 Zhaoyu Wang
Zhaoyu Wang Hong Peng
Hong Peng Yun Zhang1
Yun Zhang1 Xijing Chen
Xijing Chen Jiye A
Jiye A- 1Clinical Pharmacology Laboratory, School of Basic Medicine and Clinical Pharmacy, China Pharmaceutical University, Nanjing, China
- 2Jiangsu Provincial Key Laboratory of Drug Metabolism and Pharmacokinetics, China Pharmaceutical University, Nanjing, China
Objective: This study aimed to assess the bioequivalence and safety of two formulations of sorafenib in healthy Chinese subjects under fasting conditions.
Methods: A single-center, randomized, open, single-dose, two-formulation, four-period, crossover study was performed in 36 healthy Chinese subjects under fasting conditions. Blood samples were collected within 120 h after administration. The plasma concentrations of sorafenib were analyzed by a validated UPLC-MS/MS method, and pharmacokinetic parameters were analyzed using a non-compartmental method. Safety was assessed on the basis of the occurrence of adverse events and laboratory findings throughout the study period.
Results: The GMR point estimators of Cmax, AUC0-t, and AUC0-
Conclusion: The T formulation was bioequivalent and showed a similar safety profile to the R formulation Nexavar® (Bayer AG) in healthy Chinese subjects under fasting conditions.
Clinical trial registration: http://www.chinadrugtrials.org.cn/index.html, Identifier CTR20233578.
1 Introduction
Sorafenib (BAY 43-9006, Nexavar®) is a novel lipophilic small molecule with a biaryl urea structure that belongs to the Biopharmaceutics Classification System (BCS) II because of its low solubility and high membrane permeability (Cui et al., 2022; Ranieri et al., 2012; Ramakrishnan et al., 2012; Huh et al., 2021). After oral administration, sorafenib effectively inhibited tumor cell proliferation by targeting rapidly accelerated fibrosarcoma serine-threonine kinases (Raf). Additionally, it exhibits significant inhibitory activity against receptor tyrosine kinases such as vascular endothelial growth factor receptor (VEGFR) and platelet-derived growth factor receptor β, which help reduce tumor angiogenesis (Wilhelm et al., 2004; Wilhelm et al., 2008; Liu et al., 2006). Sorafenib tablets (200 mg) were approved by the Food and Drug Administration (FDA) in 2005 for the treatment of patients with advanced renal cell carcinoma (RCC) (Kane et al., 2006). It is also the first drug approved as a first-line treatment for advanced hepatocellular carcinoma (HCC) as a first-line treatment (Cui et al., 2022).
The pharmacokinetics of sorafenib varies greatly among individuals after a single oral dose. However, food intake does not significantly affect metabolic profiles (Strumberg et al., 2005). Sorafenib reaches its peak concentration approximately 3 h after oral administration, and its mean elimination half-life is 25–48 h (Kane et al., 2006). Sorafenib is mainly metabolized in the liver via two pathways: phase I oxidative metabolism through CYP3A4, and phase II glucuronidation via UGT1A9 (Keating and Santoro, 2009). After oral administration of a 100 mg solution formulation, 77% of the dose is excreted in the feces and 19% in the urine as glucuronidated metabolites. The prototype drug is mostly excreted in feces at 51% of the dose (Kane et al., 2006).
Common adverse drug reactions associated with sorafenib administration include gastrointestinal issues such as diarrhea, nausea, anorexia, and dermatological issues such as hand-foot syndrome (HFS), skin dryness, rash, pruritus, alopecia, stomatitis, and fatigue (Clark et al., 2005; Awada et al., 2005; Moore et al., 2005; Strumberg et al., 2005). Most patients with advanced refractory solid tumors experience mild to moderate severity of these adverse events, indicating that sorafenib is generally well-tolerated and safe (Strumberg et al., 2007).
Currently, studies on the pharmacokinetics of sorafenib in healthy Chinese subjects are limited, necessitating further investigation. This trial aimed to evaluate the pharmacokinetics, bioequivalence, and safety of two sorafenib tosylate tablets obtained from different manufacturers (Renhexidelong Pharmaceutical Co. Ltd., and Bayer AG, Germany) after single-dose administration under fasting conditions in healthy Chinese adults.
2 Methods
2.1 Study design
This study adhered to the principles of the Declaration of Helsinki (World Medical Association, 2013), Good Clinical Practice (GCP) (NMPA, National Medical Products Administration, National Health Commission of the People’s Republic of China, 2020), and the guidelines of the National Medical Products Administration (NMPA) (NMPA, The National Medical Products Administration of China, Center for Drug Evaluation, 2019) of China. The trial was registered with the number CTR20233578, conducted at the Phase I Clinical Trial Center of Zhonghui Cardiovascular Disease Hospital, Henan (Zhengzhou), from 1 December 2023, to 4 February 2024, and approved by the Institutional Ethics Committee before initiation.
This trial involved a single-center, randomized, open, single oral administration, two-formulation, four-period crossover study and featured a screening period, four treatment periods, a washout period of 14 days after each treatment period, and a follow-up period following the last treatment (Figure 1). A total of 36 healthy male and female Chinese subjects were included in the study and randomly assigned to either the K1 group (TRTR) or K2 group (RTRT) with equal sex ratios using SAS software (v9.4). The subjects were admitted to the Phase I Clinical Research Center the day before dosing and fasted overnight (10 h). All subjects received a single oral administration of 0.2 g of sorafenib tosylate tablets (T formulation,0.2 g, lot SRF1230602, Renhe Xidelong Pharmaceuticals Co., Ltd.) or Nexavar®(R formulation, 0.2 g, lot: BXJX3D1, Bayer AG) on the day of dosing with 240 mL of water. The participants were not allowed to drink additional water for 1 h before or after treatment. and forbidden from walking or eating for 4 h after administration.

Figure 1. Flow chart of the study. Abbreviations: T, test formulation; R, reference formulation; N, number of subjects.
2.2 Subjects
All participants received group and individual counseling sessions with the investigator to understand the trial process and risks, and signed informed consent forms prior to participation.The inclusion criteria are healthy male and female subjects aged 18 years and above; male subjects weighing ≥50 kg and female subjects weighing ≥45 kg with a body mass index (BMI) of 19.0–26.0 kg/m2; full understanding of the experimental procedure and risks; and no birth plan from the beginning of the experiment to 3 months after its completion.
The exclusion criteria were participation in any other clinical trial of a drug within the past 3 months, presence of any clinically relevant medical condition or history, inability to tolerate venipuncture, blood or needle sickness, blood donation or blood loss ≥400 mL within the past 3 months, abnormal and clinically significant physical examination, electrocardiogram, vital signs, laboratory tests, and hypersensitivity to the study drug. Pregnant and lactating women were excluded from this study.
2.3 Collection and preservation of blood samples
Blood samples (N = 22) were collected from before drug administration to120 h after administration for each subject at the following time points: 0 h (immediately before dosing) and 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 6, 8, 9, 10, 11, 12, 24, 48, 72, 96 and 120 h after administration. A total of 4 mL of whole blood was collected into EDTA-K2 vacuum blood collection tubes. The blood samples were centrifuged at 3,000 g for about 5 min at 2°C–8°C within 1 hour, All plasma samples were frozen at −70°C (−60 to −90°C) within 2 h of blood collection.
2.4 Analytical determinations
A methodologically validated UPLC-MS/MS method was used to determine the plasma concentration of sorafenib. Ultra-performance liquid chromatography was performed using the LC-40D system (Shimadzu, Japan), while the mass spectrometer was the AB Sciex QTRAP 6500+ (Applied Biosystems, United States). The data acquisition software was Analyst 1.7.3 (Applied Biosystems, United States).
Plasma samples containing the drug were processed using a protein precipitation method. The internal standards were sorafenib-d3 (QCSRM, lot number: 24213; purity: 97.51%) and sorafenib toluenesulfonate (China Academy of Food and Drug Control, lot number: 420138-202201; content: 99.7%). The internal standard precipitant was Sorafenib-d3 diluted with acetonitrile at a concentration of 20.00 ng/mL. The drug-containing plasma was thawed at room temperature and then precipitated by a 10-fold dilution with the internal standard precipitant. The mixture was vortexed for 5 min, centrifuged at 15,000 rpm and 4°C for 10 min, and the supernatant was used for UPLC-MS/MS analysis. The plasma concentration range of the standard curve for this analytical method was 20–3,000 ng/mL,and the LLOQ, LQC, MQC and HQC of the substances to be measured were 20.00, 60.00, 300.0 and 2400 ng/mL, respectively.The intra-lot accuracy deviations of all QC samples ranged from −5.5% to 5.2% with a precision maximum (%CV) of 1.25%; the inter-lot accuracy deviations ranged from −5.5% to 5.9% with a %CV of 1.72%.
Chromatographic separation of the samples was performed on an ACQUITY UPLC®BEH C18 column (1.7 μm, 50 × 2.1 mm). Gradient elution of sorafenib was performed using 2 mM ammonium formate-water (A): acetonitrile (B) as the mobile phase at a flow rate of 0.5 mL/min and column temperature of 40°C. Positive ionization mode was used to detect sorafenib in the plasma samples. The mass-to-charge ratios (m/z) of sorafenib and sorafenib-d3 were 465.2→252.2 and 468.1→255.2 respectively.
2.5 Pharmacokinetic and statistical analysis
The pharmacokinetic parameters of sorafenib were calculated using a non-compartmental method. Cmax and Tmax are the peak concentration and time, respectively, which were obtained directly from the measured blood concentration-time (C-T) curves. AUC0-t is the area under the blood concentration-time curve from drug administration to the last point of blood sampling, calculated using the linear trapezoidal rule. AUC0-
Statistical analysis was performed using the SAS software (V 9.4). A descriptive analysis of the subjects’ baseline characteristics, such as sex, age, height, weight, and body mass index, as well as pharmacokinetic concentration data, was performed. Means, standard deviations, maximum and minimum values, medians, and first and third quartiles (Q1,Q3) were calculated for continuous variables, while frequencies and percentages were calculated for categorical variables. The main pharmacokinetic parameters (Cmax, AUC0-t, and AUC0-
2.6 Safety evaluations
Safety analyses were performed for subjects who had received at least one dose of the test drug. The safety of sorafenib tosylate tablets was assessed based on adverse event reports, vital signs (including seated blood pressure, pulse, and temperature), physical examinations, 12-lead electrocardiograms, and clinical laboratory tests (routine blood, urine, blood biochemistry, coagulation, and virologic testing). Vital signs were measured and recorded respectively within 1 hour before and 2, 4, 6, 8, 12, 24, 48, 72, 96, and 120 h after dosing during each period. In addition, the subjects underwent clinical laboratory testing, physical examination, and 12-lead electrocardiography at screening and before withdrawal from the study. All post-dose adverse events were monitored throughout the trial and assessed according to the Common Terminology Criteria for the Evaluation of Adverse Events (CTCAE V 5.0). All adverse events were coded using the Preferred Terminology of the International Medical Terminology Dictionary (MedDRA V 26.0) and categorized and summarized according to system organ classification and preferred terminology.
3 Results
3.1 Subject characteristics
A total of 78 healthy adult subjects were screened in this study, of which 42 failed screening and 36 (29 males and 7 females) were successfully enrolled and randomly assigned to either the K1 (TRTR) or K2 (RTRT) group, which had 18 participants each. All participants received the administered drug. A total of 34 subjects completed the trial, and two subjects withdrew from the study early willingly (one in the third period and the other in the fourth period). The demographic information and baseline characteristics of the K1 and K2 groups were group-balanced. The demographic data for all subjects is summarized in Table 1, and the flow chart of the study is shown in Figure 1.
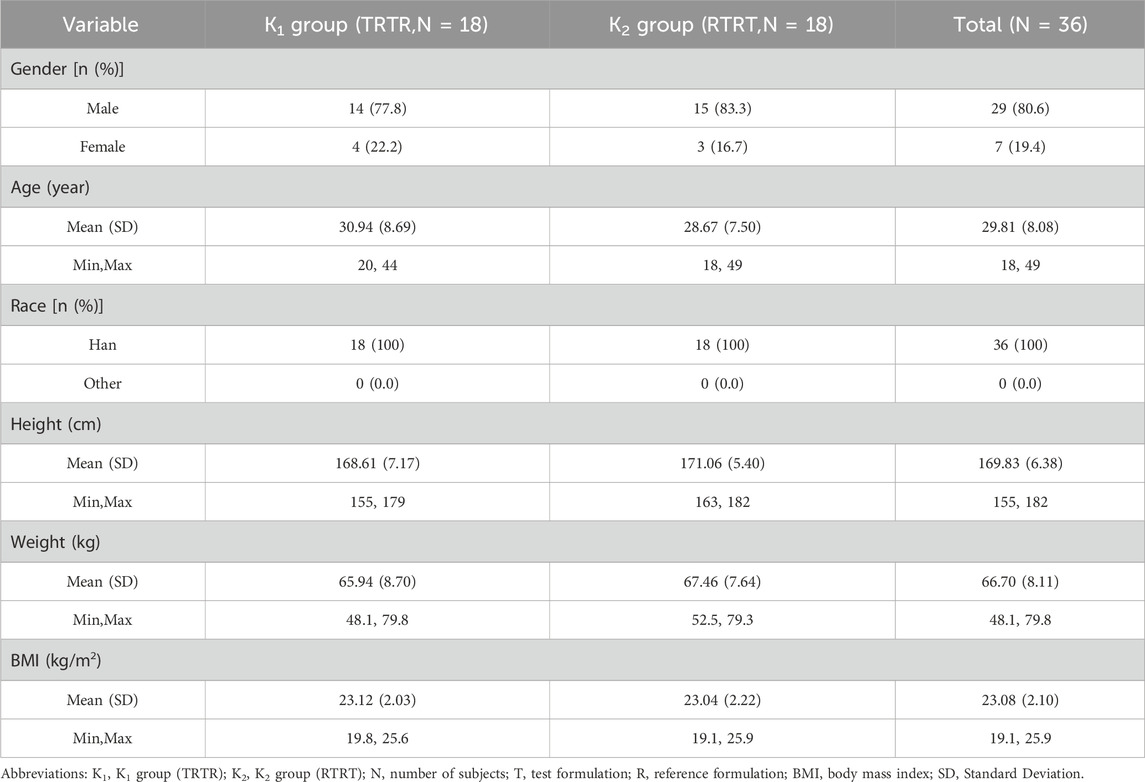
Table 1. The demographic and baseline characteristics of subjects in the clinical trials.
3.2 Pharmacokinetics of sorafenib
Data from the blood concentration studies of all 36 subjects were included in the pharmacokinetic analysis. The mean blood concentration-time curves for sorafenib after oral administration of the T formulation and R formulation under fasting conditions are shown in Figure 2. The pharmacokinetic parameters for the T and R formulations are summarized in Table 2.
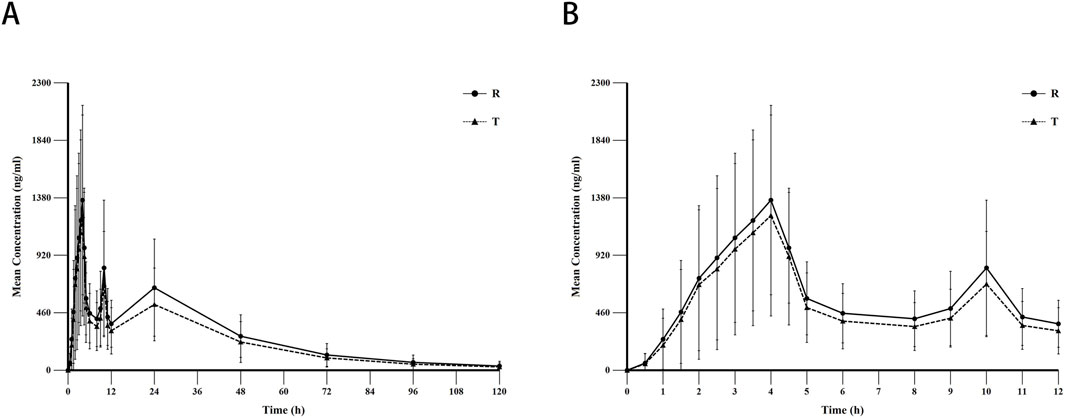
Figure 2. The mean blood concentration-time curves for sorafenib after oral administration of the test (N = 71) and reference formulation (N = 70) under fasting conditions. Notes: (A), linear scale for 120 h; (B), linear scale for first 8 h. Abbreviations: T, test formulation; R, reference formulation.
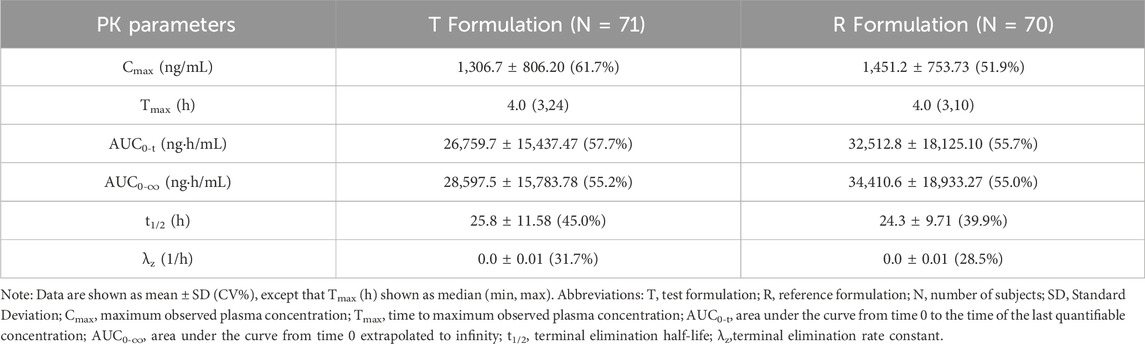
Table 2. Pharmacokinetic parameters of sorafenib after administration of T and R formulations under fasted conditions.
The mean ± SD (CV%) of the Cmax values for the T and R formulations were 1,306.7 ± 806.20 (61.7%) ng/mL and 1,451.2 ± 753.73 (51.9%) ng/mL, respectively; the AUC0-t values were 26,759.7 ± 15,437.47 (57.7%) ng·h/mL and 32,512.8 ± 18,125.10 (55.7%) ng·h/mL, respectively; the AUC0-
3.3 Bioequivalence analysis
In this study, the intra-individual standard deviation (SWR) of Cmax, AUC0-t, and AUC0-
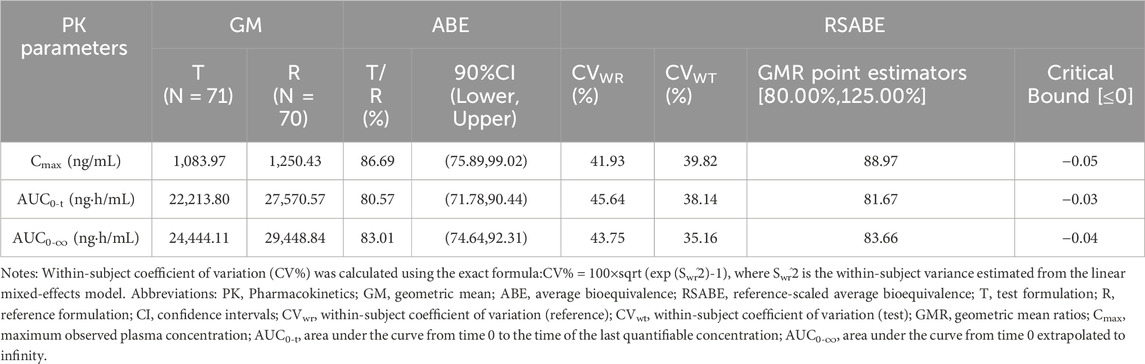
Table 3. Analysis of bioequivalence for plasma pharmacokinetic parameters of sorafenib under fasted conditions.
3.4 Safety
A total of 7 instances of TEAE were reported in 5 out of the 36 subjects after administering the T formulation. All 7 instances were possibly related to the study drug, resulting in a TEAE incidence rate of 13.89% for the T formulation. In contrast, participants experienced a total of 4 instances of TEAE in 4 subjects after receiving the R formulation with only 1 instance being possibly related to the study drug, and the remaining 3 instances being possibly unrelated to the T formulation, which resulted in a TEAE incidence rate of 11.11% for the R formulation. Table 4 displays the status and comparison of adverse events between the T and R formulations.

Table 4. Adverse events after administration of test and reference formulations under fasted conditions.
4 Discussion
The objective of this study was to evaluate the bioequivalence of the T formulation produced by Renhexidelong Pharmaceutical Co. Ltd. in comparison to Nexavar® (0.2 g) sorafenib tosylate tablets manufactured in-house by Bayer AG, which served as references.This well-designed and executed bioequivalence trial provides new data for pharmacokinetic studies of sorafenib in the Chinese population and serves as a reference for future clinical trials.
The pharmacokinetic parameters of sorafenib exhibit substantial inter-individual variability. Under oral doses of 0.2 g or 0.4 g twice daily, the variability range (%CV) of sorafenib exposure was 5%–83%, while the variability in plasma peak concentrations ranged from 33% to 88% (Jain, L. et al., 2011). A Phase I clinical trial of sorafenib demonstrated that in fasted patients with advanced solid tumors following a single 0.2 g oral dose (N = 5), the geometric mean values for AUC, Cmax, and t1/2 were 31.9 mg h/L, 1.08 mg/L, and 29.5 h, respectively (Strumberg D et al., 2005). In contrast, the geometric means observed in this study were 24.5 mg h/L for AUC, 1.10 mg/L for Cmax, and 23.9 h for t1/2, indicating lower systemic exposure, comparable Cmax, and a shorter half-life compared to advanced solid tumor patients. In a study by Huh et al., healthy male volunteers under fasting conditions (N = 8) receiving a single 0.2 g sorafenib dose showed a median Tmax of 4.0 h and t1/2 of 22.2 ± 5.1 h, consistent with our findings. However, our study revealed lower systemic exposure, which may be attributed to inter-individual variability and the limited sample size of the reference studies.
Enterohepatic circulation (EHC) refers to a physiological process whereby certain drugs excreted via biliary pathways into the intestinal lumen undergo reabsorption and re-entry into the hepatic-portal circulation. This recirculation mechanism results in prolonged systemic retention of the drug, manifesting as secondary plasma concentration peaks at delayed time intervals and an extended elimination half-life (Malik et al., 2016).The pharmacokinetic profile of sorafenib may be influenced by EHC, a phenomenon corroborated by clinical observations. Dual-peak characteristics in plasma concentration-time curves have been consistently reported in treated patients, suggesting EHC-mediated reabsorption (Jain et al., 2011). Consistent with these findings, the concentration-time profile observed in our study (Figure 2) exhibited irregular absorption patterns with substantial inter-individual variability. A primary peak occurred at 4 h post-dose, followed by secondary and tertiary concentration surges at 10 and 24 h, respectively. Furthermore, following oral administration of the T formulation, a longer elimination half-life of 25.8 h was observed, with a notably slow terminal elimination phase—a pharmacokinetic hallmark consistent with the characteristics of EHC.
High-variance drugs exhibit an intra-individual variability of ≥30% in pharmacokinetic parameters (Cmax or AUC), and sorafenib, a tyrosine kinase inhibitor, falls under this category of drugs (Huh et al., 2021; Song et al., 2021; Karalis et al., 2012; Lennernäs et al., 2024). Evaluating bioequivalence of highly variable drugs is complex and challenging, often requiring an increase in the sample size to meet the bioequivalence acceptance criteria (Davit et al., 2012), which in turn increases the cost of human resources and inputs. Both the Food and Drug Administration (FDA) (Haidar et al., 2008a; Haidar et al., 2008b) recommend the use of full-replica or half-replica designs for bioequivalence analyses of highly variable drugs; specifically, the reference product should be administered at least twice per individual (Karalis et al., 2012; Haidar et al., 2008a; Haidar et al., 2008b). Considering this characteristic, this trial utilized a full replicated crossover study design and enrolled 36 participants. The intra-individual standard deviations (SWR) of Cmax, AUC0-t, and AUC0-
It has been shown that Sorafenib binds to plasma proteins at a rate of 99.5% and has similar bioavailability when consumed with a moderate-fat diet compared to fasting. However, when consumed with a high-fat diet, the absorption of sorafenib was reduced by 30% compared with that in the fasted state (Clark et al., 2005). To accurately reflect the pharmacokinetic process of absorption, distribution, and elimination of sorafenib toluenesulfonate in vivo, this trail was conducted in the fasted state.
A total of 11 adverse events were reported in 9 subjects during this study, where 7 events were classified as mild and 4 as moderate in severity. 3 cases were likely unrelated to the test drug, while the remaining 8 events could not be ruled out as connected to the test drug and were all deemed adverse reactions to the drug. Fortunately, all 11 adverse events resolved without any subsequent effects by the end of the trial. There were no serious adverse events or withdrawals due to adverse events during the oral T and R formulations phases, indicating that 0.2 g sorafenib tosylate tablets are safe and well-tolerated in healthy Chinese subjects.
This study has several limitations that should be acknowledged. First, the sample size, although meeting the minimum regulatory requirements for bioequivalence trials, was relatively small. This may limit the ability to fully characterize inter-individual variability in pharmacokinetic parameters and safety profiles. Second, the exclusion of special populations (e.g., adolescents, children, elderly individuals) precludes extrapolation of the current findings to these groups. Future studies are warranted to assess the safety and efficacy of the T formulation in populations with distinct metabolic or physiological conditions. Furthermore, the safety assessment was confined to adverse drug reactions observed within a short-term period (≤120 h post-dose). Notably, the characteristic toxicities of sorafenib, such as hand-foot syndrome and hypertension, are typically associated with cumulative exposure and emerge following chronic administration (Boudou-Rouquette et al., 2012). Consequently, the transient monitoring window in this single-dose study may underestimate the clinically relevant risks of the T formulation in real-world settings where prolonged therapeutic use is required.
5 Conclusion
This randomized, open, single-dose, four-period crossover trial confirmed that sorafenib tosylate tablets (0.2 g), manufactured by Renhexidelong Pharmaceuticals Co., Ltd., were bioequivalent to the control drug Nexavar® (Bayer AG, Germany), and were safe and well tolerated in healthy adult Chinese subjects under fasting conditions.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Henan (Zhengzhou) Zhonghui Cardiovascular Ethics Committee. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
ZW: Data curation, Formal Analysis, Validation, Writing – original draft, Writing – review and editing. HP: Validation, Writing – original draft, Writing – review and editing. YZ: Data curation, Writing – review and editing. XC: Supervision, Writing – review and editing. JA: Supervision, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Awada, A., Hendlisz, A., Gil, T., Bartholomeus, S., Mano, M., de Valeriola, D., et al. (2005). Phase I safety and pharmacokinetics of BAY 43-9006 administered for 21 days on/7 days off in patients with advanced, refractory solid tumours. Br. J. Cancer 92 (10), 1855–1861. doi:10.1038/sj.bjc.6602584
Boudou-Rouquette, P., Ropert, S., Mir, O., Coriat, R., Billemont, B., Tod, M., et al. (2012). Variability of sorafenib toxicity and exposure over time: a pharmacokinetic/pharmacodynamic analysis. Oncologist 17 (9), 1204–1212. doi:10.1634/theoncologist.2011-0439
Clark, J. W., Eder, J. P., Ryan, D., Lathia, C., and Lenz, H. J. (2005). Safety and pharmacokinetics of the dual action Raf kinase and vascular endothelial growth factor receptor inhibitor, BAY 43-9006, in patients with advanced, refractory solid tumors. Clin. Cancer Res. 11 (15), 5472–5480. doi:10.1158/1078-0432.CCR-04-2658
Cui, Y., Li, Y., Guo, C., Li, Y., Ma, Y., and Dong, Z. (2022). Pharmacokinetic interactions between canagliflozin and sorafenib or lenvatinib in rats. Molecules 27 (17), 5419. doi:10.3390/molecules27175419
Davit, B. M., Chen, M. L., Conner, D. P., Haidar, S. H., Kim, S., Lee, C. H., et al. (2012). Implementation of a reference-scaled average bioequivalence approach for highly variable generic drug products by the US Food and Drug Administration. AAPS J. 14 (4), 915–924. doi:10.1208/s12248-012-9406-x
Haidar, S. H., Davit, B., Chen, M. L., Conner, D., Lee, L., Li, Q. H., et al. (2008a). Bioequivalence approaches for highly variable drugs and drug products. Pharm. Res. 25 (1), 237–241. doi:10.1007/s11095-007-9434-x
Haidar, S. H., Makhlouf, F., Schuirmann, D. J., Hyslop, T., Davit, B., Conner, D., et al. (2008b). Evaluation of a scaling approach for the bioequivalence of highly variable drugs. AAPS J. 10 (3), 450–454. doi:10.1208/s12248-008-9053-4
Huh, K. Y., Hwang, S., Park, S. Y., Lim, H. J., Jin, M., Oh, J., et al. (2021). Population pharmacokinetic modelling and simulation to determine the optimal dose of nanoparticulated sorafenib to the reference sorafenib. Pharmaceutics 13 (5), 629. doi:10.3390/pharmaceutics13050629
Jain, L., Woo, S., Gardner, E. R., Dahut, W. L., Kohn, E. C., Kummar, S., et al. (2011). Population pharmacokinetic analysis of sorafenib in patients with solid tumours. Br. J. Clin. Pharmacol. 72 (2), 294–305. doi:10.1111/j.1365-2125.2011.03963.x
Kane, R. C., Farrell, A. T., Saber, H., Tang, S., Williams, G., Jee, J. M., et al. (2006). Sorafenib for the treatment of advanced renal cell carcinoma. Clin. Cancer Res. 12 (24), 7271–7278. doi:10.1158/1078-0432.CCR-06-1249
Karalis, V., Symillides, M., and Macheras, P. (2012). Bioequivalence of highly variable drugs: a comparison of the newly proposed regulatory approaches by FDA and EMA. Pharm. Res. 29 (4), 1066–1077. doi:10.1007/s11095-011-0651-y
Keating, G. M., and Santoro, A. (2009). Sorafenib: a review of its use in advanced hepatocellular carcinoma. Drugs 69 (2), 223–240. doi:10.2165/00003495-200969020-00006
Lennernäs, H., Brisander, M., Liljebris, C., Jesson, G., and Andersson, P. (2024). Enhanced bioavailability and reduced variability of dasatinib and sorafenib with a novel amorphous solid dispersion technology platform. Clin. Pharmacol. Drug Dev. 13, 985, 999. doi:10.1002/cpdd.1416
Liu, L., Cao, Y., Chen, C., Zhang, X., McNabola, A., Wilkie, D., et al. (2006). Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 66 (24), 11851–11858. doi:10.1158/0008-5472.CAN-06-1377
Malik, M. Y., Jaiswal, S., Sharma, A., Shukla, M., and Lal, J. (2016). Role of enterohepatic recirculation in drug disposition: cooperation and complications. Drug Metab. Rev. 48 (2), 281–327. doi:10.3109/03602532.2016.1157600
Moore, M., Hirte, H. W., Siu, L., Oza, A., Hotte, S. J., Petrenciuc, O., et al. (2005). Phase I study to determine the safety and pharmacokinetics of the novel Raf kinase and VEGFR inhibitor BAY 43-9006, administered for 28 days on/7 days off in patients with advanced, refractory solid tumors. Ann. Oncol. 16 (10), 1688–1694. doi:10.1093/annonc/mdi310
National Medical Products Administration, National Health Commission of the People’s Republic of China (2020). Good clinical Practice.
Ramakrishnan, V., Timm, M., Haug, J. L., Kimlinger, T. K., Halling, T., Wellik, L. E., et al. (2012). Sorafenib, a multikinase inhibitor, is effective in vitro against non-Hodgkin lymphoma and synergizes with the mTOR inhibitor rapamycin. Am. J. Hematol. 87 (3), 277–283. doi:10.1002/ajh.22263
Ranieri, G., Gadaleta-Caldarola, G., Goffredo, V., Patruno, R., Mangia, A., Rizzo, A., et al. (2012). Sorafenib (BAY 43-9006) in hepatocellular carcinoma patients: from discovery to clinical development. Curr. Med. Chem. 19 (7), 938–944. doi:10.2174/092986712799320736
Song, E., Lee, W., and Kim, B. H. (2021). Model-based approach for designing an efficient bioequivalence study for highly variable drugs. Pharm. (Basel). 14 (11), 1101. doi:10.3390/ph14111101
Strumberg, D., Clark, J. W., Awada, A., Moore, M. J., Richly, H., Hendlisz, A., et al. (2007). Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumors. Oncologist 12 (4), 426–437. doi:10.1634/theoncologist.12-4-426
Strumberg, D., Richly, H., Hilger, R. A., Schleucher, N., Korfee, S., Tewes, M., et al. (2005). Phase I clinical and pharmacokinetic study of the Novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43-9006 in patients with advanced refractory solid tumors. J. Clin. Oncol. 23 (5), 965–972. doi:10.1200/JCO.2005.06.124
The National Medical Products Administration of China, Center for Drug Evaluation (2019). Guideline for bioavailability and bioequivalence studies of generic drug products.
Wilhelm, S. M., Adnane, L., Newell, P., Villanueva, A., Llovet, J. M., and Lynch, M. (2008). Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 7 (10), 3129–3140. doi:10.1158/1535-7163.MCT-08-0013
Wilhelm, S. M., Carter, C., Tang, L., Wilkie, D., McNabola, A., Rong, H., et al. (2004). BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 64 (19), 7099–7109. doi:10.1158/0008-5472.CAN-04-1443
Keywords: sorafenib, bioequivalence, pharmacokinetic, UPLC-MS/MS, safety
Citation: Wang Z, Peng H, Zhang Y, Chen X and A J (2025) Bioequivalence and safety assessment of sorafenib tosylate tablets in healthy Chinese subjects under fasting conditions. Front. Pharmacol. 16:1470095. doi: 10.3389/fphar.2025.1470095
Received: 18 October 2024; Accepted: 01 April 2025;
Published: 14 April 2025.
Edited by:
Yurong Lai, Gilead, United StatesReviewed by:
Ping Du, Capital Medical University, ChinaHaimiao Yang, The Affiliated Hospital of Changchun University of Traditional Chinese Medicine, China
Jim Hughes, Pfizer, United States
Copyright © 2025 Wang, Peng, Zhang, Chen and A. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xijing Chen, Y2hlbnhqLWxhYkBob3RtYWlsLmNvbQ==; Jiye A, aml5ZWFAY3B1LmVkdS5jbg==
†These authors have contributed equally to this work