Abstract
Acute lung injury (ALI) is a clinical syndrome characterized by excessive inflammatory responses. Despite the exploration of various therapeutic approaches, no effective pharmacological treatment is currently available for ALI. In the current study, we investigated the role of SIRT3 in LPS-induced ALI and the potential protective effects of dexmedetomidine (Dex), an agent that activates α2-adrenergic receptors. Histological analysis showed extensive lung damage and increased inflammatory cells in LPS-treated lung samples, with elevated TUNEL+ cells indicating apoptosis (p < 0.05). SIRT3 mRNA and protein expression were significantly downregulated following LPS treatment, both in vivo and in vitro (p < 0.05). DEX administration restored protein SIRT3 levels and reduced inflammation, while the SIRT3 inhibitor 3-TYP negated these benefits (p < 0.05). Additionally, DEX reduced pro-inflammatory cytokine levels and oxidative stress, effects that were also diminished by 3-TYP (p < 0.05). Our findings suggest that DEX exerts its protective effects against LPS-induced ALI via modulation of the SIRT3/LKB1/AMPK signaling pathway, highlighting the critical role of SIRT3 in inflammatory and oxidative stress responses in ALI.
Introduction
Acute lung injury (ALI) represents a significant burden in critical care medicine, identified by its high incidence and the profound impact on patient morbidity and mortality (Liu C. et al., 2022; Liu X. et al., 2021). ALI is a clinical syndrome associated with excessive inflammatory responses, which can lead to diffuse alveolar damage, hypoxemic respiratory failure, and significant mortality rates (Yin et al., 2021). The condition is often induced by various factors, including sepsis, pneumonia, and severe trauma, and is marked by the disruption of the alveolar-capillary barrier, leading to protein-rich fluid exudation and impaired gas exchange (Matthay and Zemans, 2011). ALI progression to acute respiratory distress syndrome (ARDS) and the associated increase in mortality rates highlight the urgent need for effective treatments (Xu et al., 2024). The economic and healthcare system burden is substantial, with ALI/ARDS patients often requiring prolonged intensive care, mechanical ventilation, and other resource-intensive interventions (Rubenfeld et al., 2005). Despite the advancements in intensive care, the available therapies for ALI/ARDS remain largely supportive, such as mechanical ventilation, and do not adequately target the underlying pathophysiological mechanisms (Bellani et al., 2016). The development of gene therapy and the use of therapeutic nucleic acids, such as those discussed in the review by Zhuang et al. (2023), represent a promising direction in modulating the inflammatory response in ALI/ARDS. These approaches aim to target specific genes and molecular pathways involved in the disease process, offering a more precise and potentially more effective treatment modality. Thus, understanding the pathogenesis of ALI/ARDS is critically important for developing targeted therapies to effectively prevent or treat the condition.
SIRT3 has been implicated as a potential therapeutic target in various diseases, including neurodegenerative disorders and cardiovascular diseases (Cao et al., 2022). SIRT3 increases the activity of pyruvate dehydrogenase (PDH) through deacetylation, promoting glucose oxidation and thus playing a role in function and energy production (Chen et al., 2021). SIRT3 deficiency has been associated with proliferation, oxidative stress, inflammation, and fibrosis (Chelladurai et al., 2021). SIRT3/LKB1/AMPK signaling pathway is a key regulator of cellular metabolism and inflammation (Guo et al., 2022). Overexpression of SIRT3 has been shown to increase autophagy level and promote LKB1 phosphorylation, leading to the activation of AMPK and decreased phosphorylation of mTOR, suggesting a role for the LKB1-AMPK-mTOR pathway in the induction of autophagy (Zhang M. et al., 2018). SIRT3 has been reported to be associated with ALI, for example, SIRT3-p53 pathway in sepsis-associated ALI (Gao et al., 2024). However, the role of SIRT3/LKB1/AMPK signaling pathway in ALI remain to be supplemented.
Dexmedetomidine (DEX), an agonist of the α2-adrenergic receptor, is well-known for its properties that reduce inflammation, prevent apoptosis, and provide antioxidant effects. These characteristics have demonstrated considerable advantages in alleviating lung inflammation across different experimental models (Zhang H. et al., 2019; Li et al., 2018; Wang et al., 2019). DEX has the potential to mitigate sepsis-related ALI by influencing macrophage efferocytosis via the ROS/ADAM10/AXL signaling pathway (Li et al., 2024) and our previous results showed that DEX alleviates LPS-induced acute lung injury in rats (Sun et al., 2020). On the other hand, DEX has been shown to ameliorate cardiac ischemia/reperfusion injury that can be improved by promoting autophagy via the AMPK/SIRT3 signaling pathway activation (He et al., 2023), indicating the relationship between DEX and SIRT3 signals. However, whether DEX regulates SIRT3/LKB1/AMPK signaling pathway to modulate ALI needs further exploring.
Herein, the present study aimed to investigate the protective mechanisms of DEX in LPS-induced ALI. Our findings demonstrate that DEX exerts its anti-inflammatory effects through activation of the SIRT3/LKB1/AMPK signaling pathway, thereby providing novel mechanistic insights into its therapeutic potential for ALI and other inflammation-related disorders.
Methods and materials
Animals and ethics
The studies involving animal participants were reviewed and approved by the Ethics Committee of Anhui Medical University. Seven-day-old pathogen-free Sprague-Dawley (DS) rats (male, weighing 250–300 g) were sourced from the Experimental Animal Center of Anhui Medical University (license no. SCXK-2018–031). The rats were housed in standard laboratory cages under controlled conditions, maintaining a 12-h light/dark cycle at 22°C ± 2°C, with free access to food and water.
Sixteen rats were randomly divided into four groups (4 rats/group): a saline control group, a group exposed to LPS (#L2630-100 MG; Sigma Aldrich, United States), a group exposed to both LPS and Dex (#MB4091; Meilunbio Co., Ltd.; 25 μg/kg for rats according to previous reports (Sun et al., 2020; Wang et al., 2018)), and a group treated with LPS and Dex and 3-YTP, SIRT3 inhibitor (#HY-108331; Med chem express, Shanghai, China; 50 mg/kg, ip). The rats were anesthetized through an intraperitoneal injection of pentobarbital sodium at a dose of 40 mg/kg (Merck KGaA; cat. No. 1063180500). Tracheal intubation was performed using a micro-atomizer; the control group received 300 µL saline, whereas the LPS group was given 5 mg/kg LPS. Nebulized solution was given at an oxygen flow rate of 4 L per minute for 25 min. Animals were then fed normally for 30 min without intubation following every 6-hour period. Animals were euthanized by decapitation 12 h later. Lung tissues or bronchoalveolar lavage (BAL) were collected for evaluation, followed by a thoracotomy. The right lung tissue samples were isolated for further experiments.
IHC analysis for lung morphology
For the morphological evaluation of the lungs, freshly collected samples were first weighed to record their wet mass. The samples were then dried overnight at 75°C to assess their dry mass (Herriges and Morrisey, 2014). Hematoxylin/eosin (HE) staining was performed to examine the histological structure of the lungs and assess the inflammatory response after LPS exposure. At designated time points, lung tissues were harvested and fixed in 4% paraformaldehyde, then dehydrated and embedded in paraffin wax. The samples were subsequently sectioned into 5 µm slices. The sections were analyzed using a fluorescence microscope (Olympus IX50; Olympus Corporation) in conjunction with analyzing software (NIS-Elements F3.2). Airspace volume density was determined by dividing the total airspace area by the total area (Plosa et al., 2014). At each time-point, at least 3 randomly selected images from 5 samples per group were analyzed.
Biochemical analysis assay
The levels of SOD, CAT, and MDA were measured in both serum and lung tissue samples, using purchased kits (Beyotime, Shanghai, China). Serum concentrations of tumor necrosis factor-α (TNF-α, ab46070; Abcam), interleukin-1β (IL-1β, # BMS630; Thermal Fisher Scientific, Waltham, MA, United States) and IL-10 (#ERA23RB; Thermal Fisher Scientific), IL-8 (#EK720269; AFG Scientific, MA, United States) and IL-12 (#EK720274; AFG Scientific), IL-18 (#RAB1147; Merck, United States) and IL-6 (#RAB0311; Merck, United States), IL-5 (#ab267811; Abcam) and IL-17 (#ab119536; Abcam) were quantified using ELISA kits accordingly. A hydrogen peroxide assay kit (#S0051; Beyotime) was applied to measure the levels of hydrogen peroxide (H2O2) and the lucigenin chemiluminescence method was applied to determine the superoxide anion (O2-) levels in lung tissue samples. Briefly, lung tissue samples were homogenized and supernatant after centrifugation was incubated with 5 μM lucigenin in buffer, and then samples were recorded using a Tecan Infinite 200 by light emission. Addition incubation into the medium of 350 U/ml SOD were used to confirm the specificity for O2-. Protein concentrations were determined using a BCA Protein Concentration Assay Kit (Enhanced; #P0010; Beyotime).
Inflammatory cell counts of bronchoalveolar lavage fluid (BALF)
Following treatment, animals were sacrificed, and BALF was obtained by washing the lungs three times with PBS via a tracheal cannula. The samples were centrifuged at 3,000 rpm for 10 min at 4°C, and the pellet of cells was resuspended in PBS for total cell count analysis using a hemocytometer. Cytospin preparations were made for differential cell count, which involved staining with Wright-Giemsa method. The proportions of macrophages, neutrophils, and lymphocytes in the BALF samples were determined by measuring leukocytes under a light microscope.
ELISA assay
Serum samples were collected from administrated animals, and then were subjected to ELISA assay, for the antibodies: TNF-alpha, IL-6, IL-5, IL-1beta, IL-18 and IL-17.
TUNEL analysis
This assay was conducted using a one-step detection kit (#C1086; Beyotime) following the protocol. Tissue samples were deparaffinized in xylene, rehydrated by a graded series of ethanol concentrations, and then heated for antigen retrieval. Hydrogen peroxide of 3% was used to inhibit endogenous peroxidase activity. Subsequently, different dilutions of terminal deoxynucleotidyl transferase in reaction buffer, containing a constant concentration of digoxigenin-labeled nucleotides, were applied to the sections and incubated at 37°C for 1 h, followed by a 10-minute incubation in Stop/Wash buffer. After extensive washing, 50 μL TUNEL detection dilution was applied for 30 min. Then the sections were mounted, and images were captured under the laser wavelength between 450 and 500 nm. The percentage of TUNEL+ cells was quantified and normalized relative to the total number of cells for each group.
RT-qPCR assay
Total RNA was extracted from fresh samples using the Total RNA kit (Omega Bio-Tek, Inc.) and stored on ice, following the manufacturer’s protocol. First-strand cDNA synthesis and SYBR® Green qPCR were conducted with the PrimeScript™ RT Reagent kit (Takara Bio, Inc.). Specific primers used in the study for SIRT3 are listed as follows: F-5′-AAGACATACGGGTGGAGCCT -3′, R-5′ GGACTCAGAGCAAAGGACCC-3’. Real-time PCR was conducted using the SYBR Green kit (TINGEN Biotech, Beijing, China) under the following conditions: denaturation at 95°C for 20 s, annealing at 58°C for 20 s, and extension at 68°C for 30 s mRNA expression levels were measured by the 2−ΔΔCq method and normalized to β-actin level. Levels of SIRT3 in control group against β-actin were normalized to 100%. The qPCR results represent three independent experiments.
Western blotting assay
This assay was conducted using a standard protocol with polyclonal antibodies specific to SIRT3, LKB1, phosphorylated(p)-LKB1, AMPK and phosphorylated(p)-AMPK. The procedures for protein extraction and immunoblotting were previously detailed. Protein samples were extracted from lung tissue homogenates using RIPA buffer (Beyotime, Shanghai, China), supplemented with inhibitors of protease and phosphatase. Protein concentrations were then determined using the BCA assay. The proteins were separated using 10% SDS-PAGE and subsequently transferred to a PVDF membrane. Following a blocking step with 5% non-fat milk, the membrane was incubated overnight at 4°C with antibodies targeting SIRT3 (1:1,000; #ab217319; Abcam), LKB1 (1:1,000; #ab199970), phosphorylated(p)-LKB1 (1:500; #ab63473; Abcam), AMPK (1:1,000; #ab32047; Abcam) and Anti-AMPK alpha 1 (phospho T183) + AMPK alpha 2 (phospho T172) antibody (1:1,000; #ab133448; Abcam) in TBS buffer. GAPDH was used as a loading control (1:2,000; #5174; CST, MA, United States). Following incubation with secondary antibodies, either HRP goat anti-rabbit (1:1,000; # A0208; Beyotime) or anti-mouse IgG (1:1,000; #A0216; Beyotime)—the blots were developed using the BeyoECL Star ECL (P0018AM; Beyotime) and imaged. Band intensities were quantified using ImageJ (NIH, United States). Levels in control group against GAPDH were normalized to 100%. The Western blot results represent three independent experiments.
Statistical analysis
Statistical analyses and graph construction were conducted by the software package (version 10.0; GraphPad Software, Inc.). Data are expressed as mean ± standard deviation (SD) from a minimum of three independent experiments. To assess significant differences between control and treatment groups, one-way ANOVA followed by Tukey’s post hoc test or unpaired Student’s t-test were employed. A p-value of less than 0.05 was regarded as statistically significant.
Results
LPS induces acute lung injury (ALI)
We first constructed a commonly used acute lung injury model using LPS. As described in the methods, rats were treated with LPS and then samples were collected and subjected to HE staining. Histological characteristics of the sections were observed. As illustrated in Figure 1A, LPS administration significantly caused the diffuse damage in the alveoli, alveolar tubes, alveolar sacs, bronchi, and alveolar septa. The quantified data of inflammation score shown in Figure 1B revealed that numerous inflammatory cells were observed in the alveolar septa after LPS exposure. Furthermore, samples were subjected to TUNEL assay. The results in Figure 1C showed that LPS administration markedly increased the number of TUNEL-positive pulmonary cells in lung samples. The quantified data were shown in Figure 1D. Then, we conducted bronchoalveolar lavages (BAL) assay to reveal the inflammatory cell infiltration by Wright-Giemsa method. As shown in Figure 1E, LPS administration significantly resulted in the increasement of immune cells, eosinophils, macrophages, lymphocytes and neutrophils. Furthermore, in LPS treated lung samples, the BAL eotaxin level was also increased. These data indicate that LPS administration significantly induced acute lung injury.
FIGURE 1
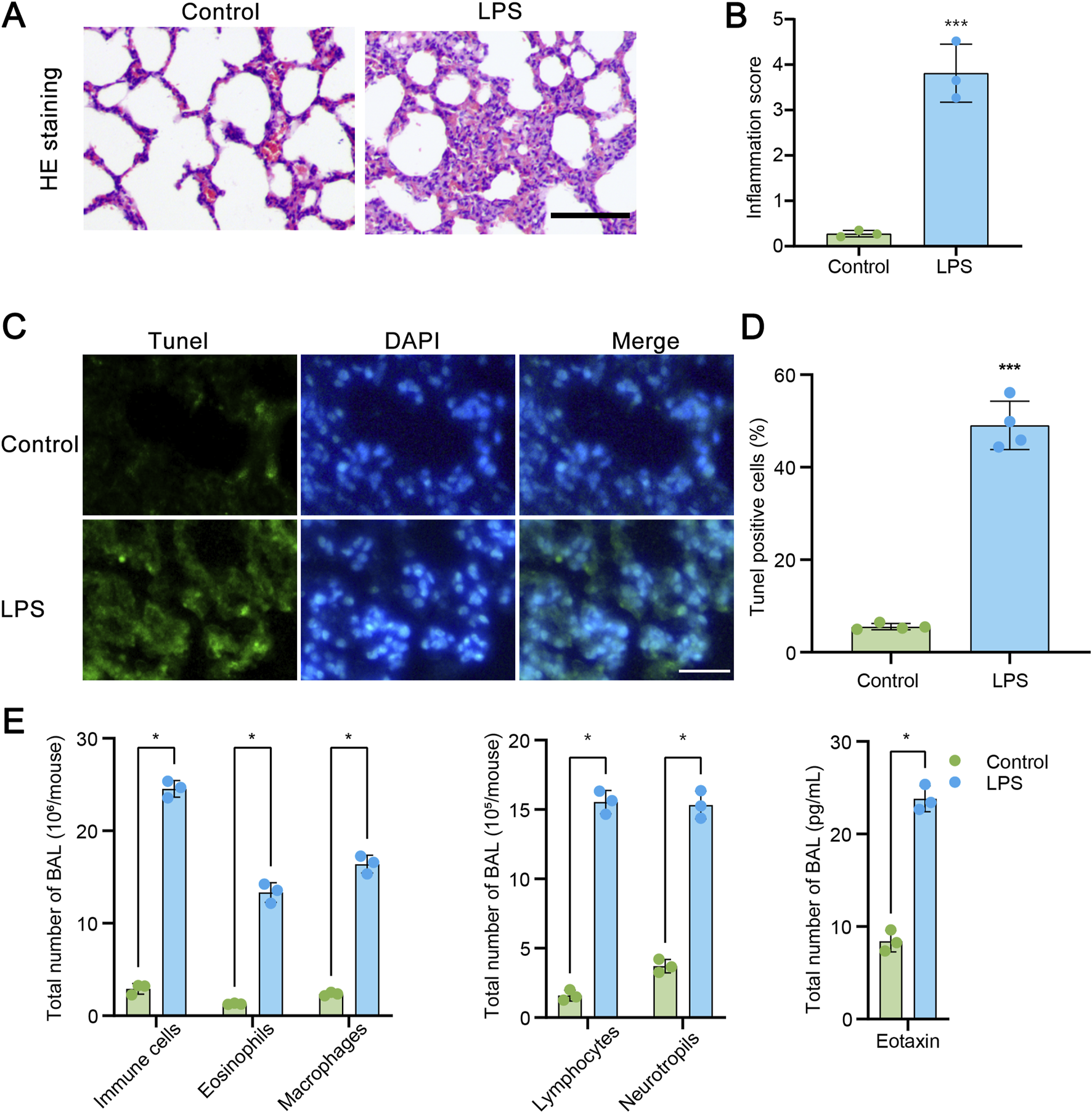
LPS administration induces to acute lung injury (ALI). (A) Hematoxylin and eosin (HE) staining of transverse lung sections from rats in the control and LPS-treated groups. (B) Quantification of the inflammatory response based on HE staining (n = 3). (C) TUNEL staining of transverse lung sections from the control and LPS-treated groups. (D) The number of TUNEL-positive cells relative to the total cells in lung tissue from the control and LPS-treated groups (n = 4). (E) Assessment of total cells, neutrophils, lymphocytes, macrophages, eosinophils, and Eotaxin levels in bronchoalveolar lavage fluid (BAL) by Wright-Giemsa (n = 3). Scale bars = 100 μm. Data are presented as mean ± SD. *P < 0.05 vs control; ***P < 0.001 vs control.
Dexmedetomidine (DEX) regulates the SIRT3/LKB1/AMPK signaling pathway in LPS-induced acute lung injury
Next, we tried to ask whether SIRT3 was involved in LPS-induced ALI. Samples from LPS treated rats were subjected to RT-qPCR and the levels of SIRT3 were measured. As shown in Figure 2A, the levels of SIRT3 were significantly reduced in LPS group, compared to control rats. Then cultured lung epithelial cells were treated with LPS of indicated time points (2, 4, 24, 48 and 72 h). The data revealed that the mRNA levels of SIRT3 were gradually decreasing along the time points (Figure 2B). Samples of LPS treated rats and cultured cells administrated with LPS were subjected to Western blotting assay. The data showed that SIRT3 protein levels were also markedly decreased upon LPS treatment (Figures 2C,D). Also, the levels of SIRT3 protein were gradually downregulated in LPS-treated lung endothelial cells, same trend as the mRNA levels (Figures 2E,F).
FIGURE 2
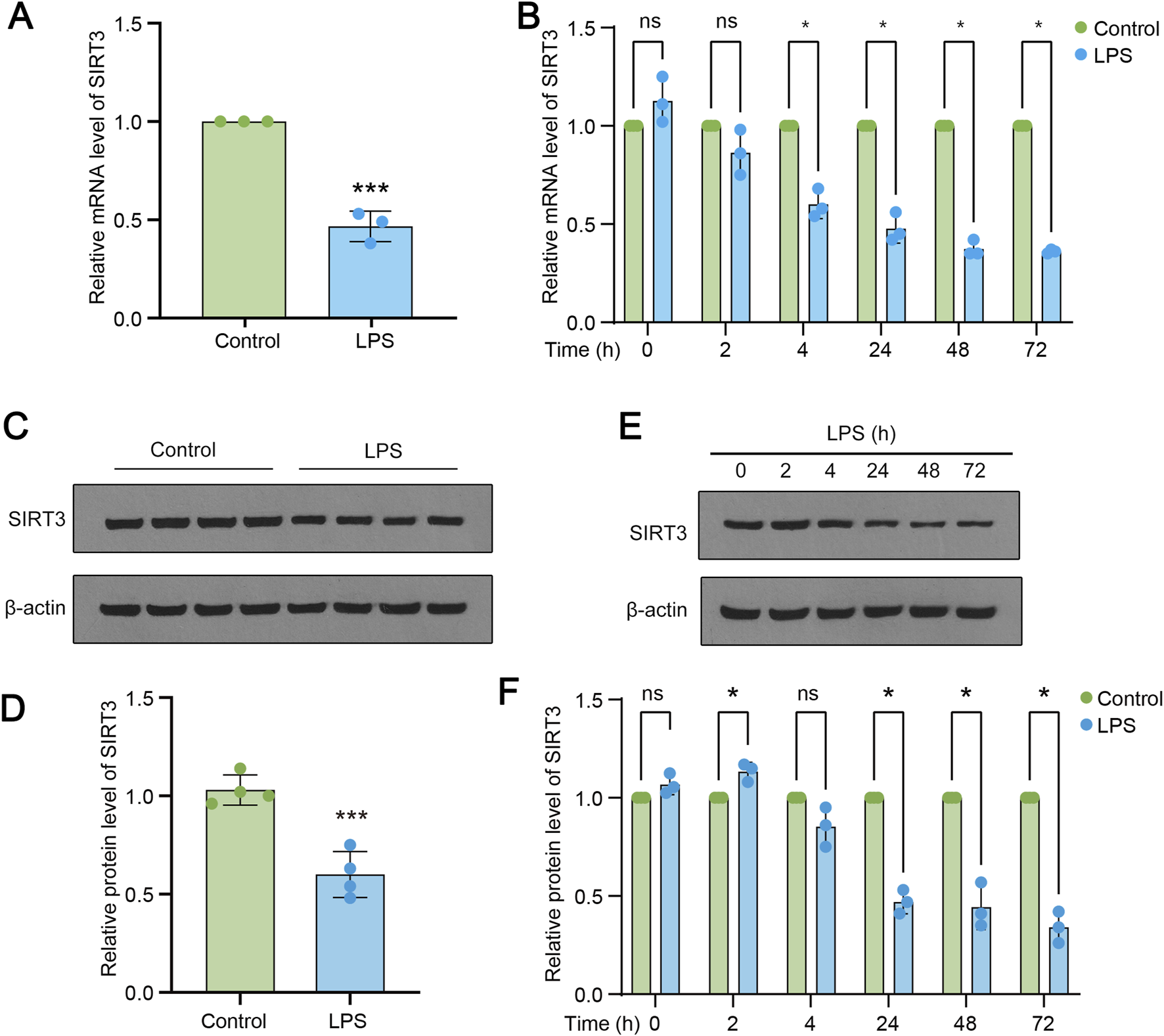
SIRT3 is downregulated during LPS induced ALI. (A) mRNA expression levels of SIRT3 in control and LPS-treated rats. (B) mRNA levels of SIRT3 in LPS-induced rat lung endothelial cells at 2, 4, 24, 48, and 72 h (C–D) Protein levels of SIRT3 in lung tissue samples (n = 4) from LPS-treated and control rats. (E–F) Protein levels of SIRT3 in lung endothelial cells at 2, 4, 24, 48, and 72 h (n = 3). Data are expressed as mean ± SD (n = 3 or 4). *P < 0.05 vs control; ***P < 0.001 vs control.
We have previously reported that DEX could alleviate LPS-induced acute lung injury (Sun et al., 2020). Since SIRT3 was involved in LPS-induced ALI, we asked whether DEX would function via the regulation of SIRT3 signaling pathway. LPS-administrated rats were treated with DEX, or the SIRT3 inhibitor 3-TYP. Firstly, we performed immunohistochemistry assay to reveal the histological characteristics of the sections under different treatment. As shown in Figure 3A, LPS-induced inflammation was significantly suppressed by DEX treatment, however, this process could be restored by SIRT3 inhibitor. The diffuse damage in the alveoli, alveolar tubes, alveolar sacs, bronchi, and alveolar septa showed the same trend as inflammation response, which was quantified in Figure 3B. Next, LPS administration significantly decreased the SIRT3 protein levels (Figures 3C,D), which could be rescued by the treatment of DEX. However, the addition of SIRT3 inhibitor 3-TYP further alleviated the restored level of SIRT3 protein (Figure 3B). Furthermore, when compared to the total level, the phosphorylated level of LKB1 and AMPK proteins were also shown the same trend as SIRT3 protein (Figures 3E,F). These results suggest that SIRT3 signaling pathway is involved in LPS-induced AKL, which could be modulated by DEX treatment.
FIGURE 3
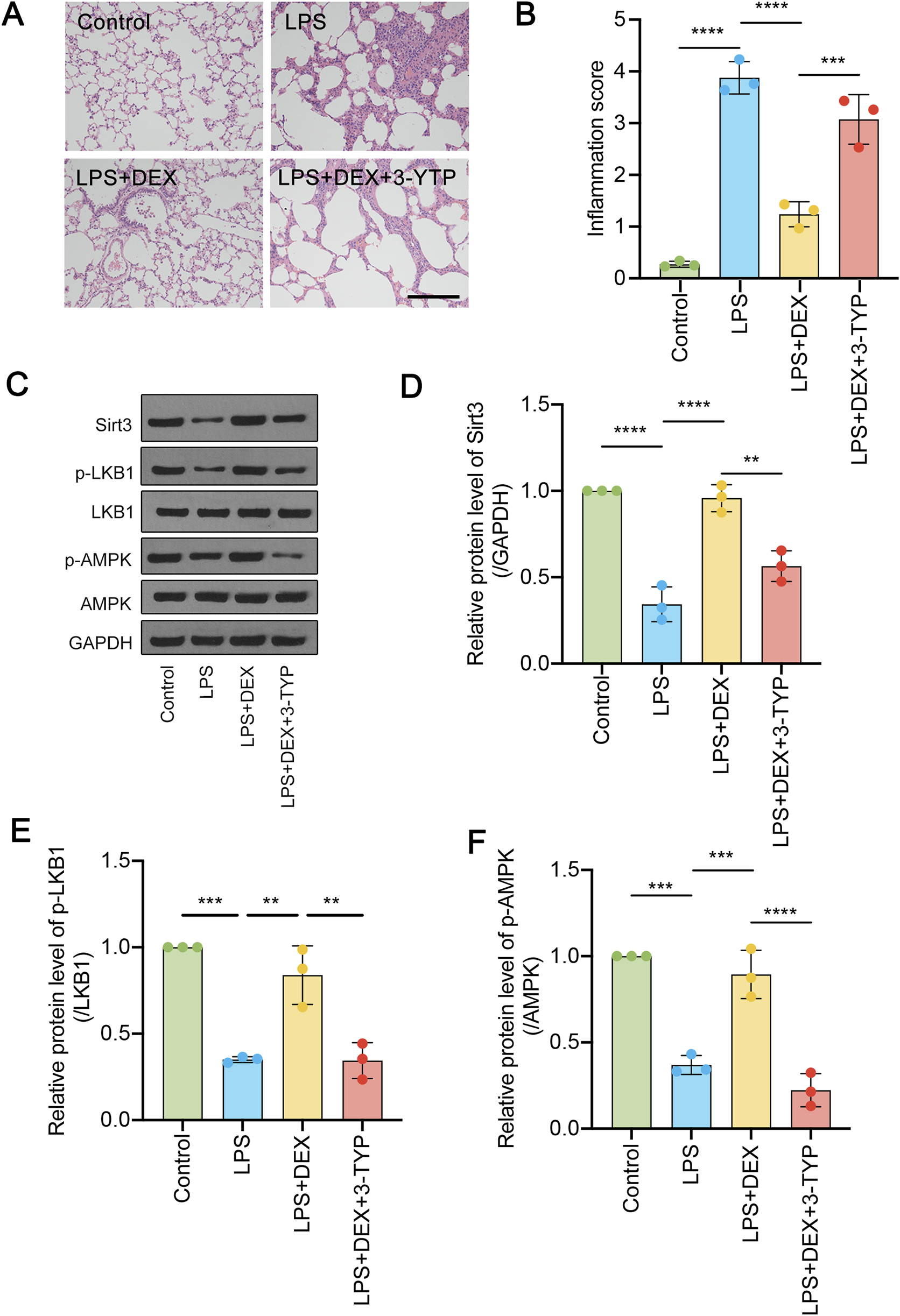
Dexmedetomidine (DEX) regulates the SIRT3/LKB1/AMPK signaling pathway in LPS-induced ALI. (A) HE-staining of transverse lung sections from rats in the control and LPS-treated groups, LPS + DEX, LPS + DEX+3-YTP (inhibitor of SIRT3) was performed. (B) The inflammation score was quantified for (A). (C) The protein levels of total SIRT3, phosphorylated and total LKB1 and phosphorylated and total AMPK were determined. (D–F) The quantified data were shown. The data are represented as mean ± SD (n = 3). **P < 0.01; ***P < 0.001; ****P < 0.0001.
Inhibition of SIRT3 signaling alleviates the protective effect of DEX against pro-inflammatory cytokine release and oxidative stress during LPS-induced ALI
To investigate whether the pro-inflammatory response evoked by LPS could be mitigated by DEX through the SIRT3 signaling pathway, we administered rats with LPS, DEX, or the SIRT3 inhibitor 3-TYP. We then assessed the levels of pro-inflammatory cytokines in their serum. As depicted in Figure 4, exposure to LPS markedly upregulated the serum levels of TNF-α (Figure 4A), IL-6 (Figure 4B), IL-5 (Figure 4C), IL-1β (Figure 4D), IL-18 (Figure 4E), and IL-17 (Figure 4F). However, the administration of DEX notably attenuated these cytokine levels, an effect that was nullified by the additional treatment with 3-TYP (Figure 4). Collectively, these findings indicate that pharmacological inhibition of the SIRT3 pathway undermines the protective role of DEX in LPS-induced inflammation in ALI.
FIGURE 4
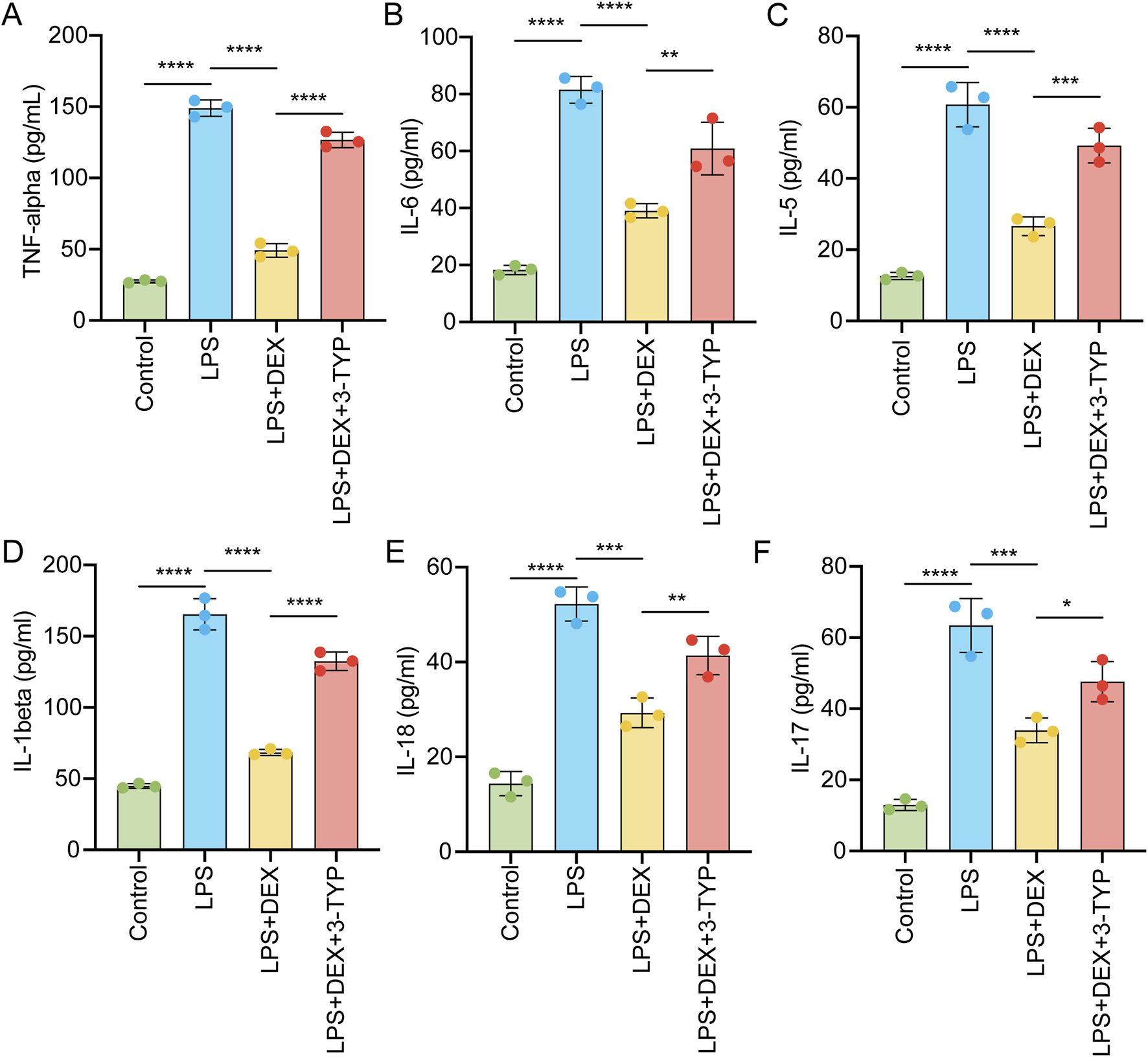
Inhibition of SIRT3 signaling abolishes the protective effect of DEX during pro-inflammatory cytokine release induced by LPS. Animals were treated as in Figure 3 and then serum pro-inflammatory cytokines were measured by ELISA, including (A) tumor necrosis factor-α (TNF-α), (B) interleukin-6 (IL-6), (C) IL-5, (D) IL-1β, (E) IL-18 and (F) IL-17. The data are presented as mean ± SD (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Oxidative stress, closely related to SIRT3 signaling pathway (Zheng et al., 2023), is critical for ALI (Zhong et al., 2024). To elucidate the role of SIRT3 in the generation of ROS in LPS-induced acute lung injury, we measured the activities of SOD and CAT in serum and lung tissue samples after LPS treatment. As depicted in Figures 5A–D, LPS induction significantly decreased the levels of SOD activity (Figures 5A,B) and CAT activity (Figures 5C,D). However, DEX administration notably reversed these reductions in SOD and CAT activities, an effect that was abrogated by the addition of the SIRT3 inhibitor 3-TYP (Figures 5A–D). In contrast, the level of MDA was upregulated by LPS treatment, but this increase was attenuated by DEX and further restored with the additional treatment of 3-TYP (Figures 5E,F). Moreover, we observed that the levels of oxidants such as O2−, H2O2, and nitrate in LPS-induced lung tissue samples were elevated following LPS treatment, but DEX treatment reduced these levels (Figures 5G–I). The addition of 3-TYP significantly inhibited the mitigating effect of DEX. Token together, these findings suggest that the inhibition of the SIRT3 pathway impairs the protective function of DEX against LPS-induced oxidative stress in ALI.
FIGURE 5
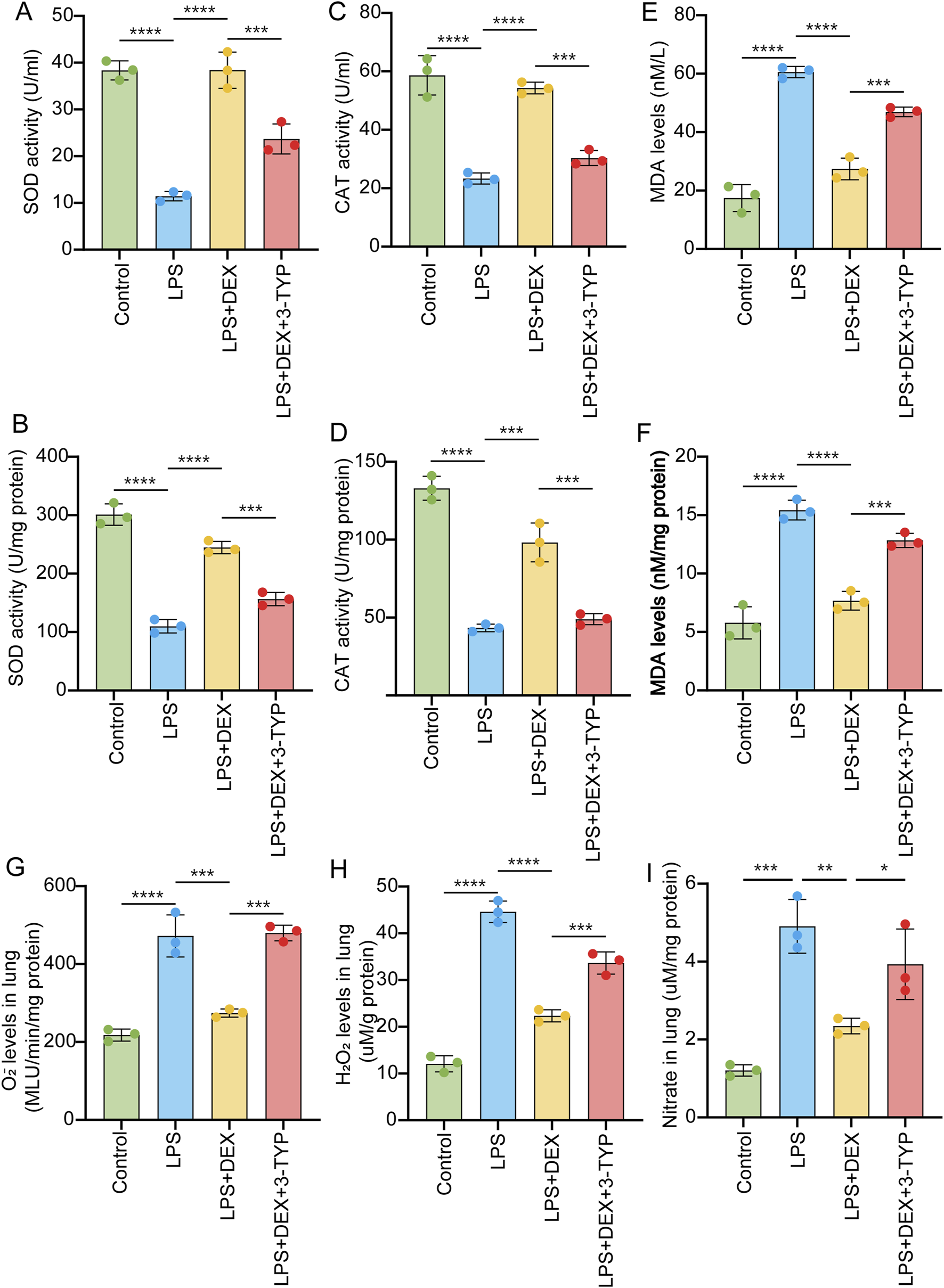
Inhibition of SIRT3 signaling abolishes the protective effect of DEX during oxidative stress induced by LPS. Rats were administered as in Figure 3. The collected samples were subjected to oxidative stress detection. SOD activity (A,B), CAT activity (C,D) and MDA (E,F) levels measured in the serum or in the lung tissue samples of LPS-treated rats, with or without DEX or 3-YTP. O2- (G) and H2O2 (H), and nitrate levels (I) in the LPS-induced lung tissue samples were measured. The data are presented as mean ± SD (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Discussion
In general, we examined the effects of LPS on acute lung injury and the role of SIRT3 in mediating these effects. We established a rat model of ALI using LPS, observing significant damage to alveolar structures, increased inflammatory cell infiltration, and heightened apoptosis as indicated by TUNEL assays. Quantitative analysis revealed a marked upregulation of pro-inflammatory cytokines and a decrease in antioxidant enzyme activities, indicative of oxidative stress. We found that LPS administration resulted in a significant downregulation of SIRT3 at both the mRNA and protein levels in lung tissues and cultured epithelial cells. Treatment with DEX effectively restored SIRT3 levels and improved lung pathology. However, the use of the SIRT3 inhibitor 3-TYP undermined DEX’s protective effects, leading to elevated pro-inflammatory cytokines and oxidative stress markers.
ALI is characterized by an intense inflammatory response within the lung tissue. A systematic review and meta-analysis has indicated a significant association between ALI and elevated levels of certain inflammatory biomarkers, including angiopoietin-2 (ANG-2), interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α (Liu Z. et al., 2022). These biomarkers are thought to play a crucial role in the pathogenesis of ALI, contributing to the severity of the condition and the subsequent development of acute respiratory distress syndrome (ARDS) (Mokra and Kosutova, 2015). The interaction between immune cells and vascular endothelial cells is another key aspect of the inflammatory process in ALI. These interactions lead to chemotaxis and adhesion of immune cells, which can exacerbate lung injury (Wu et al., 2022). For example, neutrophils, a type of white blood cell, migrating to the site of injury is a critical aspect of ALI pathophysiology, leading to further tissue damage and inflammation (Li et al., 2023). Furthermore, oxidative stress is intricately linked with acute lung injury, as it can lead to cellular damage, inflammation, and the exacerbation of lung tissue damage through the production of reactive oxygen species and the activation of inflammatory pathways (Bezerra et al., 2023). Oxidative stress not only causes direct tissue damage but also upregulates multiple inflammatory cytokines, perpetuating a cycle of damage and inflammation (Liu et al., 2023). Oxidants, such as ROS, act as inflammatory signaling molecules, activating pathways like NF-κB and NLRP3, which exacerbate ALI/ARDS (Ward, 2010). Here, we used LPS to construct the ALI model in rats. As we previously reported (Sun et al., 2020), LPS administration significantly induced severe damage in rat lungs (Figure 1). However, the detailed mechanism underlying inflammatory response during ALI need to be further explored.
The SIRT3/LKB1/AMPK signaling pathway plays a critical role in the regulation of cellular metabolism, energy homeostasis, and stress response, including its relationship with acute lung injury (ALI). SIRT3, a mitochondrial deacetylase, regulates proteins that are involved in ROS production, primarily by altering the acetylation of SOD2, which increases its activity and influences ROS homeostasis (Xi et al., 2024). SIRT3 abolishes sepsis-induced ALI via pyroptosis inhibition, which are crucial in mitigating inflammation and oxidative stress in ALI (Wu et al., 2023). Melatonin protects against ALI via SIRT3-dependent deacetylation of SOD2 (Ning et al., 2022). SIRT3 inhibition exacerbates mitochondrial dynamic imbalance and pro-inflammatory polarization, aggravating sepsis-induced ALI (Sun et al., 2024). Here, we found that LPS administration markedly downregulated the mRNA and protein levels of SIRT3, phosphorylated LKB1, and phosphorylated AMPK in lung samples. Dex treatment rescued these protein levels but could be suppressed by the additional administration of SIRT3 inhibitor 3-YTP (Figure 3). This finding suggests that the SIRT3/LKB1/AMPK signaling pathway is protective in ALI, which is consistent with above reports.
Dexmedetomidine (DEX) has been studied for its potential protective effects on ALI. Dexmedetomidine reduces systemic inflammation and lung injury by promoting Treg cells proliferation through the AMPK/SIRT1 signaling pathway (Zhang et al., 2023). DEX reduces LPS-induced ALI by targeting miR-381/NLRP3 axis (Zhang et al., 2018b). DEX mitigates IL-17-induced lung injury through a dose-dependent anti-inflammatory effect (Zhang et al., 2018c). DEX mitigates hyperoxia-induced ALI by inhibiting the activation of the NLRP3 inflammasome (Zhang et al., 2017). DEX protects against lung injury by influencing mitochondrial dynamics and promoting oxygen consumption (Zhang JR. et al., 2019), or via the PKC-alpha/HO-1 pathway (Song et al., 2022). DEX alleviates LPS-induced ALI in rats by regulating Nrf2/Keap1 and Akt signals (Yan et al., 2017). DEX alleviates pulmonary edema in LPS-induced ALI by upregulating AQP1 and AQP5 expression (Jiang et al., 2015). On the other hand, SIRT3 is involved in DEX regulated diseases. For example, DEX helps protect enteric glial cells from mitochondrial damage and cell death during experimental intestinal ischemia/reperfusion injury by utilizing a SIRT3-dependent pathway (Zhang et al., 2021). DEX safeguards the heart against ischemia-reperfusion injury by boosting autophagy via the AMPK/SIRT3 pathway, thereby reducing oxidative stress, inflammation, and improving cardiac function and mitochondrial integrity (He et al., 2023). DEX protects against nephritis by upregulating SIRT3 expression, which mitigates inflammation, oxidative stress, and apoptosis in renal cells both in vivo and in vitro (Lu et al., 2024). Here, in the current study, we found that DEX could also alleviate the LPS-induced inflammation and also DEX modulate SIRT3 signaling pathway to execute the protective function during ALI.
Dexmedetomidine is associated with several side effects, particularly hemodynamic and respiratory complications, which clinicians must monitor closely. Dexmedetomidine is associated with a significant decrease in heart rate compared to propofol, indicating potential bradycardia as a side effect (Nicholson et al., 2021). Dexmedetomidine has been noted to cause respiratory depression and hypoxia, particularly in combination with other sedatives (Liu S. et al., 2021). Despite its favorable respiratory profile, dexmedetomidine may still lead to severe circulatory complications in adults (Su and Hammer, 2011). The combination of dexmedetomidine with propofol in younger patients was associated with reduced mortality, while increasing dexmedetomidine doses correlated with increased mortality (Shehabi et al., 2023). While it shows efficacy in sedation and analgesia, its safety profile compared to other sedatives like propofol and olanzapine suggests a need for careful consideration in its use, especially in vulnerable populations.
In conclusion, the present study suggests that DEX mitigates LPS-induced ALI primarily through the SIRT3/LKB1/AMPK signaling pathway, reinforcing the importance of SIRT3 in the inflammatory and oxidative responses associated with ALI. These findings may offer insights into therapeutic strategies targeting SIRT3 in lung inflammatory diseases.
Statements
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
All animal procedures adhered to international animal welfare standards and complied with the regulations set by the committee at Anhui Medical University. Seven-day-old pathogen-free Sprague-Dawley (DS) rats (male, weighing 250–300 g) were sourced from the Experimental Animal Center of Anhui Medical University (license no. SCXK-2018–031). The rats were housed in standard laboratory cages under controlled conditions, maintaining a 12-hour light/dark cycle at 22°C ± 2°C, with free access to food and water. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
JC: Data curation, Writing – original draft. YC: Writing – original draft, Writing – review and editing. XP: Writing – review and editing. YX: Writing – review and editing. LC: Writing – review and editing. XP: Writing – review and editing. YS: Conceptualization, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This works was supported by Clinical Science Foundation from Anhui Medical University (No. 2022xkj244), Excellent Young Academic Leader Training Program of Anhui Provincial Children’s Hospital (No. eyrc014), the key project of Anhui Provincial University Scientific Research Project (No. 2023AH050654) and Clinical Medical Research Transformation Project of Anhui Provincial (No. 202304295107020068).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Bellani G. Laffey J. G. Pham T. Fan E. Brochard L. Esteban A. et al (2016). Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA315, 788–800. 10.1001/jama.2016.0291
2
Bezerra F. S. Lanzetti M. Nesi R. T. Nagato A. C. Silva C. P. E. Kennedy-Feitosa E. et al (2023). Oxidative stress and inflammation in acute and chronic lung injuries. Antioxidants (Basel)12, 548. 10.3390/antiox12030548
3
Cao M. Zhao Q. Sun X. Qian H. Lyu S. Chen R. et al (2022). Sirtuin 3: emerging therapeutic target for cardiovascular diseases. Free Radic. Biol. Med.180, 63–74. 10.1016/j.freeradbiomed.2022.01.005
4
Chelladurai P. Boucherat O. Stenmark K. Kracht M. Seeger W. Bauer U. M. et al (2021). Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br. J. Pharmacol.178, 54–71. 10.1111/bph.14932
5
Chen X. Hao B. Li D. Reiter R. J. Bai Y. Abay B. et al (2021). Melatonin inhibits lung cancer development by reversing the warburg effect via stimulating the SIRT3/PDH axis. J. Pineal Res.71, e12755. 10.1111/jpi.12755
6
Gao N. Liu X. Y. Chen J. Hu T. P. Wang Y. Zhang G. Q. (2024). Menaquinone-4 alleviates sepsis-associated acute lung injury via activating SIRT3-p53/SLC7A11 pathway. J. Inflamm. Res.17, 7675–7685. 10.2147/JIR.S486984
7
Guo Z. Tuo H. Tang N. Liu F. Y. Ma S. Q. An P. et al (2022). Neuraminidase 1 deficiency attenuates cardiac dysfunction, oxidative stress, fibrosis, inflammatory via AMPK-SIRT3 pathway in diabetic cardiomyopathy mice. Int. J. Biol. Sci.18, 826–840. 10.7150/ijbs.65938
8
He H. Liu P. Li P. (2023). Dexmedetomidine ameliorates cardiac ischemia/reperfusion injury by enhancing autophagy through activation of the AMPK/SIRT3 pathway. Drug Des. Devel Ther.17, 3205–3218. 10.2147/DDDT.S428024
9
Herriges M. Morrisey E. E. (2014). Lung development: orchestrating the generation and regeneration of a complex organ. Development141, 502–513. 10.1242/dev.098186
10
Jiang Y. X. Dai Z. L. Zhang X. P. Zhao W. Huang Q. Gao L. K. (2015). Dexmedetomidine alleviates pulmonary edema by upregulating AQP1 and AQP5 expression in rats with acute lung injury induced by lipopolysaccharide. J. Huazhong Univ. Sci. Technol. Med. Sci.35, 684–688. 10.1007/s11596-015-1490-6
11
Li F. Bai Y. Guan Z. Ji X. Zhan X. Gao Y. et al (2024). Dexmedetomidine attenuates sepsis-associated acute lung injury by regulating macrophage efferocytosis through the ROS/ADAM10/AXL pathway. Int. Immunopharmacol.142, 112832. 10.1016/j.intimp.2024.112832
12
Li J. Chen Q. He X. Alam A. Ning J. Yi B. et al (2018). Dexmedetomidine attenuates lung apoptosis induced by renal ischemia-reperfusion injury through α2AR/PI3K/Akt pathway. J. Transl. Med.16, 78. 10.1186/s12967-018-1455-1
13
Li Y. Jiang Y. Zhang H. Zhang J. Ma J. Yang Z. et al (2023). Research on acute lung injury inflammatory network. Int. J. Clin. Pharmacol. Ther.61, 394–403. 10.5414/CP204438
14
Liu C. Xiao K. Xie L. (2022a). Progress in preclinical studies of macrophage autophagy in the regulation of ALI/ARDS. Front. Immunol.13, 922702. 10.3389/fimmu.2022.922702
15
Liu S. Zhao R. Yang R. Zhao H. Ji C. Duan M. et al (2021b). Are dexmedetomidine and olanzapine suitable to control delirium in critically ill elderly patients? A retrospective cohort study. Biomed. Pharmacother.139, 111617. 10.1016/j.biopha.2021.111617
16
Liu X. Wang L. Xing Q. Li K. Si J. Ma X. et al (2021a). Sevoflurane inhibits ferroptosis: a new mechanism to explain its protective role against lipopolysaccharide-induced acute lung injury. Life Sci.275, 119391. 10.1016/j.lfs.2021.119391
17
Liu Y. Li H. Ouyang Y. Zhang Y. Pan P. (2023). Exploration of the role of oxidative stress-related genes in LPS-Induced acute lung injury via bioinformatics and experimental studies. Sci. Rep.13, 21804. 10.1038/s41598-023-49165-3
18
Liu Z. Liu D. Wang Z. Zou Y. Wang H. Li X. et al (2022b). Association between inflammatory biomarkers and acute respiratory distress syndrome or acute lung injury risk: a systematic review and meta-analysis. Wien Klin. Wochenschr134, 24–38. 10.1007/s00508-021-01971-3
19
Lu K. Li X. Wu J. (2024). Sirtuin 3 is required for the dexmedetomidine-mediated alleviation of inflammation and oxidative stress in nephritis. Immun. Inflamm. Dis.12, e1135. 10.1002/iid3.1135
20
Matthay M. A. Zemans R. L. (2011). The acute respiratory distress syndrome: pathogenesis and treatment. Annu. Rev. Pathol.6, 147–163. 10.1146/annurev-pathol-011110-130158
21
Mokra D. Kosutova P. (2015). Biomarkers in acute lung injury. Respir. Physiol. Neurobiol.209, 52–58. 10.1016/j.resp.2014.10.006
22
Nicholson C. R. Mullen C. Frazee L. A. Cucci M. D. (2021). Hemodynamic adverse effects of dexmedetomidine and propofol in a critically ill trauma and surgical population: a retrospective cohort. J. Trauma Nurs.28, 149–158. 10.1097/JTN.0000000000000576
23
Ning L. Rui X. Guorui L. Tinglv F. Donghang L. Chenzhen X. et al (2022). A novel mechanism for the protection against acute lung injury by melatonin: mitochondrial quality control of lung epithelial cells is preserved through SIRT3-dependent deacetylation of SOD2. Cell Mol. Life Sci.79, 610. 10.1007/s00018-022-04628-0
24
Plosa E. J. Young L. R. Gulleman P. M. Polosukhin V. V. Zaynagetdinov R. Benjamin J. T. et al (2014). Epithelial β1 integrin is required for lung branching morphogenesis and alveolarization. Development141, 4751–4762. 10.1242/dev.117200
25
Rubenfeld G. D. Caldwell E. Peabody E. Weaver J. Martin D. P. Neff M. et al (2005). Incidence and outcomes of acute lung injury. N. Engl. J. Med.353, 1685–1693. 10.1056/NEJMoa050333
26
Shehabi Y. Serpa Neto A. Bellomo R. Howe B. D. Arabi Y. M. Bailey M. et al (2023). Dexmedetomidine and propofol sedation in critically ill patients and dose-associated 90-Day mortality: a secondary cohort analysis of a randomized controlled trial (SPICE III). Am. J. Respir. Crit. Care Med.207, 876–886. 10.1164/rccm.202206-1208OC
27
Song K. Shi J. Zhan L. Gao Q. Yang J. Dong S. et al (2022). Dexmedetomidine modulates mitochondrial dynamics to protect against endotoxin-induced lung injury via the protein kinase C-ɑ/haem oxygenase-1 signalling pathway. Biomarkers27, 159–168. 10.1080/1354750X.2021.2023219
28
Su F. Hammer G. B. (2011). Dexmedetomidine: pediatric pharmacology, clinical uses and safety. Expert Opin. Drug Saf.10, 55–66. 10.1517/14740338.2010.512609
29
Sun M. Li Y. Xu G. Zhu J. Lu R. An S. et al (2024). Sirt3-Mediated Opa1 deacetylation protects against sepsis-induced acute lung injury by inhibiting alveolar macrophage pro-inflammatory polarization. Antioxid. Redox Signal41, 1014–1030. 10.1089/ars.2023.0322
30
Sun Y. Xia Y. Liu X. Liu J. He W. Ye H. et al (2020). Dexmedetomidine alleviates LPS-Induced acute lung injury via regulation of the p38/HO-1 pathway. Mol. Med. Rep.22, 2442–2450. 10.3892/mmr.2020.11330
31
Wang D. S. Kaneshwaran K. Lei G. Mostafa F. Wang J. Lecker I. et al (2018). Dexmedetomidine prevents excessive gamma-Aminobutyric acid type A receptor function after anesthesia. Anesthesiology129, 477–489. 10.1097/ALN.0000000000002311
32
Wang J. Yi X. Jiang L. Dong H. Feng W. Wang S. et al (2019). Protective effects of dexmedetomidine on lung in rats with one-lung ventilation. Exp. Ther. Med.17, 187–192. 10.3892/etm.2018.6952
33
Ward P. A. (2010). Oxidative stress: acute and progressive lung injury. Ann. N. Y. Acad. Sci.1203, 53–59. 10.1111/j.1749-6632.2010.05552.x
34
Wu B. Xu M. M. Fan C. Feng C. L. Lu Q. K. Lu H. M. et al (2022). STING inhibitor ameliorates LPS-Induced ALI by preventing vascular endothelial cells-mediated immune cells chemotaxis and adhesion. Acta Pharmacol. Sin.43, 2055–2066. 10.1038/s41401-021-00813-2
35
Wu Z. Wang Y. Lu S. Yin L. Dai L. (2023). SIRT3 alleviates sepsis-induced acute lung injury by inhibiting pyroptosis via regulating the deacetylation of FoxO3a. Pulm. Pharmacol. Ther.82, 102244. 10.1016/j.pupt.2023.102244
36
Xi S. Chen W. Ke Y. (2024). Advances in SIRT3 involvement in regulating autophagy-related mechanisms. Cell Div.19, 20. 10.1186/s13008-024-00124-y
37
Xu Y. Xin J. Sun Y. Wang X. Sun L. Zhao F. et al (2024). Mechanisms of sepsis-induced acute lung injury and advancements of natural small molecules in its treatment. Pharm. (Basel)17, 472. 10.3390/ph17040472
38
Yan X. Cheng X. Zhou L. He X. Zheng W. Chen H. (2017). Dexmedetomidine alleviates lipopolysaccharide-induced lung injury in wistar rats. Oncotarget8, 44410–44417. 10.18632/oncotarget.17899
39
Yin X. Zhu G. Wang Q. Fu Y. D. Wang J. Xu B. (2021). Ferroptosis, a new insight into acute lung injury. Front. Pharmacol.12, 709538. 10.3389/fphar.2021.709538
40
Zhang H. Sha J. Feng X. Hu X. Chen Y. Li B. et al (2019a). Dexmedetomidine ameliorates LPS induced acute lung injury via GSK-3β/STAT3-NF-κB signaling pathway in rats. Int. Immunopharmacol.74, 105717. 10.1016/j.intimp.2019.105717
41
Zhang J. R. Lin Q. Liang F. Q. Xie T. (2019b). Dexmedetomidine attenuates lung injury by promoting mitochondrial fission and oxygen consumption. Med. Sci. Monit.25, 1848–1856. 10.12659/MSM.913239
42
Zhang M. Deng Y. N. Zhang J. Y. Liu J. Li Y. B. Su H. et al (2018a). SIRT3 protects rotenone-induced injury in SH-SY5Y cells by promoting autophagy through the LKB1-AMPK-mTOR pathway. Aging Dis.9, 273–286. 10.14336/AD.2017.0517
43
Zhang Q. Liu X. M. Hu Q. Liu Z. R. Liu Z. Y. Zhang H. G. et al (2021). Dexmedetomidine inhibits mitochondria damage and apoptosis of enteric glial cells in experimental intestinal ischemia/reperfusion injury via SIRT3-dependent PINK1/HDAC3/p53 pathway. J. Transl. Med.19, 463. 10.1186/s12967-021-03027-6
44
Zhang Q. Wu D. Yang Y. Liu T. Liu H. (2017). Dexmedetomidine alleviates hyperoxia-induced acute lung injury via inhibiting NLRP3 inflammasome activation. Cell Physiol. Biochem.42, 1907–1919. 10.1159/000479609
45
Zhang Y. Jia S. Gao T. Zhang R. Liu Z. Wang Y. (2018c). Dexmedetomidine mitigate acute lung injury by inhibiting IL-17-induced inflammatory reaction. Immunobiology223, 32–37. 10.1016/j.imbio.2017.10.017
46
Zhang Y. Wang X. Liu Z. Yu L. (2018b). Dexmedetomidine attenuates lipopolysaccharide induced acute lung injury by targeting NLRP3 via miR-381. J. Biochem. Mol. Toxicol.32, e22211. 10.1002/jbt.22211
47
Zhang Z. T. Xie K. Luo R. J. Zhang D. Y. He Z. W. Li K. F. et al (2023). Dexmedetomidine alleviates acute lung injury by promoting tregs differentiation via activation of AMPK/SIRT1 pathway. Inflammopharmacology31, 423–438. 10.1007/s10787-022-01117-5
48
Zheng Y. Wang S. Wu J. Wang Y. (2023). Mitochondrial metabolic dysfunction and non-alcoholic fatty liver disease: new insights from pathogenic mechanisms to clinically targeted therapy. J. Transl. Med.21, 510. 10.1186/s12967-023-04367-1
49
Zhong Y. Xia S. Wang G. Liu Q. Ma F. Yu Y. et al (2024). The interplay between mitophagy and mitochondrial ROS in acute lung injury. Mitochondrion78, 101920. 10.1016/j.mito.2024.101920
50
Zhuang C. Kang M. Lee M. (2023). Delivery systems of therapeutic nucleic acids for the treatment of acute lung injury/acute respiratory distress syndrome. J. Control Release360, 1–14. 10.1016/j.jconrel.2023.06.018
Summary
Keywords
acute lung injury, sirt3, dexmedetomidine, inflammation, oxidative stress
Citation
Chen J, Cai Y, Peng X, Xu Y, Chen L, Pan X and Sun Y (2025) Dexmedetomidine reduces acute lung injury caused by LPS through the SIRT3 signaling pathway in vivo. Front. Pharmacol. 16:1524219. doi: 10.3389/fphar.2025.1524219
Received
07 November 2024
Accepted
12 June 2025
Published
25 June 2025
Volume
16 - 2025
Edited by
Lulong Bo, Navy Medical University, China
Reviewed by
Surya Prakash Pandey, University of Pittsburgh, United States
Linh Ho, California Northstate University, United States
Updates
Copyright
© 2025 Chen, Cai, Peng, Xu, Chen, Pan and Sun.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yingying Sun, liyh1081@163.com
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.