 Márta Gődény
Márta Gődény Nóra Kovács1,3
Nóra Kovács1,3 Aamir Aman
Aamir Aman Thanyada Rungrotmongkol
Thanyada Rungrotmongkol Christian Schröder
Christian Schröder- 1Department of Computational Biological Chemistry, University of Vienna, Vienna, Austria
- 2Vienna Doctoral School in Chemistry (DoSChem), University of Vienna, Vienna, Austria
- 3Institut für Chemie, Theoretische Chemie, Technische Universität, Berlin, Germany
- 4Center of Excellence in Structural and Computational Biology, Department of Biochemistry, Faculty of Science, Chulalongkorn University, Bangkok, Thailand
- 5Program in Bioinformatics and Computational Biology, Graduate School, Chulalongkorn University, Bangkok, Thailand
The M2 proton channel within the Influenza A virus constitutes an essential element for viral replication, with its functionality depending on the protonation states of four histidine residues located within the channel. This study meticulously investigates the impact of polarizability on the channel’s gating dynamics across various protonation states, employing both polarizable and non-polarizable models for water, protein, and membrane. Through a comprehensive analysis, we elucidate the nuanced role of polarizability in the channel’s operational mechanisms, differentiating between the solvent and protein polarizable effects. This investigation not only enriches our understanding of the M2 channel’s biophysical behavior but also highlights the significance of polarizability.
1 Introduction
The Influenza A virus is a major pathogen responsible for seasonal influenza epidemics. It critically depends on the M2 proton channel for replication. Thus, the channel’s transmembrane segment is a pivotal target for developing antiviral drugs, spurring extensive investigative efforts to decode its proton transfer mechanisms. A seminal study by Hu et al. (2010) employed solid-state NMR techniques to delineate a proton conduction mechanism, unveiling the structural and functional impacts of pH variations on the channel. At elevated pH levels, the formation of CH-
A comprehensive series of molecular dynamics (MD) and quantum mechanics/molecular mechanics (QM/MM) simulations conducted by Jorgensen et al. (1983), Zhong et al. (1998), Khurana et al. (2009), and Carnevale et al. (2010) have shed light on the intricate dynamics of proton transport and the structural integrity of the M2 proton channel across a spectrum of pH levels. In particular, Carnevale et al. (2010) used classical and hybrid QM/MM simulations to study different pH conditions, focusing on the role of water molecules and the four His37 residues in the proton transport mechanism. These simulations revealed distinctive configurations of water molecules within the channel, forming layered structures through strong hydrogen bonding, serving as temporary proton storage sites. Such configurations suggest a conducive free-energy landscape for translocating a classical hydronium-like entity, pinpointing specific regions within the channel that favor cation diffusion. Further investigations have examined the behavior of the channel under various protonation states, with a particular focus on the interactions between water molecules and histidine residues. These interactions profoundly impact the channel’s conformation and proton conduction capabilities. For instance, it was demonstrated (Wei and Pohorille, 2013) that the channel remains inert in the 0, +1, and +2 protonation states of the His37 residues and becomes active only upon further protonation, leading to the +3 and +4 states, attributable to electrostatic repulsion among the protonated histidines which modulates the proximity of adjacent protein
Conventional MD simulations may cover timescales for dynamical processes, however, at the cost of disregarding transient protonation of the histidine residues and the proton hopping in the channel water molecules. Constant pH MD simulations (Chen et al., 2016) have provided critical insights into the
We have developed a Python-based program, Protex (Joerg et al., 2023; Gődény et al., 2024), which efficiently handles hundreds of simultaneous proton transfers between water molecules and between water and histidine residues during polarizable MD simulations without incurring significant computational overhead (Joerg et al., 2023). This capability enables detailed investigation of the Grotthuss mechanism in the channel, as consecutive proton-hopping events can be monitored over trajectories spanning several hundreds of nanoseconds. Currently, Protex can be applied in combination with OpenMM (Eastman et al., 2017) using CHARMM (Brooks et al., 2009) force field files. Polarizable forces are a prerequisite for our pseudo-reactive simulations, as they smooth the transient Coulomb energy by reacting to changes in the local electric field when molecules get protonated or deprotonated. Water molecules approaching a histidine residue affect the electronic distribution of the amino acid, which may stabilize its solvation. Also, the molecular water dipole increases in this environment (Dang, 1998; Devereux and Popelier, 2007). Consequently, we expect stronger hydrogen bonds using polarizable force fields. However, this does not only apply to hydrogen bonds between histidine and water but also between two histidines. The current study analyzes the effect of polarizability on the stability of the M2 ion channel at various protonation states. We also dissect the effect of the polarizable solvent water and the polarizable protein. This knowledge is fundamental for a subsequent study applying Protex (Joerg et al., 2023; Gődény et al., 2024) since we can then discuss the effect of proton hopping in addition to the effects of polarizability. The free energy profiles obtained from multiscale simulations (Liang et al., 2014; 2016; Watkins et al., 2019) serve as a valuable tool for determining reaction probabilities, which are essential for parameterizing the reaction kinetics in Protex.
One of the commercial drugs used to treat influenza that functions by blocking the M2 channel is amantadine (Oxford and Galbraith, 1980). Our previous MD simulations (Intharathep et al., 2008) indicated two preferred binding positions of amantadine. The first one was deep into the channel, close to the His37 tetrad. This position can lead to a more effective inhibition, as it can directly hinder the protonation of histidines, thus keeping the channel closed. The other binding site was closer to the opening of the channel. In this case, amantadine can block the transport through the channel by physically not letting water molecules pass.
2 Methods
2.1 Setup for the M2 channel
We investigated the M2 channel embedded in a 1-palmitoyl-2-oleoylphosphatidylcholin (POPC) membrane using different protonation states of the His37 tetrad, while also varying which parts of the system are described with a polarizable or non-polarizable force field. In addition to an “empty” channel (i.e., a channel filled only with water), we also investigated the effect of an inhibitor (amantadine). The resulting ion channel systems are summarized in Table 1. In the case of the protonation state +2, two independent simulations were performed: one with adjacent protonated histidines (HSP) (2Ha) and one simulation where the two HSPs are opposite of each other (diagonal, 2Hd). Furthermore, three independent replicas for each combination of membrane/protein, water, and protonation state were simulated, and their results were averaged to increase the statistics. This resulted in 72 independent simulations each for the systems with and without the inhibitor as summarized in Table 1.
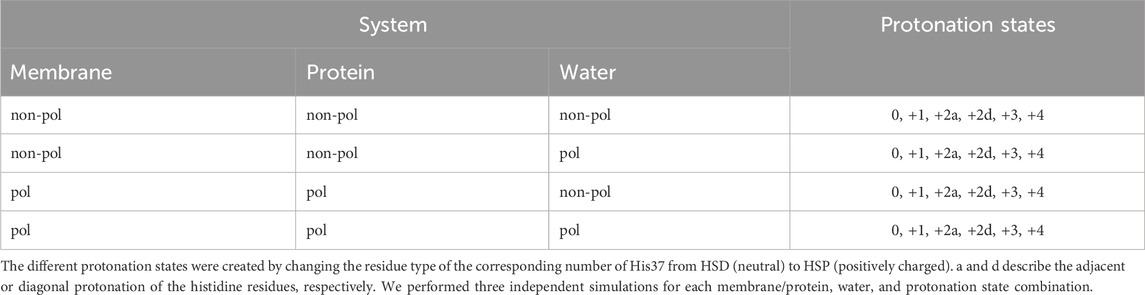
Table 1. Overview of the simulated systems.
We set up these systems using the membrane builder function (Jo et al., 2007; Wu et al., 2014) of CHARMM-GUI (Jo et al., 2008; Lee et al., 2016), starting directly from the PDBs described below. Only the four transmembrane chains and, if applicable, amantadine were selected from the PDB to build the system. Other molecules (e.g., water, ions, additional copies of the helices) were disregarded. Hydrogen coordinates and patching of the termini (NTER: positively charged N terminus and CTER: negatively charged C terminus) were added by CHARMM-GUI. No external preprocessing was conducted on the PDB structures.
The membrane comprised ca. 60 POPC molecules per bilayer (Kass and Arkin, 2005). To accurately model the biological milieu, the membrane was embedded in a rectangular simulation box flanked by 22.5 Å water layers both above and beneath the membrane (roughly 3,500 water molecules altogether), achieving a balanced 1:1 ratio between the upper and lower membrane leaflets (see Figure 1b).
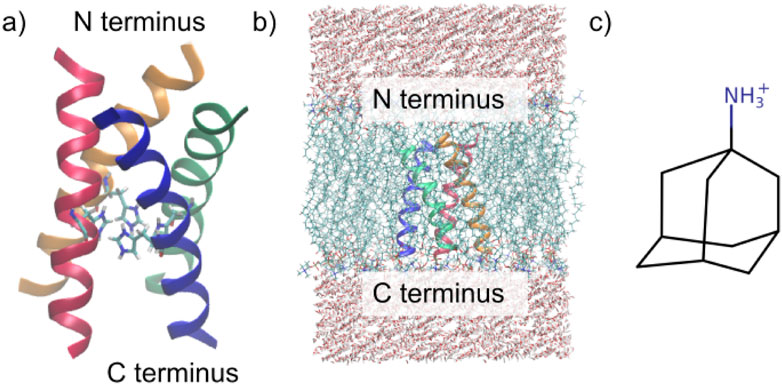
Figure 1. Investigated systems. (a) M2 channel in the 3LBW structure, including the four His37 residues. (b) Simulation box of the protein embedded in the POPC membrane surrounded by TIP3P water. (c) Structure of amantadine (Intharathep et al., 2008).
As shown in Figure 2a, the 3LBW and the 6BKK structures align very closely with the full structure suggested by AlphaFold 3 (Abramson et al., 2024). The superimposition in Figure 2b that 3LBW and 6BKK exhibit the closest alignment, whereas 1NYJ and 2L0J deviate from them (and thus from AlphaFold). These observations strongly support the use of 3LBW and 6BKK for further analysis, as their high degree of similarity facilitates direct comparison of simulations with and without amantadine.
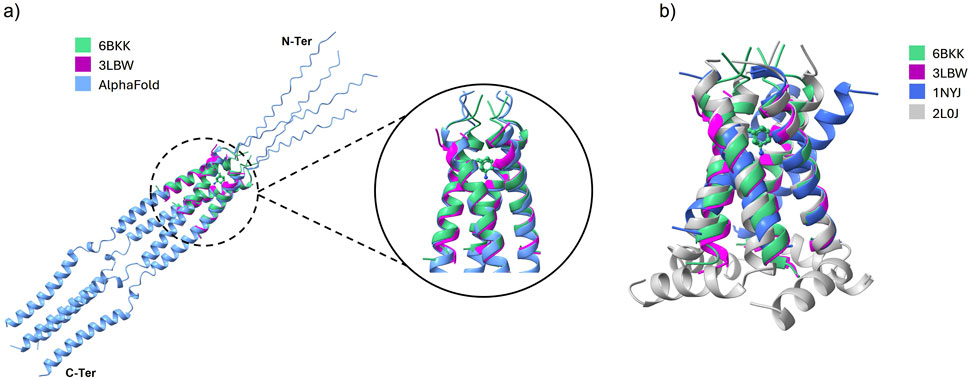
Figure 2. (a) The alignment of 3LBW (purple) and 6BKK (green) with the structure of the whole protein as suggested by AlphaFold (blue). The inset shows the binding position of amantadine in 6BKK. (b) Superimposed PDB structures of 3LBW (green), 6BKK (purple), 1NYJ (blue) and 2L0J (gray).
2.2 Trajectory production
The gating of the channel and the binding of amantadine both take place entirely in the transmembrane part of the protein (Duff and Ashley, 1992; Wang et al., 1995). Thus, we focused our investigations on this specific region of the protein, as taking a much larger structure would make the already quite resource-intensive simulations even more costly and the overall goal of this study is the comparison of polarizable and non-polarizable force fields for the protein and water.
After setting up the simulation box, CHARMM (Brooks et al., 2009) was applied to create a minimized starting structure and to make the atoms polarizable (if necessary) using Drude particles (Lamoureux et al., 2006; Baker et al., 2010; Lopes et al., 2013; Lemkul et al., 2016; Kumar et al., 2019; Lin et al., 2020). We applied the CHARMM36m force field (Huang et al., 2016) for the membrane and protein. Consequently, the water models TIP3P (Jorgensen et al., 1983; Mark and Nilsson, 2001) and SWM4 (Lamoureux et al., 2003; Sega and Schröder, 2015) were employed for the non-polarizable and polarizable water, respectively.
All MD simulations were performed in OpenMM 7.6 (Eastman et al., 2017) using a Velocity Verlet integrator (Gong and Padua, 2021). In the case of polarizable molecules, the mass of the mobile Drude particle was set to 0.4 au, and the vibrations of the Drude oscillator were kept at a temperature of 1 K. The maximum allowed distance between a Drude particle and its parent atom was 0.2 Å, the Drude force constant was 1,000 kcal mol−1 Å−2. The Particle Mesh Ewald method (Darden et al., 1993; Essmann et al., 1995), with an error tolerance of 0.0005, was selected for the electrostatic cutoff (Darden et al., 1993; Essmann et al., 1995), while van der Waals interactions were managed using the Force-switch method, applying a switch-on distance of 1.0 nm and a switch-off distance of 1.2 nm. Positional restraint force constants were applied to protein backbones, side chains, lipids, and dihedral restraint force constants for lipids, adhering to the default parameters set by CHARMM-GUI.
The simulation protocol was conducted under the NpT ensemble conditions, maintaining a temperature of 303.15 K and a pressure of 1 atm. Following a comprehensive system equilibration of 5 ns using CHARMM-GUI’s default settings, the production phase proceeded over an extensive duration of 100 ns, with a time step of 2 fs for the completely non-polarizable systems and 1 fs for the (partially) polarizable ones. This led to a total simulation period of 14.4 µs of (partially) polarizable systems, which are generally about four times as expensive as non-polarizable ones (Antila et al., 2024).
2.3 Analysis techniques
Our analysis of the trajectories is based on the Python-based library MDAnalysis (Michaud-Agrawal et al., 2011; Gowers et al., 2016), which offers standard routines for calculating the common transient RMSD values and diffusion coefficients. All RMSD values were calculated on the backbone atoms of the protein, with the first frame of the production run (after equilibration) as the reference. Using our extension (Github, 2024), the spatial structure can be analyzed in terms of the radial distribution function
A co-linear arrangement of the hydrogen bond of the protonated
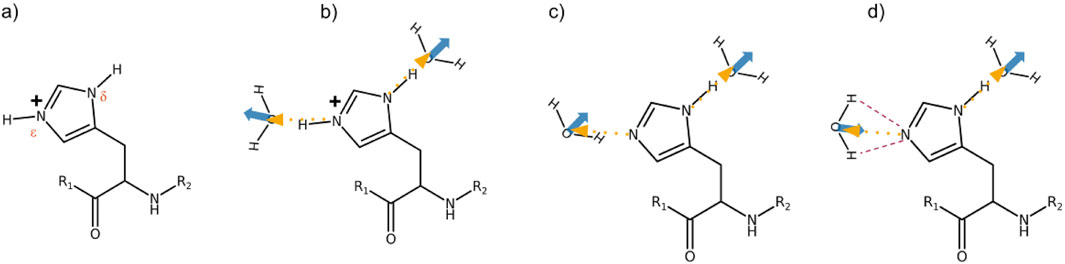
Figure 3. (a) Nomenclature of the histidine nitrogens. (b)
The channel opening and closing may be characterized by an area between the
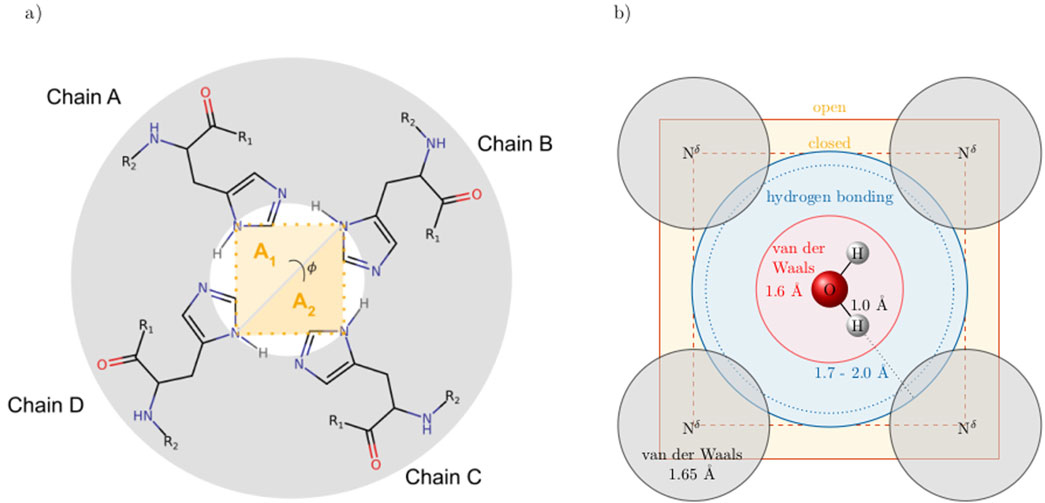
Figure 4. (a) Cross-section of the ion channel at the height of the His37 tetrad. The opening and closing of the channel can be characterized by an area
3 Results
3.1 At the level of the ion channel
All protonation states and polarizabilities resulted in stable simulations with highly conserved protein structures, which are reflected in the low root mean square deviations
where the sum of the amplitudes
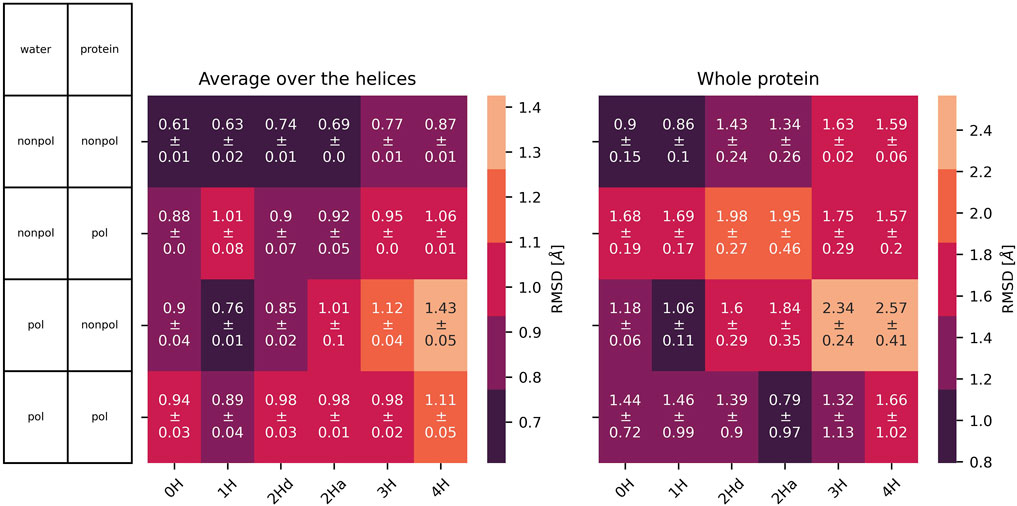
Figure 5.
In addition to the
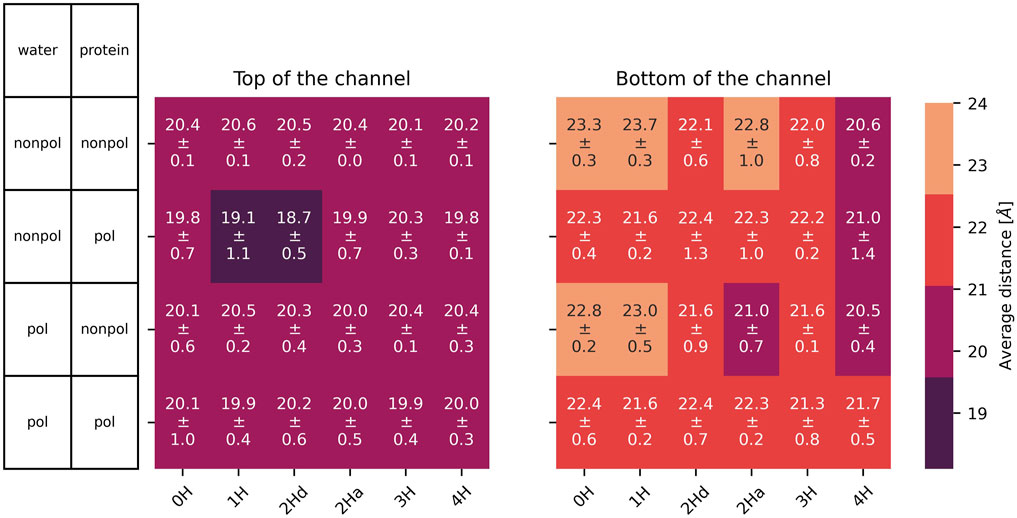
Figure 6. Average distance with standard deviation between opposite
The radius of the channel throughout the membrane was analysed with HOLE (Smart et al., 1993, Smart et al., 1996). The program seems unable to handle cases where the channel is split between primary and image cells, thus some of the results were unsatisfactory. Still, the resulting radii are shown in the supplementary material (Supplementary Figures S5–S7 for the simulations without the inhibitor and Supplementary Figures S18–20 with inhibitor). The results mainly agree with the distances shown in Figures 6, 7. We focus on these single-value distances for an easier comparison between systems.
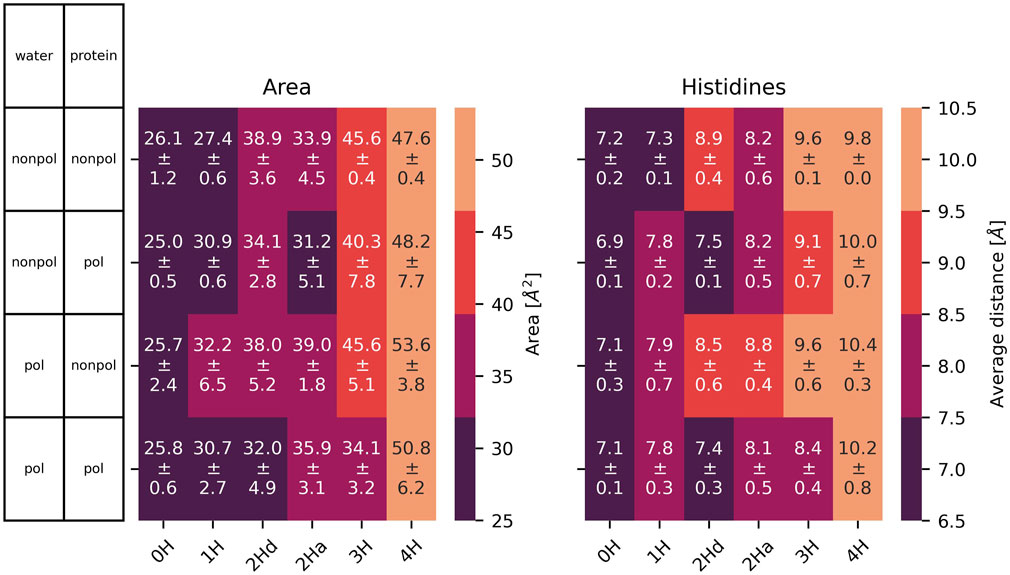
Figure 7. Left: area spanned by the
3.2 At the bottleneck
The cross distances between the
The small volume of the channel is further evidenced by the long-distance limit of
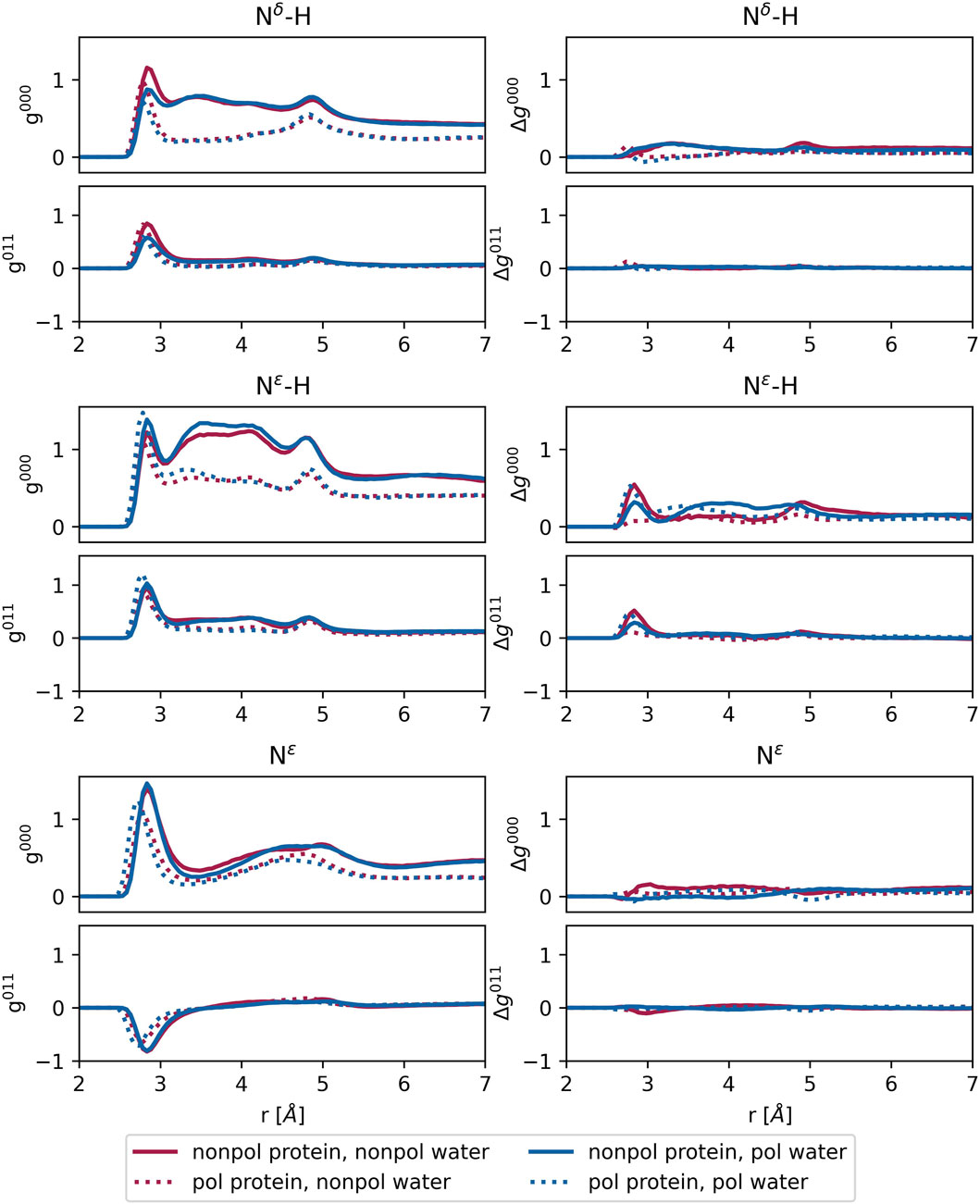
Figure 8. Left: radial distribution functions
For protonated histidine nitrogen, the
4 Discussion
4.1 Effect of the polarizability
Polarizable water models are essential for the future development of proton transfer models for water and histidines. The proton transfer within the channel may not only occur between water molecules in a Grotthus mechanism style but may also involve the channel’s histidine, as their
Overall, the water polarizability appears to have minimal influence on the
The
In summary, the protein’s polarizability induces some structural changes in the channel. However, key characteristics at the bottleneck, such as the area of the His37 tetrad and cross-distances, remain unaffected. Therefore, if our future polarizable simulations reveal discrepancies compared to behaviors observed in non-polarizable simulations from the literature, these differences are likely attributable to the enabled proton transfer rather than the polarizable forces themselves.
4.2 Effect of amantadine
The simulations that include amantadine (see Figure 1) exhibit similar trends to those without the ligand. On average, the channel diameter is slightly larger at the top, middle, and bottom of the channel compared to the ligand-free simulations, with a small deviation (
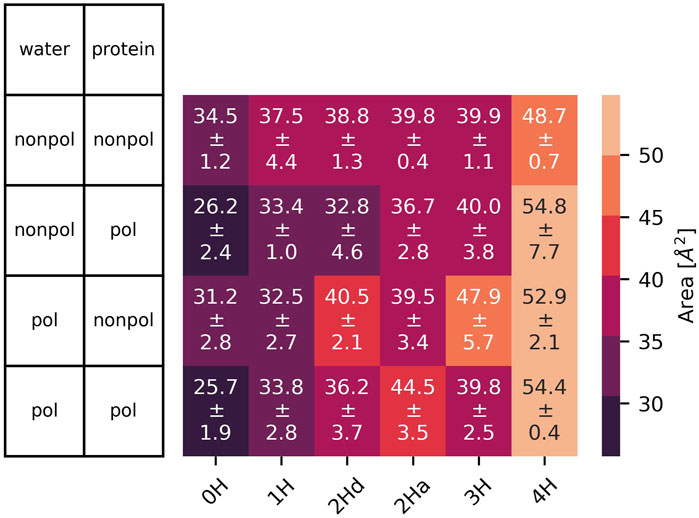
Figure 9. Area spanned by the
The impact of amantadine on the radial distribution functions between the histidine nitrogens at the bottleneck and water is minimal, as evidenced by the difference plots
All this information suggests that the ligand does not fundamentally alter the channel structure or its response to protonation. Instead, amantadine appears to block the channel mechanically, leaving no space for water molecules to pass through. This is also supported by the analysis of the hydrogen bonds shown in Figure 10. We defined a hydrogen bond with a distance of maximum 4 Å between the donor and acceptor heavy atoms and a donor-H-acceptor angle of at least 120°. In agreement with previous results (Intharathep et al., 2008), amantadine interacts in all cases with Ala30 or Ser31 the most, thus forming a blockage close to the opening of the channel. Interaction with the His37 tetrad was very rarely observed. Visual inspection shows that amantadine stays very close to its starting position, and mainly just rotates in place. This is also confirmed by the very small diffusion coefficients (Supplementary Figure S11), as well as by the overwhelmingly large number of contacts to Ala30 or Ser31, as opposed to other residues. This is the case both for hydrophobic contacts (Supplementary Figures S12–S15) and hydrogen bonds (Figure 10).
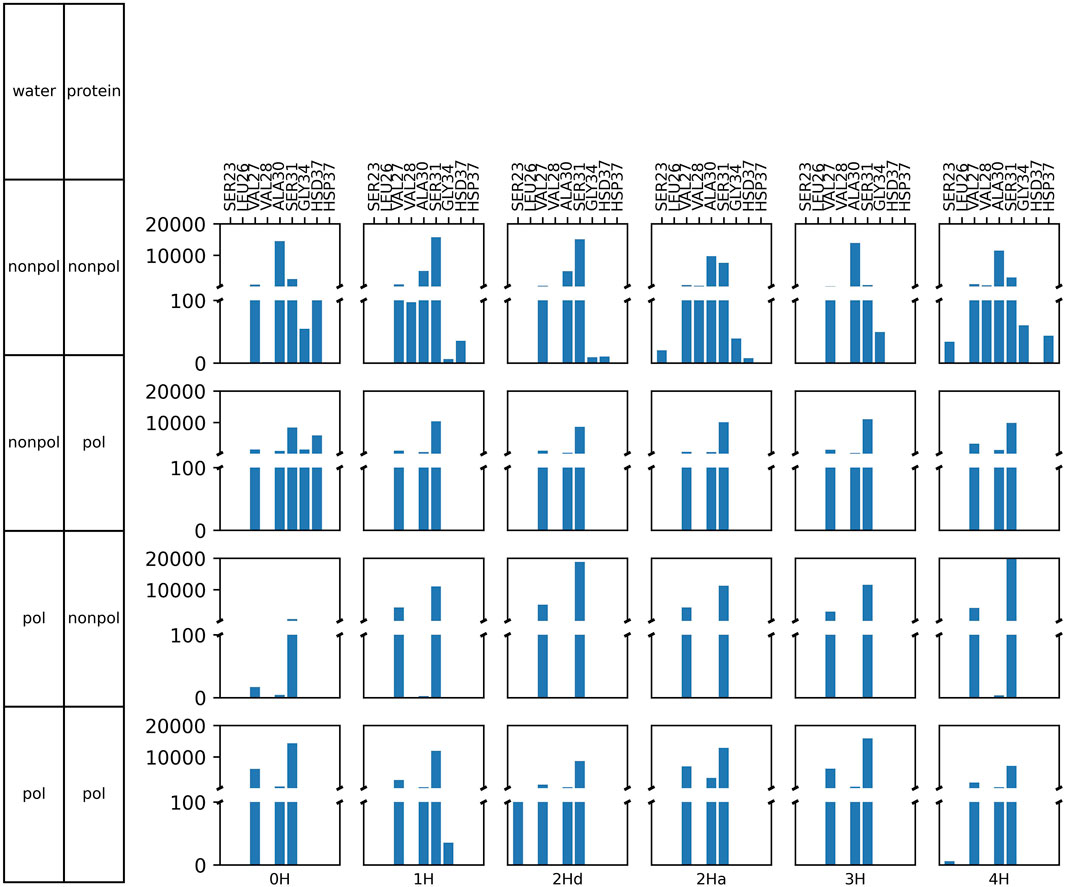
Figure 10. Frequency of hydrogen bonds between the amino hydrogens of amantadine and various amino acids of M2.
5 Conclusion
Our simulations reveal that while polarizability induces subtle changes in the channel’s structure, key features at the bottleneck, such as the cross-distances and the area spanned by the histidine residues, remain largely unaffected. This suggests that any deviations observed in the future between polarizable and non-polarizable models are likely due to proton transfer processes rather than the polarizable forces themselves. Moreover, including amantadine as a ligand did not drastically alter the channel’s overall behavior, supporting the hypothesis that amantadine mechanically blocks the channel without inducing significant structural changes. These findings provide a foundation for further investigation into the role of proton transfer in the channel’s operation. The results also offer valuable insights for future computational studies, highlighting the potential for reducing computational costs by selectively applying polarizable models only to regions of interest, such as those involved in proton transfer.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
MG: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review and editing. NK: Data curation, Formal Analysis, Investigation, Methodology, Writing – original draft. AA: Data curation, Formal Analysis, Investigation, Writing – review and editing. TR: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review and editing. CS: Conceptualization, Formal Analysis, Funding acquisition, Project administration, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. We acknowledge the support of the OeAD project ASEA UNINET/2023-2024/UniWien/4.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. Generative artificial intelligence tools were used solely to improve the clarity, grammar, and style of the language in this manuscript. No AI tools were used for data analysis, interpretation of results, or any other scientific purpose.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2025.1532697/full#supplementary-material
References
Abramson, J., Adler, J., Dunger, J., Evans, R., Green, T., Pritzel, A., et al. (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500. doi:10.1038/s41586-024-07487-w
Acharya, R., Polishchuk, A., and DeGrado, W. (2010). High resolution crystal structure of transmembrane domain of M2. doi:10.2210/pdb3lbw/pdb
Antila, H. S., Dixit, S., Kav, B., Madsen, J. J., Miettinen, M. S., and Ollila, O. H. S. (2024). Evaluating polarizable biomembrane simulations against experiments. J. Chem. Theory Comput. 20, 4325–4337. doi:10.1021/acs.jctc.3c01333
Baker, C. M., Anisimov, V. M., and MacKerell, A. D. (2010). Development of CHARMM polarizable force field for nucleic acid bases based on the classical drude oscillator model. J. Phys. Chem. B 115, 580–596. doi:10.1021/jp1092338
Blum, L., and Nartan, A. H. (1976). Diffraction by molecular liquids. Adv. Chem. Phys. 34, 203–243. doi:10.1002/9780470142530.ch4
Brooks, B. R., Brooks, C. L., Mackerell, A. D., Nilsson, L., Petrella, R. J., Roux, B., et al. (2009). CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614. doi:10.1002/jcc.21287
Carnevale, V., Fiorin, G., Levine, B. G., DeGrado, W. F., and Klein, M. L. (2010). Multiple proton confinement in the M2 channel from the influenza A virus. JPCC 114, 20856–20863. doi:10.1021/jp107431g
Chen, W., Wallace, J. A., Yue, Z., and Shen, J. K. (2013). Introducing titratable water to all-atom molecular dynamics at constant ph. Biophys. J. 105, L15–L17. doi:10.1016/j.bpj.2013.06.036
Chen, W., Huang, Y., and Shen, J. (2016). Conformational activation of a transmembrane proton channel from constant pH molecular dynamics. JPCL 7, 3961–3966. doi:10.1021/acs.jpclett.6b01853
Dang, L. X. (1998). Importance of polarization effects in modeling the hydrogen bond in water usingclassical molecular dynamics techniques. J. Phys. Chem. B 102, 620–624. doi:10.1021/jp9731258
Darden, T., York, D., and Pedersen, L. (1993). Particle mesh ewald: an N ⋅ log(N) method for ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. doi:10.1063/1.464397
Devereux, M., and Popelier, P. L. A. (2007). The effects of hydrogen-bonding environment on the polarization and electronicproperties of water molecules. J. Phys. Chem. A 111, 1536–1544. doi:10.1021/jp067922u
Dong, H., Fiorin, G., DeGrado, W. F., and Klein, M. L. (2013). Exploring histidine conformations in the M2 channel lumen of the influenza A virus at neutral pH via molecular simulations. JPCL 4, 3067–3071. doi:10.1021/jz401672h
Dong, H., Fiorin, G., DeGrado, W. F., and Klein, M. L. (2014). Proton release from the histidine-tetrad in the M2 channel of the influenza A virus. JPCB 118, 12644–12651. doi:10.1021/jp5102225
Duff, K. C., and Ashley, R. H. (1992). enThe transmembrane domain of influenza a M2 protein forms amantadine-sensitive proton channels in planar lipid bilayers. Virology 190, 485–489. doi:10.1016/0042-6822(92)91239-q
Eastman, P., Swails, J., Chodera, J. D., McGibbon, R. T., Zhao, Y., Beauchamp, K. A., et al. (2017). OpenMM 7: rapid development of high performance algorithms for molecular dynamics. PLOS Comput. Biol. 13, e1005659. doi:10.1371/journal.pcbi.1005659
Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H., and Pedersen, L. G. (1995). A smooth particle mesh ewald method. J. Chem. Phys. 103, 8577–8593. doi:10.1063/1.470117
Fu, R., Miao, Y., Qin, H., and Cross, T. A. (2020). Observation of the imidazole-imidazolium hydrogen bonds responsible for selective proton conductance in the influenza A M2 channel. JACS. doi:10.1021/jacs.9b09985
Github (2024). MD-newanalysis. Available online at: https://github.com/cbc-univie/mdy-newanalysis-package.
Gődény, M., Joerg, F., Kovar, M. P.-P., and Schröder, C. (2024). Updates to protex for simulating proton transfers in an ionic liquid. J. Phys. Chem. B 128, 3416–3426. doi:10.1021/acs.jpcb.3c07356
Gong, Z., and Padua, A. A. H. (2021). Effect of side chain modifications in imidazolium ionic liquids on the properties of the electrical double layer at a molybdenum disulfide electrode. J. Chem. Phys. 154, 084504. doi:10.1063/5.0040172
Gowers, R., Linke, M., Barnoud, J., Reddy, T., Melo, M., Seyler, S., et al. (2016). “MDAnalysis: a python package for the rapid analysis of molecular dynamics simulations,” in Proceedings of the 15th python in science conference (SciPy) (SciPy). doi:10.25080/majora-629e541a-00e
Harris, T. K., and Mildvan, A. S. (1999). High-precision measurement of hydrogen bond lengthsin proteins by nuclear magnetic resonance methods. Proteins Struct. Funct. Genet. 35, 275–282. doi:10.1002/(SICI)1097-0134(19990515)35:3⟨275::AID-PROT1⟩3.0.CO;2-V
Hong, M., Fritzsching, K. J., and Williams, J. K. (2012). Hydrogen-bonding partner of the proton-conducting histidine in the influenza M2 proton channel revealed From1H chemical shifts. JACS 134, 14753–14755. doi:10.1021/ja307453v
Hu, F., Luo, W., and Hong, M. (2010). Mechanisms of proton conduction and gating in influenza M2 proton channels from solid-state NMR. Science 330, 505–508. doi:10.1126/science.1191714
Hu, F., Schmidt-Rohr, K., and Hong, M. (2011). NMR detection of pH-Dependent histidine-water proton exchange reveals the conduction mechanism of a transmembrane proton channel. JACS 134, 3703–3713. doi:10.1021/ja2081185
Huang, J., Rauscher, S., Nawrocki, G., Ran, T., Feig, M., de Groot, B. L., et al. (2016). CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73. doi:10.1038/nmeth.4067
Intharathep, P., Laohpongspaisan, C., Rungrotmongkol, T., Loisruangsin, A., Malaisree, M., Decha, P., et al. (2008). How amantadine and rimantadine inhibit proton transport in the M2 protein channel. J. Mol. Graph. Model. 27, 342–348. doi:10.1016/j.jmgm.2008.06.002
Jansen, A., Bauer, P., Howard, R. J., Hess, B., and Lindahl, E. (2024). Constant-ph molecular dynamics simulations of closed and open states of a proton-gated ion channel. bioRχiv. doi:10.1101/2023.11.30.569372
Jeong, B., and Dyer, R. B. (2017). Proton transport mechanism of M2 proton channel studied by laser-induced pH jump. JACS 139, 6621–6628. doi:10.1021/jacs.7b00617
Jo, S., Kim, T., and Im, W. (2007). Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE 2, e880. doi:10.1371/journal.pone.0000880
Jo, S., Kim, T., Iyer, V. G., and Im, W. (2008). CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865. doi:10.1002/jcc.20945
Joerg, F., Wieder, M., and Schröder, C. (2023). Protex—A python utility for proton exchange in molecular dynamics simulations. Front. Chem. 11, 1140896. doi:10.3389/fchem.2023.1140896
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., and Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. doi:10.1063/1.445869
Kaiser, S., Yue, Z., Peng, Y., Nguyen, T. D., Chen, S., Teng, D., et al. (2024). Molecular dynamics simulation of complex reactivity with the rapid approach for proton transport and other reactions (raptor) software package. J. Phys. Chem. B 128, 4959–4974. doi:10.1021/acs.jpcb.4c01987
Kass, I., and Arkin, I. T. (2005). How pH open a H+ channel: the gating mechanism of influenza A M2. Structure 13, 1789–1798. doi:10.1016/j.str.2005.08.022
Khurana, E., Peraro, M. D., DeVane, R., Vemparala, S., DeGrado, W. F., and Klein, M. L. (2009). Molecular dynamics calculations suggest a conduction mechanism for the M2 proton channel from influenza A virus. PNAS 106, 1069–1074. doi:10.1073/pnas.0811720106
Kumar, A., Yoluk, O., and MacKerell, A. D. (2019). FFParam: standalone package for CHARMM additive and drude polarizable force field parametrization of small molecules. J. Comput. Chem. 41, 958–970. doi:10.1002/jcc.26138
Lamoureux, G., MacKerell, Jr. A. D., and Roux, B. (2003). A simplepolarizable model of water based on classical drude oscillators. J. Chem. Phys. 119, 5185–5197. doi:10.1063/1.1598191
Lamoureux, G., Harder, E., Vorobyov, I. V., Roux, B., and MacKerell, A. D. (2006). A polarizable model of water for molecular dynamics simulations of biomolecules. Chem. Phys. Lett. 418, 245–249. doi:10.1016/j.cplett.2005.10.135
Lee, J., Cheng, X., Swails, J. M., Yeom, M., Eastman, P. K., Lemkul, J. A., et al. (2016). CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 12, 405–413. doi:10.1021/acs.jctc.5b00935
Lemkul, J. A., Huang, J., Roux, B., and MacKerell, A. D. (2016). An empirical polarizable force field based on the classical drude oscillator model: development history and recent applications. Chem. Rev. 116, 4983–5013. doi:10.1021/acs.chemrev.5b00505
Liang, R., Li, H., Swanson, J. M. J., and Voth, G. A. (2014). Multiscale simulation reveals a multifaceted mechanism of proton permeation through the influenza A M2 proton channel. PNAS 111, 9396–9401. doi:10.1073/pnas.1401997111
Liang, R., Swanson, J. M. J., Madsen, J. J., Hong, M., DeGrado, W. F., and Voth, G. A. (2016). Acid activation mechanism of the influenza a m2 proton channel. PNAS 113, E6955–E6964. doi:10.1073/pnas.1615471113
Lin, F., Huang, J., Pandey, P., Rupakheti, C., Li, J., Roux, B., et al. (2020). Further optimization and validation of the classical drude polarizable protein force field. J. Chem. Theory Comput. 16, 3221–3239. doi:10.1021/acs.jctc.0c00057
Lopes, P. E. M., Huang, J., Shim, J., Luo, Y., Li, H., Roux, B., et al. (2013). Polarizable force field for peptides and proteins based on the classical drude oscillator. J. Chem. Theory Comput. 9, 5430–5449. doi:10.1021/ct400781b
Mark, P., and Nilsson, L. (2001). Structure and dynamics of the tip3p, spc, and spc/e water models at 298 k. J. Phys. Chem. A 105, 9954–9960. doi:10.1021/jp003020w
Michaud-Agrawal, N., Denning, E. J., Woolf, T. B., and Beckstein, O. (2011). MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 32, 2319–2327. doi:10.1002/jcc.21787
Nishimura, K., Kim, S., Zhang, L., and Cross, T. (2003). The closed state structure of M2 protein H+ channel by solid state NMR spectroscopy. doi:10.2210/pdb1nyj/pdb
Oxford, J. S., and Galbraith, A. (1980). Antiviral activity of amantadine: a review of laboratory and clinical data. Pharmacol. and Ther. 11, 181–262. doi:10.1016/0163-7258(80)90072-8
Sansom, M. S. P., Kerr, I. D., Smith, G. R., and Son, H. S. (1997). The influenza a virus m2 channel: a molecular modeling and simulation study. Virology 233, 163–173. doi:10.1006/viro.1997.8578
Schröder, C., Rudas, T., Neumayr, G., Gansterer, W., and Steinhauser, O. (2007). Impact of anisotropy on the structure and dynamics of ionic liquids: a computational study of 1-Butyl-3-Methyl-Imidazolium trifluoroacetate. J. Chem. Phys. 127. doi:10.1063/1.2754690
Sega, M., and Schröder, C. (2015). Dielectric and terahertz spectroscopy of polarizable andnonpolarizable water models: a comparative study. J. Phys. Chem. A 119, 1539–1547. doi:10.1021/jp507419e
Sharma, M., Yi, M., Dong, H., Qin, H., Peterson, E., Busath, D., et al. (2010). Solid state NMR structure of the M2 proton channel from influenza A virus in hydrated lipid bilayer. doi:10.2210/pdb2l0j/pdb
Smart, O. S., Goodfellow, J. M., and Wallace, B. A. (1993). The pore dimensions of gramicidin a. Biophys. J. 65, 2455–2460. doi:10.1016/S0006-3495(93)81293-1
Smart, O. S., Neduvelil, J. G., Wang, X., Wallace, B. A., and Sansom, M. S. (1996). HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 14, 354–376.
Steinhauser, O., and Bertagnolli, H. (1981). Invariant expansion coefficients of the molecular pair correlation function ofzx4-systems. Ber. Bunsenges. Phys. Chem. 85, 45–52.
Thomaston, J. L., Polizzi, N. F., Konstantinidi, A., Wang, J., Kolocouris, A., and DeGrado, W. F. (2018). Inhibitors of the m2 proton channel engage and disrupt transmembrane networks of hydrogen-bonded waters. J. Am. Chem. Soc. 140, 15219–15226. doi:10.1021/jacs.8b06741
Verkman, A. S., and Mitra, A. K. (2000). Structure and function of aquaporin water channels. Am. J. Physiol. Ren. Physiol. 278, F13–F28. doi:10.1152/ajprenal.2000.278.1.F13
Wang, C., Lamb, R. A., and Pinto, L. H. (1995). enActivation of the M2 ion channel of influenza virus: a role for the transmembrane domain histidine residue. Biophys. J. 69, 1363–1371. doi:10.1016/S0006-3495(95)80003-2
Wang, J., Ma, C., Fiorin, G., Carnevale, V., Wang, T., Hu, F., et al. (2011). Molecular dynamics simulation directed rational design of inhibitorstargeting drug-resistant mutants of influenza a virus m2. J. Am. Chem. Soc. 133, 12834–12841. doi:10.1021/ja204969m
Watkins, L. C., Liang, R., Swanson, J. M. J., DeGrado, W. F., and Voth, G. A. (2019). Proton-induced conformational and hydration dynamics in the influenza A M2 channel. JACS 141, 11667–11676. doi:10.1021/jacs.9b05136
Wei, C., and Pohorille, A. (2013). Activation and proton transport mechanism in influenza A M2 channel. Biophys. J. 105, 2036–2045. doi:10.1016/j.bpj.2013.08.030
Wu, E. L., Cheng, X., Jo, S., Rui, H., Song, K. C., Dávila-Contreras, E. M., et al. (2014). CHARMM-GUI membrane builder toward realistic biological membrane simulations. J. Comput. Chem. 35, 1997–2004. doi:10.1002/jcc.23702
Zhang, R., Skach, W., Hasegawa, H., van Hoek, A. N., and Verkman, A. S. (1993). Cloning, functional analysis and cell localization of a kidney proximal tubule water transporter homologous to chip28. J. Cell Biol. 120, 359–369. doi:10.1083/jcb.120.2.359
Keywords: influenza a virus, M2 proton channel, polarizability, channel gating dynamics, molecular dynamics simulations
Citation: Gődény M, Kovács N, Aman A, Rungrotmongkol T and Schröder C (2025) The interplay of polarizable water and protein in the activation of the M2 channel. Front. Pharmacol. 16:1532697. doi: 10.3389/fphar.2025.1532697
Received: 22 November 2024; Accepted: 11 July 2025;
Published: 02 September 2025.
Edited by:
Roope Mannikko, University College London, United KingdomCopyright © 2025 Gődény, Kovács, Aman, Rungrotmongkol and Schröder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Schröder, Y2hyaXN0aWFuLnNjaHJvZWRlckB1bml2aWUuYWMuYXQ=