 Kyung U. Hong1,2*
Kyung U. Hong1,2* David W. Hein
David W. Hein- 1Department of Pharmacology and Toxicology and Brown Cancer Center, University of Louisville School of Medicine, Louisville, KY, United States
- 2Department of Pharmaceutical and Administrative Sciences, College of Pharmacy and Health Sciences, Western New England University, Springfield, MA, United States
Introduction: Arylamine N-acetyltransferase 2 (NAT2) expresses a well-defined genetic polymorphism in humans that modifies drug and xenobiotic metabolism. Recent studies and genome wide association studies have reported that genetic variants of NAT2 are associated with differential risks of developing dyslipidemia and cardiometabolic disorders, suggesting a previously unrecognized role of NAT2 in pathophysiology of metabolic disorders. In support of this notion, we recently showed that human NAT2 expression is differentially regulated by glucose and insulin. Moreover, our in silico analysis showed that NAT2 is co-expressed with nuclear receptors enriched in the liver, e.g., NR1H4 (FXR) and NR1I2 (PXR), that have been previously implicated in regulation of hepatic glucose and lipid homeostasis. Identification of transcriptional regulator(s) of human NAT2 would aid in understanding novel functions that it may play in the liver. Thus, the present study was designed to investigate if NAT2 is transcriptionally regulated by hepatic nuclear receptors.
Methods: To test this, we treated cryopreserved human hepatocytes with agonists towards four different hepatic transcription factors/nuclear hormone receptors, namely FXR (NR1H4), PXR (NR1I2), LXR (NR1H3), and PPARα (PPARA), and measured their effects on the level of NAT2 mRNA.
Results: While the treatment with a FXR, PXR, or LXR agonist (i.e., GW-4064, SR-12813, or GW-3965) significantly induced their respective target genes, treatment with these agonists did not significantly alter the transcript level of NAT2 in human hepatocytes. PPARα agonist, GW-7647, treatment resulted in a statistically significant decrease in the NAT2 transcript level. However, its magnitude was marginal.
Conclusion: In summary, hepatic nuclear receptors we examined in the present study (FXR, PXR, LXR, and PPARα) did not significantly alter NAT2 expression in cryopreserved human hepatocytes. Additional studies are needed to identify transcriptional regulators of hepatic NAT2 expression.
1 Introduction
Arylamine N-acetyltransferase 2 (NAT2) is a phase II metabolic enzyme commonly known for its role in biotransformation of aromatic amines and hydrazines as reviewed previously (McDonagh et al., 2014). NAT2 expresses well-defined genetic polymorphisms, and previous studies have shown that single nucleotide polymorphisms (SNPs) in the NAT2 coding region often lead to changes in protein stability or altered substrate affinity of NAT2 (Hein, 2002). Combination of variant NAT2 alleles manifest as three different acetylator phenotypes in the general population based on the relative activity level they produce: slow, intermediate, and rapid acetylators (Hein, 2002; Sim et al., 2014). The acetylator phenotype has a profound impact on the rate of certain drug (e.g., isoniazid and hydralazine) and carcinogen (e.g., 4-aminobiphenyl) metabolism in individuals (Hein and Millner, 2021).
In recent years, studies have suggested that NAT2 plays unexpected roles in human pathophysiology. In a genome wide association study (GWAS), NAT2 coding SNPs, rs1208 and rs1801280, has been associated with insulin resistance (assessed by the euglycemic clamp method), independent of body-mass index (Knowles et al., 2015). Follow up studies reported that Nat1 (the functional ortholog of human NAT2) knockout mice exhibit multiple signs of metabolic dysfunction, including elevated fasting blood glucose, insulin resistance, mitochondrial dysfunction, decreased fat utilization, and marked increases in tissue fat content (Chennamsetty et al., 2016; Camporez et al., 2017), supporting the role of murine NAT1 in insulin sensitivity and energy utilization in vivo. Although the mechanism remains obscure, these studies indicate that variable levels (or activities) of NAT2 may influence the state of energy storage and utilization.
Recent reports from our group implicated NAT2 in regulation of lipid/cholesterol homeostasis in the liver. NAT2 transcript is upregulated by glucose and insulin in liver cancer cell lines (Hong et al., 2022), suggesting that its expression is regulated by the nutrient status. Moreover, according to our in silico analysis, human NAT2 is co-expressed with the genes involved in lipid and cholesterol synthesis and transport, such as APOB, ABCG8, ANGPTL3, FABP1, MOGAT2, and PLA2G12B (Hong et al., 2022). Numerous GWAS reports also implicate NAT2 in regulation of plasma lipid and cholesterol levels (Hong et al., 2023), as multiple NAT2 genetic variants have been associated with differential plasma lipid and cholesterol levels. Interestingly, the risk alleles for dyslipidemia of NAT2 gene (e.g., rs1495741-A) are associated with the rapid acetylator phenotype in humans (Hong et al., 2023), which suggests that individuals with increased levels of NAT2 activity are at a higher risk of developing dyslipidemia. The link between NAT2 acetylator phenotype and plasma lipid levels is also supported by our previous study on rats congenic for rapid or slow Nat2 acetylator genotypes. Regardless of the diet, rapid acetylator rats were more prone to develop dyslipidemia (i.e., higher triglyceride; higher LDL; and lower HDL), compared to slow acetylator rats (Hong et al., 2020).
Taken together, it appears that the level of NAT2 is not only regulated by the nutrient status (e.g., glucose and insulin) but also may play an important role in regulating lipid/cholesterol homeostasis, presumably in liver and intestines which are two tissues with relatively high expression. Although many studies have examined the functional outcomes of genetic variants of NAT2 in the context of xenobiotic metabolism, so far, few studies have explored its transcriptional regulation (Zou et al., 2020; Zhu et al., 2021; Hong et al., 2022). In our previous study, we reported that the genes that are co-expressed with NAT2 include those encoding hepatic nuclear receptors, e.g., farnesoid X receptor (FXR; NR1H4) and pregnane X receptor (PXR; NR1I2). Nuclear receptors function as ligand-activated transcription factors (Frigo et al., 2021). For example, PXR is commonly known to transactivate genes that encode proteins involved in xenobiotic metabolism, but also plays a role in glucose homeostasis and insulin sensitivity (Kliewer et al., 2002; Spruiell et al., 2014). FXR, liver X receptor alpha and beta (LXRα/β), and peroxisome proliferator activated receptor alpha (PPARα) respond to changes in cellular levels of endogenous lipid ligands by regulating the expression of genes involved in lipid metabolism (Calkin and Tontonoz, 2012; Hong and Tontonoz, 2014). This prompted us to investigate the mechanisms of transcriptional regulation of NAT2 by nuclear receptors expressed in the liver.
In the present study, we tested if FXR, PXR, LXR, or PPARα regulate NAT2 expression. We treated cryopreserved human hepatocytes with specific agonists for the nuclear receptors and compared the transcript levels of NAT2 as well as known target genes of the receptors.
2 Materials and methods
2.1 Cell culture
Cryopreserved human hepatocytes were purchased from BioIVT (http://www.bioivt.com) and stored in liquid nitrogen until use. The hepatocytes were thawed and cultured in an incubator with a humidified air (95%) and CO2 (5%) at 37°C as previously reported (Walls et al., 2023). Briefly, cells were thawed by warming at 37°C for 90 s and suspending them in InVitroGRO HT medium (BioIVT) containing TORPEDO™ Antibiotic Mix (BioIVT) and plated on Biocoat® collagen-coated plates (Corning).
2.2 Agonists for nuclear receptors
GW-4064 (FXR agonist), SR-12813 (PXR agonist), GW-3965 (LXR agonist), and GW-7647 (PPARα agonist) were purchased from Selleck. The working solutions were prepared in DMSO. The final treatment concentrations were the following: 1 μM GW-4064, 1 μM SR-12813, 2 μM GW-3965, and 10 μM GW-7647. The cryopreserved human hepatocytes, in replicates of three, were treated with DMSO (vehicle control) or an agonist for 48 h prior to the harvest.
2.3 Gene expression analysis
Following the agonist treatment, total RNA was isolated from the control and the treated cells using E.Z.N.A. Total RNA Kit 1 (Omegabiotek) according to the manufacturer’s instructions. cDNA synthesis was done using High-Capacity cDNA Reverse Transcriptase PCR kit (Thermo Scientific). Gene-specific cDNA was amplified and detected using iTaq Universal SYBR Green Supermix (Bio-Rad), StepOne real-time PCR system (Applied Biosystems), and gene-specific primers (see Table 1 for primer sequences). Results were normalized to an internal control gene, GAPDH, and the relative fold change was calculated using the delta-delta Ct (2−ΔΔCT) method. The statistical analyses were performed using GraphPad Prism v8.2.1 (GraphPad Software). Unpaired t-test was employed to compare between the control and treatment groups. The results are expressed as the mean ± standard error of the mean (SEM) and from three independent experiments (n = 3). Statistical significance was determined per the following p-values: *, p < 0.05, **, p < 0.01, ***, p < 0.001, ****, p < 0.0001.
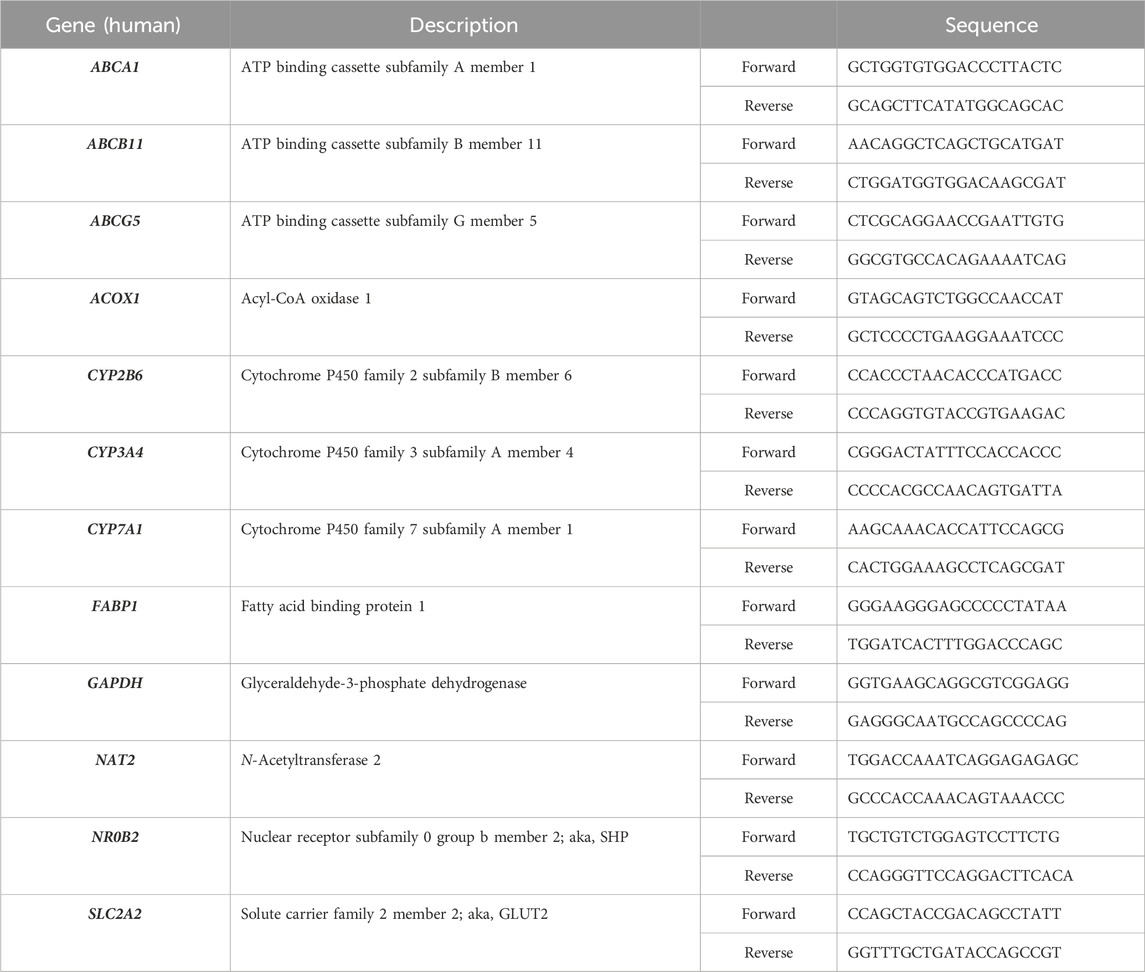
Table 1. List of PCR primer sequences used in the study.
3 Results
3.1 Gene expression changes by a FXR agonist, GW-4064
To evaluate whether NAT2 is transcriptionally regulated by FXR, we treated cryopreserved human hepatocytes with DMSO as control or a FXR agonist, GW-4064, and measured changes in mRNA expression of NAT2. Additionally, we measured changes in the mRNA expression of two known FXR target genes as positive controls: small heterodimer partner [SHP; nuclear receptor subfamily 0 group B member 2 (NR0B2)] and ATP binding cassette subfamily B member 11 (ABCB11) (Rausch et al., 2022). Expectedly, the FXR agonist treatment resulted in a statistically significant upregulation of both target genes tested [by 2.7-fold for NR0B2 (p < 0.01) and 3.4-fold for ABCB11 (p < 0.0001)], indicating activation of FXR by the agonist (Figures 1A, B). In contrast, the FXR agonist treatment failed to alter NAT2 mRNA level (Figure 1C), suggesting that NAT2 is not transcriptionally regulated by FXR in human hepatocytes.
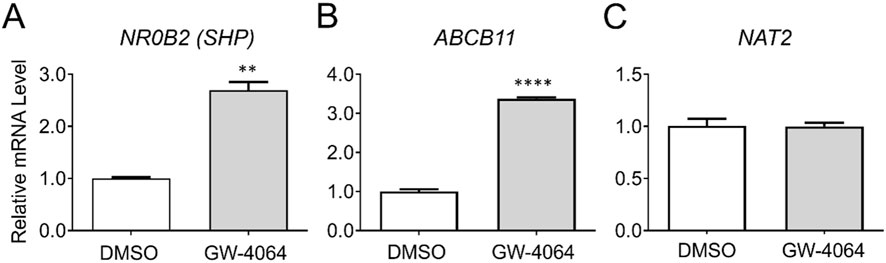
Figure 1. Gene expression changes following activation of FXR. Cryopreserved human hepatocytes were treated with a FXR agonist, GW-4064, for 2 days. The mRNA levels of the indicated genes were measured by RT-qPCR using GAPDH as an internal control. The mRNA level in the treated group was then expressed relative to that in the DMSO control. NR0B2 (A) and ABCB11 (B) represent known FXR target genes. GW-4064 did not alter the NAT2 mRNA level (C). Bars represent mean ± SEM. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
3.2 Gene expression changes by a PXR agonist, SR-12813
We also assessed if NAT2 is transcriptionally regulated by PXR. Cryopreserved human hepatocytes were treated with DMSO or a PXR agonist, SR-12813, and changes in the mRNA level of NAT2 were monitored. Along with NAT2, we measured changes in the mRNA expression of two known PXR target genes as positive controls: cytochrome P450 family 3 subfamily A member 4 (CYP3A4) and cytochrome P450 family 2 subfamily B member 6 (CYP2B6) (Hariparsad et al., 2009). The PXR agonist treatment resulted in a statistically significant upregulation of both target genes tested [by 52.9-fold for CYP3A4 (p < 0.001) and 5.2-fold for CYP2B6 (p < 0.001)], indicating activation of PXR by the agonist (Figures 2A, B). In contrast, the NAT2 mRNA level was not altered following PXR agonist treatment, suggesting NAT2 (Figure 2C) is not transcriptionally regulated by PXR in human hepatocytes.
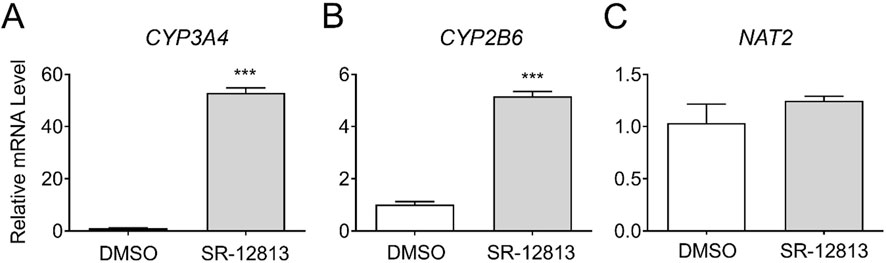
Figure 2. Gene expression changes following activation of PXR. Cryopreserved human hepatocytes were treated with a PXR agonist, SR-12813, for 2 days. The mRNA levels of the indicated genes were measured by RT-qPCR using GAPDH as an internal control. The mRNA level in the treated group was then expressed relative to that in the DMSO control. CYP3A4 (A) and CYP2B6 (B) represent known PXR target genes. SR-12813 did not alter the NAT2 mRNA level (C). Bars represent mean ± SEM. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
3.3 Gene expression changes by a LXR agonist, GW-3965
To investigate the transcriptional regulation of NAT2 by LXR, we treated cryopreserved human hepatocytes with DMSO or GW-3965, an LXR agonist, and measured changes in NAT2 mRNA expression and two positive control genes. For positive controls, we used two known target genes of LXR: ATP-binding cassette subfamily A member 1 (ABCA1) and ATP binding cassette subfamily G member 5 (ABCG5) (Fessler, 2018). While the results of the positive controls were both statistically significant, the LXR agonist treatment of ABCA1 (18.2-fold increase) had greater statistical significance (p < 0.0001) compared to ABCG5 (p < 0.01) (1.3-fold increase) (Figures 3A, B). The NAT2 mRNA level, however, showed no statistically significant changes after treatment with the LXR agonist, suggesting that NAT2 is not transcriptionally regulated by LXR in human hepatocytes (Figure 3C).
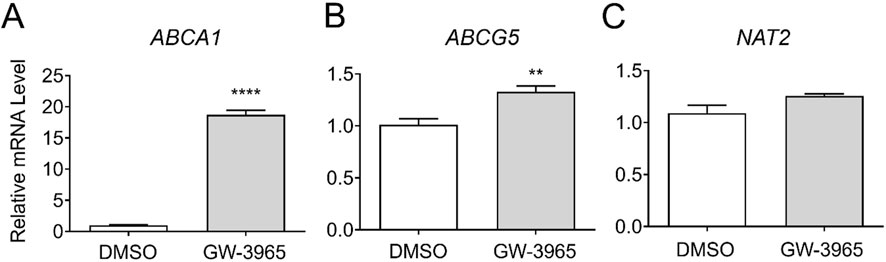
Figure 3. Gene expression changes following activation of LXR. Cryopreserved human hepatocytes were treated with a LXR agonist, GW-3965, for 2 days. The mRNA levels of the indicated genes were measured by RT-qPCR using GAPDH as an internal control. The mRNA level in the treated group was then expressed relative to that in the DMSO control. ABCA1 (A) and ABCG5 (B) represent known PXR target genes. SR-12813 did not alter the NAT2 mRNA level (C). Bars represent mean ± SEM. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
3.4 Gene expression changes by a PPARα agonist, GW-7647
To assess whether NAT2 is transcriptionally regulated by PPARα, we treated cryopreserved human hepatocytes with DMSO or a PPARα agonist, GW-7647. Additional measurements were taken investigating changes in mRNA levels of two known target genes of PPARα that served as positive controls: acyl-CoA oxidase 1 (ACOX1) and fatty acid binding protein 1 (FABP1) (Rakhshandehroo et al., 2010). Expectedly, the treatment with the PPARα agonist significantly induced the transcript levels of both ACOX1 and FABP1 [by 1.8-fold for ACOX1 (p < 0.001) and 4.0-fold for FABP1 (p < 0.001)] (Figures 4A, B). In comparison, NAT2 showed a statistically significant decrease in the mRNA expression following PPARα agonist treatment (by 0.8-fold) (p < 0.05) (Figure 4C). To further validate our data, we chose to include two additional target genes previously shown to be downregulated by PPARα: cytochrome P450 family 7 subfamily A member 1 (CYP7A1) and glucose transporter 2 (GLUT2) (Rakhshandehroo et al., 2009). The transcript level of CYP7A1 did not change (Figure 4D), but there was a statistically significant decrease in the mRNA level of GLUT2 (SLC2A2) (by 0.8-fold) (p < 0.05) (Figure 4E). Although there was a statistically significant decrease in NAT2 mRNA expression following PPARα agonist treatment, the slight downregulation of NAT2 expression may not be biologically significant.
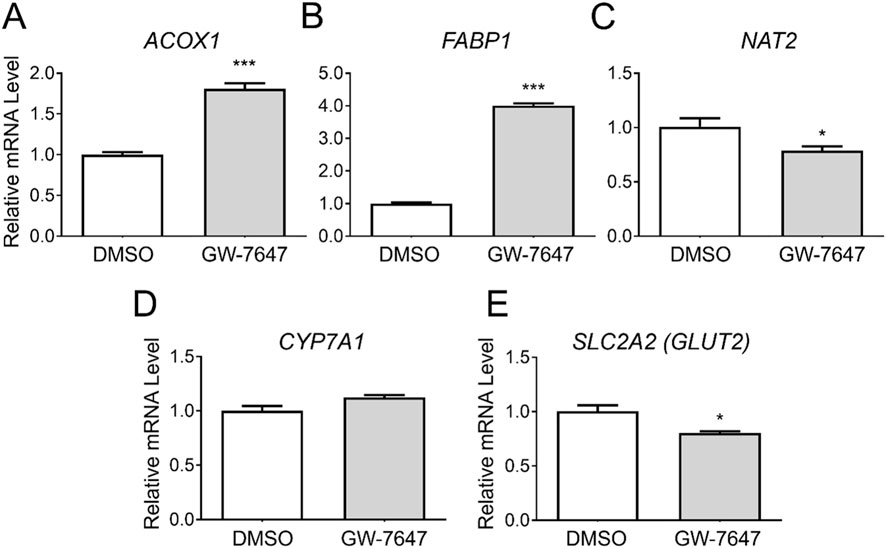
Figure 4. Gene expression changes following activation of PPARα. Cryopreserved human hepatocytes were treated with a PPARα agonist, GW-7647, for 2 days. The mRNA levels of the indicated genes were measured by RT-qPCR using GAPDH as an internal control. The mRNA level in the treated group was then expressed relative to that in the DMSO control. ACOX1 (A) and FABP1 (B) were previously shown to be upregulated by PPARα, while CYP7A1 (D) and GLUT2 (E) were reported to be suppressed by PPARα. GW-7647 significantly, yet marginally, decreased the NAT2 and GLUT2 mRNA levels (C, E). Bars represent mean ± SEM. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
4 Discussion
The activation of nuclear receptors, FXR, PXR, and LXR, by corresponding agonists did not result in significant alterations in the NAT2 mRNA level in cryopreserved human hepatocytes, which suggests that they are not major transcriptional regulators of NAT2 under the test conditions. However, it is worth noting that activation of PPARα by GW-7647 resulted in a marginal, yet significant, reduction in the NAT2 transcript level. However, it is not clear if such a minute decrease has any biological impact.
PPARα is activated by endogenous fatty acids and their derivatives (Chakravarthy et al., 2009; Pawlak et al., 2015). PPARα activation promotes fatty acid transport and metabolism (β-oxidation) as well as lipogenesis via transcriptional activation of the genes involved in these processes, including sterol regulatory element binding protein 1c (SREBP-1c; SREBF1), fatty acid synthase (FASN), and acetyl-CoA carboxylase 1 (ACC1; ACACA) (Patel et al., 2001; Fernández-Alvarez et al., 2011). While ACOX1 and FABP1 represented the target genes upregulated by PPARα (Rakhshandehroo et al., 2010), we chose to examine CYP7A1 and GLUT2 (SLC2A2) as downregulated target genes (see Figure 4). CYP7A1 encodes cholesterol 7-α-monooxygenase, the first and rate-limiting enzyme in bile acid synthesis from cholesterol (Noshiro and Okuda, 1990). It has been shown to be suppressed by Wy14643, a PPARα agonist, in HepG2 and primary hepatocytes (Marrapodi and Chiang, 2000; Rakhshandehroo et al., 2009). Rakhshandehroo and colleagues reported that the treatment with Wy14643 (another PPARα agonist) results in a significant downregulation of genes involved in glucose absorption, such as glucose transporter 2 (GLUT2; SLC2A2) and villin-1 (VIL1) in both mouse and human primary hepatocytes (Rakhshandehroo et al., 2009). In our study, GW-7647 failed to suppress CYP7A1 expression, whereas it significantly downregulated GLUT2 (SLC2A2) expression in cryopreserved human hepatocytes. It is not clear why CYP7A1 was not downregulated following PPARα activation in the present study. Marrapodi and Chiang reported that downregulation of CYP7A1 by Wy14643 is unlikely due to direct regulation by PPARα, but rather indirectly via downregulation of HNF4α (Marrapodi and Chiang, 2000). Notably, their experiments were carried out using HepG2 cells. In the study by Rakhshandehroo et al. (2009). The authors used a different agonist (Wy14643), which may account for the difference. Additionally, differences in culture condition and batch-to-batch variability between cryopreserved human hepatocytes might have contributed to this discrepancy. Differences between previous and our current results may attribute to the level of HNF4α in hepatocytes used and/or the use of different PPARα agonists.
The amino acid catabolism is one of the processes suppressed by PPARα activation. By promoting the proteasomal degradation of hepatocyte nuclear factor 4 alpha (HNF4α), PPARα indirectly downregulates genes encoding amino acid-degrading enzymes, including histidine ammonia-lyase (HAL) and serine dehydratase (SDS) (Rakhshandehroo et al., 2009; Alemán et al., 2013; Tobón-Cornejo et al., 2021). In support of this data, a global transcriptomic analysis of genes differentially regulated by Wy14643 in both mouse and human primary hepatocytes revealed that one of the biological processes that are significantly downregulated include “alpha-amino acid catabolic process” (GO:1901606) and “cellular amino acid catabolic process” (GO:0009063) (Rakhshandehroo et al., 2009). Thus, it is plausible that downregulation of the NAT2 transcript level by GW-7647 is not a direct result of PPARα activation but of subsequent downregulation of HNF4α. It would be of interest to test if NAT2 is transcriptionally regulated by HNF4α.
Our previous study showed that NAT2 mRNA levels in HepG2 cells vary according to the glucose concentration in culture media (Hong et al., 2022). Thus, downregulation of GLUT2/SLC2A2 expression by PPARα activation could potentially lead to a reduction in glucose uptake and indirectly downregulate NAT2 expression. However, despite the statistical significance, the reduction in GLUT2 expression was minute, and we question the functional significance of the change.
Although variable NAT2 activity has been linked to physiologically and clinically relevant outcomes (e.g., drug toxicity and dyslipidemia), few studies have investigated its transcriptional regulation. One recent study reported that NAT2 expression is positively regulated by the vitamin D receptor (VDR) (Zhu et al., 2021). The study showed that treatment with the biologically active form of vitamin D (1,25(OH)2D3) increases NAT2 expression and that VDR binds to the NAT2 promoter in colorectal cancer cells (Zhu et al., 2021). Whether or not NAT2 is regulated similarly by VDR in the liver remains to be tested.
In summary, we did not observe marked alterations in the NAT2 transcript level following treatment of synthetic agonists for FXR, PXR, LXR, and PPARα in cryopreserved human hepatocytes. Additional potential transcriptional regulators of hepatic NAT2 expression, among nuclear receptors, include HNF4α and VDR, which remains to be tested.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available cryopreserved human hepatocyte samples were used.
Author contributions
KH: Conceptualization, Data curation, Formal Analysis, Methodology, Project administration, Supervision, Writing–original draft, Writing–review and editing. AA: Writing–original draft, Writing–review and editing. KW: Investigation, Methodology, Writing–review and editing. DH: Funding acquisition, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. National Institutes of Health NIEHS [National Institute of Environmental Health Sciences] T32-ES011564, NIEHS P30-ES030283, NIEHS P42-ES023716, and NIGMS [National Institute of General Medical Sciences] P20-GM113226.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alemán, G., Ortiz, V., Contreras, A. V., Quiroz, G., Ordaz-Nava, G., Langley, E., et al. (2013). Hepatic amino acid-degrading enzyme expression is downregulated by natural and synthetic ligands of PPARα in rats. J. Nutr. 143, 1211–1218. doi:10.3945/jn.113.176354
Calkin, A. C., and Tontonoz, P. (2012). Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell. Biol. 13, 213–224. doi:10.1038/nrm3312
Camporez, J. P., Wang, Y., Faarkrog, K., Chukijrungroat, N., Petersen, K. F., and Shulman, G. I. (2017). Mechanism by which arylamine N-acetyltransferase 1 ablation causes insulin resistance in mice. Proc. Natl. Acad. Sci. U. S. A. 114, E11285–E11292. doi:10.1073/pnas.1716990115
Chakravarthy, M. V., Lodhi, I. J., Yin, L., Malapaka, R. R. V., Xu, H. E., Turk, J., et al. (2009). Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell. 138, 476–488. doi:10.1016/j.cell.2009.05.036
Chennamsetty, I., Coronado, M., Contrepois, K., Keller, M. P., Carcamo-Orive, I., Sandin, J., et al. (2016). Nat1 deficiency is associated with mitochondrial dysfunction and exercise intolerance in mice. Cell. Rep. 17, 527–540. doi:10.1016/j.celrep.2016.09.005
Fernández-Alvarez, A., Alvarez, M. S., Gonzalez, R., Cucarella, C., Muntané, J., and Casado, M. (2011). Human SREBP1c expression in liver is directly regulated by peroxisome proliferator-activated receptor alpha (PPARalpha). J. Biol. Chem. 286, 21466–21477. doi:10.1074/jbc.M110.209973
Fessler, M. B. (2018). The challenges and promise of targeting the liver X receptors for treatment of inflammatory disease. Pharmacol. Ther. 181, 1–12. doi:10.1016/j.pharmthera.2017.07.010
Frigo, D. E., Bondesson, M., and Williams, C. (2021). Nuclear receptors: from molecular mechanisms to therapeutics. Essays Biochem. 65, 847–856. doi:10.1042/EBC20210020
Hariparsad, N., Chu, X., Yabut, J., Labhart, P., Hartley, D. P., Dai, X., et al. (2009). Identification of pregnane-X receptor target genes and coactivator and corepressor binding to promoter elements in human hepatocytes. Nucleic Acids Res. 37, 1160–1173. doi:10.1093/nar/gkn1047
Hein, D. W. (2002). Molecular genetics and function of NAT1 and NAT2: role in aromatic amine metabolism and carcinogenesis. Mutat. Res. 506–507, 65–77. doi:10.1016/s0027-5107(02)00153-7
Hein, D. W., and Millner, L. M. (2021). Arylamine N-acetyltransferase acetylation polymorphisms: paradigm for pharmacogenomic-guided therapy-a focused review. Expert Opin. Drug Metab. Toxicol. 17, 9–21. doi:10.1080/17425255.2021.1840551
Hong, C., and Tontonoz, P. (2014). Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat. Rev. Drug Discov. 13, 433–444. doi:10.1038/nrd4280
Hong, K. U., Doll, M. A., Lykoudi, A., Salazar-González, R. A., Habil, M. R., Walls, K. M., et al. (2020). Acetylator genotype-dependent dyslipidemia in rats congenic for N-acetyltransferase 2. Toxicol. Rep. 7, 1319–1330. doi:10.1016/j.toxrep.2020.09.011
Hong, K. U., Salazar-González, R. A., Walls, K. M., and Hein, D. W. (2022). Transcriptional regulation of human arylamine N-acetyltransferase 2 gene by glucose and insulin in liver cancer cell lines. Toxicol. Sci. 190, 158–172. doi:10.1093/toxsci/kfac103
Hong, K. U., Walls, K. M., and Hein, D. W. (2023). Non-coding and intergenic genetic variants of human arylamine N-acetyltransferase 2 (NAT2) gene are associated with differential plasma lipid and cholesterol levels and cardiometabolic disorders. Front. Pharmacol. 14, 1091976. doi:10.3389/fphar.2023.1091976
Kliewer, S. A., Goodwin, B., and Willson, T. M. (2002). The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr. Rev. 23, 687–702. doi:10.1210/er.2001-0038
Knowles, J. W., Xie, W., Zhang, Z., Chennamsetty, I., Chennemsetty, I., Assimes, T. L., et al. (2015). Identification and validation of N-acetyltransferase 2 as an insulin sensitivity gene. J. Clin. Investig. 125, 1739–1751. doi:10.1172/JCI74692
Marrapodi, M., and Chiang, J. Y. (2000). Peroxisome proliferator-activated receptor α (PPARα) and agonist inhibit cholesterol 7α-hydroxylase gene (CYP7A1) transcription. J. Lipid Res. 41, 514–520. doi:10.1016/s0022-2275(20)32398-1
McDonagh, E. M., Boukouvala, S., Aklillu, E., Hein, D. W., Altman, R. B., and Klein, T. E. (2014). PharmGKB summary: very important pharmacogene information for N-acetyltransferase 2. Pharmacogenet. Genomics 24, 409–425. doi:10.1097/FPC.0000000000000062
Noshiro, M., and Okuda, K. (1990). Molecular cloning and sequence analysis of cDNA encoding human cholesterol 7 alpha-hydroxylase. FEBS Lett. 268, 137–140. doi:10.1016/0014-5793(90)80992-R
Patel, D. D., Knight, B. L., Wiggins, D., Humphreys, S. M., and Gibbons, G. F. (2001). Disturbances in the normal regulation of SREBP-sensitive genes in PPARα-deficient mice. J. Lipid Res. 42, 328–337. doi:10.1016/S0022-2275(20)31655-2
Pawlak, M., Lefebvre, P., and Staels, B. (2015). Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 62, 720–733. doi:10.1016/j.jhep.2014.10.039
Rakhshandehroo, M., Hooiveld, G., Müller, M., and Kersten, S. (2009). Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS One 4, e6796. doi:10.1371/journal.pone.0006796
Rakhshandehroo, M., Knoch, B., Müller, M., and Kersten, S. (2010). Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010, 612089. doi:10.1155/2010/612089
Rausch, M., Samodelov, S. L., Visentin, M., and Kullak-Ublick, G. A. (2022). The farnesoid X receptor as a master regulator of hepatotoxicity. Int. J. Mol. Sci. 23, 13967. doi:10.3390/ijms232213967
Sim, E., Abuhammad, A., and Ryan, A. (2014). Arylamine N-acetyltransferases: from drug metabolism and pharmacogenetics to drug discovery. Br. J. Pharmacol. 171, 2705–2725. doi:10.1111/bph.12598
Spruiell, K., Richardson, R. M., Cullen, J. M., Awumey, E. M., Gonzalez, F. J., and Gyamfi, M. A. (2014). Role of pregnane X receptor in obesity and glucose homeostasis in male mice. J. Biol. Chem. 289, 3244–3261. doi:10.1074/jbc.M113.494575
Tobón-Cornejo, S., Vargas-Castillo, A., Leyva-Martínez, A., Ortíz, V., Noriega, L. G., Velázquez-Villegas, L. A., et al. (2021). PPARα/RXRα downregulates amino acid catabolism in the liver via interaction with HNF4α promoting its proteasomal degradation. Metabolism 116, 154705. doi:10.1016/j.metabol.2021.154705
Walls, K. M., Hong, K. U., and Hein, D. W. (2023). Heterocyclic amines reduce insulin-induced AKT phosphorylation and induce gluconeogenic gene expression in human hepatocytes. Arch. Toxicol. 97, 1613–1626. doi:10.1007/s00204-023-03488-2
Zhu, C., Wang, Z., Cai, J., Pan, C., Lin, S., Zhang, Y., et al. (2021). VDR signaling via the enzyme NAT2 inhibits colorectal cancer progression. Front. Pharmacol. 12, 727704. doi:10.3389/fphar.2021.727704
Keywords: N-acetyltransferase 2, NAT2, hepatocytes, nuclear receptors, FXR, PPARα, PXR, LXR
Citation: Hong KU, Aureliano AP, Walls KM and Hein DW (2024) Investigation on regulation of N-acetyltransferase 2 expression by nuclear receptors in human hepatocytes. Front. Pharmacol. 15:1488367. doi: 10.3389/fphar.2024.1488367
Received: 29 August 2024; Accepted: 04 November 2024;
Published: 18 November 2024.
Edited by:
Luis Abel Quiñones, University of Chile, ChileReviewed by:
Tobias Sjöblom, Uppsala University, SwedenKenia El-Jaick, Rio de Janeiro State Federal University, Brazil
Copyright © 2024 Hong, Aureliano, Walls and Hein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kyung U. Hong, a3l1bmcuaG9uZ0B3bmUuZWR1; David W. Hein, ZGF2aWQuaGVpbkBsb3Vpc3ZpbGxlLmVkdQ==
†Present address: Environmental Justice, Community Health, and Environmental Review Division, US Environmental Protection Agency, Chicago, IL, United States