 J. Westley Heinle1
J. Westley Heinle1 Shannon Dalessio1
Shannon Dalessio1 Piotr Janicki2Ann Ouyang1Kent E. Vrana3
Piotr Janicki2Ann Ouyang1Kent E. Vrana3 Victor Ruiz-Velasco2
Victor Ruiz-Velasco2 Matthew D. Coates1,3*
Matthew D. Coates1,3*- 1Division of Gastroenterology and Hepatology, Penn State College of Medicine, Hershey, PA, United States
- 2Department of Anesthesiology and Perioperative Medicine, Penn State College of Medicine, Hershey, PA, United States
- 3Department of Pharmacology, Penn State College of Medicine, Hershey, PA, United States
Pain is a major issue in healthcare throughout the world. It remains one of the major clinical issues of our time because it is a common sequela of numerous conditions, has a tremendous impact on individual quality of life, and is one of the top drivers of cost in medicine, due to its influence on healthcare expenditures and lost productivity in those affected by it. Patients and healthcare providers remain desperate to find new, safer and more effective analgesics. Growing evidence indicates that the voltage-gated sodium channel Nav1.8 plays a critical role in transmission of pain-related signals throughout the body. For that reason, this channel appears to have strong potential to help develop novel, more selective, safer, and efficacious analgesics. However, many questions related to the physiology, function, and clinical utility of Nav1.8 remain to be answered. In this article, we discuss the latest studies evaluating the role of Nav1.8 in pain, with a particular focus on visceral pain, as well as the steps taken thus far to evaluate its potential as an analgesic target. We also review the limitations of currently available studies related to this topic, and describe the next scientific steps that have already been undertaken, or that will need to be pursued, to fully unlock the capabilities of this potential therapeutic target.
Introduction
Voltage-gated sodium channels (VGSCs) are part of a larger family of mammalian ion channels (Catterall et al., 2005b). VGSCs are transmembrane proteins that help to regulate the membrane potential of cells. They do this by providing a hydrophilic corridor that permits controlled movement of sodium ions through the outer hydrophobic phospholipid bilayer of eukaryotic cells. To date, nine distinct VGSC isoforms have been identified in humans, named Nav1.1 through Nav1.9 (numbered based upon the order in which each type was identified) (Goldin et al., 2000; Catterall et al., 2005a). These isoforms are structurally differentiated based upon their pore-forming alpha subunits (Yu and Catterall, 2003). A VGSC is composed of one of these alpha subunits and one or more beta subunits. The alpha subunit contains six alpha-helical transmembrane segments (S1-S6) that are folded into four domains (I-IV) (Catterall, 2000). The four homologous domains of the alpha subunit form an ion-conducting aqueous pore. This region determines the specific function of each particular VGSC, including ion selectivity, expression location and overall channel function. The beta subunits influence gating and signaling functions of each channel, and also serve to help anchor these channels within a cell membrane (Yu et al., 2003; Yu and Catterall, 2004) (Figure 1).
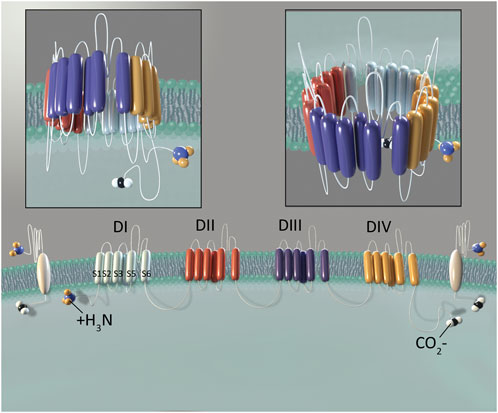
Figure 1. Structure of NaV1.8. The alpha subunit consists of four domains (DI-DIV). Domains are shown in different colors to demonstrate their independent, yet similar structure. Six transmembrane segments (S1-S6) comprise each domain. Two beta subunits are shown in white, laterally to the outside of DI and DIV. Illustration by Devon Stuart.
VGSCs are encoded by the SCN gene family. These genes are numbered 1–11A, corresponding in sequential fashion with the isoforms described above (with the exception of SCN6-7, which encode NaX, a non-selective TTX-sensitive channel (Kanellopoulos and Matsuyama, 2016; Cheng et al., 2021; Noland et al., 2022). The product of each gene is described, in part, by the prefix Nav1, while the number following the decimal point identifies a specific channel isoform. Of note, these were assigned in the approximate order in which each gene was identified (Goldin et al., 2000). A more in-depth review of the structure and physiology of each VGSC can be found in Coates et al., Neurogastroenterology and Motility, 2019 (Coates et al., 2019). While the precise functions of these channels may vary, all are important determinants of the excitability of cells, including in neurons, muscles and endocrine cells. Accordingly, they play a crucial role in muscle contractions and the initiation and propagation of action potentials (Cheng et al., 2021). These channels are expressed throughout human body. Certain subtypes appear to be preferentially expressed in either the central nervous system (CNS), or peripheral nervous system (PNS), but some may appear in both (de Lera Ruiz and Kraus, 2015). For example, Nav1.4 has been chiefly associated with skeletal muscle, and genetic variations associated with its gene, SCN4A, have been linked to several disorders of muscular function (Mannikko et al., 2018). On the other hand, Nav1.6 appears to be expressed primarily on the membranes of neural structures within the central nervous system (Caldwell et al., 2000). Nav1.7, Nav1.8, and Nav1.9 are almost exclusively expressed in the peripheral nervous system (PNS) on primary sensory neurons (Bennett et al., 2019; Cheng et al., 2021). In fact, Nav1.8 is known to be expressed in dorsal root ganglion (DRG) and trigeminal ganglion (TG) neurons (Djouhri et al., 2003), including cells that play a major role in transmission of pain-related signals. As much as any VGSC, Nav1.8 has also been implicated as a significant factor in the initiation and persistence of chronic pain in a wide range of disorders, including those related to the viscera (Beyak et al., 2004; Hillsley et al., 2006; King et al., 2009; Chen et al., 2013; Hu et al., 2013; Hu et al., 2016; Lin et al., 2017). In order to understand why Nav1.8 is so relevant to understanding visceral pain, and represents a promising therapeutic target, it is important to review what is known about its structure and physiology, and to discuss how it has been tested in this context.
The structure and physiology of Nav1.8, and its role in somatosensory pain perception
Nav1.8 is encoded by the gene SCN10A which is located on human chromosome 3p22-24 (Goldin et al., 2000). This gene has 28 exons, 27 of which are coding while one is non-coding. SCN10A is found in a gene cluster with SCN5A (which encodes NaV1.5) and SCN11A (which encodes NaV1.9). Notably, NaV1.5, NaV1.8 and NaV1.9 are all relatively resistant to the blocking effects of tetrodotoxin (TTX) when compared to other VGSCs (Kostyuk et al., 1981; Blair and Bean, 2002). This is important as TTX (a neurotoxin derived from pufferfish species) has been a critically important agent used to differentiate subtypes of VGSCs for decades (Catterall, 1980; Stevens et al., 2011).
In humans, NaV1.8 is composed of 1956 amino acids, and has a molecular weight of 220 kilodaltons (kDa), though this can vary in other species (Hameed, 2019). The structure of purified human NaV1.8 has been evaluated using cryogenic electron microscopy and there are unique aspects of the first voltage-sensing domain (VSD1) that appear to modify electrophysiological characteristics of the channel (Huang et al., 2022). Notably, compared with other VGSC isoforms, NaV1.8 exhibits several distinctive biophysical characteristics, including its activation at a more pronounced state of cellular depolarization, as well as slower inactivation. NaV1.8 remains active at voltages that inactivate other VGSCs. These features help to facilitate cellular hyperexcitability, and make NaV1.8 a major contributor to the depolarization phase in action potentials of nociceptive neurons. (Renganathan et al., 2000; Renganathan et al., 2001). These characteristics also help to differentiate NaV1.8 from NaV1.9, which (through the use of knockout rodent models) appears to be primarily involved with setting resting membrane potential and less important to the production of action potentials (Cummins et al., 1999; Dib-Hajj et al., 2002).
Since its discovery, NaV1.8 has demonstrated a strong association with nociceptors and pain perception. NaV1.8 was described as the “sensory neuron specific” (SNS) channel, as it was originally identified in the mucosal neurites and soma of small fiber DRG-associated neurons and vagal afferent neurons associated with pain in mammals. Akopian et al. first described the basic electrophysiological and pharmacological characteristics of NaV1.8, using in situ hybridization to determine that it was predominantly localized to small diameter neurons within the TG and DRG neurons (Akopian et al., 1996). Djouhri et al. demonstrated Nav1.8 immunoreactivity in small-to-medium sized DRG neurons, but not in the brain, cardiac, skeletal muscle, or a variety of other tissues (ex: liver, kidney) (Djouhri et al., 2003). Other studies performed by Akopian et al. established the importance of this channel to pain perception, when they demonstrated that mouse NaV1.8 knockout models exhibit pronounced analgesia to noxious mechanical and thermal stimuli, as well as delayed development of inflammatory hyperalgesia (Akopian et al., 1999). Other studies evaluating SCN10A genetic variants have provided further supportive evidence of the importance that NaV1.8 has in pain. For example, Faber et al. described the discovery of three polymorphisms they found associated with peripheral neuropathy, two of which resulted in apparent enhanced response of NaV1.8 channels to depolarization and the hyperexcitability of the associated neurons (L554P and A1304T) (Faber et al., 2012). This group described another polymorphism (G1662S) associated with painful small fiber neuropathy (Han et al., 2014) (Table 1).

Table 1. Clinically Relevant SCN10A (NaV1.8) Polymorphisms.
There is also evidence that NaV1.8 may influence other key factors that impact nociception and pain perception. For example, in a mouse model of psoriasis, NaV1.8-bearing sensory neurons were found to interact with antigen presenting cells to influence the expression of particular cytokines (interleukin-12/interleukin-23) (Riol-Blanco et al., 2014), which, in turn, may affect activity of the neuron itself.
However, it is important to note that these channels are not exclusively localized to nociceptive neurons. Studies of peripheral nerves in mice have demonstrated that a significant proportion of large fiber, myelinated neurons (indicative of mechanoreceptor function essential for touch sensation) expressed functioning NaV1.8 (Shields et al., 2012). Studies performed in mice have also provided immunohistochemical and electrophysiological evidence for the expression of NaV1.8 in non-nociceptive neurons within the heart (Verkerk et al., 2012). Genome-wide association studies have found links between specific SCN10A polymorphisms and arrythmias or other cardiac disorders in humans. There are also reports of increased NaV1.8 mRNA transcript and protein expression in heart tissue obtained from individuals with cardiac hypertrophy and heart failure (Dybkova et al., 2018; Ahmad et al., 2019). Interestingly, no studies to date have confirmed the presence of NaV1.8 channels in cardiomyocytes of normal human heart tissue (Casini et al., 2019). Additionally, separate immunohistochemical studies in transgenic mice have identified NaV1.8 in brain tissue (Tenza-Ferrer et al., 2022). While NaV1.8 expression has not been demonstrated in a healthy human brain, these channels have been identified in Purkinje fibers from the cerebella of rodents used in multiple sclerosis (MS) models, as well as those derived from post-mortem patients previously diagnosed with MS (Black et al., 1999; Black et al., 2000; Waxman, 2005) (Figure 2).
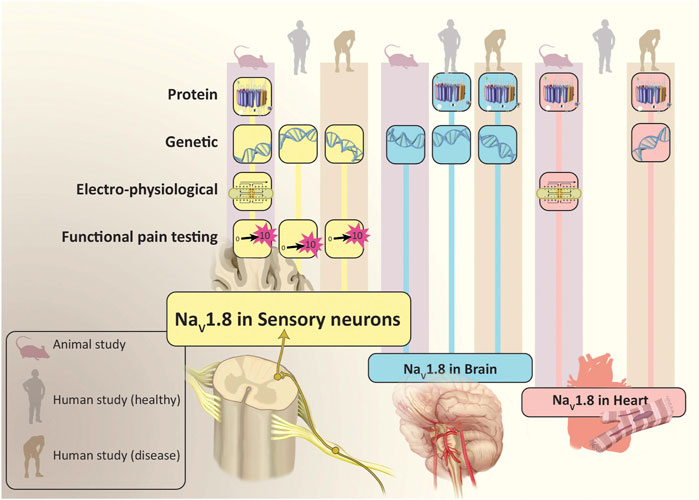
Figure 2. Evidence for Location and Function of NaV1.8. In addition to sensory neurons, studies have found that NaV1.8 is also expressed in the brain and heart. The population studied (animal, healthy human and human with disease) and evidence which demonstrated NaV1.8 (protein, genetic, electro-physiological and functional pain testing) is depicted for each location where NaV1.8 has been expressed. Illustration by Devon Stuart.
The role of NaV1.8 in visceral pain perception
There are a variety of studies that have directly and indirectly established the importance of NaV1.8 to visceral pain perception. SCN10A transcript and immunoreactivity for the NaV1.8 channel have been demonstrated throughout the gastrointestinal tract, including the stomach, small bowel and colon (Su et al., 1999; Peeters et al., 2006). Electrophysiological currents consistent with NaV1.8 have also been identified in DRGs and vagal fibers innervating these organs (Gautron et al., 2011). Additionally, a variety of animal-based studies have shown that systemic and viscerally localized genetic and/or pharmacological antagonism of Nav1.8 leads to significant deficits in several visceral pain modalities (Laird et al., 2002; Hillsley et al., 2006; Matthews et al., 2006).
Several animal-based studies have also demonstrated the important role that Nav1.8 has in pathological conditions associated with alterations in visceral pain perception. Animal models of enterocolitis have exhibited increases in Nav1.8 expression within sensory neurons (King et al., 2009). Previous studies have also suggested that NaV1.8 is required for demonstration of spontaneous activity in damaged sensory neurons (Roza et al., 2003) and for generation of enhanced intestinal nociceptor activity in rodent models of colitis (Beyak et al., 2004; Hillsley et al., 2006; Chen et al., 2013). Using a rat model of stress-induced visceral hypersensitivity, Hu et al. reported that affected animals exhibit increased expression of Nav1.8 protein and that TTX-resistant sodium channel current density in colonic afferents is increased (Hu et al., 2013). In a separate model of diabetic intestinal neuropathy, they reported that intestinal sensory afferents were hyperexcitable and that Nav1.8 expression in dorsal root ganglia was higher than what was exhibited in control animals (Hu et al., 2016). In 2022, Lima et al. investigated the role of Nav1.8 in nociception following a surgical incision. Using real time PCR, they demonstrated that Nav1.8 mRNA expression increases following surgical incisions in rats, and is reduced with antisense oligonucleotide application (Ma et al., 2008).
In humans, several lines of evidence also support the important impact that SCN10A polymorphisms have on pain perception, including that related to the viscera. Duan et al. performed multiple objective pain-related assessments in several hundred study participants who had been genotyped based upon several SCN10A gene polymorphisms. They found that homozygosity for one polymorphism (A1073V, encoding an alanine to valine switch at an intracellular loop associated with the channel) was associated with increased thresholds for reporting mechanical pain (Duan et al., 2016). Our study team performed targeted whole exome sequencing in a carefully phenotyped cohort of inflammatory bowel disease (IBD) patients, and found that individuals who were homozygous for A1073V were more likely to exhibit visceral hypoalgesia (or “silent” disease) (Gonzalez-Lopez et al., 2018). In a follow up investigation, we examined a cohort of patients who had undergone a sigmoidectomy and found that individuals who were homozygous for A1073V demonstrated significantly lower post-operative pain scores than those exhibiting the heterozygous or wild-type SCN10A genotypes (Coates et al., 2019). We also transfected rat superior cervical ganglion (SCG) with either wild-type or polymorphic cDNA constructs and found that neurons expressing the A1073V variant activated at more depolarized potentials when compared to those with wild type channels, indicating a hypoactive phenotype (Coates et al., 2019). To our knowledge, this is the only Nav1.8-associated polymorphism directly linked to a clinical condition associated with altered visceral pain perception that has also had its physiological impact on the channel described (Table 1).
In a study involving several hundred subjects, three separate SCN10A polymorphisms (e.g., A2884G, C3218T and T3275C) were associated with reduced risk for the visceral hypersensitivity disorders functional dyspepsia, epigastric pain syndrome and post-prandial distress syndrome (Arisawa et al., 2013). It is not clear what physiological effects each of these variants have on Nav1.8, or whether they impact other disorders associated with visceral pain (Table 1).
Targeting NaV1.8 for pain modulation
Considering the unique nature of NaV1.8, it is not surprising that it has served as an increasingly popular target for the development of analgesics. In Table 2, we provide an overview of the agents that have been developed for that purpose. In 2007, a small molecule pore blocker, A-803467 was discovered through a trial and error series of experiments that looked to mimic the blocking of TTX-R currents in rat DRG neurons. The authors identified this furanamide molecule and found that it selectively blocks Nav1.8 by suppressing spontaneous action potentials in vitro. A-803467 has also demonstrated the ability to reduce activity of spinal dorsal horn neurons in animal models of nerve injury (Jarvis et al., 2007). Notably, however, A-803467 exhibited relatively poor oral pharmacokinetics in this study, raising questions about how easy it would be to administer in humans. Separately, Liu et al. provided intraperitoneal injections of A-803467 to Nav1.8 wild type and knockout mice before they received an injection of atropine. The knockout mice and mice that received A-80346 exhibited reduced response to atropine (i.e., reduced rise in heart rate) compared to wild-type mice that did not receive A-803467. These findings suggested that there are potentially concomitant cardiac effects from this agent that might complicate it is application in humans (Liu et al., 2020). In 2010, A-887826, was developed (based upon the design of A-803467), and it demonstrated an ability to inactivate TTX-resistant currents and reduce neuropathic tactical allodynia in rats (Zhang et al., 2010).
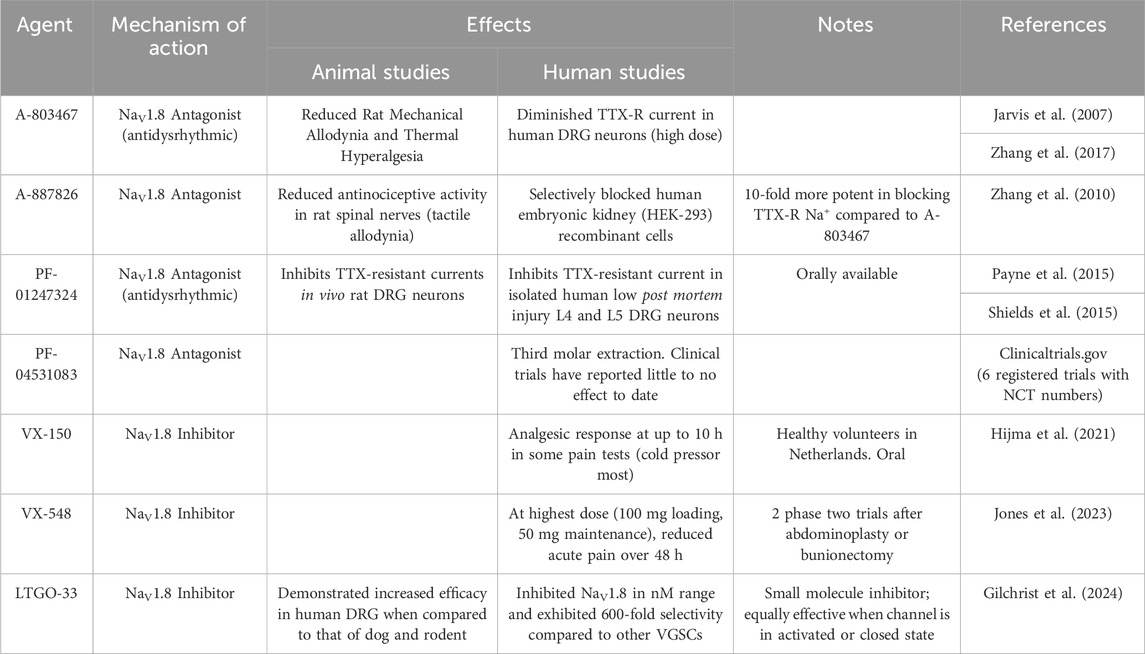
Table 2. Selective Agents for NaV1.8. DRG = dorsal root ganglion, TTX = tetrodotoxin, VGSC = voltage-gated sodium channel.
In 2015, Payne et al. reported that PF-01247324, another purported selective Nav1.8 antagonist, was able to inhibit TTX-resistant currents in isolated human DRG neurons and in vivo rat DRG neurons. They also found that this agent exhibited significant selectivity over Nav1.5 (another TTX-resistant channel) and TTX-sensitive channels. Of note, unlike studies of A-803467, PF-01247324 reduced nociception in the formalin test. These findings supported the role that Nav1.8 has in inflammatory and neuropathic pain (Payne et al., 2015). As described above, previous studies have also reported that Nav1.8 may be expressed in the cerebellum during specific disease states (e.g., MS) (Black et al., 1999; Black et al., 2000; Renganathan et al., 2001). Shields et al. demonstrated that PF-01247324 led to diminished cerebellar deficits when administered in mice with autoimmune encephalitis (a model of MS) (Shields et al., 2015). This revealed the potential for NaV1.8-directed therapies in MS and other related disorders, but also raised questions regarding its potential to result in centrally-mediated adverse effects. PF-04531083, a compound structurally similar to PF-01247324, has been the subject of a phase 2 clinical trial investigating its impact on pain after dental surgery. Thus far, no significant analgesic efficacy has been reported.
In 2021, a phase one trial was conducted which compared VX-150, an orally bioavailable pro-drug, to placebo in a two-way crossover study to evaluate the analgesic effects of VX-150 in healthy adult males. This was the first human experimental pain study using a selective NaV1.8 inhibitor. This study demonstrated that VX-150 influenced cold pressor pain thresholds, with no reported significant adverse effects to patient safety (Hijma et al., 2021). More recently, VX-548 was studied in two phase two trials (evaluating its effects on pain in patients who had undergone bunionectomy or abdominoplasty surgeries) and it was found that, at higher doses, it significantly reduced reported pain scores (Jones et al., 2023). Of note, however, there were multiple reports of headache and constipation. Most recently, Gilchrist et al. reported the development of a new selective small molecule inhibitor of NaV1.8, LTGO-33 (Gilchrist et al., 2024). This agent demonstrated high selectivity for NaV1.8 when compared to other VGSCs (reportedly 600-fold relative potency). LTGO-33 is also apparently channel state independent. demonstrating similar effects whether the channel is in an inactivated or closed state. Finally, LTGO-33 also demonstrated some degree of species specificity, working most effectively in human dorsal root ganglia when compared to those of other mammalian species.
There is also evidence that other existing medications (including some agents already utilized for their analgesic properties) may target directly or indirectly Nav1.8 as well. Tanezumab, a humanized monoclonal anti-NGF antibody, has been shown to reduce expression of Nav1.8 positive neurons and result in relative hypoalgesia in animal models, presumably through its effects on inflammatory pathways (Hoffman et al., 2011). Amitriptyline has demonstrated efficacy in reducing chemotherapy-induced neuropathic pain, and patch clamp testing suggests that, when it is applied to isolated DRG neurons, it diminishes activity of Nav1.8 (Genevois et al., 2021). The cannabinoid anandamide has been shown to reduce Nav1.8 associated currents in cell models (Okura et al., 2014). Ekins et al. used machine learning to generate a list of potential Nav1.8 inhibiting compounds from the Prestwick library, which included dihydropyridine calcium channel antagonists (Ekins et al., 2019).
Current limitations and future directions
Considering the findings shared above, there are many reasons to be hopeful when considering the potential of Nav1.8 to provide new, more effective, and potentially safer methods for managing pain, particularly visceral pain. However, before that full potential is realized, there are still multiple challenges that need to be addressed in order to better understand the physiology of Nav1.8 and to determine how best to target it in this context. For example, none of the agents outlined above have been specifically examined to assess their individual impacts on visceral pain perception. Additional pre-clinical and clinical studies dedicated to evaluating the impact of one or more of these medications on visceral pain perception will be essential in order to determine if they can be used safely and effectively in conditions associated with acute and/or chronic abdominal pain. Pending the outcomes of such studies, further investigation evaluating the optimal drug delivery methods for management of disorders associated with visceral pain may also be necessary (i.e., comparing the efficacy of oral vs. subcutaneous vs. other formulations). This issue would become particularly relevant if there are future attempts to target channels specifically associated with visceral sensory nerves (an important hypothetical to consider if further studies indicate that there are significant adverse effects related to system-wide administration). Beyond this, while several antibodies directed against epitopes on the rodent forms of the Nav1.8 channel exist, the current options result in inconsistent results. There is also a relative paucity of proven antibodies targeting the human channel. Additionally, until recently, there have been relatively few truly exclusive pharmacological agents that only target Nav1.8. This has historically made it challenging to tease out the characteristics that are truly unique to this channel, particularly when considering the other co-habitant TTX-resistant VGSCs, such as Nav1.5 and Nav1.9. Hopefully, the newer agents will help to refine our knowledge in this regard. However, this also highlights the fact that, while advancements in Nav1.8-targeting medications have been made, we are not ready to use any of them in the clinical setting. Finally, another challenge that has complicated our assessment of Nav1.8 in the past has the been the relative lack of specific models that reliably emulate the human channel and its physiology in both health and pathological conditions. Even when evaluating human versions of Nav1.8 channels, if they are not tested in human cells or tissues, significant differences in biophysical assessments may occur. Future testing should incorporate human models (e.g., iPSC-derived or cadaveric neurons) whenever possible to more accurately recapitulate human physiology.
Conclusion
Many lines of evidence, including from both animal- and human-oriented studies, reinforce the fact that Nav1.8 represents a very promising target for management of pain, including visceral pain. Nav1.8 appears to be uniquely situated to help target pain-related signal transmission, due to its relatively focused expression and critical function on dorsal root ganglion neurons. More refined studies are necessary to clarify the exact role and impact that this channel has in relation to pain, particularly in humans. Additionally, further work needs to be done to develop pharmacological agents and techniques that are more selectively targeted for Nav1.8. In time, as we improve our ability to manipulate this channel and harness its functionality, we will have an opportunity to provide patients with novel, more potent and safer analgesic options.
Author contributions
JH: Writing–review and editing, Writing–original draft. SD: Writing–review and editing. PJ: Writing–review and editing. AO: Writing–review and editing. KV: Writing–review and editing. VR-V: Writing–review and editing. MC: Writing–review and editing, Conceptualization, Writing–original draft.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by the Peter and Marshia Carlino Career Development Professorship in Medicine, the Margot E. Walrath Career Development Professorship in Gastroenterology and NIH NIDDK R01 DK122364.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahmad, S., Tirilomis, P., Pabel, S., Dybkova, N., Hartmann, N., Molina, C. E., et al. (2019). The functional consequences of sodium channel Na(V) 1.8 in human left ventricular hypertrophy. Esc. Heart Fail 6, 154–163. doi:10.1002/ehf2.12378
Akopian, A. N., Sivilotti, L., and Wood, J. N. (1996). A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature 379, 257–262. doi:10.1038/379257a0
Akopian, A. N., Souslova, V., England, S., Okuse, K., Ogata, N., Ure, J., et al. (1999). The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat. Neurosci. 2, 541–548. doi:10.1038/9195
Arisawa, T., Tahara, T., Shiroeda, H., Minato, T., Matsue, Y., Saito, T., et al. (2013). Genetic polymorphisms of SCN10A are associated with functional dyspepsia in Japanese subjects. J. Gastroenterol. 48, 73–80. doi:10.1007/s00535-012-0602-3
Bennett, D. L., Clark, A. J., Huang, J., Waxman, S. G., and Dib-Hajj, S. D. (2019). The role of voltage-gated sodium channels in pain signaling. Physiol. Rev. 99, 1079–1151. doi:10.1152/physrev.00052.2017
Beyak, M. J., Ramji, N., Krol, K. M., Kawaja, M. D., and Vanner, S. J. (2004). Two TTX-resistant Na+ currents in mouse colonic dorsal root ganglia neurons and their role in colitis-induced hyperexcitability. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G845–G855. doi:10.1152/ajpgi.00154.2004
Black, J. A., Dib-Hajj, S., Baker, D., Newcombe, J., Cuzner, M. L., and Waxman, S. G. (2000). Sensory neuron-specific sodium channel SNS is abnormally expressed in the brains of mice with experimental allergic encephalomyelitis and humans with multiple sclerosis. Proc. Natl. Acad. Sci. U. S. A. 97, 11598–11602. doi:10.1073/pnas.97.21.11598
Black, J. A., Fjell, J., Dib-Hajj, S., Duncan, I. D., O'connor, L. T., Fried, K., et al. (1999). Abnormal expression of SNS/PN3 sodium channel in cerebellar Purkinje cells following loss of myelin in the taiep rat. Neuroreport 10, 913–918. doi:10.1097/00001756-199904060-00004
Blair, N. T., and Bean, B. P. (2002). Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J. Neurosci. 22, 10277–10290. doi:10.1523/JNEUROSCI.22-23-10277.2002
Caldwell, J. H., Schaller, K. L., Lasher, R. S., Peles, E., and Levinson, S. R. (2000). Sodium channel Na(v)1.6 is localized at nodes of ranvier, dendrites, and synapses. Proc. Natl. Acad. Sci. U. S. A. 97, 5616–5620. doi:10.1073/pnas.090034797
Casini, S., Marchal, G. A., Kawasaki, M., Nariswari, F. A., Portero, V., Van Den Berg, N. W. E., et al. (2019). Absence of functional Na(v)1.8 channels in non-diseased atrial and ventricular cardiomyocytes. Cardiovasc Drugs Ther. 33, 649–660. doi:10.1007/s10557-019-06925-6
Catterall, W. (1980). Neurotoxins that act on voltage-sensitive sodium channels in excitable membranes. Ann. Rev. Pharmacol. Toxicol. 20, 15–43. doi:10.1146/annurev.pa.20.040180.000311
Catterall, W. A. (2000). From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26, 13–25. doi:10.1016/s0896-6273(00)81133-2
Catterall, W. A., Goldin, A. L., and Waxman, S. G. (2005a). International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 57, 397–409. doi:10.1124/pr.57.4.4
Catterall, W. A., Perez-Reyes, E., Snutch, T. P., and Striessnig, J. (2005b). International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 57, 411–425. doi:10.1124/pr.57.4.5
Chen, J., Winston, J. H., and Sarna, S. K. (2013). Neurological and cellular regulation of visceral hypersensitivity induced by chronic stress and colonic inflammation in rats. Neuroscience 248, 469–478. doi:10.1016/j.neuroscience.2013.06.024
Cheng, X., Choi, J. S., Waxman, S. G., and Dib-Hajj, S. D. (2021). Mini-review - sodium channels and beyond in peripheral nerve disease: modulation by cytokines and their effector protein kinases. Neurosci. Lett. 741, 135446. doi:10.1016/j.neulet.2020.135446
Coates, M. D., Vrana, K. E., and Ruiz-Velasco, V. (2019). The influence of voltage-gated sodium channels on human gastrointestinal nociception. Neurogastroenterol. Motil. 31, e13460. doi:10.1111/nmo.13460
Cummins, T. R., Dib-Hajj, S. D., Black, J. A., Akopian, A. N., Wood, J. N., and Waxman, S. G. (1999). A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J. Neurosci. 19, RC43. doi:10.1523/JNEUROSCI.19-24-j0001.1999
De Lera Ruiz, M., and Kraus, R. L. (2015). Voltage-gated sodium channels: structure, function, Pharmacology, and clinical indications. J. Med. Chem. 58, 7093–7118. doi:10.1021/jm501981g
Dib-Hajj, S., Black, J. A., Cummins, T. R., and Waxman, S. G. (2002). NaN/Nav1.9: a sodium channel with unique properties. Trends Neurosci. 25, 253–259. doi:10.1016/s0166-2236(02)02150-1
Djouhri, L., Fang, X., Okuse, K., Wood, J. N., Berry, C. M., and Lawson, S. N. (2003). The TTX-resistant sodium channel Nav1.8 (SNS/PN3): expression and correlation with membrane properties in rat nociceptive primary afferent neurons. J. Physiol. 550, 739–752. doi:10.1113/jphysiol.2003.042127
Duan, G., Han, C., Wang, Q., Guo, S., Zhang, Y., Ying, Y., et al. (2016). A SCN10A SNP biases human pain sensitivity. Mol. Pain 12, 1744806916666083. doi:10.1177/1744806916666083
Dybkova, N., Ahmad, S., Pabel, S., Tirilomis, P., Hartmann, N., Fischer, T. H., et al. (2018). Differential regulation of sodium channels as a novel proarrhythmic mechanism in the human failing heart. Cardiovasc Res. 114, 1728–1737. doi:10.1093/cvr/cvy152
Ekins, S., Gerlach, J., Zorn, K. M., Antonio, B. M., Lin, Z., and Gerlach, A. (2019). Repurposing approved drugs as inhibitors of K(v)7.1 and Na(v)1.8 to treat pitt hopkins syndrome. Pharm. Res. 36, 137. doi:10.1007/s11095-019-2671-y
Faber, C. G., Lauria, G., Merkies, I. S., Cheng, X., Han, C., Ahn, H. S., et al. (2012). Gain-of-function Nav1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. U. S. A. 109, 19444–19449. doi:10.1073/pnas.1216080109
Gautron, L., Sakata, I., Udit, S., Zigman, J. M., Wood, J. N., and Elmquist, J. K. (2011). Genetic tracing of Nav1.8-expressing vagal afferents in the mouse. J. Comp. Neurol. 519, 3085–3101. doi:10.1002/cne.22667
Genevois, A. L., Ruel, J., Penalba, V., Hatton, S., Petitfils, C., Ducrocq, M., et al. (2021). Analgesic effects of topical amitriptyline in patients with chemotherapy-induced peripheral neuropathy: mechanistic insights from studies in mice. J. Pain 22, 440–453. doi:10.1016/j.jpain.2020.11.002
Gilchrist, J. M., Yang, N. D., Jiang, V., and Moyer, B. D. (2024). Pharmacologic characterization of LTGO-33, a selective small molecule inhibitor of the voltage-gated Sodium Channel Na(V)1.8 with a unique mechanism of action. Mol. Pharmacol. 105, 233–249. doi:10.1124/molpharm.123.000789
Goldin, A. L., Barchi, R. L., Caldwell, J. H., Hofmann, F., Howe, J. R., Hunter, J. C., et al. (2000). Nomenclature of voltage-gated sodium channels. Neuron 28, 365–368. doi:10.1016/s0896-6273(00)00116-1
Gonzalez-Lopez, E., Imamura Kawasawa, Y., Walter, V., Zhang, L., Koltun, W. A., Huang, X., et al. (2018). Homozygosity for the SCN10A polymorphism rs6795970 is associated with hypoalgesic inflammatory bowel disease phenotype. Front. Med. (Lausanne) 5, 324. doi:10.3389/fmed.2018.00324
Hameed, S. (2019). Na(v)1.7 and Na(v)1.8: role in the pathophysiology of pain. Mol. Pain 15, 1744806919858801. doi:10.1177/1744806919858801
Han, C., Vasylyev, D., Macala, L. J., Gerrits, M. M., Hoeijmakers, J. G., Bekelaar, K. J., et al. (2014). The G1662S NaV1.8 mutation in small fibre neuropathy: impaired inactivation underlying DRG neuron hyperexcitability. J. Neurol. Neurosurg. Psychiatry 85, 499–505. doi:10.1136/jnnp-2013-306095
Hijma, H. J., Siebenga, P. S., De Kam, M. L., and Groeneveld, G. J. (2021). A phase 1, randomized, double-blind, placebo-controlled, crossover study to evaluate the pharmacodynamic effects of VX-150, a highly selective NaV1.8 inhibitor, in healthy male adults. Pain Med. 22, 1814–1826. doi:10.1093/pm/pnab032
Hillsley, K., Lin, J. H., Stanisz, A., Grundy, D., Aerssens, J., Peeters, P. J., et al. (2006). Dissecting the role of sodium currents in visceral sensory neurons in a model of chronic hyperexcitability using Nav1.8 and Nav1.9 null mice. J. Physiol. 576, 257–267. doi:10.1113/jphysiol.2006.113597
Hoffman, E. M., Zhang, Z., Anderson, M. B., Schechter, R., and Miller, K. E. (2011). Potential mechanisms for hypoalgesia induced by anti-nerve growth factor immunoglobulin are identified using autoimmune nerve growth factor deprivation. Neuroscience 193, 452–465. doi:10.1016/j.neuroscience.2011.06.069
Hu, J., Song, Z. Y., Zhang, H. H., Qin, X., Hu, S., Jiang, X., et al. (2016). Colonic hypersensitivity and sensitization of voltage-gated sodium channels in primary sensory neurons in rats with diabetes. J. Neurogastroenterol. Motil. 22, 129–140. doi:10.5056/jnm15091
Hu, S., Xiao, Y., Zhu, L., Li, L., Hu, C. Y., Jiang, X., et al. (2013). Neonatal maternal deprivation sensitizes voltage-gated sodium channel currents in colon-specific dorsal root ganglion neurons in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 304, G311–G321. doi:10.1152/ajpgi.00338.2012
Huang, X., Jin, X., Huang, G., Huang, J., Wu, T., Li, Z., et al. (2022). Structural basis for high-voltage activation and subtype-specific inhibition of human Nav1.8. Proc. Natl. Acad. Sci. 119, e2208211119. doi:10.1073/pnas.2208211119
Jarvis, M. F., Honore, P., Shieh, C. C., Chapman, M., Joshi, S., Zhang, X. F., et al. (2007). A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. U. S. A. 104, 8520–8525. doi:10.1073/pnas.0611364104
Jones, J., Correll, D. J., Lechner, S. M., Jazic, I., Miao, X., Shaw, D., et al. (2023). Selective inhibition of Na(V)1.8 with VX-548 for acute pain. N. Engl. J. Med. 389, 393–405. doi:10.1056/NEJMoa2209870
Kanellopoulos, A. H., and Matsuyama, A. (2016). Voltage-gated sodium channels and pain-related disorders. Clin. Sci. (Lond) 130, 2257–2265. doi:10.1042/cs20160041
King, D. E., Macleod, R. J., and Vanner, S. J. (2009). Trinitrobenzenesulphonic acid colitis alters Na 1.8 channel expression in mouse dorsal root ganglia neurons. Neurogastroenterol. Motil. 21, 880–e64. doi:10.1111/j.1365-2982.2009.01279.x
Kostyuk, P. G., Veselovsky, N. S., Fedulova, S. A., and Tsyndrenko, A. Y. (1981). Ionic currents in the somatic membrane of rat dorsal root ganglion neurons-III. Potassium currents. Neuroscience 6, 2439–2444. doi:10.1016/0306-4522(81)90090-7
Laird, J. M., Souslova, V., Wood, J. N., and Cervero, F. (2002). Deficits in visceral pain and referred hyperalgesia in Nav1.8 (SNS/PN3)-null mice. J. Neurosci. 22, 8352–8356. doi:10.1523/JNEUROSCI.22-19-08352.2002
Lin, Y. M., Fu, Y., Winston, J., Radhakrishnan, R., Sarna, S. K., Huang, L. M., et al. (2017). Pathogenesis of abdominal pain in bowel obstruction: role of mechanical stress-induced upregulation of nerve growth factor in gut smooth muscle cells. Pain 158, 583–592. doi:10.1097/j.pain.0000000000000797
Liu, B., Li, N., Zhang, J., Liu, Y., Zhang, M., Hong, Y., et al. (2020). The role of voltage-gated Sodium Channel 1.8 in the effect of atropine on heart rate: evidence from a retrospective clinical study and mouse model. Front. Pharmacol. 11, 1163. doi:10.3389/fphar.2020.01163
Ma, K., Zhou, Q. H., Chen, J., Du, D. P., Ji, Y., and Jiang, W. (2008). TTX-R Na+ current-reduction by celecoxib correlates with changes in PGE(2) and CGRP within rat DRG neurons during acute incisional pain. Brain Res. 1209, 57–64. doi:10.1016/j.brainres.2008.02.096
Mannikko, R., Wong, L., Tester, D. J., Thor, M. G., Sud, R., Kullmann, D. M., et al. (2018). Dysfunction of NaV1.4, a skeletal muscle voltage-gated sodium channel, in sudden infant death syndrome: a case-control study. Lancet 391, 1483–1492. doi:10.1016/S0140-6736(18)30021-7
Matthews, E. A., Wood, J. N., and Dickenson, A. H. (2006). Na(v) 1.8-null mice show stimulus-dependent deficits in spinal neuronal activity. Mol. Pain 2, 5. doi:10.1186/1744-8069-2-5
Noland, C. L., Chua, H. C., Kschonsak, M., Heusser, S. A., Braun, N., Chang, T., et al. (2022). Structure-guided unlocking of NaX reveals a non-selective tetrodotoxin-sensitive cation channel. Nat. Commun. 13, 1416. doi:10.1038/s41467-022-28984-4
Okura, D., Horishita, T., Ueno, S., Yanagihara, N., Sudo, Y., Uezono, Y., et al. (2014). The endocannabinoid anandamide inhibits voltage-gated sodium channels Nav1.2, Nav1.6, Nav1.7, and Nav1.8 in Xenopus oocytes. Anesth. Analg. 118, 554–562. doi:10.1213/ANE.0000000000000070
Payne, C. E., Brown, A. R., Theile, J. W., Loucif, A. J., Alexandrou, A. J., Fuller, M. D., et al. (2015). A novel selective and orally bioavailable Nav 1.8 channel blocker, PF-01247324, attenuates nociception and sensory neuron excitability. Br. J. Pharmacol. 172, 2654–2670. doi:10.1111/bph.13092
Peeters, P. J., Aerssens, J., Hoogt, R. D., Stanisz, A., Göhlmann, H. W., Hillsley, K., et al. (2006). Molecular profiling of murine sensory neurons in the nodose and dorsal root ganglia labeled from the peritoneal cavity. Physiol. Genomics 24, 252–263. doi:10.1152/physiolgenomics.00169.2005
Renganathan, M., Cummins, T., Hormuzdiar, W., Black, J., and Waxman, S. (2000). Nitric oxide is an autocrine regulator of Na + currents in axotomized C-type DRG neurons. J. Neurophysiol. 83, 2431–2442. doi:10.1152/jn.2000.83.4.2431
Renganathan, M., Cummins, T. R., and Waxman, S. G. (2001). Contribution of Na(v)1.8 sodium channels to action potential electrogenesis in DRG neurons. J. Neurophysiol. 86, 629–640. doi:10.1152/jn.2001.86.2.629
Riol-Blanco, L., Ordovas-Montanes, J., Perro, M., Naval, E., Thiriot, A., Alvarez, D., et al. (2014). Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature 510, 157–161. doi:10.1038/nature13199
Roza, C., Laird, J. M., Souslova, V., Wood, J. N., and Cervero, F. (2003). The tetrodotoxin-resistant Na+ channel Nav1.8 is essential for the expression of spontaneous activity in damaged sensory axons of mice. J. Physiol. 550, 921–926. doi:10.1113/jphysiol.2003.046110
Shields, S. D., Ahn, H. S., Yang, Y., Han, C., Seal, R. P., Wood, J. N., et al. (2012). Nav1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain 153, 2017–2030. doi:10.1016/j.pain.2012.04.022
Shields, S. D., Butt, R. P., Dib-Hajj, S. D., and Waxman, S. G. (2015). Oral administration of PF-01247324, a subtype-selective Nav1.8 blocker, reverses cerebellar deficits in a mouse model of multiple sclerosis. PLOS ONE 10, e0119067. doi:10.1371/journal.pone.0119067
Stevens, M., Peigneur, S., and Tytgat, J. (2011). Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol. 2, 71. doi:10.3389/fphar.2011.00071
Su, X., Wachtel, R. E., and Gebhart, G. F. (1999). Capsaicin sensitivity and voltage-gated sodium currents in colon sensory neurons from rat dorsal root ganglia. Am. J. Physiol. 277, G1180–G1188. doi:10.1152/ajpgi.1999.277.6.G1180
Tenza-Ferrer, H., Collodetti, M., Nicolau, E. S., Birbrair, A., Magno, L. a. V., and Romano-Silva, M. A. (2022). Transiently Nav1.8-expressing neurons are capable of sensing noxious stimuli in the brain. Front. Cell Neurosci. 16, 933874. doi:10.3389/fncel.2022.933874
Verkerk, A. O., Remme, C. A., Schumacher, C. A., Scicluna, B. P., Wolswinkel, R., De Jonge, B., et al. (2012). Functional Nav1.8 channels in intracardiac neurons: the link between SCN10A and cardiac electrophysiology. Circ. Res. 111, 333–343. doi:10.1161/circresaha.112.274035
Waxman, S. G. (2005). Cerebellar dysfunction in multiple sclerosis: evidence for an acquired channelopathy. Prog. Brain Res. 148, 353–365. doi:10.1016/S0079-6123(04)48028-5
Yu, F. H., and Catterall, W. A. (2003). Overview of the voltage-gated sodium channel family. Genome Biol. 4, 207. doi:10.1186/gb-2003-4-3-207
Yu, F. H., and Catterall, W. A. (2004). The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci. STKE 2004, re15. doi:10.1126/stke.2532004re15
Yu, F. H., Westenbroek, R. E., Silos-Santiago, I., Mccormick, K. A., Lawson, D., Ge, P., et al. (2003). Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. J. Neurosci. 23, 7577–7585. doi:10.1523/jneurosci.23-20-07577.2003
Zhang, X., Priest, B. T., Belfer, I, and Gold, M. S. (2017). Voltage-gated Na+ currents in human dorsal root ganglion neurons. Elife. 6, e23235. doi:10.7554/eLife.23235
Zhang, X., Priest, B. T., Belfer, I., and Gold, M. S. (2017). Voltage-gated Na+ currents in human dorsal root ganglion neurons. Elife 6. doi:10.7554/eLife.23235
Zhang, X. F., Shieh, C. C., Chapman, M. L., Matulenko, M. A., Hakeem, A. H., Atkinson, R. N., et al. (2010). A-887826 is a structurally novel, potent and voltage-dependent Na(v)1.8 sodium channel blocker that attenuates neuropathic tactile allodynia in rats. Neuropharmacology 59, 201–207. doi:10.1016/j.neuropharm.2010.05.009
Keywords: Nav1.8, pain, visceral, abdominal, voltage-gated sodium channel
Citation: Heinle JW, Dalessio S, Janicki P, Ouyang A, Vrana KE, Ruiz-Velasco V and Coates MD (2024) Insights into the voltage-gated sodium channel, NaV1.8, and its role in visceral pain perception . Front. Pharmacol. 15:1398409. doi: 10.3389/fphar.2024.1398409
Received: 12 March 2024; Accepted: 29 April 2024;
Published: 23 May 2024.
Edited by:
Sulayman D. Dib-Hajj, Yale University, United StatesReviewed by:
Shannon Shields, GenEdit Company, United StatesSidharth Tyagi, Yale University, United States
Copyright © 2024 Heinle, Dalessio, Janicki, Ouyang, Vrana, Ruiz-Velasco and Coates. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew D. Coates, bWNvYXRlc0BwZW5uc3RhdGVoZWFsdGgucHN1LmVkdQ==