 Xiaoyan Chen1,2†
Xiaoyan Chen1,2† Feng Gao
Feng Gao Zhi Li
Zhi Li- 1School of Basic Medical Sciences, Hubei University of Chinese Medicine, Wuhan, China
- 2Hubei University of Chinese Medicine, Wuhan, China
- 3Taihe Hospital, Hubei University of Medicine, Shiyan, China
- 4First School of Clinical Medicine, Hubei University of Medicine, Shiyan, China
- 5Interventional Cancer Institute of Chinese Integrative Medicine, Putuo Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China
A Commentary on
Transcription factor MEF2D regulates aberrant expression of ACSL3 and enhances sorafenib resistance by inhibiting ferroptosis in HCC
by Chen X, Xie J, Fan W, Zhang J, Gao F and Li Z (2025). Front. Pharmacol. 16:1575998. doi: 10.3389/fphar.2024.1464852
Introduction
Hepatocellular carcinoma (HCC) is a highly fatal cancer globally. Sorafenib, as a primary treatment agent, encounters substantial constraints owing to drug resistance, which significantly undermines its clinical effectiveness (Sun et al., 2022). A new study in Frontiers in Pharmacology has demonstrated for the first time that the transcription factor MEF2D inhibits ferroptosis by upregulating long-chain ACSL3, therefore facilitating sorafenib resistance in HCC. This study combines clinical data, animal models, and cellular investigations to validate the oncogenic function of ACSL3 in the transition from NAFLD to HCC. It clarifies the molecular mechanism via which MEF2D directly interacts with the ACSL3 promoter to modulate its transcription (Li et al., 2024). This article offers a comprehensive analysis of the study, addressing its scientific importance and possible translational implications (Figure 1).
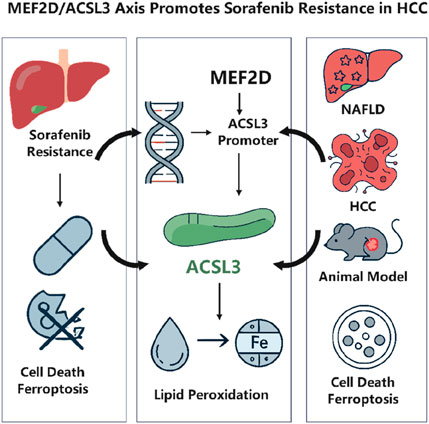
Figure 1. MEF2D drives sorafenib resistance in hepatocellular carcinoma by transcriptionally upregulating ACSL3 to suppress ferroptosis. Created with BioRender.com.
Background
The progression of HCC is intricately associated with chronic liver conditions, including viral hepatitis, alcoholic liver disease, and NAFLD (Hyun et al., 2021). Sorafenib, a multi-targeted tyrosine kinase inhibitor, prolongs patient longevity by obstructing angiogenesis and tumour growth. Nevertheless, extensive drug resistance results in unfavourable outcomes (Li et al., 2023). Recent findings indicate that sorafenib can trigger ferroptosis (Figure 2), a type of cell death driven by iron-dependent lipid peroxidation, and that drug resistance may be linked to the suppression of this process (Hao et al., 2024; Xu et al., 2023). The principal characteristic of ferroptosis is the abnormal buildup of lipid reactive oxygen species (ROS) and the deactivation of antioxidant mechanisms such as GPX4 (Chen et al., 2021). Lipid metabolism reprogramming is essential in modulating ferroptosis, as the ACSL family affects cell membrane phospholipid composition through the catalysis of fatty acid activation (Cai et al., 2023). ACSL3 has been documented to impede ferroptosis by promoting monounsaturated fatty acids and diminishing polyunsaturated fatty acid peroxidation (Cao et al., 2025); however, its precise function in HCC is yet to be elucidated. MEF2D, a member of the myocyte enhancer factor family, is aberrantly overexpressed in multiple malignancies, facilitating tumour proliferation, invasion, and drug resistance through the regulation of downstream genes (Surace et al., 2024). The role of MEF2D in regulating ferroptosis in HCC is yet uncertain.
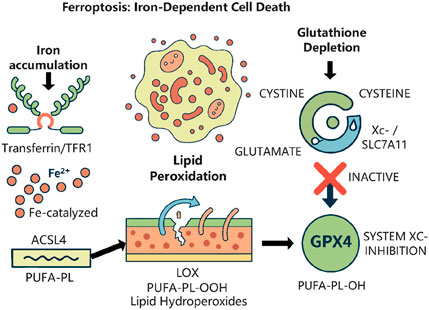
Figure 2. The core regulatory mechanism of ferroptosis. This form of regulated cell death is driven by the iron-dependent accumulation of lipid peroxides. Created with BioRender.com.
Key evidence and findings
This work examined the GSEA database and clinical samples, demonstrating that ACSL3 is markedly overexpressed in NAFLD and HCC tissues, with a positive correlation to disease severity (Li et al., 2024). Single-cell sequencing revealed that ACSL3 is primarily concentrated in hepatocytes, implying its potential role in facilitating the transition from NAFLD to HCC by modulating hepatic lipid metabolism. In animal studies, mice with high-fat diet-induced NAFLD demonstrated increased liver ACSL3 expression, alongside lipid vacuolization and intensified inflammation, hence reinforcing its pro-oncogenic function (Mossmann et al., 2023). Utilising siRNA knockdown and overexpression methodologies, researchers determined that ACSL3 mitigates sorafenib-induced lipid peroxidation and ROS generation by downregulating the ferroptosis-promoting regulator ACSL4 and upregulating the anti-ferroptotic proteins GPX4 and FTH1(12). This corresponds with ACSL3’s established role in catalysing MUFA to produce protective membrane phospholipids and, for the first time, distinctly delineates ACSL3’s inhibitory effect on ferroptosis in HCC. ChIP and luciferase reporter tests validated that MEF2D directly interacts with the MEF2 response element (MRE) in the ACSL3 promoter region, hence enhancing its transcription (Li et al., 2024). Clinical data analysis revealed a substantial positive connection between MEF2D and ACSL3 expression in HCC, with elevated levels of both correlating with unfavourable patient outcomes (Li et al., 2024). Furthermore, the suppression of MEF2D negated the ferroptosis inhibition and sorafenib resistance induced by ACSL3, underscoring the pivotal function of the MEF2D-ACSL3 axis. This study establishes a connection between the MEF2D-ACSL3 axis and the prevention of ferroptosis, as well as sorafenib resistance, for the first time, addressing a gap in the mechanisms of resistance in HCC. Utilising multi-omics analysis and experimental validation, this study elucidates ACSL3’s dual function in the evolution of NAFLD-HCC and suggests targeting MEF2D or ACSL3 as a novel approach to counteract sorafenib resistance, offering potential translational significance (Table 1).
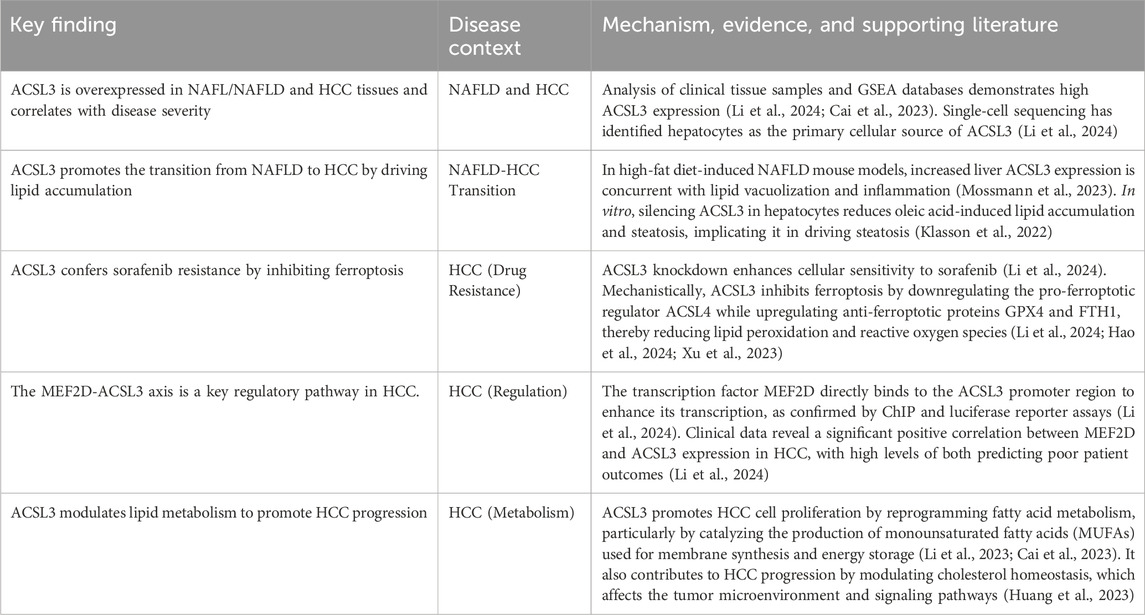
Table 1. Summary of recent findings on the role of ACSL3 in NAFLD and HCC.
Limitations and future directions
While this study provides significant insights into the MEF2D-ACSL3 axis, its limitations also illuminate critical and exciting avenues for future research. Rather than being constraints, these points represent a strategic roadmap for the scientific community to build upon our findings and develop robust therapeutic strategies against sorafenib resistance. Specifically, the reliance on a single-center cohort tempers the immediate clinical applicability of our findings, as the prevalence and importance of this axis may differ across more diverse patient populations, a critical factor for designing successful, broad-based clinical trials. Similarly, the absence of definitive genetic validation from knockout models means our therapeutic hypothesis requires more rigorous preclinical confirmation to de-risk the substantial investment required for human studies. These translational gaps underscore that moving from a compelling biological mechanism to a viable therapeutic intervention requires a dedicated phase of validation to ensure both safety and efficacy. For instance, our initial findings, based on a single-center cohort with limited NAFLD-related HCC cases, present an important opportunity for future multi-center studies to validate the generalizability of our results and dissect how the MEF2D-ACSL3 axis varies across different HCC etiologies. Similarly, advancing our in vivo work with more sophisticated models, such as hepatocyte-specific conditional knockouts for Mef2d or Acsl3 in NAFLD-HCC mice, is essential for definitively dissecting the axis’s role in a context that more accurately simulates human disease progression. Furthermore, our work invites deeper mechanistic investigations by experts in epigenetics to explore the roles of DNA methylation and histone acetylation in MEF2D’s regulation of ACSL3, as well as the precise molecular cascade by which ACSL3 modulates its downstream targets ACSL4 and GPX4. Looking ahead, a comprehensive strategy is needed to map the upstream signaling pathways regulating MEF2D, such as Hippo-YAP or MAPK, and to identify other downstream effectors of ACSL3. This presents a clear call to action for medicinal chemists and pharmacologists to develop specific MEF2D or ACSL3 inhibitors and assess their efficacy in combination with sorafenib. Moreover, critical questions remain regarding the interplay between ferroptosis and other cell death pathways like autophagy and apoptosis, the influence of lipid availability in the tumor microenvironment on ACSL3 activity, and the dynamic evolution of the MEF2D-ACSL3 axis during long-term treatment leading to acquired resistance. Collectively, these directions provide a comprehensive framework for collaborative efforts across disciplines, aimed at systematically dismantling this resistance pathway and establishing new, more effective therapeutic paradigms for HCC.
Discussion
This research elucidates the critical function of the MEF2D-ACSL3 axis in sorafenib resistance in hepatocellular carcinoma, providing novel insights into the relationship between tumour metabolism and cellular apoptosis. ACSL3, a pivotal enzyme in lipid metabolism, exhibits a dual function—facilitating cancer development while suppressing ferroptosis—highlighting the intricacies of metabolic reprogramming in tumour advancement and medication resistance. Extending the function of MEF2D into ferroptosis research establishes a foundation for exploring analogous roles of additional MEF2 family members. Targeting MEF2D or ACSL3 may represent a unique approach to counteract medication resistance, either by the application of small molecule inhibitors to disrupt MEF2D’s interaction with the ACSL3 promoter or by inhibiting ACSL3 activity using MUFA synthesis inhibitors (Henley and Koehler, 2021; Huang et al., 2025; Pisanu et al., 2018). Nonetheless, the potential toxicity to normal tissues, such as the heart and muscles, requires meticulous assessment. ACSL3 upregulation and ACSL4 downregulation correspond with ACSL4’s established pro-ferroptotic activity; however, confirmation is needed to determine if they exert functional antagonism via substrate competition (e.g., PUFA and MUFA). MEF2D has been previously identified as a promoter of HCC advancement through cell cycle regulation, and this study enhances its significance in metabolic control, indicating it may function as a “master regulator” that integrates many oncogenic signals. Future research should explore how this metabolic axis integrates with established resistance mechanisms, such as the hyperactivation of survival signaling pathways like PI3K/AKT or MAPK. For instance, MEF2D could act as a crucial downstream effector, translating oncogenic signals from these pathways into a specific metabolic rewiring program that confers resistance. This perspective positions the MEF2D-ACSL3 axis not merely as a standalone pathway but as a critical metabolic node within a larger, interconnected resistance network, thereby offering a novel therapeutic angle distinct from conventional kinase inhibitors. Notwithstanding advancements, obstacles in translational medicine, such as precision targeted delivery technologies, and the influence of the metabolic milieu on ACSL3 function persist as future problems. For example, the potential of exogenous MUFA to mitigate ferroptosis without affecting ACSL3, as well as the dynamic regulation of drug resistance by lipid supply in the tumour microenvironment, necessitate additional investigation. This study offers a theoretical foundation for the development of combination medicines; however, it requires confirmation in models that more closely resemble actual conditions to facilitate the move from mechanistic research to clinical application.
Conclusion
This study elucidates the pivotal function of the MEF2D-ACSL3 axis in sorafenib resistance in HCC using multi-dimensional tests, establishing a theoretical basis for the development of combination therapies. Future investigations should meticulously examine the regulatory network of this axis and substantiate its targeting potential in clinically pertinent models to connect mechanistic research with therapeutic application.
Author contributions
XC: Conceptualization, Writing – original draft. JX: Writing – original draft, Writing – review and editing. WF: Writing – review and editing. JZ: Writing – review and editing. FG: Writing – review and editing. ZL: Supervision, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
Thanks to the editors and reviewers for their hard work and important comments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Cai, J., Chen, T., Jiang, Z., Yan, J., Ye, Z., Ruan, Y., et al. (2023). Bulk and single-cell transcriptome profiling reveal extracellular matrix mechanical regulation of lipid metabolism reprograming through YAP/TEAD4/ACADL axis in hepatocellular carcinoma. Int. J. Biol. Sci. 19 (7), 2114–2131. doi:10.7150/ijbs.82177
Cao, Y., Li, J., Chen, Y., Wang, Y., Liu, Z., Huang, L., et al. (2025). Monounsaturated fatty acids promote cancer radioresistance by inhibiting ferroptosis through ACSL3. Cell. Death Dis. 16 (1), 184. doi:10.1038/s41419-025-07516-0
Chen, J., Ding, C., Chen, Y., Hu, W., Yu, C., Peng, C., et al. (2021). ACSL4 reprograms fatty acid metabolism in hepatocellular carcinoma via c-Myc/SREBP1 pathway. Cancer Lett. 502, 154–165. doi:10.1016/j.canlet.2020.12.019
Hao, S. H., Ma, X. D., Xu, L., Xie, J. D., Feng, Z. H., Chen, J. W., et al. (2024). Dual specific phosphatase 4 suppresses ferroptosis and enhances sorafenib resistance in hepatocellular carcinoma. Drug Resist Updat 73, 101052. doi:10.1016/j.drup.2024.101052
Henley, M. J., and Koehler, A. N. (2021). Advances in targeting 'undruggable' transcription factors with small molecules. Nat. Rev. Drug Discov. 20 (9), 669–688. doi:10.1038/s41573-021-00199-0
Huang, J., Pan, H., Sun, J., Wu, J., Xuan, Q., Wang, J., et al. (2023). TMEM147 aggravates the progression of HCC by modulating cholesterol homeostasis, suppressing ferroptosis, and promoting the M2 polarization of tumor-associated macrophages. J. Exp. Clin. Cancer Res. 42 (1), 286. doi:10.1186/s13046-023-02865-0
Huang, L., Xu, R., Chen, S., Lin, C., Li, W., Li, S., et al. (2025). Modulating lipid metabolism by nanoparticles (NPs)-mediated ACSL3 silencing to inhibit hepatocellular carcinoma growth and metastasis. Mol. Cancer 24 (1), 73. doi:10.1186/s12943-025-02274-1
Hyun, J., Han, J., Lee, C., Yoon, M., and Jung, Y. (2021). Pathophysiological aspects of alcohol metabolism in the liver. Int. J. Mol. Sci. 22 (11), 5717. doi:10.3390/ijms22115717
Klasson, T. D., LaGory, E. L., Zhao, H., Huynh, S. K., Papandreou, I., Moon, E. J., et al. (2022). ACSL3 regulates lipid droplet biogenesis and ferroptosis sensitivity in clear cell renal cell carcinoma. Cancer Metab. 10 (1), 14. doi:10.1186/s40170-022-00290-z
Li, Y., Yang, W., Zheng, Y., Dai, W., Ji, J., Wu, L., et al. (2023). Targeting fatty acid synthase modulates sensitivity of hepatocellular carcinoma to sorafenib via ferroptosis. J. Exp. Clin. Cancer Res. 42 (1), 6. doi:10.1186/s13046-022-02567-z
Li, X., Chen, S., Shi, Y., Wang, Y., Wang, X., Lin, Q., et al. (2024). Transcription factor MEF2D regulates aberrant expression of ACSL3 and enhances sorafenib resistance by inhibiting ferroptosis in HCC. Front. Pharmacol. 15, 1464852. doi:10.3389/fphar.2024.1464852
Mossmann, D., Müller, C., Park, S., Ryback, B., Colombi, M., Ritter, N., et al. (2023). Arginine reprograms metabolism in liver cancer via RBM39. Cell. 186 (23), 5068–83.e23. doi:10.1016/j.cell.2023.09.011
Pisanu, M. E., Maugeri-Saccà, M., Fattore, L., Bruschini, S., De Vitis, C., Tabbì, E., et al. (2018). Inhibition of Stearoyl-CoA desaturase 1 reverts BRAF and MEK inhibition-induced selection of cancer stem cells in BRAF-Mutated melanoma. J. Exp. Clin. Cancer Res. 37 (1), 318. doi:10.1186/s13046-018-0989-7
Sun, Y., Zhang, H., Meng, J., Guo, F., Ren, D., Wu, H., et al. (2022). S-palmitoylation of PCSK9 induces sorafenib resistance in liver cancer by activating the PI3K/AKT pathway. Cell. Rep. 40 (7), 111194. doi:10.1016/j.celrep.2022.111194
Surace, L., Wilhelm, C., and Bode, C. (2024). Mef2d: a novel transcription factor in type 2 allergic lung inflammation. Signal Transduct. Target Ther. 9 (1), 309. doi:10.1038/s41392-024-02022-9
Keywords: hepatocellular carcinoma, sorafenib resistance, Mef2d, ACSL3, ferroptosis
Citation: Chen X, Xie J, Fan W, Zhang J, Gao F and Li Z (2025) Commentary: Transcription factor MEF2D regulates aberrant expression of ACSL3 and enhances sorafenib resistance by inhibiting ferroptosis in HCC. Front. Pharmacol. 16:1575998. doi: 10.3389/fphar.2025.1575998
Received: 28 February 2025; Accepted: 29 September 2025;
Published: 13 October 2025.
Edited by:
Manish Tripathi, The University of Texas Rio Grande Valley, United StatesReviewed by:
Veerababu Nagati, The University of Texas Rio Grande Valley McAllen, United StatesSalique Hassan Shaham, University of Rio Grande, United States
Copyright © 2025 Chen, Xie, Fan, Zhang, Gao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhi Li, MTIwMjMyMDJAc2h1dGNtLmVkdS5jbg==; Feng Gao, bW91bnRhaW5AdGFpaGVob3NwaXRhbC5jb20=
†These authors have contributed equally to this work