibition of NF-κB signaling via tyrosine phosphorylation of Ymer. Biochem. Biophys. Res. Commun. 378, 744–749. doi:10.1016/j.bbrc.2008.11.102.
Kellum, E., Starr, H., Arounleut, P., Immel, D., Fulzele, S., Wenger, K., et al. (2009). Myostatin (GDF-8) deficiency increases fracture callus size, Sox-5 expression, and callus bone volume. Bone 44, 17–23. doi:10.1016/j.bone.2008.08.126.
Kular, J., Tickner, J., Chim, S. M., and Xu, J. (2012). An overview of the regulation of bone remodelling at the cellular level. Clin. Biochem. 45, 863–873. doi:10.1016/j.clinbiochem.2012.03.021.
Lee, K., Seo, I., Choi, M. H., and Jeong, D. (2018). Roles of mitogen-activated protein kinases in osteoclast biology. Int. J. Mol. Sci. 19, 3004. doi:10.3390/ijms19103004.
Lee, S.-H., Rho, J., Jeong, D., Sul, J.-Y., Kim, T., Kim, N., et al. (2006). v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat. Med. 12, 1403–1409. doi:10.1038/nm1514.
Liu, W., and Zhang, X. (2015). Receptor activator of nuclear factor-κB ligand (RANKL)/RANK/osteoprotegerin system in bone and other tissues (review). Mol. Med. Rep. 11, 3212–3218. doi:10.3892/mmr.2015.3152.
Miyazaki, T., Katagiri, H., Kanegae, Y., Takayanagi, H., Sawada, Y., Yamamoto, A., et al. (2000). Reciprocal role of ERK and nf-κb pathways in survival and activation of osteoclasts. J. Cell Biol. 148, 333–342. doi:10.1083/jcb.148.2.333.
Mizukami, J., Takaesu, G., Akatsuka, H., Sakurai, H., Ninomiya-Tsuji, J., Matsumoto, K., et al. (2002). Receptor activator of NF-κB ligand (RANKL) activates TAK1 mitogen-activated protein kinase kinase kinase through a signaling complex containing RANK, TAB2, and TRAF6. Mol. Cell. Biol. 22, 992–1000. doi:10.1128/mcb.22.4.992-1000.2002.
Morris, A. B., Farley, C. R., Pinelli, D. F., Adams, L. E., Cragg, M. S., Boss, J. M., et al. (2020). Signaling through the inhibitory Fc receptor FcγRIIB induces CD8+ T cell apoptosis to limit T cell immunity. Immunity 52, 136–150. doi:10.1016/j.immuni.2019.12.006.
Nissinen, T. A., Degerman, J., Rasanen, M., Poikonen, A. R., Koskinen, S., Mervaala, E., et al. (2016). Systemic blockade of ACVR2B ligands prevents chemotherapy-induced muscle wasting by restoring muscle protein synthesis without affecting oxidative capacity or atrogenes. Sci. Rep. 6, 32695. doi:10.1038/srep32695.
Nizon, M., Laugel, V., Flanigan, K. M., Pastore, M., Waldrop, M. A., Rosenfeld, J. A., et al. (2019). Variants in MED12L, encoding a subunit of the mediator kinase module, are responsible for intellectual disability associated with transcriptional defect. Genet. Med. 21, 2713–2722. doi:10.1038/s41436-019-0557-3.
Onuora, S. (2015). Cartilage matrix stiffness regulates chondrocyte metabolism and OA pathogenesis. Nat. Rev. Rheumatol. 11, 504. doi:10.1038/nrrheum.2015.113.
Pepene, C. E., Ilie, I. R., Marian, I., and Duncea, I. (2011). Circulating osteoprotegerin and soluble receptor activator of nuclear factor κB ligand in polycystic ovary syndrome: relationships to insulin resistance and endothelial dysfunction. Eur. J. Endocrinol. 164, 61–68. doi:10.1530/eje-10-0720.
Sakuma, T., Barry, M. A., and Ikeda, Y. (2012). Lentiviral vectors: basic to translational. Biochem. J. 443, 603–618. doi:10.1042/bj20120146.
Slominski, R. M., Tuckey, R. C., Manna, P. R., Jetten, A. M., Postlethwaite, A., Raman, C., et al. (2020). Extra-adrenal glucocorticoid biosynthesis: implications for autoimmune and inflammatory disorders. Genes Immun. 21, 150–168. doi:10.1038/s41435-020-0096-6
Sun, Y., Liu, W.-Z., Liu, T., Feng, X., Yang, N., and Zhou, H.-F. (2015). Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. Res. 35, 600–604. doi:10.3109/10799893.2015.1030412.
Tagliaferri, C., Wittrant, Y., Davicco, M.-J., Walrand, S., and Coxam, V. (2015). Muscle and bone, two interconnected tissues. Ageing Res. Rev. 21, 55–70. doi:10.1016/j.arr.2015.03.002.
Takayanagi, H. (2007). Osteoclast differentiation and activation. Clin. Calcium 17, 484–492 [in Japanese, with English summary]. doi:CliCa0704484492.
Takayanagi, H., Kim, S., Koga, T., Nishina, H., Isshiki, M., Yoshida, H., et al. (2002). Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 3, 889–901. doi:10.1016/s1534-5807(02)00369-6.
Tan, E. M., Li, L., Indran, I. R., Chew, N., and Yong, E.-L. (2017). TRAF6 mediates suppression of osteoclastogenesis and prevention of ovariectomy-induced bone loss by a novel prenylflavonoid. J. Bone Miner. Res. 32, 846–860. doi:10.1002/jbmr.3031.
Tanos, T., Marinissen, M. J., Leskow, F. C., Hochbaum, D., Martinetto, H., Gutkind, J. S., et al. (2005). Phosphorylation of c-Fos by members of the p38 MAPK family. J. Biol. Chem. 280, 18842–18852. doi:10.1074/jbc.m500620200.
Walsh, F. S., and Celeste, A. J. (2005). Myostatin: a modulator of skeletal-muscle stem cells. Biochem. Soc. Trans. 33, 1513–1517. doi:10.1042/bst0331513.
Walsh, M. C., and Choi, Y. (2014). Biology of the RANKL-RANK-OPG system in immunity, bone, and beyond. Front. Immunol. 5, 511. doi:10.3389/fimmu.2014.00511.
Wen, J., Liu, X., Qi, Y., Niu, F., Niu, Z., Geng, W., et al. (2019). BMP3 suppresses colon tumorigenesis via ActRIIB/SMAD2-dependent and TAK1/JNK signaling pathways. J. Exp. Clin. Canc. Res. 38, 428. doi:10.1186/s13046-019-1435-1.
Wrana, J. L., and Attisano, L. (2000). The Smad pathway, Cytokine Growth Factor Rev. 11, 5–13. doi:10.1016/s1359-6101(99)00024-6.
Xiao, W., Li, S., Pacios, S., Wang, Y., and Graves, D. T. (2016a). Bone remodeling under pathological conditions. Front. Oral Biol. 18, 17–27. doi:10.1159/000351896.
Xiao, W., Wang, Y., Pacios, S., Li, S., and Graves, D. T. (2016b). Cellular and molecular aspects of bone remodeling. Front. Oral Biol. 18, 9–16. doi:10.1159/000351895.
Keywords: osteoclastogenesis, myostatin, Ccdc50, Receptor activator of NF-κB ligand, NF-κB
Citation: Zhi X, Chen Q, Song S, Gu Z, Wei W, Chen H, Chen X, Weng W, Zhou Q, Cui J and Cao L (2020) Myostatin Promotes Osteoclastogenesis by Regulating Ccdc50 Gene Expression and RANKL-Induced NF-κB and MAPK Pathways. Front. Pharmacol. 11:565163. doi: 10.3389/fphar.2020.565163
Received: 24 May 2020; Accepted: 05 November 2020;
Published: 26 November 2020.
Edited by:
Nathan Pavlos, University of Western Australia, AustraliaReviewed by:
Jiake Xu, University of Western Australia, AustraliaFeng Xu, University of Alabama at Birmingham, United States
Copyright © 2020 Zhi, Chen, Song, Gu, Wei, Chen, Chen, Weng, Zhou, Cui and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liehu Cao, dHJhdW1haHVAMTYzLmNvbQ==; Jin Cui, Y3VpamluNjE2M0AxNjMuY29t
†These authors have contributed equally to this work