Abstract
Background:
Pulmonary arterial hypertension (PAH) is a severe cardiopulmonary disorder characterized by progressive elevation of pulmonary vascular resistance, resulting to right ventricular dysfunction and premature mortality. Although genetic mutations are increasingly recognized in PAH pathogenesis, cases involving digenic mutations remain exceptionally rare.
Case presentation:
We report the case of a 47-year-old female presenting with a 5-year history of exertional dyspnea, which progressively worsened over the preceding 2 months. Diagnostic imaging revealed pulmonary artery dilatation and right heart enlargement, and right heart catheterization confirmed PAH with a mean pulmonary arterial pressure of 43 mmHg. Whole exome sequencing identified a novel heterozygous mutation in FLNA (c.4754C>T, p.Thr1585Met) and a known heterozygous mutation in MMACHC (c.609G>A, p.Trp203Ter). The patient was initiated on PAH-specific therapy and pulmonary artery denervation (PADN) treatment. Over a 2-year follow-up period, her symptoms significantly improved, with no evidence of heart failure progression.
Conclusion:
This case highlights a rare instance of PAH associated with digenic mutations in FLNA and MMACHC. The patient demonstrated a favorable response to targeted PAH therapy and PADN treatment, highlighting the importance of genetic screening and personalized treatment strategies in PAH management.
Background
Pulmonary arterial hypertension (PAH) is a progressive and life-threatening disorder defined by a mean pulmonary artery pressure (mPAP) exceeding 20 mmHg at rest, accompanied by elevated pulmonary vascular resistance and right ventricular dysfunction (Humbert et al., 2023). The disease is driven by pathological remodeling of the pulmonary vasculature, ultimately leading to right heart failure and premature death. While the etiology of PAH is multifactorial, involving genetic, inflammatory, metabolic, and hemodynamic factors, the role of genetic mutations has garnered increasing attention. Mutations in genes such as BMPR2, ALK1, and ENG are well-documented in heritable PAH (Cuthbertson et al., 2023; Welch et al., 2023; Al Tabosh et al., 2024). However, cases involving digenic mutations are exceedingly rare. Here, we present a unique case of PAH associated with a novel heterozygous mutation in FLNA and a known heterozygous mutation in MMACHC. We observed that PAH-targeted therapies and pulmonary artery denervation (PADN) treatment were well tolerated by our patient and did not lead to heart failure during a 2-year follow-up period.
Case presentation
A 47-year-old female was referred to our center with a 5-year history of progressive exertional dyspnea, which had significantly worsened over the preceding 2 months. The patient denied any family history of pulmonary hypertension or cardiopulmonary diseases. Her medical history included intellectual disability, schizophrenia, epilepsy, dysaudia, and hypothyroidism. Her father passed away due to an illness in his early years, with the exact cause remaining unknown. She was a teetotaler, nonsmoker and had no history of addictive drug use or other PH-associated risk factors. Her preliminary examination revealed the following physical examination parameters: blood pressure of 103/70 mmHg, heart rate of 71 bpm, respiratory rate of 19 breaths/min, oxygen saturation of 96% on room air, and New York Heart Association functional class III heart failure. Cardiac auscultation revealed an accentuated second pulmonic sound and diminished breath sounds bilaterally. Mild pitting edema was noted in the lower extremities. The laboratory examination results were as follows: cardiac troponin I (cTn I) 145 pg/mL, D-dimer 642 ng/mL, and NT-proBNP 3,026 pg/mL. Other laboratory values, such as erythrocyte sedimentation rate, HIV, C reactive protein, rheumatic factor, antinuclear antibody, antideoxyribonuclease, complements, lipoprotein A, factors II (prothrombin), VII, VIII, IX, XI, and XII, protein C, protein S, antiphos-pholipid antibody antibodies, antiphospholipid antibody, and anti-vasculitis antibody, were within the normal range.
To exclude pulmonary embolism, we performed cardiac MR image (MRI) and pulmonary ventilation perfusion imaging, and revealed pulmonary artery dilatation and right heart enlargement, which may indicate pulmonary arterial hypertension. Transthoracic echocardiography confirmed severe pulmonary hypertension, with an estimated pulmonary artery systolic pressure of 102 mmHg, preserved LVEF (62%), reduced right ventricular wall motion, and severe tricuspid regurgitation (Figure 1). The initial MR and MR angiography of the brain showed no significant abnormalities. A subsequent right heart catheterization at rest confirmed the diagnosis of PAH: the patient had a pulmonary artery pressure of 66/31/43 mmHg, a pulmonary artery wedge pressure of 11 mmHg, a cardiac output of 4.25 L/min and a calculated pulmonary vascular resistance (PVR) of 5.56 Wood units, total pulmonary resistance (TPR) of 7.49 Wood units. Because our patient demonstrated a high pulmonary artery pressure and the cause is unknown. Hence, whole exome sequencing was carried out, and we identified a novel heterozygous mutation in FLNA (c.4754C>T, p.Thr1585Met) and a known heterozygous mutation in MMACHC (c.609G>A, p.Trp203Ter) (Figure 2). In addition, Pedigree analysis of the patient’s son revealed no evidence of these mutations (Figure 2). Following multidisciplinary discussion and being fully informed about the risks to the patient, the patient was initiated on PAH-specific therapy, including tadalafil (20 mg daily), macitentan (10 mg daily), and pulmonary artery denervation (PADN) treatment (Figure 3). Her therapeutic regimens were continued for the next 2 years with periodic follow-up. The timeline of treatment course for this patient was shown in (Table 1). Surprisingly, she showed a satisfactory clinical response to PAH therapies, her disease symptoms were alleviated, and no sign of heart failure was observed during the follow-up period.
FIGURE 1
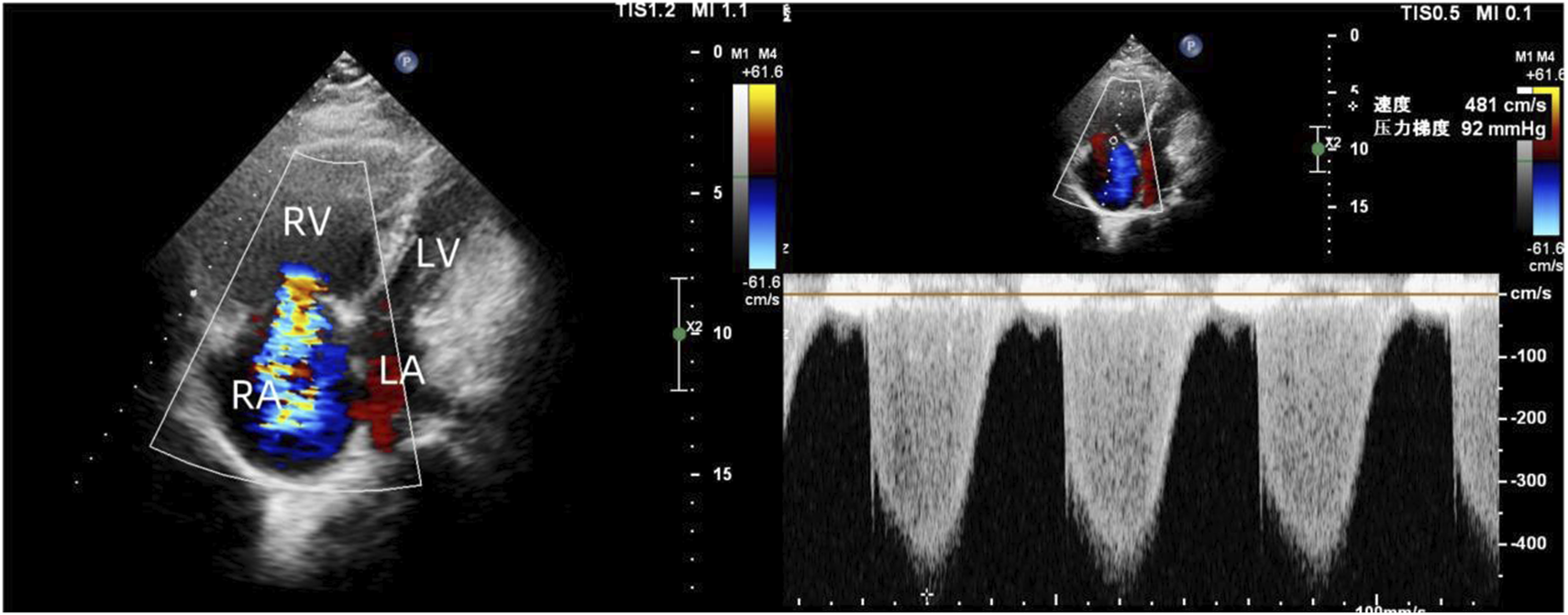
Transthoracic echocardiography revealed enlarged right heart, reduced right ventricular wall motion, and severe tricuspid regurgitation.
FIGURE 2
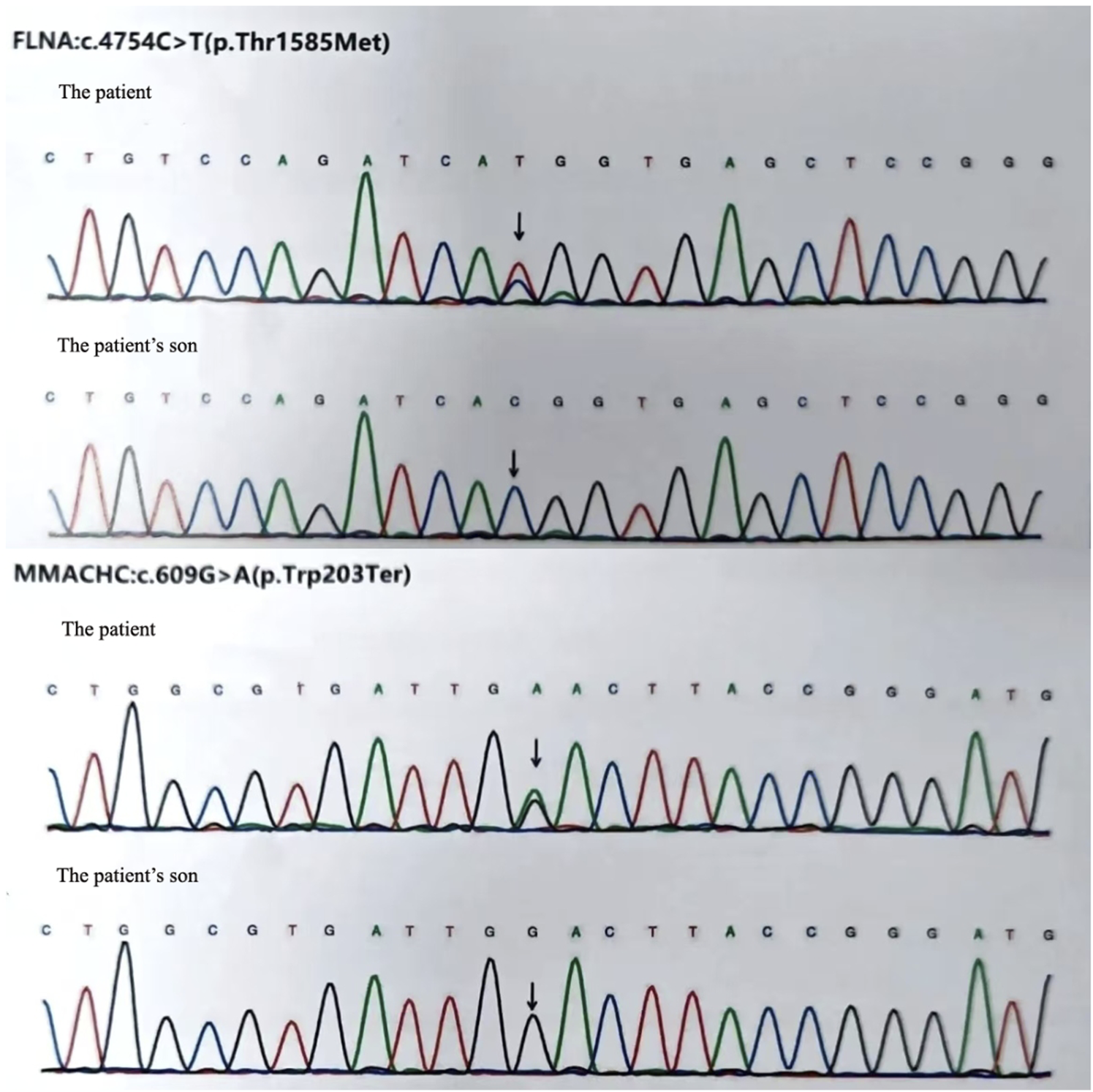
We identified a novel heterozygous mutation in FLNA (c.4754C>T, p.Thr1585Met) and a known heterozygous mutation in MMACHC (c.609G>A, p.Trp203Ter) in our patient. But the patient’s son revealed no evidence of these mutations.
FIGURE 3
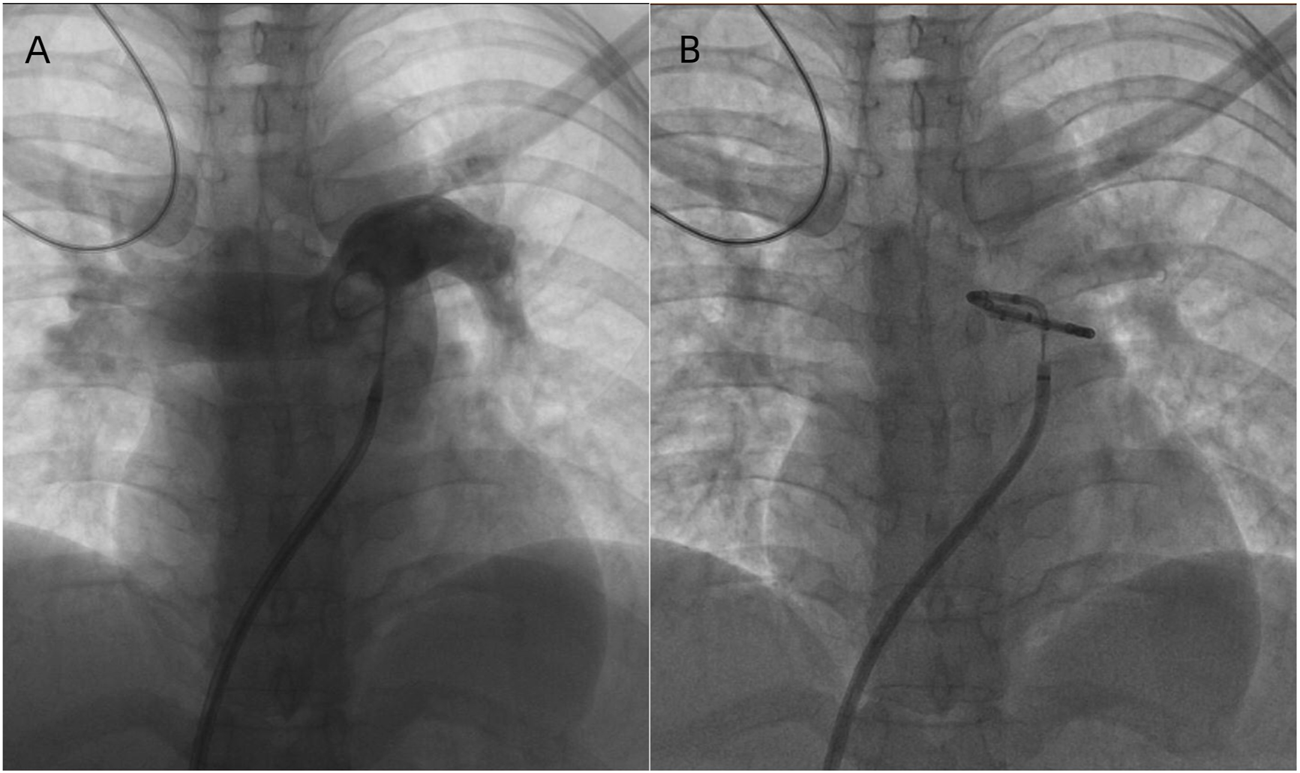
(A) Pulmonary arterial angiography and Identifying the ablation sites. (B) PA ablation is performed in the periconjunctional area between the distal main trunk and the ostial left branch.
TABLE 1
| Time of admission | Before PAH-targeted therapies | PAH-targeted therapies for 1 year | PAH-targeted therapies for 2 years |
|---|---|---|---|
| LA (cm) | 3 | 3.1 | 3 |
| LV (cm) | 2.7 | 3.6 | 3.8 |
| RA (cm) | 4.9 | 4.8 | 4.7 |
| RV (cm) | 4.8 | 4.6 | 4.5 |
| EF (%) | 53 | 69 | 68 |
| TAPSE (cm) | 1.4 | 1.6 | 1.9 |
| FAC (%) | 19 | 20 | 22 |
| TR (m/s) | 4.8 | 4.2 | 3.8 |
| RAP (mmHg) | 8 | 5 | 3 |
| PAP (mmHg) | 62/31/43 | 53/28/36 | 46/17/27 |
| RVSP (mmHg) | 58 | 50 | 47 |
| PCWP (mmHg) | 12 | 10 | 10 |
| TPR (WU) | 7.49 | 4.31 | 3.63 |
| PVR (WU) | 5.56 | 3.12 | 2.27 |
| CO (L/min) | 4.25 | 9.89 | 10.1 |
| CI (L/min/m2) | 2.53 | 5.73 | 6.5 |
| 6MWD (m) | 338 | 370 | 420 |
| NT-proBNP (pg/mL) | 3,358 | 1,025 | 336 |
| Functional class (NYHA) | III | II | II |
| PAH-target therapies | tadalafil (20 mg, qd), macitentan (10 mg, qd), and PADN | tadalafil (20 mg, qd), macitentan (10 mg, qd) | tadalafil (20 mg, qd), macitentan (10 mg, qd) |
The timeline of treatment course for this patient.
EF, was measured for the left ventricle; RA, RV, LA, LV, were determined by subxiphoid four chamber section; EF, ejection function; NYHA, New York heart association; NT-proBNP N-terminal pro-brain natriuretic peptide; PAH, pulmonary artery hypertension; RA, right atria; RV, right ventricular; LA, left atria; LV, left ventricular; RVSP, right ventricular systolic pressure; TAPSE, tricuspid annular plane systolic excursion; FAC, fractional area change; RAP, right atrial pressure; PAP, pulmonary arterial pressure; PCWP, pulmonary capillary wedge pressure; TPR, total pulmonary resistance; PVR, pulmonary vascular resistance; CO, cardiac output; CI, cardiac index; 6MWD, 6-min walk distance.
Discussion
PAH is a fatal cardiovascular disease, often referred to as malignancy of the cardiovascular system. It is a progressive and life-threatening disease driven by the remodeling of peripheral pulmonary arteries, leading to arterial occlusion, increased pulmonary vascular resistance, and elevated mPAP (Grünig et al., 2020). Its hallmark is sustained elevated pulmonary arterial pressure, ultimately leading to right ventricular hypertrophy, heart failure, and eventually death (Eichstaedt et al., 2023). Epidemiological studies have shown that the incidence and prevalence of PAH are 6 and 48–55 cases per million adults, respectively (Leber et al., 2021). The prevalence of PAH is higher in women between the ages of 30 and 60 years (Levine, 2021). The incidence rate among male patients is relatively low, yet their prognosis is often poor. Exercise intolerance is usually the main symptom in those with PAH, seriously affecting their quality of life. Untreated, the median survival is approximately 2.5 years. Although the pathogenesis of pulmonary arterial hypertension is complex, with the development of molecular genetics technology in recent years, the role of gene mutations in PAH has gradually been confirmed, and more and more PAH related genes are becoming well-known. While mutations in BMPR2 account for the majority of heritable PAH cases, emerging evidence suggests that digenic or oligogenic mutations may contribute to disease severity and phenotypic variability. We are introducing a 47 year old middle-aged female patient who was admitted to the hospital due to long-term chest tightness and shortness of breath. Through genetic testing, it was found that severe pulmonary arterial hypertension was caused by digenic mutations in FLNA and MMACHC. After standardized drug treatment, the patient’s condition improved significantly.
FLNA encodes filamin A, a cytoskeletal protein plays a critical role in cell migration, signaling, and vascular remodeling. FLNA gene mutations can cause periventricular nodular heterotopia, which is an X-linked dominant genetic disease in which neurons fail to migrate to the cerebral cortex (Zhou et al., 2021). As well as neurological manifestations, other abnormalities, particularly cardiovascular symptoms and lung diseases, especially pulmonary hypertension, are also common in patients with FLNA mutations (Bandaru et al., 2021). Its main clinical manifestations include: bicuspid aortic valve, patent ductus arteriosus, refractory epileptic seizures, intellectual disability, neuronal migration disorders, etc. Previous case reports have also found that a family with heritable PAH carried a novel heterozygous splicing mutation in the FLNA gene (Hirashiki et al., 2017). Zheng et al. revealed that FLNA knockdown reduced PASMc proliferation and migration, whereas FLNA overexpression promoted cell proliferation and migration (Zheng et al., 2023). These indicated that FLNA was involved in SMc proliferation and migration, and served a key role in maintaining arterial myogenic tone and vascular remodeling in PAH (Zheng et al., 2023). The novel FLNA mutation identified in this patient (c.4754C>T, p.Thr1585Met) likely disrupted protein function, contributing to pulmonary vascular remodeling and PAH progression. Her medical history included intellectual disability, schizophrenia, epilepsy, hearing impairment, and hypothyroidism, which is consistent with the clinical manifestations of FLNA gene mutations. MMACHC gene deficiency leads to autosomal recessive methylmalonic aciduria combined with hypercystinuria (Zhang et al., 2022). The MMACHC gene is located on chromosome 1p34.1, more than 75 mutations have been reported, but the exact function of the MMACHC protein is still unclear (Lerner-Ellis et al., 2006). The common clinical manifestations of MMACHC gene mutations include developmental delay, intellectual disability, hypotonia, visual impairment, and hematological manifestations. But there are also literature reports that this gene defect may lead to cardiac and pulmonary dysfunction, thereby causing pulmonary arterial hypertension (Hao et al., 2024). Since the discovery of the MMACHC gene, mutations have been identified in over 300 patients. The most common genetic abnormality is the c.271dupA, which accounts for more than 40% of mutant alleles (Gündüz et al., 2014). While the MMACHC mutation (c.609G>A, p.Trp203Ter) identified in our patient is not classically linked to PAH, its potential role in pulmonary vascular dysfunction warrants further investigation. Our patient exhibits PAH resulting from a heterozygous mutation in two genes. Furthermore, the patient’s favorable response to PAH-specific therapy underscores the importance of early diagnosis and targeted treatment. Genetic screening not only aids in identifying mutation carriers but also provides insights into disease mechanisms, enabling personalized therapeutic approaches. Based on this, we also conducted whole exome sequencing on the patient’s son, but did not identify any carriers of the aforementioned variant genes.
Sympathetic overactivation plays a critical role in elevating PAP. By impairing neural transmission through targeted damage to pulmonary arterial sympathetic nerves, PAP subsequently decreases. This substantial evidence suggests that PADN represents a promising therapeutic approach for pulmonary hypertension. Current indications for PADN treatment are limited to Group 1, 2, and 4 PH according to the World Health Organization (WHO) clinical classification system. Preclinical studies indicated that PADN treatment can significantly reduce mPAP, significantly reduce NTproBNP, significantly improve RV function, and significantly increase 6-MWD (Leopold, 2022). This is consistent with the follow-up results of our patients after treatment. The pulmonary vasculature is densely innervated by sympathetic, parasympathetic, and sensory nerve fibers (Zhang et al., 2022). PAH is associated with hyperactive sympathetic nervous system (Zhang et al., 2022). Excessive activation of the sympathetic nervous system can lead to pulmonary arteriolar vasoconstriction and vascular remodeling. Therefore, PADN treatment for patients with PAH can significantly reduce PAP and improve patient prognosis.
Limitations and future directions
As a single-case report, our findings require validation in larger cohorts. Multicenter studies screening PAH patients for FLNA/MMACHC mutations are warranted to confirm this digenic association.The mechanistic interplay between FLNA (cytoskeletal remodeling) and MMACHC (metabolic dysfunction) mutations remains speculative. We hypothesize that FLNA-mediated vascular instability may synergize with MMACHC-related mitochondrial dysfunction (impaired energy metabolism in pulmonary artery smooth muscle cells), but this needs experimental verification. PADN was selected over triple therapy due to the patient’s suboptimal response to dual agents (persistent NYHA III after 1 month) and right heart strain on repea echocardiography. While PADN showed efficacy here, its application should be limited to experienced centers until randomized trials establish safety profiles across PAH subtypes.
Conclusion
This case highlights the rare occurrence of PAH associated with digenic mutations in FLNA and MMACHC. The patient demonstrated significant clinical improvement following a combination of targeted PAH therapy and PADN, underscoring the therapeutic potential of PADN in managing severe PAH. The reduction in mean mPAP and improvement in right ventricular function observed in this case align with preclinical evidence supporting PADN as a promising intervention for PAH. These findings emphasize the importance of integrating genetic screening with advanced therapeutic strategies, such as PADN, to optimize outcomes in PAH management. Further research is warranted to elucidate the mechanisms underlying PADN’s efficacy and its potential role in broader PAH patient populations.
Statements
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by The study was approved by the Ethics Committee of the Central People’s Hospital of Yichang. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
Z-qZ: Writing – original draft, Writing – review and editing. Z-qQ: Writing – review and editing, Investigation. S-kZ: Writing – review and editing, Data curation. Y-hZ: Data curation, Writing – review and editing. J-wD: Data curation, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (Grant no. 81770456) and the Medical and Health Research Project of Yichang city, China (Grant no. A21-2-015).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Al Tabosh T. Liu H. Koça D. Al Tarrass M. Tu L. Giraud S. et al (2024). Impact of heterozygous ALK1 mutations on the transcriptomic response to BMP9 and BMP10 in endothelial cells from hereditary hemorrhagic telangiectasia and pulmonary arterial hypertension donors. Angiogenesis27, 211–227. 10.1007/s10456-023-09902-8
2
Bandaru S. Ala C. Zhou A. X. Akyürek L. M. (2021). Filamin A regulates cardiovascular remodeling. Int. J. Mol. Sci.22, 6555. 10.3390/ijms22126555
3
Cuthbertson I. Morrell N. W. Caruso P. (2023). BMPR2 mutation and metabolic reprogramming in pulmonary arterial hypertension. Circ. Res.132, 109–126. 10.1161/CIRCRESAHA.122.321554
4
Eichstaedt C. A. Belge C. Chung W. K. Gräf S. Grünig E. Montani D. et al (2023). Genetic counselling and testing in pulmonary arterial hypertension: a consensus statement on behalf of the international consortium for genetic studies in PAH. Eur. Respir. J.61, 2201471. 10.1183/13993003.01471-2022
5
Grünig E. Eichstaedt C. A. Seeger R. Benjamin N. (2020). Right heart size and right ventricular reserve in pulmonary hypertension: impact on management and prognosis. Diagn. (Basel)10, 1110. 10.3390/diagnostics10121110
6
Gündüz M. Ekici F. Özaydın E. Ceylaner S. Perez B. (2014). Reversible pulmonary arterial hypertension in cobalamin-dependent cobalamin C disease due to a novel mutation in the MMACHC gene. Eur. J. Pediatr.173, 1707–1710. 10.1007/s00431-014-2330-6
7
Hao L. Liang L. Gao X. Zhan X. Ji W. Chen T. et al (2024). Screening of 1.17 million newborns for inborn errors of metabolism using tandem mass spectrometry in Shanghai, China: a 19-year report. Mol. Genet. Metab.141, 108098. 10.1016/j.ymgme.2023.108098
8
Hirashiki A. Adachi S. Nakano Y. Kamimura Y. Ogo T. Nakanishi N. et al (2017). Left main coronary artery compression by a dilated main pulmonary artery and left coronary sinus of valsalva aneurysm in a patient with heritable pulmonary arterial hypertension and FLNA mutation. Pulm. Circ.7, 734–740. 10.1177/2045893217716107
9
Humbert M. Kovacs G. Hoeper M. M. Badagliacca R. Berger R. Brida M. et al (2023). 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J.61, 2200879. 10.1183/13993003.00879-2022
10
Leber L. Beaudet A. Muller A. (2021). Epidemiology of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: identification of the Most accurate estimates from a systematic literature review. Pulm. Circ.11, 2045894020977300. 10.1177/2045894020977300
11
Leopold J. A. (2022). Pulmonary artery denervation: a new therapeutic for pulmonary arterial hypertension. JACC Cardiovasc Interv.15, 2424–2426. 10.1016/j.jcin.2022.10.004
12
Lerner-Ellis J. P. Tirone J. C. Pawelek P. D. Doré C. Atkinson J. L. Watkins D. et al (2006). Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat. Genet.38, 93–100. 10.1038/ng1683
13
Levine D. J. (2021). Pulmonary arterial hypertension: updates in epidemiology and evaluation of patients. Am. J. Manag. Care27, S35–S41. 10.37765/ajmc.2021.88609
14
Welch C. L. Aldred M. A. Balachandar S. Dooijes D. Eichstaedt C. A. Gräf S. et al (2023). Defining the clinical validity of genes reported to cause pulmonary arterial hypertension. Genet. Med.25, 100925. 10.1016/j.gim.2023.100925
15
Zhang H. Wang Y. Qiu Y. Zhang C. (2022). Expanded newborn screening for inherited metabolic disorders by tandem mass spectrometry in a northern Chinese population. Front. Genet.13, 801447. 10.3389/fgene.2022.801447
16
Zhang H. Wei Y. Zhang C. Yang Z. Kan J. Gu H. et al (2022). Pulmonary artery denervation for pulmonary arterial hypertension: a sham-controlled randomized PADN-CFDA trial. JACC Cardiovasc Interv.15, 2412–2423. 10.1016/j.jcin.2022.09.013
17
Zheng Y. Ma H. Yan Y. Ye P. Yu W. Lin S. et al (2023). Deficiency of filamin A in smooth muscle cells protects against hypoxia-mediated pulmonary hypertension in mice. Int. J. Mol. Med.51, 22. 10.3892/ijmm.2023.5225
18
Zhou J. Kang X. An H. Lv Y. Liu X. (2021). The function and pathogenic mechanism of filamin A. Gene784, 145575. 10.1016/j.gene.2021.145575
Summary
Keywords
pulmonary arterial hypertension, digenic mutations, genetic pathogenesis, heart failure, case report
Citation
Zhang Z-q, Qin Z-q, Zhu S-k, Zuo Y-h and Ding J-w (2025) A case report of ‘Two-Hit’ digenic mutations in PAH: role of PADN in management. Front. Pharmacol. 16:1601777. doi: 10.3389/fphar.2025.1601777
Received
28 March 2025
Accepted
04 July 2025
Published
15 July 2025
Volume
16 - 2025
Edited by
Indira Pokkunuri, University of Houston–Downtown, United States
Reviewed by
Silvio Zaina, University of Guanajuato, Mexico
Tengteng Zhu, Central South University, China
Updates
Copyright
© 2025 Zhang, Qin, Zhu, Zuo and Ding.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia-wang Ding, dingjiawang@163.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.