 Jiwei Shen1,2,3†Huiqin Shen1,2,3†
Jiwei Shen1,2,3†Huiqin Shen1,2,3† Shujun Sun1,2,3†Shiwen Fan1,2,3
Shujun Sun1,2,3†Shiwen Fan1,2,3 Tianhao Zhang1,2,3
Tianhao Zhang1,2,3 Guobin Song1,2,3Ning An1,2,3*
Guobin Song1,2,3Ning An1,2,3* Xiangdong Chen1,2,3*
Xiangdong Chen1,2,3* Yafen Gao1,2,3*
Yafen Gao1,2,3*- 1Department of Anesthesiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Institute of Anesthesia and Critical Care Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3Key Laboratory of Anesthesiology and Resuscitation (Huazhong University of Science and Technology), Ministry of Education, Wuhan, China
Sepsis-associated lung injury (SALI) is a critical condition with high mortality. Current therapies are limited, necessitating novel approaches. This review highlights the potential of exogenous Specialized Pro-resolving Mediators (SPMs), including lipoxins, resolvins, protectins, and maresins, in mitigating SALI. SPMs, derived from polyunsaturated fatty acids, exert protective effects through multiple mechanisms: enhancing alveolar fluid clearance by upregulating ENaC, Na,K-ATPase, CFTR, and AQP5; reducing alveolar epithelial cell apoptosis and epithelial-mesenchymal transition; preserving endothelial glycocalyx integrity via modulating heparanase and exostosin-1 expression; alleviating oxidative stress and mitochondrial dysfunction by scavenging ROS and activating Nrf2; and immunomodulation by limiting neutrophil infiltration, promoting macrophage efferocytosis and M2 polarization, and dampening pro-inflammatory cytokine production. Notably, SPMs like RvD2 remain effective even during sepsis’ immunosuppressive phase. While significant debates persist regarding endogenous SPM generation, receptor mechanisms, and critically the reliable detection and physiological relevance of specific SPMs in biological samples like lung tissue (with earlier reports often misidentifying analytical artifacts or failing LOD/LOQ validation), recent evidence suggests exogenous SPMs act via biased allosteric modulation of the EP4 receptor to stimulate phagocytosis and resolution. Extensive preclinical evidence underscores SPMs’ promise in restoring immune homeostasis in SALI, though pharmacokinetic limitations of high-dose exogenous administration require consideration. Future high-quality clinical trials are essential to translate this resolution pharmacology approach into clinical practice.
1 Introduction
Sepsis is a life-threatening organ dysfunction syndrome due to dysregulation of the body’s response to infection. It is a major risk factor for acute lung injury (ALI) and acute respiratory distress syndrome (ARDS), accounting for 31% of the etiology (Bersten et al., 2002). The mortality of sepsis-induced ALI/ARDS is significantly higher than that induced by non-sepsis factors. Moreover, phenotype for sepsis that patients had more inflammation and pulmonary dysfunction have relatively higher 28-day mortality compared to some other phenotypes (Seymour et al., 2019). The pathophysiological mechanisms involved in sepsis-associated lung injury (SALI) include uncontrolled inflammatory immunity, endothelial-alveolar epithelial barrier damage, pulmonary oedema and respiratory failure. Currently, supportive therapies such as anti-infective, lung protective ventilation and fluid management strategies remain the mainstay of clinical management of SALI, which, despite significant advances over the past few years, remains a serious clinical problem with a high mortality rate (Xu et al., 2023). It is imperative to explore new strategies for modulating inflammatory-immune disorders in order to improve the prognosis of patients with SALI.
The resolution of inflammation is increasingly recognized as an active and potentially modulatable process. A substantial body of experimental research demonstrates that exogenously administered specialized pro-resolving mediators (SPMs) can promote pro-resolving effects in preclinical models (Serhan, 2017; Fredman et al., 2016; Chattopadhyay et al., 2017; Wang L. et al., 2014). SPMs, a category encompassing lipoxins (LX), resolvins (Rv), protectins (PD), and maresins (MaR) based on their proposed structural classifications, and this category also includes conjugated tissue regenerators (CTRs), proposed to form during tissue regeneration (Serhan et al., 2008; Fredman and Serhan, 2024). Critically, the physiological relevance of endogenous SPM generation, their typical in vivo concentrations, and their specific receptor interactions remain active topics of scientific debate and uncertainty. Significant analytical controversies exist regarding reliable detection of SPMs in biological samples. Numerous publications have erroneously presented chromatographic artifacts (e.g., solvent peaks, contaminants) as evidence for SPMs like RvD2 or RvE1 in sepsis or lung injury contexts, often due to failures in applying standard limit-of-detection (LOD) or limit-of-quantitation (LOQ) criteria (O'Donnell et al., 2023). Consequently, internationally agreed technical standards for oxylipin analysis by LC-MS/MS have now been established to ensure rigorous identification and quantification (Schebb et al., 2025). Notably, no study to date has definitively demonstrated the presence of SPMs (e.g., RvD2, RvE1) in SALI samples using validated methods adhering to these standards. Despite these unresolved questions regarding endogenous biology, the exogenous application of certain SPMs has shown promise in models. Unlike broad immunosuppressive agents, exogenously administered SPMs are proposed to offer a potential therapeutic advantage for conditions like sepsis by reportedly stimulating the resolution of inflammation without globally compromising host defense mechanisms, thereby promoting a return to homeostasis (Buckley et al., 2014). Numerous studies have investigated the protective effects of exogenously administered SPMs in experimental SALI (Chattopadhyay et al., 2017; Jin et al., 2007; El Kebir et al., 2009). However, a systematic review comprehensively summarizing these findings, their underlying mechanisms, and the associated scientific controversies is lacking. This article aims to review the reported protective roles of exogenously administered SPMs in SALI, examining perspectives including lung epithelial and endothelial injury, oxidative stress and mitochondria-related mechanisms, and immunology, while critically evaluating their therapeutic potential in light of ongoing debates.
2 A brief overview of the biological role of SPMs in inflammation-resolution
Traditional understanding posits that SPMs are biosynthesized endogenously from polyunsaturated fatty acids (PUFAs) like arachidonic acid (AA), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA). This process is described as involving key enzymes such as cyclooxygenase-2 (COX-2), particularly in its aspirin-acetylated form, arachidonate 5-lipoxygenase (5-LOX), and 12/15-LOX. Multiple cell types, including leukocytes, platelets, epithelial cells, and endothelial cells, have been implicated in this biosynthesis, potentially occurring within single cells or through transcellular pathways (Serhan and Petasis, 2011; Basil and Levy, 2016). Furthermore, specific G protein-coupled receptors (GPCRs) such as ALX/FPR2 for lipoxins (e.g., LXA4), ChemR23 and BLT1 for RvE1, GPR32/ALX for RvD1, GPR18 for RvD2, GPR37 for PD1, and LGR6 for maresin 1 (MaR1) have been proposed as mediators of SPM actions (Dyall et al., 2022; Jordan and Werz, 2022; Serhan and Chiang, 2023; Serhan et al., 2022; Serhan and Levy, 2018; Basil and Levy, 2016). Based on this framework, SPMs were suggested to orchestrate inflammation resolution by modulating immune cell functions: reducing neutrophil recruitment, adhesion, and activation (Gong et al., 2014); promoting neutrophil apoptosis and macrophage efferocytosis/phagocytosis; shifting macrophage polarization towards an M2 phenotype (Godson et al., 2000); dampening dendritic cell pro-inflammatory cytokine production (Haworth et al., 2008); enhancing natural killer (NK) cell cytotoxicity (Barnig et al., 2013); regulating innate lymphoid cell type 2 (ILC2) responses (Krishnamoorthy et al., 2015); and influencing B and T cell differentiation, particularly promoting regulatory T cell (Treg) activity (Ramon et al., 2012; Chiurchiù et al., 2016; Oner et al., 2021).
However, significant controversies surround the endogenous formation, physiological relevance, and receptor specificity of SPMs. Critiques highlight challenges in reliably detecting endogenous SPMs at physiologically relevant concentrations using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Concerns have been raised about the validity of data in numerous publications, including reports of unusual chromatographic features and methodological issues. Studies investigating the aspirin-triggered pathway specifically found that while acetylated COX-2 inhibits prostanoids, generation of significant amounts of 15-epi-LXA4 was undetectable in cell models even under high substrate (AA) and supratherapeutic aspirin conditions (Hofling et al., 2022). Furthermore, human studies have largely failed to demonstrate consistent increases in SPM levels in response to dietary ω-3 PUFA supplementation or during the resolution phase of evoked inflammation (Schebb et al., 2022). The proposed specific SPM receptors have also been questioned, with recent high-profile evidence suggesting that several SPMs (e.g., protectins, maresins, D-series resolvins) may function primarily as biased positive allosteric modulators of the prostaglandin E2(PGE2) receptor EP4 at concentrations higher than those typically detected endogenously, enhancing cAMP signaling and phagocytosis via EP4 rather than acting through their originally proposed cognate GPCRs. In the absence of EP4, these SPMs lose their activity (Alnouri et al., 2024).
Despite these ongoing debates regarding endogenous generation and specific receptor mechanisms, a substantial body of preclinical research utilizing exogenous administration of synthetic SPMs demonstrates compelling biological effects relevant to inflammation resolution. Numerous studies consistently report that adding defined SPMs in vitro or administering them in vivo in various disease models, including sepsis, elicits potent anti-inflammatory and pro-resolving activities (Hu et al., 2020; Tan et al., 2018; Yang et al., 2013; Li et al., 2014; Fredman and Serhan, 2024). These effects manifest as reduced neutrophil infiltration and activation, enhanced macrophage phagocytosis and efferocytosis, modulation of macrophage polarization, suppression of pro-inflammatory cytokine (e.g., TNF-ɑ, IL-1β, IL-6) and chemokine production, promotion of neutrophil apoptosis, and regulation of lymphocyte responses. The mechanisms underlying these observed effects of exogenous SPMs, explored in detail in the subsequent sections of this review, offer significant therapeutic promise, irrespective of the resolution of the controversies surrounding their precise endogenous origins and signaling.
3 Protective effects of exogenous SPMs in SALI toward lung epithelial cells
3.1 Improve alveolar fluid clearance (AFC)
Alveolar epithelial cells (AECs) are composed of type I (AT I) and type II (AT II) cells. In addition to gas exchange, AFC is also a major function of AECs. The pathologic features of SALI mainly include impaired vascular capillary barrier and impaired clearance of edema fluid, and the ability to clear edema fluid is closely related to the prognosis (Morty et al., 2007). AFC is associated with the active transport of sodium ions in the alveolar epithelium through the apical epithelial sodium channel (ENaC) and basolateral Na,K-ATPase (Eaton et al., 2009; Sznajder et al., 2002). LXA4 could increase the expression of ENaC α and ENaCγ subunit proteins and Na,K-ATPase activity in primary rat AT II cells stimulated with lipopolysaccharide (LPS), thereby activating AFC (Wang et al., 2013). RVD1, PDX, MaR1, MCTR1 and PCTR1 all had similar effects, and the mechanism partially involved the ALX/cAMP/PI3K pathway (Wang Q. et al., 2014; Zhuo X. J. et al., 2018; Zhang et al., 2017; Han et al., 2020; Zhang et al., 2020). Nedd4-2, an E3 ubiquitin protein ligase, is crucial for negative regulation of Na + transport. The above SPMs inhibited the LPS-induced elevation of Nedd4-2 protein expression. Similarly, RvE1 promoted AFC by activating the PI3K/AKT/SGK1 (serum- and glucocorticoid-induced kinase 1) pathway to promote phosphorylation of Nedd4-2 and upregulate the expression of ENaC and Na,K-ATPase (Luo et al., 2022). In addition, PCTR1 promoted the expression of lymphatic vessel endothelial receptor-1 (LYVE-1), restored lymphatic drainage, and led to increasing the clearance of pulmonary interstitial fluid (Zhang et al., 2020). Some researchers had further investigated the mechanism by which LXA4 regulates ENaC-γ in LPS-induced lung injury. In A549 cells, miR-21 upregulated phosphorylation of AKT activation through inhibition of phosphatase and tensin homolog (PTEN), thereby decreasing ENaC-γ expression. Conversely, LXA4 reversed LPS-inhibited ENaC-γ expression through inhibition of activator protein 1 (AP-1), a conserved enhancer element in the miR-21 promoter region, and activation of PTEN (Qi et al., 2015).
In addition to ENaC and Na,K-ATPase, chloride channels and aquaporins (AQPs) also play an important role in the integrity of barrier function during fluid transport. Cystic fibrosis transmembrane conductance regulator (CFTR) is a chloride ion channel expressed in both alveolar AT-I and AT-II cells. Lack or inhibition of CFTR results in a deficient increase in AFC in response to β-adrenergic agonists (Folkesson and Matthay, 2006). LXA4 enhanced CFTR protein expression and increased AFC via the PI3K/Akt pathway (Yang et al., 2013). AQP5 is specifically expressed in the apical membrane of submucosal gland cells and AT-I cells. LPS stimulation was found to increase the permeability of lung epithelial cells by altering the expression and distribution of AQP5 on the cell surface. In the LPS-induced ALI model in rats, LXA4 upregulated AQP5 expression by inhibiting phosphorylation of p38 and JNK, restored AFC function, and exerted a protective effect against SALI (Ba et al., 2019).
To summarize, in sepsis, SPMs can ameliorate AFC and attenuate pulmonary edema from multiple targets of ENaC, Na,K-ATPase, CFTR, and AQP5, suggesting their potential application as anti-inflammatory treatment for the impairment of alveolar fluid transport in SALI (Figure 1).
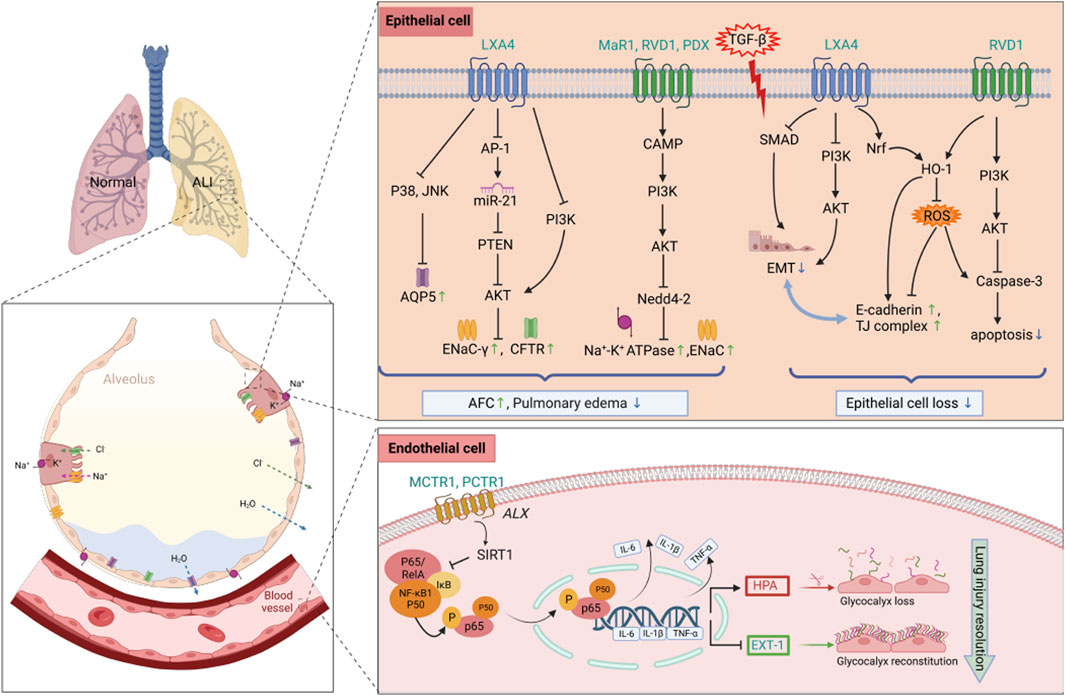
Figure 1. Protective mechanisms of SPMs against epithelial and endothelial cells in septic lung injury. SPMs play a protective role in septic lung injury by restoring the function of AFC to attenuate pulmonary edema, as well as reducing apoptosis and EMT in alveolar epithelial cells, meanwhile, maintaining the homeostasis of glycocalyx through the downregulation of HPA and the upregulation of EXT-1 expression. Abbreviations: SPMs, specialized pro-resolving mediators; AFC, alveolar fluid clearance; EMT, epithelial-mesenchymal transition; HPA, heparanase; EXT-1, exostosin-1; ALI, acute lung injury; AQP5, Aquaporin; TJ complex, tight junction complex.
3.2 Attenuate AECs injury/loss
In ARDS, the degree of alveolar epithelial destruction is closely related to the prognosis. Extensive apoptosis of AECs is the main result of alveolar epithelial destruction, leading to increased alveolar capillary permeability (Matthay and Zemans, 2011). Repair of alveolar epithelium is necessary for the repair of lung function. Abnormal regulation of repair mechanisms, such as epithelial-mesenchymal transition (EMT), can lead to fibroblast/myofibroblast differentiation, resulting in clinically significant pulmonary fibrosis (Andersson-Sjöland et al., 2008; Omenetti et al., 2008). Thus, apoptosis of AECs and EMT play a crucial role in the development of ARDS. In LPS-induced lung injury, LXA4 promoted the proliferation of AT-II cells, inhibited apoptosis, and decreased the expression of cleaved caspase-3. In addition, LXA4 was found to promote the expression of E-cadherin, an epithelial cell marker, and downregulate the expression of mesenchymal cell markers (including N-cadherin, α-SMA, and vimentin). Mechanistically, LXA4 inhibited EMT, in part through the SMAD and PI3K/AKT signaling pathways in an ALX receptor-dependent manner (Yang et al., 2019). Notably, LXA4 also mediated E-cadherin expression to protect respiratory epithelium from LPS damage by phosphorylating NF-E2-related factor 2 (Nrf2) on Ser40 and triggering its nuclear translocation to activate Nrf2 and reduce reactive oxygen species (ROS) (Cheng et al., 2016). Similarly, RvD1 reduced apoptosis (Xie et al., 2016), promoted epithelial wound repair and inhibited TGF-β-induced EMT, while reducing fiberproliferation and collagen production in primary human alveolar epithelial type 2 cells (Zheng et al., 2018). In addition, RvD1 significantly attenuated the degradation of tight junction (TI) proteins occludin and zona occludin-1 (ZO-1), thereby reducing LPS-induced permeability edema in mice (Figure 1). The heme oxygenase-1(HO-1) inhibitor, zinc protoporphyrin-9 (Znpp IX), reversed this effect, suggesting that HO-1 mediates, at least in part, the lung-protecting effects of RVD1 (Xie et al., 2013). Ocdcludin and ZO-1 are expressed by AECs as part of the TJ complex. TJ proteins establish a regulated paracellular barrier between epithelial and endothelial cells that prevents the movement of water, solutes, and immune cells, and their dysfunction can lead to barrier damage and pulmonary edema in SALI.
In conclusion, these results suggest to us a potentially interesting therapeutic strategy in which SPMs might be used to prevent and treat the fibroproliferative phase of sepsis-induced ARDS. The underlying mechanisms of the antifibrotic effects of SPMs require further experimentation.
4 Protective effects of exogenous SPMs in SALI toward lung endothelial cells
Endothelial cells play a regulatory role in the pathophysiology of lung injury. The endothelial glycocalyx layer (EGL) covers the vascular endothelial cells, forming a large vascular endothelial surface layer (ESL), which consists mainly of glycosaminoglycans and proteoglycans, and is degraded to produce heparan sulfate (HS), hyaluronic acid (HA), and syndecan-1 (SDC-1) by the activation of heparanase (HPA) (Chappell et al., 2008). The ESL also inhibits microvascular thrombosis and helps regulate leukocyte adhesion to the endothelium. In sepsis-associated acute lung injury, the breakdown of the ESL results in barrier disruption and leads to lung injury by promoting pulmonary edema and neutrophil adhesion. MCTR1 upregulated SIRT1 expression, decreased NF-κB p65 phosphorylation and downregulated HPA protein expression through the ALX/SIRT1/NF-κB/HPA pathway, which significantly inhibited HS degradation and thus reduced endothelial glycocalyx damage, thereby improving the survival of SALI mice (Li et al., 2020). Exostosin-1 (EXT-1) participates in the synthesis and reconstruction of glycocalyx, but this process is delayed in sepsis. PCTR1 could also upregulate the expression of EXT-1 to promote glycocalyx reconstruction and protect endothelial cells (Wang et al., 2021). Figure 1 summarizes the protective mechanisms of SPMs against lung epithelial and endothelial cells in SALI.
5 Reduce oxidative stress and mitochondrial dysfunction
After endotoxins and microorganisms invade lung tissue, large amounts of inflammatory factors released by phagocytic cells and endothelial cells can activate effector cells such as alveolar macrophages and neutrophils, leading to the release of a large number of ROS that damage AECs and vascular endothelial cells, affecting gas exchange in the alveoli and ultimately leading to severe lung injury. Oxidative stress reactions develop in the progression of inflammation, and have a positive feedback effect on the inflammation itself. Oxidative stress and mitochondrial dysfunction are considered to be one of the important pathogenic mechanisms in sepsis (Wen et al., 2015). Direct inhibition of the respiratory chain complex by microbial toxins and pro-inflammatory mediators is thought to be a major factor contributing to the decline in mitochondrial energy production and the initiation of the intrinsic apoptotic pathway in various cells and tissues. In severe inflammation, damaged mitochondria release a number of danger-associated molecular patterns (DAMP), such as ROS, while excessive production of ROS may also lead to structural damage of mitochondrial DNA and proteins, thereby causing mitochondrial dysfunction.
SPMs have anti-oxidative stress effects and can protect cells from oxidative damage and mitochondrial dysfunction by scavenging free radicals, inhibiting oxidative enzyme activity, and regulating redox homeostasis (Figure 2). Post-treatment of Aspirin-Triggered LXA4 inhibited malondialdehyde (MDA) production and was associated with upregulation of heme oxygenase-1 formation (Jin et al., 2007). Nrf2 is an important transcription factor that regulates the redox state of cells and the production of ROS by modulating phase II detoxifying/antioxidant enzymes. LXA4 activated Nrf2 and triggered its nuclear translocation to attenuate ROS production, thereby reducing LPS-induced acute lung injury in mice (Cheng et al., 2016). Moreover, LXA4 modulated the redox status of mitochondria in the lungs of mice, which were less oxidized in the LXA4 group compared to controls (Cheng et al., 2016). In a cecum ligation and perforation (CLP) mouse model, MaR1 downregulated mitochondrial NADPH oxidase (NOX) activity and increased catalases (CAT) and SOD activities, thereby inhibiting excessive ROS production and attenuating mitochondrial dysfunction in mice, which in consequence attenuated lung injury (Gu et al., 2018). Interestingly, it was found that the effect of MaR1 on ROS generation in the blood of endotoxin-stimulated healthy volunteers or septic patients was associated with ALX and cAMP, which is consistent with animal experiments. Consistent with this, a 1H NMR-based metabolomics analysis showed that MaR1 increased lung tissue taurine levels in CLP-induced septic mice, which could enhance the antioxidant defence system by protecting antioxidant enzymes (Hao et al., 2019). In addition, the protective effect of MCTR3 against LPS-induced ALI was partially mediated through the inactivation of the mitophagy pathway mediated by ALX/PTEN-induced putative kinase 1 (PINK1) (Zhuang et al., 2021). A recent study also found that PCTR1 treatment ameliorated mitochondrial ultrastructural damage, dampened ferroptosis through activation of ALX/protein kinase A (PKA)/cAMP-response element binding protein (CREB) and thereby ameliorated LPS-induced ALI (Lv et al., 2023).
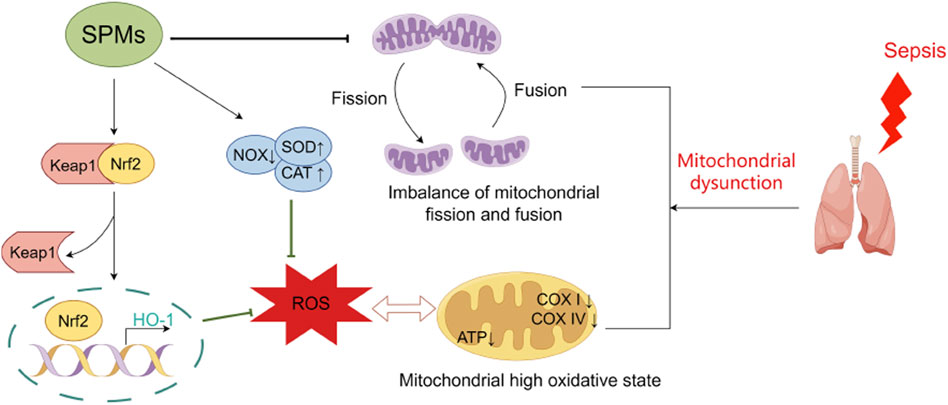
Figure 2. Protective mechanisms of SPMs against oxidative stress and mitochondrial dysfunction in septic lung injury. SPMs play a protective role in septic lung injury by attenuating the impaired mitochondrial respiratory function induced by excess reactive oxygen species, correcting the hyperoxidative state of mitochondria, and ameliorating the imbalance in mitochondrial fission-fusion kinetics. Abbreviations: SPMs, specialized pro-resolving mediators; NOX, NADPH oxidase; CAT, catalase; SOD, superoxide dismutase; ROS, reactive oxygen species.
Mitochondrial fission and fusion are critical for mitochondrial biogenesis and dynamics, and serve as new perspectives for a better understanding of the mitochondrial stress response. Inflammation-stimulated mitochondrial dysfunction in the human AEC line is reflected in impaired mitochondrial respiration, and altered fission and fusion are detected in these cells. Under TNF-α stimulation, the cellular maximal respiratory capacity was significantly reduced, and the expression of fission-regulating gene FIS1 was up-regulatedand while the fusion-associated gene MFN1 was down-regulatedand. The use of the mitochondrial fission inhibitor Mdivi-1 reduced the concentrations of the pro-inflammatory cytokines IL-6 and IL-8. RvE1 restored the impairment of mitochondrial respiratory capacity and imbalance of mitochondrial fission and fusion induced by inflammation, suggesting a novel functional mechanism for the beneficial role of RvE1 in pulmonary inflammatory responses (Mayer et al., 2019).
6 Immunologic perspectives on the protective role of exogenous SPMs in SALI
6.1 Immune cells
Circulatory inflammatory overload associated with sepsis leads to endothelial damage to pulmonary capillaries and inflammatory lung injury, which involves a variety of immune cells. As we mentioned before, SPMs can act on a variety of innate and adaptive immune cells to achieve its pro-resolving effects. The biological mechanisms behind the protective effects of SPM-targeted immune cells in SALI are beginning to be elucidated, focusing primarily on neutrophils and macrophages.
Activation of alveolar space neutrophils is a common feature of ALI in humans and animal models. During septic episodes, circulating neutrophils undergo defomation, adherence, aggregation, migration, and infiltration into lung tissue, and their own secretion of cytotoxic products and neutrophil extracellular traps (NETs) production can also lead to alveolar epithelial and endothelial damage. PD1 protects against LPS-induced ALI by inhibiting neutrophil infiltration and NETs formation in lung tissue (Wu et al., 2021). Similarly, the newly identified 13-series (T-series) resolvins (RvTs) reduced NETosis and enhanced NETs clearance by macrophages through a cyclic adenosine monophosphate/PKA/AMPK axis (Chiang et al., 2022). MaR1 reduced the expression of intercellular a dhesion molecule (ICAM)-1, P-selectin and CD24, and attenuated LPS-induced lung injury by inhibiting neutrophil adhesion and the production of pro-inflammatory mediators (Gong et al., 2014). When the vasculature is damaged, endothelial cells release a range of signalling molecules to attract surrounding neutrophils, while platelets aggregate at the site of damage and interact with endothelial cells and neutrophils, further enhancing neutrophil activation and recruitment. AT-15-Epi-LXA4 and PDX have also been shown to alleviate ALI in mice by regulating neutrophil-platelet aggregation (Ortiz-Muñoz et al., 2014; Tan et al., 2018). RvD1 could regulate bronchoalveolar lavage fluid (BALF) neutrophils accumulation through CXCL-12/CXCR4 pathway (Yaxin et al., 2014). Lung tissue expressed more CXCL-12, also known as stromal cell derived factor 1 (SDF-1),in ALI (Petty et al., 2007) and CXCR4 plays an important role in homing of aged neutrophils to bone marrow for clearance (Allen et al., 2004). RvD1 reduced the expression of CXCL-12 mRNA in lung tissue, and promoted the expression of CXCR4 in neutrophils in the early stage of inflammation (24 h) while decreased the expression of CXCR4 in the later stage of inflammation (72 h), thereby regulating neutrophil aggregation in BALF and alleviating LPS-induced ALI (Yaxin et al., 2014). Apoptosis is considered the main mechanism for clearing neutrophils from the site of inflammation (Moon et al., 2010). In sepsis, neutrophil apoptosis is suppressed, and the apoptosis process of neutrophils becomes one of the control points for the resolution of inflammation (Gong et al., 2015). Myeloperoxidase (MPO) is expressed in large quantities in neutrophils, not only producing cytotoxic oxidants but also signaling through β2 integrin CD11b/CD18 (Mac-1) to rescue neutrophils from apoptosis, thereby prolonging inflammation (El Kebir et al., 2008). In an E. coli SALI model, 15-Epi-LXA4 promoted neutrophil caspase-3-mediated apoptosis by inhibiting MPO-induced extracellular signal-regulated kinase (ERK) and Akt-mediated phosphorylation of the pro-apoptotic protein Bad and reducing expression of the anti-apoptotic protein Mcl-1 (El Kebir et al., 2009). Notably, Wang et al. applied single-cell sequencing to reveal, at the single-cell level, that maresin1 significantly reduced neutrophil infiltration in SALI, and that this regulatory effect was more focused in the neutrophil-Cxcl3 subpopulation (Wang et al., 2022).
Inflammatory lungs contain two groups of alveolar macrophages: resident alveolar macrophages (RAMs) and recruited macrophages. During the acute inflammatory phase of SALI, RAMs are immediately activated and switched to M1 macrophages, which release cytokines and chemokines to recruit neutrophils, monocytes, etc., to trigger lung inflammation (Arango Duque and Descoteaux, 2014). However, in the late rehabilitation phase, both resident and recruited macrophages shift to M2 phenotype, which exert anti-inflammatory and pro-resolving effects mainly through phagocytosis and efferocytosis to avoid excessive inflammatory damage (Herold et al., 2011). Macrophages possess a unique role and amazing adaptability that renders them a promising target for the management of SALI. CXCL2, also known as macrophage inflammatory protein 2 (MIP-2), mainly functions to induce neutrophil chemotaxis to inflammatory tissues. Monocyte chemoattractant protein-1 (MCP-1) is a widely expressed monocytes and macrophages chemokine. It was found that in lungs, MIP-2 and MCP-1 were predominantly present on RAMs (Ye et al., 2020). In LPS-induced ALI, RvD1 inhibited MIP-2 expression on RAMs, thereby suppressing neutrophil infiltration (Zhang H. W. et al., 2019). Similarly, PDX attenuated LPS-induced lung injury by inhibiting neutrophil and recruited macrophage recruitment through suppression of MIP-2 and MCP-1 expression, respectively (Ye et al., 2020). In this study, it was also suggested that tumor necrosis factor-ɑ (TNF-ɑ) was expressed mainly on recruited macrophages, and that the mechanism by which PDX inhibited neutrophil infiltration and transmembranes may be related to TNF-ɑ/MIP-2/MMP-9 signaling pathway (Ye et al., 2020). Thus, both RAMs and recruited macrophages serve as targets for PDX to reduce inflammatory cell infiltration. Moreover, in a CLP model, PDX was able to promote M2 polarization of peritoneal macrophages and enhance their phagocytic activity to reduce the bacterial load and accelerate the resolution of inflammation, thus improving the survival of septic mice and reducing multiple organ damage, including lungs (Xia et al., 2017). Also in the LPS-induced lung injury model, Mar1 was shown to promote M2 macrophage polarization and accelerate the resolution of ALI by a mechanism related to activation of PPAR-γ(Qiao et al., 2020). Another study explored the effects of MCTR1 on M2 polarization of resident and recruited macrophages and found that MCTR1 enhanced inflammatory resolution and attenuated LPS-induced lung injury primarily by promoting resident M2 macrophage polarization, which was mediated through the STAT6 pathway (Wang et al., 2020). A recent study also found that RvD1 promoted RAMs self-renewal and phagocytosis through the ALX/MAPK14/S100A8/A9 signaling pathway in a mouse model of aerosolized inhaled LPS and/or E. coli (Ye et al., 2025). Notably, autophagy is also one of the mechanisms by which SPMs target and regulate macrophage function. In a study using BML-111, an LXA4 receptor agonist, the authors found that BML-111 targeted the MAPK pathway to stimulate autophagy in AMs, significantly reducing LPS-induced AMs apoptosis, thereby suppressing inflammation and ameliorating lung injury (Liu et al., 2018).
6.2 Inflammatory cytokines and inflammatory signal pathways
Several SPMs have been shown to reduce LPS-induced pro-inflammatory cytokines (such as TNF-ɑ, IL-1β, and IL-6), chemokines (such as keratinocyte-derived chemokine, MCP-1, and MIP-1), thereby attenuating LPS-induced ALI and improving survival in septic mice (Gong et al., 2014; Jin et al., 2007; Walker et al., 2011; Wang et al., 2011; Xia et al., 2020). Signaling pathways involved are related to NF-κB, signal transducer and activator of transcription (STAT), MAPKs (Wang et al., 2011; Hu et al., 2020; de Oliveira et al., 2017; Li et al., 2016). It is well known that NF-κB, MAPKs, STAT are important transcription factors that regulate the expression of inflammatory genes and are thought to be involved in the pathogenesis of sepsis. The MAPKs pathway has three major subfamilies, including the ERK cascade, c-Jun NH2-terminal/stress-activated protein kinase (JNK/SAPK) cascade, and p38-MAPK cascade. In a CLP septic mouse model, RvD1 promoted the resolution of inflammation by upregulating SIRT1 expression and inhibiting the activation of STAT3, ERK, p38, and NF-κB in lung tissue, thereby reducing the severity of pulmonary inflammation (Zhuo Y. et al., 2018). In an in vitro experiment using LPS-stimulated human bronchial epithelial cells (BEAS-2B), AT-RvD1 reduced the concentration of the chemokine CCL-2 in dependence on FPR2/ALX, mechanistically related to its downregulation of the phosphorylation of NF-κB, STAT1 and STAT6 (de Oliveira et al., 2017). In a mouse model of E. coli pneumonia, 15-epi-LXA4-ALX/FPR2 interaction selectively upregulated the NF-κB negative regulators A20 and SIGIRR, thereby decreasing NF-κB activity, which may inhibit pulmonary neutrophil infiltration while increasing bacterial clearance and improving sepsis survival (Sham et al., 2018). Furthermore, RvD1-and PDX-induced IκBα degradation and p65 nuclear translocation inhibition were dependent on PPARγ (Liao et al., 2012; Xia et al., 2020).
Of note is the dual effect of LXA4 on LPS-stimulated COX2 expression in lung fibroblasts (Zheng et al., 2011). Exogenous LXA4 inhibited the first peak of LPS-induced COX-2 expression as well as PGE2 production in a dose-dependent manner. In contrast, LXA4 increased the second peak of LPS-induced COX-2 expression and prostaglandin D2 (PGD2) production in a dose-dependent manner. Notably, PGE2 and PGD2 are responsible for regulating the switch of lipid mediator classes from proinflammation to proresolution (Levy et al., 2001). RvD1 effectively promoted p50 homodimer nuclear translocation and upregulated COX-2 expression. The absence of p50 in knockout mice prevented RvD1 from promoting COX-2 and PGD2 expression, leading to excessive lung inflammation. It suggestd that RvD1 accelerated the reslution of inflammation through the NF-kB p50/p50-cox-2 signaling pathway (Gao et al., 2017).
6.3 Effects on microorganisms
SPMs can reduce the bacterial load in mouse serum or lungs, such as E. coli (Chiang et al., 2012; Abdulnour et al., 2016), Streptococcus pneumoniae (Wang et al., 2017), Pseudomonas aeruginosa (Abdulnour et al., 2016), and Nontypeable Haemophilus influenzae (Croasdell et al., 2016) by enhancing phagocytosis by macrophages and neutrophils. Remarkably, LXA4 not only regulates the host response, but also influences bacterial virulence and enhances antibiotic efficacy. Pseudomonas aeruginosa is a gram-negative, opportunistic bacterium that often attacks immunocompromised patients such as those with sepsis. LXA4 inhibited the quorum sensing receptor LasR, thereby reducing P. aeruginosa exotoxin release (Wu et al., 2016). In addition, LxA4 reduced the formation of P. aeruginosa biofilm and the expression of virulence genes, and enhanced the inhibitory effect of ciprofloxacin on the formation of biofilm and its bactericidal ability, both in a static biofilm-forming system and under hydrodynamic conditions (Thornton et al., 2021; Thornton et al., 2023). In mice chronically infected with P. aeruginosa, RvD1 promoted phagocytosis of P. aeruginosa by lung macrophages and regulated the expression of Toll-like receptors, downstream genes, and microRNAs (MiR)-21 and 155, which reducesd inflammatory signals (Codagnone et al., 2018). Furthermore, in the context of co-infection with Streptococcus pneumoniae and influenza A virus, AT-RvD1 promoted faster clearance of pneumococcus from the lungs while reducing the severity of pneumonia by limiting excessive leukocyte chemotaxis from the infected bronchioles to the distal regions of the lungs (Wang et al., 2017). Taken together, these findings suggest that the prosolvency of SPMs represents a new host-directed therapeutic strategy to complement current antibiotic-centered approaches to fighting infections and lays the groundwork for further investigation of SPMs as an alternative to immunosuppressive therapies such as steroids.
SPMs have also been shown to exert effects in influenza virus infection. It has been reported that PD1 significantly inhibits influenza virus replication through an RNA export mechanism, which prevents fatal influenza virus infection and improves the survival of mice with severe influenza (Morita et al., 2013). 17-HDHA enhanced antibody-mediated anti-influenza virus immune response and is important for the development of new potential influenza vaccine adjuvants (Ramon et al., 2014).
6.4 Protective role of exogenous SPMs in the immunosuppression phase
Most studies have demonstrated the pro-resolving role of SPM in early sepsis, and more encouragingly there are also some studies suggesting that SPM may play a beneficial role in late sepsis. In the development of sepsis, the early manifestation is an inflammatory burst followed by a phase of immunosuppression characterized by lymphocyte apoptosis, monocyte/macrophage exhaustion, and increased migration of myeloid-derived suppressor cells (MDSCs), leading to host vulnerability to secondary infections (Padovani and Yin, 2024). The two-hit mouse model of CLP and secondary P. aeruginosa lung infection is a commonly used model of late polymicrobial sepsis. In studies using this mouse preclinical model, administration of RvD2 in the later stages of the CLP model (48 h postoperatively) demonstrated multiple beneficial effects:1) decreased blood and lung bacterial load; 2) increased phagocytosis by alveolar macrophages/monocytes 3) splenic T-cell counts were also increased 4) increased the number of non-inflammatory alveolar macrophages 5) increased mature neutrophils and MDSC accumulation in the spleen; 6) significantly decreased lung lavage levels of IL-23; and 7) decreased mortality (Walker et al., 2022; Sundarasivarao et al., 2022). These studies provide evidence that RvD2 promotes host defense mechanisms in sepsis and secondary lung infections and may have therapeutic value in the treatment of sepsis in the immunosuppressed phase.
7 Reflections on the results of human studies
Emerging clinical studies have reported associations between SPM levels and sepsis outcomes; however, it is critical to recognize that the analytical methodologies employed in many of these studies are now known to be inadequate for reliable SPM quantification, casting doubt on the validity of these reported associations. Multiple investigations report reduced systemic SPM concentrations (e.g., LXA4, MaR1) in septic patients compared to healthy controls, with lower levels correlating with increased disease severity, ARDS development, and mortality (Tsai et al., 2013; Jundi et al., 2021; Tejera et al., 2020). Importantly, these findings must be interpreted with extreme caution due to the methodological limitations discussed below. Parallel observations in COVID-19 indicate that SPM downregulation coincides with phagocyte dysfunction, while therapeutic interventions like dexamethasone may partially exert protection through SPM upregulation (Koenis et al., 2021). The clinical relevance of SPM receptors is further supported by findings that reduced GPR18 expression on neutrophils predicts poorer sepsis outcomes (Zhang L. et al., 2019). However, contradictory evidence exists: some studies observed elevated SPMs (RvE1, RvD5, 17R-PD1) alongside pro-inflammatory lipids in non-survivors, while others documented increased LXA4 and D-series resolvins in severe COVID-19 (Dalli et al., 2017; Archambault et al., 2021). These paradoxical reports likely arise primarily from methodological artifacts rather than biological phenomena, given the analytical shortcomings pervasive in this literature. Critically, it must be emphasized that no validated evidence exists for endogenous SPMs (e.g., RvD2, RvE1) in SALI samples, as earlier reports claiming their presence often misidentified analytical artifacts or failed LOD/LOQ validation (Schebb et al., 2025; O'Donnell et al., 2023).
These analytical limitations fundamentally undermine the reliability of purported clinical associations. Substantial analytical challenges complicate SPM quantification. Specifically, earlier clinical studies reporting SPM levels in sepsis patients frequently utilized methods now recognized as insufficiently rigorous (e.g., inadequate chromatographic resolution, failure to apply LOD/LOQ validation), rendering their correlative findings unsubstantiated. Concentrations in biological samples are frequently near detection limits (<10 pg/mL), with significant inter-study variability attributed to methodological differences in sample processing, storage, and LC-MS/MS analysis (Calder, 2020). Concerns regarding the validity of endogenous SPM measurements have been raised, particularly surrounding chromatographic data quality and inappropriate analytical practices in earlier literature. Consequently, rigorous adherence to internationally standardized protocols is essential for future biomarker studies.
Despite unresolved questions about endogenous biology, SPMs retain therapeutic promise. Clinical trials demonstrate that synthetic analogs (e.g., LXA4-stabilized mouthwash for periodontitis and RvE1 derivatives for dry eye disease) exhibit safety and efficacy in reducing inflammation (Hasturk et al., 2021; OphthalmologyWeb, 2024). However, the translational gap for life-threatening conditions like SALI partly stems from pharmacokinetic challenges: preclinical studies use highly variable SPM doses (0.1 ng/kg–5 μg/kg, Table 1) via intravenous/intraperitoneal routes, yet none measure systemic or lung tissue concentrations—a critical omission given SPMs’ rapid metabolism, poor tissue penetration, and enzymatic degradation. Recent mechanistic insights revealing SPMs require ∼100 nM concentrations for EP4 allosteric modulation (Alnouri et al., 2024)—orders of magnitude above endogenous levels—justify high-dose regimens but necessitate advanced delivery strategies. Promisingly, polycarbonate micelles prolong SPM release to 20 days while reducing oxidation (de Prinse et al., 2023), offering potential solutions to these bioavailability barriers. It is important to critically address the observation that clinical trials of SPM-based drugs have thus far primarily focused on less life-threatening conditions like periodontitis and dry eye disease, rather than severe systemic illnesses such as SALI. This strategic choice likely reflects several key considerations. Firstly, the safety threshold for initial human testing is understandably lower for localized, non-fatal inflammatory conditions. Topical or localized delivery (e.g., ocular administration for dry eye, oral rinse for periodontitis) offers practical advantages and minimizes systemic exposure concerns. Secondly, the pharmacokinetic limitations of natural SPMs pose a significant hurdle for systemic applications in critical illness. Natural SPMs are rapidly inactivated and cleared in vivo, demanding high doses for efficacy in preclinical sepsis models, which raises challenges related to drug formulation, stability, cost, and potential off-target effects at supraphysiological concentrations. These PK challenges necessitate the development of stable analogs or novel delivery systems before advancing into large-scale, resource-intensive trials for life-threatening conditions like sepsis. Furthermore, defining clinically relevant endpoints and patient populations for sepsis trials is inherently more complex than for conditions like dry eye. Nevertheless, the compelling preclinical evidence summarized herein, particularly their efficacy even in the immunosuppressive phase of sepsis and their potential to enhance host defense without broad immunosuppression, strongly motivates the imperative for future high-quality clinical trials directly evaluating SPM therapeutics in sepsis-associated lung injury. Such trials are essential to establish effective dosing paradigms and validate the translational potential of this resolution pharmacology approach. Notably, ω-3 fatty acid supplementation reduces sepsis mortality and ARDS incidence (Lei et al., 2023; Chen et al., 2018), though whether this reflects SPM generation remains contested (Souza et al., 2020; Sobrino et al., 2020; Marchand et al., 2023). Recent mechanistic insights reveal that supraphysiological SPM concentrations act via biased allosteric modulation of the EP4 receptor, converting anti-phagocytic signaling to pro-phagocytic activity—a critical pathway for inflammation resolution (Alnouri et al., 2024). This supports exploration of high-dose exogenous SPM administration to circumvent endogenous production limitations. Clinical trials directly evaluating SPM therapeutics in sepsis-associated lung injury are warranted to establish dosing paradigms and validate their translational potential.
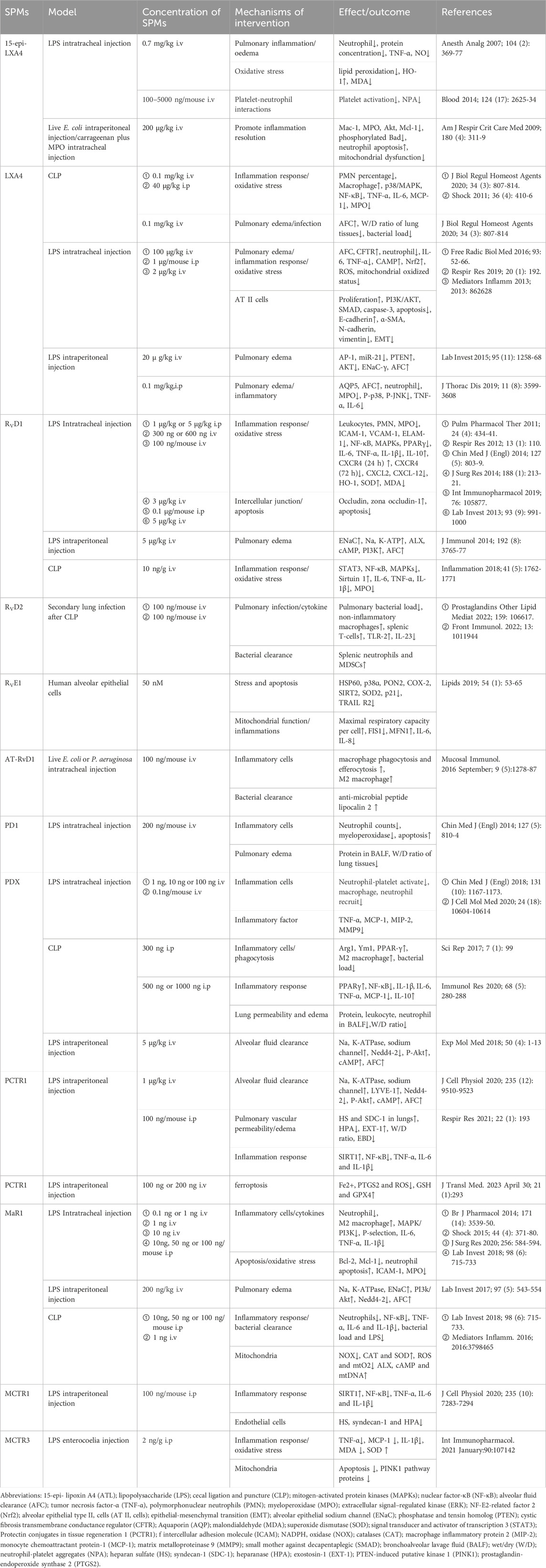
Table 1. SPMs in septic lung injury.
8 Conclusion
While current supportive therapies (anti-infectives, fluid resuscitation, mechanical ventilation) have improved survival in SALI, they fail to address the underlying immune dysregulation driving the pathology. This review synthesizes compelling preclinical evidence demonstrating that exogenously administered SPMs represent a promising therapeutic strategy targeting these fundamental mechanisms. As detailed in Table 1, we comprehensively summarize the multi-faceted protective actions of diverse SPMs in SALI: ① Epithelial & Endothelial Protection: SPMs restore AFC by enhancing ENaC, Na,K-ATPase, CFTR, and AQP5 function, reducing pulmonary edema. They attenuate AECs apoptosis and inhibit EMT. Concurrently, SPMs maintain endothelial glycocalyx homeostasis by downregulating HPA and upregulated EXT-1 expression, preserving vascular barrier integrity. ② Mitigation of Oxidative Stress & Mitochondrial Dysfunction: SPMs reduce ROS overproduction, enhance antioxidant defenses (e.g., via Nrf2 activation), restore inflammation-impaired mitochondrial respiratory capacity, and correct mitochondrial fission/fusion imbalances. ③ Pro-Resolving Immunomodulation: Critically, and distinct from broad immunosuppression, SPMs actively promote resolution by: Reducing neutrophil infiltration and NETosis, enhancing macrophage efferocytosis and phagocytic clearance. Promoting macrophage polarization towards an M2 phenotype. Dampening pro-inflammatory cytokine/chemokine production (e.g., TNF-α, IL-1β, IL-6, MIP-2, MCP-1) and modulating key signaling pathways (NF-κB, MAPK, STAT, PPARγ). Modulating microbial virulence and enhancing antibiotic efficacy. Significantly, exerting protective effects even during the immunosuppressive phase of sepsis.
It is essential to note significant controversies regarding the physiological relevance of endogenous SPM generation and their proposed cognate GPCR signaling pathways. Critically, and as emphasized throughout this review, robust detection of physiologically relevant concentrations of specific SPMs (e.g., RvD2, RvE1) in biological matrices, particularly SALI samples, remains unvalidated. Earlier studies frequently misattributed analytical artifacts (e.g., solvent peaks, contaminants) to SPMs due to methodological flaws, including failures to apply standard LOD or LOQ criteria (O'Donnell et al., 2023). Consequently, internationally standardized LC-MS/MS protocols have now been established to ensure rigorous identification and quantification (Schebb et al., 2025). To date, no study adhering to these validated standards has definitively demonstrated the presence of endogenous SPMs in human SALI samples. Recent mechanistic insights reveal that SPMs (e.g., protectins, maresins, D-series resolvins) function as biased positive allosteric modulators of the EP4 receptor at supraphysiological concentrations. This EP4-dependent action converts anti-phagocytic signaling to pro-phagocytic activity-a central mechanism underpinning the therapeutic efficacy of exogenously administered SPMs observed in preclinical models (Alnouri et al., 2024). The requirement for supraphysiological concentrations (EC50 ∼100 nM) to activate this pathway highlights the pharmacokinetic challenges of natural SPMs, including rapid clearance and undetectable lung tissue levels in existing studies. Future formulations must therefore prioritize sustained-release systems (e.g., oxidation-protective micelles) to maintain therapeutic exposure.
The robust preclinical evidence summarized herein strongly supports the therapeutic potential of exogenously delivered SPMs or their stable analogs for SALI. Translating this promise requires: ① High-quality clinical trials evaluating SPMs/analogs in SALI patients, building on positive safety/efficacy signals in other inflammatory conditions (e.g., LXA4 analog in periodontitis (Hasturk et al., 2021); RvE1 analog in dry eye OphthalmologyWeb (2024). ② Exploration of optimized delivery strategies, informed by the EP4 allosteric modulation mechanism, to overcome potential drug-likeness challenges (e.g., high-dose regimens, stable analog development, targeted delivery systems). ③ Continued mechanistic refinement to fully elucidate optimal dosing paradigms and molecular targets, integrated with pharmacokinetic assessments of lung tissue exposure; and ④ Development of delivery platforms that counteract rapid clearance, such as polymeric micelles for sustained SPM release (de Prinse et al., 2023).
In conclusion, while questions regarding endogenous SPM physiology persist, exogenous SPMs represent a novel resolution pharmacology-based approach to actively restore immune homeostasis in SALI. Focused efforts on clinical translation hold significant potential to open new therapeutic avenues for this devastating condition.
Author contributions
JS: Conceptualization, Methodology, Writing – original draft. HS: Project administration, Validation, Writing – original draft. SS: Methodology, Project administration, Writing – original draft, Funding acquisition. SF: Writing – review and editing, Investigation, Validation, Resources. TZ: Formal Analysis, Data curation, Writing – review and editing. GS: Methodology, Writing – original draft, Resources. NA: Writing – review and editing, Methodology, Formal Analysis. XC: Funding acquisition, Writing – review and editing, Resources. YG: Writing – review and editing, Supervision, Software.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the Fundamental Research Funds for the Central Universities (HUST: 2024JYCXJJ021) and the National Natural Science Foundation (No. 82471251). The sponsor will not be involved in study design; collection, management, analysis, or interpretation of data; writing of the report; or the decision to submit the report for publication.
Acknowledgments
Pictures are prepared by Figdraw (www.figdraw.com) and BioRender (https://biorender.com/).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdulnour, R. E., Sham, H. P., Douda, D. N., Colas, R. A., Dalli, J., Bai, Y., et al. (2016). Aspirin-triggered resolvin D1 is produced during self-resolving gram-negative bacterial pneumonia and regulates host immune responses for the resolution of lung inflammation. Mucosal Immunol. 9, 1278–1287. doi:10.1038/mi.2015.129
Allen, C. D., Ansel, K. M., Low, C., Lesley, R., Tamamura, H., Fujii, N., et al. (2004). Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat. Immunol. 5, 943–952. doi:10.1038/ni1100
Alnouri, M. W., Roquid, K. A., Bonnavion, R., Cho, H., Heering, J., Kwon, J., et al. (2024). SPMs exert anti-inflammatory and pro-resolving effects through positive allosteric modulation of the prostaglandin EP4 receptor. Proc. Natl. Acad. Sci. U. S. A. 121, e2407130121. doi:10.1073/pnas.2407130121
Andersson-Sjöland, A., de Alba, C. G., Nihlberg, K., Becerril, C., Ramírez, R., Pardo, A., et al. (2008). Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int. J. Biochem. Cell. Biol. 40, 2129–2140. doi:10.1016/j.biocel.2008.02.012
Arango Duque, G., and Descoteaux, A. (2014). Macrophage cytokines: involvement in immunity and infectious diseases. Front. Immunol. 5, 491. doi:10.3389/fimmu.2014.00491
Archambault, A. S., Zaid, Y., Rakotoarivelo, V., Turcotte, C., Doré, É., Dubuc, I., et al. (2021). High levels of eicosanoids and docosanoids in the lungs of intubated COVID-19 patients. Faseb J. 35, e21666. doi:10.1096/fj.202100540R
Ba, F., Zhou, X., Zhang, Y., Wu, C., Xu, S., Wu, L., et al. (2019). Lipoxin A4 ameliorates alveolar fluid clearance disturbance in lipopolysaccharide-induced lung injury via aquaporin 5 and MAPK signaling pathway. J. Thorac. Dis. 11, 3599–3608. doi:10.21037/jtd.2019.08.86
Barnig, C., Cernadas, M., Dutile, S., Liu, X., Perrella, M. A., Kazani, S., et al. (2013). Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci. Transl. Med. 5, 174ra26. doi:10.1126/scitranslmed.3004812
Basil, M. C., and Levy, B. D. (2016). Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 16, 51–67. doi:10.1038/nri.2015.4
Bersten, A. D., Edibam, C., Hunt, T., and Moran, J.Australian and New Zealand Intensive Care Society Clinical Trials Group (2002). Incidence and mortality of acute lung injury and the acute respiratory distress syndrome in three Australian states. Am. J. Respir. Crit. Care Med. 165, 443–448. doi:10.1164/ajrccm.165.4.2101124
Buckley, C. D., Gilroy, D. W., and Serhan, C. N. (2014). Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 40, 315–327. doi:10.1016/j.immuni.2014.02.009
Calder, P. C. (2020). Eicosapentaenoic and docosahexaenoic acid derived specialised pro-resolving mediators: concentrations in humans and the effects of age, sex, disease and increased omega-3 fatty acid intake. Biochimie 178, 105–123. doi:10.1016/j.biochi.2020.08.015
Chappell, D., Jacob, M., Rehm, M., Stoeckelhuber, M., Welsch, U., Conzen, P., et al. (2008). Heparinase selectively sheds heparan sulphate from the endothelial glycocalyx. Biol. Chem. 389, 79–82. doi:10.1515/bc.2008.005
Chattopadhyay, R., Raghavan, S., and Rao, G. N. (2017). Resolvin D1 via prevention of ROS-Mediated SHP2 inactivation protects endothelial adherens junction integrity and barrier function. Redox Biol. 12, 438–455. doi:10.1016/j.redox.2017.02.023
Chen, H., Wang, S., Zhao, Y., Luo, Y., Tong, H., and Su, L. (2018). Correlation analysis of omega-3 fatty acids and mortality of sepsis and sepsis-induced ARDS in adults: data from previous randomized controlled trials. Nutr. J. 17, 57. doi:10.1186/s12937-018-0356-8
Cheng, X., He, S., Yuan, J., Miao, S., Gao, H., Zhang, J., et al. (2016). Lipoxin A4 attenuates LPS-Induced mouse acute lung injury via Nrf2-mediated E-cadherin expression in airway epithelial cells. Free Radic. Biol. Med. 93, 52–66. doi:10.1016/j.freeradbiomed.2016.01.026
Chiang, N., Fredman, G., Bäckhed, F., Oh, S. F., Vickery, T., Schmidt, B. A., et al. (2012). Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 484, 524–528. doi:10.1038/nature11042
Chiang, N., Sakuma, M., Rodriguez, A. R., Spur, B. W., Irimia, D., and Serhan, C. N. (2022). Resolvin T-series reduce neutrophil extracellular traps. Blood 139, 1222–1233. doi:10.1182/blood.2021013422
Chiurchiù, V., Leuti, A., Dalli, J., Jacobsson, A., Battistini, L., Maccarrone, M., et al. (2016). Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci. Transl. Med. 8, 353ra111. doi:10.1126/scitranslmed.aaf7483
Codagnone, M., Cianci, E., Lamolinara, A., Mari, V. C., Nespoli, A., Isopi, E., et al. (2018). Resolvin D1 enhances the resolution of lung inflammation caused by long-term Pseudomonas aeruginosa infection. Mucosal Immunol. 11, 35–49. doi:10.1038/mi.2017.36
Croasdell, A., Lacy, S. H., Thatcher, T. H., Sime, P. J., and Phipps, R. P. (2016). Resolvin D1 dampens pulmonary inflammation and promotes clearance of nontypeable Haemophilus influenzae. J. Immunol. 196, 2742–2752. doi:10.4049/jimmunol.1502331
Dalli, J., Colas, R. A., Quintana, C., Barragan-Bradford, D., Hurwitz, S., Levy, B. D., et al. (2017). Human sepsis eicosanoid and proresolving lipid mediator temporal profiles: correlations with survival and clinical outcomes. Crit. Care Med. 45, 58–68. doi:10.1097/ccm.0000000000002014
de Oliveira, J. R., da Silva, P. R., and Rogério, A. P. (2017). AT-RvD1 modulates the activation of bronchial epithelial cells induced by lipopolysaccharide and Dermatophagoides pteronyssinus. Eur. J. Pharmacol. 805, 46–50. doi:10.1016/j.ejphar.2017.03.029
de Prinse, M., Qi, R., and Amsden, B. G. (2023). Polymer micelles for the protection and delivery of specialized pro-resolving mediators. Eur. J. Pharm. Biopharm. 184, 159–169. doi:10.1016/j.ejpb.2023.01.020
Dyall, S. C., Balas, L., Bazan, N. G., Brenna, J. T., Chiang, N., da Costa Souza, F., et al. (2022). Polyunsaturated fatty acids and fatty acid-derived lipid mediators: recent advances in the understanding of their biosynthesis, structures, and functions. Prog. Lipid Res. 86, 101165. doi:10.1016/j.plipres.2022.101165
Eaton, D. C., Helms, M. N., Koval, M., Bao, H. F., and Jain, L. (2009). The contribution of epithelial sodium channels to alveolar function in health and disease. Annu. Rev. Physiol. 71, 403–423. doi:10.1146/annurev.physiol.010908.163250
El Kebir, D., József, L., Pan, W., and Filep, J. G. (2008). Myeloperoxidase delays neutrophil apoptosis through CD11b/CD18 integrins and prolongs inflammation. Circ. Res. 103, 352–359. doi:10.1161/01.RES.0000326772.76822.7a
El Kebir, D., József, L., Pan, W., Wang, L., Petasis, N. A., Serhan, C. N., et al. (2009). 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am. J. Respir. Crit. Care Med. 180, 311–319. doi:10.1164/rccm.200810-1601OC
Folkesson, H. G., and Matthay, M. A. (2006). Alveolar epithelial ion and fluid transport: recent progress. Am. J. Respir. Cell. Mol. Biol. 35, 10–19. doi:10.1165/rcmb.2006-0080SF
Fredman, G., Hellmann, J., Proto, J. D., Kuriakose, G., Colas, R. A., Dorweiler, B., et al. (2016). An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat. Commun. 7, 12859. doi:10.1038/ncomms12859
Fredman, G., and Serhan, C. N. (2024). Specialized pro-resolving mediators in vascular inflammation and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 21, 808–823. doi:10.1038/s41569-023-00984-x
Gao, Y., Zhang, H., Luo, L., Lin, J., Li, D., Zheng, S., et al. (2017). Resolvin D1 improves the resolution of inflammation via activating NF-κB p50/p50-Mediated Cyclooxygenase-2 expression in acute respiratory distress syndrome. J. Immunol. 199, 2043–2054. doi:10.4049/jimmunol.1700315
Godson, C., Mitchell, S., Harvey, K., Petasis, N. A., Hogg, N., and Brady, H. R. (2000). Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol. 164, 1663–1667. doi:10.4049/jimmunol.164.4.1663
Gong, J., Liu, H., Wu, J., Qi, H., Wu, Z. Y., Shu, H. Q., et al. (2015). Maresin 1 prevents lipopolysaccharide-induced neutrophil survival and accelerates resolution of acute lung injury. Shock 44, 371–380. doi:10.1097/shk.0000000000000434
Gong, J., Wu, Z. Y., Qi, H., Chen, L., Li, H. B., Li, B., et al. (2014). Maresin 1 mitigates LPS-Induced acute lung injury in mice. Br. J. Pharmacol. 171, 3539–3550. doi:10.1111/bph.12714
Gu, J., Luo, L., Wang, Q., Yan, S., Lin, J., Li, D., et al. (2018). Maresin 1 attenuates mitochondrial dysfunction through the ALX/cAMP/ROS pathway in the cecal ligation and puncture mouse model and sepsis patients. Lab. Investig. 98, 715–733. doi:10.1038/s41374-018-0031-x
Han, J., Li, H., Bhandari, S., Cao, F., Wang, X. Y., Tian, C., et al. (2020). Maresin conjugates in tissue regeneration 1 improves alveolar fluid clearance by up-regulating alveolar ENaC, Na, K-ATPase in lipopolysaccharide-induced acute lung injury. J. Cell. Mol. Med. 24, 4736–4747. doi:10.1111/jcmm.15146
Hao, Y., Zheng, H., Wang, R. H., Li, H., Yang, L. L., Bhandari, S., et al. (2019). Maresin1 alleviates metabolic dysfunction in septic mice: a (1)H NMR-based metabolomics analysis. Mediat. Inflamm. 2019, 2309175. doi:10.1155/2019/2309175
Hasturk, H., Schulte, F., Martins, M., Sherzai, H., Floros, C., Cugini, M., et al. (2021). Safety and preliminary efficacy of a novel host-modulatory therapy for reducing gingival inflammation. Front. Immunol. 12, 704163. doi:10.3389/fimmu.2021.704163
Haworth, O., Cernadas, M., Yang, R., Serhan, C. N., and Levy, B. D. (2008). Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat. Immunol. 9, 873–879. doi:10.1038/ni.1627
Herold, S., Mayer, K., and Lohmeyer, J. (2011). Acute lung injury: how macrophages orchestrate resolution of inflammation and tissue repair. Front. Immunol. 2, 65. doi:10.3389/fimmu.2011.00065
Hofling, U., Tacconelli, S., Contursi, A., Bruno, A., Mucci, M., Ballerini, P., et al. (2022). Characterization of the acetylation of cyclooxygenase-isozymes and targeted lipidomics of eicosanoids in serum and Colon cancer cells by the new aspirin formulation IP1867B versus aspirin in vitro. Front. Pharmacol. 13, 1070277. doi:10.3389/fphar.2022.1070277
Hu, X. H., Situ, H. L., Chen, J. P., and Yu, R. H. (2020). Lipoxin A4 alleviates lung injury in sepsis rats through p38/MAPK signaling pathway. J. Biol. Regul. Homeost. Agents 34, 807–814. doi:10.23812/20-108-a-20
Jin, S. W., Zhang, L., Lian, Q. Q., Liu, D., Wu, P., Yao, S. L., et al. (2007). Posttreatment with aspirin-triggered lipoxin A4 analog attenuates lipopolysaccharide-induced acute lung injury in mice: the role of heme oxygenase-1. Anesth. Analg. 104, 369–377. doi:10.1213/01.ane.0000252414.00363.c4
Jordan, P. M., and Werz, O. (2022). Specialized pro-resolving mediators: biosynthesis and biological role in bacterial infections. Febs J. 289, 4212–4227. doi:10.1111/febs.16266
Jundi, B., Lee, D. H., Jeon, H., Duvall, M. G., Nijmeh, J., Abdulnour, R. E., et al. (2021). Inflammation resolution circuits are uncoupled in acute sepsis and correlate with clinical severity. JCI Insight 6, e148866. doi:10.1172/jci.insight.148866
Koenis, D. S., Beegun, I., Jouvene, C. C., Aguirre, G. A., Souza, P. R., Gonzalez-Nunez, M., et al. (2021). Disrupted resolution mechanisms favor altered phagocyte responses in COVID-19. Circ. Res. 129, e54–e71. doi:10.1161/circresaha.121.319142
Krishnamoorthy, N., Burkett, P. R., Dalli, J., Abdulnour, R. E., Colas, R., Ramon, S., et al. (2015). Cutting edge: Maresin-1 engages regulatory T cells to limit type 2 innate lymphoid cell activation and promote resolution of lung inflammation. J. Immunol. 194, 863–867. doi:10.4049/jimmunol.1402534
Lei, P., Xu, W., Wang, C., Lin, G., Yu, S., and Guo, Y. (2023). Mendelian randomization analysis reveals causal associations of polyunsaturated fatty acids with sepsis and mortality risk. Infect. Dis. Ther. 12, 1797–1808. doi:10.1007/s40121-023-00831-z
Levy, B. D., Clish, C. B., Schmidt, B., Gronert, K., and Serhan, C. N. (2001). Lipid mediator class switching during acute inflammation: signals in resolution. Nat. Immunol. 2, 612–619. doi:10.1038/89759
Li, H., Hao, Y., Yang, L. L., Wang, X. Y., Li, X. Y., Bhandari, S., et al. (2020). MCTR1 alleviates lipopolysaccharide-induced acute lung injury by protecting lung endothelial glycocalyx. J. Cell. Physiol. 235, 7283–7294. doi:10.1002/jcp.29628
Li, R., Wang, Y., Ma, Z., Ma, M., Wang, D., Xie, G., et al. (2016). Maresin 1 mitigates inflammatory response and protects mice from sepsis. Mediat. Inflamm. 2016, 3798465. doi:10.1155/2016/3798465
Li, X., Li, C., Liang, W., Bi, Y., Chen, M., and Dong, S. (2014). Protectin D1 promotes resolution of inflammation in a murine model of lipopolysaccharide-induced acute lung injury via enhancing neutrophil apoptosis. Chin. Med. J. Engl. 127, 810–814. doi:10.3760/cma.j.issn.0366-6999.20131104
Liao, Z., Dong, J., Wu, W., Yang, T., Wang, T., Guo, L., et al. (2012). Resolvin D1 attenuates inflammation in lipopolysaccharide-induced acute lung injury through a process involving the PPARγ/NF-κB pathway. Respir. Res. 13, 110. doi:10.1186/1465-9921-13-110
Liu, H., Zhou, K., Liao, L., Zhang, T., Yang, M., and Sun, C. (2018). Lipoxin A4 receptor agonist BML-111 induces autophagy in alveolar macrophages and protects from acute lung injury by activating MAPK signaling. Respir. Res. 19, 243. doi:10.1186/s12931-018-0937-2
Luo, J., Zhang, W. Y., Li, H., Zhang, P. H., Tian, C., Wu, C. H., et al. (2022). Pro-resolving mediator resolvin E1 restores alveolar fluid clearance in acute respiratory distress syndrome. Shock 57, 565–575. doi:10.1097/shk.0000000000001865
Lv, Y., Chen, D., Tian, X., Xiao, J., Xu, C., Du, L., et al. (2023). Protectin conjugates in tissue regeneration 1 alleviates sepsis-induced acute lung injury by inhibiting ferroptosis. J. Transl. Med. 21, 293. doi:10.1186/s12967-023-04111-9
Marchand, N. E., Choi, M. Y., Oakes, E. G., Cook, N. R., Stevens, E., Gomelskaya, N., et al. (2023). Over-the-counter fish oil supplementation and pro-resolving and pro-inflammatory lipid mediators in rheumatoid arthritis. Prostagl. Leukot. Essent. Fat. Acids 190, 102542. doi:10.1016/j.plefa.2023.102542
Matthay, M. A., and Zemans, R. L. (2011). The acute respiratory distress syndrome: pathogenesis and treatment. Annu. Rev. Pathol. 6, 147–163. doi:10.1146/annurev-pathol-011110-130158
Mayer, K., Sommer, N., Hache, K., Hecker, A., Reiche, S., Schneck, E., et al. (2019). Resolvin E1 improves mitochondrial function in human alveolar epithelial cells during severe inflammation. Lipids 54, 53–65. doi:10.1002/lipd.12119
Moon, C., Lee, Y. J., Park, H. J., Chong, Y. H., and Kang, J. L. (2010). N-acetylcysteine inhibits RhoA and promotes apoptotic cell clearance during intense lung inflammation. Am. J. Respir. Crit. Care Med. 181, 374–387. doi:10.1164/rccm.200907-1061OC
Morita, M., Kuba, K., Ichikawa, A., Nakayama, M., Katahira, J., Iwamoto, R., et al. (2013). The lipid mediator protectin D1 inhibits influenza virus replication and improves severe influenza. Cell. 153, 112–125. doi:10.1016/j.cell.2013.02.027
Morty, R. E., Eickelberg, O., and Seeger, W. (2007). Alveolar fluid clearance in acute lung injury: what have we learned from animal models and clinical studies? Intensive Care Med. 33, 1229–1240. doi:10.1007/s00134-007-0662-7
O'Donnell, V. B., Schebb, N. H., Milne, G. L., Murphy, M. P., Thomas, C. P., Steinhilber, D., et al. (2023). Failure to apply standard limit-of-detection or limit-of-quantitation criteria to specialized pro-resolving mediator analysis incorrectly characterizes their presence in biological samples. Nat. Commun. 14, 7172. doi:10.1038/s41467-023-41766-w
Omenetti, A., Porrello, A., Jung, Y., Yang, L., Popov, Y., Choi, S. S., et al. (2008). Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J. Clin. Investig. 118, 3331–3342. doi:10.1172/jci35875
Oner, F., Alvarez, C., Yaghmoor, W., Stephens, D., Hasturk, H., Firatli, E., et al. (2021). Resolvin E1 regulates Th17 function and T cell activation. Front. Immunol. 12, 637983. doi:10.3389/fimmu.2021.637983
OphthalmologyWeb (2024). Resolvyx pharmaceuticals, inc. Announces positive data from phase 2 clinical trial of the resolvin RX-10045 in patients with dry eye syndrome Available online at: https://www.ophthalmologyweb.com/1315-News/32687-Resolvyx-Pharmaceuticals-Inc-Announces-Positive-Data-From-Phase-2-Clinical-Trial-Of-The-Resolvin-RX-10045-In-Patients-With-Dry-Eye-Syndrome/[Accessed].
Ortiz-Muñoz, G., Mallavia, B., Bins, A., Headley, M., Krummel, M. F., and Looney, M. R. (2014). Aspirin-triggered 15-epi-lipoxin A4 regulates neutrophil-platelet aggregation and attenuates acute lung injury in mice. Blood 124, 2625–2634. doi:10.1182/blood-2014-03-562876
Padovani, C. M., and Yin, K. (2024). Immunosuppression in sepsis: biomarkers and specialized pro-resolving mediators. Biomedicines 12, 175. doi:10.3390/biomedicines12010175
Petty, J. M., Sueblinvong, V., Lenox, C. C., Jones, C. C., Cosgrove, G. P., Cool, C. D., et al. (2007). Pulmonary stromal-derived factor-1 expression and effect on neutrophil recruitment during acute lung injury. J. Immunol. 178, 8148–8157. doi:10.4049/jimmunol.178.12.8148
Qi, W., Li, H., Cai, X. H., Gu, J. Q., Meng, J., Xie, H. Q., et al. (2015). Lipoxin A4 activates alveolar epithelial sodium channel gamma via the microRNA-21/PTEN/AKT pathway in lipopolysaccharide-induced inflammatory lung injury. Lab. Investig. 95, 1258–1268. doi:10.1038/labinvest.2015.109
Qiao, N., Lin, Y., Wang, Z., Chen, J. Y., Ge, Y. Y., Yao, S. L., et al. (2020). Maresin1 promotes M2 macrophage polarization through peroxisome proliferator-activated Receptor-γ activation to expedite resolution of acute lung injury. J. Surg. Res. 256, 584–594. doi:10.1016/j.jss.2020.06.062
Ramon, S., Baker, S. F., Sahler, J. M., Kim, N., Feldsott, E. A., Serhan, C. N., et al. (2014). The specialized proresolving mediator 17-HDHA enhances the antibody-mediated immune response against influenza virus: a new class of adjuvant? J. Immunol. 193, 6031–6040. doi:10.4049/jimmunol.1302795
Ramon, S., Gao, F., Serhan, C. N., and Phipps, R. P. (2012). Specialized proresolving mediators enhance human B cell differentiation to antibody-secreting cells. J. Immunol. 189, 1036–1042. doi:10.4049/jimmunol.1103483
Schebb, N. H., Kampschulte, N., Hagn, G., Plitzko, K., Meckelmann, S. W., Ghosh, S., et al. (2025). Technical recommendations for analyzing oxylipins by liquid chromatography-mass spectrometry. Sci. Signal 18, eadw1245. doi:10.1126/scisignal.adw1245
Schebb, N. H., Kühn, H., Kahnt, A. S., Rund, K. M., O'Donnell, V. B., Flamand, N., et al. (2022). Formation, signaling and occurrence of specialized pro-resolving lipid mediators-what is the evidence so far? Front. Pharmacol. 13, 838782. doi:10.3389/fphar.2022.838782
Serhan, C. N. (2017). Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. Faseb J. 31, 1273–1288. doi:10.1096/fj.201601222R
Serhan, C. N., and Chiang, N. (2023). Resolvins and cysteinyl-containing pro-resolving mediators activate resolution of infectious inflammation and tissue regeneration. Prostagl. Other Lipid Mediat 166, 106718. doi:10.1016/j.prostaglandins.2023.106718
Serhan, C. N., Chiang, N., and Van Dyke, T. E. (2008). Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8, 349–361. doi:10.1038/nri2294
Serhan, C. N., and Levy, B. D. (2018). Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 128, 2657–2669. doi:10.1172/jci97943
Serhan, C. N., Libreros, S., and Nshimiyimana, R. (2022). E-series resolvin metabolome, biosynthesis and critical role of stereochemistry of specialized pro-resolving mediators (SPMs) in inflammation-resolution: preparing SPMs for long COVID-19, human clinical trials, and targeted precision nutrition. Semin. Immunol. 59, 101597. doi:10.1016/j.smim.2022.101597
Serhan, C. N., and Petasis, N. A. (2011). Resolvins and protectins in inflammation resolution. Chem. Rev. 111, 5922–5943. doi:10.1021/cr100396c
Seymour, C. W., Kennedy, J. N., Wang, S., Chang, C. H., Elliott, C. F., Xu, Z., et al. (2019). Derivation, validation, and potential treatment implications of novel clinical phenotypes for sepsis. Jama 321, 2003–2017. doi:10.1001/jama.2019.5791
Sham, H. P., Walker, K. H., Abdulnour, R. E., Krishnamoorthy, N., Douda, D. N., Norris, P. C., et al. (2018). 15-epi-Lipoxin A(4), resolvin D2, and resolvin D3 induce NF-κB regulators in bacterial pneumonia. J. Immunol. 200, 2757–2766. doi:10.4049/jimmunol.1602090
Sobrino, A., Walker, M. E., Colas, R. A., and Dalli, J. (2020). Protective activities of distinct omega-3 enriched oils are linked to their ability to upregulate specialized pro-resolving mediators. PLoS One 15, e0242543. doi:10.1371/journal.pone.0242543
Souza, P. R., Marques, R. M., Gomez, E. A., Colas, R. A., De Matteis, R., Zak, A., et al. (2020). Enriched marine oil supplements increase peripheral blood specialized pro-resolving mediators concentrations and reprogram host immune responses: a randomized double-blind placebo-controlled study. Circ. Res. 126, 75–90. doi:10.1161/circresaha.119.315506
Sundarasivarao, P. Y. K., Walker, J. M., Rodriguez, A., Spur, B. W., and Yin, K. (2022). Resolvin D2 induces anti-microbial mechanisms in a model of infectious peritonitis and secondary lung infection. Front. Immunol. 13, 1011944. doi:10.3389/fimmu.2022.1011944
Sznajder, J. I., Factor, P., and Ingbar, D. H. (2002). Invited review: lung edema clearance: role of Na(+)-K(+)-ATPase. J. Appl. Physiol. 93, 1860–1866. doi:10.1152/japplphysiol.00022.2002
Tan, W., Chen, L., Wang, Y. X., Hu, L. S., Xiong, W., Shang, Y., et al. (2018). Protectin DX exhibits protective effects in mouse model of lipopolysaccharide-induced acute lung injury. Chin. Med. J. Engl. 131, 1167–1173. doi:10.4103/0366-6999.227618
Tejera, P., Abdulnour, R. E., Zhu, Z., Su, L., Levy, B. D., and Christiani, D. C. (2020). Plasma levels of proresolving and prophlogistic lipid mediators: association with severity of respiratory failure and mortality in acute respiratory distress syndrome. Crit. Care Explor 2, e0241. doi:10.1097/cce.0000000000000241
Thornton, J. M., Padovani, C. M., Rodriguez, A., Spur, B. W., and Yin, K. (2023). Lipoxin A(4) promotes antibiotic and monocyte bacterial killing in established Pseudomonas aeruginosa biofilm formed under hydrodynamic conditions. Faseb J. 37, e23098. doi:10.1096/fj.202300619R
Thornton, J. M., Walker, J. M., Sundarasivarao, P. Y. K., Spur, B. W., Rodriguez, A., and Yin, K. (2021). Lipoxin A4 promotes reduction and antibiotic efficacy against Pseudomonas aeruginosa biofilm. Prostagl. Other Lipid Mediat 152, 106505. doi:10.1016/j.prostaglandins.2020.106505
Tsai, W. H., Shih, C. H., Yu, Y. B., and Hsu, H. C. (2013). Plasma levels in sepsis patients of annexin A1, lipoxin A4, macrophage inflammatory protein-3a, and neutrophil gelatinase-associated lipocalin. J. Chin. Med. Assoc. 76, 486–490. doi:10.1016/j.jcma.2013.05.004
Walker, J., Dichter, E., Lacorte, G., Kerner, D., Spur, B., Rodriguez, A., et al. (2011). Lipoxin a4 increases survival by decreasing systemic inflammation and bacterial load in sepsis. Shock 36, 410–416. doi:10.1097/SHK.0b013e31822798c1
Walker, J. M., Sundarasivarao, P. Y. K., Thornton, J. M., Sochacki, K., Rodriguez, A., Spur, B. W., et al. (2022). Resolvin D2 promotes host defense in a 2 - hit model of sepsis with secondary lung infection. Prostagl. Other Lipid Mediat 159, 106617. doi:10.1016/j.prostaglandins.2022.106617
Wang, B., Gong, X., Wan, J. Y., Zhang, L., Zhang, Z., Li, H. Z., et al. (2011). Resolvin D1 protects mice from LPS-Induced acute lung injury. Pulm. Pharmacol. Ther. 24, 434–441. doi:10.1016/j.pupt.2011.04.001
Wang, F., Chen, M., Wang, C., Xia, H., Zhang, D., and Yao, S. (2022). Single-cell sequencing reveals the regulatory role of Maresin1 on neutrophils during septic lung injury. Cells 11, 3733. doi:10.3390/cells11233733
Wang, H., Anthony, D., Yatmaz, S., Wijburg, O., Satzke, C., Levy, B., et al. (2017). Aspirin-triggered resolvin D1 reduces pneumococcal lung infection and inflammation in a viral and bacterial coinfection pneumonia model. Clin. Sci. (Lond) 131, 2347–2362. doi:10.1042/cs20171006
Wang, L., Yuan, R., Yao, C., Wu, Q., Christelle, M., Xie, W., et al. (2014a). Effects of resolvin D1 on inflammatory responses and oxidative stress of lipopolysaccharide-induced acute lung injury in mice. Chin. Med. J. Engl. 127, 803–809. doi:10.3760/cma.j.issn.0366-6999.20131044
Wang, Q., Lian, Q. Q., Li, R., Ying, B. Y., He, Q., Chen, F., et al. (2013). Lipoxin A(4) activates alveolar epithelial sodium channel, Na,K-ATPase, and increases alveolar fluid clearance. Am. J. Respir. Cell. Mol. Biol. 48, 610–618. doi:10.1165/rcmb.2012-0274OC
Wang, Q., Zhang, H. W., Mei, H. X., Ye, Y., Xu, H. R., Xiang, S. Y., et al. (2020). MCTR1 enhances the resolution of lipopolysaccharide-induced lung injury through STAT6-mediated resident M2 alveolar macrophage polarization in mice. J. Cell. Mol. Med. 24, 9646–9657. doi:10.1111/jcmm.15481
Wang, Q., Zheng, X., Cheng, Y., Zhang, Y. L., Wen, H. X., Tao, Z., et al. (2014b). Resolvin D1 stimulates alveolar fluid clearance through alveolar epithelial sodium channel, Na,K-ATPase via ALX/cAMP/PI3K pathway in lipopolysaccharide-induced acute lung injury. J. Immunol. 192, 3765–3777. doi:10.4049/jimmunol.1302421
Wang, X. Y., Li, X. Y., Wu, C. H., Hao, Y., Fu, P. H., Mei, H. X., et al. (2021). Protectin conjugates in tissue regeneration 1 restores lipopolysaccharide-induced pulmonary endothelial glycocalyx loss via ALX/SIRT1/NF-kappa B axis. Respir. Res. 22, 193. doi:10.1186/s12931-021-01793-x
Wen, X., Zhou, J., Zhang, D., Li, J., Wang, Q., Feng, N., et al. (2015). Denatonium inhibits growth and induces apoptosis of airway epithelial cells through mitochondrial signaling pathways. Respir. Res. 16, 13. doi:10.1186/s12931-015-0183-9
Wu, B., Capilato, J., Pham, M. P., Walker, J., Spur, B., Rodriguez, A., et al. (2016). Lipoxin A4 augments host defense in sepsis and reduces Pseudomonas aeruginosa virulence through quorum sensing inhibition. Faseb J. 30, 2400–2410. doi:10.1096/fj.201500029R
Wu, Z., Zhang, L., Zhao, X., Li, Z., Lu, H., Bu, C., et al. (2021). Protectin D1 protects against lipopolysaccharide-induced acute lung injury through inhibition of neutrophil infiltration and the formation of neutrophil extracellular traps in lung tissue. Exp. Ther. Med. 22, 1074. doi:10.3892/etm.2021.10508
Xia, H., Chen, L., Liu, H., Sun, Z., Yang, W., Yang, Y., et al. (2017). Protectin DX increases survival in a mouse model of sepsis by ameliorating inflammation and modulating macrophage phenotype. Sci. Rep. 7, 99. doi:10.1038/s41598-017-00103-0
Xia, H., Ge, Y., Wang, F., Ming, Y., Wu, Z., Wang, J., et al. (2020). Protectin DX ameliorates inflammation in sepsis-induced acute lung injury through mediating PPARγ/NF-κB pathway. Immunol. Res. 68, 280–288. doi:10.1007/s12026-020-09151-7
Xie, W., Wang, H., Liu, Q., Li, Y., Wang, J., Yao, S., et al. (2016). ResolvinD1 reduces apoptosis and inflammation in primary human alveolar epithelial type 2 cells. Lab. Investig. 96, 526–536. doi:10.1038/labinvest.2016.31
Xie, W., Wang, H., Wang, L., Yao, C., Yuan, R., and Wu, Q. (2013). Resolvin D1 reduces deterioration of tight junction proteins by upregulating HO-1 in LPS-Induced mice. Lab. Investig. 93, 991–1000. doi:10.1038/labinvest.2013.80
Xu, H., Sheng, S., Luo, W., Xu, X., and Zhang, Z. (2023). Acute respiratory distress syndrome heterogeneity and the septic ARDS subgroup. Front. Immunol. 14, 1277161. doi:10.3389/fimmu.2023.1277161
Yang, J. X., Li, M., Chen, X. O., Lian, Q. Q., Wang, Q., Gao, F., et al. (2019). Lipoxin A(4) ameliorates lipopolysaccharide-induced lung injury through stimulating epithelial proliferation, reducing epithelial cell apoptosis and inhibits epithelial-mesenchymal transition. Respir. Res. 20, 192. doi:10.1186/s12931-019-1158-z
Yang, Y., Cheng, Y., Lian, Q. Q., Yang, L., Qi, W., Wu, D. R., et al. (2013). Contribution of CFTR to alveolar fluid clearance by lipoxin A4 via PI3K/Akt pathway in LPS-Induced acute lung injury. Mediat. Inflamm. 2013, 862628. doi:10.1155/2013/862628
Yaxin, W., Shanglong, Y., Huaqing, S., Hong, L., Shiying, Y., Xiangdong, C., et al. (2014). Resolvin D1 attenuates lipopolysaccharide induced acute lung injury through CXCL-12/CXCR4 pathway. J. Surg. Res. 188, 213–221. doi:10.1016/j.jss.2013.11.1107
Ye, Y., Yang, Q., Wei, J., Shen, C., Wang, H., Zhuang, R., et al. (2025). RvD1 improves resident alveolar macrophage self-renewal via the ALX/MAPK14/S100A8/A9 pathway in acute respiratory distress syndrome. J. Adv. Res. 67, 289–299. doi:10.1016/j.jare.2024.01.017
Ye, Y., Zhang, H. W., Mei, H. X., Xu, H. R., Xiang, S. Y., Yang, Q., et al. (2020). PDX regulates inflammatory cell infiltration via resident macrophage in LPS-Induced lung injury. J. Cell. Mol. Med. 24, 10604–10614. doi:10.1111/jcmm.15679
Zhang, H. W., Wang, Q., Mei, H. X., Zheng, S. X., Ali, A. M., Wu, Q. X., et al. (2019a). RvD1 ameliorates LPS-Induced acute lung injury via the suppression of neutrophil infiltration by reducing CXCL2 expression and release from resident alveolar macrophages. Int. Immunopharmacol. 76, 105877. doi:10.1016/j.intimp.2019.105877
Zhang, J. L., Zhuo, X. J., Lin, J., Luo, L. C., Ying, W. Y., Xie, X., et al. (2017). Maresin1 stimulates alveolar fluid clearance through the alveolar epithelial sodium channel Na,K-ATPase via the ALX/PI3K/Nedd4-2 pathway. Lab. Investig. 97, 543–554. doi:10.1038/labinvest.2016.150
Zhang, L., Qiu, C., Yang, L., Zhang, Z., Zhang, Q., Wang, B., et al. (2019b). GPR18 expression on PMNs as biomarker for outcome in patient with sepsis. Life Sci. 217, 49–56. doi:10.1016/j.lfs.2018.11.061
Zhang, P. H., Han, J., Cao, F., Liu, Y. J., Tian, C., Wu, C. H., et al. (2020). PCTR1 improves pulmonary edema fluid clearance through activating the sodium channel and lymphatic drainage in lipopolysaccharide-induced ARDS. J. Cell. Physiol. 235, 9510–9523. doi:10.1002/jcp.29758
Zheng, S., Wang, Q., D'Souza, V., Bartis, D., Dancer, R., Parekh, D., et al. (2018). ResolvinD(1) stimulates epithelial wound repair and inhibits TGF-β-induced EMT whilst reducing fibroproliferation and collagen production. Lab. Investig. 98, 130–140. doi:10.1038/labinvest.2017.114
Zheng, S., Wang, Q., He, Q., Song, X., Ye, D., Gao, F., et al. (2011). Novel biphasic role of LipoxinA(4) on expression of cyclooxygenase-2 in lipopolysaccharide-stimulated lung fibroblasts. Mediat. Inflamm. 2011, 745340. doi:10.1155/2011/745340
Zhuang, R., Yang, X., Cai, W., Xu, R., Lv, L., Sun, Y., et al. (2021). MCTR3 reduces LPS-Induced acute lung injury in mice via the ALX/PINK1 signaling pathway. Int. Immunopharmacol. 90, 107142. doi:10.1016/j.intimp.2020.107142
Zhuo, X. J., Hao, Y., Cao, F., Yan, S. F., Li, H., Wang, Q., et al. (2018a). Protectin DX increases alveolar fluid clearance in rats with lipopolysaccharide-induced acute lung injury. Exp. Mol. Med. 50, 1–13. doi:10.1038/s12276-018-0075-4
Keywords: specialized pro-resolving mediators (SPMs), sepsis-associated lung injury, lipoxin, resolvin, protectin, maresins
Citation: Shen J, Shen H, Sun S, Fan S, Zhang T, Song G, An N, Chen X and Gao Y (2025) The potential of exogenous specialized pro-resolving mediators in protecting against sepsis-associated lung injury: a review. Front. Pharmacol. 16:1622754. doi: 10.3389/fphar.2025.1622754
Received: 04 May 2025; Accepted: 07 July 2025;
Published: 29 July 2025.
Edited by:
Dieter Steinhilber, Goethe University Frankfurt, GermanyReviewed by:
Paola Patrignani, University of Studies G.d’Annunzio Chieti and Pescara, ItalyNils Helge Schebb, University of Wuppertal, Germany
Copyright © 2025 Shen, Shen, Sun, Fan, Zhang, Song, An, Chen and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ning An, YW5uaW5nMTAwNUAxNjMuY29t; Xiangdong Chen, eGlhbmdkb25nY2hlbjIwMTNAMTYzLmNvbQ==; Yafen Gao, Z2FveWFmZW4xMDMwQGh1c3QuZWR1LmNu
†These authors have contributed equally to this work