 Roberta Leone
Roberta Leone Cecilia Zuglian
Cecilia Zuglian Riccardo Brambilla1,2*
Riccardo Brambilla1,2* Ilaria Morella
Ilaria Morella- 1Università di Pavia, Dipartimento di Biologia e Biotecnologie “Lazzaro Spallanzani”, Pavia, Italy
- 2Cardiff University, School of Biosciences, Neuroscience and Mental Health Innovation Institute, Cardiff, United Kingdom
Neurodevelopmental disorders (NDDs) include a broad spectrum of pathological conditions that affect >4% of children worldwide, share common features and present a variegated genetic origin. They include clinically defined diseases, such as autism spectrum disorders (ASD), attention-deficit/hyperactivity disorder (ADHD), motor disorders such as Tics and Tourette’s syndromes, but also much more heterogeneous conditions like intellectual disability (ID) and epilepsy. Schizophrenia (SCZ) has also recently been proposed to belong to NDDs. Relatively common causes of NDDs are copy number variations (CNVs), characterised by the gain or the loss of a portion of a chromosome. In this review, we focus on deletions and duplications at the 16p11.2 chromosomal region, associated with NDDs, ID, ASD but also epilepsy and SCZ. Some of the core phenotypes presented by human carriers could be recapitulated in animal and cellular models, which also highlighted prominent neurophysiological and signalling alterations underpinning 16p11.2 CNVs-associated phenotypes. In this review, we also provide an overview of the genes within the 16p11.2 locus, including those with partially known or unknown function as well as non-coding RNAs. A particularly interesting interplay was observed between MVP and MAPK3 in modulating some of the pathological phenotypes associated with the 16p11.2 deletion. Elucidating their role in intracellular signalling and their functional links will be a key step to devise novel therapeutic strategies for 16p11.2 CNVs-related syndromes.
Introduction
Neurodevelopmental disorders (NDDs) include conditions with a wide range of neuropsychiatric symptoms that are now believed to originate from alterations at the cortical and subcortical level during both prenatal and early postnatal brain development. Symptoms are highly variable, especially in the predominant idiopathic forms, but the core components are normally associated to intellectual disability (ID), autism spectrum disorder (ASD), attention-deficit/hyperactivity disorder (ADHD) and epilepsy. Associated psychiatric symptoms may include depression and anxiety, speech delays and in the most severe cases, schizophrenia and/or bipolar disorder. In syndromic forms, non-psychiatric symptoms may also occur, including metabolic dysfunctions, alterations in brain size, and cardio-facial-cutaneous malformations (Mitchell, 2011).
We know from previous literature (revised in (Backhausen et al., 2022) that cortical and subcortical grey matter formation follows a structured developmental pattern, presenting an initial increase in childhood that is followed by a decrease during adolescence. This developmental trajectory supports the mechanisms of reinforcement of important connection through learning and the elimination of redundant synapses during maturation, and it is differently affected in NDDs. For instance, while ADHD is characterised by a general reduction in the volume and surface area of basal ganglia and prefrontal cortex (PFC) (Shaw et al., 2014; Hoogman et al., 2017), children with ID present a general decrease in brain size throughout childhood and adolescence, but prefrontal and cingulate areas present higher volumes than healthy peers (Ma et al., 2021). A generalized increase in frontal cortical volumes has also been described in ASD patients within the first 2 years of age (Courchesne et al., 2011): a tendency that reverts in adulthood, when ASD brains show a higher rate of structural decline (Wallace and Rogers, 2010; Courchesne et al., 2011; Lange et al., 2015), although maintaining an abnormal growth rate in the basal ganglia (Langen et al., 2009; Wegiel et al., 2014).
Interestingly, in Schizophrenia (SCZ), a reduction in cortical volume is observed, mostly in the frontal, prefrontal and temporal lobes, accompanied by a reduction in the volume of basal ganglia and a more peculiar enlargement of the ventricles (Shenton et al., 2001; Fornito et al., 2009; Kuo and Pogue-Geile, 2019; Cai et al., 2022). While the onset of the symptoms is delayed in SCZ compared to other NDDs, morphological abnormalities have been described prior to symptoms appearance and become more severe over time, thus supporting the neurodevelopmental origin of this disease (Rund, 2009; Owen et al., 2011).
Although brain development differently deviates from its physiological developmental trajectory in different NDDs, affected areas recurrently belong to the thalamus-striatal-prefrontal axis (Di Martino et al., 2011; Prat et al., 2016; Roy et al., 2021). Morphological brain abnormalities constitute an endophenotype common to all NDDs, accompanied by a strong comorbidity of other symptoms, including cognitive, motor and social impairments (Eberhard et al., 2022), as well as seizures (Chow et al., 2019; Watkins et al., 2022) and sex biases (Santos et al., 2022). An endophenotypes can be caused by different genetic variations which affect one or more neural circuits, independently leading to an overall effect that is common to multiple clinical entities (Cannon and Keller, 2006). Results from genome-wide association studies described a strong correlation between NDDs and single genomic variations in functionally related sets of genes involved in neurodevelopmental processes, synaptic plasticity, learning and memory (Cross-Disorder Group of the Psychiatric Genomics, 2013; Ripke et al., 2013; Doherty and Owen, 2014; Fromer et al., 2014). These shared risk factors are responsible not only for symptoms comorbidity among patients with different diagnoses of NDDs, but they also increase the risk of developing NDDs in families where the same or a different NDD is already present (Larsson et al., 2005; Daniels et al., 2008; Sullivan et al., 2012; Larsson et al., 2013). However, it is very difficult to ascribe all the clinical manifestations of different NDDs to single gene variants, although several cases of direct causality do exist, such as FMR1 as a genetic cause of ASD (Fyke and Velinov, 2021), NRG1 and DISC1 for SCZ (Dahoun et al., 2017; Zhang et al., 2017) and FOXP2 in ADHD (Faraone and Larsson, 2019). Most likely, single genes found associated with NDDs should play major roles in one or more biological processes critical for brain development and function, including connectivity, synaptic transmission, and neuronal signalling.
Genetic causes of NDDs also include copy number variations (CNVs), rare genetic variants in which either deletion or duplication (less often triplication) of an entire chromosomal portion may occur (for a general review, see (Grayton et al., 2012). These chromosomal rearrangements may originate from genetic transmission to offspring or may be de-novo mutations and constitute a significant burden in the onset of NDDs (Sebat et al., 2007; Walsh et al., 2008; Sonderby et al., 2022). They include several genes of both known and unknown function, potentially interfering with multiple, interlinked, molecular pathways. Although CNVs are less common than single gene variations, they constitute a much stronger risk factor in the development of NDDs, with a penetrance from 10% to 100% in some cases (Kirov, 2015). Interestingly, penetrance of symptoms can be highly variable within the same CNV carrier population, with subjects with no notable phenotypes while others severely affected. Gene dosage does not usually help in understanding symptom severity, since opposite variations on the same genomic region often lead to similar phenotypes (Niarchou et al., 2019; Zarrei et al., 2019).
This genomic complexity represents a formidable challenge to understand the relative contribution of each gene, and its likely interactions with the nearby CNV genes. However, it strongly limits our understanding of the pathological mechanisms as well as the devising of effective therapies. Furthermore, non-coding RNAs that are commonly found in CNVs may also be relevant for the onset of NDD phenotypes.
In this review, we will focus our attention on the 16p11.2 CNV (Rein and Yan, 2020). Deletions (DEL) and duplications (DUP) at the human 16p11.2 breakpoints (BP) four to five chromosomal region account for leading causes of neurodevelopmental disorders and intellectual disabilities worldwide, with an estimated of three in 10,000 people for each syndrome. Individuals with deletions or duplications are diagnosed with intellectual disabilities and psychiatric disorders with a likelihood of 50% and 60%, respectively (Niarchou et al., 2019).
CNVs on 16p11.2 are most frequently associated with ASD and other NDDs than do other CNVs. Data pooled from different studies found a frequency of 0.35%, 0.21%, 0.17%, and 0.17% for 16p11.2 deletions, 16p11.2 duplications, 1q21.1 duplications, and 15q11.2–13.1 duplications, respectively, in the onset of ASD, ID, ADHD (Mollon et al., 2023). It is noteworthy that deletions on the chromosomal region 22q11.2 also constitute a strong risk factor for SCZ and, to a less extent, ASD (Bassett et al., 2003). Interestingly, some genes on 16p11.2 and 22q11.2 regions belong to common molecular pathways. For instance, ERK1 is located on 16p11.2, ERK2 on 22q11.2, and the reciprocal differential expression level for these two effectors of the MAPK signalling cascade has previously been associated with cognitive impairments and neurodevelopmental deficits (Mazzucchelli et al., 2002; Pucilowska et al., 2018; Indrigo et al., 2023). Other examples include the presence of different members of the T-Box family (TBX6 in 16p11.2, TBX1 in 22q11.2), implicated in embryogenesis and development, and members of the H3.3 histone chaperone complex HIRA (HIRA on 22q11.2, HIRIP3 on 16p11.2) responsible for chromatin remodelling and gene transcription.
Behavioural phenotypes, often observed in both DEL and DUP carriers, are in the domain of speech, intellectual disability, and autistic traits, with DUP carriers bearing greater cognitive impairments in full-scale IQ, verbal IQ, and performance IQ compared with DEL carriers (Chawner et al., 2021). Importantly, both DEL and DUP carriers have a significant risk of developing seizures, suggesting that an altered brain development may lead to changes in the excitation/inhibition balance. In addition, DUP carriers may be susceptible to psychosis and bipolar disorder that are generally absent in DEL carriers (Rein and Yan, 2020).
Here, we describe the pathological phenotypes, and we review the recent literature on cellular and animal models of 16p11.2 CNVs. We subsequently hypothesize a link between the genes in the 16p11.2 region and their effect on specific molecular pathways. We also suggest potential interventions to rescue the deficits affecting 16p11.2 CNVs carriers.
Clinical profile of 16p11.2 CNVs
CNVs on 16p11.2 chromosomic region were first correlated to autism spectrum disorder after a large study on a patients’ database in 2008, with a frequency of three in 10000 (Weiss et al., 2008). Deletions (DEL) occur de novo in most cases (71%), while duplications (DUP) are mainly familial (D'Angelo et al., 2016). Variations in the 16p11.2 locus lead to heterogeneous clinical effects, including intellectual disability (ID), autism (ASD), attention deficit hyperactivity disorder (ADHD), epilepsy, language and motor delays (Weiss et al., 2008; Shinawi et al., 2010; Hanson et al., 2015; D'Angelo et al., 2016; Green Snyder et al., 2016; Steinman et al., 2016; Niarchou et al., 2019; Rein and Yan, 2020), which appear in different proportions between DEL and DUP patients (reviewed in (Oliva-Teles et al., 2020)). Moreover, duplications constitute an additional risk factor for schizophrenia (SCZ) (McCarthy et al., 2009; Shinawi et al., 2010; Niarchou et al., 2019; Zarrei et al., 2019; Rein and Yan, 2020).
In the context of 16p11.2 CNVs, ASD and SCZ are often considered as opposite conditions of dosage-dependent modifications of gene expression (Crespi et al., 2010). In addition, cognitive studies showed that the 16p11.2 deletion is strongly associated with impaired verbal IQ, deficits in verbal letter and category fluency tests, consistently to autism symptomatology, while duplication affects spatial working memory and executive functions (Stefansson et al., 2014), as observed in schizophrenic patients (Park and Holzman, 1992). In other cases, 16p11.2 deletion and duplication similarly affect cognition, but with different severity degrees. For instance, duplications are usually characterised by higher variance, suggesting the possible contribution of additional familial factors (D'Angelo et al., 2016; Niarchou et al., 2019). The body mass index (BMI) and the brain size are also differently affected in DEL and DUP carriers, and negatively correlate with gene dosage (McCarthy et al., 2009; Jacquemont et al., 2011; Zufferey et al., 2012; Qureshi et al., 2014; Martin-Brevet et al., 2018), as well as facial dysmorphisms (Shinawi et al., 2010). From a neuroanatomical point of view, patients affected by 16p11.2 CNVs display structural abnormalities similar to those described in NDDs. More specifically, magnetic resonance imaging (MRI) and diffusion tensor imaging (DTI) studies on DEL children reported increased white matter volume and fibre density, with reduced orientation dispersion in the callosum and internal/external capsules, compared to healthy controls (Owen et al., 2014). The opposite effect was observed in DUP carriers (Chang et al., 2016). Altogether, these observations are consistent with the ASD phenotype and other NDDs (Owen et al., 2014; Chang et al., 2016). Importantly, the observed changes in fibre density affect the development and function of connections between brain areas involved in language, locomotion, and socio-emotional behaviours (Maillard et al., 2024).
However, 16p11.2 DEL and DUP carriers also share common pathological phenotypes, such as epilepsy. This is typically observed during the first year of life, easily responds to antiepileptic medications, and usually decreases in severity or disappears during childhood (Shinawi et al., 2010).
Animal models of 16p11.2 deletion and duplication
The clinical profile associated with 16p11.2 CNVs is very heterogeneous and suggests the presence of multifactorial effects in determining the pathological phenotypes of 16p11.2 DEL and DUP carriers. The systematic evaluation of animal models carrying the 16p11.2 CNVs has allowed a deeper investigation of the cellular and molecular pathways involved in the pathophysiology of 16p11.2 CNVs, as well as the specific functions of the genes within the 16p11.2 locus. In the human genome, the 16p11.2 locus is a region of approximately 600kb, defined by breakpoints 4 and 5 (BP4-BP5), which is conserved on mouse syntenic region located on chromosome 7F3 (Rein and Yan, 2020). The BP4-BP5 common rearrangements encompass 27 unique protein coding-genes (see Table 1; Figure 1) and multiple copies of BOLA2/2B, SLX1A/1B, SULT1A3/4 and NPIP.
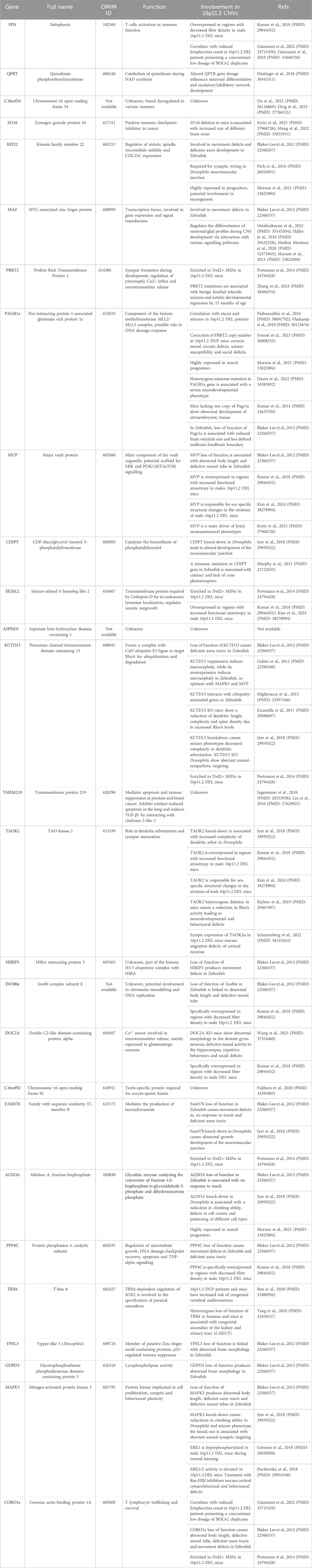
Table 1. List of genes within the 16p11.2 chromosomal region, relative functions, and involvement in the 16p11.2 CNVs.
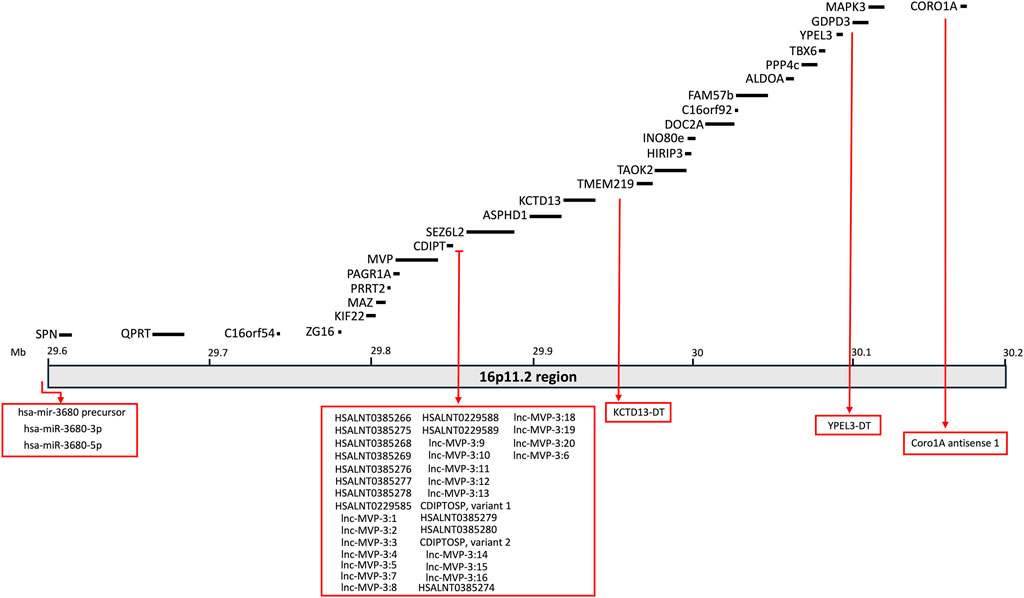
Figure 1. Schematic representation of the 16p11.2 locus containing 27 protein-coding genes. Non-coding RNAs mapping on this region are also listed in the rex boxes.
So far, 3 mouse models for 16p11.2 deletion (DEL) have been generated, carrying the heterozygous deletion in the Slx1b-Sept1 region (Mills model) (Horev et al., 2011), in the Coro1a-Spn region (Dolmetsch model) (Portmann et al., 2014) or in the Sult1a1-Spn region (Herault model) (Arbogast et al., 2016). Two mouse models for 16p11.2 duplication (DUP) are currently available, carrying the heterozygous duplication in the Slx1b-Sept1 region (Mills model) (Horev et al., 2011) or in the Sult1a1-Spn region (Herault model) (Arbogast et al., 2016). Despite the presence of similar phenotypes, these mouse models also show some differences which might be partly explained by the different targeting regions on chromosome 7, as highlighted in (Arbogast et al., 2016). Here and in Table 2, we summarize the main phenotypes observed in DEL and DUP animal models.
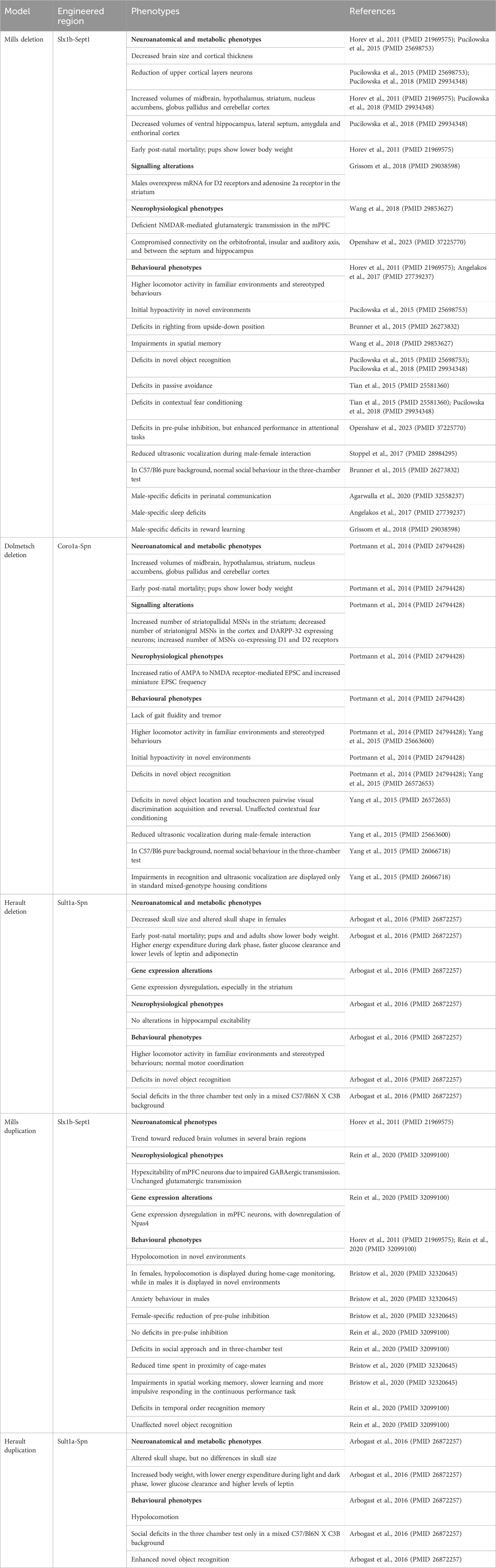
Table 2. Main phenotypes of 16p11.2 CNVs mouse models.
16p11.2 DEL mouse models
16p11.2 DEL mice display metabolic and neuroanatomical alterations
All DEL mouse models are affected by early post-natal mortality and their body weight is significantly lower than wild-type mice (Horev et al., 2011; Portmann et al., 2014; Arbogast et al., 2016). In Dolmetsch model, this effect could be corrected by improved nutrition and separation from wild-type littermates (Portmann et al., 2014). As adults, the body weight of DEL mice is similar to wild-type littermates (Horev et al., 2011) or reduced (Portmann et al., 2014; Pucilowska et al., 2015; Arbogast et al., 2016), showing decreased adiposity (Arbogast et al., 2016). This is in contrast with human findings where the 16p11.2 deletion has been associated with a highly penetrant type of obesity (Jacquemont et al., 2011). Herault model also displays a higher energy expenditure during the dark phase, a faster glucose clearance and lower blood levels of leptin and adiponectin (Arbogast et al., 2016).
Craniofacial dysmorphisms have been reported by (Arbogast et al., 2016), with decreased skull size and altered skull shape in the Herault DEL females. In contrast with humans (Qureshi et al., 2014), the brain size is modestly reduced in Mills DEL mice during early post-natal development (Pucilowska et al., 2015). This phenotype was observed also in Mills DEL adults (Horev et al., 2011; Pucilowska et al., 2015), with no significant changes in the grey matter (Kumar et al., 2018). In addition, cortical thickness is decreased in Mills DEL mice (Pucilowska et al., 2015), similarly to human carriers (Maillard et al., 2015). In particular, this model shows an aberrant cortical cytoarchitecture, with a reduction of upper cortical layer neurons at embryonic day 14.5, probably due to aberrant progenitor proliferation and premature cell cycle exit, leading to depletion of progenitor pools (Pucilowska et al., 2015; Pucilowska et al., 2018). Despite the general reduction in brain size, Mills and Dolmetsch DEL mice show increases in the relative volumes of several brain areas, including the midbrain, hypothalamus, striatum, nucleus accumbens, globus pallidus and cerebellar cortex (Horev et al., 2011; Portmann et al., 2014; Pucilowska et al., 2018). Mills DEL mice also exhibit decreased volumes in the ventral hippocampus, lateral septum, amygdala and entorhinal cortex (Pucilowska et al., 2018).
16p11.2 DEL mice show dopaminergic signalling dysregulation
Interestingly, data obtained in both Dolmetsch and Mills models suggest major alterations in dopaminergic signalling. For instance, Portmann et al. observed a significant increase in medium spiny neurons expressing dopamine receptor 2 (Drd2+ MSNs) in the striatum of 16p11.2 DEL neonates, with no changes in medium spiny neurons expressing dopamine receptor 1 (Drd1+ MSNs) (Portmann et al., 2014). The observed increase in striatopallidal MSNs in the striatum resulted in a reduced sensitivity to sedation induced by risperidone, a D2 receptor antagonist (Portmann et al., 2014). Interestingly, an increased number of cells co-expressing Drd1 and Drd2 was observed, suggesting that the 16p11.2 deletion may also affect the process of MSNs specification. In the deep layers of the cortex, Drd1+ MSNs were significantly decreased as well as DARPP-32 expressing neurons. Moreover, tyrosine-hydroxylase (TH), a rate limiting enzyme in the dopamine (DA) synthesis pathway, was decreased in mesodiencephalic DA cells (Portmann et al., 2014). Imbalances in the ratio of D1 and D2-expressing MSNs were also observed in adult brains by Grissom et al. In particular, Mills DEL males, but not females, overexpressed the mRNA for D2 receptor and adenosine 2a receptor in the striatum (Grissom et al., 2018). Interestingly, transcriptomic analysis in Herault DEL model revealed dysregulations in gene expression in DEL mice in different brain regions, with the striatum being more severely impacted (Arbogast et al., 2016). This finding may explain some of the motor and cognitive deficits displayed by these animals, dependent on basal ganglia circuitry.
16p11.2 DEL mice display significant changes in the excitability profile of MSNs and cortical pyramidal neurons, as well as compromised connectivity between different brain regions
The presence of major alterations in dopamine-mediated circuits is further supported by electrophysiological recordings on striatal MSNs from Dolmetsch DEL mice. These studies revealed an increased ratio of AMPA to NMDA receptor-mediated excitatory post synaptic currents (EPSC) and an increased miniature EPSC (mEPSC) frequency. Conversely, the paired-pulse ratios (PPRs) across multiple interstimulus intervals (ISIs) were significantly decreased (Portmann et al., 2014). These results suggest that the release probability of excitatory synapses on MSNs may be augmented.
In the hippocampus, no alterations were found in the excitability profile in Herault DEL model (Arbogast et al., 2016). However, in the medial prefrontal cortex (mPFC) of Mills DEL mice, pyramidal neurons displayed deficient NMDA-receptor-mediated glutamatergic transmission and reduced frequency of action potential (AP) firing (Wang et al., 2018). Compromised functional connectivity on the orbitofrontal, insular and auditory axis, and between the septum and the hippocampal regions, has been also reported in Mills DEL mice (Openshaw et al., 2023).
16p11.2 DEL mice recapitulates some of the behavioural deficits affecting human carriers
At the behavioural level, mild motor impairments have been observed in 16p11.2 DEL mice, such as deficits in righting from upside-down position (Mills DEL model) (Brunner et al., 2015) and lack of gait fluidity and tremor (Dolmetsch DEL model) (Portmann et al., 2014). However, normal motor coordination in the rotarod test was reported in Herault DEL model (Arbogast et al., 2016). Higher locomotor activity in familiar environments and stereotyped behaviours have also been broadly reported in all models (Horev et al., 2011; Portmann et al., 2014; Yang et al., 2015c; Arbogast et al., 2016; Angelakos et al., 2017). However, when tested in novel environments, such as in the open field test, both Dolmetsch and Mills DEL mice showed initial hypoactivity that gradually disappeared over the course of the first 10 min (Portmann et al., 2014; Pucilowska et al., 2015) which might reflect deficits in motor initiation (Portmann et al., 2014) or increased anxiety, as shown by Pucilowska et al., in the elevated plus maze test (Pucilowska et al., 2015).
DEL mice also display a wide range of cognitive deficits, including impairments in spatial memory (Mills DEL model) (Wang et al., 2018), novel object recognition (Mills, Dolmetsch and Herault DEL models) (Portmann et al., 2014; Yang et al., 2015b; Pucilowska et al., 2015; Arbogast et al., 2016; Pucilowska et al., 2018), novel object location (Dolmetsch DEL model) (Yang et al., 2015b) and passive avoidance (Mills DEL model) (Tian et al., 2015). Dolmetsch DEL model also exhibited prominent cognitive impairments in the touchscreen pairwise visual discrimination acquisition and reversal (Yang et al., 2015b), while deficits in contextual fear conditioning have been observed in Mills DEL model (Tian et al., 2015; Pucilowska et al., 2018), but not in Dolmetsch DEL model (Yang et al., 2015b). Consistently with the impaired fronto-temporal connectivity and GABAergic dysfunction observed by Openshaw et al., Mills DEL mice showed deficits in pre-pulse inhibition (PPI), a measure of sensorimotor gating, but enhanced performance in attentional tasks (Openshaw et al., 2023).
In terms of social behaviour, ultrasonic vocalizations during male-female interactions are significantly reduced in DEL mice (Mills and Dolmetsch DEL models) (Yang et al., 2015c; Stoppel et al., 2018), that could be rescued upon chronic activation of GABAB receptors (Stoppel et al., 2018). Unexpectedly, all DEL models display normal behaviour in the three-chamber social preference test, in contrast with the social deficits affecting human carriers (Yang et al., 2015a; Brunner et al., 2015; Arbogast et al., 2016). This could be partially due to the aberrant increase of oxytocin levels exhibited by the Mills DEL mice (Pucilowska et al., 2018) that may mask potential social deficits. In addition, the genetic background can profoundly influence the manifestation of social impairments. As demonstrated by Arbogast et al., a significant decrease in social preference for the second stranger in the three-chamber test can be observed in Herault DEL model with a hybrid C57/Bl6N X C3B background, but not in mice with a pure C57/Bl6N background (Arbogast et al., 2016). Housing conditions also seem to affect the emergence of social and cognitive deficits. For instance, impairments in recognition memory and ultrasonic vocalisation are displayed by Dolmetsch DEL mice only in standard mixed-genotypes housing conditions, but not by animals housed with individuals of the same genotype (Yang et al., 2015a).
Despite the higher prevalence of autism spectrum disorder in males, sex-specific phenotypes have not been systematically investigated in DEL mice. Kumar et al. found prominent male-specific structural changes in medial fibre tracts proximate to the striatum, overlapping with specific gene expression patterns associated with neurite outgrowth and MAPK pathway (Mills DEL model) (Kumar et al., 2018). At the behavioural level, male-specific deficits in perinatal communication have been observed (Mills DEL model) (Agarwalla et al., 2020). Consistently with ASD and ADHD patients’ phenotypes, male-specific sleep/wake decrements in total sleep time and longer bouts of continuous wakefulness have been reported (Mills DEL model) (Angelakos et al., 2017). Moreover, Mills DEL males display reduced motivation and impaired reward learning, which is consistent with the observed increase in the mRNA coding for dopamine D2 receptors associated with behavioural inhibition (Grissom et al., 2018).
16p11.2 DUP mouse models
16p11.2 DUP mice show opposite metabolic and neuroanatomical phenotypes in comparison with the 16p11.2 DEL mice
In comparison with DEL mice, DUP mice show opposite phenotypes in terms of body weight and metabolism, with increased body size, lower energy expenditure during the light and dark phase, a lower glucose clearance and higher blood levels of leptin (Herault model) (Arbogast et al., 2016). Craniofacial dysmorphisms have also been observed in DUP mice, showing altered skull shape but no changes in the skull size (Herault model) (Arbogast et al., 2016). At the neurostructural level, DUP mice are not significantly different from wild types, although a trend toward reduced volumes in several brain regions can be observed (Mills model) (Horev et al., 2011).
16p11.2 DUP mice display some behavioural deficits reminiscent of those observed in human carriers
In contrast with DEL mice, both Herault and Mills DUP mice display hypolocomotion (Horev et al., 2011; Arbogast et al., 2016; Bristow et al., 2020; Rein et al., 2020) with potential sex-differences. Hypo locomotion was observed in Mills DUP females only during home cage monitoring whereas males exhibited this behaviour in the novel environment of the open field arena. This observation may indicate that additional factors, such as the levels of stress, may have an impact on sex-specific phenotypes. These results are also consistent with the increased anxiety behaviour observed only in males, possibly linked to the reduced hippocampal-orbitofrontal-amygdala connectivity (Bristow et al., 2020). Importantly, this circuitry has been implicated in thought disorder, a hallmark of schizophrenia (Sumner et al., 2018). In Mills DUP mice, typical phenotypes linked to schizophrenia, such as MK-801-induced hyperlocomotion and deficits in pre-pulse inhibition, were not observed by Rein et al. (Rein et al., 2020), although there is a report of female-specific reduction of pre-pulse inhibition (Bristow et al., 2020). Mills DUP model also displays social impairments reminiscent of ASD, including deficits in social approach and in the three-chamber test (Rein et al., 2020) as well as reduced time spent in proximity with cage mates (Bristow et al., 2020). In contrast, in Herault DUP model, social deficits in the three-chamber test could be observed only in the hybrid C57/Bl6N X C3B background, similarly to the DEL mice (Arbogast et al., 2016).
Consistently with the observed deficits in hippocampal-orbitofrontal-amygdala connectivity, Mills DUP mice display impairments in spatial working memory (Bristow et al., 2020), that have been also reported in DUP human carriers (Schobel et al., 2009). Similarly to the deficits observed in patients in the continuous performance task (Fleck et al., 2001), Mills DUP mice show slower learning and more impulsive responding (Bristow et al., 2020), as well as prefrontal cortex-dependent cognitive impairments in the temporal order recognition memory (Rein et al., 2020). Novel object recognition memory was either found unaffected by the CNV (Rein et al., 2020) or significantly enhanced (Arbogast et al., 2016), possibly due to the different models and experimental protocols used in these studies.
16p11.2 DUP mice display major GABAergic dysfunctions, mediated by the transcription factor Npas4
In contrast with DEL mice showing hypoactivity in the mPFC neurons, Mills DUP mice display hyperexcitability due to a significant impairment in GABAergic synaptic transmission, while glutamatergic transmission was found unchanged (Rein et al., 2020). This is consistent to the excitatory/inhibitory imbalance observed in ASD patients (Nelson and Valakh, 2015).
In order to determine the effect of the 16p11.2 duplication on gene expression, Rein et al. performed RNA-sequencing on mPFC and identified 388 differentially expressed genes, most of which were downregulated, including epigenetic markers, ASD/ID risk genes and the sodium ion channel SCN9a. A significant downregulation was detected for Npas4, a transcription factor promoting the formation of GABAergic synapses, that was found reduced also in post-mortem PFCs from idiopathic ASD patients (Rein et al., 2020). Restoration of Npas4 levels in Mills 16p11.2 DUP mice was sufficient to rescue the synaptic and behavioural deficits, thus suggesting the pathogenic role of Npas4 in the GABAergic dysfunction underlying the 16p11.2 DUP phenotype (Rein et al., 2020).
Other animal models
16p11.2 DEL and DUP rat models
Rat models for 16p11.2 CNVs have also been generated recapitulating craniofacial phenotypes, with mirror effects between the DEL and the DUP conditions (Qiu et al., 2019). Converging and male-specific deficits in social behaviour and novel object recognition have been also observed in 16p11.2 DEL and DUP rats in two different genetic backgrounds (Martin Lorenzo et al., 2023). RNA sequencing analysis in the hippocampus found 267 differentially expressed genes dysregulated in DEL and DUP rat models. Among these genes, 100 were downregulated and 120 upregulated in both models, which could explain some overlapping phenotypes, independent from gene dosage. Differential functional analysis revealed 23 upregulated pathways in both DEL and DUP rats, associated with morphogenesis of the primary cilium. However, pathways related to synaptic function and metabolism were mostly deregulated in DEL rats, while pathways associated with transcription, epigenomic regulation and hormone regulation were mostly affected in DUP animals (Martin Lorenzo et al., 2023).
Recently, Yang et al. carried out anatomical and electrophysiological analysis of developing interneurons in 16p11.2 DEL rats, after the identification of a subset of interneurons in human foetal cerebral cortex potentially vulnerable to genetic autism risk factors. In 16p11.2 DEL rats at P21, the number or position of INs was unchanged in either CA1 or somatosensory cortex. However, somatostatin-expressing INs in CA1 display hyperexcitability, with an enlarged axon initial segment. This finding, although limited to a single developmental stage and one type of INs, supports the idea that the 16p11.2 deletion may perturb the electrophysiological properties of developing INs, thereby affecting the excitation/inhibition (E/I) balance (Yang et al., 2023).
Zebrafish and Drosophila melanogaster models
A deeper characterization of the effects of 16p11.2 CNVs at the cellular level has been achieved using more simplified animal models, allowing a high-throughput analysis of the interaction between genetic and phenotypic effects. For instance, the first 5 days of development in Zebrafish recapitulate the first weeks of development in mice and the first couple of years in humans, thus allowing to detect abnormalities in brain structures or functions that become obvious only after birth (Blaker-Lee et al., 2012). Among the 27 protein-coding genes in the 16p11.2 region, 21 are also present in Zebrafish. This model has been used to discover dosage-sensitive genes within the 16p11.2 locus by selective induced loss of function (LOF) or overexpression experiments. By performing LOF studies from 24 h post-fertilization to post-natal day 5, covering a period from 5-week gestation to toddlerhood in humans, Blaker-Lee et al. revealed that most of the 16p11.2 genes are highly active during early development and are involved in brain and body development. Selective LOF of 16p11.2 homologs was associated with spontaneous movement defects and reduced or no response to touch, possibly linked to the observed abnormalities in axonal development (Blaker-Lee et al., 2012). Using similar approaches, KCTD13 (see Table 1) was identified as a major driver of head size phenotypes, which were consistent with those observed in DEL and DUP human carriers. In particular, KCTD13 suppression induced macrocephaly in Zebrafish embryos resembling the 16p11.2 DEL condition, whereas its overexpression caused microcephaly with concomitant defects in neurogenesis, in epistasis with two other genes in the locus, MAPK3 and MVP (Golzio et al., 2012). In addition, a genetic interaction was demonstrated between KCTD13 and ciliopathy-associated genes (Migliavacca et al., 2015).
Drosophila melanogaster, which has at least 14 homologs of human 16p11.2 genes, has also been employed to test the role of these individual genes and their combinatorial effects in determining the variegated phenotypes observed in DEL and DUP human carriers (Park et al., 2016; Iyer et al., 2018). By genetic screening and RNA interference approaches, KIF22, a member of kinesin family (see Table 1), was identified as a key factor required for the establishment of synaptic connectivity in Drosophila neuromuscular junction (Park et al., 2016). By performing knock-down of single homologs and 564 pairwise knockdowns, Iyer et al. identified 24 interactions between 16p11.2 homologs and 46 interactions between 16p11.2 homologs and neurodevelopmental genes. In particular, they observed impaired motor functions and spontaneous seizures, as well as alterations in the architecture of Drosophila neuromuscular junction and dendritic arborization, consistently with the phenotypes observed in human carriers. Moreover, several homologs contributed in different proportion to the cellular composition of the fly eye, probably intervening at different timepoints during cellular proliferation and differentiation. In general, the data from Iyer et al. suggest that several genes within the 16p11.2 region are involved in neurodevelopment and their reciprocal interaction is responsible for the heterogeneous phenotypes observed in patients (Iyer et al., 2018).
Cellular models of 16p11.2 deletion and duplication
Recent advances in stem cell technologies opened the possibility to investigate neurodevelopmental disorders using patient-derived induced pluripotent stem cells (iPSCs). For instance, Deshpande et al. used iPSCs-derived forebrain cortical neurons to investigate the cellular mechanisms underlying differences in brain size associated with 16p11.2 CNVs. DEL neural progenitors display increased soma size and dendritic length as well as a more extensive arborization. In contrast, DUP neurons show opposing phenotypes. The larger neuronal size in the DEL condition was associated to altered functional properties, such as reduced excitability and membrane resistance, while the DUP neurons did not significantly deviate from controls. Interestingly, DUP neurons displayed increased outward potassium current, to stabilize their intrinsic excitability. Both DEL and DUP neurons showed less excitatory synapses with increased synaptic strength, that may underlie the altered network activity and behavioural deficits in human carriers (Deshpande et al., 2017).
Neuronal firing and synchrony have been found reduced in iPSCs-derived excitatory neurons harbouring the 16p11.2 duplication, in later stages of development, along with reduced dendrite length and impaired calcium homeostasis (Parnell et al., 2023). These findings were recapitulated in excitatory neurons derived from DUP patients with schizophrenia, thus linking excitatory neurons dysfunctions with schizophrenia pathogenesis. Transcriptomic analysis carried out on excitatory neurons after 7 weeks maturation identified 62 upregulated and 133 downregulated genes, associated with calcium ion binding and neuron projections development (Parnell et al., 2023).
Similarly to cortical neurons, iPSCs-derived dopaminergic neurons from DEL patients also displayed increased soma size. However, in contrast with the cortical neurons’ phenotype, these morphological changes correlated with increased hyperexcitability of DEL dopaminergic neurons. Interestingly, DEL dopaminergic neurons show reduced levels of KCDT13 and overexpression of RHOA, a molecular pathway also upregulated in KCDT13 heterozygous and in 16p11.2 DEL mice (Escamilla et al., 2017; Martin Lorenzo et al., 2021). Treatment with RHOA inhibitor could rescue the cell size and hyperexcitability of DEL dopaminergic neurons, thus implicating RHOA pathway in dopaminergic network excitability (Sundberg et al., 2021).
Macrocephaly in DEL carriers was recently found associated with hyperproliferation of iPSCs-derived neural progenitors, that was inversely correlated with ERK1/2 phosphorylation and response to basic fibroblast growth factor (bFGF), a mitogen that activates ERK pathway (Connacher et al., 2022). In contrast, two previous studies did not detect any differences in cell proliferation at the early stage of cortical progenitors (Deshpande et al., 2017; Sundberg et al., 2021). Brain overgrowth in the 16p11.2 deletion syndrome has been potentially linked to overexpression of CD47 in both neural and oligodendrocyte progenitor cells. CD47 is a “do not eat me” signal protein, thus preventing cells from getting engulfed or phagocytosed by macrophages and microglia (Li et al., 2021).
To date, two studies employed cerebral organoids to investigate the effects of the 16p11.2 deletion on brain development (Urresti et al., 2021; Fetit et al., 2023). Importantly, DEL and DUP cortical organoids could recapitulate the brain size phenotypes. In addition, DEL cortical organoids exhibited increased neuronal maturation, soma size and neurite length as well as depletion of neural progenitors, in comparison with control and DUP organoids. However, neuronal migration was significantly impaired in both DEL and DUP organoids. When looking at KCTD3 and total RHOA levels, the authors found decreased KCTD3 and increased RHOA levels in DEL organoids, while the DUP organoids showed opposite trends. However, the active GTP-bound form of RHOA was consistently upregulated in both CNVs, that was previously linked with impaired neuronal migration (Cappello et al., 2012). Consistently, the observed defects in neuronal migration in both DEL and DUP organoids could be rescued by RHOA inhibition (Urresti et al., 2021).
The effects of 16p11.2 CNVs on interneurons development was recently investigated by Fetit et al. Ventral organoids harbouring the 16p11.2 deletion were more variable in size compared with the isogenic controls (Fetit et al., 2023). This variability could be relevant when considering the clinical heterogenicity of DEL human carriers (Fetit et al., 2020). In addition, the authors found a substantial acceleration of subpallial development in DEL organoids, potentially leading to premature differentiation (Fetit et al., 2023).
Role of the 16p11.2 genes
The purpose of this section is to review the specific functions of the genes within the 16p11.2 region and how they might interact in specific molecular pathways, thus determining the phenotypic effects observed in patients, animal, and cellular models. As listed in Table 1, 27 protein-coding genes are involved in 16p11.2 CNVs. Some of them have been well characterised in animal and cellular models created to recapitulate the phenotypes observed in DEL and DUP patients. Other 16p11.2 genes still have unknown function, and their role in the pathogenesis of 16p11.2-associated diseases is obscure. We will describe a small subset of genes clearly involved in the 16p11.2 associated phenotypes (MAPK3, KDCT13, MVP, TAOK2 and SEZ6l2) and thus potential therapeutic targets. Subsequently, we will describe the 16p11.2 genes with known cellular functions, but not yet linked to the 16p11.2 DEL and DUP pathologies. Then, we will briefly list the limited information available for the least characterised genes. Finally, we will consider non-coding RNAs that are located within the 16p11.2 region but have not yet been linked to specific cellular functions (see Table 3; Figure 1).
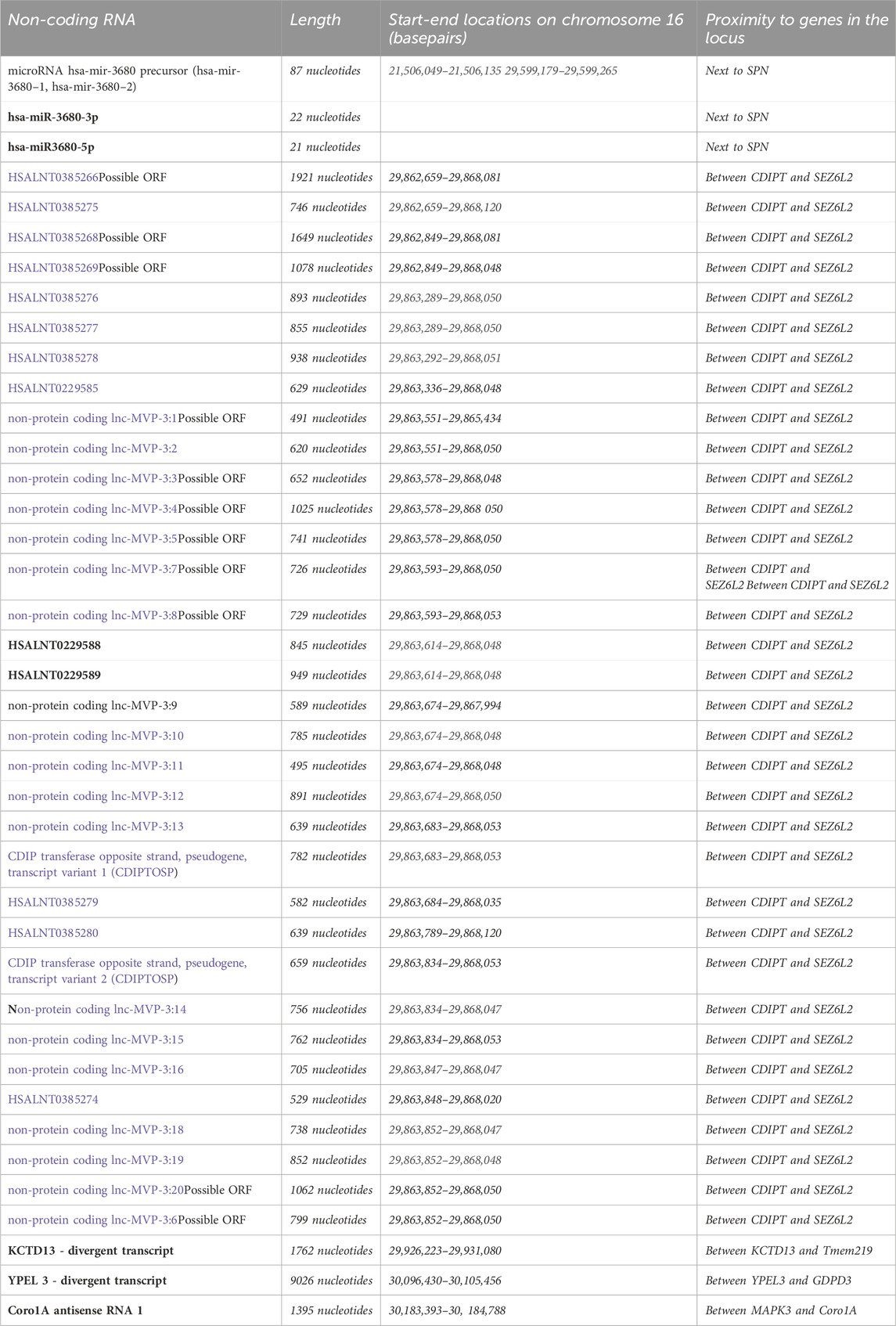
Table 3. List of non-coding RNAs mapping on the 16p11.2 region.
MAPK3, MVP, Sez6l2, TAOK2, KCDT13 all potentially modulate cell signalling in 16p11.2 deletion models
MAPK3 codes for the extracellular signal-regulated kinase 1 (ERK1), a p44 protein kinase acting as a major signal transduction component of the Ras-Raf-Mek-ERK cascades (Figure 2). Interestingly, the MAPK1 gene, coding for p42 ERK2 kinase, is found in the distal portion of the 22q11.2 CNV region, another common chromosomal rearrangement implicated in NDD (Vithayathil et al., 2018; More et al., 2020).
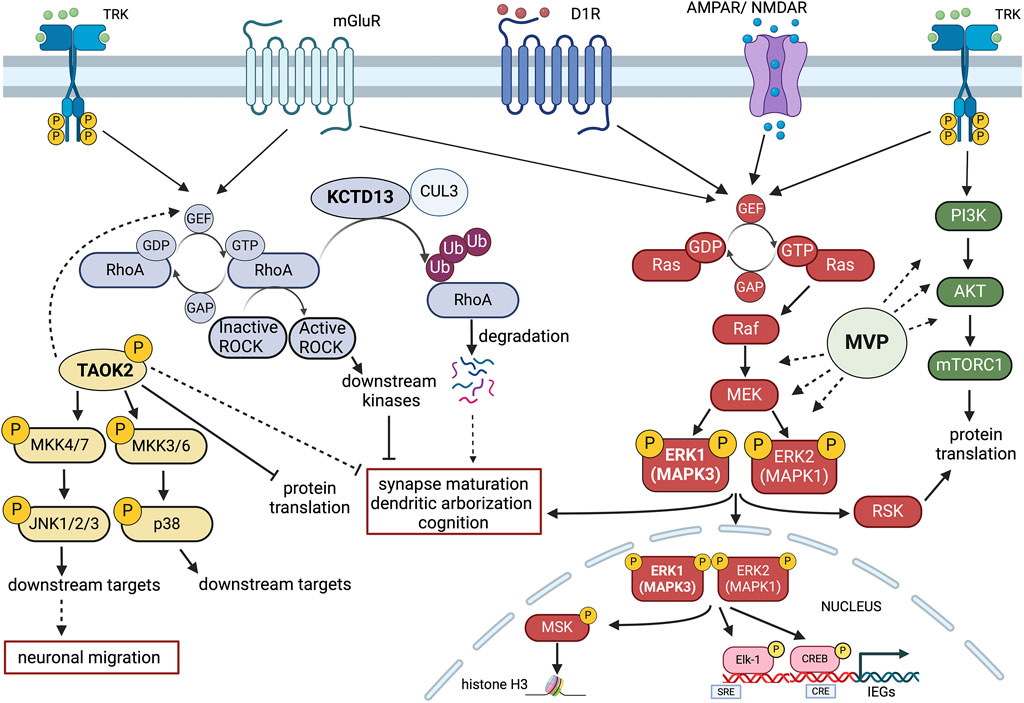
Figure 2. Schematic representation of potential signalling Interactions among four key 16p11.2 CNV genes. MAPK3 (ERK1), TAOK2, KCDT13 and MVP may interact at the intracellular level in modulating neuroanatomical, synaptic and cognitive functions. Gene dosage alterations present in either 16p11.2 deletion or duplication syndromes could imbalance the coordinated cellular activities of these genes. Multiple receptor systems can activate the Ras-Raf-MEK-ERK1/2 signalling pathway that controls both gene expression and chromatin remodelling. ERK1 (MAPK3) and ERK2 (MAPK1) also interact with the PI3K-AKT-mTORC1 pathway in modulating protein translation. One important aspect of MAPK3/MAPK1 signalling modulation is that MAPK3 gene dosage may shift the balance between the two kinase activities, resulting in different signalling intensities with consequences at the physio-pathological level. Major Vault Protein (MVP) may act as a scaffold protein for both ERK1/2 and mTORC1 signalling, thereby providing additional modulatory control. TAOK2 kinase stimulates multiple cytosolic and nuclear targets, including JNK1/2 and p38 MAP kinases that may interact with the ERK1/ERK2 pathway and the RhoA-ROCK kinase cascade. The action of TAOK2 may inhibit synaptic maturation and protein translation, potentially in opposition to ERK1/2. KCDT13 may facilitate RhoA degradation via CULLIN 3 (CUL3) interactions, also potentially antagonising some of TAOK2 functions. Figure has been created using Biorender.com.
The Ras-ERK signalling cascade has been implicated in a variety of cellular processes, from cell proliferation and cell survival to synaptic and behavioural plasticity (Fasano and Brambilla, 2011). However, unravelling its wide roles in development and in the adult brain is beyond the scope of this review.
Recent evidence indicates that targeting this pathway via pharmacological intervention may be a way forward to treat at least certain symptoms associated to 16p11.2 deletion and duplication. In order to understand the rationale of the potential therapeutic approaches based on ERK signalling modulation, we need to refer to the competitive model of ERK1 and ERK2 interaction, developed over the years by our laboratory (Mazzucchelli et al., 2002; Vantaggiato et al., 2006; Indrigo et al., 2010; Indrigo et al., 2023). Based on this model, ERK1 and ERK2 MAP kinases do not signal with the same intensity and, most importantly, they do not translocate into the nucleus at the same rate (Marchi et al., 2008). In fact, as recently described (Indrigo et al., 2023), ERK1 delays the entry of ERK2 MAPK, the most abundant of the two kinases, by specifically binding to a class of importins, the α1/KPNA2 group. This interaction occurs via the unique N-terminal domain of ERK1 to KPNA2 since this binding can be prevented by the administration of a cell penetrating TAT peptide coupled with the same ERK1 N-term domain. Either the downregulation of ERK1 expression/activity, via gene knock-out, viral mediated silencing, or via in vivo administration of the RB5 peptide results in neuronal survival, cognitive enhancement and increased structural and synaptic plasticity. Those remarkable effects are the direct cause of a global enhancement of ERK signalling in the brain (Indrigo et al., 2023).
Gene dosage of ERK1/MAPK3 is therefore a crucial determinant of the overall ERK1/2 activity. Following this reasoning, we originally speculated that ERK1/2 activity would be increased in a hemideleted condition, such as the 16p11.2 DEL. Using mouse models of the DEL condition, this prediction has been confirmed (Pucilowska et al., 2015). Importantly, in the DEL model, changes in cortical development and behavioural impairments have been rescued by treatments during embryonic development with inhibitory peptides of the Ras-ERK cascade (Papale et al., 2016; Pucilowska et al., 2018). The pharmacological treatment during gestation not only fully rescue those functional alterations but also brings back ERK1/2 activity to normal levels, as expected (Pucilowska et al., 2018). Interestingly, a later treatment during adulthood only partially improves behavioural deficits in the DEL model, suggesting that an earlier intervention may be preferable to maximise the therapeutic outcome. This evidence indicates that a manipulation of the activity of a single gene within the 16p11.2 locus may be an effective way to treat the deletion syndrome. Ras-ERK inhibitors are among the best characterised drugs available and they have been already tested in clinical trials for cancer therapy and in experimental models, also to rescue aberrant ERK activity in NDDs such as RASopathies (Papale et al., 2017).
As a mirrored situation, the 16p11.2 duplication syndrome may be characterised by a globally reduced ERK1/2 activity, due to the presence of three copies of MAPK3/ERK1. Currently, there is no published evidence supporting this claim but preliminary evidence in our laboratory suggests that this may be the case.
Additional evidence of the importance of ERK signalling in the 16p11.2 deletion syndrome comes from the observations on striatal dependent reward learning, in which ERK1 MAPK phosphorylation appears to be aberrantly elevated during acquisition of operant behaviour in response to sucrose as natural reward. Importantly, this effect was seen exclusively in males, highlighting the effect in sex-specific effects in NDDs. However, no attempts have been made to rescue this change (Grissom et al., 2018). Interestingly, in a parallel study on DEL mice linking spatial transcriptomic data and brain structural changes (MRI scans), three genes loosely linked to MAPK signalling were upregulated in DEL male mice only: MVP; Sez6l2; and TAOK2 (Kumar et al., 2018). To further investigate the relevance of such male-specific transcriptional changes, the same research group generated a triple MVP/Sez6l2/TAOK2 hemizygous mutant, using CRISPR/Cas9 technology (Kim et al., 2024). Interestingly, the triple hemideleted mutant mouse line shows male-specific behavioural alterations, such as hyperlocomotion and reduced reward learning in a progressive (but not fixed) ratio paradigm, largely overlapping with the phenotypes observed in the 16p11.2 DEL mutants. The same triple mutants did show a less pronounced phenotype, with less hyperlocomotion and no reward learning phenotype, when backcrossed in a MAPK3 hemideleted background. This observation is important but is subjected to multiple interpretations, considering the prominent role of MAPK3 and ERK signaling in striatal-dependent operant conditioning and reward learning (Mazzucchelli et al., 2002; Ferguson et al., 2006; Engel et al., 2009; Fasano and Brambilla, 2011; Shiflett and Balleine, 2011; Indrigo et al., 2023). Although ERK1/MAPK3 KO male mice show an enhancement of ERK1/2 signalling and striatal-dependent learning, neither the hemideleted nor the full ERK1/MAPK3 KO mice have been formally tested in the same protocol used by Kim et al. (Kim et al., 2024). Therefore, it is possible that the MAPK3 partial ablation may counterbalance the effect of the triple MVP/Sez6l2/TAOK2 mutation, effectively “rescuing” their phenotype by interfering with cell signalling changes. On the other hand, we recently showed that ERK signalling potentiation via pharmacological manipulation only enhances reward-based learning in WT females but not males (Indrigo et al., 2023). Considering that the treatment mimics ERK1/MAPK3 mutation, ERK1 MAP kinase may indeed play a little role in this specific form of learning in males. As a final note on this aspect, it is important to stress that differences in MAPK3 levels may lead to distinct phenotypes that may be more associated to the 16p11.2 duplication condition, including the higher propensity of the DUP carriers to develop schizophrenia. Indeed, MAPK3 mRNA increase in recent TWAS studies has been recently correlated to higher risk of psychosis and its mRNA levels have been found elevated in the prefrontal cortex of schizophrenic patients (Gandal et al., 2018; Gusev et al., 2018; Hall et al., 2020).
As further evidence supporting the central role of Sez6l2, TAOK2 and MVP, all three proteins have been loosely connected to ERK signalling. Major Vault protein (MVP) was originally discovered as a main component of the vault organelle, a ribonucleoprotein complex found in most eukaryotes (Berger et al., 2009). MVP is highly abundant in the CNS and considerable evidence has linked its function to growth factor receptors responses, serving as a potential scaffold protein for ERK and PI3K/AKT/mTOR signalling pathways (Figure 2) (Kolli et al., 2004; Kim et al., 2006; Dong et al., 2021; Wakatsuki et al., 2021).
In a recent report, we investigated how MVP may interact with other 16p11.2 genes in the definition of neuroanatomical phenotypes (NAPs), that are major determinants of brain structural changes and neuronal morphology (Kretz et al., 2023). Interestingly we found that MVP is the top driver among a set of additional 12 genes within the 16p11.2 region (PPP4C, ZG16, TAOK2, SLX1B, MAZ, Fam57b, BOLA2, TBX6, QPRT, SPN, HIRIP3, and DOC2A) to modulate NAPs in males. Remarkably, neither MAPK3 nor KCTD13 were implicated in NAPs, despite previous work for KCTD13 in Zebrafish suggesting a different scenario (Golzio et al., 2012). However, MVP and MAPK3 did show interaction in modulating anxiety-like behaviour and drug induced epilepsy, suggesting that these two proteins may work together in some of the pathological behaviours observed in the DEL carriers (Kretz et al., 2023).
Concerning the TAOK2 gene, accumulating evidence indicates that the Thousand and one amino-acid kinase 2 (TAOK2) gene product plays a central role in neurodevelopment and more specifically in the 16p11.2 deletion syndrome. TAOK2 gene is listed as a category 2-risk gene (strong association) in the SFARI GENE Scoring list (https://gene.sfari.org/database/human-gene/TAOK2). Moreover, whole-genome and exome sequencing of ASD families identified 24 different variants in TAOK2 associated to autism. Importantly, TAOK2 KO mice and their hemideleted counterpart show dose-dependent cognitive deficits, anxiety and social behaviour impairments, as well as abnormalities in brain morphology, cortical development, connectivity, dendrite and synapse formation, all resulting from a reduced excitatory activity (Richter et al., 2019). These in vivo data are all consistent with previous in vitro observations. Further evidence suggests that TAOK2 action may require the downstream JNK1 and p38 MAPK signalling activation and induces PSD95 stability and dendritic spine maturation via Septin7 phosphorylation. (Moore et al., 2000; Zhou et al., 2004; Yasuda et al., 2007; de Anda et al., 2012; Ultanir et al., 2014; Yadav et al., 2017). Recent evidence demonstrated that TAOK2 acts as translational repressor by inhibiting the eukaryotic elongation factor eEF2 (Henis et al., 2024) (Figure 2).
Importantly, overexpression of mutated TAOK2α variants, but not the TAOK2β variants, impaired neuronal migration by destabilising microtubules and reduced JNK1 activation. This effect could be replicated in TAOK2 KO brains but also in the heterozygous 16p11.2 DEL animals, that displayed reduced levels of phosphorylated JNK1 and neuronal migration deficits, that could be partially rescued by ectopic expression of TAOK2α in in the developing cortex (Scharrenberg et al., 2022).
However, another signalling pathway is also central to TAOK2 function in spine formation and stability, at least in the cortex. In fact, RHOA GTPAse activity is significantly reduced in TAOK2 KO and HET mice. Importantly, incubation with RHOA activators increases spine formation in the TAOK2 KO preparations, suggesting a link between the two molecules and indicating that TAOK2 may exploit multiple signalling pathways (i.e., JNK1 and RHOA) to control distinct neurodevelopmental processes (Richter et al., 2019).
Albeit interesting, the potential role of RHOA signalling in promoting brain function is not entirely consistent with the observation that another 16p11.2 gene, KCTD13, may exert an opposite function. KCTD13 is a member of a superfamily of at least 20 genes forming a complex with CULLIN 3 (CUL3) ubiquitin ligase. These genes have been implicated in several neuropsychiatric conditions (Teng et al., 2019). Importantly, a major target of the KCTD13-CUL3 complex is indeed RHOA, that is degraded, and its downstream signalling is attenuated in WT condition (Figure 2) (Lin et al., 2015).
Accordingly, the KCTD13 KO mouse model shows a reduction of the number of functional synapses, with a decrease of dendritic length, complexity, and dendritic spine density due to increased levels of RHOA. These alterations may be reversed by RHOA inhibition rather than RHOA activation (Escamilla et al., 2017). Additional evidence supports the negative role of RHOA signalling in behavioural deficits in a different KCTD13 KO mouse model, displaying no major differences between homozygous and heterozygous deleted animals and showing strong similarities with the 16p11.2 DEL model (Arbogast et al., 2019). In this KCTD13 KO model, as well as in the 16p11.2 DEL model, learning and memory deficits could indeed be rescued by an inhibitor of the Rho-associated protein kinase ROCK (Martin Lorenzo et al., 2021).
It remains to be established whether these contrasting observations about RHOA signalling are real or due to the fact that this molecular pathway is differentially implicated in distinct aspects of the pathology. Certainly, these findings underscore the complexity of the mechanisms associated with the 16p11.2 deletion.
The last gene that has been shown to play a significant role in some of the 16p11.2 deletion phenotypes is Sez6l2. This is a transport receptor transmembrane protein, required by the aspartic protease Cathepsin D for its endosome/lysosome localisation. Inactivating mutations or mislocalization of cathepsin D lead to neuronal dysfunctions including microcephaly, seizures, and cognitive and psychomotor defects. SEZ6L2, also known as BSRP-A (Brain-Specific Receptor A), is predominantly expressed in the brain and has been associated to febrile seizures, bipolar disorder and autism, together with the other two members of the seizure-related gene six family, SEZ6, SEZ6L. Functional studies on SEZ6L2 have also implicated this protein in neurite outgrowth. In addition, the triple mutant of SEZ6, SEZ6L and SEZ6L2 showed cerebellar deficits and a potential link with PKCα signalling activation, thus providing a possible functional link with other genes in the 16p11.2 locus, such as MAPK3, MVP, KCDT13 and TAOK2 (Miyazaki et al., 2006; Kumar et al., 2009; Boonen et al., 2016).
Genes within the 16p11.2 region with limited functional information available
SPN
The sialophorin, or CD43, is a glycoprotein expressed on the surface of T lymphocytes promoting adhesion and activation during immune responses. This gene, together with CORO1A and KIF22 have been suggested to play a role in reduced lymphocytes count in 16p11.2 DEL patient presenting a concomitant low dosage of BOLA2 duplicone, located on 16p11.2 BP4-BP5 flanking region (Giannuzzi et al., 2019; Giannuzzi et al., 2022). In male 16p11.2 DEL mice, SPN protein is overexpressed mainly in telencephalic and cerebellar regions presenting decreased fiber density (Kumar et al., 2018).
QPRT
The quinolinate phosphoribosyltransferase is a key enzyme involved in the catabolism of quinolinate, an intermediate in the synthesis of nicotinamide adenine dinucleotide (NAD). QPRT-mediated NAD biosynthesis plays a fundamental role in neuronal differentiation and neurite growth during development (Haslinger et al., 2018; Neves et al., 2022). QPRT expression is reduced in cell lines derived from 16p11.2 DEL patients, and its knock-down in neuroblastoma cells during differentiation significantly altered neuritic growth and complexity, whereas QPRT knock-out (KO) increased cell death during differentiation (Haslinger et al., 2018). Furthermore, QPRT KO leads to the downregulation of several genes, including GABRB3, SNTG2, KCNQ, CNTNAP2. These genes are implicated in the formation of GABAergic synapses formation and are associated with ASD and epilepsy (Haslinger et al., 2018). QPRT also interacts with neuroligin 3 (NLGN3), a postsynaptic transmembrane protein involved in synapse formation and neuron-glia connections which is often found mutated in ASD patients (Shen et al., 2015).
ZG16
Human zymogen granule protein 16 is highly expressed in mucus-secreting cells its overexpression significantly suppresses tumour growth through T cells-mediated immune response activation (Meng et al., 2022). Although its role in 16p11.2 CNVs is unclear, it has been correlated with increased size of several brain areas in mice selectively ablated for this single gene (Kretz et al., 2023).
KIF22
Kinesin family member 22 (KIF22) has been shown to mediate cell proliferation by regulating mitotic spindle, microtubules stability (Tokai et al., 1996; Tokai-Nishizumi et al., 2005), as well as the expression of CDC25C, a cell cycle regulator (Nilsson and Hoffmann, 2000).
Among all genes of 16p11.2 region, KIF22 is specifically enriched in neural progenitors in G2/M phase, while its expression levels significantly decline as cells become post-mitotic (Morson et al., 2021). During CNS development, KIF22 transcripts are clearly segregated in the ventricular and subventricular zones, where cortical progenitors arise. KIF22, together with ALDOA, HIRIP3, PAGR1, and MAZ are also expressed in the ventricular zone of the ganglionic eminences where interneuron progenitors reside, supporting the hypothesis that 16p11.2 CNVs may disrupt the excitatory and inhibitory components of the developing brain. KIF22 dosage seems to regulate neurogenesis by affecting the length of the cell cycle, thereby determining the proliferative or neurogenic fate of the progenitors. Kif22 gene dosage variations linked to 16p11.2 CNVs would thus affect cell-cycle kinetics and perturb neuronal formation during development (Morson et al., 2021). In Zebrafish, KIF22 and ALDOA are the only deletion “dosage sensor” genes for 16p11.2 CNVs, meaning that their functional levels are sensitive to hemizygosity and thus lead to a pathological phenotype. In particular, reduction of KIF22 is associated with abnormal brain morphology and bent tail, as well as deficient axonal development (Blaker-Lee et al., 2012). The same effect on axonal development was observed in Drosophila, in which silencing of KIF22 homologs leads to the development of ectopic innervation and off-target recurrent axon branches in the neuromuscular junction (Park et al., 2016). Interestingly, these results are in agreement with nerve regeneration studies conducted in rats, where Kif22 has been identified as a potential therapeutic target for promoting peripheral nerve injury repair via Schwann cell proliferation and migration (Lu et al., 2022).
MAZ
The MYC-associated zinc finger protein is a transcription factor involved in several, highly specific, molecular processes related to gene expression and cell cycle, leading to differentiation and developmental effects. During development, MAZ is a cofactor of CTCF during chromatin insulation of active and repressed genes within the Hox clusters (Ortabozkoyun et al., 2022), specifically involved in the differentiation of motor neurons in vertebrates. Among its targets, MAZ regulates the expression of several WNT morphogens, involved in the correct morphological development of different organs. For instance, MAZ has been related to birth defects in the genitourinary tract, that are commonly observed in the context of 16p11.2 CNVs (Haller et al., 2018) as well as in eye development, also presenting comorbidity in 16p11.2 DEL and DUP patients (Medina-Martinez et al., 2020). As KIF22 and ALDOA, MAZ is also enriched in the ventricular and subventricular zones compared to post-mitotic cells, suggesting a role in neurogenesis (Morson et al., 2021). As a transcription factor, MAZ takes part in the correct synchronization between neurogenesis and NOTCH1 mediated gliogenesis. It is well known that during development, NOTCH1 signalling is downregulated during neurogenesis and activated in gliogenesis, following a specific timing. The upregulation of ADAM10 activates NOTCH1-mediated gliogenesis. Liu and colleagues have demonstrated that MAZ enhances ADAM10 transcription in response to activation factors such as CT-1 in cultured neuronal progenitor cells (NPC) (Liu et al., 2016). In neuronal stem cells (NSC), MAZ regulates the expression of Rho-GDIγ, which in turn modulates the activity of Rho GTPases during neuritogenesis, axon formation and dendritic development in neuronal differentiation (Wang et al., 2013). Moreover, MAZ activity has been reported to enhance NMDA receptor subunit type 1 (NR1) promoter activity during neuronal differentiation of P19 cells, contributing to the assignment of the correct functional profile to differentiated excitatory neurons (Okamoto et al., 2002). Thus, changes in MAZ dosage related to 16p11.2 CNVs might be related to structural changes in neuronal/glial tissue balance.
PRTT2
Proline-rich transmembrane protein type 2 (PPRT2) is a membrane protein, located at synaptic contacts, playing a role in synapse formation during development. It is also a crucial component of the neurotransmitters’ release machinery, by interacting with SNARE proteins and synaptotagmins 1 and 2 (Valtorta et al., 2016). PRTT2 has been found enriched in Drd2+ MSNs in 16p11.2 deletion mice, together with KCTD13, Fam57b, Sez6l2 and CORO1A (Portmann et al., 2014). Mutations in PPRT2 have been associated with paroxysmal movement disorders (Heron and Dibbens, 2013), benign infantile familial seizures and autistic developmental regression (Zhang et al., 2024), as well ataxia and seizures in 16p11.2 DEL patients (Vlaskamp et al., 2019; Padmanabha et al., 2024). Importantly, correction of PRTT2 copy number in 16p11.2 DUP mice could rescue hypersynchronous activity and enhanced glutamate release in cortical circuits, seizure susceptibility and social deficits (Forrest et al., 2023).
PAGR1a
Pax-interacting protein 1 is a component of the histone methyltransferase MLL2/MLL3 complex, with possible role in DNA damage response (Cho et al., 2007; Gong et al., 2009). This protein, highly expressed in neural progenitors (Morson et al., 2021), appears to have a role in early embryonal development, as suggested by animal studies. For instance, loss of function of PAGR1a in Zebrafish is associated with reduced brain ventricle size and less defined midbrain-hindbrain boundaries (Blaker-Lee et al., 2012). In addition, null mice for PAGR1a are not viable, while the loss of one copy leads to abnormal development of extraembryonic tissues, such as the amnion, chorion, and visceral yolk sac (Kumar et al., 2014). A recent clinical report by Daum et al. identified three individuals, carrying a homozygous missense mutation in PAGR1a gene and showing microcephaly, severe developmental delay, dysmorphism, neurological deficits and death in infancy (Daum et al., 2022).
CDIPT
CDP-Diacylglycerol-Inositol 3-Phosphatidyltransferase (CDIPT) catalyses the biosynthesis of phosphatidylinositol and is highly expressed during foetal and neonatal brain development in rats (Nyquist and Helmkamp, 1989). CDIPT knock-down in Drosophila displays altered growth development of the neuromuscular junction, a model for studying synapse growth defects (Iyer et al., 2018). In addition, a missense mutation in CDIPT gene in Zebrafish causes photoreceptor cells death and cataract (Murphy et al., 2011).
DOC2A
Double C2-like domain-containing protein alpha (DOC2A) is a cytosolic protein interacting with SNARE complex and phospholipids, acting as a Ca2+ sensor and mainly expressed in glutamatergic neurons, triggering glutamate release (Courtney et al., 2018). In the context of neurodevelopmental disorder, it has been found specifically overexpressed in 16p11.2 DEL male mice in brain regions with decreased fiber density (Kumar et al., 2018). Interestingly, DOC2A-KO mice display abnormal morphology of hippocampal neurons in the dentate gyrus, defective hippocampal activity as well as social deficits and repetitive behaviours. DOC2A functions appear to be regulated by the Ca2+ binding protein Secretagogin (Wang et al., 2023).
ALDOA
Aldolase A (ALDOA) is a glycolytic enzyme which catalyses the conversion of fructose 1,6-biphosphate into glyceraldehyde 3-phosphate and dihydroxyacetone phosphate. ALDOA is highly expressed in neural progenitors and its expression declines as the cells become post-mitotic (Morson et al., 2021). It is also part of ERK1/2 interactome during epidermal and neuronal differentiation (von Kriegsheim et al., 2009). Knock-down studies in Drosophila demonstrated the involvement of this gene in the locomotor function, specifically in climbing activity. In addition, ALDOA knock-down produces altered cell counts and patterning of different cell types, including increased photoreceptors neurons and misplaced bristle cells (Iyer et al., 2018). In Zebrafish embryos, ALDOA loss of function is associated with no response to touch (Blaker-Lee et al., 2012).
PPP4C
Protein phosphatase 4, catalytic subunit (PPP4C) plays a role in several cellular processes, such as microtubule growth and organization, DNA damage checkpoint recovery, apoptosis, and TNF-alpha signalling (Chen et al., 2008). However, its role in the context of 16p11.2 syndromes has not been completely elucidated. It has been found overexpressed in 16p11.2 DEL mice specifically in regions with decreased fiber density (Kumar et al., 2018), while studies in Zebrafish suggest the role of PPP4C in locomotor function and axon growth (Blaker-Lee et al., 2012).
TBX6
T-box transcription factor 6 (TBX6) is a putative DNA-binding protein involved in the specification of paraxial mesoderm via SOX2 regulation (Takemoto et al., 2011). Heterozygous loss of function of TBX6 gene in human and mice has been associated with congenital anomalies of the kidney and urinary tract (CAKUT), thus relating this gene with the kidney defects often observed in 16p11.2 DEL carriers (Yang et al., 2020). In addition, the increased incidence of congenital vertebral malformations in 16p11.2 DUP patients could be ascribed to TBX6 dosage, as also suggested by mouse studies (Ren et al., 2020).
YPEL3
Yippee-like three is a member of putative Zinc-finger motif-coding genes and p53-regulated tumour suppressor (Kelley et al., 2010). Its role in 16p11.2 syndromes is unknown, although YPEL3 loss of function Zebrafish display abnormal brain morphology (Blaker-Lee et al., 2012).
GDPD3
Glycerophosphodiester phosphodiesterase domain-containing protein 3 (GDPD3) has lysophospholipase activity against several lisophospholipids (Ohshima et al., 2015). Loss of function Zebrafish for this gene is characterised by abnormalities in brain morphology (Blaker-Lee et al., 2012). To date, no other information is available regarding its specific role in neurodevelopmental disorders.
CORO1A
Coronin 1A (CORO1A) is an actin-binding protein previously associated with lymphocyte trafficking and homeostasis (Foger et al., 2006). Mutations in this protein have been found in patients with severe combined immunodeficiency (SCID) (Khoreva et al., 2024). Its function in the context of 16p11.2 CNVs has not been determined yet, although it is associated with low lymphocytes count in 16p11.2 DEL patients (Giannuzzi et al., 2022). Zebrafish carrying CORO1A loss of functions display abnormal body length, defective neural tubes, deficient axon tracts and movement defects (Blaker-Lee et al., 2012). In 16p11.2 DEL mice, CORO1A is enriched in Drd2+ MSNs (Portmann et al., 2014).
Genes with unknown function
C16orf54
Chromosome 16 open reading frame 54 (C16orf54) is a protein-coding gene expressed in 11 normal tissues and mainly located in the cell membrane. Although its function is unknown, overall C16orf54 appears as a potential marker of the poor prognosis of various tumours, suggesting that C16orf54 may participate in the progression of tumours (Du et al., 2022). C16orf54 expression is significantly associated with various immune-related pathways, such as PI3K/Akt/mTOR and Wnt/Beta catenin signalling pathways, inflammatory response, and interferon-gamma response (Ding et al., 2023). However, its specific biological function is still obscure and its role in 16p11.2 CNVs has not been investigated yet.
C16orf92
Chromosome 16 open reading frame 92 (C16orf92) encodes for a testis-specific protein necessary for the fusion between the oocyte and the sperm (Fujihara et al., 2020). To date, no other information is available regarding additional biological functions or potential association of this protein with neurodevelopmental disorders.
Fam57b
Fam57b consists of three variants upregulated during adipogenesis although at different degrees/timing. Fam57b mediates the production of lactosylceramide upon binding with a circulating metabolite of vitamin D with possible role in bone fracture healing (Martineau et al., 2018). Ceramides are precursors of sphingolipids, and their dysregulation can affect vesicle fusions and endocytic recycling. Ceramide derivatives interact with SNARE docking complex and recruit Munc13, the binding partner of Doc2A (calcium sensitive exocytosis regulator) thus enhancing calcium intracellular flow (Camoletto et al., 2009). McCammon et al. identified a subset of interacting 16p11.2 pairs of genes affecting brain and ventricle morphology in Zebrafish. In particular, DOC2A and Fam57a interacts to affect propensity to seizures and body size (McCammon et al., 2017). Its loss of function causes movement defects in Zebrafish, no response to touch and axonal defects (Blaker-Lee et al., 2012). In addition, Fam57b knock-down in Drosophila causes altered growth development of the neuromuscular junction (Iyer et al., 2018). Fam57b has been found enriched in Drd2+ MSNs in 16p11.2 DEL mice (Portmann et al., 2014), however its specific function in the context of neurodevelopmental disorders is currently unknown.
TMEM219
TMEM219 encodes for a transmembrane protein also known as IGFBP-3R (Insulin-like growth factor binding protein-3 receptor). It interacts with IGFBP-3 to mediate caspase-8-mediated apoptosis and tumour suppression in prostate and breast cancer xenografts (Ingermann et al., 2010). TMEM219 interacts with chitinase 3-like-1 (Chi3l1) and the multimeric complex formed by TMEM219/Chi3l1/IL-13Rα2 activates MAPK, AKT and Wnt signalling, inhibits oxidant-induced apoptosis in the lung, induces TGF- β1 and promotes melanoma metastasis (Lee et al., 2016). However, its role in 16p11.2 CNVs is currently unknown.
HIRIP3
HIRIP3 is highly expressed in human and mice embryos, in both cortex excitatory and striatal medium spiny neuron progenitors, as well as in the adult human brain (Morson et al., 2021). Early evidence suggested that HIRIP3 may be part of a histone H3.3 chaperone complex along with HIRA (Magnaghi et al., 1998; Ricketts et al., 2019), while HIRIP3 seems to partner with histone H2A/H2B (Assrir et al., 2007). HIRIP3 loss of function is known to produce movement defects in Zebrafish (Blaker-Lee et al., 2012).
Ino80e
Ino80e is a component of Ino80, an ATP-dependent chromatin remodelling complex. Although the specific role of Ino80e subunit is unknown, studies in yeast support the role of Ino80 complex in chromatin remodelling, DNA replication, stabilization of replication fork and resumption of replication after stress (Papamichos-Chronakis et al., 2006; van Attikum et al., 2007; Papamichos-Chronakis et al., 2011). Loss of function of Ino80e in Zebrafish is associated with defective neural tubes and abnormal body length (Blaker-Lee et al., 2012). This gene has been also found overexpressed in regions with decreased fiber density in 16p11.2 DEL male mice (Kumar et al., 2018).
Asphd1
The role of aspartate beta-hydroxylase domain-containing protein (Asphd1) is still unknown. In general, aspartate beta hydroxylases are rarely expressed in normal adult tissues, while they are overexpressed in several malignancies thus mediating cell proliferation and metastasis. They also appear to be a downstream target of ERK/MAPK and PI3K pathways (Hou et al., 2018).
Non-coding RNAs in the 16p11.2 region
Non-coding RNAs (ncRNAs) are a class of RNAs that do not encode for proteins and include ribosomal RNAs, transfer RNAs and regulatory non-coding RNAs, such as long non-coding RNAs, micro-RNAs, PIWI-interacting RNAs and circular RNAs.
Regulatory ncRNAs are classified by their size and subdivided in long nc-RNAs (>200 nucleotides) and short ncRNAs (<200 nucleotides). Regulatory ncRNAs can modulate the expression of homeotic genes and target chromatin remodelling complexes. In addition, they can regulate mRNA stability and protein translation (Amaral et al., 2008).
MicroRNAs, a class of small single-stranded RNAs (∼21 nucleotides), are particularly abundant in the brain. They can regulate gene expression via RNA-induced silencing complex (RISC)-mediated translational inhibition or, very rarely, via mRNA cleavage by binding to the 3′untranslated region (3′UTR) of target mRNAs (Vaishnavi et al., 2013).
ncRNAs have been involved in development and in differentiation (Amaral et al., 2008) and their altered expression has been linked with several neurodevelopmental disorders, such as ASD, Fragile X syndrome and intellectual disability (Zhang et al., 2019). Several miRNAs also play an important role in controlling gene expression programs during development by targeting genes like Notch, Nodal, and Hedgehog. In addition, miRNAs are known to be involved in brain development and dendritic spine morphology (Tekin et al., 2022). Interestingly, peripheral microRNAs could represent potential biomarkers for brain disorders due to their association with the neuroendocrine and neuroimmune system as well as potential therapeutic targets (Tekin et al., 2022).
By using RNAcentral (https://rnacentral.org), we identified ∼30 ncRNAs within the 16p11.2 region (see Table 3; Figure 1), including miRNAs, long ncRNAs and three are short ncRNAs, mostly with unknown functions.
Interestingly, has-miR-3680-3p has been recently found upregulated in the peripheral blood of bipolar disorders patients compared to controls. This miRNA targets MAOA gene, encoding for the enzyme responsible for the degradation of biogenic amines, and thus associated with mood disorders (Tekin et al., 2022). In addition, has-miR-3680-3p has been also involved in oesophageal squamous cell carcinoma and in osteoarthritis (OA) (Shi et al., 2019; Xie et al., 2022).
By investigating the presence of miRNAs in five groups of CNVs, Qiao et al., 2013 demonstrated an increased number of miRNAs in de novo CNVs compared to familial CNVs and common CNVs found in subjects with idiopathic or syndromic ID and neurotypical individuals. Although the number of miRNAs in familial CNVs is lower than in de novo CNVs, it is higher than in common CNVs. Within the 16p11.2 locus, they found two miRNAs (miR-3680-3p and miR-3680-5p) associated with the 16p11.2 paternal duplication, whereas the 16p11.2 de novo deletion had no miRNA content. The role of these miRNAs and their targets in CNVs has not been identified yet (Qiao et al., 2013).
Potential therapeutic approaches beyond the 16p11.2 genes
The focus of this review is the role of the genes located within 16p11.2 CNV in the pathophysiology of these NDDs. However, accumulating evidence indicates that therapeutic approaches to treat either 16p11.2 deletion or duplication may be effective without directly targeting genes located within this CNV region. Most of those approaches are based on the correction of excitation/inhibition (E/I) imbalances, frequently observed in different animal models of NDDs (Lee et al., 2017).
The most promising approach for 16p11.2 deletion is based on the use of GABA-B receptor agonists, such as R-Baclofen (Arbaclofen). This is a safe off-patent drug, and its racemic version has already been approved by both the FDA (Food and Drug Administration) and the EMA (European Medicines Agency) for the treatment of spasticity in multiple sclerosis and cerebral palsy. It is also commonly prescribed to children and adolescents with cerebral palsy (Dario and Tomei, 2004).
Most notably, Arbaclofen has been proven effective in reversing symptoms of models of Fragile X and has already been tested in clinical trials, although without definitive evidence of clinical efficacy (Berry-Kravis et al., 2012; Henderson et al., 2012; Zeidler et al., 2018).
More recently, the same drug has been tested in three different models of 16p11.2 deletion and the data generally support its efficacy in reverting some, but not all, behavioural symptoms (Stoppel et al., 2018; Gundersen et al., 2023).
This has led SFARI to initiate a recruitment for a clinical trial on 16p11.2 deletion carriers (https://clinicaltrials.gov/study/NCT04271332). Despite the obvious excitement about the first potential effective treatment for DEL carriers, it is important to highlight that very little is known about the mechanisms allowing Arbaclofen to change E/I in 16p11.2 deletion syndrome, a state that is apparently shared with Fragile X Syndrome.
Additional proposed treatments, already tested for Fragile X Syndrome, are based on the inhibition of metabotropic mGLUR5 receptors by using negative allosteric modulators (NAMs), such as mavoglurant and basimglurant (see for reference (Stoppel et al., 2017)). This observation is interesting and points to a potential dysregulation of protein synthesis downstream mGLUR5 receptors. However, it may need to be entirely reconciled with the additional evidence that glutamatergic function seems to be disrupted in a mouse model of 16p11.2 deletion and that chemogenetic activation of the prefrontal cortex pyramidal neurons may ameliorate cognitive symptoms (Tian et al., 2015; Berry-Kravis et al., 2017; Wang et al., 2018).
A final potentially relevant observation is that serotoninergic signalling via 5-HT1B receptors in the nucleus accumbens is disrupted in 16p11.2 deletion mice as well as in other ASD models. Both optogenetic or pharmacological stimulation of the serotonin pathway can reverse social deficits (Walsh et al., 2018; Walsh et al., 2021).
All the above approaches have been devised and tested to treat the 16p11.2 deletion syndrome. Translational research on the mirror condition, the 16p11.2 duplication syndrome, is lagging behind. However, in a recent paper, Npas4 has been identified as a potential therapeutic target. This GABAergic specific transcription factor has been found downregulated in the cortex of the DUP mouse model, in conjunction with an inhibitory transmission deficit and enhanced excitability. Remarkably, restoration of Npas4 expression in the PFC was able to rescue both synaptic and behavioural deficits in DUP mice (Rein et al., 2020). In addition, since Npas4 expression is negatively regulated by the epigenetic enzyme histone deacetylase 5 (HDAC5), administration of a HDAC5 inhibitor could restore GABAergic signalling and rescue behavioural deficits (Rein et al., 2022).
These studies are important, also because they suggest that pharmacological approaches aiming at ameliorating GABAergic activity, including the use of Arbaclofen, may be a potential way forward, as already evaluated for the 16p11.2 deletion syndrome. This evidence may also indicate that changes in E/I and circuitry disruption at the system level may converge onto the two conditions, despite the difference in gene dosage.
Conclusion
16p11.2 deletion and duplications are amongst the most intensively studied neurodevelopmental syndromes. Over the last decade, considerable evidence has accumulated on these two genetically mirrored conditions, most notably in the domain of patient phenotyping and genotyping. Few hypotheses have been proposed to explain the reason why deletion and duplication carriers display opposite phenotypes (e.g., BMI, brain size), or at least different symptoms (e.g., schizophrenia and bipolar disorder are present only in duplication carriers). One possibility is that disease-specific symptoms may harbour from gene dosage differences within a subset of the 27 genes or non-coding RNAs located in the 16p11.2 CNV. However, no convincing experimental evidence in animal or cellular models is present to support this plausible, but difficult to test, hypothesis. Genetic approaches inducing mutations of the individual genes or pairs/trios of genes from a DEL to a DUP condition (and vice versa) could be employed to demonstrate that different behavioural or cellular phenotypes “appear” to be consistent with the mirror NDD. This would create “chimeric” models that are neither DEL nor DUP but do bear unique phenotypes of each state. We believe that the technology is now mature to perform this set of experiments, even in mammalian animal models, thanks to the advent of genome editing. However, the poor characterisation of the available deletion and duplication models, especially at the behavioural level, is still a major problem in the field. Before attempting complex but feasible genetic experiments, it will be imperative to characterise “at best” the available models, also considering the genetic background which is known to be remarkably important in modifying behaviour. In the context of 16p11.2 CNVs, see for instance (Arbogast et al., 2016) where deficits in social behaviour could be unmasked in a different genetic background. It is possible that, in the next years, more progress will be made using 2D and especially 3D cellular models generated from iPSCs, potentially “simplified” systems than in vivo models to study multiple gene function. However, even those approaches still have significant drawbacks (i.e., the availability of proper isogenic controls and the significant differences in differentiation protocols among different laboratories).
An even more complex problem is that the “core symptoms” of 16p11.2 deletion and duplication syndromes, such as ID and ASD, cannot be explained by differences in gene dosage. Maybe we have so far overestimated this aspect, considering that ID and ASD, or even epilepsy, arise from hundreds of different mutations, from “monogenic” forms (e.g., FXS or various RASopathies) to a variety of CNVs associated to NDDs. If developmental and behavioural biologists have learned any common lesson, it is clear that behavioural deficits may result from a large and potentially non-overlapping set of molecular and cellular process all impacting on the relevant circuitry. The prevalent idea that E/I imbalance may be at the core of any NDDs is important at the theoretical level but may not help much from the practical point of view. Very often in the past, translational approaches have been based on limited experimental evidence supporting E/I change, and clinical trials have been undertaken without substantial support drawn from work on patients. We desperately need more imaging and neurophysiological studies in patients, before considering applying drugs in a clinical setting. We believe that a better understanding of the patient’s endophenotypes and the identification of brain biomarkers for 16p11.2 deletion and duplication syndromes will better guide also translational research in experimental models. Thus, collaborative projects including cell, mice and patients’ studies will be the key to success in devising novel treatments.
One final note is about the syndromic nature of most NDDs, including 16p11.2 CNVs. The research is still too “neuro-centric” and most scientists, including us, have overlooked metabolic dysfunctions. These are instead likely to play a central role in syndromic NDDs, such as the 16p11.2 CNVs, and can clearly impact on the patients’ quality of life and their life expectancy. For instance, the 16p11.2 deletion syndrome is also associated with obesity and hyper insulinemic hypoglycaemia (Kostopoulou et al., 2019). A recent study highlighted unique and sex-specific metabolic signatures, also related to mitochondrial function, in three different mouse models of NDDs, including the 16p11.2 DEL (Menzies et al., 2021). It might be plausible that some genes in the CNV may be directly responsible for “peripheral” phenotypes, although this aspect has not been fully addressed in our review. However, we recognise that it is no longer sustainable to study neurodevelopmental disorders without considering the body as a whole. Investigating peripheral alterations in 16p11.2 CNVs, together with dissecting “brain-specific” phenotypes, will be crucial to inform and guide the development of future clinical interventions for these disabling conditions.
Author contributions
RL: Writing–original draft, Writing–review and editing. CZ: Writing–original draft, Writing–review and editing. RB: Conceptualization, Funding acquisition, Supervision, Writing–original draft, Writing–review and editing. IM: Conceptualization, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. IM was financially supported by the Medical Research Council Award (MR/S037667/1) of the “Therapeutic Target Validation in Mental Health” Programme (to RB). Work was supported by #NEXTGENERATIONEU (NGEU) and funded by the Ministry of University and Research (MUR), National Recovery and Resilience Plan (NRRP), project MNESYS (PE0000006)–A Multiscale integrated approach to the study of the nervous system in health and disease (DN. 1553 11.10.2022).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agarwalla, S., Arroyo, N. S., Long, N. E., O'Brien, W. T., Abel, T., and Bandyopadhyay, S. (2020). Male-specific alterations in structure of isolation call sequences of mouse pups with 16p11.2 deletion. Genes. Brain Behav. 19 (7), e12681. doi:10.1111/gbb.12681
Amaral, P. P., Dinger, M. E., Mercer, T. R., and Mattick, J. S. (2008). The eukaryotic genome as an RNA machine. Science 319 (5871), 1787–1789. doi:10.1126/science.1155472
Angelakos, C. C., Watson, A. J., O'Brien, W. T., Krainock, K. S., Nickl-Jockschat, T., and Abel, T. (2017). Hyperactivity and male-specific sleep deficits in the 16p11.2 deletion mouse model of autism. Autism Res. 10 (4), 572–584. doi:10.1002/aur.1707
Arbogast, T., Ouagazzal, A. M., Chevalier, C., Kopanitsa, M., Afinowi, N., Migliavacca, E., et al. (2016). Reciprocal effects on neurocognitive and metabolic phenotypes in mouse models of 16p11.2 deletion and duplication syndromes. PLoS Genet. 12 (2), e1005709. doi:10.1371/journal.pgen.1005709
Arbogast, T., Razaz, P., Ellegood, J., McKinstry, S. U., Erdin, S., Currall, B., et al. (2019). Kctd13-deficient mice display short-term memory impairment and sex-dependent genetic interactions. Hum. Mol. Genet. 28 (9), 1474–1486. doi:10.1093/hmg/ddy436
Assrir, N., Filhol, O., Galisson, F., and Lipinski, M. (2007). HIRIP3 is a nuclear phosphoprotein interacting with and phosphorylated by the serine-threonine kinase CK2. Biol. Chem. 388 (4), 391–398. doi:10.1515/BC.2007.045
Backhausen, L. L., Herting, M. M., Tamnes, C. K., and Vetter, N. C. (2022). Best practices in structural neuroimaging of neurodevelopmental disorders. Neuropsychol. Rev. 32 (2), 400–418. doi:10.1007/s11065-021-09496-2
Bassett, A. S., Chow, E. W., AbdelMalik, P., Gheorghiu, M., Husted, J., and Weksberg, R. (2003). The schizophrenia phenotype in 22q11 deletion syndrome. Am. J. Psychiatry. 160 (9), 1580–6. doi:10.1176/appi.ajp.160.9.1580
Berger, W., Steiner, E., Grusch, M., Elbling, L., and Micksche, M. (2009). Vaults and the major vault protein: novel roles in signal pathway regulation and immunity. Cell. Mol. Life Sci. 66 (1), 43–61. doi:10.1007/s00018-008-8364-z
Berry-Kravis, E., Hagerman, R., Visootsak, J., Budimirovic, D., Kaufmann, W. E., Cherubini, M., et al. (2017). Arbaclofen in fragile X syndrome: results of phase 3 trials. J. Neurodev. Disord. 9, 3. doi:10.1186/s11689-016-9181-6
Berry-Kravis, E. M., Hessl, D., Rathmell, B., Zarevics, P., Cherubini, M., Walton-Bowen, K., et al. (2012). Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci. Transl. Med. 4 (152), 152ra127. doi:10.1126/scitranslmed.3004214
Blaker-Lee, A., Gupta, S., McCammon, J. M., De Rienzo, G., and Sive, H. (2012). Zebrafish homologs of genes within 16p11.2, a genomic region associated with brain disorders, are active during brain development, and include two deletion dosage sensor genes. Dis. Model. Mech. 5 (6), 834–851. doi:10.1242/dmm.009944
Boonen, M., Staudt, C., Gilis, F., Oorschot, V., Klumperman, J., and Jadot, M. (2016). Cathepsin D and its newly identified transport receptor SEZ6L2 can modulate neurite outgrowth. J. Cell. Sci. 129 (3), 557–568. doi:10.1242/jcs.179374
Bristow, G. C., Thomson, D. M., Openshaw, R. L., Mitchell, E. J., Pratt, J. A., Dawson, N., et al. (2020). 16p11 duplication disrupts hippocampal-orbitofrontal-amygdala connectivity, revealing a neural circuit endophenotype for schizophrenia. Cell. Rep. 31 (3), 107536. doi:10.1016/j.celrep.2020.107536
Brunner, D., Kabitzke, P., He, D., Cox, K., Thiede, L., Hanania, T., et al. (2015). Comprehensive analysis of the 16p11.2 deletion and null Cntnap2 mouse models of autism spectrum disorder. PLoS One 10 (8), e0134572. doi:10.1371/journal.pone.0134572
Cai, M., Wang, R., Liu, M., Du, X., Xue, K., Ji, Y., et al. (2022). Disrupted local functional connectivity in schizophrenia: an updated and extended meta-analysis. Schizophr. (Heidelb) 8 (1), 93. doi:10.1038/s41537-022-00311-2
Camoletto, P. G., Vara, H., Morando, L., Connell, E., Marletto, F. P., Giustetto, M., et al. (2009). Synaptic vesicle docking: sphingosine regulates syntaxin1 interaction with Munc18. PLoS One 4 (4), e5310. doi:10.1371/journal.pone.0005310
Cannon, T. D., and Keller, M. C. (2006). Endophenotypes in the genetic analyses of mental disorders. Annu. Rev. Clin. Psychol. 2, 267–290. doi:10.1146/annurev.clinpsy.2.022305.095232
Cappello, S., Bohringer, C. R., Bergami, M., Conzelmann, K. K., Ghanem, A., Tomassy, G. S., et al. (2012). A radial glia-specific role of RhoA in double cortex formation. Neuron 73 (5), 911–924. doi:10.1016/j.neuron.2011.12.030
Chang, Y. S., Owen, J. P., Pojman, N. J., Thieu, T., Bukshpun, P., Wakahiro, M. L., et al. (2016). Reciprocal white matter alterations due to 16p11.2 chromosomal deletions versus duplications. Hum. Brain Mapp. 37 (8), 2833–2848. doi:10.1002/hbm.23211
Chawner, S. J., Watson, C. J., and Owen, M. J. (2021). Clinical evaluation of patients with a neuropsychiatric risk copy number variant. Curr. Opin. Genet. Dev. 68, 26–34. doi:10.1016/j.gde.2020.12.012
Chen, G. I., Tisayakorn, S., Jorgensen, C., D'Ambrosio, L. M., Goudreault, M., and Gingras, A. C. (2008). PP4R4/KIAA1622 forms a novel stable cytosolic complex with phosphoprotein phosphatase 4. J. Biol. Chem. 283 (43), 29273–29284. doi:10.1074/jbc.M803443200
Cho, Y. W., Hong, T., Hong, S., Guo, H., Yu, H., Kim, D., et al. (2007). PTIP associates with MLL3-and MLL4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 282 (28), 20395–20406. doi:10.1074/jbc.M701574200
Chow, J., Jensen, M., Amini, H., Hormozdiari, F., Penn, O., Shifman, S., et al. (2019). Dissecting the genetic basis of comorbid epilepsy phenotypes in neurodevelopmental disorders. Genome Med. 11 (1), 65. doi:10.1186/s13073-019-0678-y
Connacher, R., Williams, M., Prem, S., Yeung, P. L., Matteson, P., Mehta, M., et al. (2022). Autism NPCs from both idiopathic and CNV 16p11.2 deletion patients exhibit dysregulation of proliferation and mitogenic responses. Stem Cell. Rep. 17 (6), 1380–1394. doi:10.1016/j.stemcr.2022.04.019
Courchesne, E., Campbell, K., and Solso, S. (2011). Brain growth across the life span in autism: age-specific changes in anatomical pathology. Brain Res. 1380, 138–145. doi:10.1016/j.brainres.2010.09.101
Courtney, N. A., Briguglio, J. S., Bradberry, M. M., Greer, C., and Chapman, E. R. (2018). Excitatory and inhibitory neurons utilize different Ca(2+) sensors and sources to regulate spontaneous release. Neuron 98 (5), 977–991. doi:10.1016/j.neuron.2018.04.022
Crespi, B., Stead, P., and Elliot, M. (2010). Evolution in health and medicine Sackler colloquium: comparative genomics of autism and schizophrenia. Proc. Natl. Acad. Sci. U. S. A. 107 (Suppl. 1), 1736–1741. doi:10.1073/pnas.0906080106
Cross-Disorder Group of the Psychiatric Genomics, C. (2013). Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381 (9875), 1371–1379. doi:10.1016/S0140-6736(12)62129-1
Dahoun, T., Trossbach, S. V., Brandon, N. J., Korth, C., and Howes, O. D. (2017). The impact of Disrupted-in-Schizophrenia 1 (DISC1) on the dopaminergic system: a systematic review. Transl. Psychiatry 7 (1), e1015. doi:10.1038/tp.2016.282
D'Angelo, D., Lebon, S., Chen, Q., Martin-Brevet, S., Snyder, L. G., Hippolyte, L., et al. (2016). Defining the effect of the 16p11.2 duplication on cognition, behavior, and medical comorbidities. JAMA Psychiatry 73 (1), 20–30. doi:10.1001/jamapsychiatry.2015.2123
Daniels, J. L., Forssen, U., Hultman, C. M., Cnattingius, S., Savitz, D. A., Feychting, M., et al. (2008). Parental psychiatric disorders associated with autism spectrum disorders in the offspring. Pediatrics 121 (5), e1357–e1362. doi:10.1542/peds.2007-2296
Dario, A., and Tomei, G. (2004). A benefit-risk assessment of baclofen in severe spinal spasticity. Drug Saf. 27 (11), 799–818. doi:10.2165/00002018-200427110-00004
Daum, H., Ganapathi, M., Hirsch, Y., Griffin, E. L., LeDuc, C. A., Hagen, J., et al. (2022). Bi-allelic PAGR1 variants are associated with microcephaly and a severe neurodevelopmental disorder: genetic evidence from two families. Am. J. Med. Genet. A 188 (1), 336–342. doi:10.1002/ajmg.a.62513
de Anda, F. C., Rosario, A. L., Durak, O., Tran, T., Graff, J., Meletis, K., et al. (2012). Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat. Neurosci. 15 (7), 1022–1031. doi:10.1038/nn.3141
Deshpande, A., Yadav, S., Dao, D. Q., Wu, Z. Y., Hokanson, K. C., Cahill, M. K., et al. (2017). Cellular phenotypes in human iPSC-derived neurons from a genetic model of autism spectrum disorder. Cell. Rep. 21 (10), 2678–2687. doi:10.1016/j.celrep.2017.11.037
Di Martino, A., Kelly, C., Grzadzinski, R., Zuo, X. N., Mennes, M., Mairena, M. A., et al. (2011). Aberrant striatal functional connectivity in children with autism. Biol. Psychiatry 69 (9), 847–856. doi:10.1016/j.biopsych.2010.10.029
Ding, T., Zhang, Y., Ren, Z., Cong, Y., Long, J., Peng, M., et al. (2023). EBV-associated hub genes as potential biomarkers for predicting the prognosis of nasopharyngeal carcinoma. Viruses 15 (9), 1915. doi:10.3390/v15091915
Doherty, J. L., and Owen, M. J. (2014). Genomic insights into the overlap between psychiatric disorders: implications for research and clinical practice. Genome Med. 6 (4), 29. doi:10.1186/gm546
Dong, X., Akuetteh, P. D. P., Song, J., Ni, C., Jin, C., Li, H., et al. (2021). Major vault protein (MVP) associated with BRAF (V600E) mutation is an immune microenvironment-related biomarker promoting the progression of papillary thyroid cancer via MAPK/ERK and PI3K/AKT pathways. Front. Cell. Dev. Biol. 9, 688370. doi:10.3389/fcell.2021.688370
Du, X., Xia, W., Fan, W., Shen, X., Wu, H., and Zhang, H. (2022). Integrated analysis of C16orf54 as a potential prognostic, diagnostic, and immune marker across pan-cancer. Dis. Markers 2022, 9365046. doi:10.1155/2022/9365046
Eberhard, D., Billstedt, E., and Gillberg, C. (2022). Neurodevelopmental disorders and comorbidity in young adults attending a psychiatric outpatient clinic. Psychiatry Res. 313, 114638. doi:10.1016/j.psychres.2022.114638
Engel, S. R., Creson, T. K., Hao, Y., Shen, Y., Maeng, S., Nekrasova, T., et al. (2009). The extracellular signal-regulated kinase pathway contributes to the control of behavioral excitement. Mol. Psychiatry 14 (4), 448–461. doi:10.1038/sj.mp.4002135
Escamilla, C. O., Filonova, I., Walker, A. K., Xuan, Z. X., Holehonnur, R., Espinosa, F., et al. (2017). Kctd13 deletion reduces synaptic transmission via increased RhoA. Nature 551 (7679), 227–231. doi:10.1038/nature24470
Faraone, S. V., and Larsson, H. (2019). Genetics of attention deficit hyperactivity disorder. Mol. Psychiatry 24 (4), 562–575. doi:10.1038/s41380-018-0070-0
Fasano, S., and Brambilla, R. (2011). Ras-ERK signaling in behavior: old questions and new perspectives. Front. Behav. Neurosci. 5, 79. doi:10.3389/fnbeh.2011.00079
Ferguson, S. M., Fasano, S., Yang, P., Brambilla, R., and Robinson, T. E. (2006). Knockout of ERK1 enhances cocaine-evoked immediate early gene expression and behavioral plasticity. Neuropsychopharmacology 31 (12), 2660–2668. doi:10.1038/sj.npp.1301014
Fetit, R., Barbato, M. I., Theil, T., Pratt, T., and Price, D. J. (2023). 16p11.2 deletion accelerates subpallial maturation and increases variability in human iPSC-derived ventral telencephalic organoids. Development 150 (4), dev201227. doi:10.1242/dev.201227
Fetit, R., Price, D. J., Lawrie, S. M., and Johnstone, M. (2020). Understanding the clinical manifestations of 16p11.2 deletion syndrome: a series of developmental case reports in children. Psychiatr. Genet. 30 (5), 136–140. doi:10.1097/YPG.0000000000000259
Fleck, D. E., Sax, K. W., and Strakowski, S. M. (2001). Reaction time measures of sustained attention differentiate bipolar disorder from schizophrenia. Schizophr. Res. 52 (3), 251–259. doi:10.1016/s0920-9964(01)00170-0
Foger, N., Rangell, L., Danilenko, D. M., and Chan, A. C. (2006). Requirement for coronin 1 in T lymphocyte trafficking and cellular homeostasis. Science 313 (5788), 839–842. doi:10.1126/science.1130563
Fornito, A., Yucel, M., Dean, B., Wood, S. J., and Pantelis, C. (2009). Anatomical abnormalities of the anterior cingulate cortex in schizophrenia: bridging the gap between neuroimaging and neuropathology. Schizophr. Bull. 35 (5), 973–993. doi:10.1093/schbul/sbn025
Forrest, M. P., Dos Santos, M., Piguel, N. H., Wang, Y. Z., Hawkins, N. A., Bagchi, V. A., et al. (2023). Rescue of neuropsychiatric phenotypes in a mouse model of 16p11.2 duplication syndrome by genetic correction of an epilepsy network hub. Nat. Commun. 14 (1), 825. doi:10.1038/s41467-023-36087-x
Fromer, M., Pocklington, A. J., Kavanagh, D. H., Williams, H. J., Dwyer, S., Gormley, P., et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506 (7487), 179–184. doi:10.1038/nature12929
Fujihara, Y., Lu, Y., Noda, T., Oji, A., Larasati, T., Kojima-Kita, K., et al. (2020). Spermatozoa lacking Fertilization Influencing Membrane Protein (FIMP) fail to fuse with oocytes in mice. Proc. Natl. Acad. Sci. U. S. A. 117 (17), 9393–9400. doi:10.1073/pnas.1917060117
Fyke, W., and Velinov, M. (2021). FMR1 and autism, an intriguing connection revisited. Genes. (Basel) 12 (8), 1218. doi:10.3390/genes12081218
Gandal, M. J., Zhang, P., Hadjimichael, E., Walker, R. L., Chen, C., Liu, S., et al. (2018). Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362 (6420), eaat8127. doi:10.1126/science.aat8127
Giannuzzi, G., Chatron, N., Mannik, K., Auwerx, C., Pradervand, S., Willemin, G., et al. (2022). Possible association of 16p11.2 copy number variation with altered lymphocyte and neutrophil counts. NPJ Genom Med. 7 (1), 38. doi:10.1038/s41525-022-00308-x
Giannuzzi, G., Schmidt, P. J., Porcu, E., Willemin, G., Munson, K. M., Nuttle, X., et al. (2019). The human-specific BOLA2 duplication modifies iron homeostasis and anemia predisposition in chromosome 16p11.2 autism individuals. Am. J. Hum. Genet. 105 (5), 947–958. doi:10.1016/j.ajhg.2019.09.023
Golzio, C., Willer, J., Talkowski, M. E., Oh, E. C., Taniguchi, Y., Jacquemont, S., et al. (2012). KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 485 (7398), 363–367. doi:10.1038/nature11091
Gong, Z., Cho, Y. W., Kim, J. E., Ge, K., and Chen, J. (2009). Accumulation of Pax2 transactivation domain interaction protein (PTIP) at sites of DNA breaks via RNF8-dependent pathway is required for cell survival after DNA damage. J. Biol. Chem. 284 (11), 7284–7293. doi:10.1074/jbc.M809158200
Grayton, H. M., Fernandes, C., Rujescu, D., and Collier, D. A. (2012). Copy number variations in neurodevelopmental disorders. Prog. Neurobiol. 99 (1), 81–91. doi:10.1016/j.pneurobio.2012.07.005
Green Snyder, L., D'Angelo, D., Chen, Q., Bernier, R., Goin-Kochel, R. P., Wallace, A. S., et al. (2016). Autism spectrum disorder, developmental and psychiatric features in 16p11.2 duplication. J. Autism Dev. Disord. 46 (8), 2734–2748. doi:10.1007/s10803-016-2807-4
Grissom, N. M., McKee, S. E., Schoch, H., Bowman, N., Havekes, R., O'Brien, W. T., et al. (2018). Male-specific deficits in natural reward learning in a mouse model of neurodevelopmental disorders. Mol. Psychiatry 23 (3), 544–555. doi:10.1038/mp.2017.184
Gundersen, B. B., O'Brien, W. T., Schaffler, M. D., Schultz, M. N., Tsukahara, T., Lorenzo, S. M., et al. (2023). Towards preclinical validation of arbaclofen (R-baclofen) treatment for 16p11.2 deletion syndrome. bioRxiv, 538987. doi:10.1101/2023.05.01.538987
Gusev, A., Mancuso, N., Won, H., Kousi, M., Finucane, H. K., Reshef, Y., et al. (2018). Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat. Genet. 50 (4), 538–548. doi:10.1038/s41588-018-0092-1
Hall, L. S., Medway, C. W., Pain, O., Pardinas, A. F., Rees, E. G., Escott-Price, V., et al. (2020). A transcriptome-wide association study implicates specific pre- and post-synaptic abnormalities in schizophrenia. Hum. Mol. Genet. 29 (1), 159–167. doi:10.1093/hmg/ddz253
Haller, M., Au, J., O'Neill, M., and Lamb, D. J. (2018). 16p11.2 transcription factor MAZ is a dosage-sensitive regulator of genitourinary development. Proc. Natl. Acad. Sci. U. S. A. 115 (8), E1849–E1858. doi:10.1073/pnas.1716092115
Hanson, E., Bernier, R., Porche, K., Jackson, F. I., Goin-Kochel, R. P., Snyder, L. G., et al. (2015). The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biol. Psychiatry 77 (9), 785–793. doi:10.1016/j.biopsych.2014.04.021
Haslinger, D., Waltes, R., Yousaf, A., Lindlar, S., Schneider, I., Lim, C. K., et al. (2018). Loss of the Chr16p11.2 ASD candidate gene QPRT leads to aberrant neuronal differentiation in the SH-SY5Y neuronal cell model. Mol. Autism 9, 56. doi:10.1186/s13229-018-0239-z
Henderson, C., Wijetunge, L., Kinoshita, M. N., Shumway, M., Hammond, R. S., Postma, F. R., et al. (2012). Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci. Transl. Med. 4 (152), 152ra128. doi:10.1126/scitranslmed.3004218
Henis, M., Rucker, T., Scharrenberg, R., Richter, M., Baltussen, L., Hong, S., et al. (2024). The autism susceptibility kinase, TAOK2, phosphorylates eEF2 and modulates translation. Sci. Adv. 10 (15), eadf7001. doi:10.1126/sciadv.adf7001
Heron, S. E., and Dibbens, L. M. (2013). Role of PRRT2 in common paroxysmal neurological disorders: a gene with remarkable pleiotropy. J. Med. Genet. 50 (3), 133–139. doi:10.1136/jmedgenet-2012-101406
Hoogman, M., Bralten, J., Hibar, D. P., Mennes, M., Zwiers, M. P., Schweren, L. S. J., et al. (2017). Subcortical brain volume differences in participants with attention deficit hyperactivity disorder in children and adults: a cross-sectional mega-analysis. Lancet Psychiatry 4 (4), 310–319. doi:10.1016/S2215-0366(17)30049-4
Horev, G., Ellegood, J., Lerch, J. P., Son, Y. E., Muthuswamy, L., Vogel, H., et al. (2011). Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc. Natl. Acad. Sci. U. S. A. 108 (41), 17076–17081. doi:10.1073/pnas.1114042108
Hou, G., Xu, B., Bi, Y., Wu, C., Ru, B., Sun, B., et al. (2018). Recent advances in research on aspartate β-hydroxylase (ASPH) in pancreatic cancer: a brief update. Bosn. J. Basic Med. Sci. 18 (4), 297–304. doi:10.17305/bjbms.2018.3539
Indrigo, M., Morella, I., Orellana, D., d'Isa, R., Papale, A., Parra, R., et al. (2023). Nuclear ERK1/2 signaling potentiation enhances neuroprotection and cognition via Importinα1/KPNA2. EMBO Mol. Med. 15 (11), e15984. doi:10.15252/emmm.202215984
Indrigo, M., Papale, A., Orellana, D., and Brambilla, R. (2010). Lentiviral vectors to study the differential function of ERK1 and ERK2 MAP kinases. Methods Mol. Biol. 661, 205–220. doi:10.1007/978-1-60761-795-2_12
Ingermann, A. R., Yang, Y. F., Han, J., Mikami, A., Garza, A. E., Mohanraj, L., et al. (2010). Identification of a novel cell death receptor mediating IGFBP-3-induced anti-tumor effects in breast and prostate cancer. J. Biol. Chem. 285 (39), 30233–30246. doi:10.1074/jbc.M110.122226
Iyer, J., Singh, M. D., Jensen, M., Patel, P., Pizzo, L., Huber, E., et al. (2018). Pervasive genetic interactions modulate neurodevelopmental defects of the autism-associated 16p11.2 deletion in Drosophila melanogaster. Nat. Commun. 9 (1), 2548. doi:10.1038/s41467-018-04882-6
Jacquemont, S., Reymond, A., Zufferey, F., Harewood, L., Walters, R. G., Kutalik, Z., et al. (2011). Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature 478 (7367), 97–102. doi:10.1038/nature10406
Kelley, K. D., Miller, K. R., Todd, A., Kelley, A. R., Tuttle, R., and Berberich, S. J. (2010). YPEL3, a p53-regulated gene that induces cellular senescence. Cancer Res. 70 (9), 3566–3575. doi:10.1158/0008-5472.CAN-09-3219
Khoreva, A., Butov, K. R., Nikolaeva, E. I., Martyanov, A., Kulakovskaya, E., Pershin, D., et al. (2024). Novel hemizygous CORO1A variant leads to combined immunodeficiency with defective platelet calcium signaling and cell mobility. J. Allergy Clin. Immunol. Glob. 3 (1), 100172. doi:10.1016/j.jacig.2023.100172
Kim, E., Lee, S., Mian, M. F., Yun, S. U., Song, M., Yi, K. S., et al. (2006). Crosstalk between Src and major vault protein in epidermal growth factor-dependent cell signalling. FEBS J. 273 (4), 793–804. doi:10.1111/j.1742-4658.2006.05112.x
Kim, J., Vanrobaeys, Y., Kelvington, B., Peterson, Z., Baldwin, E., Gaine, M. E., et al. (2024). Dissecting 16p11.2 hemi-deletion to study sex-specific striatal phenotypes of neurodevelopmental disorders. Mol. Psychiatry. doi:10.1038/s41380-024-02411-0
Kirov, G. (2015). CNVs in neuropsychiatric disorders. Hum. Mol. Genet. 24 (R1), R45–R49. doi:10.1093/hmg/ddv253
Kolli, S., Zito, C. I., Mossink, M. H., Wiemer, E. A., and Bennett, A. M. (2004). The major vault protein is a novel substrate for the tyrosine phosphatase SHP-2 and scaffold protein in epidermal growth factor signaling. J. Biol. Chem. 279 (28), 29374–29385. doi:10.1074/jbc.M313955200
Kostopoulou, E., Dastamani, A., Caiulo, S., Antell, H., Flanagan, S. E., and Shah, P. (2019). Hyperinsulinaemic hypoglycaemia: a new presentation of 16p11.2 deletion syndrome. Clin. Endocrinol. (Oxf) 90 (5), 766–769. doi:10.1111/cen.13951
Kretz, P. F., Wagner, C., Mikhaleva, A., Montillot, C., Hugel, S., Morella, I., et al. (2023). Dissecting the autism-associated 16p11.2 locus identifies multiple drivers in neuroanatomical phenotypes and unveils a male-specific role for the major vault protein. Genome Biol. 24 (1), 261. doi:10.1186/s13059-023-03092-8
Kumar, A., Lualdi, M., Loncarek, J., Cho, Y. W., Lee, J. E., Ge, K., et al. (2014). Loss of function of mouse Pax-Interacting Protein 1-associated glutamate rich protein 1a (Pagr1a) leads to reduced Bmp2 expression and defects in chorion and amnion development. Dev. Dyn. 243 (7), 937–947. doi:10.1002/dvdy.24125
Kumar, R. A., Marshall, C. R., Badner, J. A., Babatz, T. D., Mukamel, Z., Aldinger, K. A., et al. (2009). Association and mutation analyses of 16p11.2 autism candidate genes. PLoS One 4 (2), e4582. doi:10.1371/journal.pone.0004582
Kumar, V. J., Grissom, N. M., McKee, S. E., Schoch, H., Bowman, N., Havekes, R., et al. (2018). Linking spatial gene expression patterns to sex-specific brain structural changes on a mouse model of 16p11.2 hemideletion. Transl. Psychiatry 8 (1), 109. doi:10.1038/s41398-018-0157-z
Kuo, S. S., and Pogue-Geile, M. F. (2019). Variation in fourteen brain structure volumes in schizophrenia: a comprehensive meta-analysis of 246 studies. Neurosci. Biobehav Rev. 98, 85–94. doi:10.1016/j.neubiorev.2018.12.030
Lange, N., Travers, B. G., Bigler, E. D., Prigge, M. B., Froehlich, A. L., Nielsen, J. A., et al. (2015). Longitudinal volumetric brain changes in autism spectrum disorder ages 6-35 years. Autism Res. 8 (1), 82–93. doi:10.1002/aur.1427
Langen, M., Schnack, H. G., Nederveen, H., Bos, D., Lahuis, B. E., de Jonge, M. V., et al. (2009). Changes in the developmental trajectories of striatum in autism. Biol. Psychiatry 66 (4), 327–333. doi:10.1016/j.biopsych.2009.03.017
Larsson, H., Ryden, E., Boman, M., Langstrom, N., Lichtenstein, P., and Landen, M. (2013). Risk of bipolar disorder and schizophrenia in relatives of people with attention-deficit hyperactivity disorder. Br. J. Psychiatry 203 (2), 103–106. doi:10.1192/bjp.bp.112.120808
Larsson, H. J., Eaton, W. W., Madsen, K. M., Vestergaard, M., Olesen, A. V., Agerbo, E., et al. (2005). Risk factors for autism: perinatal factors, parental psychiatric history, and socioeconomic status. Am. J. Epidemiol. 161 (10), 916–925. ; discussion 926-918. doi:10.1093/aje/kwi123
Lee, C. M., He, C. H., Nour, A. M., Zhou, Y., Ma, B., Park, J. W., et al. (2016). IL-13Rα2 uses TMEM219 in chitinase 3-like-1-induced signalling and effector responses. Nat. Commun. 7, 12752. doi:10.1038/ncomms12752
Lee, E., Lee, J., and Kim, E. (2017). Excitation/inhibition imbalance in animal models of autism spectrum disorders. Biol. Psychiatry 81 (10), 838–847. doi:10.1016/j.biopsych.2016.05.011
Li, J., Brickler, T., Banuelos, A., Marjon, K., Shcherbina, A., Banerjee, S., et al. (2021). Overexpression of CD47 is associated with brain overgrowth and 16p11.2 deletion syndrome. Proc. Natl. Acad. Sci. U. S. A. 118 (15), e2005483118. doi:10.1073/pnas.2005483118
Lin, G. N., Corominas, R., Lemmens, I., Yang, X., Tavernier, J., Hill, D. E., et al. (2015). Spatiotemporal 16p11.2 protein network implicates cortical late mid-fetal brain development and KCTD13-Cul3-RhoA pathway in psychiatric diseases. Neuron 85 (4), 742–754. doi:10.1016/j.neuron.2015.01.010
Liu, B., Ma, A., Zhang, F., Wang, Y., Li, Z., Li, Q., et al. (2016). MAZ mediates the cross-talk between CT-1 and NOTCH1 signaling during gliogenesis. Sci. Rep. 6, 21534. doi:10.1038/srep21534
Lu, Y., Shan, Q., Ling, M., Ni, X. A., Mao, S. S., Yu, B., et al. (2022). Identification of key genes involved in axon regeneration and Wallerian degeneration by weighted gene co-expression network analysis. Neural Regen. Res. 17 (4), 911–919. doi:10.4103/1673-5374.322473
Ma, L., Mazidi, M., Li, K., Li, Y., Chen, S., Kirwan, R., et al. (2021). Prevalence of mental health problems among children and adolescents during the COVID-19 pandemic: a systematic review and meta-analysis. J. Affect Disord. 293, 78–89. doi:10.1016/j.jad.2021.06.021
Magnaghi, P., Roberts, C., Lorain, S., Lipinski, M., and Scambler, P. J. (1998). HIRA, a mammalian homologue of Saccharomyces cerevisiae transcriptional co-repressors, interacts with Pax3. Nat. Genet. 20 (1), 74–77. doi:10.1038/1739
Maillard, A. M., Romascano, D., Villalon-Reina, J. E., Moreau, C. A., Almeida Osorio, J. M., Richetin, S., et al. (2024). Pervasive alterations of intra-axonal volume and network organization in young children with a 16p11.2 deletion. Transl. Psychiatry 14 (1), 95. doi:10.1038/s41398-024-02810-5
Maillard, A. M., Ruef, A., Pizzagalli, F., Migliavacca, E., Hippolyte, L., Adaszewski, S., et al. (2015). The 16p11.2 locus modulates brain structures common to autism, schizophrenia and obesity. Mol. Psychiatry 20 (1), 140–147. doi:10.1038/mp.2014.145
Marchi, M., D'Antoni, A., Formentini, I., Parra, R., Brambilla, R., Ratto, G. M., et al. (2008). The N-terminal domain of ERK1 accounts for the functional differences with ERK2. PLoS One 3 (12), e3873. doi:10.1371/journal.pone.0003873
Martin-Brevet, S., Rodriguez-Herreros, B., Nielsen, J. A., Moreau, C., Modenato, C., Maillard, A. M., et al. (2018). Quantifying the effects of 16p11.2 copy number variants on brain structure: a multisite genetic-first study. Biol. Psychiatry 84 (4), 253–264. doi:10.1016/j.biopsych.2018.02.1176
Martineau, C., Naja, R. P., Husseini, A., Hamade, B., Kaufmann, M., Akhouayri, O., et al. (2018). Optimal bone fracture repair requires 24R,25-dihydroxyvitamin D3 and its effector molecule FAM57B2. J. Clin. Investig. 128 (8), 3546–3557. doi:10.1172/JCI98093
Martin Lorenzo, S., Muniz Moreno, M. D. M., Atas, H., Pellen, M., Nalesso, V., Raffelsberger, W., et al. (2023). Changes in social behavior with MAPK2 and KCTD13/CUL3 pathways alterations in two new outbred rat models for the 16p11.2 syndromes with autism spectrum disorders. Front. Neurosci. 17, 1148683. doi:10.3389/fnins.2023.1148683
Martin Lorenzo, S., Nalesso, V., Chevalier, C., Birling, M. C., and Herault, Y. (2021). Targeting the RHOA pathway improves learning and memory in adult Kctd13 and 16p11.2 deletion mouse models. Mol. Autism 12 (1), 1. doi:10.1186/s13229-020-00405-7
Mazzucchelli, C., Vantaggiato, C., Ciamei, A., Fasano, S., Pakhotin, P., Krezel, W., et al. (2002). Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 34 (5), 807–820. doi:10.1016/s0896-6273(02)00716-x
McCammon, J. M., Blaker-Lee, A., Chen, X., and Sive, H. (2017). The 16p11.2 homologs fam57ba and doc2a generate certain brain and body phenotypes. Hum. Mol. Genet. 26 (19), 3699–3712. doi:10.1093/hmg/ddx255
McCarthy, S. E., Makarov, V., Kirov, G., Addington, A. M., McClellan, J., Yoon, S., et al. (2009). Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 41 (11), 1223–1227. doi:10.1038/ng.474
Medina-Martinez, O., Haller, M., Rosenfeld, J. A., O'Neill, M. A., Lamb, D. J., and Jamrich, M. (2020). The transcription factor Maz is essential for normal eye development. Dis. Model. Mech. 13 (8), dmm044412. doi:10.1242/dmm.044412
Meng, H., Yao, W., Yin, Y., Li, Y., Ding, Y., Wang, L., et al. (2022). ZG16 promotes T-cell mediated immunity through direct binding to PD-L1 in colon cancer. Biomark. Res. 10 (1), 47. doi:10.1186/s40364-022-00396-y
Menzies, C., Naz, S., Patten, D., Alquier, T., Bennett, B. M., and Lacoste, B. (2021). Distinct basal metabolism in three mouse models of neurodevelopmental disorders. eNeuro 8 (2), ENEURO.0292–20.2021. doi:10.1523/ENEURO.0292-20.2021
Migliavacca, E., Golzio, C., Mannik, K., Blumenthal, I., Oh, E. C., Harewood, L., et al. (2015). A potential contributory role for ciliary dysfunction in the 16p11.2 600 kb BP4-BP5 pathology. Am. J. Hum. Genet. 96 (5), 784–796. doi:10.1016/j.ajhg.2015.04.002
Mitchell, K. J. (2011). The genetics of neurodevelopmental disease. Curr. Opin. Neurobiol. 21 (1), 197–203. doi:10.1016/j.conb.2010.08.009
Miyazaki, T., Hashimoto, K., Uda, A., Sakagami, H., Nakamura, Y., Saito, S. Y., et al. (2006). Disturbance of cerebellar synaptic maturation in mutant mice lacking BSRPs, a novel brain-specific receptor-like protein family. FEBS Lett. 580 (17), 4057–4064. doi:10.1016/j.febslet.2006.06.043
Mollon, J., Almasy, L., Jacquemont, S., and Glahn, D. C. (2023). The contribution of copy number variants to psychiatric symptoms and cognitive ability. Mol. Psychiatry. 28 (4), 1480–1493. doi:10.1038/s41380-023-01978-4
Moore, T. M., Garg, R., Johnson, C., Coptcoat, M. J., Ridley, A. J., and Morris, J. D. (2000). PSK, a novel STE20-like kinase derived from prostatic carcinoma that activates the c-Jun N-terminal kinase mitogen-activated protein kinase pathway and regulates actin cytoskeletal organization. J. Biol. Chem. 275 (6), 4311–4322. doi:10.1074/jbc.275.6.4311
More, L., Lauterborn, J. C., Papaleo, F., and Brambilla, R. (2020). Enhancing cognition through pharmacological and environmental interventions: examples from preclinical models of neurodevelopmental disorders. Neurosci. Biobehav Rev. 110, 28–45. doi:10.1016/j.neubiorev.2019.02.003
Morson, S., Yang, Y., Price, D. J., and Pratt, T. (2021). Expression of genes in the 16p11.2 locus during development of the human fetal cerebral cortex. Cereb. Cortex 31 (9), 4038–4052. doi:10.1093/cercor/bhab067
Murphy, T. R., Vihtelic, T. S., Ile, K. E., Watson, C. T., Willer, G. B., Gregg, R. G., et al. (2011). Phosphatidylinositol synthase is required for lens structural integrity and photoreceptor cell survival in the zebrafish eye. Exp. Eye Res. 93 (4), 460–474. doi:10.1016/j.exer.2011.06.010
Nelson, S. B., and Valakh, V. (2015). Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87 (4), 684–698. doi:10.1016/j.neuron.2015.07.033
Neves, D., Goodfellow, B. J., Vieira, S. I., and Silva, R. M. (2022). The role of NAD metabolism in neuronal differentiation. Neurochem. Int. 159, 105402. doi:10.1016/j.neuint.2022.105402
Niarchou, M., Chawner, S., Doherty, J. L., Maillard, A. M., Jacquemont, S., Chung, W. K., et al. (2019). Psychiatric disorders in children with 16p11.2 deletion and duplication. Transl. Psychiatry 9 (1), 8. doi:10.1038/s41398-018-0339-8
Nilsson, I., and Hoffmann, I. (2000). Cell cycle regulation by the Cdc25 phosphatase family. Prog. Cell. Cycle Res. 4, 107–114. doi:10.1007/978-1-4615-4253-7_10
Nyquist, D. A., and Helmkamp, G. M. (1989). Developmental patterns in rat brain of phosphatidylinositol synthetic enzymes and phosphatidylinositol transfer protein. Biochim. Biophys. Acta 987 (2), 165–170. doi:10.1016/0005-2736(89)90539-7
Ohshima, N., Kudo, T., Yamashita, Y., Mariggio, S., Araki, M., Honda, A., et al. (2015). New members of the mammalian glycerophosphodiester phosphodiesterase family: GDE4 and GDE7 produce lysophosphatidic acid by lysophospholipase D activity. J. Biol. Chem. 290 (7), 4260–4271. doi:10.1074/jbc.M114.614537
Okamoto, S., Sherman, K., Bai, G., and Lipton, S. A. (2002). Effect of the ubiquitous transcription factors, SP1 and MAZ, on NMDA receptor subunit type 1 (NR1) expression during neuronal differentiation. Brain Res. Mol. Brain Res. 107 (2), 89–96. doi:10.1016/s0169-328x(02)00440-0
Oliva-Teles, N., de Stefano, M. C., Gallagher, L., Rakic, S., Jorge, P., Cuturilo, G., et al. (2020). Rare pathogenic copy number variation in the 16p11.2 (BP4-BP5) region associated with neurodevelopmental and neuropsychiatric disorders: a review of the literature. Int. J. Environ. Res. Public Health 17 (24), 9253. doi:10.3390/ijerph17249253
Openshaw, R. L., Thomson, D. M., Bristow, G. C., Mitchell, E. J., Pratt, J. A., Morris, B. J., et al. (2023). 16p11.2 deletion mice exhibit compromised fronto-temporal connectivity, GABAergic dysfunction, and enhanced attentional ability. Commun. Biol. 6 (1), 557. doi:10.1038/s42003-023-04891-2
Ortabozkoyun, H., Huang, P. Y., Cho, H., Narendra, V., LeRoy, G., Gonzalez-Buendia, E., et al. (2022). CRISPR and biochemical screens identify MAZ as a cofactor in CTCF-mediated insulation at Hox clusters. Nat. Genet. 54 (2), 202–212. doi:10.1038/s41588-021-01008-5
Owen, J. P., Chang, Y. S., Pojman, N. J., Bukshpun, P., Wakahiro, M. L., Marco, E. J., et al. (2014). Aberrant white matter microstructure in children with 16p11.2 deletions. J. Neurosci. 34 (18), 6214–6223. doi:10.1523/JNEUROSCI.4495-13.2014
Owen, M. J., O'Donovan, M. C., Thapar, A., and Craddock, N. (2011). Neurodevelopmental hypothesis of schizophrenia. Br. J. Psychiatry 198 (3), 173–175. doi:10.1192/bjp.bp.110.084384
Padmanabha, H., Raghavendra, K., Arunachal, G., Nagaraj, A. R., Harishma, R. S., Nashi, S., et al. (2024). Episodic ataxia in child with 16p11.2 deletion including PRRT2. Park. Relat. Disord. 118, 105955. doi:10.1016/j.parkreldis.2023.105955
Papale, A., d'Isa, R., Menna, E., Cerovic, M., Solari, N., Hardingham, N., et al. (2017). Severe intellectual disability and enhanced gamma-aminobutyric acidergic synaptogenesis in a novel model of rare RASopathies. Biol. Psychiatry 81 (3), 179–192. doi:10.1016/j.biopsych.2016.06.016
Papale, A., Morella, I. M., Indrigo, M. T., Bernardi, R. E., Marrone, L., Marchisella, F., et al. (2016). Impairment of cocaine-mediated behaviours in mice by clinically relevant Ras-ERK inhibitors. Elife 5, e17111. doi:10.7554/eLife.17111
Papamichos-Chronakis, M., Krebs, J. E., and Peterson, C. L. (2006). Interplay between Ino80 and Swr1 chromatin remodeling enzymes regulates cell cycle checkpoint adaptation in response to DNA damage. Genes. Dev. 20 (17), 2437–2449. doi:10.1101/gad.1440206
Papamichos-Chronakis, M., Watanabe, S., Rando, O. J., and Peterson, C. L. (2011). Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell. 144 (2), 200–213. doi:10.1016/j.cell.2010.12.021
Park, S., and Holzman, P. S. (1992). Schizophrenics show spatial working memory deficits. Arch. Gen. Psychiatry 49 (12), 975–982. doi:10.1001/archpsyc.1992.01820120063009
Park, S. M., Littleton, J. T., Park, H. R., and Lee, J. H. (2016). Drosophila homolog of human KIF22 at the autism-linked 16p11.2 loci influences synaptic connectivity at larval neuromuscular junctions. Exp. Neurobiol. 25 (1), 33–39. doi:10.5607/en.2016.25.1.33
Parnell, E., Culotta, L., Forrest, M. P., Jalloul, H. A., Eckman, B. L., Loizzo, D. D., et al. (2023). Excitatory dysfunction drives network and calcium handling deficits in 16p11.2 duplication schizophrenia induced pluripotent stem cell-derived neurons. Biol. Psychiatry 94 (2), 153–163. doi:10.1016/j.biopsych.2022.11.005
Portmann, T., Yang, M., Mao, R., Panagiotakos, G., Ellegood, J., Dolen, G., et al. (2014). Behavioral abnormalities and circuit defects in the basal ganglia of a mouse model of 16p11.2 deletion syndrome. Cell. Rep. 7 (4), 1077–1092. doi:10.1016/j.celrep.2014.03.036
Prat, C. S., Stocco, A., Neuhaus, E., and Kleinhans, N. M. (2016). Basal ganglia impairments in autism spectrum disorder are related to abnormal signal gating to prefrontal cortex. Neuropsychologia 91, 268–281. doi:10.1016/j.neuropsychologia.2016.08.007
Pucilowska, J., Vithayathil, J., Pagani, M., Kelly, C., Karlo, J. C., Robol, C., et al. (2018). Pharmacological inhibition of ERK signaling rescues pathophysiology and behavioral phenotype associated with 16p11.2 chromosomal deletion in mice. J. Neurosci. 38 (30), 6640–6652. doi:10.1523/JNEUROSCI.0515-17.2018
Pucilowska, J., Vithayathil, J., Tavares, E. J., Kelly, C., Karlo, J. C., and Landreth, G. E. (2015). The 16p11.2 deletion mouse model of autism exhibits altered cortical progenitor proliferation and brain cytoarchitecture linked to the ERK MAPK pathway. J. Neurosci. 35 (7), 3190–3200. doi:10.1523/JNEUROSCI.4864-13.2015
Qiao, Y., Badduke, C., Mercier, E., Lewis, S. M., Pavlidis, P., and Rajcan-Separovic, E. (2013). miRNA and miRNA target genes in copy number variations occurring in individuals with intellectual disability. BMC Genomics 14, 544. doi:10.1186/1471-2164-14-544
Qiu, Y., Arbogast, T., Lorenzo, S. M., Li, H., Tang, S. C., Richardson, E., et al. (2019). Oligogenic effects of 16p11.2 copy-number variation on craniofacial development. Cell. Rep. 28 (13), 3320–3328. doi:10.1016/j.celrep.2019.08.071
Qureshi, A. Y., Mueller, S., Snyder, A. Z., Mukherjee, P., Berman, J. I., Roberts, T. P., et al. (2014). Opposing brain differences in 16p11.2 deletion and duplication carriers. J. Neurosci. 34 (34), 11199–11211. doi:10.1523/JNEUROSCI.1366-14.2014
Rein, B., Conrow-Graham, M., Frazier, A., Cao, Q., and Yan, Z. (2022). Inhibition of histone deacetylase 5 ameliorates abnormalities in 16p11.2 duplication mouse model. Neuropharmacology 204, 108893. doi:10.1016/j.neuropharm.2021.108893
Rein, B., Tan, T., Yang, F., Wang, W., Williams, J., Zhang, F., et al. (2020). Reversal of synaptic and behavioral deficits in a 16p11.2 duplication mouse model via restoration of the GABA synapse regulator Npas4. Mol. Psychiatry 26, 1967–1979. doi:10.1038/s41380-020-0693-9
Rein, B., and Yan, Z. (2020). 16p11.2 copy number variations and neurodevelopmental disorders. Trends Neurosci. 43 (11), 886–901. doi:10.1016/j.tins.2020.09.001
Ren, X., Yang, N., Wu, N., Xu, X., Chen, W., Zhang, L., et al. (2020). Increased TBX6 gene dosages induce congenital cervical vertebral malformations in humans and mice. J. Med. Genet. 57 (6), 371–379. doi:10.1136/jmedgenet-2019-106333
Richter, M., Murtaza, N., Scharrenberg, R., White, S. H., Johanns, O., Walker, S., et al. (2019). Altered TAOK2 activity causes autism-related neurodevelopmental and cognitive abnormalities through RhoA signaling. Mol. Psychiatry 24 (9), 1329–1350. doi:10.1038/s41380-018-0025-5
Ricketts, M. D., Dasgupta, N., Fan, J., Han, J., Gerace, M., Tang, Y., et al. (2019). The HIRA histone chaperone complex subunit UBN1 harbors H3/H4- and DNA-binding activity. J. Biol. Chem. 294 (23), 9239–9259. doi:10.1074/jbc.RA119.007480
Ripke, S., O'Dushlaine, C., Chambert, K., Moran, J. L., Kahler, A. K., Akterin, S., et al. (2013). Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 45 (10), 1150–1159. doi:10.1038/ng.2742
Roy, D. S., Zhang, Y., Aida, T., Choi, S., Chen, Q., Hou, Y., et al. (2021). Anterior thalamic dysfunction underlies cognitive deficits in a subset of neuropsychiatric disease models. Neuron 109 (16), 2590–2603.e13. doi:10.1016/j.neuron.2021.06.005
Rund, B. R. (2009). Is there a degenerative process going on in the brain of people with Schizophrenia? Front. Hum. Neurosci. 3, 36. doi:10.3389/neuro.09.036.2009
Santos, S., Ferreira, H., Martins, J., Goncalves, J., and Castelo-Branco, M. (2022). Male sex bias in early and late onset neurodevelopmental disorders: shared aspects and differences in Autism Spectrum Disorder, Attention Deficit/hyperactivity Disorder, and Schizophrenia. Neurosci. Biobehav Rev. 135, 104577. doi:10.1016/j.neubiorev.2022.104577
Scharrenberg, R., Richter, M., Johanns, O., Meka, D. P., Rucker, T., Murtaza, N., et al. (2022). TAOK2 rescues autism-linked developmental deficits in a 16p11.2 microdeletion mouse model. Mol. Psychiatry 27 (11), 4707–4721. doi:10.1038/s41380-022-01785-3
Schobel, S. A., Kelly, M. A., Corcoran, C. M., Van Heertum, K., Seckinger, R., Goetz, R., et al. (2009). Anterior hippocampal and orbitofrontal cortical structural brain abnormalities in association with cognitive deficits in schizophrenia. Schizophr. Res. 114 (1-3), 110–118. doi:10.1016/j.schres.2009.07.016
Sebat, J., Lakshmi, B., Malhotra, D., Troge, J., Lese-Martin, C., Walsh, T., et al. (2007). Strong association of de novo copy number mutations with autism. Science 316 (5823), 445–449. doi:10.1126/science.1138659
Shaw, P., Stringaris, A., Nigg, J., and Leibenluft, E. (2014). Emotion dysregulation in attention deficit hyperactivity disorder. Am. J. Psychiatry 171 (3), 276–293. doi:10.1176/appi.ajp.2013.13070966
Shen, C., Huo, L. R., Zhao, X. L., Wang, P. R., and Zhong, N. (2015). Novel interactive partners of neuroligin 3: new aspects for pathogenesis of autism. J. Mol. Neurosci. 56 (1), 89–101. doi:10.1007/s12031-014-0470-9
Shenton, M. E., Dickey, C. C., Frumin, M., and McCarley, R. W. (2001). A review of MRI findings in schizophrenia. Schizophr. Res. 49 (1-2), 1–52. doi:10.1016/s0920-9964(01)00163-3
Shi, N., Shan, B., Gu, B., Song, Y., Chu, H., and Qian, L. (2019). Circular RNA circ-PRKCI functions as a competitive endogenous RNA to regulate AKT3 expression by sponging miR-3680-3p in esophageal squamous cell carcinoma. J. Cell. Biochem. 120 (6), 10021–10030. doi:10.1002/jcb.28285
Shiflett, M. W., and Balleine, B. W. (2011). Contributions of ERK signaling in the striatum to instrumental learning and performance. Behav. Brain Res. 218 (1), 240–247. doi:10.1016/j.bbr.2010.12.010
Shinawi, M., Liu, P., Kang, S. H., Shen, J., Belmont, J. W., Scott, D. A., et al. (2010). Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J. Med. Genet. 47 (5), 332–341. doi:10.1136/jmg.2009.073015
Sonderby, I. E., Ching, C. R. K., Thomopoulos, S. I., van der Meer, D., Sun, D., Villalon-Reina, J. E., et al. (2022). Effects of copy number variations on brain structure and risk for psychiatric illness: large-scale studies from the ENIGMA working groups on CNVs. Hum. Brain Mapp. 43 (1), 300–328. doi:10.1002/hbm.25354
Stefansson, H., Meyer-Lindenberg, A., Steinberg, S., Magnusdottir, B., Morgen, K., Arnarsdottir, S., et al. (2014). CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 505 (7483), 361–366. doi:10.1038/nature12818
Steinman, K. J., Spence, S. J., Ramocki, M. B., Proud, M. B., Kessler, S. K., Marco, E. J., et al. (2016). 16p11.2 deletion and duplication: characterizing neurologic phenotypes in a large clinically ascertained cohort. Am. J. Med. Genet. A 170 (11), 2943–2955. doi:10.1002/ajmg.a.37820
Stoppel, L. J., Kazdoba, T. M., Schaffler, M. D., Preza, A. R., Heynen, A., Crawley, J. N., et al. (2018). R-baclofen reverses cognitive deficits and improves social interactions in two lines of 16p11.2 deletion mice. Neuropsychopharmacology 43 (3), 513–524. doi:10.1038/npp.2017.236
Stoppel, L. J., Osterweil, E. K., and Bear, M. F. (2017). The mGluR theory of fragile X: from mice to men. Fragile X Syndrome Genet. Target. Treat., 173–204. doi:10.1016/B978-0-12-804461-2.00009-3
Sullivan, P. F., Magnusson, C., Reichenberg, A., Boman, M., Dalman, C., Davidson, M., et al. (2012). Family history of schizophrenia and bipolar disorder as risk factors for autism. Arch. Gen. Psychiatry 69 (11), 1099–1103. doi:10.1001/archgenpsychiatry.2012.730
Sumner, P. J., Bell, I. H., and Rossell, S. L. (2018). A systematic review of the structural neuroimaging correlates of thought disorder. Neurosci. Biobehav Rev. 84, 299–315. doi:10.1016/j.neubiorev.2017.08.017
Sundberg, M., Pinson, H., Smith, R. S., Winden, K. D., Venugopal, P., Tai, D. J. C., et al. (2021). 16p11.2 deletion is associated with hyperactivation of human iPSC-derived dopaminergic neuron networks and is rescued by RHOA inhibition in vitro. Nat. Commun. 12 (1), 2897. doi:10.1038/s41467-021-23113-z
Takemoto, T., Uchikawa, M., Yoshida, M., Bell, D. M., Lovell-Badge, R., Papaioannou, V. E., et al. (2011). Tbx6-dependent Sox2 regulation determines neural or mesodermal fate in axial stem cells. Nature 470 (7334), 394–398. doi:10.1038/nature09729
Tekin, S. S., Erdal, M. E., Asoglu, M., Ay, O. I., Ay, M. E., and Yilmaz, S. G. (2022). Biomarker potential of hsa-miR-145-5p in peripheral whole blood of manic bipolar I patients. Braz J. Psychiatry 40 (44), 378–387. doi:10.47626/1516-4446-2021-2260
Teng, X., Aouacheria, A., Lionnard, L., Metz, K. A., Soane, L., Kamiya, A., et al. (2019). KCTD: a new gene family involved in neurodevelopmental and neuropsychiatric disorders. CNS Neurosci. Ther. 25 (7), 887–902. doi:10.1111/cns.13156
Tian, D., Stoppel, L. J., Heynen, A. J., Lindemann, L., Jaeschke, G., Mills, A. A., et al. (2015). Contribution of mGluR5 to pathophysiology in a mouse model of human chromosome 16p11.2 microdeletion. Nat. Neurosci. 18 (2), 182–184. doi:10.1038/nn.3911
Tokai, N., Fujimoto-Nishiyama, A., Toyoshima, Y., Yonemura, S., Tsukita, S., Inoue, J., et al. (1996). Kid, a novel kinesin-like DNA binding protein, is localized to chromosomes and the mitotic spindle. EMBO J. 15 (3), 457–467. doi:10.1002/j.1460-2075.1996.tb00378.x
Tokai-Nishizumi, N., Ohsugi, M., Suzuki, E., and Yamamoto, T. (2005). The chromokinesin Kid is required for maintenance of proper metaphase spindle size. Mol. Biol. Cell. 16 (11), 5455–5463. doi:10.1091/mbc.e05-03-0244
Ultanir, S. K., Yadav, S., Hertz, N. T., Oses-Prieto, J. A., Claxton, S., Burlingame, A. L., et al. (2014). MST3 kinase phosphorylates TAO1/2 to enable Myosin Va function in promoting spine synapse development. Neuron 84 (5), 968–982. doi:10.1016/j.neuron.2014.10.025
Urresti, J., Zhang, P., Moran-Losada, P., Yu, N. K., Negraes, P. D., Trujillo, C. A., et al. (2021). Cortical organoids model early brain development disrupted by 16p11.2 copy number variants in autism. Mol. Psychiatry 26 (12), 7560–7580. doi:10.1038/s41380-021-01243-6
Vaishnavi, V., Manikandan, M., Tiwary, B. K., and Munirajan, A. K. (2013). Insights on the functional impact of microRNAs present in autism-associated copy number variants. PLoS One 8 (2), e56781. doi:10.1371/journal.pone.0056781
Valtorta, F., Benfenati, F., Zara, F., and Meldolesi, J. (2016). PRRT2: from paroxysmal disorders to regulation of synaptic function. Trends Neurosci. 39 (10), 668–679. doi:10.1016/j.tins.2016.08.005
van Attikum, H., Fritsch, O., and Gasser, S. M. (2007). Distinct roles for SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J. 26 (18), 4113–4125. doi:10.1038/sj.emboj.7601835
Vantaggiato, C., Formentini, I., Bondanza, A., Bonini, C., Naldini, L., and Brambilla, R. (2006). ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. J. Biol. 5 (5), 14. doi:10.1186/jbiol38
Vithayathil, J., Pucilowska, J., and Landreth, G. E. (2018). ERK/MAPK signaling and autism spectrum disorders. Prog. Brain Res. 241, 63–112. doi:10.1016/bs.pbr.2018.09.008
Vlaskamp, D. R. M., Callenbach, P. M. C., Rump, P., Giannini, L. A. A., Brilstra, E. H., Dijkhuizen, T., et al. (2019). PRRT2-related phenotypes in patients with a 16p11.2 deletion. Eur. J. Med. Genet. 62 (4), 265–269. doi:10.1016/j.ejmg.2018.08.002
von Kriegsheim, A., Baiocchi, D., Birtwistle, M., Sumpton, D., Bienvenut, W., Morrice, N., et al. (2009). Cell fate decisions are specified by the dynamic ERK interactome. Nat. Cell. Biol. 11 (12), 1458–1464. doi:10.1038/ncb1994
Wakatsuki, S., Takahashi, Y., Shibata, M., Adachi, N., Numakawa, T., Kunugi, H., et al. (2021). Small noncoding vault RNA modulates synapse formation by amplifying MAPK signaling. J. Cell. Biol. 220 (2), e201911078. doi:10.1083/jcb.201911078
Wallace, K. S., and Rogers, S. J. (2010). Intervening in infancy: implications for autism spectrum disorders. J. Child. Psychol. Psychiatry 51 (12), 1300–1320. doi:10.1111/j.1469-7610.2010.02308.x
Walsh, J. J., Christoffel, D. J., Heifets, B. D., Ben-Dor, G. A., Selimbeyoglu, A., Hung, L. W., et al. (2018). 5-HT release in nucleus accumbens rescues social deficits in mouse autism model. Nature 560 (7720), 589–594. doi:10.1038/s41586-018-0416-4
Walsh, J. J., Llorach, P., Cardozo Pinto, D. F., Wenderski, W., Christoffel, D. J., Salgado, J. S., et al. (2021). Systemic enhancement of serotonin signaling reverses social deficits in multiple mouse models for ASD. Neuropsychopharmacology 46 (11), 2000–2010. doi:10.1038/s41386-021-01091-6
Walsh, T., McClellan, J. M., McCarthy, S. E., Addington, A. M., Pierce, S. B., Cooper, G. M., et al. (2008). Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320 (5875), 539–543. doi:10.1126/science.1155174
Wang, J., Cheng, H., Li, X., Lu, W., Wang, K., and Wen, T. (2013). Regulation of neural stem cell differentiation by transcription factors HNF4-1 and MAZ-1. Mol. Neurobiol. 47 (1), 228–240. doi:10.1007/s12035-012-8335-0
Wang, Q. W., Qin, J., Chen, Y. F., Tu, Y., Xing, Y. Y., Wang, Y., et al. (2023). 16p11.2 CNV gene Doc2α functions in neurodevelopment and social behaviors through interaction with Secretagogin. Cell. Rep. 42 (7), 112691. doi:10.1016/j.celrep.2023.112691
Wang, W., Rein, B., Zhang, F., Tan, T., Zhong, P., Qin, L., et al. (2018). Chemogenetic activation of prefrontal cortex rescues synaptic and behavioral deficits in a mouse model of 16p11.2 deletion syndrome. J. Neurosci. 38 (26), 5939–5948. doi:10.1523/JNEUROSCI.0149-18.2018
Watkins, L. V., Linehan, C., Brandt, C., Snoeijen-Schouwenaars, F., McGowan, P., and Shankar, R. (2022). Epilepsy in adults with neurodevelopmental disability - what every neurologist should know. Epileptic Disord. 24 (1), 9–25. doi:10.1684/epd.2021.1366
Wegiel, J., Flory, M., Kuchna, I., Nowicki, K., Ma, S. Y., Imaki, H., et al. (2014). Stereological study of the neuronal number and volume of 38 brain subdivisions of subjects diagnosed with autism reveals significant alterations restricted to the striatum, amygdala and cerebellum. Acta Neuropathol. Commun. 2, 141. doi:10.1186/s40478-014-0141-7
Weiss, L. A., Shen, Y., Korn, J. M., Arking, D. E., Miller, D. T., Fossdal, R., et al. (2008). Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 358 (7), 667–675. doi:10.1056/NEJMoa075974
Xie, Y., Li, J., Suo, Y., Li, Y., Li, Q., and Chen, X. (2022). Downregulation of miR-3680-3p inhibits the progression of osteoarthritis via targeting OGG1. Arch. Gerontol. Geriatr. 100, 104626. doi:10.1016/j.archger.2022.104626
Yadav, S., Oses-Prieto, J. A., Peters, C. J., Zhou, J., Pleasure, S. J., Burlingame, A. L., et al. (2017). TAOK2 kinase mediates PSD95 stability and dendritic spine maturation through Septin7 phosphorylation. Neuron 93 (2), 379–393. doi:10.1016/j.neuron.2016.12.006
Yang, M., Lewis, F., Foley, G., and Crawley, J. N. (2015a). In tribute to Bob Blanchard: divergent behavioral phenotypes of 16p11.2 deletion mice reared in same-genotype versus mixed-genotype cages. Physiol. Behav. 146, 16–27. doi:10.1016/j.physbeh.2015.04.023
Yang, M., Lewis, F. C., Sarvi, M. S., Foley, G. M., and Crawley, J. N. (2015b). 16p11.2 Deletion mice display cognitive deficits in touchscreen learning and novelty recognition tasks. Learn Mem. 22 (12), 622–632. doi:10.1101/lm.039602.115
Yang, M., Mahrt, E. J., Lewis, F., Foley, G., Portmann, T., Dolmetsch, R. E., et al. (2015c). 16p11.2 deletion syndrome mice display sensory and ultrasonic vocalization deficits during social interactions. Autism Res. 8 (5), 507–521. doi:10.1002/aur.1465
Yang, N., Wu, N., Dong, S., Zhang, L., Zhao, Y., Chen, W., et al. (2020). Human and mouse studies establish TBX6 in Mendelian CAKUT and as a potential driver of kidney defects associated with the 16p11.2 microdeletion syndrome. Kidney Int. 98 (4), 1020–1030. doi:10.1016/j.kint.2020.04.045
Yang, Y., Booker, S. A., Clegg, J. M., Quintana-Urzainqui, I., Sumera, A., Kozic, Z., et al. (2023). Identifying foetal forebrain interneurons as a target for monogenic autism risk factors and the polygenic 16p11.2 microdeletion. BMC Neurosci. 24 (1), 5. doi:10.1186/s12868-022-00771-3
Yasuda, S., Tanaka, H., Sugiura, H., Okamura, K., Sakaguchi, T., Tran, U., et al. (2007). Activity-induced protocadherin arcadlin regulates dendritic spine number by triggering N-cadherin endocytosis via TAO2beta and p38 MAP kinases. Neuron 56 (3), 456–471. doi:10.1016/j.neuron.2007.08.020
Zarrei, M., Burton, C. L., Engchuan, W., Young, E. J., Higginbotham, E. J., MacDonald, J. R., et al. (2019). A large data resource of genomic copy number variation across neurodevelopmental disorders. NPJ Genom Med. 4, 26. doi:10.1038/s41525-019-0098-3
Zeidler, S., Pop, A. S., Jaafar, I. A., de Boer, H., Buijsen, R. A. M., de Esch, C. E. F., et al. (2018). Paradoxical effect of baclofen on social behavior in the fragile X syndrome mouse model. Brain Behav. 8 (6), e00991. doi:10.1002/brb3.991
Zhang, L., Wan, Z. X., Zhu, J. Y., Liu, H. J., Sun, J., Zou, X. H., et al. (2024). A girl with PRRT2 mutation presenting with benign familial infantile seizures followed by autistic regression. Case Rep. Pediatr. 2024, 5539799. doi:10.1155/2024/5539799
Zhang, S. F., Gao, J., and Liu, C. M. (2019). The role of non-coding RNAs in neurodevelopmental disorders. Front. Genet. 10, 1033. doi:10.3389/fgene.2019.01033
Zhang, Z., Huang, J., Shen, Y., and Li, R. (2017). BACE1-Dependent neuregulin-1 signaling: an implication for schizophrenia. Front. Mol. Neurosci. 10, 302. doi:10.3389/fnmol.2017.00302
Zhou, T., Raman, M., Gao, Y., Earnest, S., Chen, Z., Machius, M., et al. (2004). Crystal structure of the TAO2 kinase domain: activation and specificity of a Ste20p MAP3K. Structure 12 (10), 1891–1900. doi:10.1016/j.str.2004.07.021
Keywords: 16p11.2 CNV, basal ganglia, MAPK3, neurodevelopmental disorders, animal models, iPSC (induced pluripotent stem cell), excitation/inhibition (E/I) imbalance, intellectual disability
Citation: Leone R, Zuglian C, Brambilla R and Morella I (2024) Understanding copy number variations through their genes: a molecular view on 16p11.2 deletion and duplication syndromes. Front. Pharmacol. 15:1407865. doi: 10.3389/fphar.2024.1407865
Received: 27 March 2024; Accepted: 16 May 2024;
Published: 14 June 2024.
Edited by:
Harry Pantazopoulos, University of Mississippi Medical Center, United StatesReviewed by:
Luigi Balasco, University of Trento, ItalySimon Trent, Keele University, United Kingdom
Copyright © 2024 Leone, Zuglian, Brambilla and Morella. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Riccardo Brambilla, cmljY2FyZG8uYnJhbWJpbGxhQHVuaXB2Lml0; Ilaria Morella, bW9yZWxsYWlAY2FyZGlmZi5hYy51aw==