 Kexin Wang
Kexin Wang Haoge Luo
Haoge Luo Liping Liu
Liping Liu Hang Gao
Hang Gao Yanyan Song
Yanyan Song Dong Li
Dong Li- 1The Second Hospital of Jilin University, Jilin University, Changchun, China
- 2Department of Immunology, College of Basic Medical Sciences, Jilin University, Changchun, China
- 3Department of Bone and Joint Surgery, The First Hospital of Jilin University, Jilin University, Changchun, China
- 4Department of Nephrology, The Second Hospital of Jilin University, Jilin University, Changchun, China
Rheumatoid arthritis (RA), a chronic autoimmune disorder, imposes a substantial global health burden through elevated disability rates, systemic complications, and socioeconomic consequences. Chronic synovitis and progressive joint destruction characterize this disease, driven by dysregulated innate and adaptive immune responses that amplify synovial inflammation, osteoclastogenesis, and irreversible tissue damage. Aberrant activation of interleukin (IL) -1 family cytokines critically contributes to RA pathogenesis. These cytokines mediate dual mechanisms: pro-inflammatory agonists like IL-1β, IL-18, and IL-36 accelerate disease progression, whereas insufficient levels of anti-inflammatory antagonists such as IL-1Ra and IL-37 disrupt the balance required to suppress pathogenic cascades. Clinical trials evaluating IL-1-targeting biologics—including anakinra and canakinumab—have demonstrated robust early efficacy. However, late-stage interventions exhibit diminished therapeutic returns, largely due to irreversible joint damage and compensatory activation of redundant cytokine networks. These findings emphasize the need for precise patient stratification. Single-pathway IL-1 inhibition faces inherent limitations, driving the development of multi-target strategies to counteract cytokine redundancy and reduce therapeutic resistance. This review systematically analyzes the mechanistic roles of IL-1 family cytokines in RA, evaluates clinical outcomes and safety profiles of IL-1-targeted therapies, and proposes innovative strategies to advance RA treatment.
1 Introduction
Rheumatoid arthritis (RA) is a multifactorial chronic autoimmune disease primarily characterized by joint involvement in the hands, wrists, feet, ankles, knees, shoulders, and elbows. Common symptoms include chronic joint pain, stiffness, tenderness, fever, and swelling, leading to limitations in daily activities and low quality of life (Long et al., 2022). According to statistics, 18 million people worldwide were affected by RA in 2019 (Collaborators, 2020). Approximately 70% of RA patients are female, 55% are over the age of 55, and 13 million RA patients can achieve improvement through treatment (Cieza et al., 2021).
The pathogenesis of RA involves a multistep cascade of immune dysregulation, stemming from aberrant interactions between innate and adaptive immunity. This process is mediated by genetic polymorphisms, epigenetic modifications, and environmental triggers, which collectively induce synovial inflammation, hyperplasia, angiogenesis, and cartilage degradation, ultimately leading to irreversible bone destruction (Jang et al., 2022). Cytokines, particularly pro-inflammatory mediators, such as tumor necrosis factor-alpha (TNF-α), interleukin (IL) -1, IL-6, and IL-17 are central to RA progression by amplifying inflammatory responses and tissue damage (Kugler et al., 2023). Targeted therapies, including TNF-α inhibitors and IL-6 receptor antagonists, effectively reduce inflammation and improve outcomes through cytokine blockade (Smolen, 2020). However, treatment resistance persists in part of patients despite these advances. Notably, IL-1 has emerged as a critical driver of bone erosion via nucleotide- binding oligomerization domain 3 (NLRP3) inflammasome activation, underscoring its therapeutic potential for refractory RA and structural damage mitigation.
Cytokines of the IL-1 family are crucial factors in regulating immune responses (Broderick and Hoffman, 2022). This family comprises both pro-inflammatory mediators—including IL-1α, IL-1β, IL-18 and IL-33—and anti-inflammatory counterparts, such as IL-37 and IL-38 (Vincent, 2019). The intricate interplay between these cytokines and their contributions, relative to the well-established actions of TNF-α and IL-6, highlights a unique pathogenic axis that may not be fully addressed by current therapeutic strategies. The IL-1 family cytokines have mechanisms that are not covered by TNF and IL-6 (for example, IL-1β drives synovial inflammation and bone erosion by activating NLRP3 inflammasomes, while IL-37 exerts its anti-inflammatory effects by inhibiting NLRP3 activity) (Tseng et al., 2022). Research on IL-1 antagonists fills the gap left by TNF-α/IL-6 inhibitors, especially in controlling structural damage, treating refractory cases, and managing comorbidities, offering unique value. Consequently, targeting IL-1 family members offers a complementary approach that could mitigate the inflammatory cascade in RA more comprehensively.
This review provides a detailed overview of recent investigations into the roles of IL-1 family cytokines in RA pathogenesis and critically examines the current status of drugs targeting these cytokines in clinical trials, thereby offering novel perspectives on the treatment of rheumatoid diseases.
2 Etiological factors of RA
2.1 Genetic factors
Genetic factors are important risk factors. Over 100 genetic loci have been identified to influence the development of RA through regulating immunity and other pathways (Padyukov, 2022). The genetic predisposition to RA is strongly linked to HLA-DRB1 alleles, particularly in anti-citrullinated protein antibody (ACPA)-positive subtypes. HLA-DRB1*04 and HLA-DRB1*10, the primary risk alleles for ACPA-positive RA, encode valine (Val) at position 11, forming the “shared epitope” (SE) (Raychaudhuri et al., 2012). These alleles confer a markedly elevated risk (odds ratio, OR = 3.88–10) by binding citrullinated peptides to activate T cells and initiate autoimmune cascades (Irigoyen et al., 2005; van der Woude et al., 2010). Smoking interacts synergistically with HLA genes, amplifying genetic risk in SE allele carriers (Stolt et al., 2003). Conversely, HLA-DRB1*13 (encoding glutamic acid [Glu] at position 71) demonstrates a protective effect against ACPA-positive RA (Terao et al., 2019). In contrast, ACPA-negative RA exhibits weaker genetic associations, primarily linked to the presence of leucine (Leu) or serine (Ser) at position 11 of HLA-DRB1 (Bossini-Castillo et al., 2015). Secondary HLA loci also contribute: HLA-B (Asp at position 9; OR = 2.12), HLA-DPB1 (Phe at position 9; OR = 1.40), and HLA-A (Asn at position 77; OR = 0.85) show modest effects, with HLA-A suggesting a protective trend (Regueiro et al., 2021).
What’s more, the genetic risk of RA shows heterogeneity across different populations. HLA-DRB1 alleles exhibit distinct risk profiles across ethnic groups. European ACPA-positive RA predominantly associates with HLA-DRB104 and HLA-DRB110 (OR = 3.88), whereas Japanese populations show HLA-DRB109 (Asp at position 11) as the primary risk allele, with independent contributions from HLA-DQ loci (Raychaudhuri et al., 2012; Okada et al., 2016). In Han Chinese, HLA-DQA1 (Asp at position 160) independently correlates with ACPA-positive RA, underscoring HLA-II molecules’ role in antigen presentation (Guo et al., 2019). The HLA-DQB103:02 allele reduces ACPA-positive RA risk in Malaysians (OR = 0.85), likely via T-cell receptor signaling modulation (Tan et al., 2021). Among African Americans, HLA-DRB104 demonstrates strong RA risk association (OR = 2.12), though its population frequency and epistatic interactions require validation (Danila et al., 2017). These findings collectively highlight HLA-driven RA pathogenesis through antigen presentation and immune dysregulation. Future cross-population multi-omics studies should clarify genetic heterogeneity’s implications for precision medicine.
The genetic architecture of RA extends beyond HLA genes to encompass over 150 non-HLA loci that orchestrate immune regulation, inflammatory signaling, and cellular activation pathways (Padyukov, 2022). Rong et al. demonstrated that specific IL1B polymorphisms differentially modulate RA risk in Han Chinese populations: the rs1143643 variant confers protection, whereas rs16944 elevates susceptibility (Rong et al., 2020). Zhu et al. further identified significant associations between IL-1A +4845G/T polymorphisms and RA susceptibility across populations (dominant model: p = 0.02; recessive model: p = 0.05; allelic model: p = 0.04), while IL-1B +3954C/T polymorphisms showed population-specific effects, demonstrating global significance in general populations (recessive model: p = 0.03; allelic model: p = 0.01) and enhanced association in Asian cohorts (recessive model: p = 0.007; allelic model: p = 0.002) (Braga et al., 2024). Beyond these loci, multiple genes critically contribute to RA pathogenesis. The PTPN22 rs2476601 variant (OR = 1.81, as reflected in pooled meta-analyses) in European populations augments autoreactive T-cell activation through impaired T-cell receptor signaling via lymphocyte tyrosine phosphatase (Begovich et al., 2004). CTLA4 mitigates autoimmune risk (OR = 0.88) by suppressing T-cell co-stimulation, particularly in ACPA-positive subtypes (Stahl et al., 2010). STAT4 rs7574865 potentiates pro-inflammatory cytokine signaling (IL-12/IL-23) with risk alleles (OR = 1.16) showing trans-ethnic associations (Okada et al., 2012). TNFAIP3 rs10499194 (OR = 1.33) drives inflammation through dysregulated NF-κB inhibition, strongly correlating with ACPA-positive RA (Eyre et al., 2012). PADI4 rs2240336 facilitates protein citrullination and subsequent ACPA production (OR = 0.88), exhibiting heightened significance in Asian populations (Eyre et al., 2012). The IL6R rs2228145 protective variant (OR = 0.93) modulates inflammatory cascades through IL-6 receptor regulation (Okada et al., 2014). Additional contributors include TRAF1-C5 (inflammatory signaling), CCR6 (T-cell chemotaxis), and IRF5 (interferon response), collectively underscoring the polygenic nature of RA susceptibility (Stahl et al., 2010).
2.2 Environmental factors
The pathogenesis of RA is closely related to genetic factors and involves the participation of various environmental factors. Among these environmental factors, smoking is central, significantly interacting with genetic susceptibility (especially HLA genes) (Wouters et al., 2022). Other factors, such as infections, hormones, and occupational exposures, contribute to disease onset through different mechanisms.
Tobacco smoking remains the most well-characterized environmental risk factor for RA, demonstrating particular association with APCA-positive subtypes. Cigarette smoking synergistically interacts with HLA-DRB1 SE alleles (*04, *10) to promote RA risk, with smokers carrying a single SE allele exhibiting an OR of approximately 3.8 (Padyukov et al., 2004). Tobacco-derived chemicals including nicotine and tar induce protein citrullination through enzymatic modification, thereby initiating ACPA production and subsequent autoimmune cascades. These compounds directly stimulate immune cell activation, enhancing TNF-α and IL-17 secretion while impairing Treg cell functionality, ultimately disrupting immune homeostasis. Occupational hazards, notably chronic silica dust and formaldehyde exposure, exert pro-inflammatory effects through alveolar macrophage activation (Klareskog et al., 2020). This process triggers IL-1β and TNF-α release, propagating systemic inflammatory responses that elevate RA susceptibility. Environmental pollutants such as PM2.5 and NOx compounds demonstrate dose-dependent RA risk enhancement, particularly in genetically predisposed populations (Zhang et al., 2023). These particulates primarily activate the NF-κB signaling pathway, stimulating synovial cells to secrete inflammatory mediators including IL-1β and TNF-α, which accelerate joint inflammation and potentiate osteodestructive processes.
Dietary modifications alter gut microbiota composition and dynamics, inducing dysbiosis that sustains systemic pro-inflammatory states (Lin et al., 2023). Vitamin D deficiency impairs immune tolerance through disrupted Treg cell regulation, elevating autoimmune disease susceptibility (Charoenngam, 2021). Elevated sodium chloride intake drives the polarization of pathogenic TH17 cells, particularly under conditions mimicking interstitial sodium concentrations, which amplifies IL-17A and GM-CSF production to exacerbate inflammatory cascades. Adipose tissue dysfunction elevates reactive oxygen species (ROS) generation while stimulating adipocyte-derived pro-inflammatory cytokines including TNF-α and IL-6, thereby accelerating systemic inflammation and RA progression (Braga et al., 2024).
Pathogen-induced RA manifests through gene-environment interactions, where PTPN22 or STAT4 genetic variants lower infection response thresholds to specific microbes (Jang et al., 2022). Periodontal pathogens like Porphyromonas gingivalis induce protein citrullination via peptidylarginine deiminase enzymes, generating autoantigens that cross-react with ACPA (Kobayashi and Bartold, 2023). While Epstein-Barr virus (EBV) exhibits epidemiological associations with RA, mechanistic evidence remains limited; proposed pathways include EBV nuclear antigen-1 molecular mimicry of citrullinated fibrinogen, which may activate autoreactive T-cell clones and synovial antibody production (Costenbader and Karlson, 2006).
2.3 Immune factors
The aberrant activation of the immune system plays a central role in the pathological progression of RA. Immune dysregulation is predominantly characterized by an imbalance in CD4+/CD8+ T-cell subset ratios and disruption of the cytokine network. In most patients, CD4+ T cells predominate over CD8+ T cells and secrete cytokines that influence the differentiation and activation of other immune cells, including CD8+ T and B cells. While CD8+ T cells contribute to the inflammatory milieu, their role is generally considered secondary to that of CD4+ T cells in driving immune dysregulation in RA (Jang et al., 2022). Within the synovial membrane in RA, CD4+ T cells serve as the primary drivers of inflammatory responses and disease relapse, while CD8+ T cells play a secondary role in disease pathogenesis. CD4+ T cells can produce IL-17, activate Th17 cells and inhibit Treg cells. CD8+ T cells secrete IFN-γ, CCL5 and other cytokines that enhance the inflammatory microenvironment (Jonsson et al., 2022). These cytokines act on CD14 mononuclear precursor cells of osteoclasts, influencing the differentiation or maturation of osteoclasts via the receptor activator of nuclear factor κB (RANK)/ receptor activator of nuclear factor κB ligand (RANKL)/osteoprotegerin (OPG) signaling pathway (Aletaha and Smolen, 2018). Under inflammatory conditions, myeloid lineage cells such as macrophages release substantial TNF-α upon stimulation by IL-17 and IFN-γ, a pivotal cytokine driving bone erosion through direct activation of osteoclast differentiation and concurrent suppression of osteoblast functionality (Wang and He, 2020). Clinically employed anti-TNF-α biologics like infliximab demonstrate efficacy in mitigating osteoclast-mediated joint destruction and attenuating inflammatory responses in RA. However, persistent impairment of osteoblast-driven bone formation remains unresolved (Malviya et al., 2009; Kwon et al., 2017). This therapeutic limitation underscores the necessity to explore complementary pathways. Notably, IL-1 family cytokines (IL-1α/IL-1β) exhibit potent osteoclastogenic effects via the RANK/RANKL/OPG signaling axis, operating independently of TNF-α-mediated pathways (Son et al., 2020). Mechanistically, IL-1 enhances RANKL expression in osteoblasts and stromal cells, amplifying osteoclast precursor differentiation through NF-κB and MAPK activation. IL-1Ra inhibits osteoclastogenesis (Izawa et al., 2014; Zou et al., 2021). While TNF-α inhibitors primarily address inflammatory osteolysis, IL-1 blockade concurrently enhances osteoanabolic processes and suppresses bone resorption. This multi-target strategy could overcome the limitations of monotherapy by simultaneously protecting against structural joint damage and facilitating bone repair in RA.
B cells play a pivotal role in the pathogenesis and progression of RA. Antigen presenting cells (APCs) can recognize and present immune complexes, activating CD4+ T helper cells and B cells via co-stimulation. Activated B cells secrete both inflammatory and regulatory cytokines, form ectopic germinal centers and activate T cells via antigen presentation and co-stimulatory molecules (Wu et al., 2021). In parallel, B cells produce various antibodies, including rheumatoid factor (RF) antibodies targeting the Fc portion of immunoglobulin G (IgG), and anti-citrullinated protein antibodies (ACPA) targeting citrullinated proteins (Xiao et al., 2021). Immune complexes formed through antigen-antibody binding accumulate in the synovial fluid, activating osteoclasts to cause bone damage. They also release cytokines such as tumor TNF-α and IL-8, inducing immune cell infiltration and exacerbating chronic inflammation in the joints (Maeda et al., 2022). The IL-1 family modulates B-cell activity and antibody production (Boraschi et al., 2018). Pro-inflammatory cytokines within the IL-1 family, such as IL-1 and IL-18, directly stimulate B-cell activation and promote the production of antibodies like RF and ACPA (Mateen et al., 2016). Current Phase III clinical trial results indicate that IL-1 blockade therapy significantly reduces ACPA and RF levels in patients’ serum and is positively correlated with the normalization of B-cell subset ratios, suggesting that targeting the IL-1 family may be a strategy for regulating abnormal B-cell activation and improving the course of RA (Winkler et al., 2021).
RA extends beyond localized joint pathology to manifest as a systemic autoimmune disorder. RA patients exhibit immune system dysregulation characterized by self-reactive tissue destruction. While hallmark rheumatic features including joint swelling, pain, and stiffness dominate clinical presentation, multi-organ involvement frequently develops. Cutaneous manifestations range from subcutaneous rheumatoid nodules to vasculitic lesions; pulmonary complications include progressive interstitial lung disease; cardiovascular risks escalate through accelerated atherosclerosis. Secondary Sjögren’s syndrome, characterized by xerophthalmia and xerostomia, develops in approximately 19.5% of RA patients (Alani et al., 2018). Additionally, hematologic disturbances (e.g., anemia of chronic disease) and peripheral neuropathies frequently accompany advanced disease (Conforti et al., 2021). This multi-system pathophysiology necessitates comprehensive management strategies targeting both synovial inflammation and extra-articular sequelae. Modern therapeutic paradigms combine immunomodulatory biologics/JAK inhibitors with proactive monitoring for cardiopulmonary and ocular complications to optimize long-term outcomes.
3 The role of IL-1 family cytokines in RA
The IL-1 family of cytokines may have overlapping and complementary effects in promoting or inhibiting the development of RA. Within the IL-1 family, IL-1α, IL-1β, IL-18, IL-33, IL-36α, IL-36β, and IL-36γ can promote the occurrence and development of RA, while IL-1Ra, IL-36Ra, IL-37, and IL-38 can suppress inflammatory responses. The specific mechanisms of each cytokine in the IL-1 family in RA are as follows:
3.1 IL-1
The IL-1 cytokine family features three principal subtypes: IL-1α, IL-1β, and the endogenous antagonist IL-1Ra. Membrane-bound IL-1α drives early localized inflammation through paracrine activation of RANKL expression in adjacent stromal cells, inducing osteoclastogenesis via RANK/RANKL/OPG-independent mechanisms (Kim et al., 2009). In contrast, caspase-1-activated IL-1β exerts systemic effects by disrupting RANKL/OPG equilibrium through dual pathways - directly upregulating osteoclast RANKL production while suppressing osteoblast OPG secretion. IL-1Ra competitively blocks IL-1 receptor binding, effectively inhibiting NF-κB and MAPK signaling cascades. Chronic TLR-mediated NF-κB activation in RA synovium induces NLRP3 and pro-IL-1β co-expression. Potassium efflux, ROS overproduction, or lysosomal damage triggers NLRP3 inflammasome assembly, enabling caspase-1-mediated proteolytic cleavage and extracellular release of mature IL-1β. This bioactive cytokine binds IL-1R to amplify NF-κB/MAPK signaling in osteoclast precursors, driving NFATc1-dependent differentiation and bone resorption (Kong et al., 1999; Roux and Orcel, 2000). IL-1β simultaneously reprograms Tregs into osteoclastogenic O-Tregs through RANKL induction, effectively converting immunosuppressive cells into bone-destructive effectors (Nasi et al., 2017; Levescot et al., 2021). IL-1α and IL-1β can stimulate the proliferation of synovial fibroblasts and the release of matrix metalloproteinases (MMPs), leading to the breakdown of collagen in the cartilage matrix (Reboul et al., 1996).
IL-1β triggers inflammatory responses in fibroblast-like synoviocytes, including MH7A cells, while upregulating GATA4 expression. GATA4 directly binds to gene promoters to enhance transcriptional activation of angiogenic factors VEGFA and VEGFC (Jia et al., 2018). Through activation of the VEGFR2/PI3K/AKT pathway, VEGF stimulates synovial fibroblast proliferation and invasion, exemplified by MH7A cells, ultimately driving pathological synovial tissue thickening. Newly formed vasculature combines with hyperplastic synovium to generate an erosive pannus that progressively degrades articular cartilage and subchondral bone, culminating in irreversible joint damage (Chen et al., 2024). Furthermore, IL-1α and IL-1β can also induce articular chondrocytes to produce nitric oxide (NO) and prostaglandin E2 (PGE2), inhibiting the synthesis of matrix proteins and contributing to cartilage destruction (Pelletier et al., 1996). Additionally, IL-1β, through its ability to activate signaling pathways such as MAPK upon binding to cellular receptors, can trigger the release of other cytokines and drive the polarization of Th17 cells. This, in turn, promotes the release of IL-17 from CD4+ T cells, ultimately enhancing the inflammatory response (Iwahashi et al., 2004).
3.2 IL-18
IL-18, a pro-inflammatory cytokine of the IL-1 superfamily, predominantly exists as an inactive precursor (pro-IL-18) in macrophages, dendritic cells, and epithelial lineages. Canonical activation requires caspase-1-mediated proteolytic cleavage within the NLRP3 inflammasome, enabling secretion of mature IL-18 via gasdermin D (GSDMD) pores. During Gram-negative bacterial infections, non-immune cells bypass this pathway by directly activating caspase-4/5 to process pro-IL-18, expanding its functional repertoire in innate immunity (Landy et al., 2024).
Upon binding to its heterodimeric receptor (IL-18Rα/β), IL-18 triggers MyD88-dependent signaling cascades that activate NF-κB and MAPK pathways. These pathways drive transcriptional upregulation of IL-6, IL-8, and CXCL10 while promoting Th17 differentiation and IL-17 production (Rex et al., 2020). Synergy with IL-12 amplifies IFN-γ secretion in Th1 and NK cells, establishing a feedforward loop that sustains synovial inflammation through TNF-α, IL-1β, and MMP-mediated cartilage degradation (Baggio et al., 2023).
In RA, IL-18 exacerbates bone erosion through dual mechanisms. First, it directly modulates osteoclastogenesis by skewing the RANKL/OPG balance toward bone resorption (Zhang et al., 2013). Second, IL-18 synergizes with TNF-α and IL-6 to activate fibroblast-like synoviocytes (FLS), which secrete matrix metalloproteinases (MMPs) that degrade cartilage and subchondral bone (Puren et al., 1998; Morel et al., 2001). T cells in RA synovium further amplify osteoclast formation by increasing RANKL production under IL-18 stimulation. Preliminary evidence indicates that IL-18 bypasses RANKL dependence in certain contexts, directly inducing osteoclast differentiation and bone destruction (Kim et al., 2015). The cytokine also recruits inflammatory cells to joints via CCL2 upregulation, creating a self-perpetuating cycle of inflammation. Concurrently, IL-18 enhances Th17-mediated IL-17 and IL-22 release, accelerating synovitis and osteoclast activation (Vasilev et al., 2021).
3.3 IL-33
IL-33 has dual pro-inflammatory and anti-inflammatory regulatory roles in RA (Chen et al., 2019). In the early acute injury phase, it exerts protective effects by activating Tregs and type II immune responses, while in the chronic phase, pro-inflammatory signals dominate and exacerbate pathological damage.
During the acute phase, IL-33 primarily exhibits anti-inflammatory effects. Early low-dose IL-33 intervention can inhibit the progression of collagen-induced arthritis models. IL-33 enhances the proliferation and immune suppressive function of Treg cells via the STAT6 pathway, inhibits Th17 differentiation and osteoclast generation, reduces the RANKL/OPG ratio, and participates in the restoration of immune tolerance mediated by Breg cells (Biton et al., 2016). IL-33 can also induce ILC2 cells to secrete IL-4/IL-13, promoting M2 macrophage polarization to inhibit NF-κB activity (Lott et al., 2015).
In the chronic phase, IL-33 binds dimers of ST2 receptor and Interleukin 1 receptor-associated protein (IL-1RacP), which recruits IL-1RacP, MyD88, Interleukin 1 receptor associated kinase (IRAK) 1, IRAK4, and TNF receptor associated factor (TRAF) 6. This complex activates NF-κB and MAPK signaling pathways, leading to the phosphorylation and activation of extracellular signal-regulated kinase (ERK) 1/2, c-Jun N-terminal kinase (JNK), p38, and phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB/AKT) signaling modules. Consequently, the expression of pro-inflammatory cytokines is enhanced (Cayrol and Girard, 2009; Yuan et al., 2011). In Th2 cells, IL-33 promotes the transcription of IL-4, IL-5, and IL-13 (Schmitz et al., 2005; Verri et al., 2010). IL-33 upregulates the expression of CXC motif chemokine ligand 1(CXCL1), C-C Motif Chemokine Ligand 3(CCL3), TNF-α, and IL-1β in macrophages, as well as IL-6, IL-13, IL-1β, GM-CSF, Monocyte chemoattractant protein-1 (MCP-1), and Macrophage inflammatory protein-1alpha (MIP-1α) in mast cells (Xu et al., 2008; Yuan et al., 2011). It also increases the expression of IL-6, IL-8, MCP-1, MMP-1, and MMP-3 in synovial fibroblasts, thereby promoting synovial inflammation and cartilage destruction (Kunisch et al., 2012).
Therefore, targeting the IL-33/ST2 axis in therapy requires a precise balance of its dual functions, such as developing sST2 antagonists to block pro-inflammatory signals while retaining the activation of membrane-bound ST2 on Tregs, providing a new direction for personalized treatment of RA.
3.4 IL-36
IL-36 has multiple subtypes, including IL-36α, IL-36β, and IL-36γ, which bind to the receptor comprising specific chain IL-36R (IL-1Rrp2) and co-receptor IL-1R accessory protein (IL-1RAcP). IL-36 can activate a cascade of downstream effectors, including IκB, NF-κB, PI3K/AKT, and ERK, leading to the regulation of inflammatory cytokine, chemokine, and growth factor production through transcription factors mTORC and Wnt5a in the cell nucleus (Zhou et al., 2018). This promotes neutrophil infiltration, dendritic cell activation, and the polarization of Th1 cells and IL-17-producing T cells (αβ T cells and γδ T cells), ultimately exacerbating the development of RA inflammation (Vigne et al., 2012; Gabay and Towne, 2015). Furthermore, IL-36 stimulates fibroblasts to produce pro-inflammatory factors (Boutet et al., 2016), and targets autophagy to regulate the proliferation, migration, and invasion of RA synovial cells (Hao and Liu, 2021). In contrast, IL-36RA antagonizes IL-36α, IL-36β, and IL-36γ by binding to IL-1Rrp2 (Towne et al., 2011; Vigne et al., 2011). IL-36RA blocks IL-1RAcP recruitment and reduces the downstream activation of NF-κB or MAPK pathways (Towne et al., 2004).
3.5 IL-37
The binding of IL-37 to its receptors, IL-1R8 and IL-18Rα, triggers a cascade of signaling pathways, including PI3K/AKT, ERK, JNK, and p38. This, in turn, modulates the activation of Notch1 and Nuclear factor erythroid 2-related factor 2 (NRF2) through signal transducer and activator of transcription 3 (STAT3) and single immunoglobulin interleukin-1-related receptor (SIGIRR), thereby conferring anti-inflammatory properties (Zhou et al., 2020). Furthermore, IL-37 has been shown to attenuate TNF-α-induced apoptosis of synovial fibroblasts by suppressing the NF-κB/Gasdermin-D (GSDMD) signaling axis (Ren et al., 2023).
The anti-inflammatory effects of IL-37 are attributed to its ability to modulate immune cell function. IL-37 can suppress the expression of inducible nitric oxide synthase (iNOS), IL-6, MCP-1 and reactive oxygen species (ROS) in macrophages, improve the expression of IL-10, glutathione peroxidase 4 (GPX4), NRF2, and CD206 (Qi et al., 2019; Zhao et al., 2022). Additionally, IL-37 can impede dendritic cell maturation by down-regulating the expression of MHC class II (MHCII), CD40, CD86, and CD80 (Liu et al., 2019). Notably, IL-37 can also inhibit IgG production in B cells, eosinophil infiltration, Th17 and Tfh cell differentiation and proliferation, while fostering Th1 and Treg cell differentiation (Liu et al., 2020; Li et al., 2021).
3.6 IL-38
IL-38 shares receptors with IL-36 and antagonizing IL-36. Binding to IL-36R or IL-1RAPL1 receptors, IL-38 triggers a signaling cascade involving Toll and interleukin-1 receptor (TIR), RohA, ERK, JNK, and p38, which in turn activates transcription factors AP-1 and the silent information regulator sirtuin 1 (SIRT1), ultimately resulting in anti-inflammatory effects in RA (Boutet et al., 2017). Notably, low levels of IL-38 can form an IL-38/36R axis, either by binding to IL-36R and blocking IL-1RAcP recruitment or by recruiting inhibitory receptors to prevent MyD88 recruitment, thereby inhibiting NF-κB or MAPK and eliciting anti-inflammatory effects (Mora et al., 2016). The IL-38/IL-1RAPL1 axis, comprising IL-38 and IL-1RAPL1, can exert anti-inflammatory or pro-inflammatory effects depending on the length of IL-38 acting on JNK/AP-1 (Mora et al., 2016; Xie et al., 2019). Additionally, IL-33 can modulate the SIRT1/HIF-1α signaling pathway to suppress inflammatory responses (Pei et al., 2020).
4 The receptor of IL-1 family
The IL-1 receptor family comprises IL-1R1 (IL-1RI), IL-1R2 (IL-1RII), IL-1R3 (IL-1RAcP), IL-1R4 (the suppression of tumorigenicity 2 receptor, ST2), IL-1R5 (IL-18Rα), IL-1R6 (IL-1Rrp2, IL-36R), IL-1R7 (IL-18Rβ), IL-1R8 (TIR8, also known as SIGIRR), IL-1R9 (TIGIRR-2), and IL-1R10 (TIGIRR-1) (Kim and Lee, 2024). Although different cytokines bind to distinct receptors, these receptors can activate the TIR pathway upon stimulation (Dinarello, 2018). Following TIR stimulation, MyD88 recruits IRAK to TIRs through the interaction of their death domains. IRAK is activated by phosphorylation and then associates with TRAF6, leading to the activation of two distinct signaling pathways, and finally to the activation of JNK and NF-κB (Zhou et al., 2022). This cascade of events culminates in the upregulation of IL-1, IL-6, IL-8, IL-12, TNF, RANKL, MMP, iNOS, VEGF, PGHS-2, and adhesion molecules, exacerbating joint inflammation (Narayanan and Park, 2015). Details are shown in Figure 1 and Table 1.
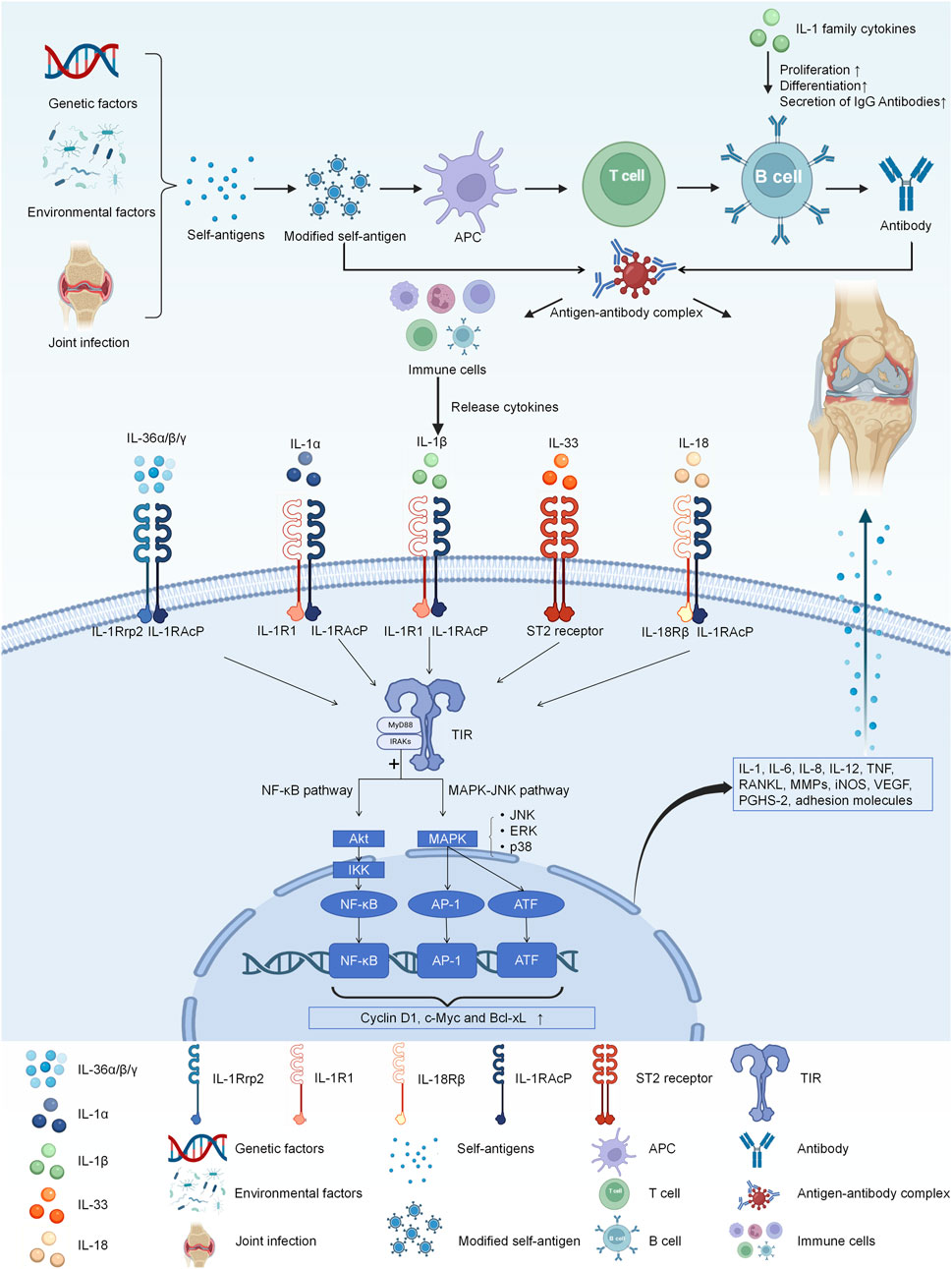
Figure 1. The IL-1 receptor family comprises IL-1R1, IL-1R2, IL-1RAcP, ST2, IL-18Rα, IL-1Rrp2, IL-18Rβ. Different cytokines bind to distinct receptors, and they all activate the TIR pathway. And then, MyD88 recruits IRAK to TIRs through the interaction of their death domains. IRAK is activated by phosphorylation and then associates with TRAF6, leading to the activation of two distinct signaling pathways, and finally to the activation of JNK and NF-κB. Finally they eventually have the influence on the expression of IL-1, IL-6, IL-8, IL-12, TNF, RANKL, MMP, iNOS, VEGF, PGHS-2, and adhesion molecules, exacerbating joint inflammation.
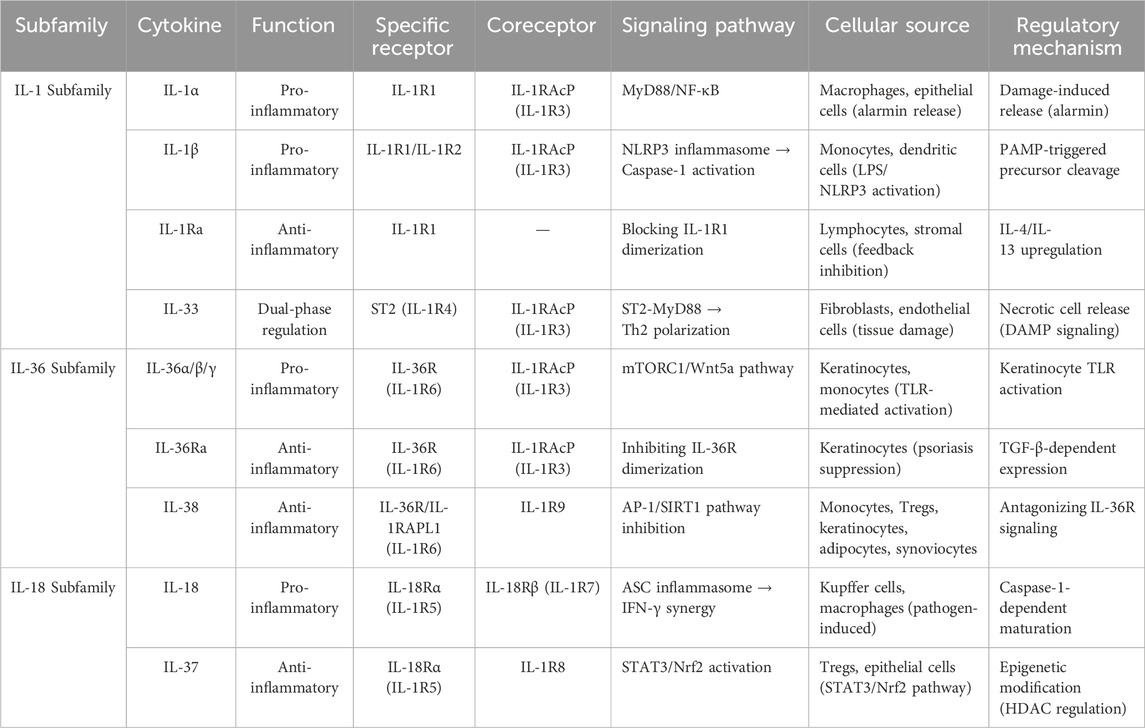
Table 1. Classification and characteristics of IL-1 family cytokines.
5 Clinical pre-study of monoclonal antibody targeting IL-1 family cytokines for the treatment of RA
There are numerous therapeutic interventions that target the IL-1 family, with a significant number having advanced into clinical research stages. The details are shown in Tables 2, 3.
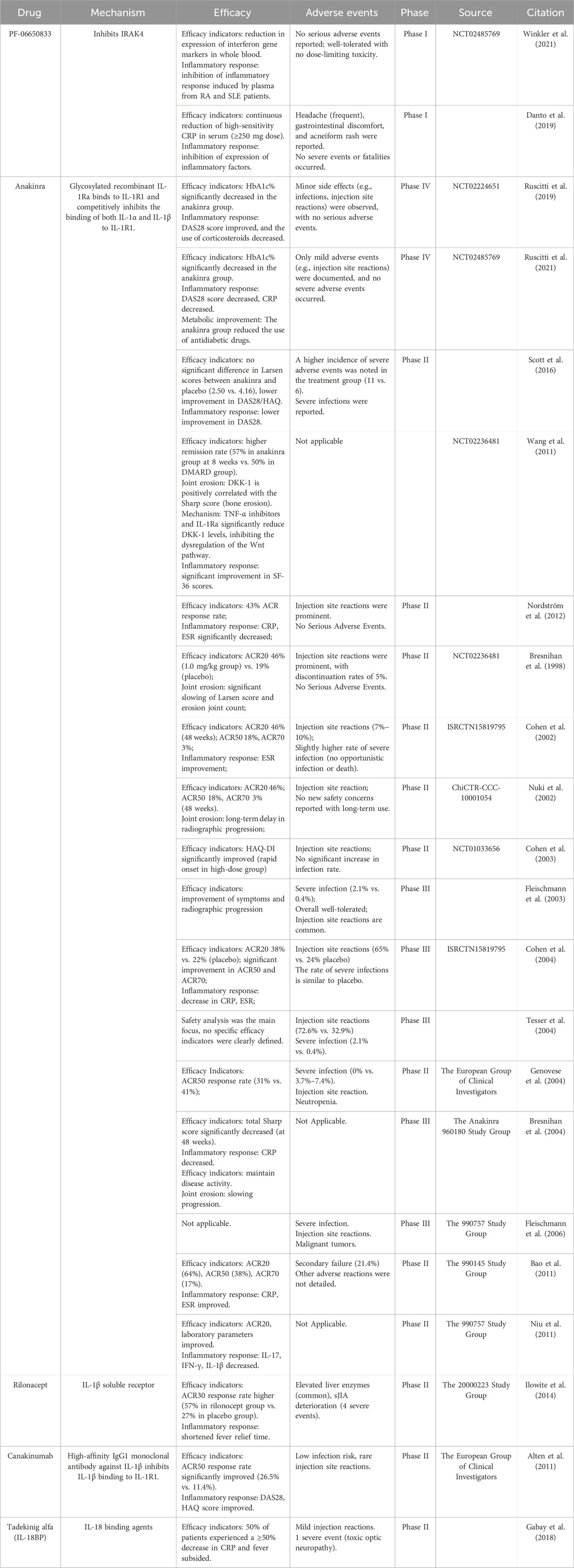
Table 2. Clinical trials of biological agents targeting IL-1 family cytokines.
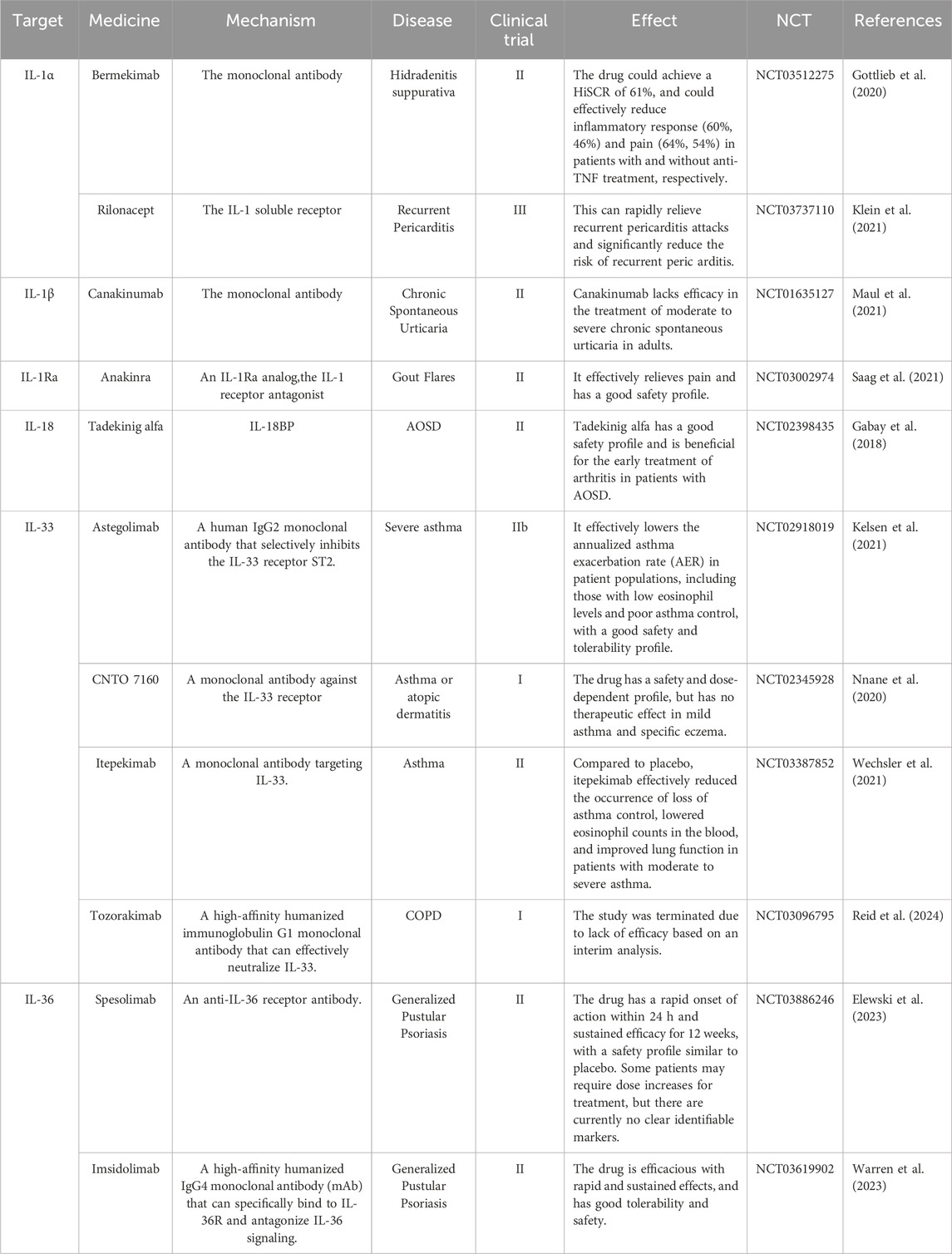
Table 3. Targeting IL-1 family cytokines with monoclonal antibodies for the treatment of autoimmune diseases in clinical trials.
5.1 IL-1
Early administration of specific anti-IL-1β antibodies blocks IL-1β, thus inhibiting the expression of RANKL on regulatory T cells and suppressing osteoclast differentiation. This improves joint swelling and bone erosion in arthritis (Levescot et al., 2021). siRNA that binds to IL-1βsilences pro-inflammatory genes, which alleviate ankle joint swelling, bone erosion and cartilage destruction (Song et al., 2019).
hIL-1RA-Fc is an IL-1 receptor antagonist (IL-1Ra) analog that inhibits Th17 cell differentiation through blocking the STAT3 signaling pathway and induces Treg cell differentiation through the STAT5 signaling pathway, exerting anti-rheumatic arthritis effects. hIL-1RA-Fc can reduce the expression of IL-17, TNF-α, RANKL and VEGF, increase forkhead box P3 (Foxp3) gene expression, and thus suppress osteoclast genesis and angiogenesis (Lee S. Y. et al., 2016).
In a collagen-induced RA model, the soluble IL-1 receptor 2 receptor blocks IL-1α and IL-1β and significantly inhibits IL-1 signaling transduction in macrophages (Shimizu et al., 2015). Moreover, IL-1R2 inhibits Th17 cell activation by blocking IL-1β signaling transduction (Kim et al., 2021).
IgG26AW has high affinity, high neutralization ability and occupies a new binding epitope that binds both to IL-1RI and IL-1RAcP, which has been validated in tumors and may be a potential drug for RA in the future (Kuo et al., 2021).
5.2 IL-18
Currently, in research treating RA by targeting IL-18, inhibitors of IL-18 such as IL-18 binding protein (IL-18BP) are a major focus. IL-18BP can correct the imbalance of Th17/Treg cells in peripheral blood mononuclear cells from RA patients and reduce osteoclast genesis induced by IL-17 (Min et al., 2021). Consequently, IL-18BP promotes apoptosis of fibroblast-like synoviocytes while reducing apoptosis of chondrocytes, which is beneficial for the treatment of RA (Min et al., 2023).
Soluble IL-18 receptor beta (sIL-18Rβ) modulates Treg cells and Th17 cells similarly to IL-18BP, which suppresses collagen-induced arthritis (Veenbergen et al., 2010). This potential reduction in IL-18 expression has been associated with alleviation of RA symptoms in experimental models, though further investigation is needed to fully establish this pathway (Guo et al., 2022).
5.3 IL-33
Treatment with IL-33 neutralizing antibodies decreases levels of IFN-γ, IL-6, IL-12, IL-33, and TNF-α. This decrease significantly reduces the severity of joint damage (Li et al., 2020). The IL-33 specific receptor ST2 is also a therapeutic target. Blocking the ST2 receptor while raising IL-37 levels cooperatively dampens inflammation. This cooperation involves lowering expression of IL-6, TNF-α, toll-like receptors, and MMPs. It also inhibits M1 phenotypic polarization. Use of ST2 inhibitors to oppose IL-33 suppresses the stimulation of synoviocytes (Rai et al., 2022). It also decreases mRNA expression of RANKL and IP-10. Concurrently, ST2 inhibition elevates proinflammatory factor and MMP levels. It further boosts NF-κB activity, thereby inhibiting bone resorption (Lee E. J. et al., 2016).
5.4 IL-36 and IL-38
In an STIA mouse model, injection of IL-38 encoding was able to reduce the production of proinflammatory factors (including IL-17, IL-23, IL-22, TNF-α) by macrophages and synovial fibroblasts, lowering the inflammatory response in arthritic mice (Boutet et al., 2017). In rat models, overexpression of IL-38 and injection of IL-36 was able to target autophagy, regulating the proliferation, migration and invasion of synovial cells in RA (Hao and Liu, 2021).
However, some studies have also found that prophylactic treatment of mice with an IL-36R blocking antibody did not change clinical onset or disease patterns. Additionally, blocking IL-36 signal transduction did not alter the histopathological features of arthritis induced by TNF (Derer et al., 2014).
5.5 IL-37
IL-37 has intrinsic anti-inflammatory effects. IL-37 alleviates RA by inhibiting the production of IL-17 and IL-17-induced cytokines, and restricting the proliferation of Th17 cells (Ye et al., 2015). In an arthritis mouse model, low doses of recombinant IL-37 were able to inhibit 51.7% of arthritic inflammation, promote IL-1R expression, and reduce the synovial levels of IL-1β, IL-6, TNF-α, CXCL1, CXCR3, macrophage inflammatory protein 1-alpha (MIP-1α), IL-1α, and myeloperoxidase (MPO), thereby decreasing the recruitment of neutrophils to the joints (Cavalli et al., 2016).
5.6 Cytokine redundancy and therapeutic strategies targeting IL-1RAcP
Cytokine redundancy refers to the phenomenon where distinct cytokines bind to identical or structurally similar receptors, activating overlapping signaling pathways to elicit analogous biological functions. This redundancy enhances the adaptability and robustness of the immune system during pathogen invasion or tissue damage, ensuring core immune responses are maintained even if one cytokine pathway is blocked. Within the IL-1 family, proinflammatory cytokines (e.g., IL-1α, IL-1β, IL-18, IL-33, IL-36α/β/γ) rely on IL-1RAcP as a shared co-receptor to form a signal-transducing complex, thereby mediating common downstream pathways.
Studies in atherosclerosis highlight the functional overlap between IL-1α and IL-1β, which jointly regulate extracellular matrix-remodeling enzymes. Single-target inhibition of either cytokine has shown limited efficacy in this context (Beltrami-Moreira et al., 2016). Similarly, cytokine redundancy complicates therapeutic outcomes in sepsis, where IL-1 and IL-6 both activate STAT3, and TNF-α synergizes with IL-1 via the NF-κB pathway (Cooney and Yumet, 2002). These observations suggest that monotherapies targeting individual cytokines may yield suboptimal results in diseases like RA, necessitating strategies to disrupt shared receptor signaling.
Addressing this challenge, Fields et al. demonstrated the feasibility of targeting IL-1RAcP to broadly block signaling by all IL-1 family cytokines (Figure 2) (Fields et al., 2024). Antibodies CAN10 and 3G5 specifically bind distinct epitopes (the c2d2 loop of the D2 domain and the D3 domain) on IL-1RAcP, effectively inhibiting cytokines dependent on this co-receptor, including IL-1α, IL-1β, IL-33, and IL-36α/β/γ. In acute peritonitis models, IL-1RAcP-targeted antibodies markedly reduced inflammatory cell infiltration and proinflammatory mediators (e.g., IL-6, G-CSF) compared to IL-1Ra, a natural antagonist of IL-1 signaling. Notably, CAN10 exhibited >10-fold higher inhibitory potency than IL-1Ra in suppressing IL-1α/β pathways. These findings underscore the therapeutic potential of targeting IL-1RAcP for IL-1-driven inflammatory disorders, particularly RA.
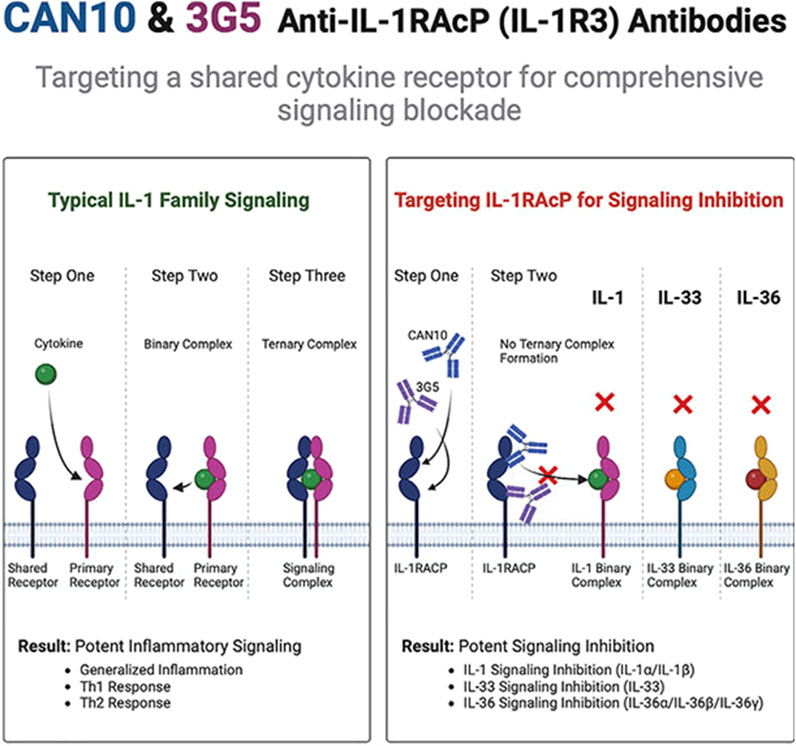
Figure 2. CAN10 and 3G5, two anti-IL-1RAcP antibodies, target distinct epitopes on this shared receptor and potently block IL-1α, IL-1β, IL-33, IL-36α, IL-36β, and IL-36γ signaling (Fields et al., 2024).
6 Clinical efficacy and adverse effects of IL-1 inhibitors
6.1 Current clinical trial results of IL-1 inhibitors
As previously established, IL-1 family cytokines play a central role in inflammatory responses and joint destruction in RA by activating synovial cells, promoting osteoclast activity, and inhibiting cartilage repair. Based on these mechanisms, multiple IL-1 inhibitors have been developed and tested in clinical trials. Table 1 summarizes the current biologics targeting IL-1 family cytokines. Figure 3 illustrates the therapeutic strategies for RA treatment using these agents.
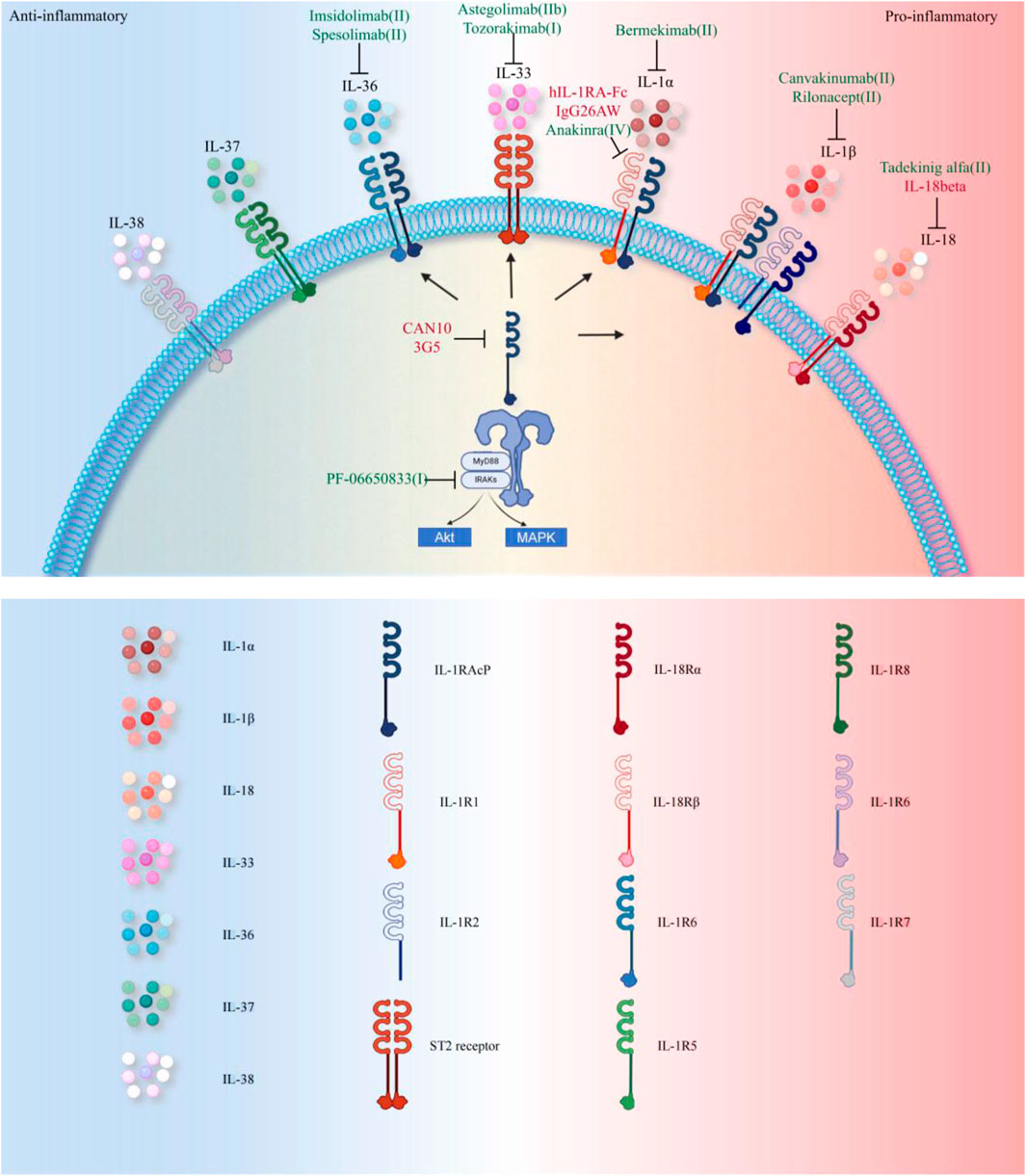
Figure 3. Therapeutic Strategies Targeting the IL-1 Family in RA. Currently, FDA-approved therapies primarily target the IL-1 pathway (annotated in green in the figure, with specific reference to RA clinical trial phases). Approved agents include: Anakinra (IL-1R1 antagonist), Canakinumab (IL-1β specific monoclonal antibody), Rilonacept (IL-1β trapping fusion protein), and Spesolimab (IL-36 receptor targeting monoclonal antibody approved for psoriasis). Investigational drugs in clinical trials address broader inflammatory targets: Bermekimab (IL-1α neutralization), PF-06650833 (IRAK4 inhibitor blocking downstream signaling), Tadekinig alfa (Th17 balance modulation via IL-18BP), Astegolimab and Tozorakimab (targeting IL-33 receptor ST2 and IL-33, respectively), Imsidolimab (IL-36R antagonism, analogous to Spesolimab). Preclinical studies (highlighted in red in the figure) explore novel mechanisms: hIL-1RA-Fc fusion protein enhances Treg/Th17 balance; CAN10 and 3G5 inhibit IL-1RAcP, a shared receptor subunit for IL-1 family cytokines; IL-38 gene therapy suppresses inflammation by inhibiting the AP-1/SIRT1 pathway; Recombinant IL-37 activates the Nrf2-mediated anti-inflammatory cascade; IgG26AW blocks IL-1α activity.
To date, the IL-1 inhibitors approved for RA treatment include PF-06650833, Anakinra, Rilonacept, and Canakinumab. Among these, Anakinra—a recombinant IL-1 receptor antagonist—remains the first and only IL-1 inhibitor specifically approved for RA. Clinical trials since 1998 have yielded mixed results regarding its efficacy and safety. For instance, Scott et al. reported that short-term Anakinra therapy (12 months) only improved ACR20 and ACR50 response rates compared to placebo, with no significant differences in other efficacy endpoints (Scott et al., 2016). However, multiple studies demonstrated that Anakinra reduces DAS28 scores and lowers CRP/ESR levels (Nordström et al., 2012; Ruscitti et al., 2019; Ruscitti et al., 2021). Long-term use of Anakinra also slows joint erosion, as evidenced by decreased Sharp scores, though it shows no significant improvement in Larsen scores (Bresnihan et al., 2004). These findings collectively suggest that Anakinra exhibits moderate anti-inflammatory effects with long-term joint protection, making it a viable option for patients with inadequate responses to TNF-α inhibitors or those requiring infection-safe therapies (Scott et al., 2016).
The primary limitation of Anakinra is its high injection frequency (100 mg/day), leading to frequent injection-site reactions and poor patient compliance. Nevertheless, its infection risk remains low, with no reports of severe opportunistic infections, offering a safety advantage over TNF-α inhibitors (Fleischmann et al., 2006). Future directions include developing sustained-release formulations to reduce dosing frequency and identifying biomarkers (e.g., IL-1β levels or genetic polymorphisms) to predict treatment responses and optimize patient selection (Akash et al., 2013; Hong et al., 2024). Prolonged IL-1 blockade disrupts innate immune defenses, increasing bacterial infection susceptibility through impaired neutrophil homeostasis. Clinical surveillance data from long-term anakinra therapy demonstrate cancer incidence rates comparable to age-matched general population estimates, with no causative association identified between anakinra exposure and malignancy development (Fleischmann et al., 2006).
Canakinumab, a monoclonal antibody targeting IL-1β, has shown superior efficacy in early trials, with an ACR50 response rate of 26.5% and rapid improvements in DAS28 and HAQ scores (Alten et al., 2011). Its dosing regimen (every 4–8 weeks) and low incidence of injection-site reactions make it suitable for long-term maintenance therapy. However, clinical data on Canakinumab in RA remain limited, warranting further investigation. Other agents like Rilonacept and Tadekinig alfa lack robust RA-specific data but show potential in systemic inflammatory diseases and animal models, necessitating additional human trials.
Long-term administration of IL-1 inhibitors in RA clinical trials primarily manifests as injection site reactions and elevated infection risks. Emerging evidence from other immune-mediated conditions suggests additional class-related adverse effects that warrant consideration in RA treatment paradigms. Hepatotoxicity represents a notable concern, with 3%–8% of anakinra recipients developing transient liver enzyme elevations that typically resolve spontaneously (Ruperto et al., 2012). The lower incidence of hepatic dysfunction observed with canakinumab and rilonacept may reflect reduced metabolic stress from their prolonged half-lives (Grevich and Shenoi, 2017). Hematologic monitoring proves crucial as 2%–5% of anakinra-treated patients develop reversible leukopenia, potentially mediated by bone marrow suppression mechanisms (Minoia et al., 2018). While rare, macrophage activation syndrome (MAS) requires particular vigilance given its association with all three IL-1 blockers (Ravelli et al., 2016). This life-threatening complication, though inherent to AOSD pathophysiology, may be precipitated by therapeutic immune modulation (Junge et al., 2017). Serious infections including sepsis and pneumonia necessitate immediate drug cessation and antimicrobial therapy (Shimizu et al., 2013). Immunogenicity profiles vary significantly among agents. Anakinra’s recombinant structure predisposes to neutralizing antibody formation with consequent efficacy loss (Wikén et al., 2018), whereas the fully human canakinumab demonstrates no such immunogenic propensity (Pascual et al., 2005). Current literature remains inconclusive regarding long-term immunosuppression risks such as opportunistic infections or malignancy development. Nevertheless, cumulative biological effects mandate sustained surveillance to ensure therapeutic safety.
6.2 Comparative analysis of IL-1 inhibitors with other biologics
To evaluate the efficacy and safety of IL-1 receptor antagonists (e.g., Anakinra) (Vasconcelos et al., 2020; Kazmi et al., 2024), TNF-α inhibitors (Hu et al., 2023), and JAK inhibitors (Ho Lee and Gyu Song, 2020), we conducted a meta-analysis comparing outcomes such as ACR20/50/70 response rates and adverse drug reactions (ADRs, serious ADRs, and treatment discontinuation due to adverse events). Key findings are summarized in Figures 4–8.
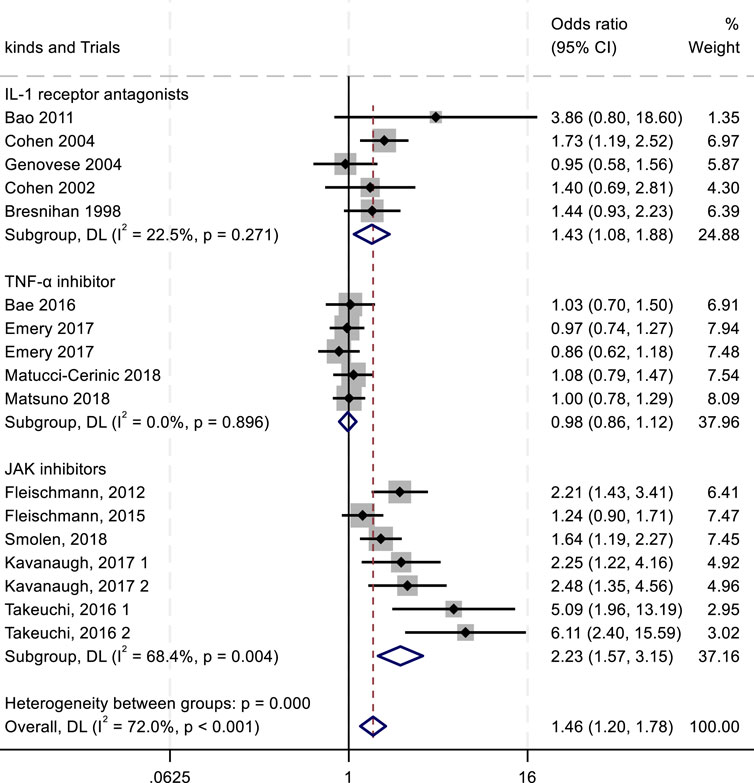
Figure 4. Results of a meta-analysis of ACR20 after 24 weeks of drug therapy.

Figure 5. Results of a meta-analysis of ACR50 after 24 weeks of drug therapy.
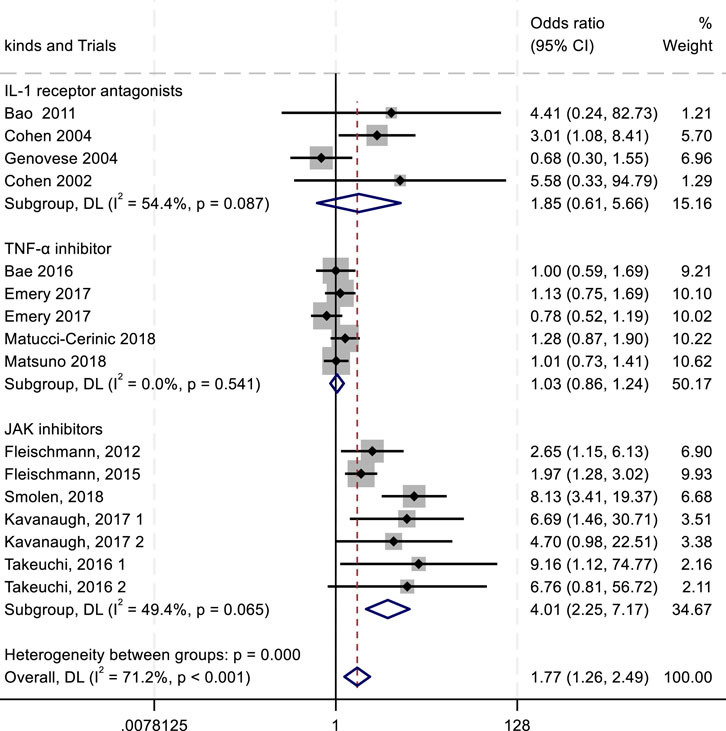
Figure 6. Results of a meta-analysis of ACR70 after 24 weeks of drug therapy.
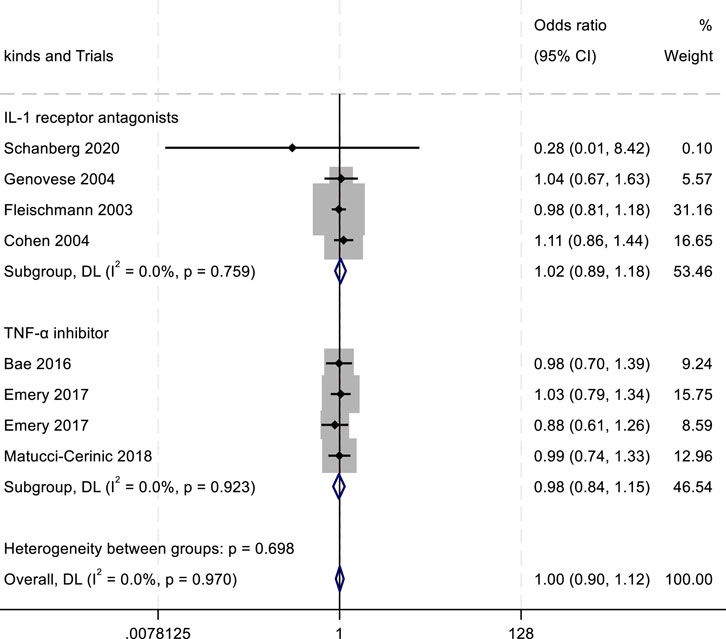
Figure 7. Results of a meta-analysis of the outcomes of adverse drug reactions after drug therapy.
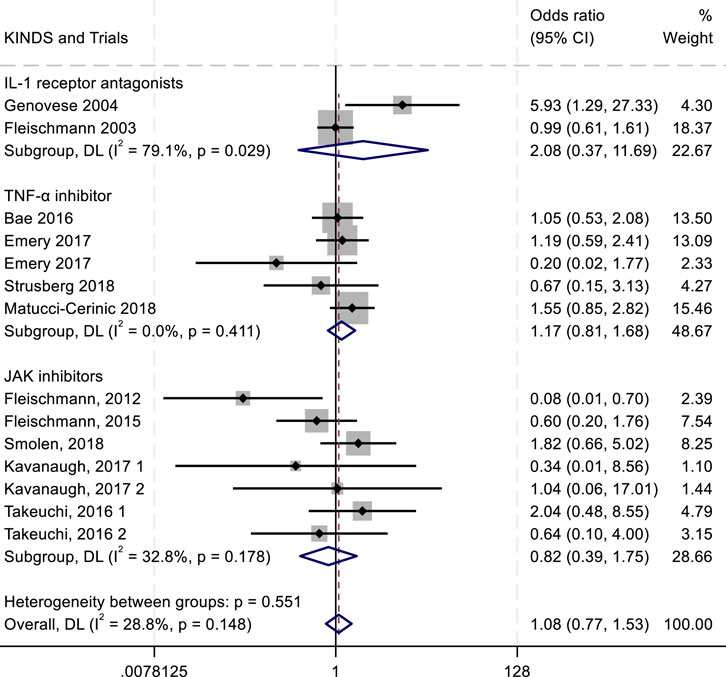
Figure 8. Results of a meta-analysis of the outcomes of serious adverse drug reactions after drug therapy.
The ACR criteria response rates were utilized for efficacy evaluation. Overall, IL-1 inhibitors, TNF-α inhibitors, and JAK inhibitors demonstrated therapeutic benefits in reducing RA activity, showing associations with higher ACR20, ACR50, and ACR70 response rates. Among all treatment groups, JAK inhibitors exhibited the highest ACR20 response rate (RR 2.23; 95% CI 1.57–3.15; P < 0.004), with moderate heterogeneity (I2 = 68.4%; P = 0.004). IL-1 inhibitors followed, showing an ACR20 response rate (RR 1.43; 95% CI 1.08–1.88; P = 0.271) with low heterogeneity (I2 = 22.5%; P = 0.271). TNF-α inhibitors displayed the lowest ACR20 response rate (RR 0.98; 95% CI 0.86–1.12; P = 0.896) and minimal heterogeneity (I2 = 0.00%; P = 0.896) (Figure 4). In the analysis of ACR50 response rates, the RR values were 2.75 for IL-1 inhibitors, 2.45 for TNF-α inhibitors, and 1.08 for JAK inhibitors (Figure 5). Similarly, for ACR70 response rates, the RR values were 1.85 for IL-1 inhibitors, 1.03 for TNF-α inhibitors, and 4.01 for JAK inhibitors (Figure 6). These patterns aligned with the ACR20 response rate trends. The findings suggest that JAK inhibitors demonstrate superior therapeutic efficacy, followed by IL-1 inhibitors, with TNF-α inhibitors showing the least effectiveness.
In the assessment of adverse events, the outcomes of adverse drug reactions (ADRs), serious adverse drug reactions (SADRs), and withdrawal due to adverse events were analyzed. For ADRs, the highest incidence was observed with IL-1 inhibitors (RR = 1.02; 95% confidence interval CI, 0.89–1.18; P = 0.759; I2 = 0.00%), followed by TNF-α antagonists (RR = 0.98; 95% CI, 0.84–1.15; P = 0.923; I2 = 0.00%) (Figure 7). Both drug classes showed numerically higher ADR rates compared with the placebo group. For SADRs, IL-1 inhibitors demonstrated the highest incidence (RR = 2.08; 95% CI, 0.37–11.69; P = 0.029; I2 = 79.1%), with substantial heterogeneity across studies. In contrast, TNF-α inhibitors (RR = 1.17; 95% CI, 0.81–1.68; P = 0.411; I2 = 0.00%) and JAK inhibitors (RR = 0.82; 95% CI, 0.39–1.75; P = 0.178; I2 = 32.8%) exhibited lower SADR rates (Figure 8). The withdrawal outcomes followed a similar trend. IL-1 inhibitors again showed the highest withdrawal rates (RR = 1.26; 95% CI, 0.80–1.98; P = 0.068; I2 = 54.2%), while TNF-α antagonists (RR = 0.77; 95% CI, 0.50–1.19; P = 0.654; I2 = 0.00%) and JAK inhibitors (RR = 0.87; 95% CI, 0.42–1.79; P = 0.174; I2 = 33.2%) had lower withdrawal rates (Figure 9). In summary, IL-1 inhibitors were associated with a higher incidence of adverse drug reactions compared with JAK inhibitors and TNF-α antagonists.
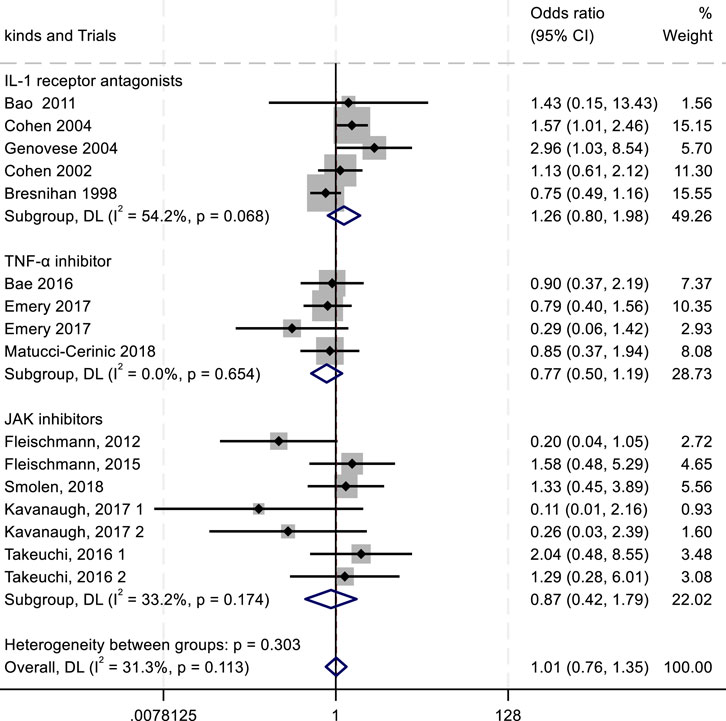
Figure 9. Results of a meta-analysis of withdrawal after drug therapy.
IL-1 inhibitors exhibit unique advantages in RA subtypes with autoinflammatory features. Their superior ACR50/70 response rates compared to TNF-α inhibitors may stem from selective blockade of innate immune pathways (e.g., IL-1β), particularly benefiting subgroups with periodic fever or NLRP3 mutations. However, their overall efficacy lags behind JAK inhibitors (notably in ACR20/70 responses), and safety concerns—including higher rates of severe adverse events and treatment discontinuation—may relate to suppression of IL-1β’s broad physiological roles (e.g., infection defense). Future strategies should focus on biomarker-guided patient selection, optimized combination therapies, and targeted drug design. Integration of real-world data and AI-driven models will advance personalized treatment paradigms, enabling high-efficacy, low-toxicity outcomes in specific RA populations.
6.3 Therapeutic potential of IL-1 inhibitors
While IL-1-targeted therapies have limitations, they remain a critical component of RA treatment. For some patients, IL-1 inhibitors are used as adjunctive therapy after inadequate responses to methotrexate (MTX) or TNF-α inhibitors, or as part of combination regimens. However, IL-1 inhibitors may be prioritized as first-line therapy in the following patient subgroups:
6.3.1 Early intervention treatment for RA
Current evidence demonstrates superior clinical efficacy of IL-1 inhibitors when administered during early-stage RA. In treatment-naïve patients with active disease, IL-1 receptor antagonists like anakinra achieve substantial symptom improvement, with 43% of recipients attaining ACR20 response criteria and 44% meeting Paulus remission standards within initial therapeutic windows. Clinical trials document significant reductions in joint swelling counts, tender joint indices, and CRP levels following early intervention (Bresnihan et al., 1998). Combination therapy with methotrexate enhances treatment response, yielding 38% ACR20 achievement versus 22% in placebo controls (p < 0.001) after 24 weeks (Cohen et al., 2004). Radiographic assessments reveal dose-dependent protection against structural damage, with 150 mg/day anakinra regimens demonstrating significantly reduced modified Sharp scores compared to placebo (p = 0.015) (Bresnihan et al., 2004). Advanced imaging analyses confirm therapeutic advantages through diminished Larsen scores and fewer eroded joints in early-treatment cohorts, supporting the critical window for IL-1 blockade in disease modification (Bresnihan et al., 1998).
The therapeutic landscape shifts dramatically in established RA, where irreversible joint damage and complex inflammatory networks limit IL-1 inhibitor efficacy. Radiographic studies show persistent elevation of modified Sharp scores (p = 0.015) despite 48-week anakinra treatment in patients with baseline structural damage, indicating irreversible osteochondral destruction (Bresnihan et al., 2004). The cytokine redundancy characteristic of late-stage disease undermines monotherapy effectiveness, as evidenced by equivalent ACR50 response rates between anakinra/etanercept combination therapy (31%) and TNF inhibitor monotherapy (41%) in methotrexate-refractory cases (Genovese et al., 2004). This therapeutic plateau reflects TNF-α and IL-6 pathway dominance that bypasses IL-1 blockade mechanisms. Combination strategies introduce heightened safety concerns, with severe infection rates escalating to 3.7%–7.4% in dual biologic regimens compared to null events in monotherapy controls (Genovese et al., 2004). Longitudinal data reveal persistent disease progression (67.80 events/100 patient-years) despite sustained IL-1 inhibition, suggesting cumulative immune cell exhaustion mechanisms (Fleischmann et al., 2006). The compromised bone marrow reserve in advanced RA may further diminish therapeutic response through altered leukocyte dynamics.
6.3.2 RA subtypes with high IL-1β expression or IL-1RN2 allele mutations
Yuan et al. employed Mendelian randomization to analyze the role of IL-1 signaling in RA. They found that seropositive RA was more closely associated with IL-1β, IL-1 receptor antagonist (IL-1Ra), and IL-6, whereas seronegative RA correlated with IL-2ra, IL-8, and IL-18 (Yuan et al., 2022). The association between IL-1β and seropositive RA was primarily driven by single nucleotide polymorphisms (SNPs) in the HLA-DQA1 region. Sensitivity analyses confirmed robust results with no horizontal pleiotropy, supporting the direct pro-inflammatory role of IL-1β and the protective antagonism of IL-1Ra. These findings suggest that sustained IL-1 inhibition or enhanced IL-1ra activity may reduce RA risk, particularly in seropositive subtypes. Reverse MR analysis further indicated that RA may promote downstream IL-6 signaling, providing a rationale for combined targeting of IL-1 and IL-6.
6.3.3 RA patients with autoinflammatory syndromes
IL-1 inhibitors demonstrate favorable safety and efficacy in managing autoinflammatory conditions such as Kawasaki disease, idiopathic recurrent pericarditis, Behçet’s disease, monogenic autoinflammatory diseases (AIDs), undifferentiated AIDs, chronic non-bacterial osteomyelitis, macrophage activation syndrome, and febrile infection-related epilepsy (Maniscalco et al., 2020; Del Giudice et al., 2022; Alexeeva et al., 2023). For severe or recurrent cases, IL-1 inhibitors should be considered a valuable therapeutic option in pediatric populations with rare inflammatory disorders.
6.3.4 Patients with metabolic comorbidities
Clinical studies comparing Anakinra and TNF-α inhibitors (TNFi) revealed distinct benefits in RA patients with type 2 diabetes. Anakinra significantly improved metabolic parameters (e.g., HbA1c) and inflammatory markers (e.g., DAS28), with sustained effects during long-term follow-up. These improvements reduced the need for antidiabetic medications, whereas TNFi showed no such metabolic benefits (Ruscitti et al., 2021). This positions Anakinra as a superior choice for RA patients with concurrent metabolic disorders.
6.3.5 Patients at high cardiovascular risk
Systemic inflammation in RA contributes to endothelial dysfunction and atherosclerosis, underscoring the importance of inflammation control for reducing cardiovascular risk (Weber et al., 2023). IL-1 inhibitors have demonstrated efficacy in lowering inflammation and improving cardiovascular outcomes, making them a promising option for high-risk RA patients (Dragoljevic et al., 2020). Although JAK inhibitors are effective in RA, their association with elevated venous thromboembolism risk limits their use in this population (Goldman et al., 2024). While TNF inhibitors reduce cardiovascular events in RA, their efficacy varies, and some patients exhibit inadequate responses (Desai et al., 2014; Sattin and Towheed, 2016; Hsieh et al., 2020). IL-6 inhibitors also show potential in cardiovascular risk reduction, though long-term data remain limited (Gerasimova et al., 2024).
7 Discussion
Existing drugs for the treatment of RA face challenges related to drug tolerance, incomplete efficacy, and patient heterogeneity. As a result, research has increasingly focused on cytokine-targeted therapies that align with the immune mechanisms underlying RA (Cavalli and Dinarello, 2015). Among these, the IL-1 family plays a significant role in disease pathogenesis by engaging distinct receptors and activating TIR-mediated Akt and MAPK signaling pathways, ultimately influencing inflammatory factor expression (Dinarello, 2018). The complex interplay within this cytokine family includes both pro-inflammatory members—such as IL-1α, IL-1β, IL-18, IL-33, IL-36α, IL-36β, and IL-36γ—and anti-inflammatory members—including IL-1Ra, IL-36Ra, IL-37, and IL-38. This intricate balance suggests that targeted modulation of IL-1 family cytokines, using soluble cytokine receptors, cytokine antagonists, and cytokine analogues, holds promise for RA treatment.
However, current research targeting the IL-1 family is relatively limited. In clinical trials, multiple antibodies have not been studied, and the studies that have been conducted have not shown ideal therapeutic effects on RA, often being used for RA that is ineffective to other drug treatments or in combination with other drugs to treat RA. The reasons for the poor efficacy may include the following:
7.1 Cytokine redundancy and compensation mechanisms
One of the primary challenges in targeting a single cytokine within the IL-1 family is the presence of functional redundancy. Many IL-1 family cytokines have overlapping roles, and inhibiting one member can lead to compensatory upregulation of others, thereby sustaining inflammation (Voronov et al., 2013). For example, blockade of IL-1β may lead to increased activity of IL-α, potentially undermining the efficacy of IL-1β-targeted therapies. This compensatory response has been observed in other inflammatory conditions, such as atherosclerosis, where cytokine inhibitors inadvertently modulate multiple inflammatory pathways, sometimes reducing treatment effectiveness (Beltrami-Moreira et al., 2016).
7.2 Limited clinical success of IL-1 inhibitors
While IL-1 inhibition has demonstrated efficacy in conditions such as systemic juvenile idiopathic arthritis and autoinflammatory syndromes, its impact in RA has been less pronounced (Pardeo et al., 2021; Arnold et al., 2022). Clinical trials investigating IL-1-targeting agents—such as anakinra (IL-1 receptor antagonist) and canakinumab (anti-IL-1β monoclonal antibody)—have not shown superiority over existing RA treatments like TNF-α inhibitors or IL-6 blockade (Singh et al., 2016). Consequently, IL-1 inhibitors are often reserved for patients who do not respond to other therapies or are used in combination regimens. Furthermore, IL-1 is a key mediator of innate immunity, and blocking its activity may weaken the body’s defense against pathogens. For instance, IL-1β activates neutrophils and macrophages to clear pathogens, and inhibiting its function may increase the risk of bacterial or viral infections. Treatment with IL-1 inhibitors may induce serious infections such as pneumonia and tuberculosis, especially more pronounced in patients with immunosuppression (Selmi et al., 2015). This may be a reason limiting the clinical application of IL-1.
7.3 Anti-drug antibodies (ADA) and immunogenicity
The human immune system can generate ADA against therapeutic monoclonal antibodies, potentially leading to treatment failure or hypersensitivity reactions. For instance, monoclonal antibodies such as infliximab, adalimumab, anakinra, and tocilizumab have been associated with ADA formation, reducing their therapeutic efficacy. Similarly, heterologous monoclonal antibodies, such as the rabbit-derived TNF-α antibody SSS07, have been reported to trigger ADA responses, limiting their clinical utility (Wang and Jin, 2020; Iwaszko et al., 2021).
7.4 Inter-individual variability and disease heterogeneity
RA exhibits significant inter-individual variability, influenced by genetic background, environmental factors, and disease subtypes. This variability affects cytokine expression patterns, making a one-size-fits-all therapeutic approach challenging. For example, racial differences have been shown to influence the efficacy of certain biologics, such as golimumab in ulcerative colitis (Greywoode et al., 2023). Personalised medicine approaches—such as gene and protein-level analyses—may be necessary to optimise IL-1-targeted therapies in RA (Lim et al., 2022).
7.5 Therapeutic potential of combined IL-1 family antagonists with other cytokine inhibitors in RA
The primary challenge of IL-1 family inhibitors in RA treatment lies in the limited efficacy of single-target inhibition due to cytokine network redundancy and compensatory mechanisms. Combining these inhibitors with other cytokine blockers (e.g., TNF-α or IL-6 antagonists) or developing multi-target drugs may enhance therapeutic outcomes through the following mechanisms: 1) IL-1 family cytokines primarily activate the TIR-MyD88-Akt/MAPK pathway, driving inflammatory responses in innate immune cells. In contrast, TNF-α and IL-6 mediate adaptive immune changes and systemic inflammation via the NF-κB and JAK-STAT pathways, respectively. Combination therapy inhibits these pathways synergistically, reducing cross-induction of pro-inflammatory factors and improving disease control. Notably, IL-1 inhibitors and TNF-α/IL-6 antagonists share overlapping pathways; for example, combining IL-1 inhibitors with TNF-α inhibitorss can cooperatively suppress synovial fibroblast activation and bone erosion, potentially achieving deeper remission. 2) Single-agent IL-1β blockade may trigger compensatory increases in IL-18 or IL-36γ. However, combining IL-1 inhibitors with TNF-α blockers or IL-6 receptor antagonists can counteract these compensatory signals and prolong clinical remission. 3) Targeting two IL-1 family cytokines simultaneously neutralizes dual pro-inflammatory isoforms, minimizing therapeutic resistance caused by functional redundancy and enabling comprehensive suppression of inflammation. This integrated approach highlights the potential of IL-1-targeted combination therapies to address RA heterogeneity while emphasizing the need for rigorous safety evaluation to balance efficacy and infection risks.
8 Prospects
Targeting the IL-1 family has improved symptoms of RA, but some issues remain to be addressed. In the future, we can conduct in-depth research from the following perspectives: First, develop multi-specific antibodies that can target multiple IL-1 family cytokines simultaneously, to enhance efficacy. Second, use human monoclonal antibodies to replace existing murine antibodies in order to mitigate immune-related adverse reactions. Third, explore the combined application of IL-1 inhibitors with conventional therapies such as DMARDs, which may significantly enhance treatment effects and shorten treatment cycles. Last but not least, develop personalized treatment regimens tailored to different subtypes of patients based on their genetic profiles and protein expression characteristics, to achieve precision medicine. In the future, further research is believed to be able to improve the level of IL-1 targeted therapy for RA and bring about better clinical outcomes for patients.
Author contributions
KW: Writing – original draft, Writing – review and editing. HL: Investigation, Writing – original draft. LL: Investigation, Writing – original draft, Writing – review and editing. HG: Investigation, Writing – review and editing. YS: Investigation, Writing – review and editing. DL: Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This project is supported by Undergraduate Innovation and Entrepreneurship Training Program of Jilin University (X202410183437).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akash, M. S., Rehman, K., and Chen, S. (2013). IL-1Ra and its delivery strategies: inserting the association in perspective. Pharm. Res. 30 (11), 2951–2966. doi:10.1007/s11095-013-1118-0
Alani, H., Henty, J. R., Thompson, N. L., Jury, E., and Ciurtin, C. (2018). Systematic review and meta-analysis of the epidemiology of polyautoimmunity in Sjögren’s syndrome (secondary Sjögren’s syndrome) focusing on autoimmune rheumatic diseases. Scand. J. Rheumatol. 47 (2), 141–154. doi:10.1080/03009742.2017.1324909
Aletaha, D., and Smolen, J. S. (2018). Diagnosis and management of rheumatoid arthritis: a review. JAMA 320 (13), 1360–1372. doi:10.1001/jama.2018.13103
Alexeeva, E., Shingarova, M., Dvoryakovskaya, T., Lomakina, O., Fetisova, A., Isaeva, K., et al. (2023). Safety and efficacy of canakinumab treatment for undifferentiated autoinflammatory diseases: the data of a retrospective cohort two-centered study. Front. Med. (Lausanne) 10, 1257045. doi:10.3389/fmed.2023.1257045
Alten, R., Gomez-Reino, J., Durez, P., Beaulieu, A., Sebba, A., Krammer, G., et al. (2011). Efficacy and safety of the human anti-IL-1β monoclonal antibody canakinumab in rheumatoid arthritis: results of a 12-week, Phase II, dose-finding study. BMC Musculoskelet. Disord. 12, 153. doi:10.1186/1471-2474-12-153
Arnold, D. D., Yalamanoglu, A., and Boyman, O. (2022). Systematic review of safety and efficacy of IL-1-targeted biologics in treating immune-mediated disorders. Front. Immunol. 13, 888392. doi:10.3389/fimmu.2022.888392
Baggio, C., Bindoli, S., Guidea, I., Doria, A., Oliviero, F., and Sfriso, P. (2023). IL-18 in autoinflammatory diseases: focus on adult onset still disease and macrophages activation syndrome. Int. J. Mol. Sci. 24 (13), 11125. doi:10.3390/ijms241311125
Bao, J., Yue, T., Liu, W., Zhang, Q., Zhou, L., Xu, H. J., et al. (2011). Secondary failure to treatment with recombinant human IL-1 receptor antagonist in Chinese patients with rheumatoid arthritis. Clin. Rheumatol. 30 (5), 697–701. doi:10.1007/s10067-010-1654-5
Begovich, A. B., Carlton, V. E., Honigberg, L. A., Schrodi, S. J., Chokkalingam, A. P., Alexander, H. C., et al. (2004). A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am. J. Hum. Genet. 75 (2), 330–337. doi:10.1086/422827
Beltrami-Moreira, M., Vromman, A., Sukhova, G. K., Folco, E. J., and Libby, P. (2016). Redundancy of IL-1 isoform signaling and its implications for arterial remodeling. PLoS One 11 (3), e0152474. doi:10.1371/journal.pone.0152474
Biton, J., Khaleghparast Athari, S., Thiolat, A., Santinon, F., Lemeiter, D., Hervé, R., et al. (2016). In vivo expansion of activated Foxp3+ regulatory T cells and establishment of a type 2 immune response upon IL-33 treatment protect against experimental arthritis. J. Immunol. 197 (5), 1708–1719. doi:10.4049/jimmunol.1502124
Boraschi, D., Italiani, P., Weil, S., and Martin, M. U. (2018). The family of the interleukin-1 receptors. Immunol. Rev. 281 (1), 197–232. doi:10.1111/imr.12606
Bossini-Castillo, L., de Kovel, C., Kallberg, H., van 't Slot, R., Italiaander, A., Coenen, M., et al. (2015). A genome-wide association study of rheumatoid arthritis without antibodies against citrullinated peptides. Ann. Rheum. Dis. 74 (3), e15. doi:10.1136/annrheumdis-2013-204591
Boutet, M. A., Bart, G., Penhoat, M., Amiaud, J., Brulin, B., Charrier, C., et al. (2016). Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn’s disease. Clin. Exp. Immunol. 184 (2), 159–173. doi:10.1111/cei.12761
Boutet, M. A., Najm, A., Bart, G., Brion, R., Touchais, S., Trichet, V., et al. (2017). IL-38 overexpression induces anti-inflammatory effects in mice arthritis models and in human macrophages in vitro. Ann. Rheum. Dis. 76 (7), 1304–1312. doi:10.1136/annrheumdis-2016-210630
Braga, G. C., Simões, J. L. B., Teixeira Dos Santos, Y. J., Filho, J. C. M., and Bagatini, M. D. (2024). The impacts of obesity in rheumatoid arthritis and insights into therapeutic purinergic modulation. Int. Immunopharmacol. 136, 112357. doi:10.1016/j.intimp.2024.112357
Bresnihan, B., Alvaro-Gracia, J. M., Cobby, M., Doherty, M., Domljan, Z., Emery, P., et al. (1998). Treatment of rheumatoid arthritis with recombinant human interleukin-1 receptor antagonist. Arthritis Rheum. 41 (12), 2196–2204. doi:10.1002/1529-0131(199812)41:12<2196::Aid-art15>3.0.Co;2-2
Bresnihan, B., Newmark, R., Robbins, S., and Genant, H. K. (2004). Effects of anakinra monotherapy on joint damage in patients with rheumatoid arthritis. Extension of a 24-week randomized, placebo-controlled trial. J. Rheumatol. 31 (6), 1103–1111. Available online at: https://www.jrheum.org/content/31/6/1103.tab-article-info
Broderick, L., and Hoffman, H. M. (2022). IL-1 and autoinflammatory disease: biology, pathogenesis and therapeutic targeting. Nat. Rev. Rheumatol. 18 (8), 448–463. doi:10.1038/s41584-022-00797-1
Cavalli, G., and Dinarello, C. A. (2015). Treating rheumatological diseases and co-morbidities with interleukin-1 blocking therapies. Rheumatol. (Oxford) 54 (12), 2134–2144. doi:10.1093/rheumatology/kev269
Cavalli, G., Koenders, M., Kalabokis, V., Kim, J., Tan, A. C., Garlanda, C., et al. (2016). Treating experimental arthritis with the innate immune inhibitor interleukin-37 reduces joint and systemic inflammation. Rheumatol. (Oxford) 55 (12), 2220–2229. doi:10.1093/rheumatology/kew325
Cayrol, C., and Girard, J. P. (2009). The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc. Natl. Acad. Sci. U. S. A. 106 (22), 9021–9026. doi:10.1073/pnas.0812690106
Charoenngam, N. (2021). Vitamin D and rheumatic diseases: a review of clinical evidence. Int. J. Mol. Sci. 22 (19), 10659. doi:10.3390/ijms221910659
Chen, X., Liu, C., Deng, J., Xia, T., Zhang, X., Xue, S., et al. (2024). Schisandrin B ameliorates adjuvant-induced arthritis in rats via modulation of inflammatory mediators, oxidative stress, and HIF-1α/VEGF pathway. J. Pharm. Pharmacol. 76 (6), 681–690. doi:10.1093/jpp/rgae020
Chen, Z., Bozec, A., Ramming, A., and Schett, G. (2019). Anti-inflammatory and immune-regulatory cytokines in rheumatoid arthritis. Nat. Rev. Rheumatol. 15 (1), 9–17. doi:10.1038/s41584-018-0109-2
Cieza, A., Causey, K., Kamenov, K., Hanson, S. W., Chatterji, S., and Vos, T. (2021). Global estimates of the need for rehabilitation based on the global burden of disease study 2019: a systematic analysis for the global burden of disease study 2019. Lancet 396 (10267), 2006–2017. doi:10.1016/s0140-6736(20)32340-0
Cohen, S., Hurd, E., Cush, J., Schiff, M., Weinblatt, M. E., Moreland, L. W., et al. (2002). Treatment of rheumatoid arthritis with anakinra, a recombinant human interleukin-1 receptor antagonist, in combination with methotrexate: results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 46 (3), 614–624. doi:10.1002/art.10141
Cohen, S. B., Moreland, L. W., Cush, J. J., Greenwald, M. W., Block, S., Shergy, W. J., et al. (2004). A multicentre, double blind, randomised, placebo controlled trial of anakinra (Kineret), a recombinant interleukin 1 receptor antagonist, in patients with rheumatoid arthritis treated with background methotrexate. Ann. Rheum. Dis. 63 (9), 1062–1068. doi:10.1136/ard.2003.016014
Cohen, S. B., Woolley, J. M., and Chan, W.Anakinra 960180 Study Group (2003). Interleukin 1 receptor antagonist anakinra improves functional status in patients with rheumatoid arthritis. J. Rheumatol. 30 (2), 225–231. Available online at: https://www.jrheum.org/content/30/2/225.tab-article-info
Collaborators, G. D. A.I. (2020). Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 396 (10258), 1204–1222. doi:10.1016/s0140-6736(20)30925-9
Conforti, A., Di Cola, I., Pavlych, V., Ruscitti, P., Berardicurti, O., Ursini, F., et al. (2021). Beyond the joints, the extra-articular manifestations in rheumatoid arthritis. Autoimmun. Rev. 20 (2), 102735. doi:10.1016/j.autrev.2020.102735
Cooney, R. N., and Yumet, G. (2002). Cytokine signaling in sepsis: redundancy, crosstalk, and regulatory mechanisms. Crit. Care Med. 30 (1), 262–263. doi:10.1097/00003246-200201000-00048
Costenbader, K. H., and Karlson, E. W. (2006). Epstein-Barr virus and rheumatoid arthritis: is there a link? Arthritis Res. Ther. 8 (1), 204. doi:10.1186/ar1893
Danila, M. I., Laufer, V. A., Reynolds, R. J., Yan, Q., Liu, N., Gregersen, P. K., et al. (2017). Dense genotyping of immune-related regions identifies loci for rheumatoid arthritis risk and damage in African Americans. Mol. Med. 23, 177–187. doi:10.2119/molmed.2017.00081
Danto, S. I., Shojaee, N., Singh, R. S. P., Li, C., Gilbert, S. A., Manukyan, Z., et al. (2019). Safety, tolerability, pharmacokinetics, and pharmacodynamics of PF-06650833, a selective interleukin-1 receptor-associated kinase 4 (IRAK4) inhibitor, in single and multiple ascending dose randomized phase 1 studies in healthy subjects. Arthritis Res. Ther. 21 (1), 269. doi:10.1186/s13075-019-2008-6
Del Giudice, E., Sota, J., Orlando, F., Picciano, L., Cimaz, R., Cantarini, L., et al. (2022). Off-label use of canakinumab in pediatric rheumatology and rare diseases. Front. Med. (Lausanne) 9, 998281. doi:10.3389/fmed.2022.998281
Derer, A., Groetsch, B., Harre, U., Böhm, C., Towne, J., Schett, G., et al. (2014). Blockade of IL-36 receptor signaling does not prevent from TNF-induced arthritis. PLoS One 9 (8), e101954. doi:10.1371/journal.pone.0101954
Desai, R. J., Rao, J. K., Hansen, R. A., Fang, G., Maciejewski, M., and Farley, J. (2014). Tumor necrosis factor-α inhibitor treatment and the risk of incident cardiovascular events in patients with early rheumatoid arthritis: a nested case-control study. J. Rheumatol. 41 (11), 2129–2136. doi:10.3899/jrheum.131464
Dinarello, C. A. (2018). Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 281 (1), 8–27. doi:10.1111/imr.12621
Dragoljevic, D., Lee, M. K. S., Louis, C., Shihata, W., Kraakman, M. J., Hansen, J., et al. (2020). Inhibition of interleukin-1β signalling promotes atherosclerotic lesion remodelling in mice with inflammatory arthritis. Clin. Transl. Immunol. 9 (11), e1206. doi:10.1002/cti2.1206
Elewski, B. E., Lebwohl, M. G., Anadkat, M. J., Barker, J., Ghoreschi, K., Imafuku, S., et al. (2023). Rapid and sustained improvements in Generalized Pustular Psoriasis Physician Global Assessment scores with spesolimab for treatment of generalized pustular psoriasis flares in the randomized, placebo-controlled Effisayil 1 study. J. Am. Acad. Dermatol 89 (1), 36–44. doi:10.1016/j.jaad.2023.02.040
Eyre, S., Bowes, J., Diogo, D., Lee, A., Barton, A., Martin, P., et al. (2012). High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat. Genet. 44 (12), 1336–1340. doi:10.1038/ng.2462
Fields, J. K., Gyllenbäck, E. J., Bogacz, M., Obi, J., Birkedal, G. S., Sjöström, K., et al. (2024). Antibodies targeting the shared cytokine receptor IL-1 receptor accessory protein invoke distinct mechanisms to block all cytokine signaling. Cell. Rep. 43 (5), 114099. doi:10.1016/j.celrep.2024.114099
Fleischmann, R. M., Schechtman, J., Bennett, R., Handel, M. L., Burmester, G. R., Tesser, J., et al. (2003). Anakinra, a recombinant human interleukin-1 receptor antagonist (r-metHuIL-1ra), in patients with rheumatoid arthritis: a large, international, multicenter, placebo-controlled trial. Arthritis Rheum. 48 (4), 927–934. doi:10.1002/art.10870
Fleischmann, R. M., Tesser, J., Schiff, M. H., Schechtman, J., Burmester, G. R., Bennett, R., et al. (2006). Safety of extended treatment with anakinra in patients with rheumatoid arthritis. Ann. Rheum. Dis. 65 (8), 1006–1012. doi:10.1136/ard.2005.048371
Gabay, C., Fautrel, B., Rech, J., Spertini, F., Feist, E., Kötter, I., et al. (2018). Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still’s disease. Ann. Rheum. Dis. 77 (6), 840–847. doi:10.1136/annrheumdis-2017-212608
Gabay, C., and Towne, J. E. (2015). Regulation and function of interleukin-36 cytokines in homeostasis and pathological conditions. J. Leukoc. Biol. 97 (4), 645–652. doi:10.1189/jlb.3RI1014-495R
Genovese, M. C., Cohen, S., Moreland, L., Lium, D., Robbins, S., Newmark, R., et al. (2004). Combination therapy with etanercept and anakinra in the treatment of patients with rheumatoid arthritis who have been treated unsuccessfully with methotrexate. Arthritis Rheum. 50 (5), 1412–1419. doi:10.1002/art.20221
Gerasimova, E. V., Popkova, T. V., Kirillova, I. G., Gerasimova, D. A., and Nasonov, E. L. (2024). Dynamics of modified cardiovascular risk factors in patients with rheumatoid arthritis on the background of 5-year therapy with an interleukin 6 receptor inhibitor. Nauchno-Prakticheskaya Revmatol. 62 (1), 81–89. doi:10.47360/1995-4484-2024-81-89
Goldman, A., Galper, B. L., Druyan, A., Grossman, C., Sharif, K., Shechtman, L., et al. (2024). Adverse cardiovascular events in rheumatoid arthritis patients treated with JAK inhibitors: an analysis of postmarketing spontaneous safety reports. Semin. Arthritis Rheum. 67, 152461. doi:10.1016/j.semarthrit.2024.152461
Gottlieb, A., Natsis, N. E., Kerdel, F., Forman, S., Gonzalez, E., Jimenez, G., et al. (2020). A phase II open-label study of Bermekimab in patients with hidradenitis suppurativa shows resolution of inflammatory lesions and pain. J. Invest. Dermatol. 140 (8), 1538–1545.e2. doi:10.1016/j.jid.2019.10.024
Grevich, S., and Shenoi, S. (2017). Update on the management of systemic juvenile idiopathic arthritis and role of IL-1 and IL-6 inhibition. Adolesc. Health Med. Ther. 8, 125–135. doi:10.2147/ahmt.S109495
Greywoode, R., Petralia, F., Ullman, T. A., Frederic Colombel, J., and Ungaro, R. C. (2023). Racial difference in efficacy of golimumab in ulcerative colitis. Inflamm. Bowel Dis. 29 (6), 843–849. doi:10.1093/ibd/izac161
Guo, J., Zhang, T., Cao, H., Li, X., Liang, H., Liu, M., et al. (2019). Sequencing of the MHC region defines HLA-DQA1 as the major genetic risk for seropositive rheumatoid arthritis in Han Chinese population. Ann. Rheum. Dis. 78 (6), 773–780. doi:10.1136/annrheumdis-2018-214725
Guo, T., Xing, Y., Chen, Z., Zhu, H., Yang, L., Xiao, Y., et al. (2022). Long non-coding RNA NEAT1 knockdown alleviates rheumatoid arthritis by reducing IL-18 through p300/CBP repression. Inflammation 45 (1), 100–115. doi:10.1007/s10753-021-01531-x
Hao, Z., and Liu, Y. (2021). IL-38 and IL-36 target autophagy for regulating synoviocyte proliferation, migration, and invasion in rheumatoid arthritis. Dis. Markers 2021, 7933453. doi:10.1155/2021/7933453
Ho Lee, Y., and Gyu Song, G. (2020). Comparative efficacy and safety of tofacitinib, baricitinib, upadacitinib, filgotinib and peficitinib as monotherapy for active rheumatoid arthritis. J. Clin. Pharm. Ther. 45 (4), 674–681. doi:10.1111/jcpt.13142
Hong, Y., Duan, Y., Zhu, Z., Yu, Q., Mo, Z., Wang, H., et al. (2024). IL-1ra loaded chondroitin sulfate-functionalized microspheres for minimally invasive treatment of intervertebral disc degeneration. Acta Biomater. 185, 336–349. doi:10.1016/j.actbio.2024.06.048
Hsieh, M. J., Lee, C. H., Tsai, M. L., Kao, C. F., Lan, W. C., Huang, Y. T., et al. (2020). Biologic agents reduce cardiovascular events in rheumatoid arthritis not responsive to tumour necrosis factor inhibitors: a national cohort study. Can. J. Cardiol. 36 (11), 1739–1746. doi:10.1016/j.cjca.2020.01.003
Hu, R., Yuan, T., Wang, H., Zhao, J., Shi, L., Li, Q., et al. (2023). Efficacy, safety and immunogenicity of etanercept biosimilars versus reference biologics in patients with rheumatoid arthritis: a meta-analysis. Front. Pharmacol. 14, 1089272. doi:10.3389/fphar.2023.1089272
Ilowite, N. T., Prather, K., Lokhnygina, Y., Schanberg, L. E., Elder, M., Milojevic, D., et al. (2014). Randomized, double-blind, placebo-controlled trial of the efficacy and safety of rilonacept in the treatment of systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 66 (9), 2570–2579. doi:10.1002/art.38699
Irigoyen, P., Lee, A. T., Wener, M. H., Li, W., Kern, M., Batliwalla, F., et al. (2005). Regulation of anti-cyclic citrullinated peptide antibodies in rheumatoid arthritis: contrasting effects of HLA-DR3 and the shared epitope alleles. Arthritis Rheum. 52 (12), 3813–3818. doi:10.1002/art.21419
Iwahashi, M., Yamamura, M., Aita, T., Okamoto, A., Ueno, A., Ogawa, N., et al. (2004). Expression of Toll-like receptor 2 on CD16+ blood monocytes and synovial tissue macrophages in rheumatoid arthritis. Arthritis Rheum. 50 (5), 1457–1467. doi:10.1002/art.20219
Iwaszko, M., Wielińska, J., Świerkot, J., Kolossa, K., Sokolik, R., Bugaj, B., et al. (2021). IL-33 gene polymorphisms as potential biomarkers of disease susceptibility and response to TNF inhibitors in rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis patients. Front. Immunol. 12, 631603. doi:10.3389/fimmu.2021.631603
Izawa, A., Ishihara, Y., Mizutani, H., Kobayashi, S., Goto, H., Okabe, E., et al. (2014). Inflammatory bone loss in experimental periodontitis induced by Aggregatibacter actinomycetemcomitans in interleukin-1 receptor antagonist knockout mice. Infect. Immun. 82 (5), 1904–1913. doi:10.1128/iai.01618-13
Jang, S., Kwon, E. J., and Lee, J. J. (2022). Rheumatoid arthritis: pathogenic roles of diverse immune cells. Int. J. Mol. Sci. 23 (2), 905. doi:10.3390/ijms23020905
Jia, W., Wu, W., Yang, D., Xiao, C., Huang, M., Long, F., et al. (2018). GATA4 regulates angiogenesis and persistence of inflammation in rheuma toid arthritis. Cell. Death and Dis. 9 (5), 503. doi:10.1038/s41419-018-0570-5
Jonsson, A. H., Zhang, F., Dunlap, G., Gomez-Rivas, E., Watts, G. F. M., Faust, H. J., et al. (2022). Granzyme K(+) CD8 T cells form a core population in inflamed human tissue. Sci. Transl. Med. 14 (649), eabo0686. doi:10.1126/scitranslmed.abo0686
Junge, G., Mason, J., and Feist, E. (2017). Adult onset Still's disease-The evidence that anti-interleukin-1 treatment is effective and well-tolerated (a comprehensive literature review). Semin. Arthritis Rheum. 47 (2), 295–302. doi:10.1016/j.semarthrit.2017.06.006
Kazmi, S., Salehi-Pourmehr, H., Sadigh-Eteghad, S., and Farhoudi, M. (2024). The efficacy and safety of interleukin-1 receptor antagonist in stroke patients: a systematic review. J. Clin. Neurosci. 120, 120–128. doi:10.1016/j.jocn.2024.01.009
Kelsen, S. G., Agache, I. O., Soong, W., Israel, E., Chupp, G. L., Cheung, D. S., et al. (2021). Astegolimab (anti-ST2) efficacy and safety in adults with severe asthma: a randomized clinical trial. J. Allergy Clin. Immunol. 148 (3), 790–798. doi:10.1016/j.jaci.2021.03.044
Kim, D. H., Kim, H. Y., Cho, S., Yoo, S. J., Kim, W. J., Yeon, H. R., et al. (2021). Induction of the IL-1RII decoy receptor by NFAT/FOXP3 blocks IL-1β-dependent response of Th17 cells. Elife 10, e61841. doi:10.7554/eLife.61841
Kim, D. H., and Lee, W. W. (2024). IL-1 receptor dynamics in immune cells: Orchestrating immune precision and balance. Immune Netw. 24 (3), e21. doi:10.4110/in.2024.24.e21
Kim, J. H., Jin, H. M., Kim, K., Song, I., Youn, B. U., Matsuo, K., et al. (2009). The mechanism of osteoclast differentiation induced by IL-1. J. Immunol. 183 (3), 1862–1870. doi:10.4049/jimmunol.0803007
Kim, K. W., Kim, H. R., Kim, B. M., Cho, M. L., and Lee, S. H. (2015). Th17 cytokines regulate osteoclastogenesis in rheumatoid arthritis. Am. J. Pathol. 185 (11), 3011–3024. doi:10.1016/j.ajpath.2015.07.017
Klareskog, L., Rönnelid, J., Saevarsdottir, S., Padyukov, L., and Alfredsson, L. (2020). The importance of differences; on environment and its interactions with genes and immunity in the causation of rheumatoid arthritis. J. Intern Med. 287 (5), 514–533. doi:10.1111/joim.13058
Klein, A. L., Imazio, M., Cremer, P., Brucato, A., Abbate, A., Fang, F., et al. (2021). Phase 3 trial of interleukin-1 trap rilonacept in recurrent pericarditis. N. Engl. J. Med. 384 (1), 31–41. doi:10.1056/NEJMoa2027892
Kobayashi, T., and Bartold, P. M. (2023). Periodontitis and periodontopathic bacteria as risk factors for rheumatoid arthritis: a review of the last 10 years. Jpn. Dent. Sci. Rev. 59, 263–272. doi:10.1016/j.jdsr.2023.08.002
Kong, Y. Y., Feige, U., Sarosi, I., Bolon, B., Tafuri, A., Morony, S., et al. (1999). Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 402 (6759), 304–309. doi:10.1038/46303
Kugler, M., Dellinger, M., Kartnig, F., Müller, L., Preglej, T., Heinz, L. X., et al. (2023). Cytokine-directed cellular cross-talk imprints synovial pathotypes in rheumatoid arthritis. Ann. Rheum. Dis. 82 (9), 1142–1152. doi:10.1136/ard-2022-223396
Kunisch, E., Chakilam, S., Gandesiri, M., and Kinne, R. W. (2012). IL-33 regulates TNF-α dependent effects in synovial fibroblasts. Int. J. Mol. Med. 29 (4), 530–540. doi:10.3892/ijmm.2012.883
Kuo, W. C., Lee, C. C., Chang, Y. W., Pang, W., Chen, H. S., Hou, S. C., et al. (2021). Structure-based development of human interleukin-1β-specific antibody that simultaneously inhibits binding to both IL-1RI and IL-1RAcP. J. Mol. Biol. 433 (4), 166766. doi:10.1016/j.jmb.2020.166766
Kwon, S. R., Jung, K. H., Lim, M. J., Son, M. J., Choi, B. H., Park, S. G., et al. (2017). The effect of anti-TNF treatment on osteoblastogenesis in ankylosing spondylitis: the number of circulating osteoblast-lineage cells in peripheral blood decreased after infliximab therapy in patients with ankylosing spondylitis. Clin. Exp. Rheumatol. 35 (5), 837–843. Available online at: https://www.clinexprheumatol.org/abstract.asp?a=11297
Landy, E., Carol, H., Ring, A., and Canna, S. (2024). Biological and clinical roles of IL-18 in inflammatory diseases. Nat. Rev. Rheumatol. 20 (1), 33–47. doi:10.1038/s41584-023-01053-w
Lee, E. J., So, M. W., Hong, S., Kim, Y. G., Yoo, B., and Lee, C. K. (2016a). Interleukin-33 acts as a transcriptional repressor and extracellular cytokine in fibroblast-like synoviocytes in patients with rheumatoid arthritis. Cytokine 77, 35–43. doi:10.1016/j.cyto.2015.10.005
Lee, S. Y., Min, H. K., Lee, S. H., Shin, H. J., Lee, W. Y., Cho, Y. G., et al. (2016b). IL-1 receptor antagonist (IL-1Ra)-Fc ameliorate autoimmune arthritis by regulation of the Th17 cells/Treg balance and arthrogenic cytokine activation. Immunol. Lett. 172, 56–66. doi:10.1016/j.imlet.2016.02.011
Levescot, A., Chang, M. H., Schnell, J., Nelson-Maney, N., Yan, J., Martínez-Bonet, M., et al. (2021). IL-1β-driven osteoclastogenic Tregs accelerate bone erosion in arthritis. J. Clin. Invest. 131 (18), e141008. doi:10.1172/jci141008
Li, L., Liao, Z., Ye, M., and Jiang, J. (2021). Recombinant human IL-37 inhibited endometriosis development in a mouse model through increasing Th1/Th2 ratio by inducing the maturation of dendritic cells. Reprod. Biol. Endocrinol. 19 (1), 128. doi:10.1186/s12958-021-00811-3
Li, Y., Fu, Y., Chen, H., Liu, X., and Li, M. (2020). Blocking interleukin-33 alleviates the joint inflammation and inhibits the development of collagen-induced arthritis in mice. J. Immunol. Res. 2020, 4297354. doi:10.1155/2020/4297354
Lim, S. H., Kim, K., and Choi, C. I. (2022). Pharmacogenomics of monoclonal antibodies for the treatment of rheumatoid arthritis. J. Pers. Med. 12 (8), 1265. doi:10.3390/jpm12081265
Lin, L., Zhang, K., Xiong, Q., Zhang, J., Cai, B., Huang, Z., et al. (2023). Gut microbiota in pre-clinical rheumatoid arthritis: from pathogenesis to preventing progression. J. Autoimmun. 141, 103001. doi:10.1016/j.jaut.2023.103001
Liu, T., Liu, J., Lin, Y., Que, B., Chang, C., Zhang, J., et al. (2019). IL-37 inhibits the maturation of dendritic cells through the IL-1R8-TLR4-NF-κB pathway. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 1864 (10), 1338–1349. doi:10.1016/j.bbalip.2019.05.009
Liu, Z., Zhu, L., Lu, Z., Chen, H., Fan, L., Xue, Q., et al. (2020). IL-37 represses the autoimmunity in myasthenia gravis via directly targeting follicular Th and B cells. J. Immunol. 204 (7), 1736–1745. doi:10.4049/jimmunol.1901176
Long, H., Liu, Q., Yin, H., Wang, K., Diao, N., Zhang, Y., et al. (2022). Prevalence trends of site-specific osteoarthritis from 1990 to 2019: findings from the global burden of disease study 2019. Arthritis Rheumatol. 74 (7), 1172–1183. doi:10.1002/art.42089
Lott, J. M., Sumpter, T. L., and Turnquist, H. R. (2015). New dog and new tricks: evolving roles for IL-33 in type 2 immunity. J. Leukoc. Biol. 97 (6), 1037–1048. doi:10.1189/jlb.3RI1214-595R
Maeda, K., Yoshida, K., Nishizawa, T., Otani, K., Yamashita, Y., Okabe, H., et al. (2022). Inflammation and bone metabolism in rheumatoid arthritis: molecular mechanisms of joint destruction and pharmacological treatments. Int. J. Mol. Sci. 23 (5), 2871. doi:10.3390/ijms23052871
Malviya, A., Kuiper, J. H., Makwana, N., Laing, P., and Ashton, B. (2009). The effect of newer anti-rheumatic drugs on osteogenic cell proliferation: an in-vitro study. J. Orthop. Surg. Res. 4, 17. doi:10.1186/1749-799x-4-17
Maniscalco, V., Abu-Rumeileh, S., Mastrolia, M. V., Marrani, E., Maccora, I., Pagnini, I., et al. (2020). The off-label use of anakinra in pediatric systemic autoinflammatory diseases. Ther. Adv. Musculoskelet. Dis. 12, 1759720x20959575. doi:10.1177/1759720x20959575
Mateen, S., Zafar, A., Moin, S., Khan, A. Q., and Zubair, S. (2016). Understanding the role of cytokines in the pathogenesis of rheumatoid arthritis. Clin. Chim. Acta 455, 161–171. doi:10.1016/j.cca.2016.02.010
Maul, J. T., Distler, M., Kolios, A., Maul, L. V., Guillet, C., Graf, N., et al. (2021). Canakinumab lacks efficacy in treating adult patients with moderate to severe chronic spontaneous urticaria in a phase II randomized double-blind placebo-controlled single-center study. J. Allergy Clin. Immunol. Pract. 9 (1), 463–468.e3. doi:10.1016/j.jaip.2020.07.058
Min, H. K., Kim, S., Lee, J. Y., Kim, K. W., Lee, S. H., and Kim, H. R. (2021). IL-18 binding protein suppresses IL-17-induced osteoclastogenesis and rectifies type 17 helper T cell/regulatory T cell imbalance in rheumatoid arthritis. J. Transl. Med. 19 (1), 392. doi:10.1186/s12967-021-03071-2
Min, H. K., Kim, S. H., Lee, J. Y., Lee, S. H., and Kim, H. R. (2023). Interleukin-18 binding protein regulates the apoptosis and necroptosis of fibroblast-like synoviocytes and chondrocytes in rheumatoid arthritis. Clin. Exp. Rheumatol. 41 (11), 2207–2215. doi:10.55563/clinexprheumatol/hjvvf1
Minoia, F., Consolaro, A., and Ravelli, A. (2018). Filling the gap: toward a disease activity tool for systemic juvenile idiopathic arthritis. J. Rheumatol. 45 (1), 3–5. doi:10.3899/jrheum.170703
Mora, J., Schlemmer, A., Wittig, I., Richter, F., Putyrski, M., Frank, A. C., et al. (2016). Interleukin-38 is released from apoptotic cells to limit inflammatory macrophage responses. J. Mol. Cell. Biol. 8 (5), 426–438. doi:10.1093/jmcb/mjw006
Morel, J. C., Park, C. C., Kumar, P., and Koch, A. E. (2001). Interleukin-18 induces rheumatoid arthritis synovial fibroblast CXC chemokine production through NFkappaB activation. Lab. Invest. 81 (10), 1371–1383. doi:10.1038/labinvest.3780351
Narayanan, K. B., and Park, H. H. (2015). Toll/interleukin-1 receptor (TIR) domain-mediated cellular signaling pathways. Apoptosis 20 (2), 196–209. doi:10.1007/s10495-014-1073-1
Nasi, S., Ea, H. K., So, A., and Busso, N. (2017). Revisiting the role of interleukin-1 pathway in osteoarthritis: interleukin-1α and -1β, and NLRP3 inflammasome are not involved in the pathological features of the murine menisectomy model of osteoarthritis. Front. Pharmacol. 8, 282. doi:10.3389/fphar.2017.00282
Niu, X., He, D., Deng, S., Li, W., Xi, Y., Xie, C., et al. (2011). Regulatory immune responses induced by IL-1 receptor antagonist in rheumatoid arthritis. Mol. Immunol. 49 (1-2), 290–296. doi:10.1016/j.molimm.2011.08.020
Nnane, I., Frederick, B., Yao, Z., Raible, D., Shu, C., Badorrek, P., et al. (2020). The first-in-human study of CNTO 7160, an anti-interleukin-33 receptor monoclonal antibody, in healthy subjects and patients with asthma or atopic dermatitis. Br. J. Clin. Pharmacol. 86 (12), 2507–2518. doi:10.1111/bcp.14361
Nordström, D., Knight, A., Luukkainen, R., van Vollenhoven, R., Rantalaiho, V., Kajalainen, A., et al. (2012). Beneficial effect of interleukin 1 inhibition with anakinra in adult-onset Still’s disease. An open, randomized, multicenter study. J. Rheumatol. 39 (10), 2008–2011. doi:10.3899/jrheum.111549
Nuki, G., Bresnihan, B., Bear, M. B., and McCabe, D.European Group of Clinical Investigators (2002). Long-term safety and maintenance of clinical improvement following treatment with anakinra (recombinant human interleukin-1 receptor antagonist) in patients with rheumatoid arthritis: extension phase of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 46 (11), 2838–2846. doi:10.1002/art.10578
Okada, Y., Suzuki, A., Ikari, K., Terao, C., Kochi, Y., Ohmura, K., et al. (2016). Contribution of a non-classical HLA gene, HLA-DOA, to the risk of rheumatoid arthritis. Am. J. Hum. Genet. 99 (2), 366–374. doi:10.1016/j.ajhg.2016.06.019
Okada, Y., Terao, C., Ikari, K., Kochi, Y., Ohmura, K., Suzuki, A., et al. (2012). Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the Japanese population. Nat. Genet. 44 (5), 511–516. doi:10.1038/ng.2231
Okada, Y., Wu, D., Trynka, G., Raj, T., Terao, C., Ikari, K., et al. (2014). Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506 (7488), 376–381. doi:10.1038/nature12873
Padyukov, L. (2022). Genetics of rheumatoid arthritis. Semin. Immunopathol. 44 (1), 47–62. doi:10.1007/s00281-022-00912-0
Padyukov, L., Silva, C., Stolt, P., Alfredsson, L., and Klareskog, L. (2004). A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum. 50 (10), 3085–3092. doi:10.1002/art.20553
Pardeo, M., Rossi, M. N., Pires Marafon, D., Sacco, E., Bracaglia, C., Passarelli, C., et al. (2021). Early treatment and IL1RN single-nucleotide polymorphisms affect response to anakinra in systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 73 (6), 1053–1061. doi:10.1002/art.41612
Pascual, V., Allantaz, F., Arce, E., Punaro, M., and Banchereau, J. (2005). Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J. Exp. Med. 201 (9), 1479–1486. doi:10.1084/jem.20050473
Pei, B., Chen, K., Zhou, S., Min, D., and Xiao, W. (2020). IL-38 restrains inflammatory response of collagen-induced arthritis in rats via SIRT1/HIF-1α signaling pathway. Biosci. Rep. 40 (5). doi:10.1042/bsr20182431
Pelletier, J. P., Mineau, F., Ranger, P., Tardif, G., and Martel-Pelletier, J. (1996). The increased synthesis of inducible nitric oxide inhibits IL-1ra synthesis by human articular chondrocytes: possible role in osteoarthritic cartilage degradation. Osteoarthr. Cartil. 4 (1), 77–84. doi:10.1016/s1063-4584(96)80009-4
Puren, A. J., Fantuzzi, G., Gu, Y., Su, M. S., and Dinarello, C. A. (1998). Interleukin-18 (IFNgamma-inducing factor) induces IL-8 and IL-1beta via TNFalpha production from non-CD14+ human blood mononuclear cells. J. Clin. Invest. 101 (3), 711–721. doi:10.1172/jci1379
Qi, F., Liu, M., Li, F., Lv, Q., Wang, G., Gong, S., et al. (2019). Interleukin-37 ameliorates influenza pneumonia by attenuating macrophage cytokine production in a MAPK-dependent manner. Front. Microbiol. 10, 2482. doi:10.3389/fmicb.2019.02482
Rai, V., Dilisio, M. F., Samadi, F., and Agrawal, D. K. (2022). Counteractive effects of IL-33 and IL-37 on inflammation in osteoarthritis. Int. J. Environ. Res. Public Health 19 (9), 5690. doi:10.3390/ijerph19095690
Ravelli, A., Minoia, F., Davì, S., Horne, A., Bovis, F., Pistorio, A., et al. (2016). 2016 classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Ann. Rheum. Dis. 75 (3), 481–489. doi:10.1136/annrheumdis-2015-208982
Raychaudhuri, S., Sandor, C., Stahl, E. A., Freudenberg, J., Lee, H. S., Jia, X., et al. (2012). Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 44 (3), 291–296. doi:10.1038/ng.1076
Reboul, P., Pelletier, J. P., Tardif, G., Cloutier, J. M., and Martel-Pelletier, J. (1996). The new collagenase, collagenase-3, is expressed and synthesized by human chondrocytes but not by synoviocytes. A role in osteoarthritis. J. Clin. Invest. 97 (9), 2011–2019. doi:10.1172/jci118636
Regueiro, C., Casares-Marfil, D., Lundberg, K., Knevel, R., Acosta-Herrera, M., Rodriguez-Rodriguez, L., et al. (2021). HLA-B*08 identified as the most prominently associated major histocompatibility complex locus for anti-carbamylated protein antibody-positive/anti-cyclic citrullinated peptide-negative rheumatoid arthritis. Arthritis Rheumatol. 73 (6), 963–969. doi:10.1002/art.41630
Reid, F., Singh, D., Albayaty, M., Moate, R., Jimenez, E., Sadiq, M. W., et al. (2024). A randomized phase I study of the anti-interleukin-33 antibody Tozorakimab in healthy adults and patients with chronic obstructive pulmonary disease. Clin. Pharmacol. Ther. 115 (3), 565–575. doi:10.1002/cpt.3147
Ren, C., Chen, J., Che, Q., Jia, Q., Lu, H., Qi, X., et al. (2023). IL-37 alleviates TNF-α-induced pyroptosis of rheumatoid arthritis fibroblast-like synoviocytes by inhibiting the NF-κB/GSDMD signaling pathway. Immunobiology 228 (3), 152382. doi:10.1016/j.imbio.2023.152382
Rex, D. A. B., Agarwal, N., Prasad, T. S. K., Kandasamy, R. K., Subbannayya, Y., and Pinto, S. M. (2020). A comprehensive pathway map of IL-18-mediated signalling. J. Cell. Commun. Signal 14 (2), 257–266. doi:10.1007/s12079-019-00544-4
Rong, H., He, X., Wang, L., Bai, M., Jin, T., Wang, Y., et al. (2020). Association between IL1B polymorphisms and the risk of rheumatoid arthritis. Int. Immunopharmacol. 83, 106401. doi:10.1016/j.intimp.2020.106401
Roux, S., and Orcel, P. (2000). Bone loss. Factors that regulate osteoclast differentiation: an update. Arthritis Res. 2 (6), 451–456. doi:10.1186/ar127
Ruperto, N., Quartier, P., Wulffraat, N., Woo, P., Ravelli, A., Mouy, R., et al. (2012). A phase II, multicenter, open-label study evaluating dosing and preliminary safety and efficacy of canakinumab in systemic juvenile idiopathic arthritis with active systemic features. Arthritis Rheum. 64 (2), 557–567. doi:10.1002/art.33342
Ruscitti, P., Berardicurti, O., Cipriani, P., and Giacomelli, R.TRACK study group (2021). Benefits of anakinra versus TNF inhibitors in rheumatoid arthritis and type 2 diabetes: long-term findings from participants furtherly followed-up in the TRACK study, a multicentre, open-label, randomised, controlled trial. Clin. Exp. Rheumatol. 39 (2), 403–406. doi:10.55563/clinexprheumatol/phsqg7
Ruscitti, P., Masedu, F., Alvaro, S., Airò, P., Battafarano, N., Cantarini, L., et al. (2019). Anti-interleukin-1 treatment in patients with rheumatoid arthritis and type 2 diabetes (TRACK): a multicentre, open-label, randomised controlled trial. PLoS Med. 16 (9), e1002901. doi:10.1371/journal.pmed.1002901
Saag, K. G., Khanna, P. P., Keenan, R. T., Ohlman, S., Osterling Koskinen, L., Sparve, E., et al. (2021). A randomized, phase II study evaluating the efficacy and safety of anakinra in the treatment of gout flares. Arthritis Rheumatol. 73 (8), 1533–1542. doi:10.1002/art.41699
Sattin, M., and Towheed, T. (2016). The effect of TNFα-inhibitors on cardiovascular events in patients with rheumatoid arthritis: an updated systematic review of the literature. Curr. Rheumatol. Rev. 12 (3), 208–222. doi:10.2174/1573397112666160404124655
Schmitz, J., Owyang, A., Oldham, E., Song, Y., Murphy, E., McClanahan, T. K., et al. (2005). IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23 (5), 479–490. doi:10.1016/j.immuni.2005.09.015
Scott, I. C., Ibrahim, F., Simpson, G., Kowalczyk, A., White-Alao, B., Hassell, A., et al. (2016). A randomised trial evaluating anakinra in early active rheumatoid arthritis. Clin. Exp. Rheumatol. 34 (1), 88–93. Available online at: https://www.clinexprheumatol.org/abstract.asp?a=9346
Selmi, C., Ceribelli, A., Naguwa, S. M., Cantarini, L., and Shoenfeld, Y. (2015). Safety issues and concerns of new immunomodulators in rheumatology. Expert Opin. Drug Saf. 14 (3), 389–399. doi:10.1517/14740338.2015.993605
Shimizu, K., Nakajima, A., Sudo, K., Liu, Y., Mizoroki, A., Ikarashi, T., et al. (2015). IL-1 receptor type 2 suppresses collagen-induced arthritis by inhibiting IL-1 signal on macrophages. J. Immunol. 194 (7), 3156–3168. doi:10.4049/jimmunol.1402155
Shimizu, M., Nakagishi, Y., and Yachie, A. (2013). Distinct subsets of patients with systemic juvenile idiopathic arthritis based on their cytokine profiles. Cytokine 61 (2), 345–348. doi:10.1016/j.cyto.2012.11.025
Singh, J. A., Hossain, A., Tanjong Ghogomu, E., Kotb, A., Christensen, R., Mudano, A. S., et al. (2016). Biologics or tofacitinib for rheumatoid arthritis in incomplete responders to methotrexate or other traditional disease-modifying anti-rheumatic drugs: a systematic review and network meta-analysis. Cochrane Database Syst. Rev. 2016 (5), Cd012183. doi:10.1002/14651858.Cd012183
Smolen, J. S. (2020). Insights into the treatment of rheumatoid arthritis: a paradigm in medicine. J. Autoimmun. 110, 102425. doi:10.1016/j.jaut.2020.102425
Son, H. S., Lee, J., Lee, H. I., Kim, N., Jo, Y. J., Lee, G. R., et al. (2020). Benzydamine inhibits osteoclast differentiation and bone resorption via down-regulation of interleukin-1 β expression. Acta Pharm. Sin. B 10 (3), 462–474. doi:10.1016/j.apsb.2019.11.004
Song, P., Yang, C., Thomsen, J. S., Dagnæs-Hansen, F., Jakobsen, M., Brüel, A., et al. (2019). Lipidoid-siRNA nanoparticle-mediated IL-1β gene silencing for systemic arthritis therapy in a mouse model. Mol. Ther. 27 (8), 1424–1435. doi:10.1016/j.ymthe.2019.05.002
Stahl, E. A., Raychaudhuri, S., Remmers, E. F., Xie, G., Eyre, S., Thomson, B. P., et al. (2010). Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 42 (6), 508–514. doi:10.1038/ng.582
Stolt, P., Bengtsson, C., Nordmark, B., Lindblad, S., Lundberg, I., Klareskog, L., et al. (2003). Quantification of the influence of cigarette smoking on rheumatoid arthritis: results from a population based case-control study, using incident cases. Ann. Rheum. Dis. 62 (9), 835–841. doi:10.1136/ard.62.9.835
Tan, L. K., Too, C. L., Diaz-Gallo, L. M., Wahinuddin, S., Lau, I. S., Heselynn, H., et al. (2021). The spectrum of association in HLA region with rheumatoid arthritis in a diverse Asian population: evidence from the MyEIRA case-control study. Arthritis Res. Ther. 23 (1), 46. doi:10.1186/s13075-021-02431-z
Terao, C., Brynedal, B., Chen, Z., Jiang, X., Westerlind, H., Hansson, M., et al. (2019). Distinct HLA associations with rheumatoid arthritis subsets defined by serological subphenotype. Am. J. Hum. Genet. 105 (3), 616–624. doi:10.1016/j.ajhg.2019.08.002
Tesser, J., Fleischmann, R., Dore, R., Bennett, R., Solinger, A., Joh, T., et al. (2004). Concomitant medication use in a large, international, multicenter, placebo controlled trial of anakinra, a recombinant interleukin 1 receptor antagonist, in patients with rheumatoid arthritis. J. Rheumatol. 31 (4), 649–654. Available online at: https://www.clinexprheumatol.org/abstract.asp?a=9346
Towne, J. E., Garka, K. E., Renshaw, B. R., Virca, G. D., and Sims, J. E. (2004). Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J. Biol. Chem. 279 (14), 13677–13688. doi:10.1074/jbc.M400117200
Towne, J. E., Renshaw, B. R., Douangpanya, J., Lipsky, B. P., Shen, M., Gabel, C. A., et al. (2011). Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J. Biol. Chem. 286 (49), 42594–42602. doi:10.1074/jbc.M111.267922
Tseng, H. W., Samuel, S. G., Schroder, K., Lévesque, J. P., and Alexander, K. A. (2022). Inflammasomes and the IL-1 family in bone homeostasis and disease. Curr. Osteoporos. Rep. 20 (3), 170–185. doi:10.1007/s11914-022-00729-8
van der Woude, D., Lie, B. A., Lundström, E., Balsa, A., Feitsma, A. L., Houwing-Duistermaat, J. J., et al. (2010). Protection against anti-citrullinated protein antibody-positive rheumatoid arthritis is predominantly associated with HLA-DRB1*1301: a meta-analysis of HLA-DRB1 associations with anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis in four European populations. Arthritis Rheum. 62 (5), 1236–1245. doi:10.1002/art.27366
Vasconcelos, L. B., Silva, M. T., and Galvao, T. F. (2020). Reduction of biologics in rheumatoid arthritis: a systematic review and meta-analysis. Rheumatol. Int. 40 (12), 1949–1959. doi:10.1007/s00296-020-04651-z
Vasilev, G., Manolova, I., Ivanova, M., Stanilov, I., Miteva, L., and Stanilova, S. (2021). The role of IL-18 in addition to Th17 cytokines in rheumatoid arthritis development and treatment in women. Sci. Rep. 11 (1), 15391. doi:10.1038/s41598-021-94841-x
Veenbergen, S., Smeets, R. L., Bennink, M. B., Arntz, O. J., Joosten, L. A., van den Berg, W. B., et al. (2010). The natural soluble form of IL-18 receptor beta exacerbates collagen-induced arthritis via modulation of T-cell immune responses. Ann. Rheum. Dis. 69 (1), 276–283. doi:10.1136/ard.2008.100867
Verri, W. A., Souto, F. O., Vieira, S. M., Almeida, S. C., Fukada, S. Y., Xu, D., et al. (2010). IL-33 induces neutrophil migration in rheumatoid arthritis and is a target of anti-TNF therapy. Ann. Rheum. Dis. 69 (9), 1697–1703. doi:10.1136/ard.2009.122655
Vigne, S., Palmer, G., Lamacchia, C., Martin, P., Talabot-Ayer, D., Rodriguez, E., et al. (2011). IL-36R ligands are potent regulators of dendritic and T cells. Blood 118 (22), 5813–5823. doi:10.1182/blood-2011-05-356873
Vigne, S., Palmer, G., Martin, P., Lamacchia, C., Strebel, D., Rodriguez, E., et al. (2012). IL-36 signaling amplifies Th1 responses by enhancing proliferation and Th1 polarization of naive CD4+ T cells. Blood 120 (17), 3478–3487. doi:10.1182/blood-2012-06-439026
Vincent, T. L. (2019). IL-1 in osteoarthritis: time for a critical review of the literature. F1000Res 8. doi:10.12688/f1000research.18831.1
Voronov, E., Dotan, S., Krelin, Y., Song, X., Elkabets, M., Carmi, Y., et al. (2013). Unique versus redundant functions of IL-1α and IL-1β in the tumor microenvironment. Front. Immunol. 4, 177. doi:10.3389/fimmu.2013.00177
Wang, S. Y., Liu, Y. Y., Ye, H., Guo, J. P., Li, R., Liu, X., et al. (2011). Circulating Dickkopf-1 is correlated with bone erosion and inflammation in rheumatoid arthritis. J. Rheumatol. 38 (5), 821–827. doi:10.3899/jrheum.100089
Wang, T., and He, C. (2020). TNF-Α and IL-6: the link between immune and bone system. Curr. Drug Targets 21 (3), 213–227. doi:10.2174/1389450120666190821161259
Wang, W. M., and Jin, H. Z. (2020). Biologics in the treatment of pustular psoriasis. Expert Opin. Drug Saf. 19 (8), 969–980. doi:10.1080/14740338.2020.1785427
Warren, R. B., Reich, A., Kaszuba, A., Placek, W., Griffiths, C. E. M., Zhou, J., et al. (2023). Imsidolimab, an anti-interleukin-36 receptor monoclonal antibody, for the treatment of generalized pustular psoriasis: results from the phase II GALLOP trial. Br. J. Dermatol 189 (2), 161–169. doi:10.1093/bjd/ljad083
Weber, B. N., Giles, J. T., and Liao, K. P. (2023). Shared inflammatory pathways of rheumatoid arthritis and atherosclerotic cardiovascular disease. Nat. Rev. Rheumatol. 19 (7), 417–428. doi:10.1038/s41584-023-00969-7
Wechsler, M. E., Ruddy, M. K., Pavord, I. D., Israel, E., Rabe, K. F., Ford, L. B., et al. (2021). Efficacy and safety of itepekimab in patients with moderate-to-severe asthma. N. Engl. J. Med. 385 (18), 1656–1668. doi:10.1056/NEJMoa2024257
Wikén, M., Hallén, B., Kullenberg, T., and Koskinen, L. O. (2018). Development and effect of antibodies to anakinra during treatment of severe CAPS: sub-analysis of a long-term safety and efficacy study. Clin. Rheumatol. 37 (12), 3381–3386. doi:10.1007/s10067-018-4196-x
Winkler, A., Sun, W., De, S., Jiao, A., Sharif, M. N., Symanowicz, P. T., et al. (2021). The interleukin-1 receptor-associated kinase 4 inhibitor PF-06650833 blocks inflammation in preclinical models of rheumatic disease and in humans enrolled in a randomized clinical trial. Arthritis Rheumatol. 73 (12), 2206–2218. doi:10.1002/art.41953
Wouters, F., Maurits, M. P., van Boheemen, L., Verstappen, M., Mankia, K., Matthijssen, X. M. E., et al. (2022). Determining in which pre-arthritis stage HLA-shared epitope alleles and smoking exert their effect on the development of rheumatoid arthritis. Ann. Rheum. Dis. 81 (1), 48–55. doi:10.1136/annrheumdis-2021-220546
Wu, F., Gao, J., Kang, J., Wang, X., Niu, Q., Liu, J., et al. (2021). B cells in rheumatoid arthritis:pathogenic mechanisms and treatment prospects. Front. Immunol. 12, 750753. doi:10.3389/fimmu.2021.750753
Xiao, Z. X., Miller, J. S., and Zheng, S. G. (2021). An updated advance of autoantibodies in autoimmune diseases. Autoimmun. Rev. 20 (2), 102743. doi:10.1016/j.autrev.2020.102743
Xie, L., Huang, Z., Li, H., Liu, X., Zheng, S., and Su, W. (2019). IL-38: a new player in inflammatory autoimmune disorders. Biomolecules 9 (8), 345. doi:10.3390/biom9080345
Xu, D., Jiang, H. R., Kewin, P., Li, Y., Mu, R., Fraser, A. R., et al. (2008). IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc. Natl. Acad. Sci. U. S. A. 105 (31), 10913–10918. doi:10.1073/pnas.0801898105
Ye, L., Jiang, B., Deng, J., Du, J., Xiong, W., Guan, Y., et al. (2015). IL-37 alleviates rheumatoid arthritis by suppressing IL-17 and IL-17-triggering cytokine production and limiting Th17 cell proliferation. J. Immunol. 194 (11), 5110–5119. doi:10.4049/jimmunol.1401810
Yuan, F. L., Li, X., Lu, W. G., Li, C. W., Xu, R. S., and Dong, J. (2011). IL-33: a promising therapeutic target for rheumatoid arthritis? Expert Opin. Ther. Targets 15 (5), 529–534. doi:10.1517/14728222.2011.560838
Yuan, S., Li, X., Lin, A., and Larsson, S. C. (2022). Interleukins and rheumatoid arthritis: bi-directional Mendelian randomization investigation. Semin. Arthritis Rheum. 53, 151958. doi:10.1016/j.semarthrit.2022.151958
Zhang, J., Fang, X. Y., Wu, J., Fan, Y. G., Leng, R. X., Liu, B., et al. (2023). Association of combined exposure to ambient air pollutants, genetic risk, and incident rheumatoid arthritis: a prospective cohort study in the UK biobank. Environ. Health Perspect. 131 (3), 37008. doi:10.1289/ehp10710
Zhang, W., Cong, X. L., Qin, Y. H., He, Z. W., He, D. Y., and Dai, S. M. (2013). IL-18 upregulates the production of key regulators of osteoclastogenesis from fibroblast-like synoviocytes in rheumatoid arthritis. Inflammation 36 (1), 103–109. doi:10.1007/s10753-012-9524-8
Zhao, L., Zhao, T., Yang, X., Cao, L., Xu, R., Liu, J., et al. (2022). IL-37 blocks gouty inflammation by shaping macrophages into a non-inflammatory phagocytic phenotype. Rheumatol. (Oxford) 61 (9), 3841–3853. doi:10.1093/rheumatology/keac009
Zhou, J., Xiao, Y., Ren, Y., Ge, J., and Wang, X. (2022). Structural basis of the IL-1 receptor TIR domain-mediated IL-1 signaling. iScience 25 (7), 104508. doi:10.1016/j.isci.2022.104508
Zhou, L., Todorovic, V., Kakavas, S., Sielaff, B., Medina, L., Wang, L., et al. (2018). Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation. J. Biol. Chem. 293 (2), 403–411. doi:10.1074/jbc.M117.805739
Zhou, P., Li, Q., Su, S., Dong, W., Zong, S., Ma, Q., et al. (2020). Interleukin 37 suppresses M1 macrophage polarization through inhibition of the Notch1 and nuclear factor kappa B pathways. Front. Cell. Dev. Biol. 8, 56. doi:10.3389/fcell.2020.00056
Keywords: rheumatoid arthritis (RA), IL-1, autoimmune diseases, cytokines, immunotherapy, monoclonal antibody
Citation: Wang K, Luo H, Liu L, Gao H, Song Y and Li D (2025) Blockade of IL-1 family cytokines in the treatment of rheumatoid arthritis. Front. Pharmacol. 16:1577628. doi: 10.3389/fphar.2025.1577628
Received: 16 February 2025; Accepted: 09 May 2025;
Published: 30 May 2025.
Edited by:
Pier Maria Fornasari, Regen Health Solutions, ItalyCopyright © 2025 Wang, Luo, Liu, Gao, Song and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanyan Song, eWFueWFuczEyMTlAamx1LmVkdS5jbg==; Dong Li, bGlkb25nMUBqbHUuZWR1LmNu