 Jennifer C. Wong1*
Jennifer C. Wong1* Kameryn M. Butler1,2
Kameryn M. Butler1,2 Lindsey Shapiro1Jacquelyn T. Thelin1
Lindsey Shapiro1Jacquelyn T. Thelin1 Kari A. Mattison1Kathryn B. Garber1
Kari A. Mattison1Kathryn B. Garber1 Paula C. Goldenberg3Shobana Kubendran4
Paula C. Goldenberg3Shobana Kubendran4 G. Bradley Schaefer5
G. Bradley Schaefer5 Andrew Escayg1
Andrew Escayg1- 1Department of Human Genetics, Emory University, Atlanta, GA, United States
- 2Greenwood Genetic Center, Greenwood, SC, United States
- 3Department of Pediatrics and Medical Genetics, Harvard Medical School, Boston, MA, United States
- 4Department of Pediatrics, Kansas University School of Medicine-Wichita, Wichita, KS, United States
- 5University of Arkansas for Medical Sciences, Little Rock, AR, United States
Numerous SCN8A mutations have been identified, of which, the majority are de novo missense variants. Most mutations result in epileptic encephalopathy; however, some are associated with less severe phenotypes. Mouse models generated by knock-in of human missense SCN8A mutations exhibit seizures and a range of behavioral abnormalities. To date, there are only a few Scn8a mouse models with in-frame deletions or insertions, and notably, none of these mouse lines exhibit increased seizure susceptibility. In the current study, we report the generation and characterization of two Scn8a mouse models (ΔIRL/+ and ΔVIR/+) carrying overlapping in-frame deletions within the voltage sensor of domain 4 (DIVS4). Both mouse lines show increased seizure susceptibility and infrequent spontaneous seizures. We also describe two unrelated patients with the same in-frame SCN8A deletion in the DIV S5-S6 pore region, highlighting the clinical relevance of this class of mutations.
Introduction
Voltage-gated sodium channel (VGSC) alpha subunits are comprised of four homologous domains, annotated DI-DIV, and each domain contains six highly conserved transmembrane segments (S1-S6, Figure 1A (Noda et al., 1984; Yu and Catterall, 2003; Catterall, 2012; Catterall, 2000). The S4 segments of VGSCs are enriched with positively charged residues at every third position, and these residues are critical for voltage-dependent gating (Catterall, 2000; Wisedchaisri et al., 2019; Yang et al., 1996; Sokolov et al., 2005). The inactivation gate located between DIII-DIV, and the residues between S5-S6 which form the channel pore, are also highly conserved (Yu and Catterall, 2003; Meisler et al., 2021). Mutations that affect residues in functional domains can impact channel function and alter neuronal excitability (Hodgkin and Huxley, 1952; Catterall, 2000; Yu and Catterall, 2003). Greater variability is observed in the sequence of the intracellular loops between the DI-DII and DII-DIII domains (Figure 1A) (Yu and Catterall, 2003; Meisler et al., 2021).
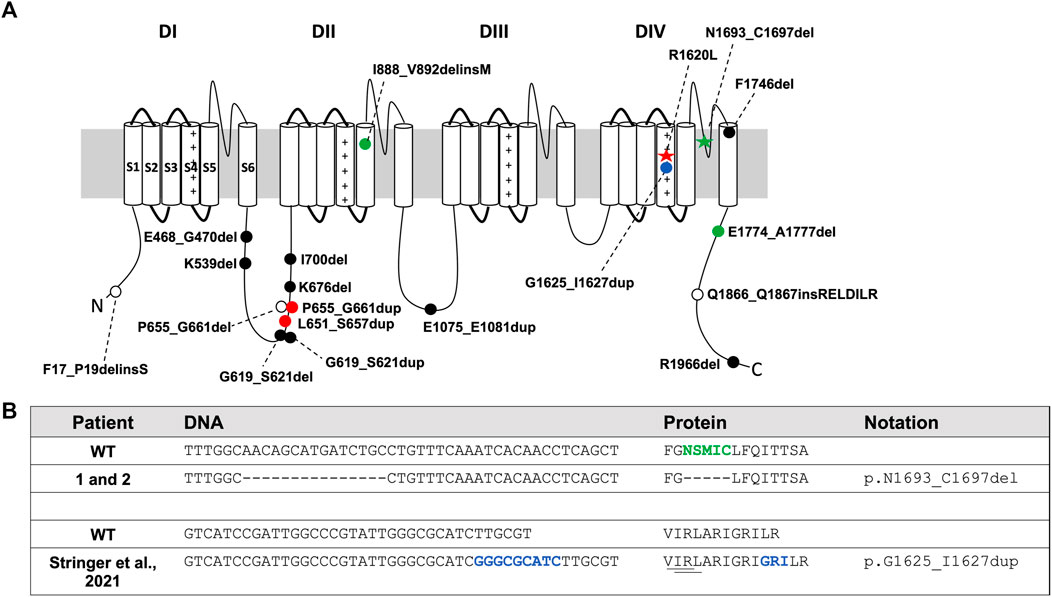
FIGURE 1. Patient SCN8A variants and in-frame variants from gnomAD and ClinVar. (A) SCN8A channel with variants denoted by: red star, R1620L mutation; green star, in-frame deletion in Patients 1 and 2; filled blue circle, in-frame duplication in proband from Stringer et al., 2021; filled green circle, in-frame variants from Johannesen et al., 2021; open circles, in-frame deletions from ClinVar (July 2021); filled black circle, in-frame deletions and duplications from the gnomAD database (v2.1.1 and v3.1.1; July 2021); filled red circle, in-frame duplication in both gnomAD and ClinVar. (B) DNA and protein sequence alignment for Patients 1 and 2 and the proband identified in Stringer et al., 2021. Green indicates amino acids deleted in Patients 1 and 2. Dash (-) indicates a deleted nucleotide or amino acid compared to WT. Blue indicates inserted DNA nucleotides or amino acids. Underline shows close proximity of amino acids deleted in ΔVIR and ΔIRL mouse lines to the variant identified in Stringer et al., 2021.
VGSCs play a critical role in the initiation and propagation of action potentials (Catterall, 2000; Yu and Catterall, 2003; Hu et al., 2009; Catterall, 2012), and mutations in the different members of this gene family are responsible for a wide range of disorders (Brunklaus et al., 2020; Brunklaus and Lal, 2020; Meisler et al., 2021; Wang and Frankel, 2021). Four VGSC alpha subunits are highly expressed in the brain: SCN1A, SCN2A, SCN3A and SCN8A. Mutations in each of these genes are responsible for different forms of epilepsy (Brunklaus et al., 2020; Brunklaus and Lal, 2020; Meisler et al., 2021). For example, SCN1A mutations are the main cause of genetic epilepsy with febrile seizures plus and Dravet syndrome (Escayg et al., 2000; Lossin, 2009; Escayg and Goldin, 2010). SCN2A mutations lead to benign familial neonatal infantile seizures and epileptic encephalopathy (Meisler et al., 2010; Allen et al., 2016; Fukasawa et al., 2015; Saitoh et al., 2015), and SCN3A is associated with focal epilepsy (Vanoye et al., 2014; Lamar et al., 2017). The first human SCN8A epilepsy-associated mutation (c.5302A > G, p.N1768D) was identified in a proband who presented with spontaneous seizures, behavioral deficits, and sudden unexpected death in epilepsy (SUDEP) (Veeramah et al., 2012). Since this discovery, numerous primarily de novo missense SCN8A mutations have been identified (Blanchard et al., 2015; Larsen et al., 2015; Wagnon et al., 2016; Butler et al., 2017; Gardella et al., 2018; Johannesen et al., 2018; Brunklaus et al., 2020). Most of the pathogenic SCN8A variants are associated with severe epileptic encephalopathy (Blanchard et al., 2015; Larsen et al., 2015; Wagnon et al., 2016; Butler et al., 2017; Gardella et al., 2018; Johannesen et al., 2018; Brunklaus et al., 2020); however, milder phenotypes associated with missense variants have also been reported (Gardella et al., 2016; Rossi et al., 2017; Liu et al., 2019). Truncating mutations (nonsense, frameshift, and splice site alterations) or in-frame deletions/insertions in SCN8A are not as frequently observed, and patients with such variants may present with less severe phenotypes (Trudeau et al., 2006; Berghuis et al., 2015). For example, Trudeau et al. identified a patient with a maternally inherited heterozygous truncation in SCN8A who presented with cognitive and motor deficits (Trudeau et al., 2006), and Berghuis et al. identified an individual with absence epilepsy, developmental delay, aggression, and attention problems who had a paternally inherited deletion of SCN8A exons 2–14 and a maternally inherited missense variant (p.I1583T) (Berghuis et al., 2015).
The first mouse model of SCN8A epileptic encephalopathy was generated by knock-in of the SCN8A p.N1768D mutation (Wagnon et al., 2015). Heterozygous mice expressing the N1768D mutation recapitulated several phenotypes observed in the original patient, including spontaneous seizures and SUDEP (35). A conditional mouse model with the SCN8A R1872W mutation also exhibited spontaneous seizures and early mortality when the mutation was expressed globally in the brain or selectively in excitatory neurons (Bunton-Stasyshyn et al., 2019). Recently, using CRISPR/Cas9 technology, we generated a mouse line expressing the SCN8A R1620L mutation (Figure 1A), which was identified in a patient with relatively mild epilepsy and behavioral deficits (Wong et al., 2021a). Mice heterozygous for this mutation exhibit increased seizure susceptibility, spontaneous seizures, impaired learning and memory, social deficits, and altered neuronal excitability (Wong et al., 2021a).
Prior to the development of the mouse lines expressing human SCN8A epilepsy mutations, most of the published Scn8a mouse models carried loss-of-function missense or truncating Scn8a mutations (Martin et al., 2007; Papale et al., 2009; Hawkins et al., 2011; Makinson et al., 2014). Mice heterozygous for loss-of-function Scn8a mutations display increased resistance to induced seizures (Martin et al., 2007; Hawkins et al., 2011; Makinson et al., 2014), and depending on the genetic background, some lines also exhibit spike-wave discharges (Papale et al., 2009). There are currently only a few Scn8a mouse models with in-frame deletions or insertions (Jones et al., 2016; Inglis et al., 2020), and notably, none of these mouse lines show increased seizure susceptibility. Jones et al., 2016 reported an in-frame deletion (p.I1750del) in the DIVS6 domain, and homozygous mutants with this mutation exhibited motor impairments and early mortality (Jones et al., 2016). We previously generated two Scn8a mouse lines, one with an in-frame deletion (p.R848_F850del, Δ9) and the other with an in-frame insertion (p.R848_V849insD, ∇3) in the DIIS4 (Inglis et al., 2020). Heterozygous mutants from both of these lines exhibit increased seizure resistance (Inglis et al., 2020). In the current study, we report the generation and characterization of two novel Scn8a mouse models with overlapping in-frame deletions in the DIVS4 that exhibit increased seizure susceptibility and spontaneous seizures. We also describe two unrelated patients with the same in-frame SCN8A deletion in the DIV S5-S6 pore region, highlighting the clinical relevance of in-frame SCN8A mutations.
Materials and Methods
Animals
Using CRISPR/Cas9, we knocked in the human SCN8A p.R1620L mutation into the mouse Scn8a gene on the C57BL/6J background (corresponding to R1618L in the mouse, Figure 2A (Wong et al., 2021a). As a result of nonhomologous end joining during this process, we generated two additional Scn8a mouse lines (Scn8a9Δ_IRL and Scn8a9Δ_VIR) with overlapping 9 base pair in-frame deletions which include removal of the positively charged R1618 residue (Figure 2A). Heterozygous mutants from the Scn8a9Δ_IRL (ΔIRL/+) and Scn8a9Δ_VIR (ΔVIR/+) lines were backcrossed to C57BL/6J mice (Strain: 000664, Jackson Laboratories) for four generations. We performed Sanger sequencing on mutant mice from each line to confirm that the in-frame deletions were the only changes in Scn8a exon 26 and that the conserved exon 26 of the other brain sodium channels, Scn1a, Scn2a, and Scn3a, were unaltered. The 9 bp deletions were the only alterations observed in Scn8a exon 26 and no off-target CRISPR editing was observed in exon 26 of the other sodium channels. Male and female ΔIRL/+ and ΔVIR/+ mutants and respective wild-type (WT) littermates at the N4 generation were used for all experiments. To examine weight gain and survival of ΔIRL mice, male and female heterozygous mutants were bred to generate homozygous ΔIRL/ΔIRL mutants, heterozygous ΔIRL/+ mutants, and WT littermates. We tried to similarly breed the ΔVIR mouse line, however an insufficient number of litters were generated. Therefore, for the ΔVIR mouse line, heterozygous male mutants were bred with C57BL/6J females to generate heterozygous ΔVIR/+ mutants and WT littermates.
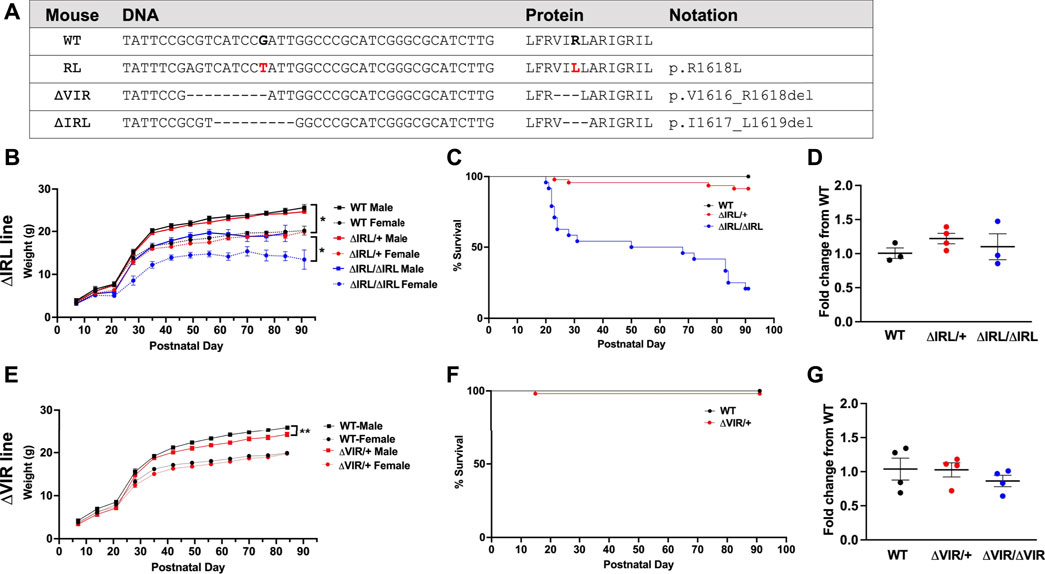
FIGURE 2. Generation and characterization of the ΔIRL and ΔVIR mouse lines. (A) DNA and protein sequence alignment. WT, wild-type mouse sequence, RL, the human R1620L mutation (red) in the RL mouse line, ΔVIR, the 3 amino acid deletion in the ΔVIR mouse line, ΔIRL, the 3 amino acid deletion in the ΔIRL mouse line. Dash (-) indicates a deleted nucleotide or amino acid when compared to the WT sequence. (B) Homozygous ΔIRL/ΔIRL mutants weigh significantly less than same-sex heterozygous ΔIRL/+ mutants and WT littermates. Kruskal-Wallis test followed by Dunn’s multiple comparisons. (C) Approximately 25% of ΔIRL/ΔIRL mutants and 90% of ΔIRL/+ mutants survive to postnatal day 90 (P90). Log-rank Mantel Cox test. (B and C) WT male N = 10; WT female N = 11; ΔIRL/+ male N = 29; ΔIRL/+ female N = 17, ΔIRL/ΔIRL male N = 13; ΔIRL/ΔIRL female N = 11. (D) Scn8a mRNA expression levels were comparable for the three genotypes in the ΔIRL mouse line. Kruskal-Wallis test. N = 3–4/genotype. (E) Heterozygous ΔVIR/+ male mutants weigh significantly less than same-sex WT littermates. Female ΔVIR/+ mutants and WT littermates had comparable weights. Paired Student's t test. (F) Approximately 95% of ΔVIR/+ mutants survive to P90. Log-rank Mantel Cox test. (E and F) WT male N = 27; WT female N = 22; ΔVIR/+ male N = 18; ΔVIR/+ female N = 36. (G) Scn8a mRNA expression levels were comparable for the three genotypes in the ΔVIR mouse line. Kruskal-Wallis test. N = 4/genotype. *p ≤ 0.05, **p ≤ 0.01.
To generate biallelic mice expressing the R1620L and ΔVIR mutations, male ΔVIR/+ mutants were crossed with female RL/+ mutants to generate the following genotypes: ΔVIR/+, RL/+, ΔVIR/RL, and WT. This breeding scheme allowed us to examine littermates expressing the R1620L and ΔVIR mutations. Survival and weights were recorded. Mice were housed on a 12 h light/dark cycle with food and water ad libitum. All experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of Emory University.
Genotyping
DNA was isolated from tail biopsies. Mice with the 9Δ_IRL or 9Δ_VIR allele were identified by PCR amplification using primer R1620L_R1: TACGCGAAGTTGGACATCCC and forward primers that span the respective 9 bp deletions: Scn8a_9del_IRL_F1: CCCTATTCCGCGTGGCCC and Scn8a_9del_VIR_F2: TCTCCCCGACCCTATTCCGATT. The 9Δ_IRL and 9Δ_VIR alleles generated 175 bp and 184 bp PCR products, respectively. The WT allele was identified using the primers R1620L_R1 and Scn8a_WT_F: CTATTCCGCGTCATCCGATTGG, which generated a 183 bp product. The RL/+ mutants were genotyped as previously described (Wong et al., 2021a).
qRT-PCR
Whole brains were extracted from P20-P21 WT, heterozygous, and homozygous mutants of both sexes from each mouse line. RNA extraction and cDNA synthesis were performed as previously described (Lamar et al., 2017). The following Scn8a primer pair was used: Scn8a_F: AGATTTAGCGCCACTCCTGC and Scn8a_R: GGACCATTCGGGAGGGTTAC. Analyses were conducted in technical triplicates using the Real-Time PCR Detection System and SYBR Green (BioRad). Expression levels were normalized to beta-actin (F: CAGCTTCTTTGCAGCTCCTT and R: ACGATGGAGGGGAATACAGC). Relative expression between genotypes was compared using the ΔΔct method.
6 Hz Seizure Induction
Seizures were induced using the 6 Hz seizure induction paradigm as previously described (Barton et al., 2001; Devinsky et al., 2016; Wong et al., 2016; Lamar et al., 2017; Shapiro et al., 2019; Wong et al., 2019; Wong et al., 2021b). Briefly, ΔIRL/+ and ΔVIR/+ mutants and their WT littermates of both sexes (2 months old) were subjected to corneal stimulation (6 Hz, 0.2 ms pulse width, 3 s) using a constant current device (ECT unit, 57800; Ugo Basile, Comerio, Italy). The ΔIRL and ΔVIR mouse lines were tested at 14 and 16 mA, respectively. Behavioral seizures were scored using a modified Racine scale (RS): RS0, no abnormal behavior; RS1, immobile ≥3 s; RS2, forelimb clonus, head bobbing, paw waving; and RS3, rearing and falling. A greater RS value indicates a more severe behavioral seizure.
Flurothyl Seizure Induction
ΔIRL/+, ΔVIR/+, RL/+, and their WT littermates (2 months old) of both sexes were individually placed into a clear acrylic chamber and flurothyl (2,2,2-trifluroethylether, Sigma-Aldrich) was introduced into the chamber at 20 μL/min as previously described (Prichahd et al., 1969; Wong et al., 2016; Shapiro et al., 2019). The latencies to the first myoclonic jerk (MJ) and generalized tonic-clonic seizure (GTCS) were recorded for each mouse.
EEG Surgery and Analyses
Cortical electrodes were implanted into adult (2–4 months old) male ΔIRL/+ and male ΔVIR/+ mutants as previously described (Lamar et al., 2017; Wong et al., 2018; Inglis et al., 2020; Shapiro et al., 2019). Four cortical screw electrodes were implanted into the skull at the following coordinates relative to bregma: anterior-posterior(AP) +2.0 mm and medial-lateral(ML) +1.2 mm; AP −1.5 mm and ML +1.2 mm; AP +0.5 mm and ML −2.2 mm; and AP −3.5 mm and ML −2.2 mm. Two fine-wire electrodes were implanted into the neck muscles for EMG recordings. Each mouse was allowed 1 week to recover from the surgical procedure. Stellate Harmonie rodent software was used to obtain and analyze EEG recordings. Seizures were manually identified and characterized by high frequency and amplitude EEG signals that were at least 3 s in duration and twice the background. Simultaneous video recordings were used to confirm behavioral seizures.
Next-Generation Sequencing of Patients
Two unrelated patients (Patients 1 and 2) had targeted gene panel sequencing performed by GeneDx. Clinical information was provided by the corresponding clinician in each case. The corresponding author can be contacted for additional information on the patients described in this manuscript. This study was approved by the Institutional Review Board of Emory University.
Databases
Three databases were used for the identification of in-frame SCN8A variants: 1) the Genome Aggregation database (gnomAD, v2.1.1 and v3.1.1; https://gnomad.broadinstitute.org) (Karczewski et al., 2020), 2) ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) (Landrum et al., 2018), and 3) the Human Gene Mutation database (HGMD, v2021.2; http://www.hgmd.cf.ac.uk/ac/index.php). The gnomAD database is a compilation of exome and genome sequencing data from a variety of large-scale sequencing projects, and efforts have been made to remove individuals affected by severe pediatric disease. There are currently two versions of the gnomAD database: v2.1.1 contains both exome and whole genome sequences (141,456 samples, GRCh37 build), while v3.1.1 contains only whole genome sequences (76,156 samples, GRCh38 build). Approximately 20,000 genomes overlap between v2.1.1 and v3.1.1. The ClinVar database is a freely available, non-curated, public repository of human genetic variants. Interpretation of the significance of these variants to disease is provided by the submitter and does not require experimental validation. Submitters include clinical testing laboratories, research laboratories, expert panels, and other groups that must be approved by the National Center for Biotechnology Information (NCBI). The HGMD database is a curated collection of published genetic variants that are associated with human disease.
Statistical Analyses
Data are presented as mean ± SEM with p ≤ 0.05 considered as statistically significant. All statistical analyses were performed with Prism 9.0 (GraphPad Software, San Diego, CA). A Chi-square test was used to compare the observed number of offspring with the expected ratio. A log-rank Mantel Cox test was used to compare survival curves. A Kruskal-Wallis test followed by Dunn’s multiple comparisons test was used to compare weights within the ΔIRL mouse line. A paired Student’s t test was used to compare weights between ΔVIR/+ mutants and same-sex WT littermates. A Kruskal-Wallis test was used to compare Scn8a expression levels between genotypes. A Mann-Whitney test was used to compare Racine scores following 6 Hz-induced seizures between mutants and WT littermates. An unpaired Student’s t test was used to compare the latency to the first MJ and GTCS between mutants and WT littermates from the ΔVIR and ΔIRL mouse lines. A Kruskal-Wallis test followed by Dunn’s multiple comparisons test was used to compare the latency to the first MJ and GTCS between the mutants and WT littermates from the ΔVIR x RL matings.
Results
Survival
Male and female ΔIRL/+ mutants were crossed to generate homozygous ΔIRL/ΔIRL, heterozygous ΔIRL/+, and WT littermates. From a total of 9 litters, we observed 21 WT (10 males, 11 females), 46 ΔIRL/+ (29 males, 17 females), and 24 ΔIRL/ΔIRL (13 males, 11 females) offspring, which is consistent with the predicted 1:2:1 Mendelian ratio (p = 0.21). ΔIRL/ΔIRL mutants weighed significantly less than sex-matched ΔIRL/+ mutants and WT littermates (p ≤ 0.05, Figure 2B), and ΔIRL/+ and ΔIRL/ΔIRL mutants exhibited approximately 90 and 25% survival at postnatal day 90 (P90), respectively (Figure 2C). Heterozygous ΔVIR/+ female mutants did not breed efficiently, and we were unable to obtain enough offspring to generate a survival curve. As an alternative, we mated male ΔVIR/+ mutants with C57BL/6J females in order to examine the survival of heterozygous ΔVIR/+ mutants. Male ΔVIR/+ mutants weighed significantly less than sex-matched WT littermates (p ≤ 0.01, Figure 2E), and one ΔVIR/+ male did not survive to P90 (Figure 2F). In contrast, none of the ΔVIR/+ females died, and their body weights were comparable to same-sex WT littermates (Figure 2E). From 16 litters, we observed 54 ΔVIR/+ mutants (18 males, 36 females) and 49 WT littermates (27 males, 22 females), which is consistent with the expected 1:1 ratio (p = 0.78).
mRNA Expression
Quantitative real-time RT-PCR analysis was performed on whole brain samples from P20-P22 mice of each genotype from the ΔIRL and ΔVIR mouse lines. We observed similar levels of Scn8a expression within and between each mouse line (Figures 2D–G).
Heterozygous ΔIRL/+ and ΔVIR/+ Mutants Exhibit Increased Seizure Susceptibility
Susceptibility to 6 Hz- and flurothyl-induced seizures were compared between heterozygous mutants (ΔIRL/+ or ΔVIR/+) and WT littermates from each line. We did not observe any statistically significant differences in susceptibility to 6 Hz- or flurothyl-induced seizures between the sexes; therefore, data from both sexes were combined for analysis. Both ΔIRL/+ and ΔVIR/+ mutants were significantly more susceptible to 6 Hz-induced seizures when compared to their respective WT littermates (Figures 3A,D). For the ΔIRL/+ mutants, 12/20 mice seized (RS = Racine Score; 8RS0, 1 RS1, 11 RS2, 1 RS3) when compared to 6/25 WT littermates (19 RS0, 1 RS1, 5 RS2). Similarly, all of the ΔVIR/+ mutants exhibited a 6 Hz seizure (21 RS2, 2 RS3) whereas most of the WT littermates did not seize (16 RS0, 1 RS1, 3 RS2). When tested with flurothyl, ΔIRL/+ and ΔVIR/+ mutant mice exhibited significantly shorter latencies to the first myoclonic jerk (Figures 3B,E) and the first GTCS (Figures 3C,F) when compared to their WT littermates. Although we cannot directly compare the two mouse lines, the average latencies to the first MJ and GTCS in the ΔVIR/+ mutants were approximately 40% shorter than the corresponding measurements from the ΔIRL/+ mutants, suggesting that the ΔVIR/+ mutants may be more severely affected.
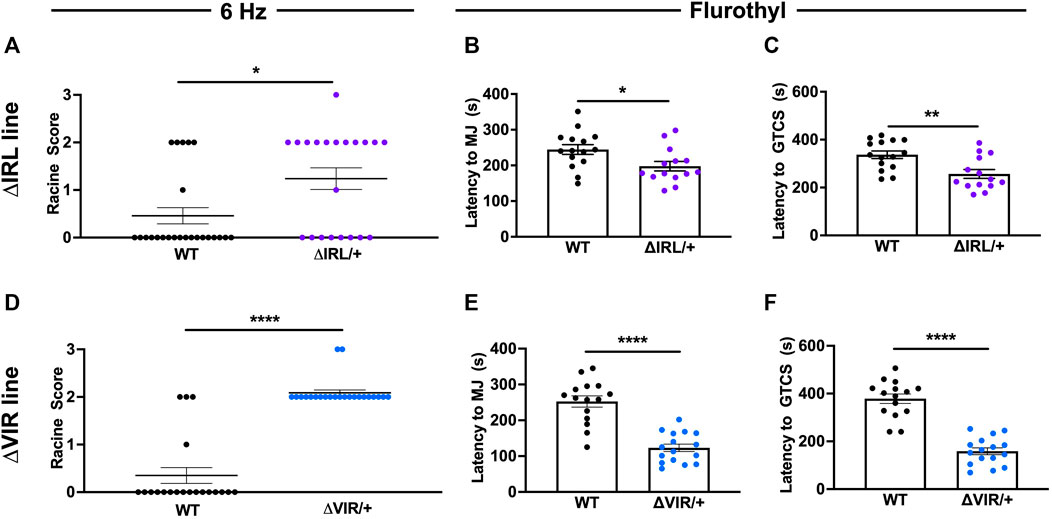
FIGURE 3. ΔIRL/+ and ΔVIR/+ mutants exhibit increased seizure susceptibility. (A) ΔIRL/+ mutants are significantly more susceptible to 6 Hz-induced seizures compared to WT littermates. Mann-Whitney test. WT N = 25; ΔIRL/+ N = 21. (B,C) ΔIRL/+ mutants exhibit a lower average latency to the first MJ (B) and GTCS (C) compared to WT littermates. Unpaired student’s t test. WT N = 15; ΔIRL/+ N = 14. (D) ΔVIR/+ mutants are more susceptible to 6 Hz-induced seizures compared to WT littermates. Mann-Whitney test. WT N = 20; ΔVIR/+: N = 23. (E,F) ΔVIR/+ mutants exhibit lower average latencies to the first MJ (E) and GTCS (F) compared to WT littermates. Unpaired Student's t test. WT N = 15; ΔVIR/+ N = 16. *p ≤ 0.05, **p ≤ 0.01, ****p ≤ 0.0001.
Heterozygous ΔIRL/+ and ΔVIR/+ Mutants Exhibit Spontaneous Seizures
Continuous EEG recordings were obtained from five male ΔIRL/+ and five male ΔVIR/+ mutants. 2/5 ΔIRL/+ mutants and 3/5 ΔVIR/+ mutants exhibited spontaneous seizures. Figure 4A provides an example of a spontaneous seizure observed in a ΔVIR/+ mutant. Spontaneous seizures were between 17–60 s in duration with the exception of one ΔVIR/+ mutant (Mouse #6, Figure 4B) that had a 4 min-long seizure, followed by 2 min of recovery, entry into status epilepticus for approximately 4 h, and ultimately, death. Spontaneous seizure frequency was infrequent, with an average seizure frequency of less than 1 seizure/day in the mice that exhibited seizures (Figure 4B). Electrographic seizures were accompanied by rearing, paw waving, loss of posture, and in some instances, wild running and bouncing.
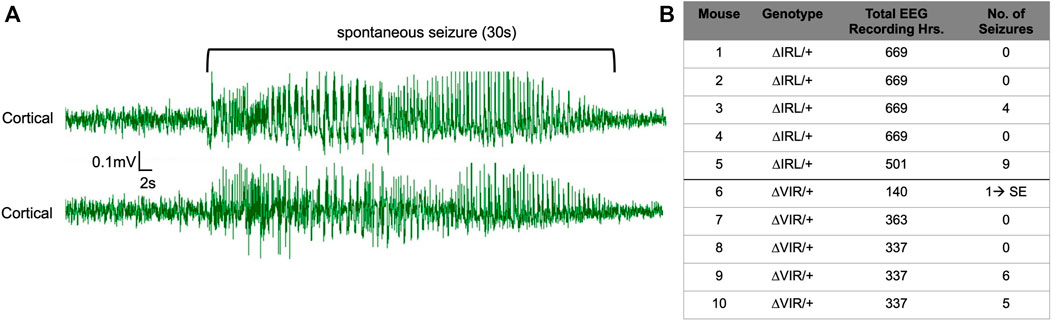
FIGURE 4. ΔIRL/+ and ΔVIR/+ mutants exhibit spontaneous seizures. (A) Representative cortical EEG traces from a ΔVIR/+ mutant before, during, and after a spontaneous seizure. (B) Number of spontaneous seizures observed in 5 male ΔIRL/+ and 5 male ΔVIR/+ mutants. SE denotes status epilepticus.
Biallelic Mutants Exhibit Premature Mortality and Increased Seizure Susceptibility
Based on our previous characterization of the RL mutants (Wong et al., 2021a), we know that homozygous RL mutants gain weight normally until postnatal day 15 (P15) and have a maximum lifespan of 22 days. Thus, to examine the relative severity of the ΔVIR mutation compared to the pathogenic human SCN8A p.R1620L mutation (Wong et al., 2021a), we crossed heterozygous male ΔVIR/+ mutants with heterozygous female RL/+ mutants to generate WT, ΔVIR/+, RL/+, and biallelic (ΔVIR/RL) littermates. Beginning at P12, ΔVIR/RL mutants of both sexes were smaller than their same-sex littermates (Figures 5A,B). Male and female ΔVIR/RL mutants exhibited premature death, and maximum lifespans of 22 and 31 days were observed, respectively (Figure 5C).
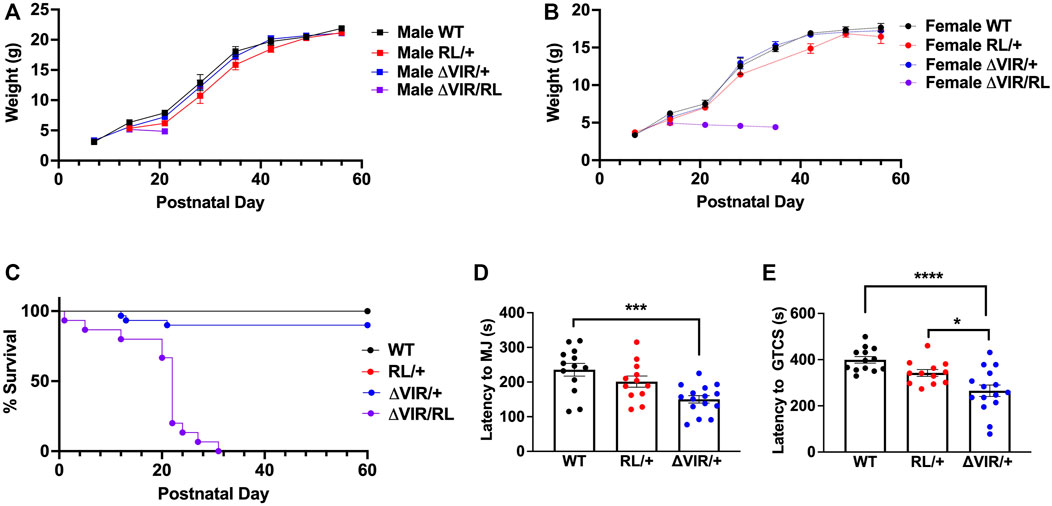
FIGURE 5. Generation and characterization of ΔVIR/RL biallelic heterozygous mice. (A,B) ΔVIR/RL mutants weighed less than same-sex littermates from P12. (C) Male and female ΔVIR/RL mutants exhibited a maximum lifespan of 22 and 31 days, respectively. Only 3/22 ΔVIR/+ mutants died prematurely. Log-rank Mantel Cox test. (A-C) WT male N = 12; WT female N = 10; RL/+ male N = 12; RL/+ female N = 3; ΔVIR/+ male N = 16; ΔVIR/+ female N = 14; ΔVIR/RL male N = 7; ΔVIR/RL female N = 8. (D) Compared to WT littermates, ΔVIR/+ mutants exhibited a significantly lower average latency to the first MJ. (E) ΔVIR/+ mutants exhibit a significantly lower average latency to the first GTCS compared to RL/+ mutants and WT littermates. (D,E) Kruskal-Wallis test followed by Dunn’s multiple comparisons. WT N = 13; RL/+ N = 12; ΔVIR/+ N = 15. *p ≤ 0.05, ***p ≤ 0.001, ****p ≤ 0.0001.
We observed no sex differences in the latency to the first MJ or GTCS following flurothyl exposure; therefore, we combined the data from male and female mice of the same genotype. Although not statistically significant, the RL/+ mutants exhibited a lower average latency to the first MJ and GTCS compared to WT littermates (Figures 5D,E). Average latency to the first MJ was significantly lower in ΔVIR/+ mutants compared to WT littermates (Figure 5D), and the average latency to the first GTCS was significantly lower in ΔVIR/+ mutants compared to RL/+ mutants and WT littermates (Figure 5E), suggesting greater severity of the ΔVIR mutation. Due to the premature death of the ΔVIR/RL mutants, we were unable to evaluate their susceptibility to flurothyl-induced seizures.
Patients With in-Frame SCN8A Variants
Patients 1 and 2: c.5077_5091del, p.Asn1693_Cys1697del
Gene panel testing of two unrelated patients (Patient 1 and Patient 2) identified the same heterozygous in-frame deletion variant, SCN8A c.5077_5091del (p.N1693_C1697del) (Figure 1B), located in the DIV S5-S6 pore region (Figure 1A, green star). The c.5077_5091del variant is currently classified as a “variant of uncertain clinical significance” (Richards et al., 2015). Patient 1 presented with autism, developmental delay, dysmorphic facial features, but no seizures. The deletion was determined to be inherited from the unaffected mother. Patient 2 presented with autism, encephalopathy with developmental delay, tremors, facial asymmetry, low muscle tone, skeletal abnormalities, and femoral torsion, but no history of seizures. The deletion was determined to be de novo for Patient 2. Patient 2 also had another de novo heterozygous variant in ANK3 c.3821C > A (p.S1274Y), which was classified as a “variant of uncertain clinical significance.”
Discussion
In the current manuscript, we describe the generation and characterization of two Scn8a mouse lines (ΔIRL and ΔVIR) with overlapping, in-frame deletions. Heterozygous ΔIRL/+ and ΔVIR/+ mutants exhibit increased seizure susceptibility and spontaneous seizures, demonstrating the potential for this class of genetic variation to contribute to the clinical burden associated with SCN8A dysfunction. Consistent with this, we also report the identification of an in-frame SCN8A variant in two unrelated patients with neurodevelopmental phenotypes, but no seizures.
We previously observed increased resistance to induced seizures in heterozygous Scn8aΔ9/+ and Scn8a∇3/+ mutant mice, expressing an in-frame 9 bp deletion (Δ9) and 3 bp insertion (∇3) in the DIIS4, respectively (Inglis et al., 2020). EEG analyses of the Scn8aΔ9/+ mutants revealed normal electrographic activity and no spontaneous seizures (Inglis et al., 2020). In contrast, ΔIRL/+ and ΔVIR/+ mutants, harboring overlapping 9 bp deletions within the DIVS4, exhibited increased seizure susceptibility and spontaneous seizures. We performed in silico analyses of the DIIS4 and DIVS4 transmembrane domains (Figure 6) and found that the Δ9 mutation in the DIIS4 causes a loss of a positive charge and shift of polar charges. The ∇3 mutation in DIIS4 causes a shift of the polar charges to one side of the helix. In contrast, the VIR and IRL deletions in DIVS4 do not significantly alter the structure and distribution of charges in the alpha helix. Differences between the phenotypes of mice expressing the Δ9 and ∇3 mutations versus the ΔVIR and ΔIRL mutations may also be due, in part, to differences in the functional properties of the domains, with DI-DIII primarily for activation (Kontis et al., 1997; Yu and Catterall, 2003) and DIV primarily involved in inactivation (Kontis and Goldin, 1997; Yu and Catterall, 2003). These contrasting observations also highlight that the phenotypic consequences of in-frame deletions or insertions may likely be difficult to predict. Additional studies are warranted to characterize the biophysical impact of the ΔIRL and ΔVIR alleles relative to other SCN8A missense and in-frame variants.
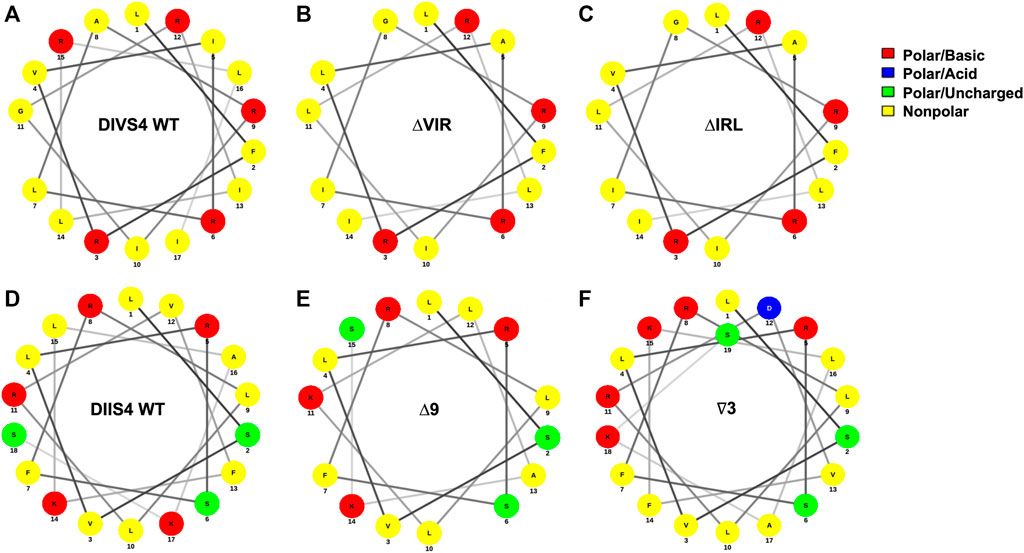
FIGURE 6. In silico analysis of in-frame deletions in the DIVS4 and DIIS4 transmembrane segments. (A–C) Alpha helix wheel diagrams of the WT DIVS4 transmembrane segment (A), the ΔVIR mutation (B), and the ΔIRL mutation (C). (D–F) Alpha helix wheel diagrams of the WT DIIS4 transmembrane segment (D), the Δ9 mutation (E), and the ∇3 mutation (F) from Inglis et al., 2020. The alpha helix wheel diagrams were generated using NetWheels (http://lbqp.unb.br/NetWheels/).
To date, most identified human SCN8A-epilepsy associated mutations have been de novo amino acid substitutions, many of which are predicted or shown to have gain-of-function properties (Veeramah et al., 2012; Estacion et al., 2014; Liu et al., 2019; Heyne et al., 2020). A small number of truncating and frameshift mutations have also been described and appear to contribute to neurodevelopmental phenotypes such as autism and intellectual disability (Brunklaus et al., 2020; Brunklaus and Lal, 2020). Of direct clinical relevance, we report two unrelated individuals with the same in-frame SCN8A variant. Patient 1 and Patient 2 both carry the same in-frame deletion (p.N1693_C1697del) which removes five amino acids (Figure 1B) from the DIV S5-S6 pore region (Figure 1A). Previous genotype-phenotype correlations found that variants in the pore region were more likely to be associated with loss-of-function rather than gain-of-function effects and epilepsy (Holland et al., 2018; Brunklaus et al., 2020). Consistent with this, Patients 1 and 2 exhibit developmental delay and autism without seizures. Although this variant was not observed in the gnomAD database, additional functional studies will be necessary to resolve its clinical significance. This is particularly relevant since the p.N1693_C1697del variant occurred de novo in Patient 2, but in Patient 1, was inherited from an unaffected parent. Patient 2 was also found to harbor a de novo substitution (p.S1274Y) in ANK3, which is important for connecting integral proteins with the spectrin-actin cytoskeleton (Kordeli and Bennett, 1991). ANK3 is associated with both autosomal dominant and recessive neurodevelopmental disorders, including intellectual disability and autism spectrum disorder (Bi et al., 2012; Iqbal et al., 2013; Kloth et al., 2017; Hu et al., 2019). In silico algorithms suggest a possible deleterious effect of the p.S1274Y variant (e.g., CADD = 26.3); however, this variant is observed twice in the gnomAD database (v3.1.1), and there are no pathogenic variants in the HGMD database that are in close proximity to this variant. Interestingly, ANK3 has been shown to associate with VGSCs via a binding site on the DII-DIII intracellular loop (Jenkins and Bennett, 2001; Lemaillet et al., 2003; Gasser et al., 2012), raising the possibility that it may act as a genetic modifier of SCN8A.
Stringer and others recently described a patient with epileptic encephalopathy who harbors a de novo heterozygous in-frame duplication in SCN8A (p.G1625_I1627dup) and an inherited heterozygous missense variant in CACNA1H (p.G318S) (Stringer et al., 2021). The p.G1625_I1627dup variant (Figure 1A, blue circle), located in the DIVS4 transmembrane domain, is proximal to several pathogenic variants, including p.R1620L (Rossi et al., 2017; Liu et al., 2019) and p.A1622D (Liu et al., 2019). This duplication results in the insertion of three amino acids, including an additional positively charged arginine residue (Figure 1B). Interestingly, while this duplication does not overlap with the amino acids altered in the ΔIRL and ΔVIR mice, it is immediately adjacent to them (Figure 1B). The p.G1625_I1627dup variant resulted in a hyperpolarizing shift of the voltage-dependence of activation of Nav1.6 but had no effect on sodium current density or gating mechanisms (Stringer et al., 2021). This observation is consistent with a gain-of-function effect on the Nav1.6 channel; however, Stringer et al. also demonstrated that the c.952G > A, p.G318S variant in CACNA1H was associated with loss-of-function effects. Further work will be required to resolve the relative contribution of each variant to the clinical presentation.
Johannesen et al. also recently reported two “likely pathogenic” in-frame SCN8A variants in a large cohort of Danish patients with SCN8A mutations (Johannesen et al., 2021). The maternally inherited p.I888_V892delinsM variant was identified in one patient with moderate intellectual disability without epilepsy (Johannesen et al., 2021). Another patient had a maternally inherited variant (E1774_A1777del) and presented with several types of seizures, including febrile, myoclonic, and atonic seizures, but normal intellect (Johannesen et al., 2021). It is unclear whether the mothers of these patients displayed similar clinical features.
There are at least six in-frame variants in the ClinVar database (Table 1; Figure 1A), including the p.N1693_C1697del variant observed in Patients 1 and 2. One ClinVar variant (p.Q1866_Q1867insRELDILR) introduces seven amino acids in the C-terminus (Figure 1A) and is classified as “likely pathogenic”. This variant is not observed in the gnomAD database (v3.1.1), and it is adjacent to several reported variants associated with epileptic encephalopathy, including p.L1865P (Trump et al., 2016), E1870D (Boerma et al., 2016), and R1872 which is one of the most frequently mutated SCN8A residues (Ohba et al., 2014; Larsen et al., 2015; Wagnon et al., 2016). Examination of the population gnomAD database identified 11 in-frame variants in SCN8A (Table 2; Figure 1A). Whether these variants alter the biophysical properties of the channel is currently unknown; however, most of these variants are located in the intracellular DI-DII and DII-DIII linkers (Figure 1A), which are generally more tolerant of variation (Yu and Catterall, 2003; Meisler et al., 2021). In contrast, pathogenic SCN8A variants are typically located in the more conserved parts of the channel, such as the transmembrane segments, inactivation gate, and pore (Wagnon and Meisler, 2015; Brunklaus et al., 2020).
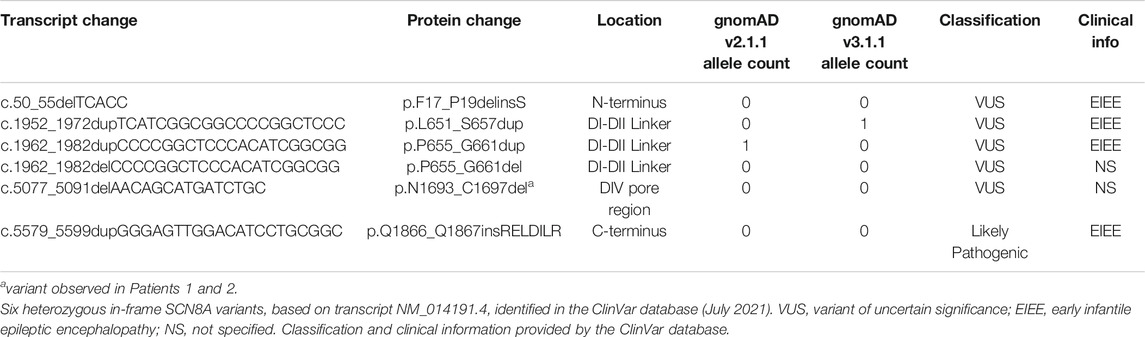
TABLE 1. In-frame SCN8A variants observed in the ClinVar database.
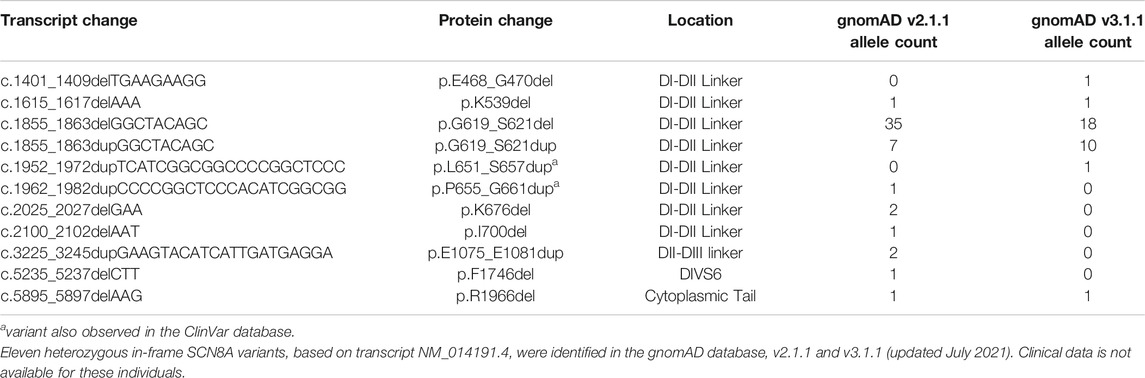
TABLE 2. In-frame SCN8A variants observed in the gnomAD database.
In summary, it is likely that additional in-frame SCN8A variants will be identified as more patients undergo whole exome and genome sequencing, further expanding the genetic landscape of SCN8A-associated disease, and potentially posing challenges for genetic counseling and precision therapy. Due to the rare nature of in-frame SCN8A variants, it is currently unclear if penetrance is reduced for this class of variants. Loss-of-function SCN8A variants (nonsense, frameshift) have been previously reported to exhibit incomplete penetrance (Trudeau et al., 2006); therefore, penetrance may depend on the functional consequence of the individual in-frame variant. The ΔIRL and ΔVIR Scn8a mouse lines will provide the opportunity to further study genotype-phenotype relationships in SCN8A-related disease and will assist in the identification of appropriate treatments for patients with this class of SCN8A mutation.
Data Availability Statement
The human datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author(s).
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Review Board of Emory University. Written informed consent to participate in this study was provided by the participant’s legal guardian/next of kin. The animal study was reviewed and approved by the Institutional Animal Care and Use Committee of Emory University.
Author Contributions
JW and AE contributed to the conception and design of the study. JW performed the experiments, statistical analyses, and wrote the first draft of the manuscript. LS and JT performed experiments and statistical analyses. KB, KM, and KG surveyed databases. KG, PG, SK, and BS performed genetic testing and clinical diagnoses. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This project was supported by the National Institutes of Health (JCW, R21NS114795; AE R03NS114791). The content is solely the author’s responsibility and does not necessarily reflect the official view of the National Institutes of Health. The authors have no financial interests related to this work and declare no competing interests.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank Dr. Deborah Cook for editorial assistance.
References
Allen, N. M., Conroy, J., Shahwan, A., Lynch, B., Correa, R. G., Pena, S. D., et al. (2016). Unexplained Early Onset Epileptic Encephalopathy: Exome Screening and Phenotype Expansion. Epilepsia 57 (1), e12–e17. doi:10.1111/epi.13250
Barton, M. E., Klein, B. D., Wolf, H. H., and White, H. S. (2001). Pharmacological Characterization of the 6 Hz Psychomotor Seizure Model of Partial Epilepsy. Epilepsy Res. 47 (3), 217–227. doi:10.1016/s0920-1211(01)00302-3
Berghuis, B., de Kovel, C. G., van Iterson, L., Lamberts, R. J., Sander, J. W., Lindhout, D., et al. (2015). Complex SCN8A DNA-Abnormalities in an Individual with Therapy Resistant Absence Epilepsy. Epilepsy Res. 115, 141–144. doi:10.1016/j.eplepsyres.2015.06.007
Bi, C., Wu, J., Jiang, T., Liu, Q., Cai, W., Yu, P., et al. (2012). Mutations of ANK3 Identified by Exome Sequencing Are Associated with Autism Susceptibility. Hum. Mutat. 33 (12), 1635–1638. doi:10.1002/humu.22174
Blanchard, M. G., Willemsen, M. H., Walker, J. B., Dib-Hajj, S. D., Waxman, S. G., Jongmans, M. C., et al. (2015). De Novo gain-of-function and Loss-Of-Function Mutations of SCN8A in Patients with Intellectual Disabilities and Epilepsy. J. Med. Genet. 52 (5), 330–337. doi:10.1136/jmedgenet-2014-102813
Boerma, R. S., Braun, K. P., van den Broek, M. P., van Berkestijn, F. M., Swinkels, M. E., Hagebeuk, E. O., et al. (2016). Remarkable Phenytoin Sensitivity in 4 Children with SCN8A-Related Epilepsy: A Molecular Neuropharmacological Approach. Neurotherapeutics 13, 192–197. doi:10.1007/s13311-015-0372-8
Brunklaus, A., Du, J., Steckler, F., Ghanty, , Johannesen, K. M., Fenger, C. D., et al. (2020). Biological Concepts in Human Sodium Channel Epilepsies and Their Relevance in Clinical Practice. Epilepsia 61 (3), 387–399. doi:10.1111/epi.16438
Brunklaus, A., and Lal, D. (2020). Sodium Channel Epilepsies and Neurodevelopmental Disorders: from Disease Mechanisms to Clinical Application. Dev. Med. child Neurol. 62 (7), 784–792. doi:10.1111/dmcn.14519
Bunton-Stasyshyn, R. K. A., Wagnon, J. L., Wengert, E. R., Barker, B. S., Faulkner, A., Wagley, P. K., et al. (2019). Prominent Role of Forebrain Excitatory Neurons in SCN8A Encephalopathy. Brain. doi:10.1093/brain/awy324
Butler, K. M., da Silva, C., Shafir, Y., Weisfeld-Adams, J. D., Alexander, J. J., Hegde, M., et al. (2017). De Novo and Inherited SCN8A Epilepsy Mutations Detected by Gene Panel Analysis. Epilepsy Res. 129, 17–25. doi:10.1016/j.eplepsyres.2016.11.002
Catterall, W. A. (2000). From Ionic Currents to Molecular Mechanisms: the Structure and Function of Voltage-Gated Sodium Channels. Neuron 26 (1), 13–25. doi:10.1016/s0896-6273(00)81133-2
Catterall, W. A. (2012). Voltage-gated Sodium Channels at 60: Structure, Function and Pathophysiology. J. Physiol. 590, 2577–2589. doi:10.1113/jphysiol.2011.224204
Devinsky, O., Marsh, E., Friedman, D., Thiele, E., Laux, L., Sullivan, J., et al. (2016). Cannabidiol in Patients with Treatment-Resistant Epilepsy: an Open-Label Interventional Trial. Lancet Neurol. 15 (3), 270–278. doi:10.1016/S1474-4422(15)00379-8
Escayg, A., and Goldin, A. L. (2010). Sodium Channel SCN1A and Epilepsy: Mutations and Mechanisms. Epilepsia 51, 1650–1658. doi:10.1111/j.1528-1167.2010.02640.x
Escayg, A., MacDonald, B. T., Meisler, M. H., Baulac, S., Huberfeld, G., An-Gourfinkel, I., et al. (2000). Mutations of SCN1A, Encoding a Neuronal Sodium Channel, in Two Families with GEFS+2. Nat. Genet. 24 (4), 343–345. doi:10.1038/74159
Estacion, M., O'Brien, J. E., Conravey, A., Hammer, M. F., Waxman, S. G., Dib-Hajj, S. D., et al. (2014). A Novel De Novo Mutation of SCN8A (Nav1.6) with Enhanced Channel Activation in a Child with Epileptic Encephalopathy. Neurobiol. Dis. 69, 117–123. doi:10.1016/j.nbd.2014.05.017
Fukasawa, T., Kubota, T., Negoro, T., Saitoh, M., Mizuguchi, M., Ihara, Y., et al. (2015). A Case of Recurrent Encephalopathy with SCN2A Missense Mutation. Brain Dev. 37 (6), 631–634. doi:10.1016/j.braindev.2014.10.001
Gardella, E., Becker, F., Møller, R. S., Schubert, J., Lemke, J. R., Larsen, L. H., et al. (2016). Benign Infantile Seizures and Paroxysmal Dyskinesia Caused by an SCN8A Mutation. Ann. Neurol. 79 (3), 428–436. doi:10.1002/ana.24580
Gardella, E., Marini, C., Trivisano, M., Fitzgerald, M. P., Alber, M., Howell, K. B., et al. (2018). The Phenotype of SCN8A Developmental and Epileptic Encephalopathy. Neurology 91 (12), e1112–e24. doi:10.1212/WNL.0000000000006199
Gasser, A., Ho, T. S., Cheng, X., Chang, K. J., Waxman, S. G., Rasband, M. N., et al. (2012). An ankyrinG-Binding Motif Is Necessary and Sufficient for Targeting Nav1.6 Sodium Channels to Axon Initial Segments and Nodes of Ranvier. J. Neurosci. 32 (21), 7232–7243. doi:10.1523/JNEUROSCI.5434-11.2012
Hawkins, N. A., Martin, M. S., Frankel, W. N., Kearney, J. A., and Escayg, A. (2011). Neuronal Voltage-Gated Ion Channels Are Genetic Modifiers of Generalized Epilepsy with Febrile Seizures Plus. Neurobiol. Dis. 41 (3), 655–660. doi:10.1016/j.nbd.2010.11.016
Heyne, H. O., Baez-Nieto, D., Iqbal, S., Palmer, D. S., Brunklaus, A., May, P., et al. (2020). Predicting Functional Effects of Missense Variants in Voltage-Gated Sodium and Calcium Channels. Sci. Transl Med. 12 (556). doi:10.1126/scitranslmed.aay6848
Hodgkin, A. L., and Huxley, A. F. (1952). A Quantitative Description of Membrane Current and its Application to Conduction and Excitation in Nerve. J. Physiol. 117 (4), 500–544. doi:10.1113/jphysiol.1952.sp004764
Holland, K. D., Bouley, T. M., and Horn, P. S. (2018). Location: A Surrogate for Personalized Treatment of Sodium Channelopathies. Ann. Neurol. 84 (1), 1–9. doi:10.1002/ana.25268
Hu, H., Kahrizi, K., Musante, L., Fattahi, Z., Herwig, R., Hosseini, M., et al. (2019). Genetics of Intellectual Disability in Consanguineous Families. Mol. Psychiatry 24 (7), 1027–1039. doi:10.1038/s41380-017-0012-2
Hu, W., Tian, C., Li, T., Yang, M., Hou, H., and Shu, Y. (2009). Distinct Contributions of Na(v)1.6 and Na(v)1.2 in Action Potential Initiation and Backpropagation. Nat. Neurosci. 12 (8), 996–1002. doi:10.1038/nn.2359
Inglis, G. A. S., Wong, J. C., Butler, K. M., Thelin, J. T., Mistretta, O. C., Wu, X., et al. (2020). Mutations in the Scn8a DIIS4 Voltage Sensor Reveal New Distinctions Among Hypomorphic and Null Nav 1.6 Sodium Channels. Genes, Brain Behav. 19 (4), e12612. doi:10.1111/gbb.12612
Iqbal, Z., Vandeweyer, G., van der Voet, M., Waryah, A. M., Zahoor, M. Y., Besseling, J. A., et al. (2013). Homozygous and Heterozygous Disruptions of ANK3: at the Crossroads of Neurodevelopmental and Psychiatric Disorders. Hum. Mol. Genet. 22 (10), 1960–1970. doi:10.1093/hmg/ddt043
Jenkins, S. M., and Bennett, V. (2001). Ankyrin-G Coordinates Assembly of the Spectrin-Based Membrane Skeleton, Voltage-Gated Sodium Channels, and L1 CAMs at Purkinje Neuron Initial Segments. J. Cel Biol 155 (5), 739–746. doi:10.1083/jcb.200109026
Johannesen, K. M., Gardella, E., Scheffer, I., Howell, K., Smith, D. M., Helbig, I., et al. (2018). Early Mortality in SCN8A-Related Epilepsies. Epilepsy Res. 143, 79–81. doi:10.1016/j.eplepsyres.2018.04.008
Johannesen, K. M., Liu, Y., Koko, M., Gjerulfsen, C. E., Sonnenberg, L., Schubert, J., et al. (2021). Genotype-phenotype Correlations in SCN8A-Related Disorders Reveal Prognostic and Therapeutic Implications. Brain 25, awab321. doi:10.1093/brain/awab321
Jones, J. M., Dionne, L., Dell'Orco, J., Parent, R., Krueger, J. N., Cheng, X., et al. (2016). Single Amino Acid Deletion in Transmembrane Segment D4S6 of Sodium Channel Scn8a (Nav1.6) in a Mouse Mutant with a Chronic Movement Disorder. Neurobiol. Dis. 89, 36–45. doi:10.1016/j.nbd.2016.01.018
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 581 (7809), 434–443. doi:10.1038/s41586-020-2308-7
Kloth, K., Denecke, J., Hempel, M., Johannsen, J., Strom, T. M., Kubisch, C., et al. (2017). First De Novo ANK3 Nonsense Mutation in a Boy with Intellectual Disability, Speech Impairment and Autistic Features. Eur. J. Med. Genet. 60 (9), 494–498. doi:10.1016/j.ejmg.2017.07.001
Kontis, K. J., and Goldin, A. L. (1997). Sodium Channel Inactivation Is Altered by Substitution of Voltage Sensor Positive Charges. J. Gen. Physiol. 110 (4), 403–413. doi:10.1085/jgp.110.4.403
Kontis, K. J., Rounaghi, A., and Goldin, A. L. (1997). Sodium Channel Activation Gating Is Affected by Substitutions of Voltage Sensor Positive Charges in All Four Domains. J. Gen. Physiol. 110 (4), 391–401. doi:10.1085/jgp.110.4.391
Kordeli, E., and Bennett, V. (1991). Distinct Ankyrin Isoforms at Neuron Cell Bodies and Nodes of Ranvier Resolved Using Erythrocyte Ankyrin-Deficient Mice. J. Cel Biol 114 (6), 1243–1259. doi:10.1083/jcb.114.6.1243
Lamar, T., Vanoye, C. G., Calhoun, J., Wong, J. C., Dutton, S. B. B., Jorge, B. S., et al. (2017). SCN3A Deficiency Associated with Increased Seizure Susceptibility. Neurobiol. Dis. 102, 38–48. doi:10.1016/j.nbd.2017.02.006
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Res. 46 (D1), D1062–D7. doi:10.1093/nar/gkx1153
Larsen, J., Carvill, G. L., Gardella, E., Kluger, G., Schmiedel, G., Barisic, N., et al. (2015). The Phenotypic Spectrum of SCN8A Encephalopathy. Neurology 84 (5), 480–489. doi:10.1212/WNL.0000000000001211
Lemaillet, G., Walker, B., and Lambert, S. (2003). Identification of a Conserved Ankyrin-Binding Motif in the Family of Sodium Channel Alpha Subunits. J. Biol. Chem. 278 (30), 27333–27339. doi:10.1074/jbc.M303327200
Liu, Y., Schubert, J., Sonnenberg, L., Helbig, K. L., Hoei-Hansen, C. E., Koko, M., et al. (2019). Neuronal Mechanisms of Mutations in SCN8A Causing Epilepsy or Intellectual Disability. Brain 142 (2), 376–390. doi:10.1093/brain/awy326
Lossin, C. (2009). A Catalog of SCN1A Variants. Brain Dev. 31, 114–130. doi:10.1016/j.braindev.2008.07.011
Makinson, C. D., Tanaka, B. S., Lamar, T., Goldin, A. L., and Escayg, A. (2014). Role of the hippocampus in Nav1.6 (Scn8a) Mediated Seizure Resistance. Neurobiol. Dis. 68, 16–25. doi:10.1016/j.nbd.2014.03.014
Martin, M. S., Tang, B., Papale, L. A., Yu, F. H., Catterall, W. A., and Escayg, A. (2007). The Voltage-Gated Sodium Channel Scn8a Is a Genetic Modifier of Severe Myoclonic Epilepsy of Infancy. Hum. Mol. Genet. 16 (23), 2892–2899. doi:10.1093/hmg/ddm248
Meisler, M. H., Hill, S. F., and Yu, W. (2021). Sodium Channelopathies in Neurodevelopmental Disorders. Nat. Rev. Neurosci. 22 (3), 152–166. doi:10.1038/s41583-020-00418-4
Meisler, M. H., O'Brien, J. E., and Sharkey, L. M. (2010). Sodium Channel Gene Family: Epilepsy Mutations, Gene Interactions and Modifier Effects. J. Physiol. 588, 1841–1848. doi:10.1113/jphysiol.2010.188482
Noda, M., Shimizu, S., Tanabe, T., Takai, T., Kayano, T., Ikeda, T., et al. (1984). Primary Structure of Electrophorus Electricus Sodium Channel Deduced from cDNA Sequence. Nature 312 (5990), 121–127. doi:10.1038/312121a0
Ohba, C., Kato, M., Takahashi, S., Lerman-Sagie, T., Lev, D., Terashima, H., et al. (2014). Early Onset Epileptic Encephalopathy Caused by De Novo SCN8A Mutations. Epilepsia 55 (7), 994–1000. doi:10.1111/epi.12668
Papale, L. A., Beyer, B., Jones, J. M., Sharkey, L. M., Tufik, S., Epstein, M., et al. (2009). Heterozygous Mutations of the Voltage-Gated Sodium Channel SCN8A Are Associated with Spike-Wave Discharges and Absence Epilepsy in Mice. Hum. Mol. Genet. 18 (9), 1633–1641. doi:10.1093/hmg/ddp081
Prichahd, J. W., Gallagher, B. B., and Glaser, G. H. (1969). Experimental Seizure-Threshold Testing with Flurothyl. J. Pharmacol. Exp. Ther. 166 (1), 170–178.
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Rossi, M., El-Khechen, D., Black, M. H., Farwell Hagman, K. D., Tang, S., and Powis, Z. (2017). Outcomes of Diagnostic Exome Sequencing in Patients with Diagnosed or Suspected Autism Spectrum Disorders. Pediatr. Neurol. 70, 34–e2. doi:10.1016/j.pediatrneurol.2017.01.033
Saitoh, M., Ishii, A., Ihara, Y., Hoshino, A., Terashima, H., Kubota, M., et al. (2015). Missense Mutations in Sodium Channel SCN1A and SCN2A Predispose Children to Encephalopathy with Severe Febrile Seizures. Epilepsy Res. 117, 1–6. doi:10.1016/j.eplepsyres.2015.08.001
Shapiro, L., Wong, J. C., and Escayg, A. (2019). Reduced Cannabinoid 2 Receptor Activity Increases Susceptibility to Induced Seizures in Mice. Epilepsia 60 (12), 2359–2369. doi:10.1111/epi.16388
Sokolov, S., Scheuer, T., and Catterall, W. A. (2005). Ion Permeation through a Voltage- Sensitive Gating Pore in Brain Sodium Channels Having Voltage Sensor Mutations. Neuron 47 (2), 183–189. doi:10.1016/j.neuron.2005.06.012
Stringer, R. N., Jurkovicova-Tarabova, B., Souza, I. A., Ibrahim, J., Vacik, T., Fathalla, W. M., et al. (2021). De Novo SCN8A and Inherited Rare CACNA1H Variants Associated with Severe Developmental and Epileptic Encephalopathy. Mol. Brain 14 (1), 126. doi:10.1186/s13041-021-00838-y
Trudeau, M. M., Dalton, J. C., Day, J. W., Ranum, L. P., and Meisler, M. H. (2006). Heterozygosity for a Protein Truncation Mutation of Sodium Channel SCN8A in a Patient with Cerebellar Atrophy, Ataxia, and Mental Retardation. J. Med. Genet. 43, 527–530. doi:10.1136/jmg.2005.035667
Trump, N., McTague, A., Brittain, H., Papandreou, A., Meyer, E., Ngoh, A., et al. (2016). Improving Diagnosis and Broadening the Phenotypes in Early-Onset Seizure and Severe Developmental Delay Disorders through Gene Panel Analysis. J. Med. Genet. 53 (5), 310–317. doi:10.1136/jmedgenet-2015-103263
Vanoye, C. G., Gurnett, C. A., Holland, K. D., George, A. L., and Kearney, J. A. (2014). Novel SCN3A Variants Associated with Focal Epilepsy in Children. Neurobiol. Dis. 62 (0), 313–322. doi:10.1016/j.nbd.2013.10.015
Veeramah, K. R., O'Brien, J. E., Meisler, M. H., Cheng, X., Dib-Hajj, S. D., Waxman, S. G., et al. (2012). De Novo pathogenic SCN8A Mutation Identified by Whole-Genome Sequencing of a Family Quartet Affected by Infantile Epileptic Encephalopathy and SUDEP. Am. J. Hum. Genet. 90 (3), 502–510. doi:10.1016/j.ajhg.2012.01.006
Wagnon, J. L., Barker, B. S., Hounshell, J. A., Haaxma, C. A., Shealy, A., Moss, T., et al. (2016). Pathogenic Mechanism of Recurrent Mutations of SCN8A in Epileptic Encephalopathy. Ann. Clin. Transl Neurol. 3 (2), 114–123. doi:10.1002/acn3.276
Wagnon, J. L., Korn, M. J., Parent, R., Tarpey, T. A., Jones, J. M., Hammer, M. F., et al. (2015). Convulsive Seizures and SUDEP in a Mouse Model of SCN8A Epileptic Encephalopathy. Hum. Mol. Genet. 24 (2), 506–515. doi:10.1093/hmg/ddu470
Wagnon, J. L., and Meisler, M. H. (2015). Recurrent and Non-recurrent Mutations of SCN8A in Epileptic Encephalopathy. Front. Neurol. 6, 104. doi:10.3389/fneur.2015.00104
Wang, W., and Frankel, W. N. (2021). Overlaps, Gaps, and Complexities of Mouse Models of Developmental and Epileptic Encephalopathy. Neurobiol. Dis. 148, 105220. doi:10.1016/j.nbd.2020.105220
Wisedchaisri, G., Tonggu, L., McCord, E., Gamal El-Din, T. M., Wang, L., Zheng, N., et al. (2019). Resting-State Structure and Gating Mechanism of a Voltage-Gated Sodium Channel. Cell 178 (4), 993–e12. doi:10.1016/j.cell.2019.06.031
Wong, J. C., Dutton, S. B., Collins, S. D., Schachter, S., and Escayg, A. (2016). Huperzine A Provides Robust and Sustained protection against Induced Seizures in Scn1a Mutant Mice. Front. Pharmacol. 7 (357), 357. doi:10.3389/fphar.2016.00357
Wong, J. C., Grieco, S. F., Dutt, K., Chen, L., Thelin, J. T., Inglis, G. A. S., et al. (2021). Autistic-like Behavior, Spontaneous Seizures, and Increased Neuronal Excitability in a Scn8a Mouse Model. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology.
Wong, J. C., Makinson, C. D., Lamar, T., Cheng, Q., Wingard, J. C., Terwilliger, E. F., et al. (2018). Selective Targeting of Scn8a Prevents Seizure Development in a Mouse Model of Mesial Temporal Lobe Epilepsy. Sci. Rep. 8 (1), 126. doi:10.1038/s41598-017-17786-0
Wong, J. C., Shapiro, L., Thelin, J. T., Heaton, E. C., Zaman, R. U., D'Souza, M. J., et al. (2021). Nanoparticle Encapsulated Oxytocin Increases Resistance to Induced Seizures and Restores Social Behavior in Scn1a-Derived Epilepsy. Neurobiol. Dis. 147, 105147. doi:10.1016/j.nbd.2020.105147
Wong, J. C., Thelin, J. T., and Escayg, A. (2019). Donepezil Increases Resistance to Induced Seizures in a Mouse Model of Dravet Syndrome. Ann. Clin. Transl Neurol. 6 (8), 1566–1571. doi:10.1002/acn3.50848
Yang, N., George, A. L., and Horn, R. (1996). Molecular Basis of Charge Movement in Voltage-Gated Sodium Channels. Neuron 16 (1), 113–122. doi:10.1016/s0896-6273(00)80028-8
Keywords: SCN8A, sodium channel, epilepsy, seizure, mouse, mutation
Citation: Wong JC, Butler KM, Shapiro L, Thelin JT, Mattison KA, Garber KB, Goldenberg PC, Kubendran S, Schaefer GB and Escayg A (2021) Pathogenic in-Frame Variants in SCN8A: Expanding the Genetic Landscape of SCN8A-Associated Disease. Front. Pharmacol. 12:748415. doi: 10.3389/fphar.2021.748415
Received: 27 July 2021; Accepted: 21 October 2021;
Published: 17 November 2021.
Edited by:
Heather Ray, Idaho State University, United StatesReviewed by:
Miriam Meisler, University of Michigan, United StatesMark R. Estacion, Yale University, United States
Copyright © 2021 Wong, Butler, Shapiro, Thelin, Mattison, Garber, Goldenberg, Kubendran, Schaefer and Escayg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jennifer C. Wong, amVubmlmZXIuYy53b25nQGVtb3J5LmVkdQ==