Abstract
Fluoropyrimidines are chemotherapeutic agents widely used for the treatment of various solid tumors. Commonly prescribed FPs include 5-fluorouracil (5-FU) and its oral prodrugs capecitabine (CAP) and tegafur. Bioconversion of 5-FU prodrugs to 5-FU and subsequent metabolic activation of 5-FU are required for the formation of fluorodeoxyuridine triphosphate (FdUTP) and fluorouridine triphosphate, the active nucleotides through which 5-FU exerts its antimetabolite actions. A significant proportion of FP-treated patients develop severe or life-threatening, even fatal, toxicity. It is well known that FP-induced toxicity is governed by genetic factors, with dihydropyrimidine dehydrogenase (DPYD), the rate limiting enzyme in 5-FU catabolism, being currently the cornerstone of FP pharmacogenomics. DPYD-based dosing guidelines exist to guide FP chemotherapy suggesting significant dose reductions in DPYD defective patients. Accumulated evidence shows that additional variations in other genes implicated in FP pharmacokinetics and pharmacodynamics increase risk for FP toxicity, therefore taking into account more gene variations in FP dosing guidelines holds promise to improve FP pharmacotherapy. In this review we describe the current knowledge on pharmacogenomics of FP-related genes, beyond DPYD, focusing on FP toxicity risk and genetic effects on FP dose reductions. We propose that in the future, FP dosing guidelines may be expanded to include a broader ethnicity-based genetic panel as well as gene*gene and gender*gene interactions towards safer FP prescription.
1 Introduction
Fluoropyrimidines (FPs) are chemotherapeutic agents belonging to the antimetabolite drug class that are widely used in the treatment of solid tumors, including gastrointestinal (colorectal, liver, and pancreatic), head and neck, and breast cancer (Ioannou et al., 2021). FPs used in clinical practice are 5-fluorouracil (5-FU), and its prodrugs, capecitabine (CAP) and tegafur. FPs inhibit the biosynthetic process of DNA and RNA synthesis by directly incorporating into nucleic acids the active nucleotides fluorouridine triphosphate (FUTP) and fluorodeoxyuridine triphosphate (FdUTP). FPs can be administered as a monotherapy or in combination with other antineoplastic medications such as irinotecan, oxaliplatin, leucovorin, or biological therapies (Longley et al., 2003).
Both FP efficacy and toxicity rely on the final concentration of FUTP and FdUTP. FP efficacy as a first line or add-on therapy in different cancers is well-documented (García-Alfonso et al., 2021; Yagi et al., 2021; Kataria et al., 2022; Khankhel et al., 2022) and will not be further discussed herein. Toxicity, however, is a serious drawback of FP therapy. Approximately 10%–40% of FP-treated patients present with severe or life-threatening, even fatal, toxicity (Barin-Le Guellec et al., 2020; Deac et al., 2020); the most common reported toxicities are hematological and gastrointestinal (nausea, vomiting, diarrhea), whereas other complications of FP treatment include hand and foot syndrome (HFS) (Thorn et al., 2011). Death rates due to FP-induced toxicity are high; in France, approximately 150 patients die per year due to FP-toxicity (Barin-Le Guellec et al., 2020), whereas in the U.S.A approximately 1.300 FP-related deaths occur (Santos et al., 2017). The incidence and severity of FP-induced adverse drug reactions (ADRs) vary depending on multiple factors, including misadjusted dosing proportion and schedule, drug interactions and individual clinicopathological characteristics (Lam et al., 2016). Their effect on FP therapy is of utmost clinical importance since ADRs result in unavoidable dose reductions, delay of chemotherapeutic scheme administration or even chemotherapy termination and need for administration of alternative agents.
Pharmacogenomics is currently applied in guiding FP dosing aiming in reducing ADR incidence. Clinical implementation consists of DPYD genotyping encoding for dihydropyrimidine dehydrogenase (DPD), the rate limiting enzyme of 5-FU activation (Knikman et al., 2021). However, it appears that apart from DPYD, several polymorphisms exist in additional genes, mainly involved in FP metabolic pathway, that can affect FP response. In pharmacogenomics, it is well established that once a pharmacogene is clearly associated with drug dose, other genes may improve both dose prediction and sensitivity to ADRs, to a variable degree, after adjusting for the strongest association (Ragia et al., 2014). The polygenic dosing algorithm is being used for other drugs and vitamin K antagonists best fit within this approach (Verhoef et al., 2013; Ragia et al., 2017a; Ragia et al., 2017b). For this drug class the polygenic dosing algorithm includes genetic variations in CYP2C9/VKORC1, the principal enzymes that affect dose requirements, and, additionally, genetic information of secondary enzymes such as CYP4F2 and GGCX, that influence interindividual dose requirements after adjusting for CYP2C9/VKORC1 (warfarindosing.org). In the present review we provide a comprehensive overview of the current data on FP pharmacogenomics, focusing on gene variants that increase risk for FP-induced toxicity and can potentially be used in FP dose decisions. We focus on genes beyond DPYD as we aim to identify additional genetic components and potential gene*gene and environment*gene interactions that contribute to FP response variability with the goal of assessing whether a polygenic FP dosing algorithm would improve targeted clinical use of the drug. The ultimate goal of this review is to propose the concept and the components of such an approach for individualized dose adjustments, in order to minimize FP-induced toxic events. The proposed components are presented in Table 1. It should be clarified, however, that the actual form and mathematical equation of such an algorithm is beyond the scope of the present work.
TABLE 1
| Gene | Affected FP | Variations | Risk for toxicity | Evidence, dose reduction recommendation | Other considerations |
|---|---|---|---|---|---|
| DPYD | 5-FU, CAP, tegafur | *2A (rs3918290), *13 (rs55886062), c.2846T>A (rs67376798), c.1129–5923C>G (rs75017182, HapB3) | Increased overall toxicity, severe toxicity | Strong, available guidelines for dose reduction or alternative therapy | High positive predictive value, low sensitivity |
| Additional deleterious and/or reduced activity alleles | Need for validation in different populations and identification of ethnicity-specific variants | ||||
| DPD phenotyping can also be used to guide FP dosing | |||||
| TYMS | 5-FU, CAP, tegafur | 2R (rs45445694); 3′UTR 6bp ins/del (rs11280056) | Increased overall toxicity, severe toxicity | Strong, need for guidelines for dose reduction | Gene*gender interaction, priority in females |
| Low expression alleles | |||||
| ENOSF1 | 5-FU, CAP, tegafur | rs2612091, additional LOF, low expression alleles | Increased overall toxicity, severe toxicity | Strong, need for guidelines for dose reduction | TYMS*ENOSF1 interaction |
| CYP2A6 | tegafur | LOF, reduced activity alleles | Lack of response | Strong, need for clinical trials for dose modification (increase), need for guidelines for alternative therapy | Increased significance in Asian populations due to increased frequency of CYP2A6 LOF alleles |
| CES1 | CAP | Reduced activity alleles | HFS, toxicity | Moderate, potential dose reduction | Application in non-Asian populations |
| The clinical significant variants need to be identified and validated in different populations | |||||
| CDA | CAP | Reduced activity alleles | HFS, toxicity | Moderate, potential dose reduction | The clinical significant variants need to be identified and validated in different populations |
| CES2 | CAP | Reduced activity alleles | HFS, toxicity | Low | The clinical significant variants need to be identified and validated in different populations |
| DPYS | 5-FU, CAP, tegafur | Reduced activity alleles | Toxicity | Low | Analysis in cases of extreme toxicity in absence of other gene polymorphisms |
| UPB1 | 5-FU, CAP, tegafur | Reduced activity alleles | Toxicity | Low | Analysis in cases of extreme toxicity in absence of other gene polymorphisms |
| MTHFR | 5-FU, CAP, tegafur | LOF, reduced activity alleles | Toxicity | Extremely low | Gene*gender interaction, potential association in females |
| TYMP | CAP | Reduced activity alleles | HFS, toxicity | Extremely low | |
| UMPS | 5-FU, CAP, tegafur | Reduced activity alleles | HFS, toxicity | Extremely low |
Genetic variations that should be considered in any polygenic dosing clinical algorithm together with other non-genetic factors discriminating individuals at risk for FP-induced toxicity and in need for dose reduction or alternative therapy.
FPs, fluoropyrimidines; DPYD, dihydropyrimidine dehydrogenase; 5-FU, 5-fluorouracil; CAP, capecitabine; LOF, loss-of-function; ΤΥΜS, thymidylate synthase; ENOSF1, enolase superfamily member 1; CYP2A6, cytochrome P450 isoenzyme 2A6; CES, carboxylesterases; HFS, hand and foot syndrome; CDA, cytidine deaminase; DPYS, dihydropyrimidinase; UPB1, β-ureidopropionase 1; MTHFR, methylene tetrahydrofolate reductase; TYMP, thymidine phosphorylase; UMPS, uridine monophosphate synthetase.
2 Fluoropyrimidine pharmacokinetic and pharmacodynamic pathways
Pharmacokinetics and pharmacodynamics of the three FPs clinically used, 5-FU, CAP, and tegafur, are complex but well characterized and have been described elsewhere in detail (Thorn et al., 2011). We briefly present herein the enzymes that are involved in each step, from drug activation to drug action, since these enzymes are further discussed as for their potential pharmacogenomic importance. FP pharmacokinetic and pharmacodynamic pathways and the key enzymes involved are presented in Figure 1.
FIGURE 1
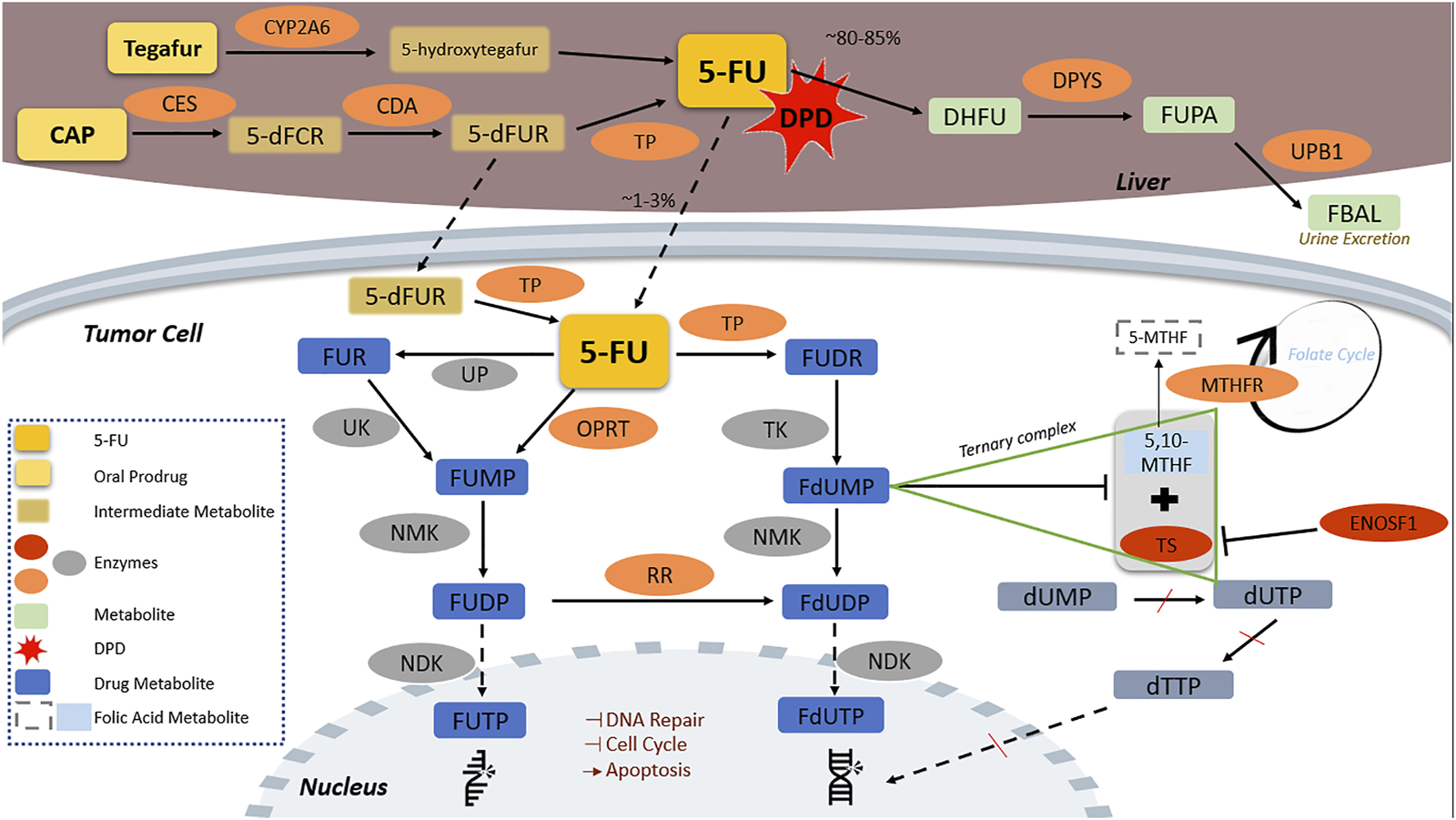
Fluoropyrimidine pharmacokinetic and pharmacodynamic pathway. CAP is transformed by CES to 5-dFCR which turns into 5-dFUR by CDA and then to its active form of 5-FU by TP. Tegafur, is metabolized in the liver by CYP2A6 to the unstable form of 5-hydroxytegafur, which spontaneously turns into 5-FU. 5-FU is catabolized by DPD to inactive metabolites in the liver; DPYS catabolizes DHFU to FUPA and UPB1 turns FUPA to FBAL which is excreted in the urine. The remaining 5-FU (approximately 1%–3% of initially administered) ends to the fluorine-substituted derivatives of uracil, FUTP and FdUTP, formed through two distinct pathways in which several enzymes are implicated. FUTP and FdUTP incorporate into RNA and DNA sequence inhibiting thus nucleic acid synthesis. FdUTP additionally inhibits the ternary complex of MTHFR substrate 5,10-MTHF with TS, which converts dUMP to dUTP and dTTP, a vital precursor for DNA replication and repair, inducing thereby cell apoptosis. ENOSF1 downregulates TS expression. 5,10-MTHF, 5,10-methylene tetrahydrofolate; 5-dFCR, 5-deoxy-5-fluorocytidine; 5-dFUR, 5-deoxy-5-fluorouridine; 5-FU, 5-fluorouracil; 5-MTHF, 5-methyltetrahydrofolate; CAP, capecitabine; CDA, cytidine deaminase; CES2, carboxylesterase 2; CYP2A6, cytochrome P450 2A6; DHFU, 5-fluoro-5,6-dihydrouracil; DPD, dihydropyrimidine dehydrogenase; DPYS, dihydropyrimidinase; dTTP, deoxythymidine triphosphate; dUMP, deoxyuridine monophosphate; dUTP, deoxyuridine triphosphate; ENOSF1, enolase superfamily member 1; FBAL, α-fluoro-β-alanine; FdUDP, fluorodeoxyuridine diphosphate; FdUMP, fluorodeoxyuridine monophosphate; FdUTP, fluorodeoxyuridine triphosphate; FUDP, fluorouridine diphosphate; FUDR, fluorodeoxyuridine; FUMP, fluorouridine monophosphate; FUPA, fluoro-β-ureidopropionate; FUR, fluorouridine; FUTP, fluorouridine triphosphate; MTHFR, methylenetetrahydrofolate reductase; NDK, nucleotide diphosphate kinase; NMK, nucleotide monophosphate kinase; OPRT, orotate phosphoribosyltransferase; RR, ribonucleotide reductase; TK, thymidine kinase; TP, thymidine phosphorylase; TS, thymidylate synthase; UK, uridine kinase; UP, uridine phosphorylase; UPB1, β-ureidopropionase 1.
In brief, CAP passes through the intestinal tract unchanged and is transformed to 5-deoxy-5-fluorocytidine (5-dFCR) by liver carboxylesterases (CES). 5-dFCR turns into 5-deoxyfluorouridine (5-dFUR) by cytidine deaminase (CDA) and then to its active form of 5-FU by thymidine-phosphorylase (TP, encoded by TYMP). The latter reaction is also catalyzed in cancer tissues (Lam et al., 2016). The second prodrug, tegafur, is metabolized in the liver by CYP2A6 to the unstable form of 5-hydroxytegafur, which spontaneously turns into 5-FU (Deac et al., 2020).
Approximately 80%–85% of the administered 5-FU is catabolized to inactive metabolites in the liver with DPD holding the key role in this critical step. 5-FU is converted to 5,6-dihydro-5-fluorouracil (DHFU) by DPD; DHFU is catabolized to fluoro-β-ureidopropionate (FUPA) and finally to α-fluoro-β-alanine (FBAL), which is excreted in the urine within 24 h (Miura et al., 2010). The enzymes involved in this process are dihydropyrimidinase (DPYS) and β-ureidopropionase 1 (UPB1). A proportion as low as 1%–3% of the administered 5-FU ends to fluorine-substituted derivatives of uracil fluorouridine triphosphate (FUTP) and fluorodeoxyuridine triphosphate (FdUTP) that mediate the therapeutic and cytotoxic effects of the drug. FUTP and FdUTP are formed through two distinct pathways (Thorn et al., 2011). Several enzymes, such as uridine kinase (UK), nucleotide monophosphate kinase (NMK), nucleotide diphosphate kinase (NDK), orotate phosphoribosyltransferase (OPRT), ribonucleotide reductase (RR), thymidine kinase (TK), and uridine phosphorylase (UP) are involved in these steps (Figure 1).
FUTP and FdUTP incorporate into RNA and DNA sequence thus inhibiting nucleic acid synthesis (Longley et al., 2003). FdUTP also inhibits thymidylate synthase (TS), the enzyme that binds deoxyuridine monophosphate (dUMP) (a precursor of dUTP) and this inhibition results in dUMP accumulation, deoxythymidine monophosphate (dTMP) suppression, and, subsequently, depletion of deoxythymidine triphosphate (dTTP), a vital precursor for DNA replication and repair. Reduced dTTPs disrupt DNA synthesis and repair, inducing thereby cell apoptosis (Lam et al., 2016; Ioannou et al., 2021). Importantly, additional enzymes, such as enolase superfamily member 1 (ENOSF1) and methylenetetrahydrofolate reductase (MTHFR) interfere with FP action, through interactions with TS and intermediate metabolites (Lam et al., 2016; De Mattia et al., 2019b). Finally, enzymes involved in DNA repair, cell cycle, and apoptosis mediate the cellular response to FPs (De Angelis et al., 2006).
As it appears, multiple enzymes are involved in FP pharmacokinetic and pharmacodynamic pathways. Identifying genetic variations affecting the activity of these enzymes and interfering with the final concentration of FUTP and FdUTP, holds promise to improve FP pharmacogenomics. Assumptions are based on published data presented in detail in this review.
3 Fluoropyrimidine pharmacogenomics
3.1 DPYD in the prime time
DPD is the primary enzyme for 5-FU breakdown, whereas DPD deficiency is known to be a leading cause of severe FP-induced toxicity (White et al., 2022). Genetic variations in the DPYD gene can severely affect DPD activity and are the cornerstone of FP pharmacogenomics. Over 160 polymorphisms have been identified in DPYD. However, four DPYD variants, namely, DPYD*2A (rs3918290), DPYD*13 (rs55886062), c.2846T>A (rs67376798), and c.1129–5923C>G (rs75017182, HapB3) currently drive FP dosing strategy. The association of these variations in DPYD both with FP-induced severe toxicity and the need for dose reductions is firmly established. Results of a recent meta-analysis showed that carriers of pathogenic DPYD gene variants have a severely increased (over 25-fold) risk of treatment-related death and a prevalence of 3.7% of treatment-related mortality (Sharma et al., 2021). Advances in clinical DPYD genotyping include guidelines for DPYD genotype and FP dosing released by Clinical Pharmacogenetics Implementation Consortium (CPIC) (Amstutz et al., 2018) and the Dutch Pharmacogenetics Working Group (DPWG) (Lunenburg et al., 2020). Both CPIC and DPWG recommend reductions in DPYD defective patients ranging from 25% to 50% based on DPYD allele combination (DPYD activity score) and, additionally, avoidance of FP use in patients with a complete lack of DPD activity (activity score 0) (Amstutz et al., 2018; Lunenburg et al., 2020).
Overall, the benefits and cost-effectiveness of implementing DPYD genotyping prior to FP dosing have been shown in several prospective trials and meta-analyses (Meulendijks et al., 2015; Henricks et al., 2018; Brooks et al., 2022; Glewis et al., 2022; van der Wouden et al., 2022). Regulatory agencies such as the Food and Drug Administration (FDA) and European Medicine Agency (EMA), have moved towards recognizing the importance of the application of DPYD genotyping in clinical practice. FDA has included drug label warnings describing the association of DPD deficiency with FP toxicity and the need for potential dose adjustments, however, no further details are provided (FDA, 2015a; FDA, 2016). EMA, since April 2020, has recommended that patients should be tested for the lack of DPD enzyme before starting FP treatment, either by measuring the level of uracil in the blood (phenotyping) or by genotyping for DPYD*2A, *13, c.2846T>A, and c.1129–5923C>G alleles (EMA, 2020). DPD phenotyping is a strategy that can be used to identify patients at risk for FP-induced toxicity (Deac et al., 2020), albeit has yet to be broadly incorporated in routine clinical practice. However, when phenotypic data is available, this information can guide FP-dosing decisions (Table 1). Following EMA recommendation, upfront DPYD genotyping has been endorsed by several countries in Europe, including Spain, Switzerland, Germany, Austria, the United Kingdom, and the Netherlands (Martens et al., 2019; Wörmann et al., 2020; Begré et al., 2022; Etienne-Grimaldi et al., 2022; García-Alfonso et al., 2022; Wang et al., 2022; White et al., 2022).
Despite the undisputable significance of DPYD variations on FP dosing decisions, DPYD polymorphisms can only explain a small percentage of FP-induced ADRs. The combined sensitivity of the major DPYD variants to predict grade 3–4 5-FU related toxicity is relatively low; results from DPYD genotyping in more than 2,500 5-FU treated patients (33.0% > 3 grade 5-FU toxicity) showed that DPYD*2A, D949V, and I560S variants resulted in 5.3% sensitivity, 99.4% specificity, 81.8% positive predictive value and 68.0% negative predictive value for grade ≥3 5-FU AE prediction (Lee et al., 2014). Considering the scarcity of the four recommended for genotyping DPYD variants [lower than 5% in European populations (Innocenti et al., 2020)] a missing heritability component exists within DPYD gene. Thus, the proportion of FP-treated patients that experience any grade of FP-induced toxicity is higher than the percentage of DPYD variant allele carriers treated with FPs, meaning that a negative DPYD test cannot exclude the possibility of experiencing drug-induced ADRs.
To improve sensitivity, DPYD gene is currently under intense study for the identification of additional, population-specific, clinically relevant variants, rare mutations, and/or copy number variations (Palles et al., 2021; De Luca et al., 2022; De Mattia et al., 2022; Kanai et al., 2022; Wigle et al., 2023). Indeed, results from a large scale genetic analysis in patients from the QUASAR2 clinical trial revealed three, additional to the CPIC-recommended, variants, namely, rs12132152, rs12022243, and p.Ala551Thr, that were associated with CAP-toxicity (Rosmarin et al., 2015). The combination of additional deleterious, albeit rare, DPYD variants can increase sensitivity (Etienne-Grimaldi et al., 2017), however, still, the estimated 10%–15% of DPD-linked FP-related AEs cannot be uniquely explained by DPYD low-frequency variants.
3.2 Beyond DPYD: most prominent associations
3.2.1 Thymidylate synthase, ΤYΜS
Thymidylate synthase (TS), encoded by TYMS gene, influences abundance of dTTPs while it is also inhibited by FdUMP (Figure 1). TYMS, therefore, is both necessary for DNA synthesis and a target of FPs (Hernando-Cubero et al., 2017). Patients overexpressing TS commonly present with 5-FU resistance, while patients with lower TS expression are better responders to the antitumor therapy being, however, also more vulnerable to drug-induced toxicity (Marsh, 2005). TYMS gene polymorphisms have been shown to alter TS expression and have been associated with FP treatment effectiveness and toxicity (Ioannou et al., 2021). Four common polymorphisms in TYMS gene located in TYMS untranslated regions (UTRs) are known to affect TS expression: TYMS-TSER 2R/3R (rs45445694), 2RG/2RC (rs183205964), 3RG/3RC (rs2853542) and a 6bp deletion allele (rs11280056) (Marsh, 2005; Lam et al., 2016; Xie et al., 2020).
Several TYMS polymorphisms have been extensively studied and are associated with FP-induced toxicity, suggesting that FP dose can be tailored to TYMS genotype. In the meta-analysis conducted by Rosmarin et al. (2014)TYMS-TSER 2R allele, 3′UTR 6bp ins/del variant and TYMS low activity genetic burden favor global CAP-induced toxicity (OR 1.36 for TYMS-TSER 2R allele, OR 1.25 for 3′UTR 6bp ins/del and OR 1.31 for TYMS genetic score). In a following meta-analysis in non-DPYD*2A carriers, low TYMS expression genotypes (3RC/2RC, 2RG/2RC and 2RC/2RC) were associated with global severe toxicity (OR 3.0) and toxicity-related hospitalization (OR 3.8) (Meulendijks et al., 2016). Recently, in the largest, so far, meta-analysis including more than 1,900 patients, TYMS 2R allele was associated with grade 3 HFS (OR 1.50), while TYMS 3′UTR 6bp ins/del was associated with overall toxicity (OR 1.21) and grade 3 HFS (OR 1.41) (Hamzic et al., 2020). Schaerer et al. (2020) have proposed that TYMS polymorphisms affect the number of upstream stimulatory factor (USF1) binding sites in the gene. The authors have shown that patients with fewer USF1-binding sites have increased risk for early-onset gastrointestinal toxicity (OR 1.66) and severe gastrointestinal toxicity (OR 1.74 after adjustment for DPYD) in response to 5-FU treatment.
TYMS is considered a gene of importance in FP pharmacogenomics, however, no predictive strategies have yet been clinically applied (Amstutz et al., 2018). It appears, therefore, that TYMS variants that reduce TS expression can improve the pharmacogenomic DPYD-guided dosing strategy and it is anticipated that soon TYMS will be the second FP-dosing associated pharmacogene.
3.2.2 Enolase superfamily member 1, ENOSF1
ENOSF1 gene encodes for the mitochondrial enolase superfamily member 1, an enzyme that catalyzes the conversion of L-fuconate to 2-keto-3-deoxy-L-fuconate. It was initially identified as a gene coding for an antisense RNA that downregulates TS expression via promoting TYMS mRNA degradation (Dolnick, 1993; Dolnick et al., 1996; Chu and Dolnick, 2002). ENOSF1 and TYMS, both located on chromosome 18, show a partial overlap in their sequences and are transcribed in opposite directions. Several polymorphisms have been identified within ENOSF1 and have been studied as for their functional effect on TYMS gene expression and consequently on FP-induced toxicity.
The seminal report on the association of ENOSF1 and CAP toxicity was provided by Rosmarin et al. (2015). The authors have sequenced 25 CAP/5-FU pathway genes in a cohort of 968 patients, participants of QUASAR2 study. They have found a significant association of ENOSF1 rs2612091A intronic variant, lying 10 kb downstream of TYMS, with CAP toxicity (OR = 1.59) (Rosmarin et al., 2015). The authors additionally proposed that ENOSF1 rs2612091 polymorphism affects TS protein activity rather than TYMS expression (Rosmarin et al., 2015). The association of ENOSF1 rs2612091 polymorphism with FP-induced toxicity was later replicated by a study in a cohort of 239 CAP-treated patients showing that ENOSF1 rs2612091 was associated with HFS (OR = 2.28) (García-González et al., 2015). Further studies have shown that ENOSF1 rs2612091 is associated with shorter overall survival (Meulendijks et al., 2017) and treatment non-response (Arjmandi et al., 2022), whereas ENOSF1/TYMS rs699517 polymorphism was associated with CAP-induced severe nausea/vomiting, anorexia and fatigue (Pellicer et al., 2017). A meta-analysis of ENOSF1 rs2612091 association with FP-induced toxicity has demonstrated that ENOSF1 rs2612091 was associated with severe HFS (OR = 1.64) independently of TYMS variants (Hamzic et al., 2020).
ENOSF1 appears as a promising marker for prediction of FP-induced toxicity. While we expect that more studies will be published on ENOSF1 and its association with dose requirements, available associations show a high degree of consistency that favors incorporation of ENOSF1 pharmacogenomics in clinical practice. It is noteworthy that the ENOSF1 rs2612091 effect appears independently of the effect of TYMS polymorphisms. More importantly, Palles et al. (2021) have shown that when ENOSF1 rs2612091 is integrated to the DPYD-based prediction model for FP-induced toxicity, it significantly improves prediction of global toxicity, hematological toxicity, HFS and diarrhea. It appears that ENOSF1 may have a place in any future polygenic algorithm for FP-dosing.
3.2.3 Methylene tetrahydrofolate reductase, MTHFR
MTHFR is also an extensively-studied pharmacogene in terms of its effects on FP treatment response. MTHFR irreversibly catalyzes 5,10 MTHF conversion to 5-methyltetrahydrofolate (5-MTHF), the primary methyl donor responsible for DNA methylation. As in the case of TS, FdUMP forms a ternary complex with MTHFR substrate, 5,10-MTHF. Therefore, decreased MTHFR activity leads to increased 5,10-MTHF concentration and can enhance TS inhibition and FP activity, increasing this way the risk for FP-induced ADRs (Lam et al., 2016; De Mattia et al., 2019b).
Two MTHFR polymorphisms, which are in linkage disequilibrium, −677C>T (rs1801133) and −1298C>A (rs1801131), lead to impaired MTHFR enzymatic activity and have been extensively studied in association with FP-induced toxicity (Ramalakshmi et al., 2016). A large number of studies focused on MTHFR pharmacogenomics have been published, however, to date, the impact of MTHFR variations on FP-induced toxicity is still not clear (Toffoli and De Mattia, 2008; De Mattia and Toffoli, 2009). Results of meta-analyses, as reviewed by Campbell et al. (2016), show isolated associations within studies rather than a universal effect of MTHFR on FP-induced toxicity. In a more recent meta-analysis, Zhong et al. (2018) also conclude that MTHFR polymorphisms could not be considered as reliable factors for predicting FP clinical response. A potential limitation in MTHFR case may rely on the folate cycle, a pathway involving several enzymes that can modulate MTHFR potency; folylpolyglutamate synthetase enzyme (FPGS) and gamma-glutamyl hydrolase (GGH) are such enzymes interfering with intracellular folate concentrations. Studies on the association of FPGS and GGH variations on chemotherapy response have started to emerge (Kim et al., 2008; Fernandes et al., 2021) and we anticipate that more information will be gathered in the near future.
Based on these findings, more research is required for the integration of MTHFR genetic information in a polygenic FP-dosing algorithm. Future research, however, should focus on limitations that may hinder the effect of MTHFR on FP response, such as heterogeneity of cancer patients recruited in different studies, variable 5-FU based regimens and interactions of gene with nutrition, ethnicity and other environmental factors.
3.3 Additional genetic variations towards a polygenic dosing algorithm
For FPs, beyond DPYD and the abovementioned extensively studied genes, several polymorphisms in other genes have been assessed as for their potential association with FP-induced toxicity. In the following sections, results of studies assessing the pharmacogenomic implication of genes participating in FP response pathways are described and critically reviewed. Genes that show a high degree of consistency in their association with FP response across different studies are summarized in Table 1.
3.3.1 Carboxylesterases, CES
Carboxylesterases (CES) belong to the serine enzyme superfamily and they metabolize and activate several drugs, including CAP. In humans, five CES enzymes are coded (CES1-CES5); among them, CES1 and CES2 have a role in CAP metabolism, catalyzing 5-DFCR formation with similar catalytic efficiency (Ramalakshmi et al., 2016). Both CES2 and CES1 polymorphisms have been found to influence CAP treatment outcomes. Results of relevant studies are described in Table 2 and Table 3, respectively.
TABLE 2
| Examined CES2 variants | Study population | Endpoints | Results | Ref |
|---|---|---|---|---|
| −823 C>G; −854 G>C; 5841 G>A; 6046 G>A; 6174 G>A; 6320 G>A | 136 CAP-treated metastatic breast or colorectal cancer patients, 39% grade 3–4 toxicity (n = 123 genotyped for CES2) | Grade 3–4 toxicity | A trend towards association between 6046 G>A and diarrhea incidence (p = 0.09) | Ribelles et al. (2008) |
| rs11075646 (−823 C>G); rs2241409; rs11568314; rs11568311 | 111 CAP-treated HER2/neu- negative metastatic breast cancer patients | Grade 3–4 toxicity | −823 C>G associated with grade 3–4 HFS (OR = 4.49, 95% CI 1.43–14.14, p = 0.01) | Martin et al. (2015) |
| rs11075646; rs2241409; rs11568314; rs11568311 | QUASAR2 trial (ISRCTN45133151) | Grade 0–2 vs. grade 3+ toxicity | No association found at the threshold set in the study | Rosmarin et al. (2014) |
| 927 post-operative stage II/III colorectal cancer patients administered CAP in monotherapy or in combination with bevacizumab | ||||
| Meta-analysis: rs11075646; rs11568314; rs2241409; rs11568311; 6046 G>A; 6320 G>A | Rs11075646 genotyped in 881 patients, while rs2241409 rs11568311 | |||
| genotyped in 442 and rs11568314 | ||||
| in 439 patients | ||||
| rs11075646; rs2241409; rs11568314; rs11568311 | 130 breast and colorectal cancer patients | Grade 3 HFS | No association found | Caronia et al. (2011) |
| rs11075646 | 188 women with HER2-negative metastatic breast cancer randomized on CAP-including or CAP-excluding schemes, n = 184 genotyped for rs11075646 | HFS, first dose reduction | CES2 rs11075646 WT genotype benefit from CAP-including scheme (progression-free survival benefit) | Lam et al. (2018) |
| No association with HFS or first dose reduction | ||||
| rs11075646 | 26 metastatic breast adenocarcinoma female patients | Grade 1–3 toxicity | No association found | Rudek et al. (2013) |
| rs11075646 | 446 patients treated with CAP monotherapy or in combination with oxaliplatin | HFS occurrence | No association found | de With et al. (2023) |
Studies examining the effect of CES2 gene polymorphisms on CAP-induced toxicity.
CES2, carboxylesterase 2; CAP, capecitabine; HER2, human epidermal growth factor receptor 2; HFS, hand and foot syndrome; OR, odds ratio; WT, wild-type.
TABLE 3
| Examined CES1 variants | Study population | Endpoints | Results | Ref |
|---|---|---|---|---|
| rs2244613; rs2244614; rs3217164; rs7187684; rs1186118; rs71647871 | 144 cancer patients, CAP monotherapy or in combination regimens | Grade 0–1 vs. 2 vs. 3–4 adverse events (HFS, diarrhea), overall toxicity | Associated with overall toxicity: 1165–41 C>T (p = 0.001), 690 + 129del (p < 0.001), rs1186118 and rs7187684 (p = 0.012 each), 1165–33 C>A (p = 0.013) | Hamzic et al. (2017) |
| A3 minor-alleles-haplotype associated with overall toxicity in additive (OR 2.18, 95% CI 1.19–4.00, p = 0.012) and recessive genetic model (OR 10.25, 95% CI 2.12–49.43, p = 0.0038) | ||||
| A1 major alleles haplotype was protective against CAP-induced toxicity (OR 0.60, 95% CI 0.36–0.99, p = 0.047) | ||||
| rs2244613; rs2244614; rs3217164 | 446 patients treated with CAP monotherapy or in combination with oxaliplatin | HFS occurrence | 1165–33 C>A variation associated with HFS ≥ grade 2 (OR 1.888; 95% CI 1.075–3.315, p = 0.027) | de With et al. (2023) |
| rs7498748 | 301 colorectal cancer patients, CAP monotherapy or in combination regimens | Grade 2 adverse events, overall toxicity, treatment administration delay | No association found | Pellicer et al. (2017) |
| rs2244613; rs2244614; rs3217164; rs7187684; rs1186118 | 36 Japanese postoperative or metastatic colorectal cancer patients | Overall toxicity (grade 3) | No association found | Matsumoto et al. (2020) |
| rs2244613; rs2244614; rs3217164; rs7187684; rs1186118 | 338 Chinese colorectal and gastric cancer administered FPs | Hematological, hepatic, gastrointestinal toxicity, and HFS | No association found | Liu et al. (2021) |
Studies examining the effect of CES1 gene polymorphisms on CAP-induced toxicity.
CES1, carboxylesterase 1; CAP, capecitabine; HFS, hand and foot syndrome; OR, odds ratio; FPs, fluoropyrimidines.
3.3.1.1 CES2
Historically, CES2 polymorphisms were studied prior to CES1 polymorphisms for their potential association with CAP response in terms of toxicity incidence. Ribelles et al. (2008) have analysed CES2 −823 C>G, −854 G>C, 5841 G>A, 6046 G>A, 6174 G>A, and 6320 G>A polymorphisms in relation to CAP response and severe toxicity in a prospective study in 123 CAP-treated patients. CES2 6046 G>A showed a non-significant trend (p = 0.09) towards increased incidence of grade 3–4 diarrhea. Martin et al. (2015) have assessed the association of CES2 rs2241409, rs11568314, rs11568311, and rs11075646 (−823 C>G) polymorphisms with grade 3–4 toxicity in 111 women with advanced breast cancer treated with CAP monotherapy. It was found that rs11075646 (5′UTR 823 C>G) minor allele was associated with increased risk for grade 3–4 HFS (OR 4.49, p = 0.01). Evidence for association of CES2 polymorphisms with CAP-induced HFS also derive from QUASAR2 clinical trial, a study on 927 CAP-treated patients. Rs2241409 showed a trend towards association with grade ≥3 HFS (p = 0.035), however, this association is not statistically significant at the threshold set in the study (Rosmarin et al., 2014). There appears to be a tendency towards a weak association of CES2 polymorphisms with HFS, however, these results were not replicated in several other studies (Caronia et al., 2011; Rudek et al., 2013; Lam et al., 2018; de With et al., 2023).
Overall, it appears that results accumulated so far do not support a role of CES2 polymorphisms on FP-induced toxicity. The potential association of CES2 variations with HFS merits further study.
3.3.1.2 CES1
CES1 variants have also been studied in relation to CAP-induced toxicity (Table 3). Hamzic et al. (2017) examined the potential role of CAP-activating genes (CES1, CES2, TYMP, UPP1, and UPP2) in severe (grade 3–4) early-onset toxicity in 144 cancer patients administered CAP. Patients were genotyped for six CES1 polymorphisms; rs2244613, rs2244614, rs3217164, rs7187684, rs1186118, and rs71647871. With the exception of rs71647871, all studied CES1 polymorphisms were significantly associated with grade 2–4 CAP-induced toxicity. In multivariate analysis (adjusted for DPYD risk variants) the haplotype encompassing rs2244613, rs2244614, rs3217164, rs7187684, and rs1186118 minor alleles was an independent predictor of CAP-induced toxicity. More recently, CES1 1165–33 C>A variation was strongly associated with HFS ≥ grade 2 (OR 1.888, p = 0.027) in a study including 446 patients of whom 32.7% developed HFS (17.3%≥ grade 2) (de With et al., 2023). It should be noted that studies conducted in Asian populations did not find an association of various CES1 polymorphisms and CAP-induced toxicity (Matsumoto et al., 2020; Liu et al., 2021).
Whether the association of CES1 with FP-response is limited in non-Asian populations needs to be further investigated. Following the example of DPYD for which it has been reported that the registered CPIC variations have only a minor role in FP-related toxicity in an Asian population (Kanai et al., 2022), we propose that CES1 could be initially incorporated in a polygenic FP dosing algorithm for other ancestries until firm conclusions are drawn on its potential effect on FP response in Asian populations. It appears that multiple CES1 variants should be considered as predictive factors to CAP-induced HFS. The exact CES1 polymorphism combination as well as the potential interaction of CES1*CES2 genes need to be verified.
3.3.2 Cytidine deaminase, CDA
Cytidine deaminase (CDA) has a crucial role in pharmacological activation of CAP to 5-FU and exhibits a highly variable enzymatic activity among individuals. This variation can be partially attributed to its different haplotypes which can lead to an increase or decrease of 5-FU concentration (Morita et al., 2003). The results of studies examining the association of CDA variants with CAP-induced toxicity are summarized in Table 4.
TABLE 4
| Examined CDA variants | Study population | Endpoints | Results | Ref |
|---|---|---|---|---|
| −943del/insC; 1052 A>C; 575 C>T; 771 C>G; 794 G>A; 942 C>G | 136 CAP-treated metastatic breast or colorectal cancer patients, 39% grade 3–4 toxicity (n = 123 genotyped for CDA) | Grade 3–4 toxicity | A trend towards association between −943insC allele and grade 3 HFS (21% of the carriers of one or two insertion alleles vs. 8% for the wild type patients, p = 0.07) | Ribelles et al. (2008) |
| rs532545 (−451 C>T); rs602950 (−92 A>G); rs2072671; rs3215400 (−943del/insC); rs603412 | 130 CAP-treated breast and colorectal cancer patients | Grade 3 HFS | −451T associated with HFS (OR 2.02, 95% CI 1.02–3.99, p = 0.039) in additive genetic model | Caronia et al. (2011) |
| −943insC associated with lower risk for HFS in additive (OR 0.51, 95% CI 0.27–0.95, p = 0.028) and recessive genetic model (OR 0.37, 95% CI 0.16–0.86, p = 0.020) | ||||
| −451C>T; −943del/insC; −92 A>G; 79 A>C | 244 patients with gastrointestinal cancer administered CAP regimens | Adverse events, overall toxicity | −451T associated with diarrhea grade 2–4 (OR 2.3, 95% CI 1.3–4.2, p = 0.0082) | Loganayagam et al. (2013) |
| −92G associated with grade 2–4 diarrhea (p = 0.002) and dehydration (p = 0.042) | ||||
| rs2072671 (79 A>C) | 239 colorectal cancer patients administered CAP in monotherapy (n = 69) or in combination regimens (n = 170) | Grade 3 toxicities (HFS, diarrhea, hematological toxicity), overall toxicity, treatment reduction/delay/withdrawal | 79A associated with overall toxicity in univariate (p = 0.008) and in multivariate analysis (OR 1.84, 95% CI 1.06–3.18, p = 0.029) | García-González et al. (2015) |
| −182 G>A; 79 A>C; 435 C>T | 301 colorectal cancer patients administered CAP in monotherapy or in combination regimens | Grade 2 adverse events, overall toxicity, and treatment administration delay | 79AA associated with risk of toxicity (OR 1.89) and HFS (OR 3.83) | Pellicer et al. (2017) |
| 435T associated with higher bilirubin (OR 8.621, 95% CI 1.058–70.247, p = 0.044) | ||||
| −182A associated with treatment administration delay (OR 2.743, 95% CI 1.346–5.588, p = 0.005) | ||||
| 1172 G>A; −451 C>T; −92 A>G; 435 C>T; 265 A>T; 266+242 A>G; 79 A>C | 144 cancer patients administered CAP in monotherapy or in combination regimens | Grade 0–1 vs. 2 vs. 3–4 toxicities (HFS, diarrhea), overall toxicity | −92G, −451T associated with grade 2–4 diarrhea (OR 4.40, 95% CI 1.34–14.5, p = 0.015, and OR 4.29, 95% CI 1.30–14.2 p = 0.017, respectively) | Hamzic et al. (2017) |
| 1172A associated with HFS (OR 3.50, 95% CI 1.13–10.9, p = 0.030) | ||||
| 266 + 242A>G associated with overall toxicity (OR 2.00, 95% CI 1.00–3.96, p = 0.048) and diarrhea (OR 3.33, 95% CI 1.26–8.81, p = 0.015) | ||||
| −451T, −92G, 33delC and 79C haplotype associated with diarrhea (OR 2.09, 95% CI 1.07–4.10, p = 0.032) | ||||
| −451 A>G; −92 C>T; 79 A>C; 1172 G>A | 322 colorectal or gastric cancer Chinese patients administered CAP in monotherapy or in combination regimens | Toxicity | −451AA genotype associated with less hepatotoxicity (OR 0.200, 95% CI 0.045–0.895, p = 0.035) and grade 3–4 hematological toxicity (OR 0.205, 95% CI 0.045–0.927, p = 0.039) | Liu et al. (2019) |
| rs2072671; rs603412; rs10916825 | 446 patients treated with CAP monotherapy or in combination with oxaliplatin | HFS occurrence | 266 + 242 A>G (rs10916825) was associated with HFS ≥ grade 2 (OR 1.865, 95% CI 1.087–3.200, p = 0.024) | de With et al. (2023) |
| −451 C>T; −92 A>G; 79 A>C; −943del/insC; rs603412 | 111 CAP treated HER2/neu- negative metastatic breast cancer patients, (50 control patients +61 experimental patients) | Grade 3–4 toxicity | No association found | Martin et al. (2015) |
| −451 C>T; −943del/insC | 188 women with HER2-negative metastatic breast cancer randomized on CAP-including (n = 93) or CAP-excluding schemes (n = 95) | HFS | No association found | Lam et al. (2018) |
| −451 C>T; 79 A>C; −205 C>G | QUASAR2 trial (ISRCTN45133151) | Grade 0–2 vs. grade ≥3 adverse events | No association found at the threshold set in the study | Rosmarin et al. (2014) |
| 927 post-operative stage II/III colorectal cancer patients administered CAP in monotherapy or in combination regimens | ||||
| −451C>T and 79A>C genotyped in 927 patients, −205C>G | ||||
| Meta analysis: 79 A>C; -943del/insC; 575 C>T; 771 C>G; −205 C>G; −92 A>G; 794 G>A | genotyped in 89 patients |
Studies examining the effect of CDA gene polymorphisms on CAP-induced toxicity.
CDA, cytidine deaminase; CAP, capecitabine; HFS, hand and foot syndrome; OR, odds ratio; ADRs, adverse drug reactions; HER2, human epidermal growth factor receptor 2; 5-FU, 5-fluoruracil.
Ribelles et al. (2008) were the first to report a trend towards increased risk for grade 3 HFS in CDA −943insC allele carriers (p = 0.07); study cohort consisted of 123 patients with metastatic breast or colorectal cancer. Caronia et al. (2011) genotyped CDA −451C>T, −92A>G, 79A>C, −943delC, and −205C>A variants in 130 cancer patients and showed that −451T allele predicted severe HFS (OR 2.02, p = 0.039). Interestingly, −943insC allele was found to have a protective effect against HFS (OR 0.51, p = 0.028). Those results were further verified through CDA expression analysis in 89 lymphoblastoid cell lines from Caucasian healthy individuals, in which −943delC homozygous cell lines had >3-fold increased CDA mRNA expression (compared to the carriers of the insertion allele). In a larger retrospective study enrolling 430 patients with gastrointestinal cancer, Loganayagam et al. (2013) genotyped CDA −451C>T, −943delC, −92A>G, and 79A>C polymorphisms in a subcohort of 244 patients receiving CAP-based chemotherapy, who did not carry DPYD defective variants. In regression analysis, −92G allele was associated with grade 2–4 diarrhea (p = 0.002) and dehydration (p = 0.042), whereas −451T was associated with grade 2–4 diarrhea (p = 0.0082).
The association of CDA 79A>C polymorphism with FP-induced toxicity was further studied by García-González et al. (2015) in a study including 239 CAP-treated patients. CDA 79A>C polymorphism was associated with overall toxicity (any toxicity grade ≥3) both in multivariate and univariate analysis (p = 0.029 and 0.008, respectively). Three CDA variants (−182G>A, 79A>C, and 435C>T) were further examined in another study, including 301 patients. CDA 435T allele was associated with hepatotoxicity (as estimated by higher bilirubin) (OR 8.62, p = 0.044), −182A with treatment administration delay due to ADRs (OR 2.743, p = 0.005) and 79A with both HFS (OR 3.83), and overall toxicity (OR 1.89) (Pellicer et al., 2017). CDA variants were also analyzed by Hamzic et al. (2017) in a cohort of 144 CAP-treated patients. After the coding and flanking areas of CDA were sequenced, seven variations (−1172G>A, −451C>T, −92A>G, 435C>T, 265A>T, 266 + 242A>G, and 79A>C) were further examined. The authors have shown that carriers of c.266 + 242A allele had increased risk for overall toxicity (OR 2.0, p = 0.048) and diarrhea (OR 3.33, p = 0.015). Additional associations with diarrhea were found for −451T, and −92G alleles (OR 4.29, p = 0.017 and OR 4.40, p = 0.015, respectively), and with the haplotype consisting of −451T, −92G, 1-33delC, and 79C alleles (OR 2.09, p = 0.032). Furthermore, −1172A was associated with HFS (OR 3.50, p = 0.030). The association of CDA -451A>G polymorphism with CAP-induced ADRs was also studied in 322 Chinese patients with gastrointestinal cancer. The authors have found that −451AA genotype has a rather protective effect on hematological (p = 0.039) and hepatological toxicity (p = 0.035) (Liu et al., 2019). More recently, in a study including 446 patients of whom 32.7% developed HFS (17.3% ≥ grade 2), CDA 266 + 242 A>G variation was strongly associated with HFS ≥ grade 2 (OR 1.865, p = 0.024) (de With et al., 2023).
Despite the extensive evidence of multiple CDA variants being associated with CAP-induced ADRs, these results were not replicated in all studies (Rosmarin et al., 2014; Martin et al., 2015; Lam et al., 2018). The low prevalence of studied variants may potentially explain this discordance in findings. For CDA, therefore, current evidence suggests that multiple gene variants exist that potentially affect CAP-induced ADRs and this gene should be considered for incorporation in a polygenic algorithm to adjust for CAP dose.
3.3.3 Thymidine phosphorylase, TYMP
Thymidine phosphorylase (TP), encoded by TYMP gene, catalyzes the conversion of CAP to 5-FU and the subsequent conversion of 5-FU to FUDR in the metabolic pathway forming FdUMP (Bonotto et al., 2013). Apart from liver, this reaction takes also place in cancer cells (Figure 1); TYMP expression is higher in tumor cells compared to healthy tissue, leading to CAP preferential activation in tumor cells (Lam et al., 2016; Liu et al., 2021). The impact of TYMP polymorphisms on FP treatment response in terms of ADRs has been extensively investigated, however, the results appear contradictory (Table 5).
TABLE 5
| Examined TYMP variants | Study population | Endpoints | Results | Ref |
|---|---|---|---|---|
| rs11479; rs112723255 | 253 colorectal cancer patients administered CAP (n = 159) or 5-FU (n = 94) in monotherapy or in combination regimens | Early dose modifications (delays or reductions), severe toxicity | rs11479 associated with early dose modifications (OR 2.02, 95% CI 1.03–4.00, p = 0.042), and severe toxicity (OR 2.70, 95% CI 1.23–5.92, p = 0.013) | Jennings et al. (2013) |
| rs11479; rs470119 | QUASAR2 trial (ISRCTN45133151) | Grade 0–2 vs. grade 3+ adverse events | No association found at the threshold set in the study | Rosmarin et al. (2014) |
| 927 post-operative stage II/III colorectal cancer patients administered CAP in monotherapy or in combination regimens; rs470119 genotyped in 927 patients and rs11479 in 857 patients | ||||
| Meta-analysis: rs11479; rs470119; rs131804 | ||||
| 92 TYMP variants | 940 post-operative stage II/III colorectal cancer patients administered CAP | Grade 0–2 vs. 3–4 HFS, diarrhea, and overall toxicity | No association found at the threshold set in the study | Rosmarin et al. (2015) |
| rs11479; rs131804; rs470119 | 130 breast and colorectal cancer patients administered CAP | Grade 3 HFS | No association found | Caronia et al. (2011) |
| rs11479; rs470119; rs131804 | 111 HER-2/neu- negative metastatic breast cancer patients administered CAP (50 control patients +61 experimental) | Grade 3–4 toxicity | No association found | Martin et al. (2015) |
| rs11479 | 185 gastric cancer patients administered CAP in combination regimens | Toxicities (hematological, gastrointestinal, HFS) | No association found | Meulendijks et al. (2017) |
| Sequencing of coding and exon-flanking regions of TYMP | Discover subset: 24 different cancer-type patients with grade 3 toxicity (without DPYD-risk variants or comedication), and 24 matched controls all administered CAP in monotherapy or in combination regimens | Grade 0–1, 2, 3–4 adverse events (HFS, diarrhea), overall toxicity (grade 0–1 vs. 2 vs. 3–4) | No association found | Hamzic et al. (2017) |
| rs470119 | 301 colorectal cancer patients administered CAP in monotherapy or in combination regimens | Grade 2 adverse events, overall toxicity, treatment administration delay | No association found | Pellicer et al. (2017) |
| rs11479 | 208 cancer patients administered CAP or placebo once daily | Grade 2 HFS | No association found | Yap et al. (2017) |
| rs470119 | 338 Chinese colorectal and gastric cancer administered FPs | Hematological, liver, gastrointestinal toxicity, and HFS | No association found | Liu et al. (2021) |
| rs11479 | 216 Brazilian gastrointestinal (n = 92) or colorectal cancer (n = 124) patients administered 5-FU in monotherapy or in combination regimens | Grade 1–4 and overall toxicity | No association found | Fernandes et al. (2021) |
Studies examining the effect of TYMP gene polymorphisms on CAP-induced adverse events.
CDA, cytidine deaminase; CAP, capecitabine; HFS, hand and foot syndrome; OR, odds ratio; ADRs, adverse drug reactions; HER2, human epidermal growth factor receptor 2; 5-FU, 5-fluoruracil.
Jennings et al. (2013) were the first to report an association between TYMP rs11479 polymorphism and FP-induced ADRs, in an observational study including 253 colorectal cancer patients. Study endpoints included severe toxicity incidence and early dose modifications. Rs11479 minor allele was associated with both overall severe toxicity (OR 2.70, p = 0.013) and a need for early dose modifications (OR 2.02, p = 0.042). Results of this seminal study, however, were not replicated in other studies (Caronia et al., 2011; Rosmarin et al., 2014; Martin et al., 2015; Rosmarin et al., 2015; Hamzic et al., 2017; Meulendijks et al., 2017; Pellicer et al., 2017; Yap et al., 2017; Fernandes et al., 2021; Liu et al., 2021). Overall, it appears unlikely that TYMP polymorphisms could have a significant effect on CAP-induced toxicity.
3.3.4 CYP2A6
Tegafur is an oral prodrug of 5-FU, predominantly administered in combination with uracil or as a combination fluoropyrimidine regimen, called S-1, consisting of tegafur, 5-chloro-2,4-dihydroxypyridine (CDHP), and potassium oxonate (Oxo) (Choi et al., 2012). Tegafur is converted to 5-FU by the cytochrome P450 isoenzyme CYP2A6. CYP2A6 is a highly polymorphic gene and its genetic variations have been previously described in detail (Tanner and Tyndale, 2017). In brief, clinically significant alleles include CYP2A6*4 that leads to CYP2A6 gene deletion, and several reduced activity alleles, including *2 and *5 to *35. There are significant differences in CYP2A6 allele frequency among ethnicities; a higher prevalence of CYP2A6 defective alleles in Asian and African American populations has been reported. Additionally, CYP2A6 is a highly inducible enzyme. Among CYP2A6 inducers, estrogens may account for gender related differences in CYP2A6 activity (Tanner and Tyndale, 2017).
The presence of CYP2A6 reduced activity alleles has been extensively studied in association with tegafur pharmacokinetics (Table 6). It appears that CYP2A6 poor metabolizer phenotype leads to lower 5-FU AUC, lower tegafur oral clearance, and higher tegafur Cmax and AUC (van Schaik, 2005; Kaida et al., 2008; Hirose et al., 2010; Ishii et al., 2010; Kim et al., 2011; Kim et al., 2016). It can be expected therefore that CYP2A6 poor metabolizers are poor responders to tegafur. Several studies have shown that carriers of CYP2A6 wild-type alleles present with higher response rates to tegafur or S-1 (Kong et al., 2009; Park et al., 2011; Kim et al., 2013; Jeong et al., 2017; Yang et al., 2017; Kim et al., 2018). Based on pharmacokinetic data, the relative risk for tegafur-induced toxicity may be decreased for CYP2A6 poor metabolizers. Indeed, the majority of the studies that assessed the effect of CYP2A6 defective alleles on tegafur-induced toxicity did not find an association (Kaida et al., 2008; Kong et al., 2009; Hirose et al., 2010; Ishii et al., 2010; Kim et al., 2011; Park et al., 2011; Choi et al., 2012; Fang et al., 2012; Kim et al., 2013; Kim et al., 2016; He et al., 2017; Jeong et al., 2017; Kim et al., 2018) (Table 6). Some sparse associations that have been published for common CYP2A6 defective alleles did not remain significant in multivariate analyses (Tsunoda et al., 2011; Kim et al., 2017) and the authors suggest that CYP2A6 defective alleles are not involved in tegafur-induced toxicity. However, it cannot be excluded that toxicity in CYP2A6 poor metabolizers may occur due to alternative tegafur-activating pathways. Exome sequencing of CYP2A6 has revealed additional variants, such as rs60823196 and rs138978736. For these variants, an association with grade 3–4 diarrhea was shown in 60 patients treated with S-1 plus oxaliplatin (OR 6.43 for rs60823196 and OR 14.86 for rs138978736, respectively), however, their functional effect on CYP2A6 is still unknown (Yang et al., 2017). CYP2A6 variations therefore could be considered as an additional genetic factor to guide the choice of FP therapy leading to avoidance of tegafur-based schemes in carriers of CYP2A6 defective alleles.
TABLE 6
| Examined CYP2A6 variants | Study population | Endpoints | Results | Ref |
|---|---|---|---|---|
| CYP2A6*1; CYP2A6*4C; CYP2A6*7; CYP2A6*9 | 46 Japanese patients with non-small-cell lung cancer administered S-1 in combination with cisplatin (n = 31) or in monotherapy (n = 15) | Tegafur and 5-FU pharmacokinetics: AUC0–10hr, Cmax, Tmax | CYP2A6*4C carriers have lower 5-FU Cmax, lower 5-FU AUC0-10 and higher tegafur AUC0-10 in No association with toxicity endpoints | Kaida et al. (2008) |
| Patients grouped into CYP2A6*4C carriers (n = 31) and non-carriers (n = 15) | Efficacy endpoints: response rate, adverse events | |||
| CYP2A6*1; CYP2A6*4A; CYP2A6*5; CYP2A6*7; CYP2A6*9; CYP2A6*10; CYP2A6*11 | 34 patients with progressive/recurrent digestive organ cancer administered S-1 | Pharmacokinetic endpoints: plasma 5-FU concentration | Lower tegafur, and higher 5-FU concentration in the carriers of one variant allele compared to the carriers of two variant alleles (p = 0.0309 and p = 0.0025, respectively) | Ishii et al. (2010) |
| Efficacy endpoints: antitumor effect, adverse events | No association with efficacy endpoints | |||
| CYP2A6*1; CYP2A6*4; CYP2A6*7; CYP2A6*9; CYP2A6*10 | 43 Asian patients with locally advanced gastric cancer administered S-1 in combination with docetaxel | Pharmacokinetic parameters: tegafur, 5-FU CDHP AUC0–12hr, Cmax | Carriage of two variant alleles associated with higher mean Cmax for tegafur (p = 0.045) and worse survival | Kim et al. (2016) |
| Efficacy endpoints; response rate, PFS, OS | ||||
| Adverse events | ||||
| CYP2A6*1; CYP2A6*4; CYP2A6*7; CYP2A6*9 | 57 Japanese patients with metastatic or recurrent solid malignancies administered S-1 | Pharmacokinetic parameters; tegafur, 5′-FU, and CDHP AUC0–24hr, Cmax, T1/2, CL | Carriers of two variant alleles had 58% less tegafur CL compared to wild-type patients | Hirose et al. (2010) |
| Adverse events | ||||
| CYP2A6*1; CYP2A6*4; CYP2A6*7; CYP2A6*9; CYP2A6*10 | 49 Korean patients with recurrent/metastatic adenocarcinoma of the biliary tract administered S-1 in combination with oxaliplatin | Pharmacokinetic parameters; tegafur, 5-FU AUC0–24hr Cmax, Tmax Efficacy endpoints; response rate, PFS, OS | 5-FU concentrations tended to be higher and tegafur’s lower in the homozygous for the wild type allele patients compared to those carrying variant alleles | Kim et al. (2011) |
| Adverse events | No association found for toxicity or efficacy endpoints | |||
| CYP2A6*1; CYP2A6*4; CYP2A6*7; CYP2A6*9; CYP2A6*10 | 50 metastatic gastric cancer Asian patients administered S-1 in combination with docetaxel | Response rate, PFS, OS, adverse events | Carriers of at least one wild type allele had higher response rate and PFS than the homozygous for variant alleles both in univariate and multivariate analysis (p = 0.04 and p = 0.001, respectively) | Kong et al. (2009) |
| No association with toxicity endpoints | ||||
| CYP2A6*4; CYP2A6*7; CYP2A6*9; CYP2A6*10 | 106 patients with metastatic gastric cancer administered S-1 in combination with cisplatin | Response rate, PFS, and OS | CYP2A6*4 associated with lower response rate | Park et al. (2011) |
| Adverse events | (OR 0.220, 95% CI 0.067–0.719, p = 0.012), shorter PFS (OR 2.288, 95% CI 1.245–4.207, p = 0.008), and shorter OS (OR 3.118, 95% CI 1.483–6.558, p = 0.003) | |||
| No association with toxicity endpoints | ||||
| CYP2A6*4; CYP2A6*7; CYP2A6*9 | 42 patients with unresectable or metastatic adenocarcinoma of the colon or rectum administered S-1 in combination with irinotecan and oxaliplatin | Response rate, PFS, OS, adverse events | Carriers of the variant alleles were associated with poorer response rate (p = 0.05) | Kim et al. (2013) |
| No association with toxicity endpoints | ||||
| CYP2A6*1; CYP2A6*4; CYP2A6*7; CYP2A6*8; CYP2A6*9; CYP2A6*10 | 200 Asian gastric cancer patients administered S-1 | RFS, OS, adverse events | Carriers of variant alleles had poorer RFS (HR 3.41, 95% CI 1.01–11.52, p = 0.049 for heterozygous, HR = 3.41, 95% CI 1.16–13.93, p = 0.028 for homozygous) | Jeong et al. (2017) |
| No association with toxicity endpoints | ||||
| Whole exome sequencing of CYP2A6, 22 SNPs identified, among them | 60 gastric cancer patients administered SOX (oxaliplatin + S-1) | PFS, OS, adverse events | rs60823196 and rs138978736 associated with grade 3–4 diarrhea (OR 4.905, 95% CI 1.38–17.45, p = 0.02 and OR 15.860, 95% CI 4.05–62.11, p = 0.0002, respectively) | Yang et al. (2017) |
| CYP2A6*5; CYP2A6*7; CYP2A6*8; CYP2A6*10; CYP2A6*11; rs60823196; rs138978736; rs150586234; rs771265125; rs58571639; rs2644907; rs60988093 | rs138978736 associated with shorter OS (p = 0.006) in the subgroup of 30 patients administered S-1 as adjuvant chemotherapy | |||
| CYP2A6*1; CYP2A6*4; CYP2A6*7; CYP2A6*8; CYP2A6*9; CYP2A6*10 | Elderly patients with recurrent or metastatic gastric cancer randomised in CAP-treated (n = 53) and S-1-treated (n = 52) groups | Response rate, disease control rate, PFS, OS, adverse events | In the S-1 subgroup, patients carrying two variant alleles (except for *8) had shorter PFS (HR 2.46, 95% CI 1.20–5.05, p = 0.015) and OS (HR 2.22, 95% CI 1.14–4.31, p = 0.019) | Kim et al. (2018) |
| No association for toxicity endpoints | ||||
| CYP2A6*1A; CYP2A6*1B; CYP2A6*4C | 77 Chinese patients with cancer of the digestive system administered S-1 | Response rate, disease progression, adverse events | No association found | Fang et al. (2012) |
| CYP2A6*4; CYP2A6*7; CYP2A6*9; CYP2A6*10 | 29 patients with metastatic or recurrent colorectal adenocarcinoma administered S-1 in combination | Response rate, survival, adverse events | No association found | Choi et al. (2012) |
| CYP2A6*1D; CYP2A6*4; CYP2A6*7; CYP2A6*9 | 99 stage II-III colorectal cancer patients administered tegafur-uracil in combination with leucovorin | Grade ≥2 toxicity, overall toxicity | Variant alleles associated with hyperbilirubinemia in univariate analysis (p = 0.039) | Tsunoda et al. (2011) |
| CYP2A6*4; CYP2A6*7; CYP2A6*9; CYP2A6*10 | 91 patients with confirmed adenocarcinoma of the rectum orally administered tegafur- uracil | Pathologic complete response, PFS, OS, adverse events | Variant alleles associated with grade 2 or higher leucopenia (p = 0.022) and stomatitis (p = 0.012) | Kim et al. (2017) |
Studies examining the effect of CYP2A6 gene polymorphisms on tegafur pharmacokinetics and response.
CYP2A6, cytochrome P450 2A6; 5-FU, 5-fluoruracil; AUC, area under the curve; C, concentration; PFS, progression free survival; OS, overall survival; CDPH, 5-chloro-2,4-dihydroxypyridine; CL, clearance; RFS, relapse free survival.
3.3.5 Uridine monophosphate synthetase, UMPS
Orotate phosphoribosyltransferase (OPRT), also known as uridine monophosphate synthetase (UMPS), is responsible for 5-FU phosphorylation into its active metabolite FUMP (Wang et al., 2014). Studies assessing the effect of UMPS polymorphisms on FP therapeutic outcomes are presented in Table 7.
TABLE 7
| Examined UMPS variants | Study population | Endpoints | Results | Ref |
|---|---|---|---|---|
| rs1801019 | 69 primary colon or rectum carcinoma patients administered bolus 5-FU in combination with leucovorin | Grade 3–4 neutropenia, and diarrhea, time to onset of toxicity | rs1801019 minor allele | Ichikawa et al. (2006) |
| associated with grade 3–4 neutropenia (p = 0.0393), diarrhea (OR 13.3, 95% CI 1.9–280.9, p = 0.026) and earlier time to toxicity onset (p < 0.0001) | ||||
| rs1801019 | 99 stage II-III colorectal cancer patients administered tegafur-uracil in combination with leucovorin | Grade ≥2 toxicity, overall toxicity | rs1801019 minor allele associated with grade 3 diarrhea (p = 0.031), grade 2–3 anorexia (p = 0.035), hyperbilirubinemia (p = 0.013) time to severe toxicity onset (p < 0.0002) In multivariate analysis: rs1801019 homozygous for the minor allele associated with grade 3 diarrhea (OR 19.84, 95% CI 1.82–215.90, p = 0.014) and overall toxicity (OR 17.60, 95% CI 1.58–195.89, p = 0.020) | Tsunoda et al. (2011) |
| rs1801019 | 91 patients with confirmed adenocarcinoma of the rectum orally administered tegafur- uracil | Toxicity | rs1801019 associated with grade ≥2 diarrhea (p = 0.018) and modesty associated with grade ≥2 abdominal pain (p = 0.067) | Kim et al. (2017) |
| rs2279199; rs4678145; rs1139538; rs9844948; rs3772804 | 301 colorectal cancer patients administered CAP in monotherapy or in combination regimens | Grade 2 adverse events, overall toxicity, treatment administration delay | rs2279199 protective for grade >2 nausea/vomiting (OR 0.210, 95% CI 0.049–0.900, p = 0.036) | Pellicer et al. (2017) |
| rs4678145 associated with grade >2 fatigue (OR 4.542, 95% CI 1.557–13.243, p = 0.006) | ||||
| 34 UMPS SNPs | 940 post-operative stage II/III | Grade 0–2 vs. 3–4 HFS, diarrhea, overall toxicity | No association found at the threshold set in the study | Rosmarin et al. (2015) |
| colorectal cancer | ||||
| patients | ||||
| administered CAP | ||||
| rs1801019, Meta-analysis: rs1801019; rs3772809 | QUASAR2 trial (ISRCTN45133151) 927 post-operative stage II/III colorectal cancer patients administered CAP in monotherapy or in combination regimens |
Grade 0–2 vs. grade 3+ adverse events | No association found at the threshold set in the study | Rosmarin et al. (2014) |
| rs1801019; rs2279199; rs4678145 | 338 Chinese colorectal and gastric cancer administered FPs | Hematological, liver and gastrointestinal toxicity, HFS | No association found | Liu et al. (2021) |
| rs1801019 | 253 colorectal cancer patients administered CAP (n = 159) or 5-FU (n = 94) in monotherapy or in combination regimens | Early dose modifications (delays or reductions), severe toxicity | No association found | Jennings et al. (2013) |
| rs1801019 | 216 Brazilian gastrointestinal (n = 92) or colorectal cancer (n = 124) patients administered 5-FU in monotherapy or in combination regimens | Grade 1–4 toxicity | No association found | Fernandes et al. (2021) |
| rs1801019; rs3772809 | Exploration and validation cohort administered 5-FU in combination regimens (160 and 340 patients, respectively) | Gastrointestinal toxicity (stomatitis/pharyngitis, nausea/vomiting, diarrhea) | No association found | Afzal et al. (2011) |
| rs1801019 | 93 gastric cancer patients treated with FP-based chemotherapy | Adverse events | No association found | Cordova-Delgado et al. (2021) |
Studies examining the effect of UMPS gene polymorphisms on FP-induced adverse events.
UMPS, uridine monophosphate synthetase; OR, odds ratio; CAP, capecitabine; HFS, hand and foot syndrome; FPs, fluoropyrimidines; 5-FU, 5-fluoruracil.
In the seminal study that assessed the potential association of UMPS polymorphisms with FP-toxicity, Ichikawa et al. (2006) reported a significant association between UMPS 638G>A variant (rs1801019) and 5-FU induced grade 3–4 toxicity in a cohort consisting of 69 5-FU treated colon or rectum cancer patients. The missense UMPS 638G>A variant was associated with increased risk for severe neutropenia (p = 0.039), diarrhea (OR 13.3, p < 0.0001) and an earlier onset of toxicity (p < 0.0001) in carriers of the minor allele. These results were replicated by Tsunoda et al. (2011) in a cohort of 99 colorectal cancer patients treated with tegafur-uracil. In this study, UMPS 638G>A variant was associated with grade 3 toxicity (OR 17.60, p = 0.02), grade 3 diarrhea (p = 0.031), grade 2–3 anorexia (p = 0.035), hyperbilirubinemia (p = 0.013) and a shorter time of toxicity onset (p < 0.0002) (Tsunoda et al., 2011). The association of the multiallelic 638G>C allele with diarrhea was also replicated in another study on 91 tegafur-treated rectum cancer patients (p = 0.018) (Kim et al., 2017). Pellicer et al. (2017) reported the association of two additional UMPS variants, rs2279199 and rs4678145, with nausea/vomiting (OR 0.21, p = 0.036) and fatigue (OR 4.54, p = 0.006), respectively.
These results are really promising in identifying an additional genetic marker for FP-induced ADRs that can be included in a polygenic dosing algorithm. However, several studies did not identify an association of UMPS variants with FP-response (Afzal et al., 2011; Jennings et al., 2013; Rosmarin et al., 2014; Cordova-Delgado et al., 2021; Fernandes et al., 2021; Liu et al., 2021). Therefore, UMPS polymorphisms in clinical decisions on FPs is still under investigation.
3.3.6 Other genes in FP metabolic pathway
Several studies examined the potential significance of additional, less investigated genes, encoding enzymes implicated in the metabolic or catabolic pathway of FPs. These enzymes are dihydropyrimidinase (DPYS) and β-ureidopropionase 1 (UPB1) participating in 5-FU excretion pathway (Lam et al., 2016) and the regulatory component of ribonucleotide reductase (RR), encoded by RRM1, that is involved in 5-FU metabolism to active metabolites (Aoki et al., 2013). Results of FP pharmacogenomic studies on these genes are shown in Table 8.
TABLE 8
| Examined variants | Study population | Endpoints | Results | Ref |
|---|---|---|---|---|
| Sequencing of entire coding sequence and flanking intronic regions | 113 cancer patients (67 with grade 3–4 toxicity) treated with FPs and 69 non-cancer individuals as control group | Grade 3–4 hematological toxicity | Homozygous -1CC patients were at increased risk for mucositis (OR 4.13, 95% CI 1.51–11.31, p = 0.006), diarrhea (OR 2.12, 95% CI 0.94–4.76, p = 0.007) and gastrointestinal toxicity (OR 3.54, 95% CI 1.59–7.88, p = 0.002) | Fidlerova et al. (2010) |
| DPYS | DPYS −1C was more frequent in the high-toxicity group (66%) compared to the well-tolerance-group (57%) and healthy individuals (54%), p = 0.06 | |||
| −1 T>C; −58 T>C | −58C allele was rarer in the high-toxicity group (47%) compared to the well-tolerance-group (59%) and the non-cancerous individuals (57%), p = 0.02 | |||
| −58C allele was associated with lower risk for overall gastrointestinal toxicity (OR 0.40, 95% CI 0.17–0.93, p = 0.03) and leukopenia (OR 0.29, 95% CI 0.08–1.01, p = 0.05) | ||||
| DPYS rs61758444; rs36027551; rs34895123; rs2669429 | 430 FP-treated patients with gastrointestinal cancer | Adverse events, overall toxicity | No association found | Loganayagam et al. (2013) |
| DPYS rs2669429 | 338 Chinese colorectal and gastric cancer administered FPs | Hematological, liver and gastrointestinal toxicity, HFS | No association found | Liu et al. (2021) |
| Sequencing of entire coding sequence and flanking intronic regions | 113 different cancer type patients (67 with grade 3–4 toxicity) treated with FPs and 69 individuals as control group | Grade 3–4 hematological toxicity and/or gastrointestinal toxicity | −80 GG patients were at risk for grade 3–4 mucositis (OR 7.5, 95% CI 2.60–21.60, p = 0.0002) and at decreased risk for severe diarrhea (OR 0.379, 95% CI 0.15–0.98, p = 0.044) | Fidlerova et al. (2012) |
| UPB1 | ||||
| −80 C>G; −17 A>T; 105–61 A>G | ||||
| DPYS | 514 cancer patients administered 5-FU or CAP regimens (164 in the discovery cohort, 85 patients with severe toxicity, 79 matched controls) | Overall toxicity | Several associations present for DPYS −1C, −58C and UPB1 −80G alleles and overall toxicity, gastrointestinal toxicity and nausea | Kummer et al. (2015) |
| −1 T>C; −58 T>C | When adjusted for DPYD variants: | |||
| UPB1 −80 C>G | DPYS -1C and UPB1 –80G associated with FP toxicity (OR 1.78, 95% CI 1.03–3.08, p = 0.039 and OR 1.77, 95% CI 1.08–2.92, p = 0.047, respectively) | |||
| For DPYS, results potentially driven by 5-FU group | ||||
| 69 DPYS variants; 30 UPB1 variants; 29 RRM1 variants | 940 post-operative stage II/III | Grade 0–2 vs. 3–4 HFS, diarrhea, and overall toxicity | No association found at the threshold set in the study | Rosmarin et al. (2015) |
| colorectal cancer | ||||
| patients | ||||
| administered CAP | ||||
| RRM1 rs12806698; rs1042927 | 216 Brazilian gastrointestinal (n = 92) or colorectal cancer (n = 124) patients administered 5-FU in monotherapy or in combination regimens | Grade 1–4 adverse events, overall toxicity | No association found | Fernandes et al. (2021) |
Studies examining the effect of DPYS, UPB1 and RRM1 gene variants on FP-induced adverse events.
DPYS, dihydropyrimidinase; UPB1, β-ureidopropionase 1; RRM1, regulatory component of ribonucleotide reductase; FPs, fluoropyrimidines; OR, odds ratio; CAP, capecitabine; 5-FU, 5-fluoruracil; HFS, hand and foot syndrome.
Fidlerova et al. (2010) were the first to analyze genes downstream of DPD. In two sequential studies, hematological and gastrointestinal toxicity rates were analyzed within DPYS and UPB1 genotypes in a cohort of 113 cancer patients (67 of whom experienced severe toxicity, and 46 with good tolerance to FP) and 69 non-cancer individuals. DPYS −1CC genotype was more frequent within the patient group who experienced diarrhea (OR 2.12, p = 0.007), gastrointestinal toxicity (OR 3.54, p = 0.002), and severe mucositis (OR 4.13, p = 0.006), whereas −58C allele was associated with a lower risk for overall gastrointestinal toxicity (OR 0.4, p = 0.03) and leucopenia (OR 0.29, p = 0.05) (Fidlerova et al., 2010). Similar results for DPYS −1C were reported in a study including 514 patients; DPYS -1C allele was associated with overall toxicity (OR 1.78, p = 0.039), gastrointestinal toxicity (OR 3.06, p = 0.006) and nausea (OR 4.46, p = 0.016), while −58C had a protective effect against gastrointestinal toxicity (OR 0.55, p = 0.024) and nausea (OR 0.45, p = 0.014) (Kummer et al., 2015). Interestingly, those associations were stronger in patients receiving 5-FU based regimens.
For UPB1, Fidlerova et al. (2012) have found that −80C>G variant minor allele was an independent predictor of severe mucositis (OR 7.5, p = 0.0002). Additionally, a borderline association with leukopenia (p = 0.076) and hematologic toxicities (p = 0.061) has been reported for UPB1 −80C>G allele (Kummer et al., 2015). For RRM1 variants, no association with FP response or toxicity has been found (Fernandes et al., 2021).
In conclusion, DPYS and UPB1 appear as attractive candidates of FP variable response. The possibility of their inclusion in a polygenic dosing algorithm is worthy of further consideration.
3.3.7 Genes affecting response to FP-based chemotherapeutic schemes
FPs can be administered as monotherapy or in combination with other chemotherapeutics such as oxaliplatin (FOLFOX, XELOX), irinotecan (FOLFILIRI, XELIRI), taxanes (paclitaxel, docetaxel), and monoclonal antibodies. In chemotherapeutic combination schemes, incidence of ADRs or treatment efficiency can be induced by other drugs beyond FPs. Therefore, in patients treated with FP-including chemotherapeutic schemes, additionally to polymorphisms of genes encoding enzymes involved in FP pathway, variations in genes encoding membrane transporters (De Mattia et al., 2015), nuclear receptors (De Mattia et al., 2013; Cecchin et al., 2016; De Mattia et al., 2018; De Mattia et al., 2019a), transcription factors and molecules mediating downstream therapeutic pathways (De Mattia et al., 2021) may be associated with therapy resistance or ADR incidence.
Currently, established pharmacogenomic markers for chemotherapeutic regimens including irinotecan exist; both FDA and EMA suggest dose reductions for the homozygous carriers of UGT1A1*28 allele (FDA, 2015b; EMA, 2015). Other gene associations include variations in the genes coding for rate-limiting enzymes of the nucleotide excision DNA repair system ERCC1 and ERCC2 in association with response and survival to oxaliplatin (Di Francia et al., 2013; De Mattia et al., 2015). CYP3A4, CYP3A5, and CYP1B1 genetic variants as well as variations in ABCB1 transporter have been associated with response to taxanes (De Iuliis et al., 2015).
The specific review concentrates on the idea of a potential polygenic dosing algorithm for FPs, however, precision medicine in oncology relies on onco-omics (Ragia and Manolopoulos, 2022), thus, a broad spectrum of genes should be implemented to predict response to combination chemotherapeutic schemes.
4 A polygenic algorithm for FP dosing: New challenges in oncology
FP pharmacogenomics has entered clinical practice via the well-documented association of DPYD with FP-induced severe toxicity, there is still, however, an unmet medical need to predict and reduce severe FP-induced toxicity in cancer patients receiving chemotherapy. We anticipate that eventually there will be advances in FP genotype-based clinical decisions in oncology. Firstly, we anticipate that DPYD will be globally accepted as for its impact on FP dose requirements. Though in Europe EMA has endorsed DPYD genotyping prior to FP administration, FDA is still rather modest on the modifications of drug label. Most likely, DPYD will still be at the center of attention regarding FP pharmacogenomics since there is still much to understand on the clinical significance of DPYD rare variants, on the impact of the identified variants on FP dosing and toxicity in different populations, and on the optimal ethnicity-based variant combination for preemptive genotyping. DPYD is on the prime time due to its dominant role in 5-FU metabolism, irrespectively of the administered FP. As important as this step for FP pharmacogenomics may be, we suggest that this may not be the whole story but just the beginning of it. We believe, therefore, that DPYD will not remain for much longer as the sole genetic factor affecting FP dosing decisions.
Polygenic scores are primarily been applied for disease risk prediction, but they have yet to be broadly adopted in the field of pharmacogenomics. Drug dosing requirements based on a multigenic model are applied for vitamin K antagonists and this consists the most well characterized example of the application of a pharmacogenomic dosing algorithm in clinical practice. Hitherto, in oncology, such an approach has not been developed, however, the majority of pharmacogenomic drug safety studies have been conducted in antineoplastics (Siemens et al., 2022) suggesting that we are approaching to the era of a FP polygenic dosing algorithm.
Towards this direction, accumulated evidence shows that TYMS and ENOSF1 low expression variants are strongly associated with FP toxicity and can improve the DPYD-pharmacogenomic guided dosing. Therefore, DPYD, TYMS and ENOSF1 can form the core of a polygenic algorithm. In patients with normal DPD activity, TYMS and ENOSF1 polymorphisms may guide the appropriate dose reductions. For ENOSF1 rs2612091 it has been recently shown that when it is integrated to the DPYD-based prediction model for FP-induced toxicity, it significantly improves prediction of global toxicity, hematological toxicity, HFS, and diarrhea (Palles et al., 2021). These results, therefore, support our idea and we anticipate that more studies will move from single DPYD based FP-dosing to a multigenic dosing approach.
Depending on the FP used, additional gene variations can improve the outcome and safety of therapy. For CAP, it appears that multiple genes associated with reduced CAP catabolism can help in predicting HFS. Following liver metabolism, CAP is further catabolized to 5-FU by the enzymes encoded by CES2/1, CDA and TYMP. Polymorphisms in these genes have been associated with CAP-induced HFS. For CES1 and CDA a more prominent association appears, suggesting that they merit further investigation for their contribution to reducing CAP-induced toxicity and overall incidence of ADRs. For the second 5-FU prodrug, tegafur, CYP2A6 is crucial for its bioactivation. CYP2A6 defective alleles seriously impact tegafur pharmacokinetics and 5-FU generation. CYP2A6 poor metabolizers can potentially benefit from alternative treatment.
Downstream enzymes that may affect FP toxicity include UMPS, DPYS, UPB1, and RR participating either in 5-FU phosphorylation into its active metabolites or in 5-FU excretion. These genes are relatively less studied, however, several promising associations with increased risk for FP toxicity have been reported. Currently, there is no clear role for these genes in a FP polygenic algorithm, however, in the case of FP-induced toxicity in patients who do not carry any other variation, UMPS, DPYS, UPB1, and RR genotyping may help in elucidating rare cases of toxicity.
Therapeutic response to FP involves several pharmacodynamic aspects. Though pharmacodynamic genes downstream to FP active nucleotides are not expected to strongly affect FP dose requirements, it should be acknowledged that enzymes involved in DNA repair, cell cycle and apoptosis may drive tumor resistance to chemotherapy and in synergy with pharmacokinetic genes may increase risk for ADRs (Tecza et al., 2018; Boige et al., 2019; Varma et al., 2020).
Beyond the effect of single genetic variations to FP-induced toxicity and dose requirements, several other hurdles exist until a polygenic dosing algorithm could be applied for FPs. Firstly, for most genes, an increased number of variations has been identified; the role of these variations in enzyme activity has yet to be determined. In addition, the existence of additional deleterious variants, albeit rare in frequency, cannot be excluded. Consequently, the current panel of variations is rather broad, potentially affecting the sensitivity of such an approach. To overcome this limitation, advances in genotyping technologies can be helpful. The process of building a FP polygenic dosing algorithm will benefit from (very) high throughput genotyping platforms, in multiple ways. Next-generation sequencing (NGS) technology can be used both to screen the genetic variations in all pharmacogenes of an individual, as well as to identify rare variants in pharmacogenes, thus adding important information to predict drug response (Ingelman-Sundberg et al., 2018; Tafazoli et al., 2021). It should be acknowledged that rare variants in pharmacogenes can shed light on toxicity mechanisms and have emerged as clinically useful markers in predicting ADRs; not only they contribute significantly to pharmacogenomic variability, but also their frequency varies within populations of different ethnicity (Ingelman-Sundberg et al., 2018; Runcharoen et al., 2021). Recently, it was shown that polygenic risk scores adjusted for common variants can efficiently improve power in rare variant association discovery (Jurgens et al., 2023). Therefore, a polygenic dosing algorithm might help in the identification of additional pathways associated with FP response.
Ethnicity could be a major factor that should be taken into account for the selection of genetic variations. It has been shown for different pharmacogenomic applications that even for common variations within established pharmacogenes, current dosing algorithms should be adjusted per ethnicity. Such an example is the CYP2C9/VKORC1 pharmacogenomic dosing algorithm for vitamin K antagonists (Ragia et al., 2017b). For FPs, it appears that the frequency of pharmacogene polymorphisms varies within populations of different ancestry; in Asians, DPYD variations have only a minor role in FP-related toxicity (Kanai et al., 2022), whereas a higher prevalence of CYP2A6 defective alleles has been reported (Tanner and Tyndale, 2017). Therefore, we expect that different variants will be incorporated in a FP polygenic algorithm and that, in each case, the predictive value of the algorithm should be tested and validated within populations. Moreover, even though a 100% overall concordance has been shown for DPYD genotyping in blood and tissue samples (Morelli et al., 2022), it should be considered that in tumor tissue genetic alterations are possible in other FP-associated genes that may affect tissue-specific FP catabolism and action. Additionally, the expression of genes regulating FP bioavailability can be influenced by transcriptional factors activated by the tumor-related characteristics (Wu et al., 2016; Suenaga et al., 2021; Delhorme et al., 2022). Therefore, genetic variation of these factors might also play a role in FP dose requirements.
In pharmacogenomics, multiple interactions exist within genes and it cannot be excluded that several genetic associations are masked (Ragia et al., 2014) raising thus the need for analyses in sub-phenotypes (Ragia et al., 2017a). In cancer, gene interactions are expected in different layers, not only within pharmacogenes, but additionally within cancer predisposing genes (Capellini et al., 2021). Additionally, gene*environment interactions exist both in cancer development (Mbemi et al., 2020) and in response to chemotherapy (Cui et al., 2017); once such interactions are identified, they can be implemented in therapeutic strategies. More importantly, in cancer, gender interactions are well established; for 5-FU, it has been reported that women are a priori at increased ADR risk due to reduced drug elimination (Mueller et al., 2013). Additionally, gender-related biological factors can influence the (pharmaco)genetic associations (Mezzalira and Toffoli, 2021). The pharmacogenomic impact of gene*gender interactions is currently less studied, however, data previously published indicate that in FP response pharmacogenomic associations in genes beyond DPYD are gender dependent (Ioannou et al., 2021, 2022). In a recent research article published by our team, a gender dependent association of low TYMS expression alleles has been found: TYMS-TSER 2R/2R genotype was associated with FP dose reduction due to ADRs in female patients, a finding potentially attributed to estrogen receptor regulation of TS expression (Ioannou et al., 2021). Similarly, we have found a gender*MTHFR interaction possibly interfering with FP response: in gender stratification analysis MTHFR -677C>T polymorphism increased both need for FP dose reduction (OR 5.05) and percentage of dose reduction (β = 3.318) in female patients, whereas no differences were present in pooled sample analyses (Ioannou et al., 2022). This gender dependent association is potentially driven by variable homocysteine levels between two genders and merits further investigation. Thereby, when considering a polygenic algorithm for FP-dosing, gene*gender interactions should be taken into account.
Apart from pharmacogenomics, the underlying factors predisposing patients to FP-induced toxicity are largely unknown. However, several patient-related factors exist that increase risk for FP toxicity. Advanced age is an independent predictor of severe toxicity and elderly patients should be closely monitored for FP-induced ADRs (Stein et al., 1995). Pre-existing diseases, such as cardiovascular diseases (CVD), hepatic impairment, and renal insufficiency, as well as CVD risk factors (hypertension, hyperlipidemia and smoking), have been studied as for their association with increased risk for 5-FU-induced cardiotoxicity (Brutcher et al., 2018; Sara et al., 2018). The perception of incorporating non-genetic factors into polygenic risk score models is currently being evaluated for disease risk assessment (van Dam et al., 2023). This approach holds promise to increase the discriminative power of the often low variance explained solely by the genetic factors for the predictive outcome(s). We propose, therefore, that non-genetic factors, once identified, could also be added in a FP-polygenic dosing algorithm to improve pharmacogenomic predictions.
Heterogeneity exists not only in cancer tissue genetics and in cancer stratification but also in ADR symptoms. Pharmacogenomics of FPs currently involves cohorts of patients with different solid tumors, treated often with a combination of FP-based scheme, and ADRs on different systems are grouped together to increase statistical power. Pooled analyses can indeed highlight universal associations, however, other pharmacogenomic associations, cancer- or system-specific, may be underestimated. On the other hand, in schemes that include chemotherapeutic combinations, incidence of ADRs can be induced by other drugs beyond or additionally to FPs. Therefore, future research should focus on distinct phenotypes to reduce heterogeneity, as well as on integration of pharmacogenomics of every single administered drug.
We have herein discussed extensively the genetic architecture of FP-induced toxicity and the potential role of gene*gene, gene*gender, and gene*environment interactions in FP dosing decisions. In therapeutics, however, drug-drug interactions consist a major driver of drug-induced ADRs. In FP therapy, several clinically relevant drug-drug interactions, pharmaceutical, pharmacodynamic, and pharmacokinetic, exist, either among chemotherapeutics or with supportive care drugs that increase risk for FP-induced toxicity (van Leeuwen et al., 2011; Ramsdale et al., 2022; van Leeuwen et al., 2022). Precision medicine in oncology, thus, can also have a role in polypharmacy guidelines.
While we propose herein the concept of a polygenic FP dosing algorithm, the actual form and mathematical equation of such an algorithm is beyond the scope of the present work. Several tools exist to guide this procedure. Machine learning could be of help in prioritizing the genetic variants included in the algorithm aiding to select the optimal regimen and doses, and, if employed, signatures with clinical utility can be constructed (Panagopoulou et al., 2021; Pandi et al., 2021). Artificial intelligence models have rapidly gained attention in pharmacological field. In pharmacogenomics, indicatively, studies published so far highlight the use of artificial intelligence models for warfarin dose prediction in Asian populations (Jahmunah et al., 2023) and antidepressant response (Bobo et al., 2022). Initial data for the use of machine learning and artificial intelligence in FP treatment are also available, focusing on DPYD variant classification (Shrestha et al., 2018) and on identification of genomic and transcriptomic biomarkers for 5-FU response (Kong et al., 2020). Therefore, a multi-omics integrative approach for FP-induced toxicity assisted by computational sciences can shed additional light on this complexity (Ragia and Manolopoulos, 2022).
In the present review we extensively discuss current pharmacogenomic evidence that can guide FP dosing. It should be acknowledged, however, that several hurdles exist at multiple levels until a polygenic dosing algorithm could conceivably be implemented in clinical practice. These include, the identification of the associations, the validation of the polygenic algorithm, the exact dosing recommendations, and their incorporation in routine clinical practice. Once a polygenic dosing algorithm is formed, prospective pharmacogenomic clinical trials will be required to assess whether this approach is indeed superior compared to standard dosing. Nevertheless, several limitations also exist in standard dosing procedures that currently take into account body surface area (BSA)-adjusted dosing. A large proportion of FP-treated patients (more than 50%) are in the under-exposure range while more than 10% fall in the toxicity ranges (Gamelin et al., 2008; Saam et al., 2011; Li et al., 2023). To avoid further underdosing when adding multicomponent risk variants for toxicity risk prediction, current dosing strategies should be clinically improved. Once standard dosing is improved, the predictive power of pharmacogenomic markers becomes even stronger. Implementation of preemptive FP pharmacogenomics needs the acceptance of such an approach by oncologists. Therefore, we believe that clinical implementation relies on consensus of experts from oncological societies and chemotherapy boards worldwide.
5 Conclusion
Precision medicine in oncology has rapidly expanded throughout the last 20 years and significant milestones have been reached towards translation in clinical practice of gene based therapeutic options. DPYD-based dosing approach is one of them, however, we suggest that it may be only the first step towards FP therapy personalization. The prognostic value of DPYD genotyping can stand for the opening of a new era in FP pharmacogenomics. It is time to accept the critical challenges and move from single gene approach to a polygenic dosing algorithm approach to succeed in the ultimate goal of optimal and safer chemotherapy in cancer patients.
Statements
Author contributions
AM: data acquisition, writing of the original draft. VGM: conceptualization, data evaluation, writing, review and editing, funding acquisition, final approval. GR: conceptualization, data evaluation, writing, review and editing. All authors made a significant intellectual contribution and read and approved the manuscript.
Funding
Financial support for project IMPReS (MIS 5047189) was provided to VGM by the Program “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014–2020) co-financed by Greece and the European Union (European Regional Development Fund).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Afzal S. Gusella M. Vainer B. Vogel U. B. Andersen J. T. Broedbaek K. et al (2011). Combinations of polymorphisms in genes involved in the 5-Fluorouracil metabolism pathway are associated with gastrointestinal toxicity in chemotherapy-treated colorectal cancer patients. Clin. Cancer Res.17 (11), 3822–3829. 10.1158/1078-0432.CCR-11-0304
2
Amstutz U. Henricks L. M. Offer S. M. Barbarino J. Schellens J. H. M. Swen J. J. et al (2018). Clinical pharmacogenetics implementation Consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin. Pharmacol. Ther.103 (2), 210–216. 10.1002/cpt.911
3
Aoki Y. Sakogawa K. Hihara J. Emi M. Hamai Y. Kono K. et al (2013). Involvement of ribonucleotide reductase-M1 in 5-fluorouracil-induced DNA damage in esophageal cancer cell lines. Int. J. Oncol.42 (6), 1951–1960. 10.3892/ijo.2013.1899
4
Arjmandi K. Ameli F. Salahshourifar I. Esfandbod M. Irani S. (2022). Evaluation of TS and ENOSF1 variants as a biomarker in response to neoadjuvant chemotherapy based on 5FU in gastric cancer patients. Asian Pac J. Cancer Prev.23 (9), 2983–2989. 10.31557/apjcp.2022.23.9.2983
5
Barin-Le Guellec C. Lafay-Chebassier C. Ingrand I. Tournamille J. F. Boudet A. Lanoue M. C. et al (2020). Toxicities associated with chemotherapy regimens containing a fluoropyrimidine: A real-life evaluation in France. Eur. J. Cancer124, 37–46. 10.1016/j.ejca.2019.09.028
6
Begré U. B. M. Jörger M. Aebi S. Amstutz U. Largiadèr C. R. (2022). Clinical implementation of DPYD pharmacogenetic testing to prevent early-onset fluoropyrimidine-related toxicity in cancer patients in Switzerland. Front. Pharmacol.13, 885259. 10.3389/fphar.2022.885259
7
Bobo W. V. Van Ommeren B. Athreya A. P. (2022). Machine learning, pharmacogenomics, and clinical psychiatry: Predicting antidepressant response in patients with major depressive disorder. Expert Rev. Clin. Pharmacol.15 (8), 927–944. 10.1080/17512433.2022.2112949
8
Boige V. Mollevi C. Gourgou S. Azria D. Seitz J. F. Vincent M. et al (2019). Impact of single-nucleotide polymorphisms in DNA repair pathway genes on response to chemoradiotherapy in rectal cancer patients: Results from ACCORD-12/PRODIGE-2 phase III trial. Int. J. Cancer145 (11), 3163–3172. 10.1002/ijc.32417
9
Bonotto M. Bozza C. Di Loreto C. Osa E. O. Poletto E. Puglisi F. (2013). Making capecitabine targeted therapy for breast cancer: Which is the role of thymidine phosphorylase?Clin. Breast Cancer13 (3), 167–172. 10.1016/j.clbc.2012.10.002
10
Brooks G. A. Tapp S. Daly A. T. Busam J. A. Tosteson A. N. A. (2022). Cost-effectiveness of DPYD genotyping prior to fluoropyrimidine-based adjuvant chemotherapy for colon cancer. Clin. Colorectal Cancer21 (3), e189–e195. 10.1016/j.clcc.2022.05.001
11
Brutcher E. Christensen D. Hennessey Smith M. Koutlas J. B. Sellers J. B. Timmons T. et al (2018). 5-Fluorouracil and capecitabine: Assessment and treatment of uncommon early-onset severe toxicities associated with administration. Clin. J. Oncol. Nurs.22 (6), 627–634. 10.1188/18.Cjon.627-634
12
Campbell J. M. Bateman E. Peters M. Bowen J. M. Keefe D. M. Stephenson M. D. (2016). Fluoropyrimidine and platinum toxicity pharmacogenetics: An umbrella review of systematic reviews and meta-analyses. Pharmacogenomics17 (4), 435–451. 10.2217/pgs.15.180
13
Capellini A. Williams M. Onel K. Huang K. L. (2021). The functional hallmarks of cancer predisposition genes. Cancer Manag. Res.13, 4351–4357. 10.2147/cmar.S311548
14
Caronia D. Martin M. Sastre J. de la Torre J. Garcia-Saenz J. A. Alonso M. R. et al (2011). A polymorphism in the cytidine deaminase promoter predicts severe capecitabine-induced hand-foot syndrome. Clin. Cancer Res.17 (7), 2006–2013. 10.1158/1078-0432.CCR-10-1741
15
Cecchin E. De Mattia E. Toffoli G. (2016). Nuclear receptors and drug metabolism for the personalization of cancer therapy. Expert Opin. Drug Metab. Toxicol.12 (3), 291–306. 10.1517/17425255.2016.1141196
16
Choi Y. H. Kim T. W. Kim K. P. Lee S. S. Hong Y. S. Ryu M. H. et al (2012). A phase II study of clinical outcomes of 3-week cycles of irinotecan and S-1 in patients with previously untreated metastatic colorectal cancer: Influence of the UGT1A1 and CYP2A6 polymorphisms on clinical activity. Oncology82 (5), 290–297. 10.1159/000337989
17
Chu J. Dolnick B. J. (2002). Natural antisense (rTSalpha) RNA induces site-specific cleavage of thymidylate synthase mRNA. Biochim. Biophys. Acta1587 (2-3), 183–193. 10.1016/s0925-4439(02)00081-9
18
Cordova-Delgado M. Bravo M. L. Cumsille E. Hill C. N. Munoz-Medel M. Pinto M. P. et al (2021). A case-control study of a combination of single nucleotide polymorphisms and clinical parameters to predict clinically relevant toxicity associated with fluoropyrimidine and platinum-based chemotherapy in gastric cancer. BMC Cancer21 (1), 1030. 10.1186/s12885-021-08745-0
19
Cui J. J. Wang L. Y. Zhu T. Gong W. J. Zhou H. H. Liu Z. Q. et al (2017). Gene-gene and gene-environment interactions influence platinum-based chemotherapy response and toxicity in non-small cell lung cancer patients. Sci. Rep.7 (1), 5082. 10.1038/s41598-017-05246-8
20
De Angelis P. M. Svendsrud D. H. Kravik K. L. Stokke T. (2006). Cellular response to 5-fluorouracil (5-FU) in 5-FU-resistant colon cancer cell lines during treatment and recovery. Mol. Cancer5, 20. 10.1186/1476-4598-5-20
21
De Iuliis F. Salerno G. Taglieri L. Scarpa S. (2015). Are pharmacogenomic biomarkers an effective tool to predict taxane toxicity and outcome in breast cancer patients? Literature review. Cancer Chemother. Pharmacol.76 (4), 679–690. 10.1007/s00280-015-2818-4
22
De Luca O. Salerno G. De Bernardini D. Torre M. S. Simmaco M. Lionetto L. et al (2022). Predicting dihydropyrimidine dehydrogenase deficiency and related 5-fluorouracil toxicity: Opportunities and challenges of DPYD exon sequencing and the role of phenotyping assays. Int. J. Mol. Sci.23 (22), 13923. 10.3390/ijms232213923
23
De Mattia E. Cecchin E. Montico M. Labriet A. Guillemette C. Dreussi E. et al (2018). Association of STAT-3 rs1053004 and VDR rs11574077 with FOLFIRI-related gastrointestinal toxicity in metastatic colorectal cancer patients. Front. Pharmacol.9, 367. 10.3389/fphar.2018.00367
24
De Mattia E. Cecchin E. Toffoli G. (2015). Pharmacogenomics of intrinsic and acquired pharmacoresistance in colorectal cancer: Toward targeted personalized therapy. Drug Resist Updat20, 39–70. 10.1016/j.drup.2015.05.003
25
De Mattia E. Dreussi E. Cecchin E. Toffoli G. (2013). Pharmacogenetics of the nuclear hormone receptors: The missing link between environment and drug effects?Pharmacogenomics14 (16), 2035–2054. 10.2217/pgs.13.214
26
De Mattia E. Polesel J. Roncato R. Labriet A. Bignucolo A. Dreussi E. et al (2019a). Germline polymorphisms in the nuclear receptors PXR and VDR as novel prognostic markers in metastatic colorectal cancer patients treated with FOLFIRI. Front. Oncol.9, 1312. 10.3389/fonc.2019.01312
27
De Mattia E. Polesel J. Roncato R. Labriet A. Bignucolo A. Gagno S. et al (2021). IL15RA and SMAD3 genetic variants predict overall survival in metastatic colorectal cancer patients treated with folfiri therapy: A new paradigm. Cancers (Basel)13 (7), 1705. 10.3390/cancers13071705
28
De Mattia E. Roncato R. Dalle Fratte C. Ecca F. Toffoli G. Cecchin E. (2019b). The use of pharmacogenetics to increase the safety of colorectal cancer patients treated with fluoropyrimidines. Cancer Drug Resist2 (1), 116–130. 10.20517/cdr.2019.04
29
De Mattia E. Silvestri M. Polesel J. Ecca F. Mezzalira S. Scarabel L. et al (2022). Rare genetic variant burden in DPYD predicts severe fluoropyrimidine-related toxicity risk. Biomed. Pharmacother.154, 113644. 10.1016/j.biopha.2022.113644
30
De Mattia E. Toffoli G. (2009). C677T and A1298C MTHFR polymorphisms, a challenge for antifolate and fluoropyrimidine-based therapy personalisation. Eur. J. Cancer45 (8), 1333–1351. 10.1016/j.ejca.2008.12.004
31
de With M. van Doorn L. Maasland D. C. Mulder T. A. M. Oomen-de Hoop E. Mostert B. et al (2023). Capecitabine-induced hand-foot syndrome: A pharmacogenetic study beyond DPYD. Biomed. Pharmacother.159, 114232. 10.1016/j.biopha.2023.114232
32
Deac A. L. Burz C. C. Bocse H. F. Bocsan I. C. Buzoianu A. D. (2020). A review on the importance of genotyping and phenotyping in fluoropyrimidine treatment. Med. Pharm. Rep.93 (3), 223–230. 10.15386/mpr-1564
33
Delhorme J. B. Bersuder E. Terciolo C. Vlami O. Chenard M. P. Martin E. et al (2022). CDX2 controls genes involved in the metabolism of 5-fluorouracil and is associated with reduced efficacy of chemotherapy in colorectal cancer. Biomed. Pharmacother.147, 112630. 10.1016/j.biopha.2022.112630
34
Di Francia R. Siesto R. S. Valente D. Del Buono A. Pugliese S. Cecere S. et al (2013). Current strategies to minimize toxicity of oxaliplatin: Selection of pharmacogenomic panel tests. Anticancer Drugs24 (10), 1069–1078. 10.1097/cad.0000000000000002
35
Dolnick B. J. Black A. R. Winkler P. M. Schindler K. Hsueh C. T. (1996). rTS gene expression is associated with altered cell sensitivity to thymidylate synthase inhibitors. Adv. Enzyme Regul.36, 165–180. 10.1016/0065-2571(95)00009-7
36
Dolnick B. J. (1993). Cloning and characterization of a naturally occurring antisense RNA to human thymidylate synthase mRNA. Nucleic Acids Res.21 (8), 1747–1752. 10.1093/nar/21.8.1747
37
Dosing Warfarin (2023). warfarindosing. Available at: http://www.warfarindosing.org/(Accessed February 20, 2023).
38
EMA (2020). EMA recommendations on DPD testing prior to treatment with fluorouracil, capecitabine, tegafur and flucytosine.
39
EMA (2015), onivyde-pegylated-liposomal-epar-product-information. Available at: https://www.ema.europa.eu/en/documents/product-information/onivyde-pegylated-liposomal-epar-product-information_en.pdf (Accessed February 20, 2023).
40
Etienne-Grimaldi M. C. Boyer J. C. Beroud C. Mbatchi L. van Kuilenburg A. Bobin-Dubigeon C. et al (2017). New advances in DPYD genotype and risk of severe toxicity under capecitabine. PLoS One12 (5), e0175998. 10.1371/journal.pone.0175998
41
Etienne-Grimaldi M. C. Pallet N. Boige V. Ciccolini J. Chouchana L. Barin-Le Guellec C. et al (2022). Current diagnostic and clinical issues of screening for dihydropyrimidine dehydrogenase deficiency. Eur. J. Cancer181, 3–17. 10.1016/j.ejca.2022.11.028
42
Fang W. J. Mou H. B. Jin D. Z. Zheng Y. L. Zhao P. Mao C. Y. et al (2012). Characteristic CYP2A6 genetic polymorphisms detected by TA cloning-based sequencing in Chinese digestive system cancer patients with S-1 based chemotherapy. Oncol. Rep.27 (5), 1606–1610. 10.3892/or.2012.1678
43
FDA (2015b), drugsatfda_docs/label/2015/207793lbl. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/207793lbl.pdf (Accessed March 27, 2023).
44
FDA (2016), drugsatfda_docs/label/2016/012209s040lbl. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/012209s040lbl.pdf (Accessed February 20, 2023).
45
FDA (2015a). drugsatfda_docs/label/2015/020896s037lbl.pdf. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/020896s037lbl.pdf (Accessed February 20, 2023).
46
Fernandes M. R. Rodrigues J. C. G. Dobbin E. A. F. Pastana L. F. da Costa D. F. Barra W. F. et al (2021). Influence of FPGS, ABCC4, SLC29A1, and MTHFR genes on the pharmacogenomics of fluoropyrimidines in patients with gastrointestinal cancer from the Brazilian Amazon. Cancer Chemother. Pharmacol.88 (5), 837–844. 10.1007/s00280-021-04327-w
47
Fidlerova J. Kleiblova P. Bilek M. Kormunda S. Formankova Z. Novotny J. et al (2010). Contribution of dihydropyrimidinase gene alterations to the development of serious toxicity in fluoropyrimidine-treated cancer patients. Cancer Chemother. Pharmacol.65 (4), 661–669. 10.1007/s00280-009-1071-0
48
Fidlerova J. Kleiblova P. Kormunda S. Novotny J. Kleibl Z. (2012). Contribution of the beta-ureidopropionase (UPB1) gene alterations to the development of fluoropyrimidine-related toxicity. Pharmacol. Rep.64 (5), 1234–1242. 10.1016/s1734-1140(12)70919-2
49
Gamelin E. Delva R. Jacob J. Merrouche Y. Raoul J. L. Pezet D. et al (2008). Individual fluorouracil dose adjustment based on pharmacokinetic follow-up compared with conventional dosage: Results of a multicenter randomized trial of patients with metastatic colorectal cancer. J. Clin. Oncol.26 (13), 2099–2105. 10.1200/jco.2007.13.3934
50
García-Alfonso P. Muñoz Martín A. J. Ortega Morán L. Soto Alsar J. Torres Pérez-Solero G. Blanco Codesido M. et al (2021). Oral drugs in the treatment of metastatic colorectal cancer. Ther. Adv. Med. Oncol.13, 175883592110090. 10.1177/17588359211009001
51
García-Alfonso P. Saiz-Rodríguez M. Mondéjar R. Salazar J. Páez D. Borobia A. M. et al (2022). Consensus of experts from the Spanish Pharmacogenetics and Pharmacogenomics Society and the Spanish Society of Medical Oncology for the genotyping of DPYD in cancer patients who are candidates for treatment with fluoropyrimidines. Clin. Transl. Oncol.24 (3), 483–494. 10.1007/s12094-021-02708-4
52
García-González X. Cortejoso L. García M. I. García-Alfonso P. Robles L. Grávalos C. et al (2015). Variants in CDA and ABCB1 are predictors of capecitabine-related adverse reactions in colorectal cancer. Oncotarget6 (8), 6422–6430. 10.18632/oncotarget.3289
53
Glewis S. Alexander M. Khabib M. N. H. Brennan A. Lazarakis S. Martin J. et al (2022). A systematic review and meta-analysis of toxicity and treatment outcomes with pharmacogenetic-guided dosing compared to standard of care BSA-based fluoropyrimidine dosing. Br. J. Cancer127 (1), 126–136. 10.1038/s41416-022-01779-6
54
Hamzic S. Kummer D. Froehlich T. K. Joerger M. Aebi S. Palles C. et al (2020). Evaluating the role of ENOSF1 and TYMS variants as predictors in fluoropyrimidine-related toxicities: An IPD meta-analysis. Pharmacol. Res.152, 104594. 10.1016/j.phrs.2019.104594
55
Hamzic S. Kummer D. Milesi S. Mueller D. Joerger M. Aebi S. et al (2017). Novel genetic variants in carboxylesterase 1 predict severe early-onset capecitabine-related toxicity. Clin. Pharmacol. Ther.102 (5), 796–804. 10.1002/cpt.641
56
He M. M. Zhang D. S. Wang F. Wang Z. X. Yuan S. Q. Wang Z. Q. et al (2017). Phase II trial of S-1 plus leucovorin in patients with advanced gastric cancer and clinical prediction by S-1 pharmacogenetic pathway. Cancer Chemother. Pharmacol.79 (1), 69–79. 10.1007/s00280-016-3209-1
57
Henricks L. M. Lunenburg C. de Man F. M. Meulendijks D. Frederix G. W. J. Kienhuis E. et al (2018). DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: A prospective safety analysis. Lancet Oncol.19 (11), 1459–1467. 10.1016/s1470-2045(18)30686-7
58
Hernando-Cubero J. Matos-García I. Alonso-Orduña V. Capdevila J. (2017). The role of fluoropirimidines in gastrointestinal tumours: From the bench to the bed. J. Gastrointest. Cancer48 (2), 135–147. 10.1007/s12029-017-9946-5
59
Hirose T. Fujita K. Nishimura K. Ishida H. Yamashita K. Sunakawa Y. et al (2010). Pharmacokinetics of S-1 and CYP2A6 genotype in Japanese patients with advanced cancer. Oncol. Rep.24 (2), 529–536. 10.3892/or_00000889
60
Ichikawa W. Takahashi T. Suto K. Sasaki Y. Hirayama R. (2006). Orotate phosphoribosyltransferase gene polymorphism predicts toxicity in patients treated with bolus 5-fluorouracil regimen. Clin. Cancer Res.12 (13), 3928–3934. 10.1158/1078-0432.CCR-05-2665
61
Ingelman-Sundberg M. Mkrtchian S. Zhou Y. Lauschke V. M. (2018). Integrating rare genetic variants into pharmacogenetic drug response predictions. Hum. Genomics12 (1), 26. 10.1186/s40246-018-0157-3
62
Innocenti F. Mills S. C. Sanoff H. Ciccolini J. Lenz H. J. Milano G. (2020). All you need to know about DPYD genetic testing for patients treated with fluorouracil and capecitabine: A practitioner-friendly guide. JCO Oncol. Pract.16 (12), 793–798. 10.1200/op.20.00553
63
Ioannou C. Ragia G. Balgkouranidou I. Xenidis N. Amarantidis K. Koukaki T. et al (2021). Gender-dependent association of TYMS-TSER polymorphism with 5-fluorouracil or capecitabine-based chemotherapy toxicity. Pharmacogenomics22 (11), 669–680. 10.2217/pgs-2021-0031
64
Ioannou C. Ragia G. Balgkouranidou I. Xenidis N. Amarantidis K. Koukaki T. et al (2022). MTHFR c.665C>T guided fluoropyrimidine therapy in cancer: Gender-dependent effect on dose requirements. Drug Metab. Pers. Ther.37 (3), 323–327. 10.1515/dmpt-2021-0219
65
Ishii Y. Suzuki S. Takahashi Y. Takayama T. Asai S. (2010). Can the 2-(13)C-uracil breath test be used to predict the effect of the antitumor drug S-1?Cancer Chemother. Pharmacol.66 (2), 333–343. 10.1007/s00280-009-1168-5
66
Jahmunah V. Chen S. Oh S. L. Acharya U. R. Chowbay B. (2023). Automated warfarin dose prediction for Asian, American, and Caucasian populations using a deep neural network. Comput. Biol. Med.153, 106548. 10.1016/j.compbiomed.2023.106548
67
Jennings B. A. Loke Y. K. Skinner J. Keane M. Chu G. S. Turner R. et al (2013). Evaluating predictive pharmacogenetic signatures of adverse events in colorectal cancer patients treated with fluoropyrimidines. PLoS One8 (10), e78053. 10.1371/journal.pone.0078053
68
Jeong J. H. Park S. R. Ahn Y. Ryu M. H. Ryoo B. Y. Kong S. Y. et al (2017). Associations between CYP2A6 polymorphisms and outcomes of adjuvant S-1 chemotherapy in patients with curatively resected gastric cancer. Gastric Cancer20 (1), 146–155. 10.1007/s10120-015-0586-9
69
Jurgens S. J. Pirruccello J. P. Choi S. H. Morrill V. N. Chaffin M. Lubitz S. A. et al (2023). Adjusting for common variant polygenic scores improves yield in rare variant association analyses. Nat. Genet.55, 544–548. 10.1038/s41588-023-01342-w
70
Kaida Y. Inui N. Suda T. Nakamura H. Watanabe H. Chida K. (2008). The CYP2A6*4 allele is determinant of S-1 pharmacokinetics in Japanese patients with non-small-cell lung cancer. Clin. Pharmacol. Ther.83 (4), 589–594. 10.1038/sj.clpt.6100484
71
Kanai M. Kawaguchi T. Kotaka M. Manaka D. Hasegawa J. Takagane A. et al (2022). Poor association between dihydropyrimidine dehydrogenase ( DPYD) genotype and fluoropyrimidine‐induced toxicity in an Asian population. Cancer Med. 10.1002/cam4.5541
72
Kataria S. P. Nagar M. Verma S. Purohit V. (2022). Oral tegafur-uracil combination plus leucovorin versus other fluoropyrimidine agents in colorectal cancer: A systematic review and meta-analysis. South Asian J. Cancer11 (1), 084–094. 10.1055/s-0041-1735650
73
Kerr R. S. Love S. Segelov E. Johnstone E. Falcon B. Hewett P. et al (2016). Adjuvant capecitabine plus bevacizumab versus capecitabine alone in patients with colorectal cancer (QUASAR 2): An open-label, randomised phase 3 trial. Lancet Oncol.17 (11), 1543–1557. 10.1016/s1470-2045(16)30172-3
74
Khankhel Z. S. Goring S. Bobiak S. Lamy F. X. Nayak D. Garside J. et al (2022). Second-line treatments in advanced biliary tract cancer: Systematic literature review of efficacy, effectiveness and safety. Future Oncol.18 (18), 2321–2338. 10.2217/fon-2021-1302
75
Kim K. Kang S. B. Chung H. H. Kim J. W. Park N. H. Song Y. S. (2008). XRCC1 Arginine194Tryptophan and GGH-401Cytosine/Thymine polymorphisms are associated with response to platinum-based neoadjuvant chemotherapy in cervical cancer. Gynecol. Oncol.111 (3), 509–515. 10.1016/j.ygyno.2008.08.034
76
Kim K. P. Jang G. Hong Y. S. Lim H. S. Bae K. S. Kim H. S. et al (2011). Phase II study of S-1 combined with oxaliplatin as therapy for patients with metastatic biliary tract cancer: Influence of the CYP2A6 polymorphism on pharmacokinetics and clinical activity. Br. J. Cancer104 (4), 605–612. 10.1038/bjc.2011.17
77
Kim M. J. Kong S. Y. Nam B. H. Kim S. Park Y. I. Park S. R. (2018). A randomized phase II study of S-1 versus capecitabine as first-line chemotherapy in elderly metastatic gastric cancer patients with or without poor performance status: Clinical and pharmacogenetic results. Pharmacogenet Genomics28 (1), 23–30. 10.1097/FPC.0000000000000320
78
Kim S. Y. Baek J. Y. Oh J. H. Park S. C. Sohn D. K. Kim M. J. et al (2017). A phase II study of preoperative chemoradiation with tegafur-uracil plus leucovorin for locally advanced rectal cancer with pharmacogenetic analysis. Radiat. Oncol.12 (1), 62. 10.1186/s13014-017-0800-5
79
Kim S. Y. Y S. H. E K. S. Kong S. Y. Shin A. Baek J. Y. et al (2013). S-1 plus irinotecan and oxaliplatin for the first-line treatment of patients with metastatic colorectal cancer: A prospective phase II study and pharmacogenetic analysis. Br. J. Cancer109 (6), 1420–1427. 10.1038/bjc.2013.479
80
Kim Y. W. Kim M. J. Ryu K. W. Lim H. S. Lee J. H. Kong S. Y. et al (2016). A phase II study of perioperative S-1 combined with weekly docetaxel in patients with locally advanced gastric carcinoma: Clinical outcomes and clinicopathological and pharmacogenetic predictors for survival. Gastric Cancer19 (2), 586–596. 10.1007/s10120-015-0490-3
81
Knikman J. E. Gelderblom H. Beijnen J. H. Cats A. Guchelaar H. J. Henricks L. M. (2021). Individualized dosing of fluoropyrimidine-based chemotherapy to prevent severe fluoropyrimidine-related toxicity: What are the options?Clin. Pharmacol. Ther.109 (3), 591–604. 10.1002/cpt.2069
82
Kong J. Lee H. Kim D. Han S. K. Ha D. Shin K. et al (2020). Network-based machine learning in colorectal and bladder organoid models predicts anti-cancer drug efficacy in patients. Nat. Commun.11 (1), 5485. 10.1038/s41467-020-19313-8
83
Kong S. Y. Lim H. S. Nam B. H. Kook M. C. Kim Y. W. Ryu K. W. et al (2009). Association of CYP2A6 polymorphisms with S-1 plus docetaxel therapy outcomes in metastatic gastric cancer. Pharmacogenomics10 (7), 1147–1155. 10.2217/pgs.09.48
84
Kummer D. Froehlich T. K. Joerger M. Aebi S. Sistonen J. Amstutz U. et al (2015). Dihydropyrimidinase and beta-ureidopropionase gene variation and severe fluoropyrimidine-related toxicity. Pharmacogenomics16 (12), 1367–1377. 10.2217/pgs.15.81
85
Lam S. W. Guchelaar H. J. Boven E. (2016). The role of pharmacogenetics in capecitabine efficacy and toxicity. Cancer Treat. Rev.50, 9–22. 10.1016/j.ctrv.2016.08.001
86
Lam S. W. van der Noort V. van der Straaten T. Honkoop A. H. Peters G. J. Guchelaar H. J. et al (2018). Single-nucleotide polymorphisms in the genes of CES2, CDA and enzymatic activity of CDA for prediction of the efficacy of capecitabine-containing chemotherapy in patients with metastatic breast cancer. Pharmacol. Res.128, 122–129. 10.1016/j.phrs.2017.08.005
87
Lee A. M. Shi Q. Pavey E. Alberts S. R. Sargent D. J. Sinicrope F. A. et al (2014). DPYD variants as predictors of 5-fluorouracil toxicity in adjuvant colon cancer treatment (NCCTG N0147). J. Natl. Cancer Inst.106 (12). 10.1093/jnci/dju298
88
Li M. Mindt S. Lück A. Hutzschenreuter U. Kollendt M. Lathan B. et al (2023). Drug monitoring detects under- and overdosing in patients receiving 5-fluorouracil-containing chemotherapy-results of a prospective, multicenter German observational study. ESMO Open8 (2), 101201. 10.1016/j.esmoop.2023.101201
89
Liu D. Li X. Li X. Wang H. Dong M. (2021). Carboxylesterase 1 polymorphisms are associated with clinical outcomes in gastroenteric cancer patients treated with capecitabine. Cancer Chemother. Pharmacol.87 (5), 681–687. 10.1007/s00280-021-04247-9
90
Liu D. Li X. Li X. Zhang M. Zhang J. Hou D. et al (2019). CDA and MTHFR polymorphisms are associated with clinical outcomes in gastroenteric cancer patients treated with capecitabine-based chemotherapy. Cancer Chemother. Pharmacol.83 (5), 939–949. 10.1007/s00280-019-03809-2
91
Loganayagam A. Arenas Hernandez M. Corrigan A. Fairbanks L. Lewis C. M. Harper P. et al (2013). Pharmacogenetic variants in the DPYD, TYMS, CDA and MTHFR genes are clinically significant predictors of fluoropyrimidine toxicity. Br. J. Cancer108 (12), 2505–2515. 10.1038/bjc.2013.262
92
Longley D. B. Harkin D. P. Johnston P. G. (2003). 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer3 (5), 330–338. 10.1038/nrc1074
93
Lunenburg C. van der Wouden C. H. Nijenhuis M. Crommentuijn-van Rhenen M. H. de Boer-Veger N. J. Buunk A. M. et al (2020). Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction of DPYD and fluoropyrimidines. Eur. J. Hum. Genet.28 (4), 508–517. 10.1038/s41431-019-0540-0
94
Marsh S. (2005). Thymidylate synthase pharmacogenetics. Invest. New Drugs23 (6), 533–537. 10.1007/s10637-005-4021-7
95
Martens F. K. Huntjens D. W. Rigter T. Bartels M. Bet P. M. Cornel M. C. (2019). DPD testing before treatment with fluoropyrimidines in the amsterdam UMCs: An evaluation of current pharmacogenetic practice. Front. Pharmacol.10, 1609. 10.3389/fphar.2019.01609
96
Martin M. Martinez N. Ramos M. Calvo L. Lluch A. Zamora P. et al (2015). Standard versus continuous administration of capecitabine in metastatic breast cancer (GEICAM/2009-05): A randomized, noninferiority phase II trial with a pharmacogenetic analysis. Oncologist20 (2), 111–112. 10.1634/theoncologist.2014-0379
97
Matsumoto N. Kubota Y. Ishida H. Sekido M. Ohkuma R. Ishiguro T. et al (2020). Variants of carboxylesterase 1 have no impact on capecitabine pharmacokinetics and toxicity in capecitabine plus oxaliplatin treated-colorectal cancer patients. Cancer Chemother. Pharmacol.85 (6), 1119–1128. 10.1007/s00280-020-04087-z
98
Mbemi A. Khanna S. Njiki S. Yedjou C. G. Tchounwou P. B. (2020). Impact of gene-environment interactions on cancer development. Int. J. Environ. Res. Public Health17 (21), 8089. 10.3390/ijerph17218089
99
Meulendijks D. Henricks L. M. Sonke G. S. Deenen M. J. Froehlich T. K. Amstutz U. et al (2015). Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: A systematic review and meta-analysis of individual patient data. Lancet Oncol.16 (16), 1639–1650. 10.1016/s1470-2045(15)00286-7
100
Meulendijks D. Jacobs B. A. Aliev A. Pluim D. van Werkhoven E. Deenen M. J. et al (2016). Increased risk of severe fluoropyrimidine-associated toxicity in patients carrying a G to C substitution in the first 28-bp tandem repeat of the thymidylate synthase 2R allele. Int. J. Cancer138 (1), 245–253. 10.1002/ijc.29694
101
Meulendijks D. Rozeman E. A. Cats A. Sikorska K. Joerger M. Deenen M. J. et al (2017). Pharmacogenetic variants associated with outcome in patients with advanced gastric cancer treated with fluoropyrimidine and platinum-based triplet combinations: A pooled analysis of three prospective studies. Pharmacogenomics J.17 (5), 441–451. 10.1038/tpj.2016.81
102
Mezzalira S. Toffoli G. (2021). The effects of sex on pharmacogenetically guided drug treatment. Pharmacogenomics22 (15), 959–962. 10.2217/pgs-2021-0088
103
Miura K. Kinouchi M. Ishida K. Fujibuchi W. Naitoh T. Ogawa H. et al (2010). 5-fu metabolism in cancer and orally-administrable 5-fu drugs. Cancers (Basel)2 (3), 1717–1730. 10.3390/cancers2031717
104
Morelli C. Formica V. Doldo E. Riondino S. Rofei M. Vergilii L. et al (2022). Concordance of blood-based and normal tissue-based dihydropyrimidine dehydrogenase (DPYD) genotyping. Oncologist27 (5), e420–e421. 10.1093/oncolo/oyac057
105
Morita T. Matsuzaki A. Kurokawa S. Tokue A. (2003). Forced expression of cytidine deaminase confers sensitivity to capecitabine. Oncology65 (3), 267–274. 10.1159/000074480
106
Mueller F. Büchel B. Köberle D. Schürch S. Pfister B. Krähenbühl S. et al (2013). Gender-specific elimination of continuous-infusional 5-fluorouracil in patients with gastrointestinal malignancies: Results from a prospective population pharmacokinetic study. Cancer Chemother. Pharmacol.71 (2), 361–370. 10.1007/s00280-012-2018-4
107
Palles C. Fotheringham S. Chegwidden L. Lucas M. Kerr R. Mozolowski G. et al (2021). An evaluation of the diagnostic accuracy of a panel of variants in DPYD and a single variant in ENOSF1 for predicting common capecitabine related toxicities. Cancers (Basel)13 (7), 1497. 10.3390/cancers13071497
108
Panagopoulou M. Karaglani M. Manolopoulos V. G. Iliopoulos I. Tsamardinos I. Chatzaki E. (2021). Deciphering the methylation landscape in breast cancer: Diagnostic and prognostic biosignatures through automated machine learning. Cancers (Basel)13 (7), 1677. 10.3390/cancers13071677
109
Pandi M. T. Koromina M. Tsafaridis I. Patsilinakos S. Christoforou E. van der Spek P. J. et al (2021). A novel machine learning-based approach for the computational functional assessment of pharmacogenomic variants. Hum. Genomics15 (1), 51. 10.1186/s40246-021-00352-1
110
Park S. R. Kong S. Y. Nam B. H. Choi I. J. Kim C. G. Lee J. Y. et al (2011). CYP2A6 and ERCC1 polymorphisms correlate with efficacy of S-1 plus cisplatin in metastatic gastric cancer patients. Br. J. Cancer104 (7), 1126–1134. 10.1038/bjc.2011.24
111
Pellicer M. Garcia-Gonzalez X. Garcia M. I. Robles L. Gravalos C. Garcia-Alfonso P. et al (2017). Identification of new SNPs associated with severe toxicity to capecitabine. Pharmacol. Res.120, 133–137. 10.1016/j.phrs.2017.03.021
112
Ragia G. Karantza I. M. Kelli-Kota E. Kolovou V. Kolovou G. Konstantinides S. et al (2017a). Role of CYP4F2, CYP2C19, and CYP1A2 polymorphisms on acenocoumarol pharmacogenomic algorithm accuracy improvement in the Greek population: Need for sub-phenotype analysis. Drug Metab. Pers. Ther.32 (4), 183–190. 10.1515/dmpt-2017-0034
113
Ragia G. Kolovou V. Kolovou G. Konstantinides S. Maltezos E. Tavridou A. et al (2017b). A novel acenocoumarol pharmacogenomic dosing algorithm for the Greek population of EU-PACT trial. Pharmacogenomics18 (1), 23–34. 10.2217/pgs-2016-0126
114
Ragia G. Manolopoulos V. G. (2022). The revolution of pharmaco-omics: Ready to open new avenues in materializing precision medicine?Pharmacogenomics23 (16), 869–872. 10.2217/pgs-2022-0145
115
Ragia G. Tavridou A. Elens L. Van Schaik R. H. Manolopoulos V. G. (2014). CYP2C9*2 allele increases risk for hypoglycemia in POR*1/*1 type 2 diabetic patients treated with sulfonylureas. Exp. Clin. Endocrinol. Diabetes122 (1), 60–63. 10.1055/s-0033-1361097
116
Ramalakshmi S. Kavimani S. Srineevas S. Vetriselvi V. Bhaskar L. (2016). Molecular markers for capecitabine therapy: A review. Int. J. Pharm. Sci. Res.7 (11). 10.13040/ijpsr.0975-8232.7(11).4315-26
117
Ramsdale E. Mohamed M. Yu V. Otto E. Juba K. Awad H. et al (2022). Polypharmacy, potentially inappropriate medications, and drug-drug interactions in vulnerable older adults with advanced cancer initiating cancer treatment. Oncologist27 (7), e580–e588. 10.1093/oncolo/oyac053
118
Ribelles N. López-Siles J. Sánchez A. González E. Sánchez M. J. Carabantes F. et al (2008). A carboxylesterase 2 gene polymorphism as predictor of capecitabine on response and time to progression. Curr. Drug Metab.9 (4), 336–343. 10.2174/138920008784220646
119
Rosmarin D. Palles C. Church D. Domingo E. Jones A. Johnstone E. et al (2014). Genetic markers of toxicity from capecitabine and other fluorouracil-based regimens: Investigation in the QUASAR2 study, systematic review, and meta-analysis. J. Clin. Oncol.32 (10), 1031–1039. 10.1200/JCO.2013.51.1857
120
Rosmarin D. Palles C. Pagnamenta A. Kaur K. Pita G. Martin M. et al (2015). A candidate gene study of capecitabine-related toxicity in colorectal cancer identifies new toxicity variants at DPYD and a putative role for ENOSF1 rather than TYMS. Gut64 (1), 111–120. 10.1136/gutjnl-2013-306571
121
Rudek M. A. Connolly R. M. Hoskins J. M. Garrett-Mayer E. Jeter S. C. Armstrong D. K. et al (2013). Fixed-dose capecitabine is feasible: Results from a pharmacokinetic and pharmacogenetic study in metastatic breast cancer. Breast Cancer Res. Treat.139 (1), 135–143. 10.1007/s10549-013-2516-z
122
Runcharoen C. Fukunaga K. Sensorn I. Iemwimangsa N. Klumsathian S. Tong H. et al (2021). Prevalence of pharmacogenomic variants in 100 pharmacogenes among southeast asian populations under the collaboration of the southeast asian pharmacogenomics research network (SEAPharm). Hum. Genome Var.8 (1), 7. 10.1038/s41439-021-00135-z
123
Saam J. Critchfield G. C. Hamilton S. A. Roa B. B. Wenstrup R. J. Kaldate R. R. (2011). Body surface area-based dosing of 5-fluoruracil results in extensive interindividual variability in 5-fluorouracil exposure in colorectal cancer patients on FOLFOX regimens. Clin. Colorectal Cancer10 (3), 203–206. 10.1016/j.clcc.2011.03.015
124
Santos C. Morgan B. W. Geller R. J. (2017). The successful treatment of 5-fluorouracil (5-FU) overdose in a patient with malignancy and HIV/AIDS with uridine triacetate. Am. J. Emerg. Med.35 (5), 802.e807–802. 10.1016/j.ajem.2016.11.038
125
Sara J. D. Kaur J. Khodadadi R. Rehman M. Lobo R. Chakrabarti S. et al (2018). 5-fluorouracil and cardiotoxicity: A review. Ther. Adv. Med. Oncol.10, 1758835918780140. 10.1177/1758835918780140
126
Schaerer D. Froehlich T. K. Hamzic S. Offer S. M. Diasio R. B. Joerger M. et al (2020). A novel nomenclature for repeat motifs in the thymidylate synthase enhancer region and its relevance for pharmacogenetic studies. J. Pers. Med.10 (4), 181. 10.3390/jpm10040181
127
Sharma B. B. Rai K. Blunt H. Zhao W. Tosteson T. D. Brooks G. A. (2021). Pathogenic DPYD variants and treatment-related mortality in patients receiving fluoropyrimidine chemotherapy: A systematic review and meta-analysis. Oncologist26 (12), 1008–1016. 10.1002/onco.13967
128
Shrestha S. Zhang C. Jerde C. R. Nie Q. Li H. Offer S. M. et al (2018). Gene-specific variant classifier (DPYD-Varifier) to identify deleterious alleles of dihydropyrimidine dehydrogenase. Clin. Pharmacol. Ther.104 (4), 709–718. 10.1002/cpt.1020
129
Siemens A. Anderson S. J. Rassekh S. R. Ross C. J. D. Carleton B. C. (2022). A systematic review of polygenic models for predicting drug outcomes. J. Pers. Med.12 (9), 1394. 10.3390/jpm12091394
130
Stein B. N. Petrelli N. J. Douglass H. O. Driscoll D. L. Arcangeli G. Meropol N. J. (1995). Age and sex are independent predictors of 5-fluorouracil toxicity. Analysis of a large scale phase III trial. Cancer75 (1), 11–17. 10.1002/1097-0142(19950101)75:1<11::aid-cncr2820750104>3.0.co;2-n
131
Suenaga M. Cao S. Zhang W. Matsusaka S. Okazaki S. Berger M. D. et al (2021). Role of enterocyte-specific gene polymorphisms in response to adjuvant treatment for stage III colorectal cancer. Pharmacogenet Genomics31 (1), 10–16. 10.1097/fpc.0000000000000416
132
Tafazoli A. Guchelaar H. J. Miltyk W. Kretowski A. J. Swen J. J. (2021). Applying next-generation sequencing platforms for pharmacogenomic testing in clinical practice. Front. Pharmacol.12, 693453. 10.3389/fphar.2021.693453
133
Tanner J. A. Tyndale R. F. (2017). Variation in CYP2A6 activity and personalized medicine. J. Pers. Med.7 (4), 18. 10.3390/jpm7040018
134
Tecza K. Pamula-Pilat J. Lanuszewska J. Butkiewicz D. Grzybowska E. (2018). Pharmacogenetics of toxicity of 5-fluorouracil, doxorubicin and cyclophosphamide chemotherapy in breast cancer patients. Oncotarget9 (10), 9114–9136. 10.18632/oncotarget.24148
135
Thorn C. F. Marsh S. Carrillo M. W. McLeod H. L. Klein T. E. Altman R. B. (2011). PharmGKB summary: Fluoropyrimidine pathways. Pharmacogenet Genomics21 (4), 237–242. 10.1097/FPC.0b013e32833c6107
136
Toffoli G. De Mattia E. (2008). Pharmacogenetic relevance of MTHFR polymorphisms. Pharmacogenomics9 (9), 1195–1206. 10.2217/14622416.9.9.1195
137
Tsunoda A. Nakao K. Watanabe M. Matsui N. Ooyama A. Kusano M. (2011). Associations of various gene polymorphisms with toxicity in colorectal cancer patients receiving oral uracil and tegafur plus leucovorin: A prospective study. Ann. Oncol.22 (2), 355–361. 10.1093/annonc/mdq358
138
van Dam S. Folkertsma P. Castela Forte J. de Vries D. H. Herrera Cunillera C. Gannamani R. et al (2023). The necessity of incorporating non-genetic risk factors into polygenic risk score models. Sci. Rep.13 (1), 1351. 10.1038/s41598-023-27637-w
139
van der Wouden C. H. Marck H. Guchelaar H. J. Swen J. J. van den Hout W. B. (2022). Cost-effectiveness of pharmacogenomics-guided prescribing to prevent gene-drug-related deaths: A decision-analytic model. Front. Pharmacol.13, 918493. 10.3389/fphar.2022.918493
140
van Leeuwen R. W. F. le Comte M. Reyners A. K. L. van den Tweel A. van Vlijmen B. Kwee W. et al (2022). Evidence- and consensus-based guidelines for drug-drug interactions with anticancer drugs; A practical and universal tool for management. Semin. Oncol.49 (2), 119–129. 10.1053/j.seminoncol.2022.03.002
141
van Leeuwen R. W. Swart E. L. Boven E. Boom F. A. Schuitenmaker M. G. Hugtenburg J. G. (2011). Potential drug interactions in cancer therapy: A prevalence study using an advanced screening method. Ann. Oncol.22 (10), 2334–2341. 10.1093/annonc/mdq761
142
van Schaik R. H. (2005). Cancer treatment and pharmacogenetics of cytochrome P450 enzymes. Invest. New Drugs23 (6), 513–522. 10.1007/s10637-005-4019-1
143
Varma A. Mathaiyan J. Shewade D. Dubashi B. Sunitha K. (2020). Influence of ABCB-1, ERCC-1 and ERCC-2 gene polymorphisms on response to capecitabine and oxaliplatin (CAPOX) treatment in colorectal cancer (CRC) patients of South India. J. Clin. Pharm. Ther.45 (4), 617–627. 10.1111/jcpt.13166
144
Verhoef T. I. Ragia G. de Boer A. Barallon R. Kolovou G. Kolovou V. et al (2013). A randomized trial of genotype-guided dosing of acenocoumarol and phenprocoumon. N. Engl. J. Med.369 (24), 2304–2312. 10.1056/NEJMoa1311388
145
Wang J. Wang X. Zhao M. Choo S. P. Ong S. J. Ong S. Y. et al (2014). Potentially functional SNPs (pfSNPs) as novel genomic predictors of 5-FU response in metastatic colorectal cancer patients. PLoS One9 (11), e111694. 10.1371/journal.pone.0111694
146
Wang L. Howlett S. Essapen S. (2022). Treating patients with dihydropyrimidine dehydrogenase (DPD) deficiency with fluoropyrimidine chemotherapy since the onset of routine prospective testing-The experience of a large oncology center in the United Kingdom. Semin. Oncol.49 (2), 170–177. 10.1053/j.seminoncol.2021.11.004
147
White C. Scott R. J. Paul C. Ziolkowski A. Mossman D. Fox S. B. et al (2022). Dihydropyrimidine dehydrogenase deficiency and implementation of upfront DPYD genotyping. Clin. Pharmacol. Ther.112 (4), 791–802. 10.1002/cpt.2667
148
Wigle T. J. Medwid S. Ross C. Schwarz U. I. Kim R. B. (2023). DPYD exon 4 deletion associated with fluoropyrimidine toxicity and importance of copy number variation. Curr. Oncol.30 (1), 663–672. 10.3390/curroncol30010051
149
Wörmann B. Bokemeyer C. Burmeister T. Köhne C. H. Schwab M. Arnold D. et al (2020). Dihydropyrimidine dehydrogenase testing prior to treatment with 5-fluorouracil, capecitabine, and tegafur: A consensus paper. Oncol. Res. Treat.43 (11), 628–636. 10.1159/000510258
150
Wu R. Nie Q. Tapper E. E. Jerde C. R. Dunlap G. S. Shrestha S. et al (2016). Histone H3K27 trimethylation modulates 5-fluorouracil resistance by inhibiting PU.1 binding to the DPYD promoter. Cancer Res.76 (21), 6362–6373. 10.1158/0008-5472.Can-16-1306
151
Xie P. Mo J. L. Liu J. H. Li X. Tan L. M. Zhang W. et al (2020). Pharmacogenomics of 5-fluorouracil in colorectal cancer: Review and update. Cell Oncol. (Dordr)43 (6), 989–1001. 10.1007/s13402-020-00529-1
152
Yagi S. Yamada K. Terayama M. Wake H. Enomoto N. Nohara K. et al (2021). Current status of doublet combinations of platinum and fluoropyrimidines using oxaliplatin for advanced gastric cancer. Glob. Health Med.3 (1), 31–36. 10.35772/ghm.2020.01075
153
Yang L. Zou S. Shu C. Song Y. Sun Y. K. Zhang W. et al (2017). CYP2A6 polymorphisms associate with outcomes of S-1 plus oxaliplatin chemotherapy in Chinese gastric cancer patients. Genomics Proteomics Bioinforma.15 (4), 255–262. 10.1016/j.gpb.2016.11.004
154
Yap Y. S. Kwok L. L. Syn N. Chay W. Y. Chia J. W. K. Tham C. K. et al (2017). Predictors of hand-foot syndrome and pyridoxine for prevention of capecitabine-induced hand-foot syndrome: A randomized clinical trial. JAMA Oncol.3 (11), 1538–1545. 10.1001/jamaoncol.2017.1269
155
Zhong L. He X. Zhang Y. Chuan J. L. Chen M. Zhu S. M. et al (2018). Relevance of methylenetetrahydrofolate reductase gene variants C677T and A1298C with response to fluoropyrimidine-based chemotherapy in colorectal cancer: A systematic review and meta-analysis. Oncotarget9 (58), 31291–31301. 10.18632/oncotarget.24933
Summary
Keywords
pharmacogenomics, precision medicine, fluoropyrimidines, 5-fluorouracil, capecitabine, tegafur, toxicity, polygenic dosing algorithm
Citation
Maslarinou A, Manolopoulos VG and Ragia G (2023) Pharmacogenomic-guided dosing of fluoropyrimidines beyond DPYD: time for a polygenic algorithm?. Front. Pharmacol. 14:1184523. doi: 10.3389/fphar.2023.1184523
Received
11 March 2023
Accepted
19 April 2023
Published
15 May 2023
Volume
14 - 2023
Edited by
Jessica Cusato, University of Turin, Italy
Reviewed by
Erika Cecchin, Aviano Oncology Reference Center (IRCCS), Italy
David Paez, Hospital de la Santa Cruz y San Pablo, Spain
Carlo R. Largiader, University Hospital of Bern, Switzerland
Elena De Mattia, Aviano Oncology Reference Center (IRCCS), Italy
Updates
Copyright
© 2023 Maslarinou, Manolopoulos and Ragia.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vangelis G. Manolopoulos, emanolop@med.duth.gr; Georgia Ragia, gragia@med.duth,gr
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.