 Yike Wang1
Yike Wang1 Dai Li2,3Tong Zhang1
Dai Li2,3Tong Zhang1 Sumei Xu2,3Yanxin Zhang2,3Kaijing Zhao1Shaorong Li1
Sumei Xu2,3Yanxin Zhang2,3Kaijing Zhao1Shaorong Li1 Kai Shen1Xiaomin Li2,3*
Kai Shen1Xiaomin Li2,3* Pingsheng Xu2,3*
Pingsheng Xu2,3*- 1Department of Clinical Pharmacology, Jiangsu Hengrui Pharmaceuticals Co., Ltd., Shanghai, China
- 2Phase I Clinical Trial Center, Xiangya Hospital, Central South University, Changsha, China
- 3National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, China
Aims: To investigate the potential pharmacokinetic interactions of montmorillonite powder or loperamide on pyrotinib.
Methods: This study was a single-center, open-label, single-dose, fixed-sequence clinical trial conducted with healthy volunteers. The participants were divided into two groups (A and B), each consisting of 18 subjects. Both groups received a single oral dose of 400 mg of pyrotinib on day 1. On day 9, Group A received a single dose of 400 mg of pyrotinib followed by 3 g of montmorillonite powder 2 h later, while Group B received a single dose of pyrotinib and 4 mg of loperamide after breakfast on day 9, followed by single oral doses of 2 mg of loperamide at 2 and 4 h post-administration. Blood samples were collected to determine pyrotinib blood concentrations.
Results: In Group A, the combination treatment with montmorillonite powder resulted in a decrease in Cmax, AUC0-t, and AUC0-
Conclusion: Even when montmorillonite powder was administered 2 h after pyrotinib dosing, it still reduced systemic exposure of pyrotinib by 32.4% in AUC0-
Clinical trial registration: [ClinicalTrials.gov], identifier [NCT05252546].
Highlights
What is already known about this subject:
- Pyrotinib is an irreversible dual Human Epidermal growth factor Receptor 2(HER2) and epidermal growth factor receptor(EGFR) tyrosine kinase inhibitor(TKI). Diarrhea is the most common adverse reaction observed in clinical trials of pyrotinib.
- Symptomatic management of diarrhea of pyrotinib is usually done by using either montmorillonite powder or loperamide. The population pharmacokinetics found concomitant use of montmorillonite powder could decrease the bioavailability of pyrotinib by 50.3%.
- This study investigated the interaction of montmorillonite powder or loperamide on pyrotinib.
What this study adds:
- Even when montmorillonite powder was administered 2 hours after pyrotinib dosing, it still reduced systemic exposure of pyrotinib by 32.4% in AUC0-
- Loperamide could increase pyrotinib exposure by 19% in AUC0-
- Loperamide is recommended in symptom control in pyrotinib-induced diarrhea.
1 Introduction
Receptor tyrosine kinases (RTKs) are transmembrane proteins that play a crucial role in growth factor signaling transduction. To date, nearly 60 RTKs have been identified, classified into 20 distinct families. Dysregulated expression or activation of RTKs is closely associated with tumorigenesis, tumor progression, metastasis, and resistance to chemotherapy. RTK inhibitors have demonstrated efficacy in various malignancies. For instance, EGFR inhibitors, including erlotinib and gefitinib, have significantly improved outcomes in non-small cell lung cancer (NSCLC) patients harboring epidermal growth factor receptor (EGFR) mutations (Roskoski, 2023). Vascular endothelial growth factor receptor (VEGFR) inhibitors, such as sunitinib and sorafenib, have been widely used in the treatment of renal cell carcinoma and hepatocellular carcinoma by targeting tumor angiogenesis (Roskoski, 2023). Another well-characterized example is the epidermal Human Epidermal growth factor Receptor 2 (HER2), which is overexpressed in approximately 30% of human tumors (Slamon et al., 1987), including breast cancer. HER2 overexpression serves as an independent prognostic factor for recurrence and survival in breast cancer patients, making it a critical target for therapeutic intervention. As a result, multiple HER2-targeting agents, such as trastuzumab and pertuzumab, have been approved for clinical use.
Pyrotinib is an irreversible dual inhibitor of HER2 and EGFR tyrosine kinases. In China, the combination of 400 mg pyrotinib and capecitabine is approved as a second-line treatment for HER2-positive metastatic breast cancer (Yan et al., 2020; Xu et al., 2021). In healthy volunteers, the time to peak concentration following oral administration of 400 mg pyrotinib is approximately 5 h. Pyrotinib is primarily metabolized in liver by the Cytochrome P450 3A4 (CYP3A4) enzyme. The apparent volume of distribution in healthy volunteers ranges from 2,248 to 7,870 L, and the elimination half-life is approximately 18 h (data on file).
The PHILA study demonstrated that pyrotinib, when combined with trastuzumab and docetaxel, significantly improved progression-free survival (PFS) compared to placebo, trastuzumab, and docetaxel in patients with untreated HER2-positive metastatic breast cancer (24.3 versus 10.4 months; hazard ratio 0.41) (Ma et al., 2023). Diarrhea was the most common adverse event the PHILA study, occurring in nearly all patients in the pyrotinib group (293/297, 99%), with 46% (138/297) experiencing grade 3 or higher severity. Diarrhea usually lasts for 2–3 days, and the vast majority of cases could be controlled by drug withdrawal or dose reduction and symptomatic treatment (Ma et al., 2023).
Montmorillonite powder or loperamide were allowed in secondary prevention and intervention for diarrhea in PHILA study and diarrhea was generally manageable. Loperamide can bind to intestinal wall opioid receptors, inhibit acetylcholine and prostaglandin release, reduce propulsive peristalsis, and increase intestinal transit time. Loperamide is mostly absorbed by the intestinal wall, but due to the significant first-pass effect, its bioavailability is only about 0.3% (Janssen Pharmaceutica Inc., IMODIUM package insert. (2016)). The antidiarrheal gastro-intestinal protectant activity of montmorillonite powder is due to its powerful coating property on the gastrointestinal mucosa. By interacting with the glycoprotein of mucus, it increases the resistance of the mucosal gel in response to aggressive agents. Montmorillonite powder is not absorbed or metabolized (SMECTA Consumer Medication Information Leaflet, 2019).
Ad hoc analysis showed median PFS of those who had used montmorillonite powder in diarrhea management were 27.83 months, compared to 19.5 months in loperamide (data on file). However, the population pharmacokinetics of pyrotinib found concomitant use of montmorillonite could decrease the bioavailability of pyrotinib by 50.3% (Wen et al., 2021). And real-world clinical feedback indicates that patients using montmorillonite powder to manage diarrhea may experience reduced anti-tumor efficacy of pyrotinib. Given the high incidence of diarrhea during pyrotinib treatment for HER2-positive breast cancer, questions remain regarding the recommended antidiarrheal medication in the pyrotinib prescribing information. This study would evaluate the effects of montmorillonite powder and loperamide on the pharmacokinetics of pyrotinib in healthy volunteers to the inform the drug label instructions.
2 Methods
2.1 Subjects
Eligible subjects were healthy male and female volunteers aged 18–45 years, with a minimum of three subjects per sex, and a body mass index (BMI) ranging from19∼26 kg/m2. Participants were excluded if they exhibited abnormalities in physical examinations, laboratory profiles, vital signs, or 12-lead electrocardiograms (ECGs). Participants who required bowel motility inhibitors or had taken medications that affect liver enzyme activity within 4 weeks prior to the administration of the study drug were also excluded.
2.2 Treatment protocol
The study was a single-center, open-label, single-dose, fixed-sequence clinical trial conducted in healthy adult subjects (ClinicalTrials.gov identifier: NCT05252546). All research protocols received approval from the Ethics Committee of Xiangya Hospital of Central South University.
As summarized in Figure 1, the participants were divided into two groups: Group A (montmorillonite powder) and Group B (loperamide), with 18 subjects in each group, resulting in a total of 36 subjects. On day 1, all subjects received a single oral dose of 400 mg pyrotinib after breakfast. On day 9, subjects in Group A received a single oral dose of 400 mg pyrotinib after breakfast, followed by a single oral dose of 3 g of montmorillonite powder 2 h later. Subjects in Group B received a single oral dose of 400 mg pyrotinib and 4 mg loperamide after breakfast on day 9, followed by additional doses of 2 mg loperamide at 2 h and 4 h post-administration. Subjects were instructed to have a light meal the night before the administration of the drug and to fast for 10 h overnight.
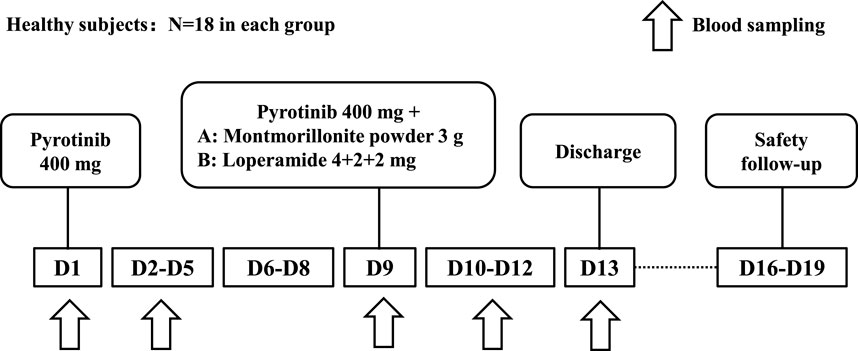
Figure 1. Study diagram.
The timing of concomitant drug administration was designed to reflect clinically relevant conditions rather than to capture the maximum drug interaction seen in index studies (U. S. Food and Drug Administration, 2020b).
2.3 Blood sampling for pharmacokinetic analyses
Blood samples were collected at baseline (within 30 min prior to drug administration) and at 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 24, 48, 72, and 96 h following pyrotinib administration on days 1 and 9 to measure the concentration of pyrotinib in the plasma. After completion of the study on day 13, subjects underwent a final examination and were subsequently discharged. A safety follow-up was conducted between days 16 and 19.
The analysis of plasma samples for pyrotinib was based on a validated and sensitive liquid chromatography-tandem mass spectrometry (LC-MS/MS) method with high specificity. The plasma samples were stored at −70°C and analyzed within a stability period of 176 days. The calibration range for the standard curve was between 1.00 and 500 ng/mL, and the regression equation used linear fitting with a weighting factor of W = 1/X2 (bioanalysis method validation report, data on file). In each batch of analysis, the number of quality control (QC) samples accounted for more than 5% of the unknown samples in that batch. The inter-batch precision of the QC samples for low, medium, and high concentrations (LQC, 3.00 ng/mL; MQC, 25.0 ng/mL; and HQC, 400 ng/mL, respectively) was 5.9%, 6.1%, and 5.3%, respectively. The inter-batch deviation between the low, medium, and high concentration QC levels and the theoretical concentrations was −2.9%, 1.5%, and −0.5%, respectively. Representative chromatograms of pyrotinib at the LLOQ (lower limit of quantification) and in a plasma sample can be found in the Supplementary Material.
2.4 Statistical analysis
The individual pharmacokinetic parameters of pyrotinib would be calculated using non-compartmental analysis (NCA) and summarized by groups and combination status with descriptive statistics. To estimate the difference in the least-square means between the combination treatment and pyrotinib alone, a mixed-effects model was used to analyze the natural log-transformed pharmacokinetic parameters of pyrotinib (including AUC0-
2.5 Safety analysis
Safety assessments included adverse events (AEs), physical examinations, vital signs, 12-lead ECGs, and clinical laboratory tests (hematology, serum chemistry, and urinalysis). All AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA®), version 25.0, and summarized by groups and combination status with loperamide or montmorillonite powder.
3 Results
3.1 Participants
The study was conducted from 2 March 2022 to 2 August 2022. A total of 36 subjects completed the screening assessments, met all eligibility criteria, and received the scheduled study drug. All subjects had completed each scheduled pharmacokinetic blood sample collection. One subject in each of Groups A and B was lost to follow-up during the monitoring of a grade 1 hypertriglyceridemia AE. Demographic characteristics are available for all 36 subjects, as shown in Table 1. The baseline characteristics are generally comparable between the two groups. Approximately one-fourth of the subjects were female, and the majority were of Han ethnicity.
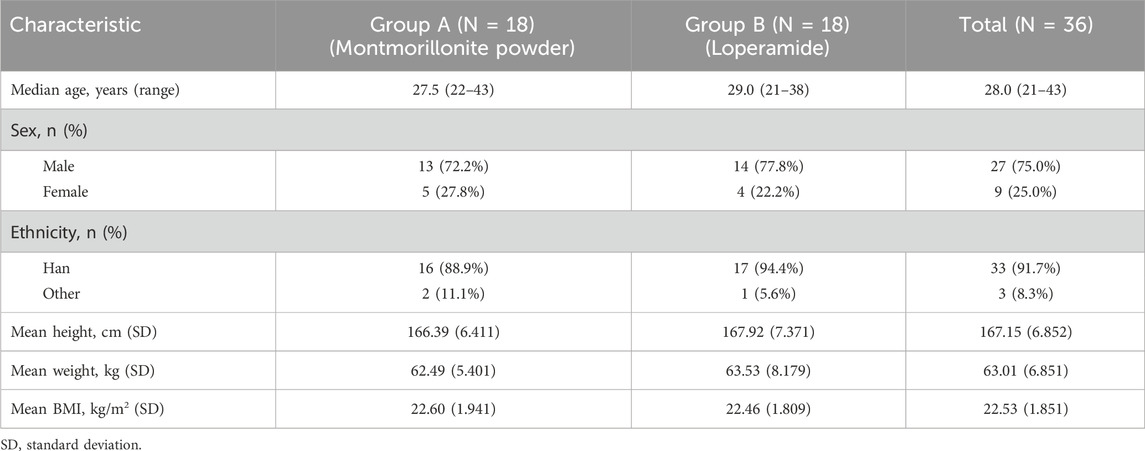
Table 1. Demographic and baseline characteristics.
3.2 Pharmacokinetics
Mean plasma concentration-time profiles in semi-log scale of pyrotinib in Group A can be found in Figure 2. The pharmacokinetic parameters and statistical comparison in Group A are presented in Tables 2, 3, respectively. The geometric mean AUC0-t, AUC0-
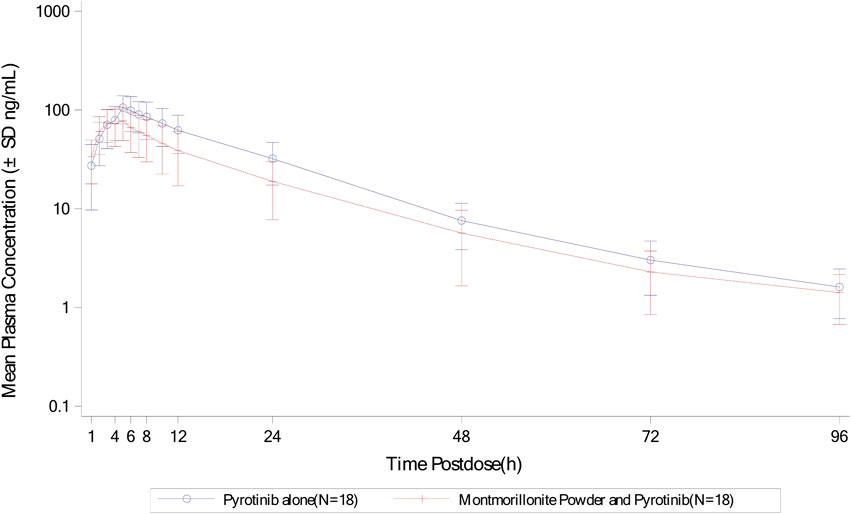
Figure 2. Mean plasma concentration-time profiles (semi-log) of plasma pyrotinib in Group A.
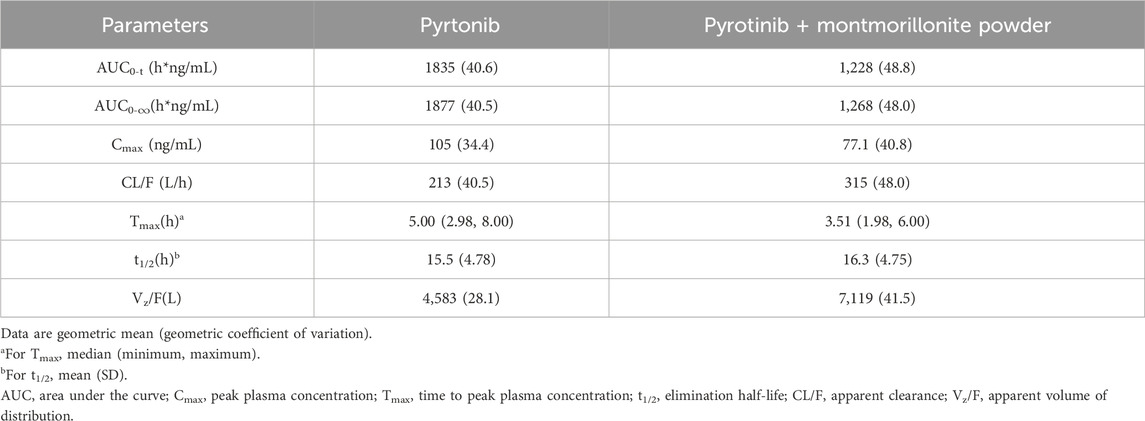
Table 2. Pyrotinib pharmacokinetic parameters in Group A (montmorillonite powder).
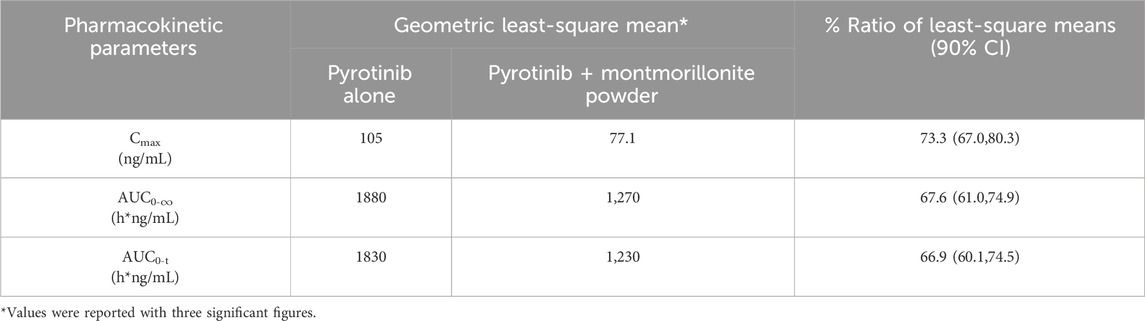
Table 3. Statistical comparison of pyrotinib plasma pharmacokinetic parameters for pyrotinib in combination with montmorillonite powder vs. pyrotinib alone.
Mean plasma concentration-time profiles in semi-log scale of pyrotinib in Group B can be found in Figure 3. The pharmacokinetic parameters and statistical comparison in Group B are presented in Tables 4, 5. The geometric mean AUC0-t, AUC0-
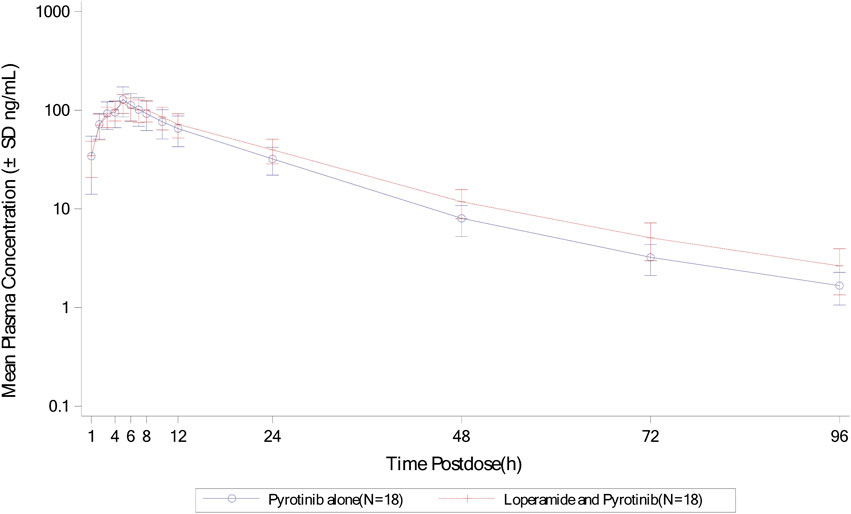
Figure 3. Mean plasma concentration-time profiles (semi-log) of plasma pyrotinib in Group B.
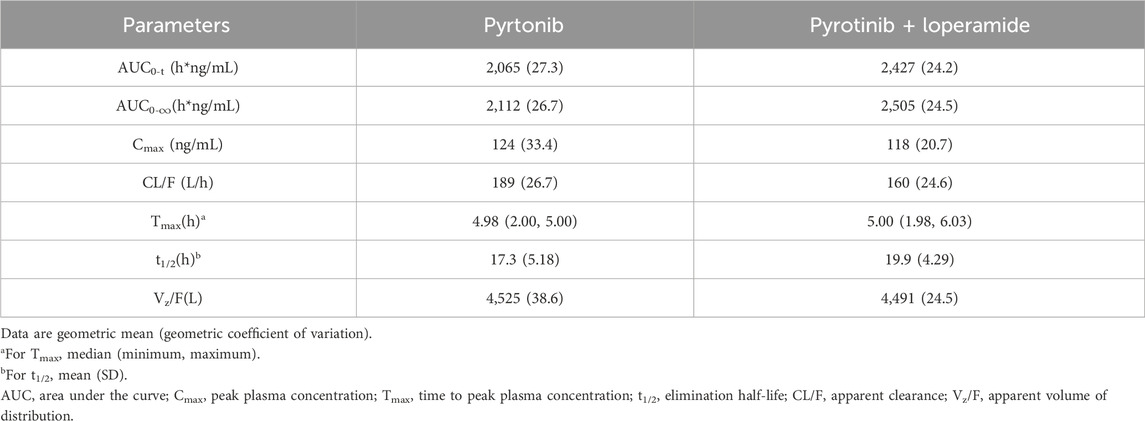
Table 4. Pyrotinib pharmacokinetic parameters in Group B (loperamide).
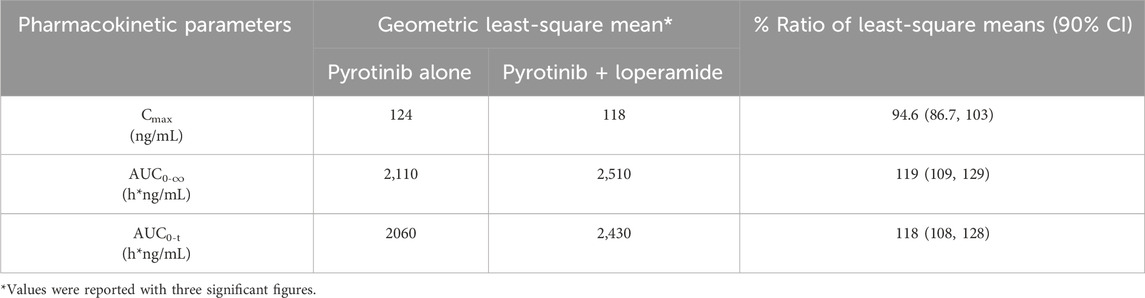
Table 5. Statistical comparison of pyrotinib plasma pharmacokinetic parameters for pyrotinib in combination with loperamide vs. pyrotinib alone.
Overall, the results of Group A showed that there was no significant change in t1/2 of pyrotinib when used in combination with montmorillonite powder compared to pyrotinib alone. Median time to maximum pyrotinib concentration was 3.51 h when combined with montmorillonite powder, which was earlier than the 5 h observed in the single treatment group. The results of Group B showed that after combination with loperamide, there was no significant change in the median Tmax compared to pyrotinib alone. The exposure of pyrotinib (Cmax) was similar, while CL/F decreased which may be explained by increased F mathematically or reduced propulsive peristalsis and increased absorption time mechanistically. Loperamide had minimal impact on the absorption rate of pyrotinib, with a decrease in Cmax of approximately 5.4%, and an increase in AUC0-t and AUC0-
3.3 Safety results
A total of eight subjects in Group A experienced 11 treatment-emergent adverse events (TEAEs), all of which were mild (Grade 1) in severity and occurred during the treatment period. Common TEAEs (experienced by ≥ 2 subjects) included hypertriglyceridemia (11.1%). Other TEAEs included hypercholesterolemia, nausea, abdominal distension, alanine aminotransferase elevation, aspartate aminotransferase elevation, bradycardia, elevated blood bilirubin, upper respiratory tract infection, and orthostatic hypotension (each occurring in 5.6% of subjects).
In Group B, a total of 12 subjects experienced 18 TEAEs. Except for one subject who experienced a Grade 2 hypertriglyceridemia event, and another subject who experienced a Grade 2 rash event, all other TEAEs were mild (Grade 1) in severity. Common TEAEs (experienced by ≥ 2 subjects) included hypertriglyceridemia (22.2%), hyperuricemia (11.1%), oral ulcer (11.1%), and rash (11.1%). Other TEAEs included nausea, abdominal distension, PR interval prolongation on electrocardiogram, increased heart rate, increased total bile acid, headache, and dizziness (each occurring in 5.6% of subjects).
No TEAEs resulted in subject withdrawal, treatment discontinuation, or death in either Group A or Group B. No serious adverse events were reported. Overall, the safety profile of a single oral dose of pyrotinib alone or in combination with montmorillonite powder or loperamide was manageable in healthy subjects.
4 Discussion
The guidance recommended that doses of the perpetrator drug in DDI studies should maximize the possibility of identifying a DDI (U. S. Food and Drug Administration, 2020a; U. S. Food and Drug Administration, 2020b). However, for concomitant-use studies, a risk-based approach to evaluate the interaction in clinical settings should be adopted (U. S. Food and Drug Administration, 2020b). The timing of drug administration should be based on the purpose. The guidance recommended that the sponsor should adjust the timing of drug administration to maximize the potential to detect an interaction in index studies) and/or to reflect the clinically relevant conditions in concomitant-use studies (U. S. Food and Drug Administration, 2020b).
The clinically relevant conditions for concomitant drugs are from clinical trial or guidance. An average of three sachets per day is recommended for montmorillonite powder in the label. The interval between administration of montmorillonite powder and pyrotinib is at least 2 h in clinical trials. Therefore, the administration method used in this protocol is to administer montmorillonite powder 2 h after pyrotinib administration. According to the “Expert consensus on the management of adverse events of ErbB family tyrosine kinase inhibitors (TKI) in breast cancer”, diarrhea caused by small-molecule TKIs targeting the HER2 receptor should be treated with an initial dose of 4 mg of loperamide, followed by 2 mg every 2 h, up to a maximum of 16 mg (Wang et al., 2020). To reduce the risk of constipation in healthy volunteers, the maximum daily dose of loperamide is restricted to 8 mg. The regimen of loperamide in the study is to administer 4 mg in combination with pyrotinib, followed by 2 mg of loperamide 2 and 4 h after pyrotinib administration.
Loperamide works by slowing down the movement of the intestines and can increase the residence time of pyrotinib (Janssen Pharmaceutica Inc., 2016). This extended residence time allows more time for pyrotinib to be absorbed, which is expected to significantly increase the AUC, reflecting the total drug exposure over time. In contrast, the maximum concentration may not be as significantly affected, as it is influenced by the rate of absorption, which can be less impacted by slowed motility than total absorption over time. Thus, a greater change in AUC is anticipated compared to Cmax, as the overall exposure to pyrotinib is prolonged, while the peak concentration might remain similar or change less markedly. This hypothesis is supported by the study results, which showed a more pronounced effect on AUC than on Cmax. Though not included in the package insert, literature has reported that loperamide was a potent inhibitor of CYP3A4 with an IC50 of 0.050 µM (Marechal et al., 2006). Interaction potential should be evaluated since pyrotinib is mainly metabolized through the CYP3A4 pathway. However, only 0.3% of orally administered loperamide is available in blood and 95% of them bind to albumin. The maximum concentration of loperamide after 4 mg oral administration was 0.62 ng/mL (Niemi et al., 2006). The calculated R1 for reversible inhibition would be less than 1.0001. Based on the available data, significant CYP3A4 inhibition by loperamide in vivo is unlikely at clinically relevant doses due to its low systemic bioavailability. However, the calculated R1,gut for loperamide is 1,342.7 based on molecular weight of 477 Dalton, which is significantly higher than the cut off of 11 from guidance. Loperamide use may need to be limited for medications that undergoes high gut CYP3A4 metabolism.
Montmorillonite powder is a type of clay mineral that belongs to the smectite group. It brings a powerful coating on the gastrointestinal mucosa and increases the resistance of the mucosal gel in response to aggressive agents (SMECTA Consumer Medication Information Leaflet, 2019). The dedicated concomitant-use study showed good consistence to the model result. The current study further verifies that montmorillonite powder influences the absorption rate and extent of pyrotinib with 1.5 h earlier in time to maximum concentration and 32.4% lower of AUC0-
5 Conclusion
Even when montmorillonite powder was administered 2 h after pyrotinib dosing, it still reduced systemic exposure of pyrotinib by 32.4% in AUC0-
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Ethics Committee of Xiangya Hospital of Central South University. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
YW: Writing – original draft. DL: Writing – original draft. TZ: Writing – original draft. SX: Project administration, Writing – review and editing. YZ: Project administration, Writing – review and editing. KZ: Methodology, Writing – review and editing. SL: Methodology, Writing – review and editing. KS: Writing – review and editing. XL: Methodology, Project administration, Writing – review and editing. PX: Methodology, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by Jiangsu Hengrui Pharmaceuticals Co., Ltd., Lianyungang, China. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Conflict of interest
Authors YW, TZ, KZ, SL, and KS were employed by Jiangsu Hengrui Pharmaceuticals Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2025.1563556/full#supplementary-material
References
Janssen Pharmaceutica Inc., (2016). IMODIUM package insert. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/017694s052lbl.pdf. (Accessed April 22, 2024).
Ma, F., Yan, M., Li, W., Ouyang, Q., Tong, Z., Teng, Y., et al. (2023). Pyrotinib versus placebo in combination with trastuzumab and docetaxel as first line treatment in patients with HER2 positive metastatic breast cancer (PHILA): randomised, double blind, multicentre, phase 3 trial. bmj 383, e076065. doi:10.1136/bmj-2023-076065
Marechal, J.-D., Yu, J., Brown, S., Kapelioukh, I., Rankin, E. M., Wolf, C. R., et al. (2006). In silico and in vitro screening for inhibition of cytochrome P450 CYP3A4 by comedications commonly used by patients with cancer. Drug metabolism Dispos. 34 (4), 534–538. doi:10.1124/dmd.105.007625
Niemi, M., Tornio, A., Pasanen, M. K., Fredrikson, H., Neuvonen, P. J., and Backman, J. T. (2006). Itraconazole, gemfibrozil and their combination markedly raise the plasma concentrations of loperamide. Eur. J. Clin. Pharmacol. 62, 463–472. doi:10.1007/s00228-006-0133-z
Roskoski, Jr (2023). Properties of FDA-approved small molecule protein kinase inhibitors: a 2023 update. Pharmacol. Res. 187, 106552. doi:10.1016/j.phrs.2022.106552
Slamon, D. J., Clark, G. M., Wong, S. G., Levin, W. J., Ullrich, A., and McGuire, W. L. (1987). Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. science 235 (4785), 177–182. doi:10.1126/science.3798106
SMECTA Consumer Medication Information Leaflet (2019). SMECTA®3 g STRAWBERRY, POWDER FOR ORAL SUSPENSION IN SACHET. Available online at: https://www.smecta.com.my/wp-content/uploads/2021/01/SmectaSB_PI.pdf. (Accessed April 22, 2024).
U. S. Food and Drug Administration. (2020a). In Vitro Drug interaction studies — Cytochrome P450 enzyme- and transporter-mediated drug interactions guidance for industry, Available online at: https://www.fda.gov/media/135587/download. (Accessed April 18, 2025).
U. S. Food and Drug Administration. (2020b). Clinical drug interaction studies —Cytochrome P450 enzyme- and transporter-mediated drug interactions guidance for industry. Available online at: https://www.fda.gov/media/135586/download. (Accessed April 18, 2025).
U. S. Food and Drug Administration. (2022). Assessing the effects of food on drugs in INDs and NDAs — clinical pharmacology considerations guidance for industry. Available online at: https://www.fda.gov/media/121313/download. (Accessed April 18, 2025).
Wang, B. Y., Ge, R., and Jiang, Z. F.Breast Cancer Expert Group of Chinese Society of Clinical Oncology (2020). Expert consensus on the management of adverse events of ErbB family tyrosine kinase inhibitors in breast cancer. Zhonghua Zhong liu za zhi Chin. J. Oncol. 42 (10), 798–806. doi:10.3760/cma.j.cn112152-20200805-00711
Wen, H.-ni, Liu, Y. X., Xu, D., Zhao, K. J., and Jiao, Z. (2021). Population pharmacokinetic modeling of pyrotinib in patients with HER2-positive advanced or metastatic breast cancer. Eur. J. Pharm. Sci. 159, 105729. doi:10.1016/j.ejps.2021.105729
Xu, B., Yan, M., Ma, F., Hu, X., Feng, J., Ouyang, Q., et al. (2021). Pyrotinib plus capecitabine versus lapatinib plus capecitabine for the treatment of HER2-positive metastatic breast cancer (PHOEBE): a multicentre, open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 22 (3), 351–360. doi:10.1016/S1470-2045(20)30702-6
Yan, M., Bian, L., Hu, X., Zhang, Q., Ouyang, Q., Feng, J., et al. (2020). Pyrotinib plus capecitabine for human epidermal growth factor receptor 2-positive metastatic breast cancer after trastuzumab and taxanes (PHENIX): a randomized, double-blind, placebo-controlled phase 3 study. Transl. Breast Cancer Res. 1, 13. doi:10.21037/tbcr-20-25
Keywords: pyrotinib, montmorillonite powder, loperamide, diarrhea, pharmacokinetics
Citation: Wang Y, Li D, Zhang T, Xu S, Zhang Y, Zhao K, Li S, Shen K, Li X and Xu P (2025) Evaluation of the pharmacokinetic interactions of montmorillonite powder or loperamide on pyrotinib in healthy volunteers. Front. Pharmacol. 16:1563556. doi: 10.3389/fphar.2025.1563556
Received: 22 January 2025; Accepted: 16 April 2025;
Published: 12 May 2025.
Edited by:
Yurong Lai, Gilead, United StatesReviewed by:
Mian Zhang, Bristol Myers Squibb, United KingdomRyan Varghese, Saint Joseph’s University, United States
Copyright © 2025 Wang, Li, Zhang, Xu, Zhang, Zhao, Li, Shen, Li and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pingsheng Xu, eHBzMjAxOTAxQGNzdS5lZHUuY24=; Xiaomin Li, bGl4aWFvbWluMDkxNEBjc3UuZWR1LmNu