 Ji Yang1†
Ji Yang1† Wei Liu
Wei Liu Xiaolin Ren
Xiaolin Ren- 1College of Laboratory Medicine, Chengdu Medical College, Chengdu, China
- 2Department of Clinical Medicine, Chengdu Medical College, Chengdu, China
- 3Pidu District Hospital of Traditional Chinese Medicine, Chengdu, China
- 4School of Bioscience and Technology, Chengdu Medical College, Chengdu, China
- 5The Second Affiliated Hospital of Chengdu Medical College·Nuclear Industry 416 Hospital, Chengdu, China
- 6Key Laboratory of Target Discovery and Protein Drug Development in Major Diseases of Sichuan Higher Education Institutes, Chengdu, China
Background: Current therapeutic options for inflammatory bowel disease (IBD) remain suboptimal due to limited efficacy, significant side effects, and high relapse rates, necessitating novel treatment strategies. Lumefantrine, a clinically established antimalarial drug, emerges as a compelling repurposing candidate based on its putative anti-inflammatory activity, though its efficacy and mechanism in IBD remain unexplored.
Methods: A murine IBD model was induced by 3% dextran sulfate sodium (DSS). Mice received oral Lumefantrine (20 mg/kg/day) for 7 days. Disease progression was monitored via disease activity index (DAI) scoring and histological analysis. Serum cytokines (IL-1β, IL-6, TNF-α) and colonic inflammatory mediators (Cox-2, iNos) were quantified by ELISA and qPCR. Tight junction proteins (Claudin-1, ZO-1) were assessed by immunohistochemistry and Western blot. Molecular targets were identified through computational docking and pull-down assays. Additionally, NF-κB signaling modulation was assessed in lipopolysaccharide (LPS)-stimulated intestinal epithelial cells (IEC-6 and NCM460) via Western blot analysis.
Results: Oral administration of Lumefantrine significantly attenuated disease activity index (DAI) scores and restored intestinal barrier integrity through upregulation of epithelial tight junction proteins Claudin-1 and ZO-1. Treated mice exhibited reduced serum levels of IL-1β, IL-6 and TNF-α, along with decreased colonic expression of inflammatory mediators cyclooxygenase-2 (Cox-2) and inducible nitric oxide synthase (iNos). Computational and experimental approaches identified FLI-1 a transcription factor upregulated in IBD colon tissues as Lumefantrine’s direct binding target. This interaction mediated suppression of NF-κB signaling, specifically downregulating phosphorylation of IκBα and p65 in LPS-stimulated intestinal epithelial cells.
Conclusion: Lumefantrine ameliorates experimental colitis through FLI-1-dependent inhibition of the NF-κB pathway, demonstrating high repurposing potential as an IBD therapeutic.
1 Introduction
Inflammatory bowel disease (IBD), encompassing ulcerative colitis (UC) and Crohn’s disease (CD), is a chronic, immune-mediated disorder characterized by relapsing gastrointestinal inflammation (Hodson, 2016; Flynn and Eisenstein, 2019). Its complex, multifactorial etiology culminates in immune dysregulation, persistent mucosal inflammation, epithelial barrier dysfunction, and increased colorectal cancer risk (Agrawal et al., 2022; Baumgart and Carding, 2007; Terzić et al., 2010).
Central to IBD pathogenesis is the aberrant activation of the NF-κB signaling pathway, a master regulator of inflammation. Constitutive NF-κB activation in IBD tissues drives the overexpression of key pro-inflammatory cytokines (e.g., IL-1β, IL-6, TNF-α) and mediators (e.g., COX-2, iNOS), perpetuating tissue damage and inflammation (Biasi et al., 2013; Mussbacher et al., 2023; Mukherjee et al., 2024). Furthermore, this inflammatory cascade disrupts intestinal barrier integrity by downregulating tight junction proteins such as Claudin-1 and ZO-1 (zona occludens protein 1) (Cui et al., 2021).
While current therapies (aminosalicylates, immunosuppressants, glucocorticoids, biologics, and antibiotics) offer moderate efficacy (Lamb et al., 2019), the relapsing-remitting nature of IBD necessitates repeated interventions, exposing patients to significant risks of progression and treatment-related complications (Herlihy and Feakins, 2022). Consequently, the development of novel therapeutic strategies remains imperative, with targeting the NF-κB axis continuing to represent a highly promising approach.
Drug repurposing offers an efficient strategy to accelerate IBD drug discovery. Lumefantrine (chemical name: β-dibutylamine-[2,7-dichloro-9-p-chlorophenylmethyl-4-fluorene] ethanol), an FDA-approved antimalarial (Wiesner et al., 2003; Ippolito et al., 2017), emerges as a compelling candidate. Beyond its primary use, recent studies indicate Lumefantrine inhibits the transcription factor FLI-1 (friend leukemia virus integration 1), modulating processes like extracellular matrix remodeling (Rajesh et al., 2020). Critically, FLI-1 has been shown to regulate NF-κB component expression (NF-κB1/p50 and RelA/p65) (Sartori et al., 2021). These findings raise the possibility that Lumefantrine might suppress pathogenic NF-κB signaling in IBD through FLI-1 inhibition, warranting experimental verification.
Therefore, we hypothesized that Lumefantrine attenuates IBD by targeting FLI-1, thereby inhibiting NF-κB activation. This study aimed to evaluate Lumefantrine’s efficacy in a DSS-induced murine colitis model and elucidate its mechanism, focusing on FLI-1 targeting and NF-κB pathway modulation in intestinal epithelial cells.
2 Materials and methods
2.1 Chemicals and antibodies
Lumefantrine was purchased from Aladdin (Shanghai, China), Camptothecin (CPT) from Macklin (Shanghai, China), YK-4-279 from MedChemExpress (Shanghai, China), dextran sulfate sodium (DSS) from MP Biomedicals (Shanghai, China). The primary antibodies used were: Claudin-1 (Abcam, ab15098, Cambridge, United Kingdom), ZO-1 (CST, 8193P, Massachusetts, United States), p-p65 (Proteintech, 82335-1-RR, Wuhan, China), p65 (CST, 8242S, Massachusetts, United States), p-IκB (Proteintech, 82349-1-RR, Wuhan, China), IκB (Proteintech, 66418-1-Ig, Wuhan, China), β-Actin (Proteintech, 66009-1-Ig, Wuhan, China), and FLI-1 (Santa Cruz, sc-365294, Shanghai, China). HRP-conjugated secondary antibodies anti-mouse/rabbit IgG were purchased from ZSGB-Bio (Beijing, China) for IHC, and CST (Massachusetts, United States) for WB.
2.2 Animal model establishment
Female C57BL/6J mice were purchased from Chengdu Dossy experimental animals Co., Ltd. All procedures were approved by the Institutional Animal Care and Use Committee of Chengdu Medical College. 8-week-old (19–21 g) mice were housed under SPF conditions (22°C ± 1°C, 55% ± 5% humidity, 12-h light/dark cycle) with a 7-day acclimatization period prior to experimentation. And then 18 mice were randomly assigned to three experimental groups: (1) control group receiving standard drinking water, (2) DSS-induced colitis group administered 3% dextran sulfate sodium (DSS) in drinking water, and (3) treatment group receiving both DSS and daily oral gavage of Lumefantrine (20 mg/kg body weight) administered as a suspension in sterile distilled water. Throughout 7-day induction and treatment phase, clinical parameters - weight loss, stool consistency, and rectal bleeding - were recorded daily and quantified via standardized scoring criteria. Terminal procedures were performed on day 7 under ether anesthesia. Blood samples collected by eyeball excision were centrifuged at 3000 rpm for 10 min to isolate serum, which was subsequently stored at −80°C. Colons were excised from the ileocecal junction to the anal verge for precise length measurement, followed by systematic tissue sampling - distal colon segments were snap-frozen in liquid nitrogen for molecular analyses while middle colon sections were processed for histological evaluation.
2.3 Disease activity index (DAI) assessment
Disease progression was quantified using a validated Disease Activity Index (DAI) scoring system (Mukherjee et al., 2024), which evaluates three parameters: percentage weight loss, fecal bleeding severity, and stool consistency. The composite DAI score represents the arithmetic mean of these three components, with higher scores indicating more severe colitis. Detailed scoring criteria are presented in Supplementary Table S1.
2.4 Histopathological analysis
Mid-colon segments were fixed in 4% paraformaldehyde, paraffin-embedded, and sectioned at 5 μm thickness for hematoxylin and eosin (H&E) staining using stain kit (Solarbio, G1120, Beijing, China). Histopathological assessment was performed by a board-certified pathologist using the standardized criteria outlined in Supplementary Table S2.
2.5 Immunohistochemical staining (IHC)
Immunohistochemistry was performed as previously described. Briefly, antigen retrieval was conducted using citrate buffer (pH 6.0) at 95°C for 20 min. Endogenous peroxidase activity was quenched with 3% H2O2 for 15 min. Sections were incubated with primary antibodies at 4°C overnight. Sequential incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies (anti-mouse/rabbit IgG; 1:200 dilution) was performed at 37°C for 30 min. Diaminobenzidine (DAB) (Beijing ZSGB-BIO, Beijing, China) chromogen was used for signal development, with hematoxylin counterstaining.
2.6 Inflammatory cytokine quantification (ELISA)
Serum levels of IL-1β, IL-6, and TNF-α were measured using commercial ELISA kits (Cusabio, Wuhan, China) according to the manufacturer’s protocol. Optical density was measured at 450 nm using a microplate reader (Molecular Devices, United States).
2.7 Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from colon tissues using TRIzol reagent (Tiangen Biotech, Beijing, China), followed by cDNA synthesis with Reverse Transcriptase (Vazyme Biotech, Nanjing, China). Primers were synthesized by Tsingke Biotech Co., Ltd. (Beijing, China), and the sequences are listed in Supplementary Table S3. Quantitative PCR amplification was conducted using the following protocol: initial denaturation at 95°C for 15 min, followed by 40 cycles of 95°C for 10 s (denaturation) and 60°C for 30 s (annealing/extension), according to the manufacturer’s instructions for SYBR Green I master mix (Roche Diagnostics, Branchburg, United States). β-Actin served as the endogenous control. Relative gene expression was calculated via 2−ΔΔCt method.
2.8 Molecular docking analysis
Computational docking was performed to predict Lumefantrine-FLI-1 interactions. The FLI-1 tertiary structure was modeled using AlphaFold2. The PDB file of FLI-1 was uploaded to the PlayMolecule platform (https://playmolecule.com/) to predict potential ligand-binding pockets. Docking simulations were performed using SailVian, and binding stability was assessed through 100 ns molecular dynamics simulations.
2.9 Pull-down assay
Cell lysates were incubated with epoxy-activated μSphere magnetic beads (Tianyan Biotech, Wuxi, China) pre-conjugated with 20 μM Lumefantrine (4°C, 12 h rotation). After washing (3× PBS), bound proteins were eluted (0.5 mol/L NaCl) and analyzed by Western blotting. Control groups received ethanolamine-blocked beads without compound.
2.10 Cell culture
The Chinese Academy of Sciences Cellular Bank (Shanghai, China) provided IEC-6 and NCM460 cell lines. Both cell lines were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), cultured in a humidified incubator (37°C, 5% CO2, 95% air). Cells were subcultured at 80%–90% confluence using 0.25% trypsin-EDTA.
2.11 Cell viability assay (CCK-8)
Cells were seeded in 96-well plates at 5,000-10,000 cells/well and allowed to adhere for 24 h. Following treatment with Lumefantrine (0–40 μM) for 24/48 h, 10 μL CCK-8 reagent (Labgic, Beijing, China) was added to each well. After 2–4 h incubation protected from light, absorbance was measured at 450 nm using a microplate reader (Molecular Devices, United States). Cell viability (%) was calculated.
2.12 Western blot analysis
IEC-6 was treated with 20 μg/mL LPS for 48 h, and then treated with Lumefantrine, Camptothecin (CPT) or YK-4-279 for another 24 h. Treated cells were lysed in RIPA buffer containing protease/phosphatase inhibitors (Roche, Penzberg, Germany). Lysates were centrifuged, and supernatants were quantified via BCA assay (Thermo Fisher Scientific, United States). Proteins with equal amounts (20 μL) were electrophoretic separated on 10% SDS-PAGE gels and transferred to PVDF membranes (Merck Millipore, Darmstadt, Germany). After blocking with 5% non-fat milk/TBST (1 h, RT), membranes were incubated with primary antibodies (1:1000) overnight at 4°C, followed by HRP-conjugated secondary antibodies (anti-mouse/rabbit IgG, 1:5,000) for 2 h at RT. Signals were developed using ECL Prime (Labgic, Beijing, China) and quantified via ImageJ software.
2.13 Statistical analysis
All experiments were repeated independently at least three times. All data are represened as mean values ±standard deviation (S.D.). GraphPad Prism 9.0 performed unpaired t-tests. Significance thresholds: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns = not significant (p ≥ 0.05).
3 Result
3.1 Lumefantrine ameliorates DSS-Induced colitis in mice
Daily monitoring revealed significant weight loss in DSS-treated mice compared to controls (Figure 1A). Lumefantrine administration markedly attenuated weight reduction. Disease activity index (DAI) scores progressively increased in model mice but were suppressed by Lumefantrine treatment (Figure 1B). Colon shortening induced by DSS was partially reversed in the treatment group (Figure 1C). Histopathological analysis demonstrated severe mucosal erosion and inflammatory infiltration in DSS mice, whereas Lumefantrine preserved epithelial continuity and reduced crypt damage (Figure 1D). Correspondingly, immunohistochemical and Western blot analyses demonstrated that DSS challenge significantly reduced colonic Claudin-1 and ZO-1 expression, indicative of impaired epithelial barrier integrity (Figures 1E,F). Lumefantrine administration restored these tight junction proteins, correlating with preserved mucosal architecture.
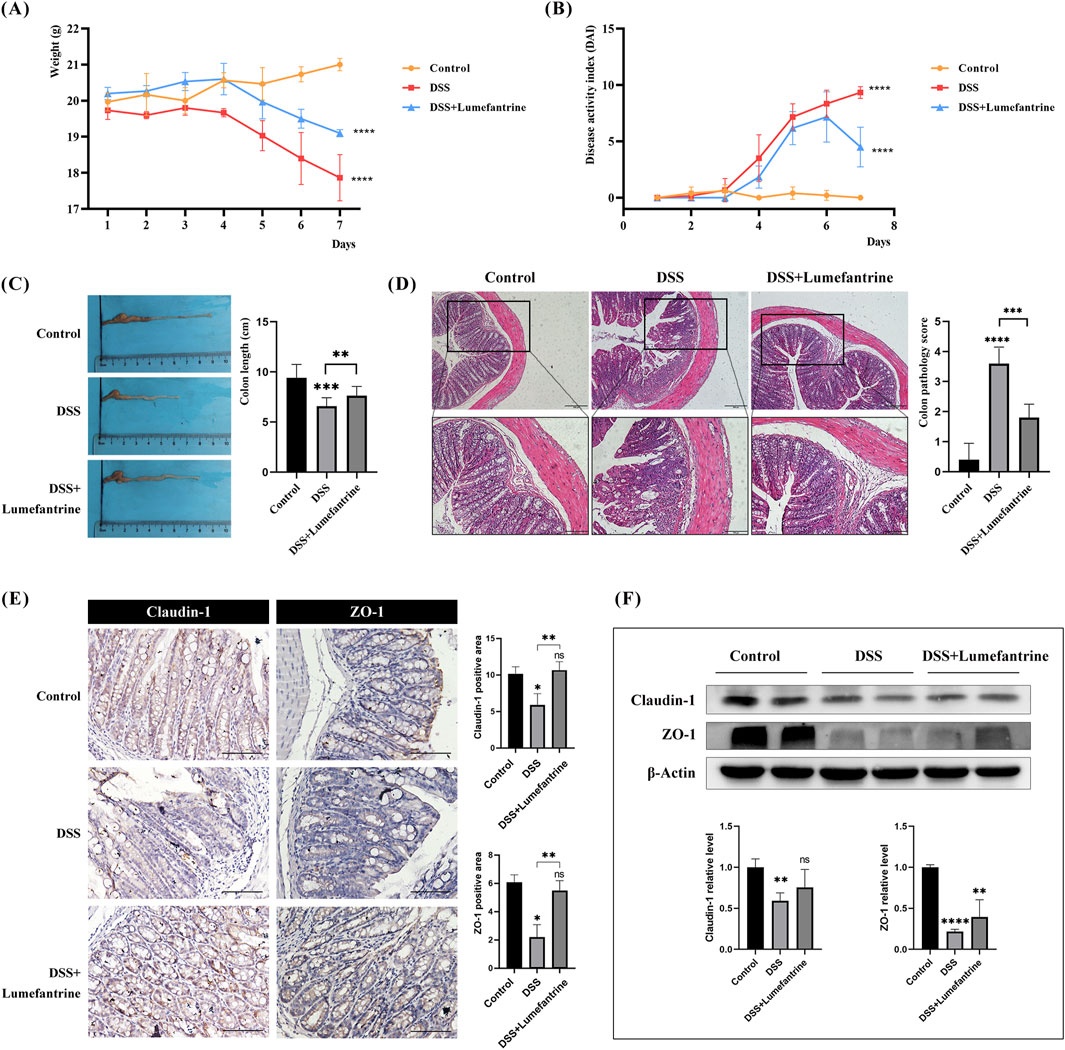
Figure 1. Lumefantrine ameliorates DSS-induced colitis in mice. (A) Body weight changes in mice. (B) Disease Activity Index (DAI) scores. (C) Colon length measurement. (D) Hematoxylin and eosin, (H&E)-stained colon sections (scale bar: 200 μm above and 100 μm below), showing the colon pathological score on the right side. (E) Immunohistochemical and (F) Western blot analysis of tight junction proteins Claudin-1 and ZO-1 expression in colonic tissues. (scale bar: 100 μm).
3.2 Anti-inflammatory effects of Lumefantrine
DSS challenge significantly upregulated colonic mRNA expression of pro-inflammatory cytokines IL-1β, IL-6, Tnf-α, along with inflammatory mediators Cox-2 and iNos. Lumefantrine administration markedly attenuated these elevations (Figures 2A–E). Serum ELISA quantification revealed corresponding decreases in protein concentrations: IL-1β, IL-6 and TNF-α all declined following Lumefantrine intervention (Figures 2F–H).
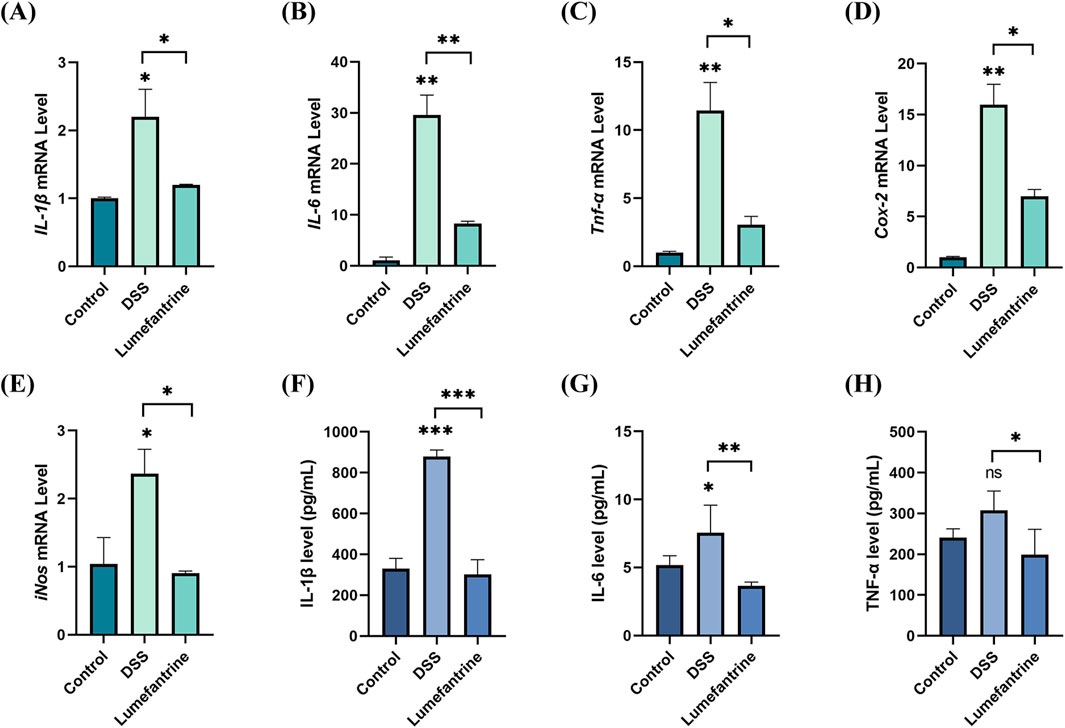
Figure 2. Anti-inflammatory effects of Lumefantrine. (A–E) mRNA expression levels of pro-inflammatory cytokines (IL-1β, IL-6, Tnf-α) and mediators (Cox-2, iNos) in murine model colon tissues. (F–H) Serum protein concentrations of IL-1β, IL-6, and TNF-α measured by ELISA.
3.3 Computational validation of Lumefantrine-FLI-1 interaction
The three-dimensional structure of FLI-1 predicted via the UniProt database (Figure 3A) and Lumefantrine (Figure 3B) were subjected to molecular docking. The 2D interaction diagram (Figure 3C) revealed that Lumefantrine binds to FLI-1 through hydrogen bonds and van der Waals forces, with the strongest binding affinity observed at arginine 123 (Arg123) (ΔEtotal = −167.90 kcal/mol). PlayMolecule combined with SailVian predicted three potential binding sites for Lumefantrine on FLI-1 (Figures 3D–F). The optimal docking pose exhibited a binding score of −7.9, comparable to positive control molecules TK216 (−7.9) and YK-4-279 (−7.6).
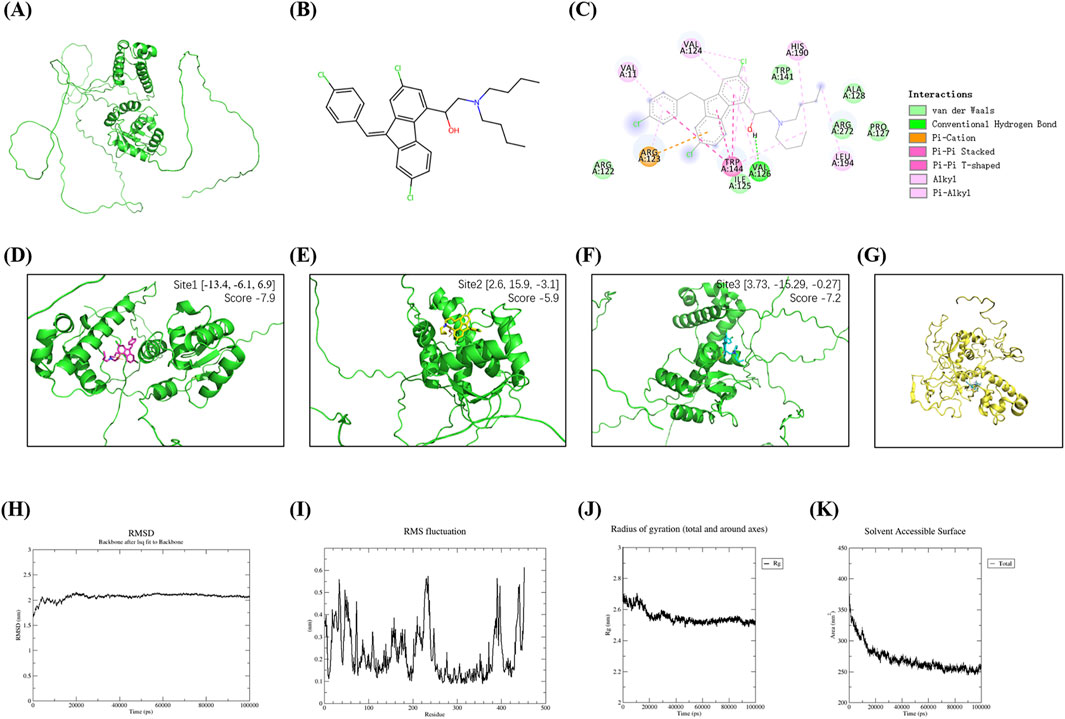
Figure 3. Molecular docking and dynamics simulations of Lumefantrine-FLI-1 interaction. (A) Predicted tertiary structure of FLI-1 (UniProt database). (B) Chemical structure of Lumefantrine. (C) 2D interaction diagram showing intermolecular force between FLI-1 and Lumefantrine. (D–F) Predicted ligand-binding pockets on FLI-1. (G) Binding conformation of the Lumefantrine-FLI-1 complex during molecular dynamics simulations. (H–K) Molecular dynamics parameters: Root mean square deviation [RMSD, (H)], root mean square fluctuation [RMSF, (I)], radius of gyration [Rg, (J)], and solvent-accessible surface area [SASA, (K)], confirming stable complex formation.
Molecular dynamics simulations revealed stable complex formation (Figure 3G): Root mean square deviation (RMSD) stabilized at 2.0–2.2 nm after 20 ns equilibration (Figure 3H). Root mean square fluctuation (RMSF) analysis indicates residue 20-60/220-240 flexibility, while active site conformation preserved (Figure 3I). The radius of gyration (Rg) of the protein stabilized between 2.4–2.8 nm, reflecting stable folding influenced by secondary structure (Figure 3J). Decreased solvent-accessible surface area (SASA) indicates a tightly packed, stable complex (Figure 3K). Total binding energy was calculated to be −45.25 kcal/mol, dominated by nonpolar energy (−38.7 kcal/mol).
3.4 Experimental validation of FLI-1 targeting
To experimentally validate the Lumefantrine-FLI-1 interaction predicted by computational analyses, we performed affinity-based pull-down assays. FLI-1 was selectively enriched in Lumefantrine-conjugated μSphere bead lysates compared to GST-tagged negative controls (Figures 4A,B), confirming direct binding between Lumefantrine and FLI-1. Western blot analysis further revealed FLI-1 suppression by Lumefantrine, comparable to the inhibitory effects of Camptothecin (CPT), a known FLI-1 antagonist (Figure 4C).
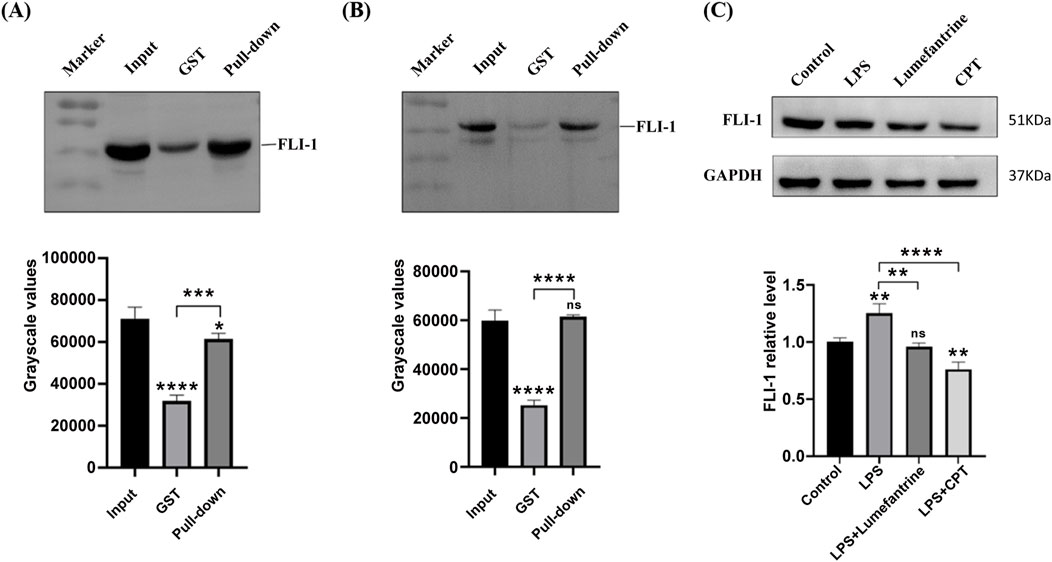
Figure 4. Binding of Lumefantrine to FLI-1 protein. Pull-down analysis of Lumefantrine conjugated magnetic beads and cell lysates from (A) IEC-6 and (B) NCM460, followed by WB detection using FLI-1 antibody. (C) Western blot was used to detect the expression of FLI-1 protein in IEC-6 after LPS modeling and treatment with Lumefantrine or Camptothecin (CPT).
3.5 NF-κB pathway modulation by Lumefantrine
CCK-8 assays demonstrated negligible cytotoxicity at concentrations below 40 μM during 24-h exposure in intestinal epithelial cells IEC-6 and NCM460 (Figures 5A,B). Thus, 20 μM Lumefantrine was employed for subsequent mechanistic investigations. Western blot analysis demonstrated that Lumefantrine significantly suppressed LPS-induced phosphorylation of both p65 and IκB (Figure 5C). Notably, Lumefantrine exhibited NF-κB inhibitory efficacy comparable to established FLI-1 inhibitors YK-4-279 and CPT, which served as positive controls in this study. Critically, when FLI-1 was inhibited by CPT, Lumefantrine’s suppression of p-p65 and p-IκBα was abolished (Figure 5D), confirming that its NF-κB inhibition operates specifically through FLI-1 targeting.
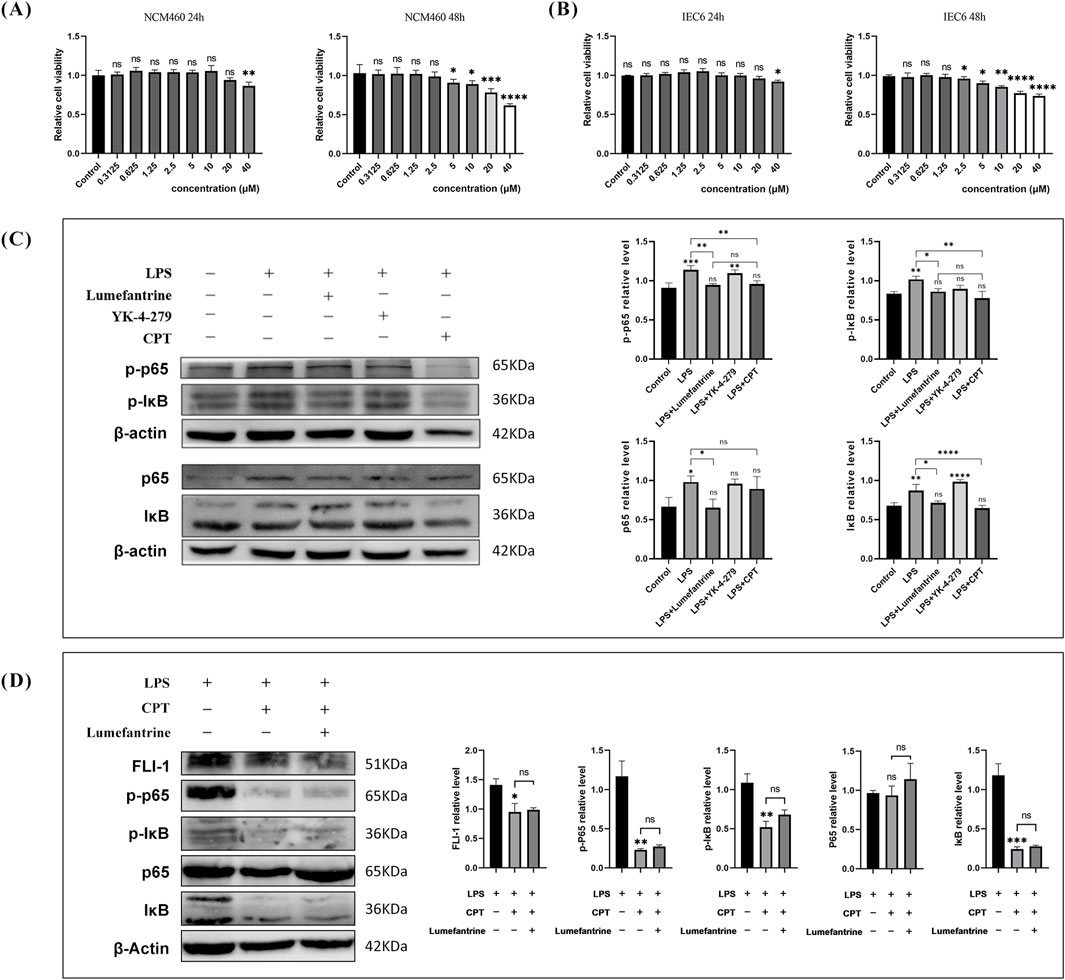
Figure 5. Lumefantrine modulates the NF-κB pathway. (A,B) CCK-8 assay showing cytotoxicity of Lumefantrine in NCM460 and IEC-6 cells after 24/48 h exposure. (C) Western blot analysis demonstrating Lumefantrine’s suppression of LPS-induced phosphorylation of p65 and IκB. FLI-1 inhibitors YK-4-279 and CPT used as positive controls. (D) Western blot assessment of Lumefantrine’s effect on NF-κB pathway proteins under CPT-mediated FLI-1 suppression.
Building upon FLI-1’s regulatory role in NF-κB signaling, we explored its therapeutic relevance in IBD. Analysis of public transcriptome datasets revealed significantly elevated FLI-1 expression in intestinal tissues from IBD patients compared to healthy controls (Figure 6A). Consistently, our DSS-induced colitis model showed increased colonic FLI-1 expression, which was effectively suppressed by Lumefantrine treatment (Figure 6B). These results establish FLI-1 as a promising therapeutic target in IBD, with Lumefantrine exerting its anti-inflammatory effects through direct FLI-1 binding and subsequent suppression of the NF-κB signaling cascade.
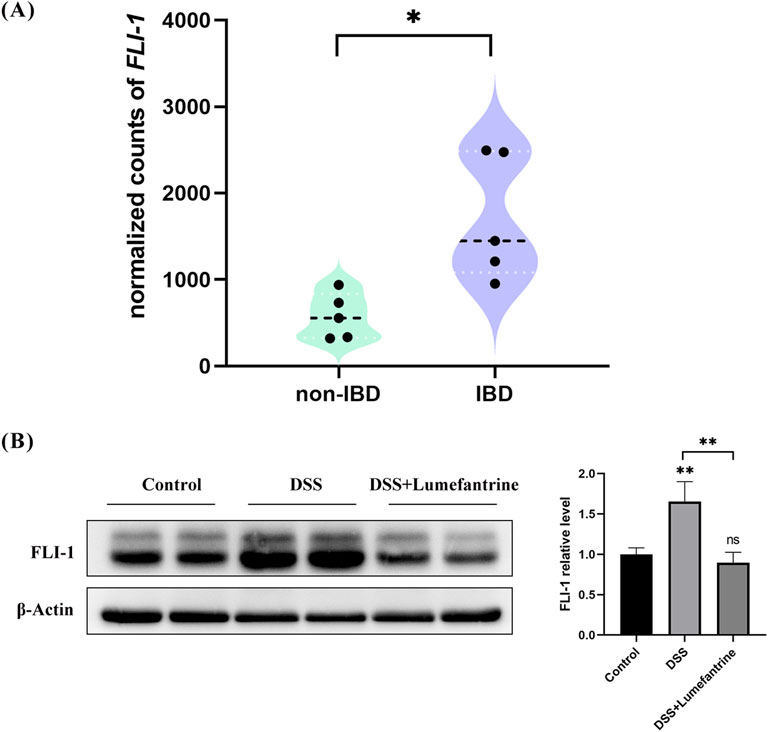
Figure 6. FLI-1 overexpression in IBD colonic tissues and its suppression by Lumefantrine. (A) Differential expression of FLI-1 in intestinal tissues between healthy individuals and IBD patients. Data sourced from the GSE255720 dataset (RNA-Seq libraries constructed from single-cell suspensions of colon biopsy tissues from 5 non-IBD healthy controls and 5 IBD patients), p = 0.011. (B) Western blot analysis of FLI-1 protein expression in normal controls, DSS-induced colitis mice and DSS mice treated with Lumefantrine.
4 Discussion
Inflammatory bowel disease (IBD) remains a clinical enigma due to its complex pathogenesis, relapsing nature, and potential for neoplastic transformation (Ko and Auyeung, 2014). This chronic immune-mediated disorder is characterized by persistent mucosal inflammation, oxidative stress, and inflammatory mediator activation. Lumefantrine, a mefloquine-class antimalarial agent developed by the Chinese Academy of Military Sciences, was approved by the U.S. Food and Drug Administration (FDA) in 2009 for treating acute non-severe malaria in adults and pediatric populations (Okwu et al., 2025). Clinically employed in combination regimens to enhance therapeutic efficacy and mitigate drug resistance, Lumefantrine demonstrates a favorable safety profile with minimal adverse effects (Ippolito et al., 2017; Omari et al., 2005). Our study revealed its previously unrecognized therapeutic potential in attenuating IBD progression through novel mechanisms.
Using a dextran sulfate sodium (DSS)-induced murine colitis model that recapitulates human IBD pathology, we demonstrated Lumefantrine’s capacity to preserve intestinal mucosal architecture and suppress inflammatory cascades. Model group mice exhibited characteristic disease manifestations including progressive body weight loss, loose stools, hematochezia, and significant colon shortening, accompanied by sustained elevation of Disease Activity Index (DAI) scores–observations consistent with established IBD murine models (Wirtz et al., 2017; Mizoguchi, 2012; He et al., 2024). In contrast, Lumefantrine-treated groups showed marked symptom alleviation, with declining DAI scores observed between days 6–7 and significantly attenuated colon shortening compared to vehicle controls.
The intestinal barrier–comprising mechanical, immunological, biological, and chemical components–crucially depends on tight junction integrity as the primary defense line of mucosal protection (Dunleavy et al., 2023). Intestinal epithelial tight junctions regulate paracellular permeability and maintain epithelial homeostasis (Parikh et al., 2019). Immunohistochemical and Western blot analysis revealed Lumefantrine-mediated restoration of Claudin-1 and ZO-1 expression at colonic tight junctions, mechanistically confirming its barrier-protective effects through structural reinforcement of the mechanical barrier.
Inflammatory cytokines IL-1β, IL-6, and TNF-α drive IBD pathogenesis via various mechanisms: IL-1β compromises epithelial permeability (Bulek et al., 2020), IL-6 potentiates adaptive immune dysregulation and carcinogenesis(Hunter and Jones, 2015; Taniguchi and Karin, 2014), while TNF-α amplifies oxidative stress and chemokine production(Wang et al., 2017; Oikonomopoulos et al., 2013). Our ELISA and qRT-PCR data concordantly showed Lumefantrine’s suppression of these cytokines in serum and colon tissue, highlighting its anti-inflammatory efficacy.
Mechanistically, Lumefantrine targeted the NF-κB pathway—a master regulator of inflammatory responses. By inhibiting IκBα and p65 phosphorylation, it attenuated NF-κB nuclear translocation and suppressed transcriptional activation of Cox-2 and iNos. COX-2, minimally expressed in normal tissues, becomes markedly upregulated during inflammation, driving prostaglandin E2 (PGE2) overproduction that amplifies inflammatory cascades (Singer et al., 1998). Similarly, while nitric oxide (NO) serves as a physiological signaling molecule in gastrointestinal homeostasis, pathological overexpression of iNOS drives excessive NO generation that damages intestinal epithelium and perpetuates inflammation (Cross and Wilson, 2003). Critically, these inflammatory mediators further activate NF-κB signaling through a pathogenic feedback loop, creating a self-reinforcing cycle of mucosal injury (Nie et al., 2023; Hassanein et al., 2022; Pan et al., 2011). Notably, multiple clinically approved IBD therapeutics exert their efficacy through NF-κB pathway inhibition (Atreya et al., 2008), underscoring the translational relevance of targeting this axis. Our findings position Lumefantrine as a promising candidate for modulating dysregulated NF-κB activation—a strategy with demonstrated therapeutic potential in inflammatory pathologies.
Through computational docking and experimental validation, we identified FLI-1, an ETS-family transcription factor, as the direct molecular target of Lumefantrine. This finding aligns with previous reports demonstrating that Lumefantrine inhibit FLI-1 expression to suppress glioblastoma multiforme (GBM) progression (Rajesh et al., 2020). While FLI-1 has been implicated as a therapeutic target in multiple malignancies such as Ewing sarcoma, hemangioma, and squamous cell carcinoma (Zhang et al., 2024; Yasir et al., 2023; Li et al., 2022; Ben-David et al., 2022), and autoimmune diseases such as systemic sclerosis (SSc) and systemic lupus erythematosus (sLE) (He et al., 2021), our work newly positions FLI-1 as a potential therapeutic target for IBD. Public transcriptomic datasets revealed elevated FLI-1 expression in IBD lesions compared to healthy intestinal tissues. Consistent with this observation, FLI-1 upregulation was recapitulated in our DSS-induced murine colitis model. FLI-1 inhibition by Lumefantrine disrupts its regulatory role in NF-κB activation, leading to reduced phosphorylation of p65 and IκBα. This aligns with prior findings in diffuse large B-cell lymphoma models, where FLI-1 knockdown attenuated NF-κB1 (p50) and RelA (p65) expression (Sartori et al., 2021). Notably, Lumefantrine’s NF-κB inhibitory efficacy mirrored that of established FLI-1 inhibitors–Camptothecin (Wang et al., 2023) and YK-4-279 (Huang et al., 2021). Critically, under CPT-mediated FLI-1 suppression, Lumefantrine failed to modulate the expression of NF-κB signaling components p-p65 and p-IκB. Collectively, Beyond validating Lumefantrine’s therapeutic potential for IBD, our findings suggest FLI-1 as a novel and actionable target warranting further exploration.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used. The animal study was approved by Chengdu Medical College. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
JY: Writing – original draft, Investigation, Formal Analysis, Data curation. PG: Visualization, Formal Analysis, Data curation, Investigation, Writing – original draft. HL: Writing – original draft, Investigation, Visualization. XT: Writing – original draft, Investigation. WL: Writing – review and editing, Conceptualization. XR: Writing – review and editing, Writing – original draft, Visualization, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Sichuan Provincial Natural Science Foundation (Grant No. 2023NSFSC0616) and Natural Science Foundation of Chengdu Medical College (Grant No. CYZ19-18).
Acknowledgments
We would like to express our gratitude to Professor Tao Zhang from Chengdu Medical College for his guidance on the research design and financial support for this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2025.1614978/full#supplementary-material
References
Agrawal, M., Allin, K. H., Petralia, F., Colombel, J. F., and Jess, T. (2022). Multiomics to elucidate inflammatory bowel disease risk factors and pathways. Nat. Rev. Gastroenterol. Hepatol. 19 (6), 399–409. doi:10.1038/s41575-022-00593-y
Atreya, I., Atreya, R., and Neurath, M. F. (2008). NF-kappaB in inflammatory bowel disease. J. Intern. Med. 263 (6), 591–596. doi:10.1111/j.1365-2796.2008.01953.x
Baumgart, D. C., and Carding, S. R. (2007). Inflammatory bowel disease: cause and immunobiology. Lancet 369 (9573), 1627–1640. doi:10.1016/S0140-6736(07)60750-8
Ben-David, Y., Gajendran, B., Sample, K. M., and Zacksenhaus, E. (2022). Current insights into the role of Fli-1 in hematopoiesis and malignant transformation. Cell. Mol. Life Sci. 79 (3), 163. doi:10.1007/s00018-022-04160-1
Biasi, F., Leonarduzzi, G., Oteiza, P. I., and Poli, G. (2013). Inflammatory bowel disease: mechanisms, redox considerations, and therapeutic targets. Antioxid. Redox Signal. 19 (14), 1711–1747. doi:10.1089/ars.2012.4530
Bulek, K., Zhao, J., Liao, Y., Rana, N., Corridoni, D., Antanaviciute, A., et al. (2020). Epithelial-derived gasdermin D mediates nonlytic IL-1β release during experimental colitis. J. Clin. Invest. 130 (8), 4218–4234. doi:10.1172/JCI138103
Cross, R. K., and Wilson, K. T. (2003). Nitric oxide in inflammatory bowel disease. Inflamm. Bowel Dis. 9 (3), 179–189. doi:10.1097/00054725-200305000-00006
Cui, L., Guan, X., Ding, W., Luo, Y., Wang, W., Bu, W., et al. (2021). Scutellaria baicalensis georgi polysaccharide ameliorates DSS-Induced ulcerative colitis by improving intestinal barrier function and modulating gut microbiota. Int. J. Biol. Macromol. 166, 1035–1045. doi:10.1016/j.ijbiomac.2020.10.259
Dunleavy, K. A., Raffals, L. E., and Camilleri, M. (2023). Intestinal barrier dysfunction in inflammatory bowel disease: underpinning pathogenesis and therapeutics. Dig. Dis. Sci. 68 (12), 4306–4320. doi:10.1007/s10620-023-08122-w
Flynn, S., and Eisenstein, S. (2019). Inflammatory bowel disease presentation and diagnosis. Surg. Clin.-North Am. 99 (6), 1051–1062. doi:10.1016/j.suc.2019.08.001
Hassanein, E. H. M., Althagafy, H. S., Atwa, A. M., Kozman, M. R., Kotb El-Sayed, M. I., and Soubh, A. A. (2022). Taurine attenuated methotrexate-induced intestinal injury by regulating NF-κB/iNOS and Keap1/Nrf2/HO-1 signals. Life Sci. 311 (Pt A), 121180. doi:10.1016/j.lfs.2022.121180
He, S., Zhang, T., Wang, Y., Yuan, W., Li, L., Li, J., et al. (2024). Isofraxidin attenuates dextran sulfate sodium-induced ulcerative colitis through inhibiting pyroptosis by upregulating Nrf2 and reducing reactive oxidative species. Int. Immunopharmacol. 128, 111570. doi:10.1016/j.intimp.2024.111570
He, Y. S., Yang, X. K., Hu, Y. Q., Xiang, K., and Pan, H. F. (2021). Emerging role of Fli1 in autoimmune diseases. Int. Immunopharmacol. 90, 107127. doi:10.1016/j.intimp.2020.107127
Herlihy, N., and Feakins, R. (2022). Gut inflammation induced by drugs: can pathology help to differentiate from inflammatory bowel disease? United european gastroenterol. J. 10 (5), 451–464. doi:10.1002/ueg2.12242
Huang, L., Zhai, Y., La, J., Lui, J. W., Moore, S. P. G., Little, E. C., et al. (2021). Targeting Pan-ETS factors inhibits melanoma progression. Cancer Res. 81 (8), 2071–2085. doi:10.1158/0008-5472.CAN-19-1668
Hunter, C. A., and Jones, S. A. (2015). IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 16 (5), 448–457. doi:10.1038/ni.3153
Ippolito, M. M., Johnson, J., Mullin, C., Mallow, C., Morgan, N., Wallender, E., et al. (2017). The relative effects of artemether-lumefantrine and non-artemisinin antimalarials on gametocyte carriage and transmission of plasmodium falciparum: a systematic review and meta-analysis. Clin. Infect. Dis. 65 (3), 486–494. doi:10.1093/cid/cix336
Ko, J. K., and Auyeung, K. K. (2014). Inflammatory bowel disease: etiology, pathogenesis and current therapy. Curr. Pharm. Des. 20 (7), 1082–1096. doi:10.2174/13816128113199990416
Lamb, C. A., Kennedy, N. A., Raine, T., Hendy, P. A., Smith, P. J., Limdi, J. K., et al. (2019). British society of gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut 68 (Suppl. 3), s1–s106. doi:10.1136/gutjnl-2019-318484
Li, L., Yu, J., Cheng, S., Peng, Z., and Luo, H. (2022). Transcription factor Fli-1 as a new target for antitumor drug development. Int. J. Biol. Macromol. 209 (Pt A), 1155–1168. doi:10.1016/j.ijbiomac.2022.04.076
Mizoguchi, A. (2012). Animal models of inflammatory bowel disease. Prog. Molec. Biol. Transl. Sci. 105, 263–320. doi:10.1016/B978-0-12-394596-9.00009-3
Mukherjee, T., Kumar, N., Chawla, M., Philpott, D. J., and Basak, S. (2024). The NF-κB signaling system in the immunopathogenesis of inflammatory bowel disease. Sci. Signal. 17 (818), eadh1641. doi:10.1126/scisignal.adh1641
Mussbacher, M., Derler, M., Basílio, J., and Schmid, J. A. (2023). NF-κB in monocytes and macrophages – an inflammatory master regulator in multitalented immune cells. Front. Immunol. 14, 1134661. doi:10.3389/fimmu.2023.1134661
Nie, C., Zou, Y., Liao, S., Gao, Q., and Li, Q. (2023). Molecular targets and mechanisms of 6,7-Dihydroxy-2,4-dimethoxyphenanthrene from Chinese yam modulating NF-κB/COX-2 signaling pathway: the application of molecular docking and gene silencing. Nutrients 15 (4), 883. doi:10.3390/nu15040883
Oikonomopoulos, A., van Deen, W. K., and Hommes, D. W. (2013). Anti-TNF antibodies in inflammatory bowel disease: do we finally know how it works? Curr. Drug Targets 14 (12), 1421–1432. doi:10.2174/13894501113149990164
Okwu, D. G., Manego, R. Z., Duparc, S., Kremsner, P. G., Ramharter, M., and Mombo-Ngoma, G. (2025). The non-artemisinin antimalarial drugs under development: a review. Clin. Microbiol. Infect. S1198--743X (25), 00127–2. doi:10.1016/j.cmi.2025.03.009
Omari, A. A. A., Gamble, C., and Garner, P. (2005). Artemether-lumefantrine (six-dose regimen) for treating uncomplicated falciparum malaria. Cochrane database Syst. Rev. 2005 (4), CD005564. doi:10.1002/14651858.CD005564
Pan, M. H., Hong, H. M., Lin, C. L., Jhang, A. Z., Tsai, J. H., Badmaev, V., et al. (2011). Se-methylselenocysteine inhibits lipopolysaccharide-induced NF-κB activation and iNOS induction in RAW 264.7 murine macrophages. Mol. Nutr. Food Res. 55 (5), 723–732. doi:10.1002/mnfr.201000481
Parikh, K., Antanaviciute, A., Fawkner-Corbett, D., Jagielowicz, M., Aulicino, A., Lagerholm, C., et al. (2019). Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 567 (7746), 49–55. doi:10.1038/s41586-019-0992-y
Rajesh, Y., Biswas, A., Kumar, U., Banerjee, I., Das, S., Maji, S., et al. (2020). Lumefantrine, an antimalarial drug, reverses radiation and temozolomide resistance in glioblastoma. Proc. Natl. Acad. Sci. U. S. A. 117 (22), 12324–12331. doi:10.1073/pnas.1921531117
Sartori, G., Napoli, S., Cascione, L., Chung, E., Priebe, V., Arribas, A. J., et al. (2021). ASB2 is a direct target of FLI1 that sustains NF-κB pathway activation in germinal center-derived diffuse large B-cell lymphoma. J. Exp. Clin. Cancer Res. 40 (1), 357. doi:10.1186/s13046-021-02159-3
Singer, I. I., Kawka, D. W., Schloemann, S., Tessner, T., Riehl, T., and Stenson, W. F. (1998). Cyclooxygenase 2 is induced in colonic epithelial cells in inflammatory bowel disease. Gastroenterology 115 (2), 297–306. doi:10.1016/s0016-5085(98)70196-9
Taniguchi, K., and Karin, M. (2014). IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin. Immunol. 26 (1), 54–74. doi:10.1016/j.smim.2014.01.001
Terzić, J., Grivennikov, S., Karin, E., and Karin, M. (2010). Inflammation and Colon cancer. Gastroenterology 138 (6), 2101–2114.e5. doi:10.1053/j.gastro.2010.01.058
Wang, K. S., Lv, Y., Wang, Z., Ma, J., Mi, C., Li, X., et al. (2017). Imperatorin efficiently blocks TNF-α-mediated activation of ROS/PI3K/Akt/NF-κB pathway. Oncol. Rep. 37 (6), 3397–3404. doi:10.3892/or.2017.5581
Wang, X., Richard, M. L., Caldwell, T. S., Sundararaj, K., Sato, S., Nowling, T. K., et al. (2023). Role of the transcription factor Fli-1 on the CXCL10/CXCR3 axis. Front. Immunol. 14, 1219279. doi:10.3389/fimmu.2023.1219279
Wiesner, J., Ortmann, R., Jomaa, H., and Schlitzer, M. (2003). New antimalarial drugs. Angew. Chem.-Int. Ed. 42 (43), 5274–5293. doi:10.1002/anie.200200569
Wirtz, S., Popp, V., Kindermann, M., Gerlach, K., Weigmann, B., Fichtner-Feigl, S., et al. (2017). Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 12 (7), 1295–1309. doi:10.1038/nprot.2017.044
Yasir, M., Park, J., and Chun, W. (2023). EWS/FLI1 characterization, activation, repression, target genes and therapeutic opportunities in ewing sarcoma. Int. J. Mol. Sci. 24 (20), 15173. doi:10.3390/ijms242015173
Keywords: Lumefantrine, Fli-1, NF-κB, inflammatory bowel disease (IBD), colitis
Citation: Yang J, Guo P, Luo H, Tang X, Liu W and Ren X (2025) Lumefantrine ameliorates DSS-induced colitis by targeting FLI-1 to suppress NF-κB signaling. Front. Pharmacol. 16:1614978. doi: 10.3389/fphar.2025.1614978
Received: 20 April 2025; Accepted: 26 June 2025;
Published: 11 July 2025.
Edited by:
Olumayokun Olajide, University of Huddersfield, United KingdomReviewed by:
Marc Christophe Karam, University of Balamand, LebanonLiming Mao, Nantong University, China
Copyright © 2025 Yang, Guo, Luo, Tang, Liu and Ren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaolin Ren, cmVueGxjaG5AMTYzLmNvbQ==
†These authors have contributed equally to this work