 Fei Lin
Fei Lin Xinhua Lin1†
Xinhua Lin1† Xuewen Wang
Xuewen Wang Bing Chen
Bing Chen Hong Yao
Hong Yao Lingyi Huang
Lingyi Huang- 1Department of Pharmaceutical Analysis, School of Pharmacy, Fujian Medical University, Fuzhou, China
- 2Department of Preventive Medicine, School of Public Health, Fujian Medical University, Fuzhou, China
Introduction: Selaginella doederleinii Hieron is a traditional Chinese herbal medicine, the ethyl acetate extract from Selaginella doederleinii (SDEA) showed favorable anticancer potentials. However, the effect of SDEA on human cytochrome P450 enzymes (CYP450) remains unclear. To predict the herb-drug interaction (HDI) and lay the groundwork for further clinical trials, the inhibitory effect of SDEA and its four constituents (Amentoflavone, Palmatine, Apigenin, Delicaflavone) on seven CYP450 isoforms were investigated by using the established CYP450 cocktail assay based on LC-MS/MS.
Methods: Appropriate substrates for seven tested CYP450 isoforms were selected to establish a reliable cocktail CYP450 assay based on LC-MS/MS. The contents of four constituents (Amentoflavone, Palmatine, Apigenin, Delicaflavone) in SDEA were determined as well. Then, the validated CYP450 cocktail assay was applied to test the inhibitory potential of SDEA and four constituents on CYP450 isoforms.
Results: SDEA showed strong inhibitory effect on CYP2C9 and CYP2C8 (IC50 ≈ 1 μg/ml), moderate inhibitory effect against CYP2C19, CYP2E1 and CYP3A (IC50 < 10 μg/ml). Among the four constituents, Amentoflavone had the highest content in the extract (13.65%) and strongest inhibitory effect (IC50 < 5 μM), especially for CYP2C9, CYP2C8 and CYP3A. Amentoflavone also showed time-dependent inhibition on CYP2C19 and CYP2D6. Apigenin and Palmatine both showed concentration-dependent inhibition. Apigenin inhibited CYP1A2, CYP2C8, CYP2C9, CYP2E1 and CYP3A. Palmatine inhibited CYP3A and had a weak inhibitory effect on CYP2E1. As for Delicaflavone, which has the potential to develop as an anti-cancer agent, showed no obvious inhibitory effect on CYP450 enzymes.
Conclusion: Amentoflavone may be one of the main reasons for the inhibition of SDEA on CYP450 enzymes, the potential HDI should be considered when SDEA or Amentoflavone were used with other clinical drugs. On the contrast, Delicaflavone is more suitable to develop as a drug for clinical use, considering the low level of CYP450 metabolic inhibition.
1 Introduction
Numerous studies have shown that drug-drug interaction (DDI) can cause serious adverse reactions, drug ineffectiveness and even death (Han et al., 2012; Kozakai et al., 2012; Al-Ramahi et al., 2015; Guengerich, 2021). Induction or inhibition of human Cytochromes P450 (CYP450) enzyme by compounds is the main cause for drug-drug metabolic interaction (Qin et al., 2013). About 60%–70% of the clinical drugs are clear by phase I reaction, where 75% of the involved metabolizing enzymes are CYP450 isoforms (Kim et al., 2005; Lee and Kim, 2013). Usually, CYP450 inhibition leads to drug accumulation and even toxicity (Lin and Lu, 1998; Guengerich, 2008; Hakkola et al., 2020). Based on that, DDI test is necessary for a chemical from pre-clinical stage into clinical trials in pharmaceutical industry. The United States Food and Drug Administration (FDA) guidelines for DDI test have recommended several major human CYP450 isoforms, including CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A. Those CYP450 isoforms have account for mostly enzymatic metabolism of clinical drugs (Prueksaritanont et al., 2013; Sudsakorn et al., 2020). Although CYP2E1 was not clearly specified in the FDA guidelines, many studies have revealed that CYP2E1 can metabolize Coumarin, Quinoline, Isoniazid, ethanol, Caffeine, acetaminophen and other substances. Therefore, it is reasonable to include CYP2E1 when conducting the DDI study (Valicherla et al., 2019).
Nowadays, many people consume herbal products for self-health or medical use in their daily life. Similar to small molecule medicine, when people take herbal products and drugs at the same time, potential herb-drug interaction (HDI) should be considered as well (van Breemen, 2015). In the past 20 years, more and more studies have shown that there are significant CYP450 enzyme interaction between herbal products and prescription medicines, which will increase patients’ risk of unnecessary and unintended adverse effect. For example, Angelica dahurica root extract inhibited CYP450 enzymes such as CYP2C, CYP3A and CYP2D1, and caused pharmacokinetic and pharmacodynamic interactions with Tolbutamide and Diazepam (Ishihara et al., 2000). Shenmai injection, one of the most popular herbal medicines in China, has reported to inhibit CYP3A1/2 and CYP2C6 (Xia et al., 2010). Researchers have found that Huanglian [Rhizoma coptidis (L.)] has obvious inhibition on CYP2D6 (Han et al., 2011). Newbouldia laevis extract inhibited the enzyme activities of CYP1A2, CYP2C9, CYP2C19, and Newbouldia laevis showed time-dependent inhibition effect on CYP1A2 (Thomford et al., 2016). Licorice is a common use dietary supplement even is consumed as a condiment, which was found to inhibit several CYP450 enzymes. What’s more, three major licorice species (G. glabra, G. uralensis and G. inflata) showed unique pattern pf enzyme inhibition (Li et al., 2017). The most reported CYP450 isoforms involved in HDI are CYP1A2, CYP2C, CYP2D6 and CYP3A, which is similar to the list of small molecules DDI test according to FDA guideline (Choi et al., 2016; Awortwe et al., 2018).
Selaginella doederleinii Hieron belongs to Selaginella genus of the Selaginellaceae family and is commonly used as a traditional Chinese herbal medicine, which was reported to have anti-hyperglycemia, anti-virus, anti-cancer and other pharmacological activities, and used in the treatment of nasopharyngeal carcinoma, lung cancer and trophoblastic tumor (Zheng et al., 2005; Chen et al., 2018). The previous in vitro and in vivo studies have revealed that the ethyl acetate extract from S. doederleinii (SDEA) had anti-cancer effect (Yao et al., 2017), especially for lung and colorectal cancer (Sui et al., 2016; Li et al., 2020). Those main ingredients from the extract, such as Amentoflavone, Palmatine, Apigenin and Delicaflavone, may account for the pharmacological activities of SDEA. Among those components, Delicaflavone was proved to induce apoptosis in cervical cancer HeLa cells (Yao et al., 2019), colorectal cancer cells (Yao et al., 2020) and lung cancer cells (Sui et al., 2017). Moreover, Delicaflavone can reverse cisplatin resistance via endoplasmic reticulum stress signaling pathway in non-small cell lung cancer cells, which may serve as a useful adjunct in treatment of cisplatin-resistant lung cancer (Wang et al., 2020). Therefore, SDEA and Delicaflavone are the most promising anti-tumor agents, whether use alone or in combination with other drugs.
Although the pharmacological activities of SDEA and its bioactive compounds have been extensively studied, there is no report on HDI of SDEA on human CYP450 enzymes. As a plant extract, the composition of SDEA is complex, whose metabolic process in human body is not fully understood. Meanwhile, the HDI study for the bioactive components of SDEA have not been conducted either. The pre-clinical study on the ADME properties of candidate compound is of great significance for improving success rate, reducing cost and toxic risk, which is crucial for further drug development. This rule also applies to herbal product drug development (Brantley et al., 2014). Regulatory agencies in most countries, such as FDA, EMA, NMPA, et al., all require that new chemicals should be clarified the possible inhibition or induction effect on human CYP450 enzyme in the investigational new drug (IND) application stage. For newly developed medicines, they are likely to be accepted on the market only if they have clear human metabolic information (Sudsakorn et al., 2020). Based on that, it is necessary for SDEA and its bioactive compounds to screen the potential HDI before moving into clinical trials stage. Thus, a reliable HDI test assay is needed.
Compared to in vivo animal test, in vitro CYP450 probe substrate approach can provide reliable context and prospective knowledge in terms of less cost and time (Bjornsson et al., 2003; Venkatakrishnan et al., 2003), which are widely used in DDI and HDI study. Moreover, since human and animals (rat, mice et al.) have essential differences in basic tissue, cell structure and metabolic types, in vitro HDI assay usually uses commercial human liver microsomes (HLM) or human recombinant CYP450 enzymes (Lu and Di, 2020). Traditional CYP450 substrate method owns low efficiency because it only uses one probe substrate to test the activity of one metabolic enzyme at a time (“one-in-one” assay), which is difficult to meet the requirements of high throughput screening (Lin and Lu, 1998). However, as the development of high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS), this problem has been solved finally. The triple quadrupole LC-MS can select specific substrate metabolites for determination by using multiple reaction monitoring (MRM), which has better selectivity and specificity compared with other analytical methods (liquid chromatography-UV, fluorescence and luminescence detections, etc.) (Youdim and Saunders, 2010). Based on the advantages of LC-MS/MS, the cocktail CYP450 assay (“N-in-one” assay) have been developed, which can simultaneously test the inhibitory effects of several CYP450 isoforms in one assay, with the effect of significantly reducing cost and time to evaluate DDI or HDI (Li et al., 2015; Liua et al., 2015; Spaggiari et al., 2016). The cocktail assay is very suitable for high-throughput screening in drug development process, especially for early drug metabolism studies, which is extremely important for dose design of those compounds with multiple metabolic pathways. It has been applied to monitor the in vitro inhibition activity of various CYP450 enzymes and time-dependent inhibition study (Chen et al., 2016). Therefore, in our study, an in vitro cocktail CYP450 assay was established with LC-MS/MS to reflect the activities of corresponding CYP450 isoforms. This assay was able to monitor the metabolic changes of specific substrates and validated by known enzymes inhibitors, so that it could achieve more accurate detection, better sensitivity and less interference.
In summary, we established an in vitro cocktail CYP450 assay by LC-MS/MS to detect the inhibitory effects of SDEA and its four constituents (Amentoflavone, Palmatine, Apigenin and Delicaflavone) on seven human CYP450 isoforms: CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A. To determine whether SDEA and its components are the potential inhibitors of any of those important human CYP450 isoforms. Our research could reveal the metabolic interactions between CYP450 enzymes and SDEA constituents, which could clarify the clinical safety issues and promote the drug development of SDEA and its constituents.
2 Materials and methods
2.1 Materials and chemicals
Pooled mixed gender human liver microsomes from 50 donors were purchased from XenoTech (Lenexa, United States), β-nicotinamide adenine dinucleotide phosphate (NADPH) were purchased from solarbiobiotech (Beijing, China), Omeprazole (HPLC purity> 99%), Taxol (HPLC purity> 99%), Tolbutamide (HPLC purity> 99%), Chlorzoxazone (HPLC purity> 99%), Dextromethorphan Hydrobromide (HPLC purity> 98%), Alpha-Naphthoflavone (HPLC purity> 98%), Fluconazole (HPLC purity> 98%), Quercetin (HPLC purity> 98%) and Ketoconazole (HPLC purity> 99%) were purchased from Dalian Meilunbio. Co. Ltd (Dalian, China), Phenaceti (HPLC purity> 99%), Sulfaphenazolum (HPLC purity> 99%), Quinidine (HPLC purity> 99%) were purchased from Shyuanye Biotechnology Co. Ltd (Shanghai, China), Testosterone (HPLC purity> 98%) were purchased from Derick Biotechnology Co. Ltd (Chengdu, China), 4-Methylpyrazole (HPLC purity> 97%) were purchased from J&K Scientific (San Jose, United States).
The SDEA extract was prepared following our previously described procedure (Sui et al., 2016; Yao et al., 2017). Delicaflavone (purity≥ 98%, determined by the peak area normalization method using HPLC-PDA) were isolated from S. doederleinii and the structure was fully elucidated by MS, UV, 1H-NMR and 13C-NMR, which was confirmed by comparison with the literatures (Li et al., 2013; Li et al., 2014; Yao et al., 2017; Chen et al., 2018). Amentoflavone (HPLC purity> 98%) and Apigenin (HPLC purity> 98%) were purchased from Dalian Meilunbio. Co. Ltd (Dalian, China), Palmatine (HPLC purity> 98%) was purchased from Shyuanye Biotechnology Co. Ltd (Shanghai, China).
Methanol and acetonitrile (HPLC grade) were purchased from Merck KGaA (Darmstadt, Germany), formic acid (HPLC grade) was purchased from Aladdin (Shanghai, China), ethanol (analytical grade) was obtained from Sinopharm Chemical Reagents (Shanghai, China), and ultrapure water was prepared by a Millpore Milli-Q system (Beddford, United States).
2.2 Quantitative analysis of four constituents in SDEA
Amentoflavone, Delicaflavone, Apigenin, Palmatine and Rutin (internal standard) were precisely weighed and dissolved in methanol to obtain a stock solution with a concentration of 1 mg/ml, which was stored in refrigerator at 4°C. Before use, the stock solution was diluted with methanol into standard working solution and quality control working solution.
The concentrations of Amentoflavone and Apigenin standard curve working solution were: 3.125, 6.25, 12.5, 25, 50, 100, 200, 400 and 800 ng/ml; the concentrations of Amentoflavone and Apigenin quality control (QC) working solution were as 30, 150 and 650 ng/ml. The concentrations of Palmatine standard curve working solution were: 0.39, 0.78, 1.56, 3.125, 6.25, 12.5, 25, 50, 100 and 200 ng/ml; three level QCs were as 3, 30, and 150 ng/ml. The concentrations of Delicaflavone standard curve working solution were: 3.125, 6.25, 12.5, 25, 50, 100 and 200 ng/ml; and three QCs were as 8, 30 and 150 ng/ml. SDEA solid was accurately weighed and dissolved with methanol, then sonicated until complete dissolution. Diluted it with methanol to 1, 5, 25, 40 μg/ml and mix with the 40 ng/ml internal standard (Rutin) respectively. The mixture was vortexed for 2 min, then centrifuged at 13000 rpm for 10 min at room temperature. The supernatant was acquired for LC-MS/MS quantitative analysis.
2.3 Establishment of cytochrome P450 cocktail inhibition assay
Potassium phosphate buffer (200 µl, 0.1 M, pH 7.4) containing 1 mM NADPH, 0.5 mg/mL human liver microsomes, and a cocktail assay of seven probe substrates (Phenacetin for CYP1A2, Paclitaxel for CYP2C8, Tolbutamide for CYP2C9, Omeprazole for CYP2C19, Dextromethorphan for CYP2D6, Chlorzoxazone for CYP2E1, Dextromethorphan and Testosterone for CYP3A) or a single substrate (≤Km) were incubated at 37°C for 15 min (Supplementary Table S1). The contents of organic solvent and DMSO in incubation mixture was under 1% (v/v) and 0.1% (v/v) respectively. Reactions were terminated by adding 200 µL of an ice-cold stop solution consisting of methanol containing Rutin (2 µg/ml) as internal standard. Samples were subsequently cooled in ice bath to precipitate proteins. Supernatants were collected into clean tubes after centrifugation at 12000 rpm at 4°C for 10 min prior to inject into LC-MS/MS.
2.3.1 Determination of linearity of metabolite formation
To determine the optimal incubation time for each CYP substrate, human liver microsomes (0.5 mg/mL) were incubated at 37°C for 0, 5, 10, 20, 30 and 60 min with each CYP substrates. All substrates concentration were 1 µM. After quantitative analysis using LC-MS/MS, the linearity of metabolite formation was evaluated.
2.3.2 Validation of direct CYP450 inhibition
For the determination of inhibition curves using single substrate and the cocktail, seven selective CYP inhibitors were used at different concentrations as follows: 0.05–1 µM α-Naphthoflavone for CYP1A2, 1–50 μM Fluconazole for CYP2C19, 0.1–15 µM Quercetin for CYP2C8, 0.01–5 µM Sulfaphenazole for CYP2C9, 0.005–2 µM Quinidine for CYP2D6, 0.1–5 µM 4-Methylpyrazole for CYP2E1, and 0.005–1 µM Ketoconazole for CYP3A (Supplementary Table S2).
2.4 Concentration-dependent inhibitory effect of SDEA on CYP450 isoforms
Inhibitory effect of SDEA on CYP450 isoforms was determined by cocktail assay. SDEA at 10 concentrations from 0 to 200 µg/ml were used to measure IC50 value. The amount of metabolites produced by incubation without SDEA (control group) was taken as 100%, then draw inhibition efficiency curve.
2.5 Concentration-dependent inhibitory effect of four components in SDEA on CYP450 isoforms
Single-point inhibition was assessed by Delicaflavone, Amentoflavone, Apigenin and Palmatine at 10 µM, using the same protocol as described in Section 2.3. Percentages of control activity were calculated to determine inhibition potency. The inhibition curves were obtained by incubating the cocktail of substrates with 10–11 different compounds’ concentrations, where Delicaflavone and Amentoflavone were 0–50 µM.
2.6 Time-dependent inhibition study of SDEA, Delicaflavone and Amentoflavone
In order to explore whether SDEA, Delicaflavone and Amentoflavone have time-dependent inhibition on CYP450 isoforms, the cocktail inhibition assay (Section 2.3.) was used. First, SDEA (0.5 µg/ml), Delicaflavone (2 µM), and Amentoflavone (0.1 µM) were incubated with HLM (0.5 mg/ml) in the presence and absence of 1 mM NADPH for 35 min. After pre-incubation, a 20 µl aliquot of the incubation mixture was transferred to 180 µl potassium phosphate buffer (0.1 M, pH 7.4) containing 1 mM NADPH and mixed probe substrates for secondary culture, then stopped reaction after 15 min. Enzymes that may be involved in time-dependent were pre-incubated for 0, 5, 10, 15 and 35 min respectively, and then re-incubated to determine the type of inhibition. Residual enzyme activity pre-incubated without NADPH was set as the control group, then drawing suppression curves based on the measured data.
2.7 LC-MS/MS conditions
All metabolites and surrogate standards were analyzed by using LC-MS/MS on a Shimadzu (Kyoto, Japan) LCMS-8040 triple quadrupole mass spectrometer. Chromatographic separation was performed using an Ultimate® XB-C18 (100 × 2.1 mm, 3 μm) with gradient elution of water (0.2% formic acid) and methanol at 0.3 mL/min (gradient B). Column maintained at 40°C. 5 μl of supernatant was injected for LC-MS/MS analysis.
The following optimized MS parameters were used: ion spray voltage: 6.0 kV; Nebulizer gas flow: 3 L/min; Drying gas flow: 12 L/min; DL temperature: 250°C; Heatblock temperature: 400°C.
2.7.1 Quantitative analysis of SDEA constituents
The gradient elution procedure is: 0–1 min, 30%–45% B; 1–3 min, 40%–75% B; 3–5 min, 75%–85% B; 5–8 min, 85%–95% B; 8–12 min, 95% B; 12–13 min, 95%–30% B; 30% B for equilibration.
Detection was carried out using electrospray with polarity switching, collision-induced dissociation, and multiple reaction monitoring (MRM). The MRM mode transition: m/z 536.90 → 375.00 (−) for Amentoflavone; m/z 538.9 → 256.95 (+) for Delicaflavone; m/z 352.00 → 336.10 (+) for Palmatine; m/z 271.15 → 153.05 (+) for Apigenin; m/z 611.00 → 302.85 (+) for Rutin (IS). The collision energies (CE) of the four components were 33 V, −45 V, −35 V, −35 V and −35 V respectively.
2.7.2 Determination of the inhibitory effects of SDEA and four constituents on CYP450 enzymes by cocktail assay
A gradient condition was applied with the following program: 0–1 min, 10%–20% B; 1–1.01 min, 20%–75% B; 1.01–3 min, 75%–77% B; 3–5 min, 77%–80% B; 5–8 min, 80%–85% B; 8–9 min, 85%–95% B; 9–12 min, 95% B; and then 10% B for equilibration. For specific MRM parameters, please refer to Supplementary Table S3.
2.8 Data analysis
Quantitative LC-MS/MS data were analyzed using Shimadzu LabSolutions software (Kyoto, Japan). The Km and IC50 values were determined by using the GraphPad Prism 5.0 software (San Diego, United States).
3 Results
3.1 The content of four constituents in SDEA
The structures of Amentoflavone, Delicaflavone, Palmatine, and Apigenin were shown in Figure 1. Amentoflavone and Apigenin were linearly related in a range of 3.125–800 ng/mL by using 1/X2 weighting. Palmatine showed a linear relationship in the range of 0.391–200 ng/ml, and Delicaflavone showed a linear relationship in the range of 3.125–200 ng/ml. The four SDEA constituents showed a good linear relationship over their concentration range with coefficient of determination r2 ≥ 0.99 (Supplementary Table S4).
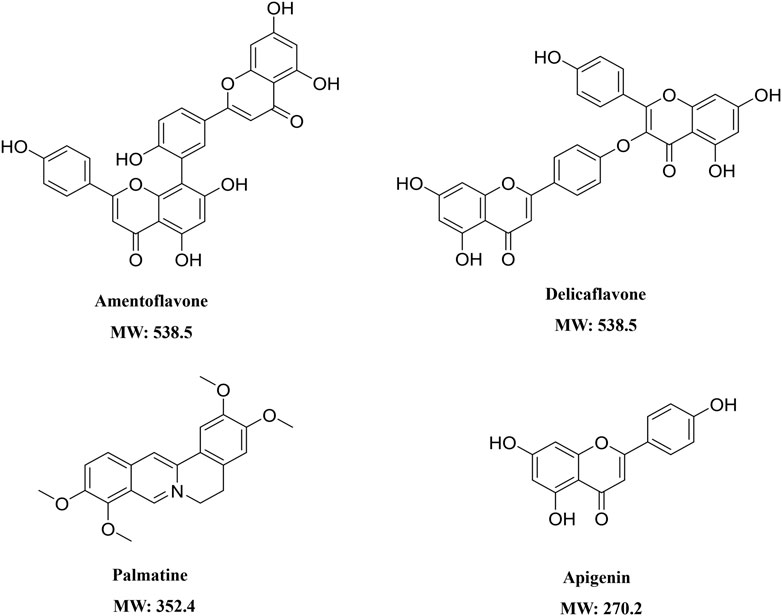
FIGURE 1. Structural formula and molecular weight of the components in SDEA.
QC samples in three levels (Low QC, Medium QC and High QC) were prepared separately and used to evaluate those quantitative methods, all samples were analyzed by three replicates. The related standard deviation (RSD) of the tested compounds ranged from −8.88% to 12.60% (Supplementary Table S4). MRM mass spectrum of blank sample and Limit of Quantity (LOQ) were displayed in Supplementary Figure S1. As well, all of them demonstrated the feasibility of this analytical methods.
1 μg/ml, 5 μg/ml, 25 μg/ml and 40 μg/ml SDEA samples were prepared and injected into LC-MS/MS respectively. Using the established quantitative method, the content of four constituents in SDEA was determined. The average w/w % (Weight compound/weight extract) showed that Delicaflavone, Palmatine, Apigenin were 2.21%, 0.019% and 0.024% respectively, and Amentoflavone reached 13.65%, which was the most abundant one in those four constituents (Table 1).
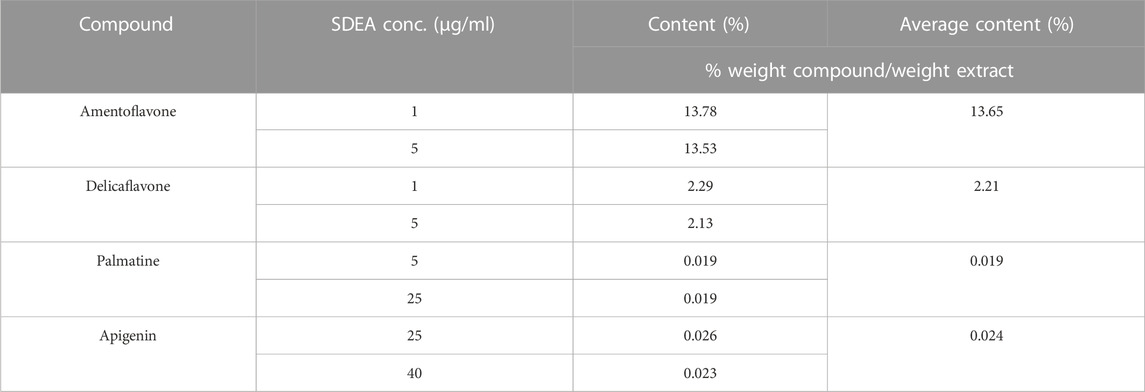
TABLE 1. Contents (% w/w) of the four constituents in SDEA. (Higher SDEA concentrations were used because Apigenin and Palmatine are less in SDEA).
3.2 Development of CYP450 cocktail assay
The probe substrates in this cocktail assay were recommended based on FDA guidance. CYP3A sub-family can bind multiple structurally different substrates (Wrighton et al., 2000; Ekroos and SjOgren, 2006; Watanabe et al., 2007), it is necessary to use more than two CYP3A in vitro marker reactions to evaluate the activity of CYP3A (Wrighton et al., 2000), so that can show more accurate results (Galetin et al., 2005). We selected Testosterone and Dextromethorphan as two different substrates of CYP3A subfamily, their specific products metabolized by CYP3A were Testosterone: 6β-hydroxytestosterone; Dextromethorphan: 3-methoxmorphine. The MRM parameters of substrates corresponding to seven human CYP450 isoforms were summarized in Supplementary Table S3. LC-MS/MS chromatography of substrates and their metabolites were included in Supplementary Figure S2.
To determine the optimist incubation time for all substrates, we measured time-dependent trends in amount of metabolites produced by seven CYP450 enzyme substrates. Although Dextromethorphan (CYP2D6), Chlorzoxazone (CYP2E1), Paclitaxel (CYP2C8) remained linear for first 20 min and metabolic rates of Tolbutamide (CYP2C9), Omeprazole (CYP2C19), Phenacetine (CYP1A2) were linear for up 15 min, Testosterone (CYP3A4) remained linear only for first 10 min and then flattened out. Trend diagram of the generation of metabolites for seven enzyme substrates with time was included in Supplementary Figure S3. Compromising sensitivity to detect metabolites formed at low substrate concentrations and high inhibitor concentrations, an incubation time of 15 min was finally determined.
Considering the linear curve of metabolic reaction, the final concentration of substrate is usually lower than its Km value in order to ensure linear relationship of metabolic rate range and high affinity. What’s moreover, enzyme-substrate affinity data illustrated that the Km value should be similar for a specific enzyme and substrate, regardless of enzyme source (Donnell et al., 2007). Since the incubated substrates could interact with each other (Spaggiari et al., 2014), reducing substrate concentration is the most direct and effective way to reduce the interference (Pillai et al., 2013). Therefore, the final incubation concentration of each substrate was ideal when it was below Km. According to the recommended data in literature (Spaggiari et al., 2014) and sensitivity of MS, final substrate concentration in assay were determined: Phenacetin (1A2), Paclitaxel (2C8) and Tolbutamide (2C9) were 10 μM, Dextromethorphan (2D6) and Testosterone (3A) were 5 μM, Omeprazole (2C19) was 2 μM, and Chlorzoxazone (2E1) was 15 μM. All data were summarized in Supplementary Table S1.
The IC50 values obtained by cocktail assay was compared with a single substrate using known inhibitors of each enzyme reaction alone, so as to verify the feasibility of this assay. a-Naphthoflavone was inhibitor of CYP1A2, our cocktail assay showed that IC50 value of a-Naphthoflavone on CYP1A2 was 0.18 µM, and the value was 0.31 µM from single substrate assay, which indicated similar results. The IC50 value of Quercetin on CYP2C8 was 5.43 µM in cocktail assay, while the number was 6.07 µM in single substrate experiment, all of them were located in desirable range by literature (Kozakai et al., 2012; Chen et al., 2016; Valicherla et al., 2019). IC50 values of other CYP450 isoforms inhibitors (Sulfaphenazole for CYP2C9, Fluconazole for CYP2C19, Quinidine for CYP2D6, 4-Methylpyrazole for CYP2E1, Ketoconazole for CYP3A) on cocktail assay and single substrate method were also performed and compared. Inhibition curves were shown in Figure 2 and IC50 values were summarized in Supplementary Table S2, all of them showed good agreement between cocktail assay and single-substrate approach, which indicated the accuracy of this cocktail inhibition assay.
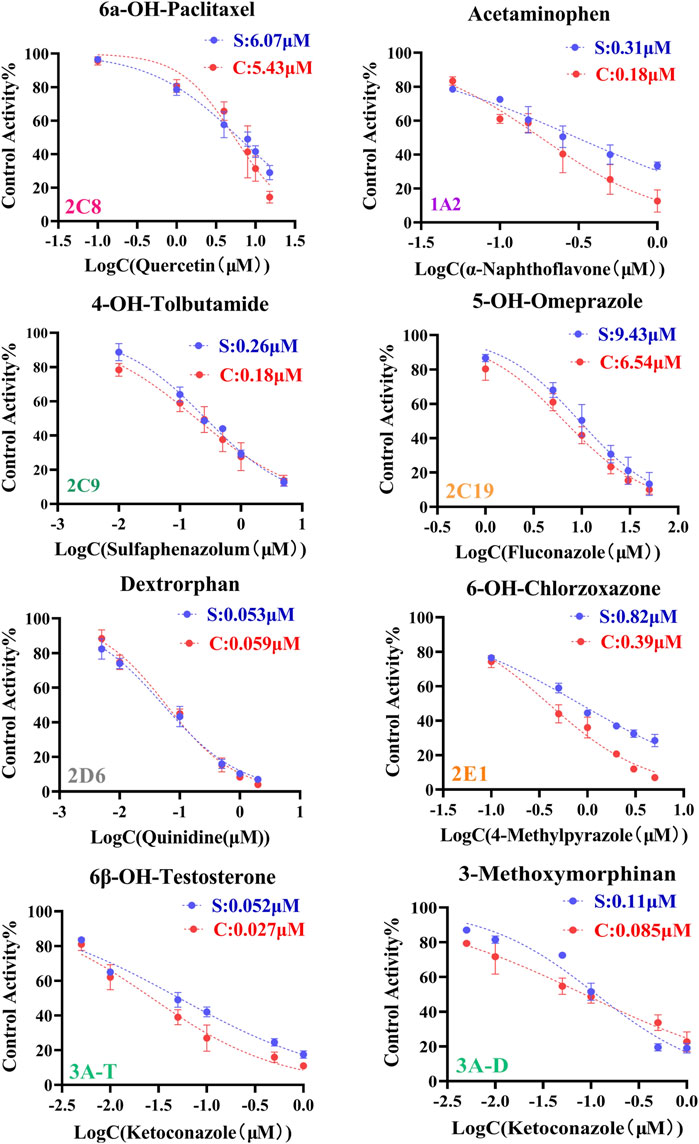
FIGURE 2. Inhibition curves each substrate incubated with cocktail assay and single substrate method. Note: 3A-T, CYP3A (Testosterone); 3A-D, CYP3A (Dextromethorphan).
3.3 Concentration-dependent inhibition effect of SDEA and four constituents on CYP450 isoforms
The established cocktail assay was used to determine potential HDI of SDEA. Results showed that SDEA has strong inhibitory effect on CYP2C8 and CYP2C9, where IC50 values were 1.04 μg/ml and 1.06 μg/ml. SDEA also showed moderate inhibitory ability on CYP2C19 (IC50 was 2.22 μg/ml), CYP2E1 (IC50 was 8.90 μg/ml) and CYP3A (IC50 was 5.18 μg/ml for Testosterone and 7.97 μg/ml for Dextromethorphan). The inhibition activities for CYP1A2, CYP2D6 were mildly, both IC50 values were above 10 μg/ml (Figure 3).
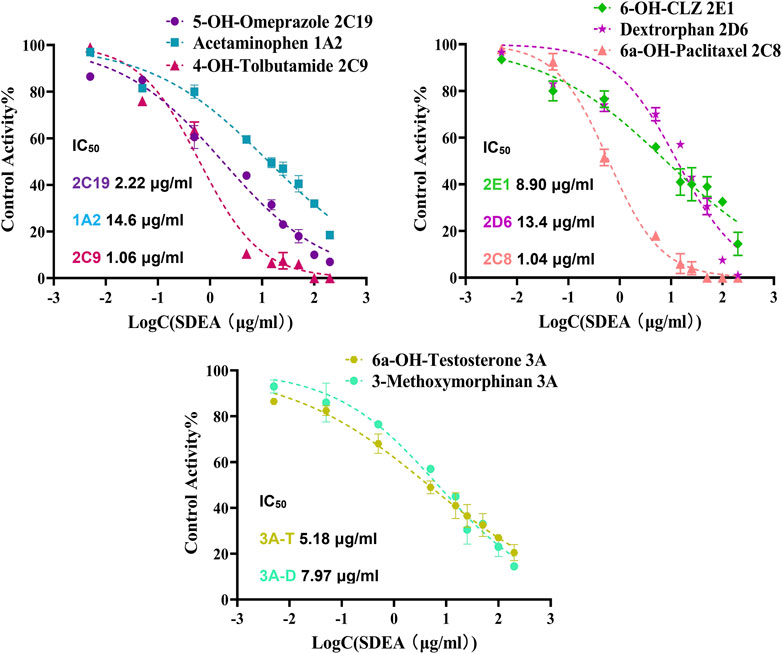
FIGURE 3. Inhibitory potency curves and IC50 values of SDEA on seven CYP450 enzyme isoforms. Note: 3A-T, CYP3A (Testosterone); 3A-D, CYP3A (Dextromethorphan).
We also determined the inhibitory effects of SDEA constituents on seven cytochrome P450 isoforms. At the concentration of 10 μM, Amentoflavone showed more than 50% inhibitory effect on seven enzyme types, the inhibitory effect of Apigenin on CYP1A2, CYP2C8, CYP2C9, CYP2E1 and CYP3A (Testosterone) was more than 50%. Palmatine inhibits CYP3A (Testosterone) by more than 50% and has a weak inhibitory effect on CYP2E1. Delicaflavone has a weak inhibitory effect on CYP2E1 and CYP3A, but it had no obvious inhibitory effect on the other CYP450 isomers at this concentration (Figure 4).
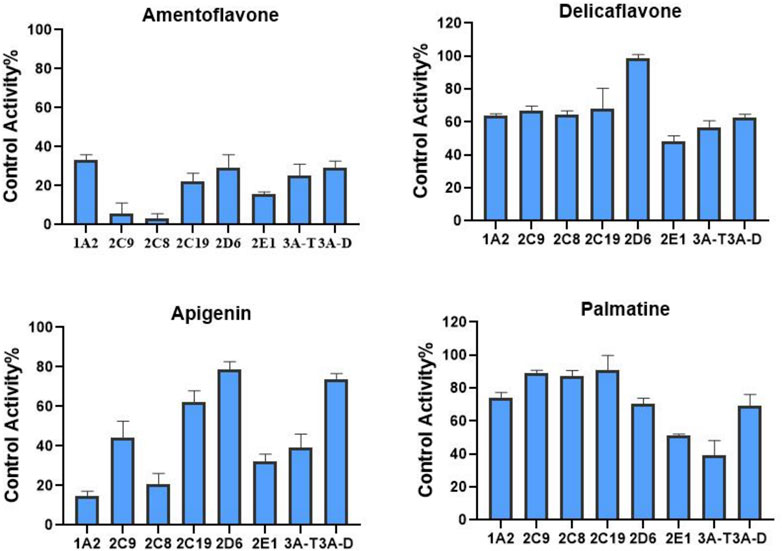
FIGURE 4. Inhibition of CYP enzymes by Amentoflavone, Delicaflavone, Apigenin, and Palmatine at 10 μM. Inhibition Activity was expressed as a percentage of remaining activity compared to a control test without enzyme inhibitor. Note: 3A-T, CYP3A (Testosterone); 3A-D, CYP3A (Dextromethorphan).
Amentoflavone were most abundant in four SDEA constituents (13.65% w/w, Table 1), which showed a broad-spectrum potent CYP enzymes inhibitory effect. The IC50 values were less than 5 μM for the seven cytochrome P450 isoforms, especially for CYP2C9, CYP2C8 and CYP3A, the IC50 values were even less than 0.5 μM (Figure 5A). In contrast, Delicaflavone only had a moderate inhibitory effect on CYP3A (Testosterone), weak inhibitory effect on CYP2C8, CYP2E1 and had almost no inhibitory effect on the other CYP isoforms (Figure 5B).
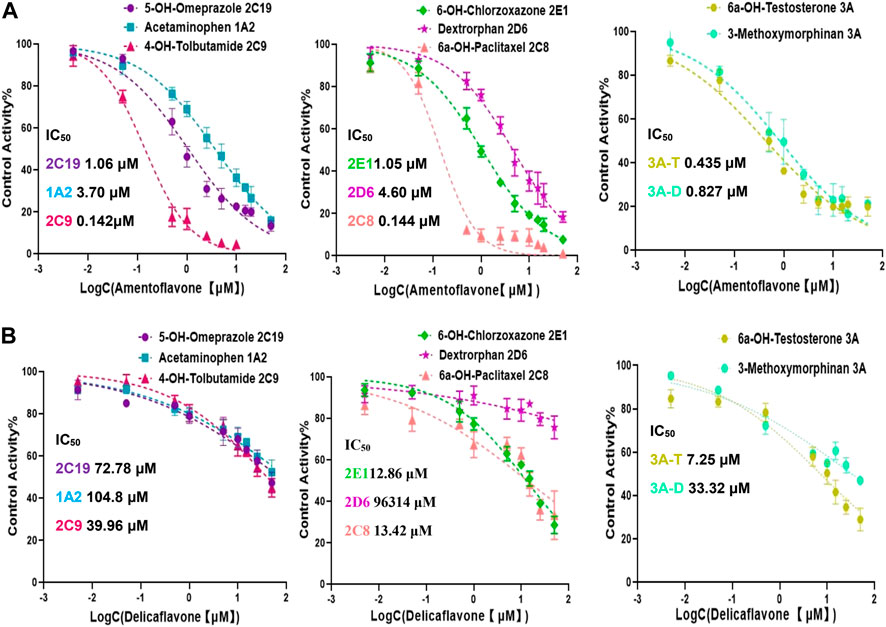
FIGURE 5. Inhibition curves and IC50 values of Amentoflavone (A) and Delicaflavone (B) against seven CYP450 isoforms. Note: 3A-T, CYP3A (Testosterone); 3A-D, CYP3A (Dextromethorphan).
3.4 Time-dependent inhibition study
Possible time-dependent inhibition of SDEA, Amentoflavone and Delicaflavone was tested using cocktail assay. Single point screening assay showed that SDEA (0.5 μg/ml) demonstrated a time-dependent inhibition on CYP2C19, CYP2D6, CYP3A (Testosterone) (Figure 6A), and Amentoflavone (0.1 μM) showed the time-dependent inhibition on CYP2C19, CYP2D6 (Figure 6B). While Delicaflavone (2 μM) did not produce time-dependent inhibition effect on those enzymes (Figure 6C).
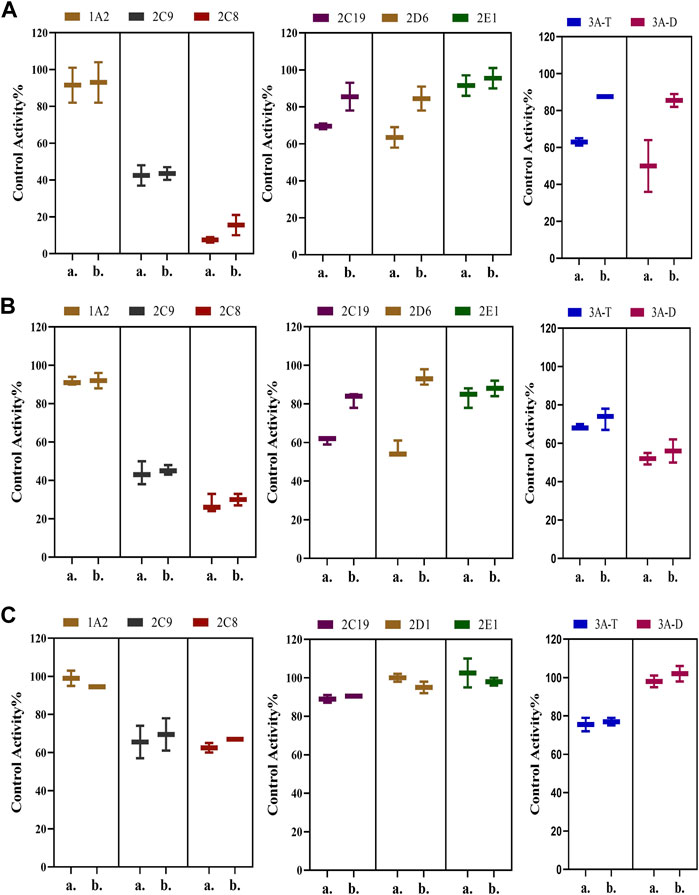
FIGURE 6. Percentage reduction in activity of seven enzymes after incubation with SDEA (A), Amentoflavone (B) and Delicaflavone (C) using a single point time-dependent inhibition screening assay. The experiment was carried out three times. Note: (1) a. presence of NADPH; b. without NADPH. (2) 3A-T, CYP3A (Testosterone); 3A-D, CYP3A (Dextromethorphan).
Based on these results, we have determined the inhibition effect of incubation time with SDEA or Amentoflavone on CYP2C19, CYP2D6, CYP3A (Testosterone) activity. When HLM was co-incubated with SDEA (0.5 μg/ml) in presence of NADPH for 0–35 min, the metabolic activity of CYP2C19 decreased from 95.5% to 69.5%; the activity of CYP2D6 decreased from 84.5% to 63.5% and CYP3A (Testosterone) decreased from 98.5% to 63% (Figure 7A). As for Amentoflavone (0.1 μM), the CYP2C19 activity decreased from 92% to 61% and CYP2D6 activity decreased from 91% to 56%, during the 0–35 min of incubation with HLM and NADPH (Figure 7B). The control group was without NADPH, after 0–35 min co-incubation of HLM with SDEA or Amentoflavone, no significant change in enzymes activity was observed. Those data indicated that SDEA and Amentoflavone may follow a time-dependent inhibition on CYP450 enzymes.
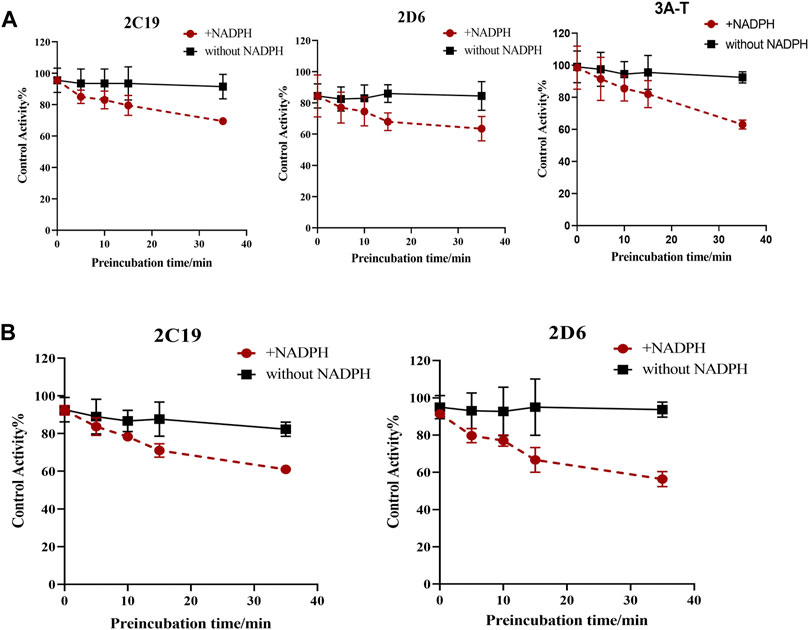
FIGURE 7. Effect of incubation time on the inhibition of CYP3A, CYP2D6 and CYP2C19, with SDEA (A) and Amentoflavone (B). Note: 3A-T, CYP3A (Testosterone).
4 Discussion
In vitro model of human CYP450 enzymes inhibition cocktail assay used in this study can discover the potential of interaction between herb and drugs, reduce the risk of using clinical drug, lay a foundation for clinical medication. Incubation time (15 min) and substrate concentration were determined by combining literature data with experimental conditions (Supplementary Table S1). The established cocktail assay was then validated with known CYP450 enzymes inhibitors, and was applied to evaluate whether SDEA and its bioactive constituents produce concentration-dependent or time-dependent inhibition on the selected CYP450 enzymes. Amentoflavone and Delicaflavone are the main active pharmacological components of SDEA and have high content in SDEA. Palmatine and Apigenin are also SDEA constituents and have been reported to show inhibitory effects on CYP450 enzymes (von Moltke et al., 2004; Kim et al., 2000). Moreover, Palmatine and Apigenin are more common in everyday life than other ingredients, the results will be more practical. Thus, those four compounds were selected for HDI test.
The experiment results showed that SDEA had a strong concentration-dependent inhibition effect on CYP2C9 and CYP2C8 (Figure 3), time-dependent inhibition effect on CYP2D6, CYP2C19 and CYP3A (Figure 7A), which may lead to some serious adverse effects in clinical use. CYP3A subfamily is a particularly important CYP metabolizing, mediating over 50% in vivo biotransformation of clinical drugs (Liu et al., 2006; Xia et al., 2015). In addition, CYP3A subfamily also plays an important role in maintaining metabolic balance of important endogenous substances such as bile acids, steroid hormones and cholesterol (Liang et al., 2015; Xia et al., 2015; Liang et al., 2016), and it is also the main metabolic enzyme of many narrow-window drugs, such as Paclitaxel, Bortezomib, and Gefitinib. SDEA not only inhibits CYP3A (IC50 < 10 μg/ml), more importantly, after 35 min co-incubation of SDEA with NADPH, CYP3A activity was reduced from 98.5% to 63%, which significantly increases the risk of herb-drug interaction with serious consequences. Some oral hypoglycemic agents, such as Glyburide, Rosiglitazone, and Repaglinide, are mainly metabolized by CYP2C8 or CYP2C9, it may increase hypoglycemia risk when those medicines are used together with SDEA. For epilepsy patients, SDEA should be used with caution when taking Phenytoin and Valproic acid (metabolized by CYP2C19) at the same time, in order to avoid serious adverse reactions.
The contents of Amentoflavone and Delicaflavone in SDEA were 13.65% w/w and 2.21% w/w respectively, which were similar to results of previous studies (Li et al., 2013) (Table 1). Amentoflavone not only shows pharmacological activities, such as anti-tumor (Ndongoa et al., 2015), antiviral (Coulerie et al., 2013), antioxidant (Arwa et al., 2015), anti-inflammatory (Abdallah et al., 2015), but also has therapeutic effects on central nervous (Zhang et al., 2015) and cardiovascular system (Yu et al., 2017). The data indicated that Amentoflavone may be the main contributor for SDEA to inhibit CYP enzymes activity. Amentoflavone showed high content in SDEA and strong or moderate inhibitory effect on CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A (Figure 5A). The inhibitory effect was even stronger than some specific inhibitors of CYP2C9, CYP2C8 and CYP3A. In previous reports, Amentoflavone had a strong inhibitory effect on various CYP450 isoforms such as CYP2C9, CYP2C19, CYP2D6 and CYP3A4 (von Moltke et al., 2004; Kimura et al., 2010), which were consistent with the experiment results. It should be noted that this paper found a strong inhibitory effect on CYP2C8 and a moderate inhibitory effect on CYP1A2 and CYP2E1 by Amentoflavone, which were not reported in previous article (von Moltke et al., 2004). The researchers did not find the inhibitory effect of Amentoflavone on CYP1A2, which may be related to the differences in enzyme sources, substrate types and concentrations.
Similar to SDEA, Amentoflavone also has time-dependent inhibitory effects on CYP2D6 and CYP2C19 (Figure 7B). Although CYP2D6 accounts for a small fraction of CYP450 expression in liver, it participates in metabolism of various drugs. As a therapeutic drug for breast cancer, Tamoxifen (TAM) has better pharmacological activity only after the formation of 4-hydroxy-N-demethylamoxifen (Endoxifen), which is a metabolite catalyzed by CYP2D6 (Kleina et al., 2013). What’s more, CYP2D6 is the most polymorphic metabolic enzyme, which is of great significance in genetics, especially in pharmacogenetics. CYP2D6 and CYP2C19 are jointly involved in the metabolism of psychotropic drugs, such as Selective Serotonin Recycle Inhibitors (SSRIs), Clozapine (CZ) (Caetano1 and Piatkov, 2016; Menkes et al., 2018) and Risperidone. For those patients who have weak CYP2D6 and CYP2C19 activities, the inhibition is more dangerous and may lead to serious consequences (Lymperopoulos et al., 2015). A recent study has found that Amentoflavone was a strong and broad-spectrum UDP-glucuronosyltransferase (UGT) inhibitor (Lv et al., 2018). Since UGT enzymes involve in the most phase II elimination in body, the inhibition of UGT would bring potential risks for those medicines mainly clearing by this pathway. Considering the relationship of SDEA and Amentoflavone, we speculate that SDEA may also have the potential to produce significant inhibition on UGT, which needs further experiments to confirm.
Because the content determination and single concentration inhibition showed that Palmatine and Apigenin contents were low (Table 1), and the inhibitory effect on enzyme type was not as strong as Amentoflavone (Figure 4), so no further time-dependent study was conducted for Palmatine and Apigenin. Unlike SDEA, Amentoflavone, Apigenin or Palmatine, Delicaflavone shown no significant inhibition effect on the selected CYP450 isomers at 10 μM, only a weak inhibitory effect on CYP2E1 and CYP3A. Although Delicaflavone is the isomer of Amentoflavone, the difference on CYP450 enzymes inhibition effect may account in the steric hindrance. There are active site (responsible for substrate biding and NADPH-CYP450 oxidase reaction) and allosteric site (responsible for outside molecules to modulate enzyme activity) in CYP450 enzymes (Deodhar et al., 2020). It is possible that Amentoflavone binds to the allosteric site on CYP450 enzymes and lead to a strong inhibition effect. Delicaflavone did not bind to or weakly work on the allosteric site on CYP450 enzymes, resulting in the different CYP450 inhibition activities for those two isomers. Delicaflavone also has excellent anti-cancer and tumor-suppressing ability, the less influence on the CYP450 enzymes activity will make it a better candidate for next step drug development than SDEA or Amentoflavone.
5 Conclusion
Through the established reliable cocktail assay, SDEA was found to have concentration-dependent inhibition on several CYP450 enzymes, and inhibited CYP2D6, CYP2C19, CYP3A in a time-dependent manner. The inhibition activity may be mainly due to the higher content of component: Amentoflavone (13.65%). Amentoflavone has inhibitory effects on all tested CYP450 enzymes, the inhibitory effects on CYP2C9, CYP2C8 and CYP3A were even greater than the corresponding specific inhibitors. Amentoflavone also has time-dependent inhibition on CYP2D6 and CYP2C19. The other two constituents from SDEA, Apigenin and Palmatine, both showed concentration-dependent inhibition. For Delicaflavone, no significant inhibitory effect on CYP450 enzymes was observed. Since Delicaflavone owns excellent anti-cancer ability and low HDI potential, it was more suitable to develop as a new anti-cancer drug.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
FL: methodology, data analysis and writing (original draft). XL: conceptualization and supervision. GM: writing (review and editing). XW, BC, and HY: methodology. LH methodology, data analysis, conceptualization, supervision and writing (review and editing). All authors have read and agreed to the published version of the manuscript.
Funding
This research work was supported by those funds: (1) The High Level Talent Foundation of Fujian Medical University (grant number XRCZX2021005); (2) The Young Teachers Education Foundation of Fujian Province, China (grant number JAT220080); (3) National Nature Science Foundation of China (grant number 22074017); (4) Key Project Supported by the Natural Science Foundation of Fujian Province, China (grant number 2021J02034).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2023.1108867/full#supplementary-material
References
Abdallah, H. M., Almowallad, F. M., Esmat, A., Shehata, I. A., and Essam, A. (2015). Anti-inflammatory activity of flavonoids from Chrozophora tinctoria. Phytochem. Lett. 13, 74–80. DOI: doi:10.1016/j.phytol.2015.05.008
Al-Ramahi, R., Jaradat, N., Shalalfeh, R., Nasir, S., Manasra, Y., Shalalfeh, I., et al. (2015). Evaluation of potential drug-herb interactions among a group of palestinian patients with chronic diseases. BMC Complement. Altern. Med. 15, 221–225. DOI: doi:10.1186/s12906-015-0764-7
Arwa, P. S., Zeraik, M. L., Ximenes, V. F., da Fonseca, L. M., da Silva Bolzani, V., and Silva, D. H. S. (2015). Redox-active biflavonoids from Garcinia brasiliensis as inhibitors of neutrophil oxidative burst and human erythrocyte membrane damage. J. Ethnopharmacol. 174, 410–418. DOI: doi:10.1016/j.jep.2015.08.041
Awortwe, C., Makiwane, M., Reuter, H., Muller, C., Louw, J., and Rosenkranz, B. (2018). Critical evaluation of causality assessment of herb–drug interactions in patients. Br. J. Clin. Pharmacol. 84 (4), 679–693. doi:10.1111/bcp.13490
Bjornsson, T. D., Callaghan, J. T., Einolf, H. J., Fischer, V., Gan, L., Grimm, S., et al. (2003). The conduct of in vitro and in vivo drug-drug interaction studies: A PhRMA perspective. J. Clin. Pharmacol. 43, 443–469. DOI: doi:10.1177/0091270003252519
Brantley, S. J., Argikar, A. A., Lin, Y. S., Nagar, S., and Paine, M. F. (2014). Herb–drug interactions: Challenges and opportunities for improved predictions. Drug Metab. Dispos. 42 (3), 301–317. doi:10.1124/dmd.113.055236
Caetano1, D., and Piatkov, I. (2016). Ultrarapid clozapine metabolism and CYP2D6 gene duplication in a patient with schizophrenia. Per. Med. 13 (2), 113–117. doi:10.2217/pme.15.56
Chen, B., Wang, X. W., Zou, Y. L., Chen, W. Y., Wang, G., Yao, W. S., et al. (2018). Simultaneous quantification of five biflavonoids in rat plasma by LC-ESI-MS/MS and its application to a comparatively pharmacokinetic study of Selaginella doederleinii Hieron extract in rats. J. Pharm. Biomed. Anal. 149, 80–88. doi:10.1016/j.jpba.2017.10.028
Chen, Z. H., Zhang, S. X., Long, N., Lin, L. S., Chen, T., Zhang, F. P., et al. (2016). An improved substrate cocktail for assessing direct inhibition and time-dependent inhibition of multiple cytochrome P450s. Acta Pharmacol. Sin. 37 (5), 708–718. doi:10.1038/aps.2016.10
Choi, J. G., Eom, S. M., Kim, J., Kim, S. H., Huh, E., Kim, H., et al. (2016). A comprehensive review of recent studies on herb-drug interaction: A focus on pharmacodynamic interaction. J. Altern. Complement. Med. 22 (4), 262–279. doi:10.1089/acm.2015.0235
Coulerie, P., Nour, M., Maciuk, A., Eydoux, C., Guillemot, J. C., Lebouvier, N., et al. (2013). Structure-activity relationship study of biflavonoids on the Dengue virus polymerase DENV-NS5 RdRp. Planta Med. 79, 1313–1318. doi:10.1055/s-0033-1350672
Deodhar, M., Rihani, S. B. A., Arwood, M. J., Darakjian, L., Dow, P., Turgeon, J., et al. (2020). Mechanisms of CYP450 inhibition: Understanding drug-drug interactions due to mechanism-based inhibition in clinical practice. Pharmaceutics 12 (9), 846. doi:10.3390/pharmaceutics12090846
Donnell, C. J., Grime, K., Courtney, P., Slee, D., and Riley, R. J. (2007). The development of a cocktail CYP2B6, CYP2C8, and CYP3A5 inhibition assay and a preliminary assessment of utility in a drug discovery setting. Drug Metab. Dispos. 35, 381–385. doi:10.1124/dmd.106.012344
Ekroos, M., and SjOgren, T. (2006). Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. U.S.A. 103, 13682–13687. doi:10.1073/pnas.0603236103
Galetin, A., Ito, K., Hallifax, D., and Houston, J. B. (2005). CYP3A4 substrate selection and substitution in the prediction of potential drug–drug interactions. J. Pharmacol. Exp. Ther. 314, 180–190. doi:10.1124/jpet.104.082826
Guengerich, P. F. (2021). A history of the roles of cytochrome P450 enzymes in the toxicity of drugs. Toxicol. Res. 37 (1), 1–23. doi:10.1007/s43188-020-00056-z
Guengerich, P. F. (2008). Cytochrome p450 and chemical toxicology. Chem. Res. Toxicol. 21 (1), 70–83. doi:10.1021/tx700079z
Hakkola, J., Hukkanen, J., Turpeinen, M., and Pelkonen, O. (2020). Inhibition and induction of CYP enzymes in humans: An update. Arch. Toxicol. 94 (11), 3671–3722. doi:10.1007/s00204-020-02936-7
Han, Y. L., Li, D., Renb, B., Jing, G. P., Menga, X. L., Zhoua, Z. Y., et al. (2012). Evaluation of impact of herba erigerontis injection, a Chinese herbal prescription, on rat hepatic cytochrome p450 enzymes by cocktail probe drugs. J. Ethnopharmacol. 139 (1), 104–109. doi:10.1016/j.jep.2011.10.019
Han, Y. L., Yu, H. L., Li, D., Meng, X. L., Zhou, Z. Y., Yu, Q., et al. (2011). In vitro inhibition of huanglian[Rhizoma coptidis (L.)] and its six active alkaloids on six cytochrome P450 isoforms in human liver microsomes. Phytother. . Res. 25, 1660–1665. doi:10.1002/ptr.3475
Ishihara, K., Kushida, H., Yuzurihara, M., WakuiYanagisawa, Y. T., Kamei, H., et al. (2000). Interaction of drugs and Chinese herbs: Pharmacokinetic changes of tolbutamide and diazepam caused by extract of Angelica dahurica. J. Pharm. Pharmacol. 52, 1023–1029. doi:10.1211/0022357001774750
Kim, H. J., Chun, H. S., and Yang, R. (2000). Inhibition of benzo[a]pyrene-induced cytotoxicity and cytochrome P450 1A activity by dietary flavonoids in human liver cell model: Structure-activity relationship. Biotechnol. Lett. 22, 1941–1946. doi:10.1023/A:1026749606363
Kim, M. J., Kim, H., Cha, I. J., Park, J. S., Shon1, J. H., Liu, K. H., et al. (2005). High throughput screening of inhibitory potential of nine cytochrome P450 enzymes in vitro using liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 19 (18), 2651–2658. doi:10.1002/rcm.2110
Kimura, Y., Ito, H. K., Ohnishi, R., and Hatano, T. (2010). Inhibitory effects of polyphenols on human cytochrome P450 3A4 and 2C9 activity. Food Chem. Toxicol. 48, 429–435. doi:10.1016/j.fct.2009.10.041
Kleina, D. J., Thorna, C. F., Destac, Z., Flockhartc, D. A., Altmana, R. B., and Kleina, T. E. (2013). PharmGKB summary: Tamoxifen pathway, pharmacokinetics. Pharmacogenet. Genomics 23, 643–647. doi:10.1097/fpc.0b013e3283656bc1
Kozakai, K., Yamada, Y., Oshikata, M., Kawase, T., Suzuki, E., Haramaki, Y., et al. (2012). Reliable high-throughput method for inhibition assay of 8 cytochrome p450 isoforms using cocktail of probe substrates and stable isotope-labeled internal standards. Drug Metab. Pharmacokinet. 27 (5), 520–529. doi:10.2133/dmpk.dmpk-12-rg-014
Lee, K. S., and Kim, S. K. (2013). Direct and metabolism-dependent cytochrome P450 inhibition assays for evaluating drug-drug interactions. J. Appl. Toxicol. 33 (2), 100–108. doi:10.1002/jat.1720
Li, G., Huang, K., Nikolic, D., and van Breemen, R. B. (2015). High-throughput cytochrome P450 cocktail inhibition assay for assessing drug-drug and drug-botanical interactions. Drug Metab. Dispos. 43 (11), 1670–1678. doi:10.1124/dmd.115.065987
Li, G., Simmler, C., ChenNikolic, L. D., Chen, S. N., Pauli, G. F., et al. (2017). Cytochrome P450 inhibition by three licorice species and fourteen licorice constituents. Eur. J. Pharm. Sci. 15 (109), 182–190. doi:10.1016/j.ejps.2017.07.034
Li, S. G., Wang, X. W., Wang, G., Shi, P. Y., Lin, S. L., Xu, D. F., et al. (2020). Ethyl acetate extract of Selaginella doederleinii hieron induces cell autophagic death and apoptosis in colorectal cancer via PI3K-Akt-mTOR and AMPKα-signaling pathways. Front. Pharmacol. 11, 565090. doi:10.3389/fphar.2020.565090
Li, S. G., Yao, H., Zhao, M. F., Li, Y. X., Huang, L. Y., and Lin, X. H. (2013). Determination of seven biflavones of Selaginella doederleinii by high performance liquid chromatography. Anal. Lett. 46, 2835–2845. doi:10.1080/00032719.2013.831426
Li, S., Zhao, M. F., Li, Y. X., Sui, Y. X., Yao, H., Huang, L. Y., et al. (2014). Preparative isolation of six anti-tumour biflavonoids from Selaginella doederleinii Hieron by high-speed counter-current chromatography. Phytochem. Anal. 25, 127–133. doi:10.1002/pca.2478
Liang, S. C., Ge, G. B., Xia, Y. L., Wei, J. Z., Qi, X. Y., Tu, C. X., et al. (2015). In vitro evaluation of the effect of 7-methyl substitution on glucuronidation of daphnetin: Metabolic stability, isoform selectivity, and bioactivity analysis. J. Pharm. Sci. 104 (10), 3557–3564. doi:10.1002/jps.24538
Liang, S. C., Xia, Y. L., Hou, J., Ge, G. B., Zhang, J. W., He, Y. Q., et al. (2016). Methylation, glucuronidation, and sulfonation of daphnetin in human hepatic preparations in vitro: Metabolic profiling, pathway comparison, and bioactivity analysis. J. Pharm. Sci. 105 (2), 808–816. doi:10.1016/j.xphs.2015.10.010
Lin, J. H., and Lu, A. Y. (1998). Inhibition and induction of cytochrome P450 and the clinical implications,Clin. Pharmacokinet 35 (5), 361–390. doi:10.2165/00003088-199835050-00003–
Liu, Y., Zhang, J. w., Li, W., Ma, H., Sun, J., Denge, M. C., et al. (2006). Ginsenoside metabolites, rather than naturally occurring ginsenosides, lead to inhibition of human cytochrome P450 enzymes. Toxicol. Sci. 91 (2), 356–364. doi:10.1093/toxsci/kfj164
Liua, L. Y., Hana, Y. L., Zhua, J. H., Yua, Q., Yang, Q. J., Lua, J., et al. (2015). A sensitive and high-throughput LC-MS/MS method for inhibition assay of seven major cytochrome P450s in human liver microsomes using an in vitro cocktail of probe substrates. Biomed. Chromatogr. 29 (3), 437–444. doi:10.1002/bmc.3294
Lu, C., and Di, L. (2020). In vitro and in vivo methods to assess pharmacokinetic drug–drug interactions in drug discovery and development. Biopharm. Drug Dispos. 41 (1-2), 3–31. doi:10.1002/bdd.2212
Lv, X., Zhang, J. B., Wang, X. X., Hu, W. Z., Shi, Y. S., Liue, S. W., et al. (2018). Amentoflavone is a potent broad-spectrum inhibitor of human UDP-glucuronosyltransferases. Chem.-Biol. Interact. 284, 48–55. doi:10.1016/j.cbi.2018.02.009
Lymperopoulos, A., McCrink, K. A., and Brill, A. (2015). Impact of CYP2D6 genetic variation on the response of the cardiovascular patient to carvedilol and metoprolol. Curr. Drug Metab. 17 (1), 30–36. doi:10.2174/1389200217666151105125425
Menkes, D. B., Glue, P., Gale, C., Lam, F., Hung, C. T., and Hung, N. (2018). Steady-state clozapine and norclozapine pharmacokinetics in Maori and European patients. EBioMedicine 27, 134–137. doi:10.1016/j.ebiom.2017.11.030
Ndongoa, J. T., Issab, M. E., Messic, A. N., Mbingc, J. N., Cuendetb, M., Pegnyemb, D. E., et al. (2015). Cytotoxic flavonoids and other constituents from the stem bark of Ochna schweinfurthiana. Nat. Prod. Res. 29 (17), 1684–1687. doi:10.1080/14786419.2014.991321
Pillai, V. C., Stromb, S. C., Caritis, S. N., and Venkataramanana, R. (2013). A sensitive and specific CYP cocktail assay for the simultaneous assessment of human cytochrome P450 activities in primary cultures of human hepatocytes using LC–MS/MS. J. Pharm. Biomed. Anal. 74, 126–132. doi:10.1016/j.jpba.2012.10.016
Prueksaritanont, T., Chu, X., Gibson, C., Cui, D., Yee, K. L., Ballard, J., et al. (2013). Drug–drug interaction studies: Regulatory guidance and an industry perspective. AAPS J. 15 (3), 629–645. doi:10.1208/s12248-013-9470-x
Qin, C. Z., Renb, X., Tana, Z. R., Chena, Y., Yina, J. Y., Yua, J., et al. (2013). A high-throughput inhibition screening of major human cytochrome P450 enzymes using an in vitro cocktail and liquid chromatography-tandem mass spectrometry. Biomed. Chromatogr. 28 (2), 197–203. doi:10.1002/bmc.300
Spaggiari, D., Daali, Y., and Rudaz, S. (2016). An extensive cocktail approach for rapid risk assessment of in vitro CYP450 direct reversible inhibition by xenobiotic exposure. Toxicol. Appl. Pharmacol. 302, 41–51. doi:10.1016/j.taap.2016.04.013
Spaggiari, D., Geiser, L., Daali, Y., and Rudaz, S. (2014). A cocktail approach for assessing the in vitro activity of human cytochrome P450s: An overview of current methodologies. J. Pharm. Biomed. Anal. 101, 221–237. doi:10.1016/j.jpba.2014.03.018
Sudsakorn, S., Bahadduri, P., Fretland, J., and Lu, C. (2020). 2020 FDA drug-drug interaction guidance: A comparison analysis and action plan by pharmaceutical industrial scientists. Curr. Drug Metab. 21 (6), 403–426. doi:10.2174/1389200221666200620210522
Sui, Y. X., Li, S. G., Shi, P. Y., Wu, Y. J., Li, Y. X., Chen, W. Y., et al. (2016). Ethyl acetate extract from Selaginella doederleinii Hieron inhibits the growth of human lung cancer cells A549 via caspase-dependent apoptosis pathway. J. Ethnopharmacol. 190, 261–271. doi:10.1016/j.jep.2016.06.029
Sui, Y. X., Yao, H., Li, S. G., Jin, L., Shi, P. Y., Li, Z. J., et al. (2017). Delicaflavone induces autophagic cell death in lung cancer via Akt/mTOR/p70S6K signaling pathway. J. Mol. Med. 95 (3), 311–322. doi:10.1007/s00109-016-1487-z
Thomford, N., Dzobo, K., Chopera, D., Wonkam, A., Maroyi, A., Blackhurst, D., et al. (2016). In vitro reversible and time-dependent CYP450 inhibition profiles of medicinal herbal plant extracts newbouldia laevis and Cassia abbreviata: Implications for herb-drug interactions. Molecules 21 (7), 891. doi:10.3390/molecules2107089
Valicherla, G. R., Mishra, A., Lenkalapelly, S., Jillela, B., Francis, F. M., Rajagopalan, L., et al. (2019). Investigation of the inhibition of eight major human cytochrome P450 isozymes by a probe substrate cocktail in vitro with emphasis on CYP2E1. Xenobiotica 49 (12), 1396–1402. doi:10.1080/00498254.2019.1581301
van Breemen, R. B. (2015). Development of safe and effective botanical dietary supplements. J. Med. Chem. 58 (21), 8360–8372. doi:10.1021/acs.jmedchem.5b00417
Venkatakrishnan, K., von Moltke, L. L., Obach1, R. S., and Greenblatt, D. J. (2003). Drug metabolism and drug interactions: Application and clinical value of in vitro models. Curr. Drug Metab. 4, 423–459. doi:10.2174/1389200033489361
von Moltke, L. L., Weemhoff, J. L., Bedir, E., Khan, I. A., Harmatz, J. S., Goldman, P., et al. (2004). Inhibition of human cytochromes P450 by components of Ginkgo biloba. J. Pharm. Pharmacol. 56 (8), 1039–1044. doi:10.1211/0022357044021
Wang, X. W., Chen, B., Xu, D. F., Li, Z. J., Sui, Y. X., and Lin, X. H. (2020). Delicaflavone reverses cisplatin resistance via endoplasmic reticulum stress signaling pathway in non-small cell lung cancer cells. Onco Targets Ther. 13, 10315–10322. doi:10.2147/ott.s255586
Watanabe, A., Nakamura, K., Okudaira, N., Okazaki, O., and Sudo, K. I. (2007). Risk assessment for drug–drug interaction caused by metabolism-based inhibition of CYP3A using automated in vitro assay systems and its application in the early drug discovery process. Drug Metab. Dispos. 35, 1232–1238. doi:10.1124/dmd.107.015016
Wrighton, S. A., Schuetz, E. G., Thummel, K. E., Shen, D. D., Korzekwa, K. R., Watkinspaul, B., et al. (2000). The human CYP3A subfamily: Practical considerations. Drug Metab. Rev. 32, 339–361. doi:10.1081/dmr-100102338
Xia, C. H., Sun, J. G., Wang, G. J., Shang, L. L., ZhangZhang, X. X. R., et al. (2010). Herb–drug interactions: In vivo and in vitro effect of shenmai injection, a herbal preparation, on the metabolic activities of hepatic cytochrome P450 3A1/2, 2C6, 1A2, and 2E1 in rats. Planta Med. 76, 245–250. doi:10.1055/s-0029-1186082
Xia, Y. L., Ge, G. B., Wang, P., Liang, S. C., He, Y. Q., Ning, J., et al. (2015). Structural modifications at the C-4 position strongly affect the glucuronidation of 6,7-dihydroxycoumarins. Drug Metab. Dispos. 43 (4), 553–560. doi:10.1124/dmd.114.060681
Xia, Y. L., Liang, S. C., Zhu, L. L., Ge, G. B., He, G. Y., Ning, J., et al. (2014). Identification and characterization of human UDP-glucuronosyltransferases responsible for the glucuronidation of fraxetin. Drug Metab. Pharmacokinet. 29 (2), 135–140. doi:10.2133/dmpk.dmpk-13-rg-059
Yao, H., Chen, B., Zhang, Y. Y., Ou, H. G., Li, Y. X., Li, S. G., et al. (2017). Analysis of the total biflavonoids extract from Selaginella doederleinii by HPLC-QTOF-MS and its in vitro and in vivo anticancer effects. Molecules 22, 325–342. doi:10.3390/molecules22020325
Yao, W. S., Lin, Z., Shi, P. Y., Chen, B., Wang, G., Huang, J. Y., et al. (2020). Delicaflavone induces ROS-mediated apoptosis and inhibits PI3K/AKT/mTOR and Ras/MEK/Erk signaling pathways in colorectal cancer cells. Biochem. Pharmacol. 171, 113680–680. doi:10.1016/j.bcp.2019.113680
Yao, W. S., Lin, Z., Wang, G., Li, S. G., Chen, B., Sui, Y. X., et al. (2019). Delicaflavone induces apoptosis via mitochondrial pathway accompanying G2/M cycle arrest and inhibition of MAPK signaling cascades in cervical cancer HeLa cells. Phytomedicine 62, 152973–973. doi:10.1016/j.phymed.2019.152973
Youdim, K. A., and Saunders, K. C. (2010). A review of LC–MS techniques and high-throughput approaches used to investigate drug metabolism by cytochrome P450s. J. Chromatogr. B 878, 1326–1336. doi:10.1016/j.jchromb.2010.02.013
Yu, S., Yan, H., Zhang, L., Shan, M., Chen, P., Ding, A., et al. (2017). A review on the phytochemistry, pharmacology, and pharmacokinetics of amentoflavone, a naturally-occurring biflavonoid. Molecules 22 (2), 299. doi:10.3390/molecules22020299
Zhang, Z., Sun, T., Niu, J. g., He, Z. q., Liu, Y., and Wang, F. (2015). Amentoflavone protects hippocampal neurons: Anti-inflammatory, antioxidative, and antiapoptotic effects. Neural Regen. Res. 10, 1125–1133. doi:10.4103/1673-5374.160109
Keywords: herb-drug interaction, the ethyl acetate extract from S. doederleinii, CYP inhibition, cocktail CYP450 assay, time--dependent inhibition, delicaflavone, amentoflavone
Citation: Lin F, Lin X, Wang X, Mei G, Chen B, Yao H and Huang L (2023) Inhibitory effect of Selaginella doederleinii hieron on human cytochrome P450. Front. Pharmacol. 14:1108867. doi: 10.3389/fphar.2023.1108867
Received: 26 November 2022; Accepted: 30 January 2023;
Published: 15 February 2023.
Edited by:
Junmin Zhang, Lanzhou University, ChinaReviewed by:
Ying-yuan Lu, Peking University, ChinaPing Wang, Shanghai University of Traditional Chinese Medicine, China
Copyright © 2023 Lin, Lin, Wang, Mei, Chen, Yao and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lingyi Huang, bGluZ3lpLmh1YW5nQGZqbXUuZWR1LmNu
†These authors have contributed equally to this work