Abstract
Introduction:
The α1-adrenoceptor (α1AR) is involved in the physiopathology of the central nervous system (CNS), but its function in the adult male rat locus coeruleus (LC) has not been fully studied. We aimed to characterize the role of the α1AR in the regulation of the firing rate (FR) of LC neurons and to describe the signaling pathways involved.
Methods:
We measured, through single-unit extracellular recordings of LC neurons from adult male rats were used to measure the effect of adrenergic agonists in the presence and absence of adrenergic antagonists or inhibitors of several signalling pathways.
Results:
Noradrenaline (NA) (100 µM) and phenylephrine (PE) (100 µM) induced a stimulatory effect in the presence of α2-adrenoceptor (α2AR) antagonist RS 79948 (0.1 µM). The α1AR agonist cirazoline (1–100 µM) also stimulated the FR of LC neurons. The stimulatory effects of NA (100 µM), PE (100 µM), and cirazoline (1 μM and 10 µM) were blocked by α1AR antagonist WB 4101 (0.5 µM). NA (100 µM)-induced stimulation was reduced in the presence of Gi/o protein inactivator pertussis toxin (PTX) (500 ng ml-1) and the transient receptor potential (TRP) channel blocker 2-APB (30 µM), but not by protein kinase C (PKC) inhibitor Go 6976 (1 µM), G protein-activated inward rectifier potassium (GIRK) channel blocker BaCl2 (300 µM), or protein kinase A (PKA) inhibitor H-89 (10 µM). The stimulatory effect of cirazoline was not reduced by any of the tested inhibitors.
Conclusions:
From α1AR activation stimulates the FR of adult rat LC neurons through a signaling pathway that involves Gi/o proteins and TRP channels.
1 Introduction
The α1-adrenoceptor (α1AR) and the α2-adrenoceptor (α2AR), which belong to the G protein-coupled receptor (GPCR) family, have been involved in brain developmental processes and constitute potential therapeutic targets for different neuropathological disorders such as drug addiction, Parkinson’s and Alzheimer’s diseases, or post-traumatic stress (Ghanemi and Hu, 2015; Perez, 2020). The α1AR predominantly couples to Gq/11, the stimulation of which leads to the activation of phospholipase C (PLC) and the production of inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 activates the release of Ca2+ into the cytoplasm, whereas DAG activates protein kinase C (PKC) (Hein and Michel, 2007). The α2AR couples to the Gi/o protein, the activation of which results in the inhibition of adenylyl cyclase and reduction of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA) activity (Aantaa et al., 1995). However, each adrenoceptor can couple to multiple signaling pathways. Thus, the α1AR can activate Gi proteins (Petitcolin et al., 2001) and the α2AR can couple to Gs proteins (Eason and Liggett, 1995). Furthermore, adrenergic agonists can display ligand-directed signaling bias (Alexander et al., 2023).
Locus coeruleus (LC) is the main noradrenergic nucleus in the central nervous system (CNS) (Foote et al., 1983). It is involved in the regulation of CNS functions, including sleep–wake cycle, attention, memory, and stress-related responses (Berridge and Waterhouse, 2003; Matt et al., 2024). The activity of LC cells is regulated, among others, by the αAR (Schwarz et al., 2015). Both α1AR and α2AR have been localized in LC neurons through quantitative autoradiography (Chamba et al., 1991) and RT-PCR (Osborne et al., 2002). In situ hybridization techniques have revealed that the α1AAR is the main subtype of the α1AR in the LC (Day et al., 1997). Moreover, a recent immunohistochemical study has shown that the α1AR colocalizes with tyrosine hydroxylase in LC dendrites, which indicates that the α1AR is expressed in NA neurons (Luyo et al., 2023). However, there are conflicting data regarding the functional role of the α1AR in LC neurons. Some authors have suggested that the α1AR decreases its activity during development and disappears in the adult male rat LC (Finlayson and Marshall, 1984; Williams and Marshall, 1987). In contrast, indirect evidence has suggested that the α1AR contributes to the excitability of LC neurons observed in the presence of the α2AR antagonist in adult male rat brain slices (Ivanov and Aston-Jones, 1995). Furthermore, the activation of the α1AR reduces outward potassium currents induced by α2AR activation in LC neurons (Osborne et al., 2002). Finally, microdialysis studies have shown that local administration of α1AR agonist cirazoline increases noradrenaline (NA) in the LC, whereas administration of an α1AR antagonist decreases it (Pudovkina et al., 2001; Pudovkina and Westerink, 2005).
Although the function of the α1AR in different brain areas of the CNS and its involvement in several neurological disorders has been studied (Ghanemi and Hu, 2015; Lemmens et al., 2015; Perez, 2020), the role of this receptor in the adult LC remains controversial. Therefore, the aim of this work was to characterize functionally the α1AR in LC neurons from adult male rats and to examine possible downstream processes linked to receptor activation through single-unit extracellular recordings in brain slices.
2 Materials and methods
2.1 Animals
A total of 99 adult male Sprague–Dawley rats (200–300 g) were used to perform electrophysiological assays. Animals were obtained from the animal facilities of the University of the Basque Country (Leioa, Spain) and housed (2–5 rats/cage) under controlled environmental conditions (22°C, 12:12 h light/dark cycles with the light phase starting at 8:00 a.m. and humidity of 65%–70%) with free access to food and water. All the experiments were carried out according to EU Directive 2010/63 on the protection of animals used for scientific purposes and reviewed and approved by the local Ethical Committee for Research and Teaching of the University of the Basque Country (UPV/EHU, Spain) and the Department of Sustainability and Natural Environment of Provincial Council from Bizkaia. All the efforts were made to minimize animal suffering and to reduce the number of animals used.
2.2 Brain slice preparation
Experiments were performed as previously described (Nazabal et al., 2023). Animals were anesthetized with chloral hydrate (400 mg kg-1, i.p.) and decapitated. The brain was rapidly removed, and a block of tissue containing the brain stem was immersed in ice-cold modified artificial cerebrospinal fluid (aCSF), where NaCl was equiosmotically substituted with sucrose to improve neuronal viability. Coronal slices of approximately 600 μM thickness containing the LC were cut using a vibratome (FHC Inc., Brunswick, GA, USA) and then allowed to recover from the slicing for 90 min in oxygenated aCSF (95% O2/5% CO2, pH = 7.34). Then, slices were placed on a nylon mesh in a modified Haas-type interface chamber continuously perfused with aCSF at a temperature of 33 °C and a flow rate of 1.5 mL min-1, as previously made in different studies performed in rat brain slices from the LC (Nazabal et al., 2023). This temperature maintains a balance between near-natural physiological functionality and adequate tissue viability suitable for stable recordings. The aCSF had the following composition: NaCl 130 mM, KCl 3 mM, NaH2PO4 1.25 mM, D-glucose 10 mM, NaHCO3 20 mM, CaCl2 2 mM, and MgSO4 2 mM. The modified aCSF, in which NaCl was equiosmotically replaced by sucrose, had the following composition: KCl 3 mM, NaH2PO4 1.25 mM, D-glucose 10 mM, NaHCO3 24 mM, sucrose 252 mM, CaCl2 2 mM, and MgSO4 2 mM.
2.3 Extracellular recordings
Single-unit extracellular recordings of LC noradrenergic neurons were made as previously described (Nazabal et al., 2023). The recording electrode consisted of an Omegadot glass micropipette (Sutter Instruments, Novato, CA, USA) pulled and filled with 50 mM NaCl, with the tip broken back to a size of 2–5 μm (3–5 MΩ). The electrode was placed in the LC, which was identified visually in the rostral pons as a dark oval area on the lateral borders of the central grey and the fourth ventricle, just anterior to the genu of the facial nerve. The extracellular signal recorded using the microelectrode was passed through a high-input impedance amplifier system (Axoclamp 2A, Axon Instruments, Foster City, CA) and monitored using an oscilloscope with an audio analyzer (Cibertec S.A., Madrid, Spain). Individual (single-unit) neuronal spikes were isolated from the background noise with a window discriminator and counted. FR was represented and analyzed using a PC-based custom-made program (HFCP®; Cibertec S.A. Madrid, Spain), which generated histogram bars representing the cumulative number of spikes in consecutive 10-s bins. Noradrenergic neurons in the LC were identified by the following electrophysiological criteria: a spontaneous and regular discharge, a slow firing rate (FR), and a positive–negative biphasic waveform of 3–4 ms duration (Andrade and Aghajanian, 1984). We only selected cells that showed stable firing rates between 0.5 and 1.5 Hz for at least 3–5 min and clear inhibitory responses to perfusion with [Met]enkephalin (ME, 0.8 μM, 1 min) or γ-aminobutyric acid (GABA, 1 mM, 1 min) according to previous studies (Nazabal et al., 2023).
2.4 Pharmacological procedures
To characterize the functional role of α1AR in LC neurons, first, the effect of nonselective AR agonist NA (100 μM, 1 min) was studied before or during the administration of α2AR antagonist RS 79948 (0.1 μM, 10 min). Next, α1AR agonists cirazoline (1, 10, 100 μM, 5–10 min) or phenylephrine (100 μM, 1 min) were perfused in the presence or absence of α1AR antagonist WB 4101 (0.5 μM, 10 min) to confirm that their effects on the FR were mediated by α1AR activation. To characterize the putative signaling pathways involved in the effects of α1AR agonists on the spontaneous FR of LC neurons, NA (100 μM, 1 min) or cirazoline (10 μM, 5–10 min) was tested during perfusion with the following drugs: Go 6976 (1 μM, 30 min; inhibitor of classical type PKC isoenzymes), chelerythrine (10 μM, 30 min; PKC inhibitor), U73122 (10 μM, 30 min; PLC inhibitor), H-89 (10 μM, 20 min; PKA inhibitor), BaCl2 [300 μM, 15 min; G protein-activated inward rectifier potassium (GIRK) channel inhibitor], or 2-APB (3, 10, and 30 μM, 10 min; blocker of TRPC5 and TRPM7 channels). Control applications of α1AR agonists were performed in the presence of the vehicle used to dissolve each inhibitor or blocker. To study the role of Gi/o proteins in the stimulatory effect induced by NA (100 μM, 1 min) or cirazoline (10 μM, 5–10 min), we treated brain slices with the catalyst of ADP-ribosylation of Gi/o protein pertussis toxin (PTX) (500 ng ml-1, 18 h). In PTX-pretreated cells, the effect of NA was tested in the same neuron both in the absence and presence of RS 79948 (0.1 μM, 10 min), whereas the effect of cirazoline was studied in different neurons because its effect was not washable. At the beginning of all the experiments, inhibitory effects of GABA (1 mM, 1 min) and ME (0.8 μM, 1 min) were measured to verify that slices were correctly perfused (inhibition magnitudes >80% of the basal firing rate). In PTX-pretreated cells, only neurons with a reduced inhibitory effect of ME (<80%) were considered.
2.5 Data analysis and statistics
The effects of AR agonists were calculated by subtracting the FR before drug perfusion from the FR after drug perfusion (FRafter−FRbefore). For the inhibitory effect of NA, FRafter was considered the average FR recorded for 60 s after agonist perfusion. For the stimulatory effect, FRafter was the average peak FR recorded for 30 s after perfusion with NA in the presence of RS 79948 or the average FR recorded for the last 120 s after perfusion with cirazoline. FRbefore was the average FR recorded for 60 s before agonist administration. Effects were normalized as the percentage change from the baseline FR before the application of adrenergic agonist. The data and statistical analysis were carried out using the computer program GraphPad Prism (version 5.0 for Windows; GraphPad Software, Inc., San Diego, CA, USA) and comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Values are expressed as the mean ± SEM of n experiments. The FR before or after drug application or the effects of drugs on the FR (% change from the baseline FR) were compared using a paired Student’s t-test within the same cell or an unpaired Student’s t-test between different groups. Comparisons between the effects of different concentrations of a drug in the same cell (cirazoline and 2-APB) were performed using the repeated-measures ANOVA, followed by Bonferroni’s multiple comparison post hoc test. Comparisons between the effect of cirazoline in the absence and presence of different inhibitors were performed using the one-way ANOVA, followed by the Dunnet post hoc test. The threshold of significance was considered as P = 0.05.
2.6 Drugs and reagents
The following drugs were purchased from Tocris Bioscience (Bristol, United Kingdom): 2-APB, chelerythrine chloride, cirazoline hydrochloride, Go 6976, H-89 dihydrochloride, PTX, PE hydrochloride, RS 79948 hydrochloride, and U73122. The following drugs were obtained from Sigma-Aldrich Química S.L. (Madrid, Spain): BaCl2 dihydrate, GABA, L-(−)-NA (+)-bitartrate salt monohydrate, and WB 4101 hydrochloride. ME acetate salt was purchased from Bachem (Weil am Rhein, Germany). Stock solutions of cirazoline, GABA, H-89, NA, PE, PTX, RS 79948, and WB4101 were first prepared in Mili-Q water and then diluted in aCSF to the final concentration before application. Go 6976, U73122, and 2-APB stock solutions were prepared in DMSO. The final concentration of DMSO was 0.2% (U73122 dilution), 0.03% (2-APB dilution), or 0.01% (Go 6976 dilution), which fail to change the FR of LC cells. BaCl2 was directly dissolved in aCSF.
3 Results
3.1 Effect of nonselective αAR NA and α1AR agonists PE and cirazoline on the firing rate of LC cells
To study the functional role of α1AR in LC neurons, we tested the effects of several α-adrenergic agonists in the presence or absence of α-adrenergic antagonists on the spontaneous FR of LC neurons. As expected, application of the nonselective α-adrenergic agonist NA (100 μM, 1 min), which also binds to the β-adrenoceptor (βAR) and dopamine D2 receptor (D2 receptor), inhibited the FR of LC neurons by 90.5% ± 5.1% (n = 5, P < 0.05). However, in the presence of α2AR antagonist RS 79948 (0.1 μM, 10 min), NA (100 μM, 1 min) stimulated the FR by 114.3% ± 23.7% (n = 5, P < 0.05) (Figures 1A,C). After the application of α1AR antagonist WB 4101 (0.5 μM, 10 min), NA (100 μM, 1 min) induced a decrease of 98.8% ± 1.0% on the FR (n = 6, P < 0.05) (Figure 1C), whereas in the presence of both α1AR and α2AR antagonists, NA (100 μM, 1 min) failed to induce any significant change in the FR (n = 5) (Figures 1B,C). To study whether other drugs mimicked the α1AR-mediated effect of NA, we studied the effects of α1AR agonists PE (100 μM, 1 min) and cirazoline (1, 10, 100 μM, 5–10 min) on the FR of LC neurons. Perfusion with the α1AR agonist PE (100 μM, 1 min), which also shows affinity for α2AR and β1AR, inhibited the FR of LC neurons by 28.7% ± 7.6% (n = 5, P < 0.05) (Figures 2A,E). In the presence of RS 79948 (0.1 μM, 10 min), PE (100 μM, 1 min) stimulated the FR by 60.7% ± 12.4% (n = 5, P < 0.05) (Figure 2C), whereas in the presence of WB 4101 (0.5 μM, 10 min), it induced an inhibition of 52.9% ± 9.0% on the FR (n = 7, P < 0.05) (Figure 2E). The simultaneous application of RS 79948 (0.1 μM, 10 min) and WB 4101 (0.5 μM, 10 min) blocked both the inhibitory and stimulatory effects of PE (100 μM, 1 min) (n = 5) (Figure 2E). Finally, the administration of α1AR agonist cirazoline (1, 10, and 100 μM, 5–10 min), which also binds to the serotonin 1A receptor (5-HT1A receptor) and α2AR receptors, induced a concentration-dependent increase in the FR of LC neurons. Thus, the stimulatory effect induced by cirazoline 1 μM was 35.5% ± 10.6% (n = 12, P < 0.05), whereas those induced by cirazoline 10 μM and 100 μM were 60.1% ± 11.2% (n = 12, P < 0.05) and 68.9% ± 11.5% (n = 12, P < 0.05), respectively (Figures 2B,F). In the presence of WB 4101 (0.5 μM, 10 min), which shows lower affinity for the D2 receptor and α2AR, the lowest concentrations of cirazoline (1 μM and 10 μM, 5–10 min each) failed to stimulate the FR of LC cells (effect of cirazoline 1 μM and 10 μM = −4.4% ± 1.4% and −7.9% ± 2.2%, respectively, n = 5 in both cases), whereas the highest concentration of the α1AR agonist (100 μM, 5–10 min) significantly stimulated the FR by 70.1% ± 22.6% (n = 5, P < 0.05) (Figures 2D,F). These results suggest that α1AR activation regulates the FR of LC neurons in a stimulatory way.
FIGURE 1
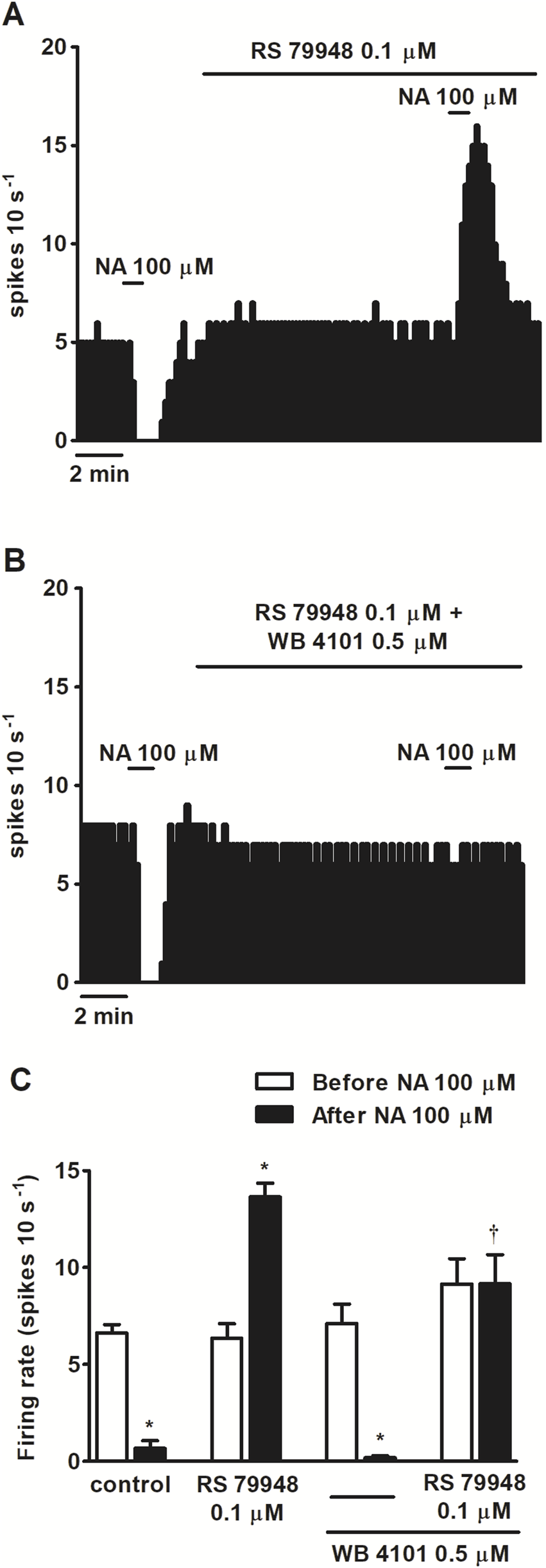
Effect of nonselective adrenergic agonist NA before and after the administration of α2AR antagonist RS 79948 or RS 79948 and α1AR antagonist WB 4101 on the spontaneous FR of LC neurons. (A,B) Representative examples of recordings of single LC neurons showing the inhibitory effect of NA (100 μM, 1 min) or its stimulatory effect in the presence of α2AR antagonist RS 79948 (0.1 μM, 10 min) on the basal FR (A) and the blockade of NA-induced effects in the presence of both RS 79948 (0.1 μM, 10 min) and α1AR antagonist WB 4101 (0.5 μM, 10 min) (B). Vertical lines represent the integrated firing rates (spikes per 10 s). Drugs were bath-applied at the concentrations and for the durations indicated by horizontal bars. (C) Bar histograms showing the mean ± SEM of LC neurons FR before and after the application of NA (100 μM, 1 min, n = 5), NA (100 μM, 1 min) + RS 79948 (0.1 μM, 10 min, n = 5), NA (100 μM, 1 min) + WB 4101 (0.5 μM, 10 min, n = 6), or NA (100 μM, 1 min) + RS 79948 (0.1 μM, 10 min) + WB 4101 (0.5 μM, 10 min, n = 5). *P < 0.05, compared with the FR before the application of NA (100 μM, 1 min) using a paired Student’s t-test. †P < 0.05, compared with the effect (normalized as the percentage change from the baseline FR) induced by NA (100 μM, 1 min) in the absence of WB 4101 (0.5 μM, 10 min) during RS 79948 (0.1 μM, 10 min) perfusion using an unpaired Student’s t-test.
FIGURE 2
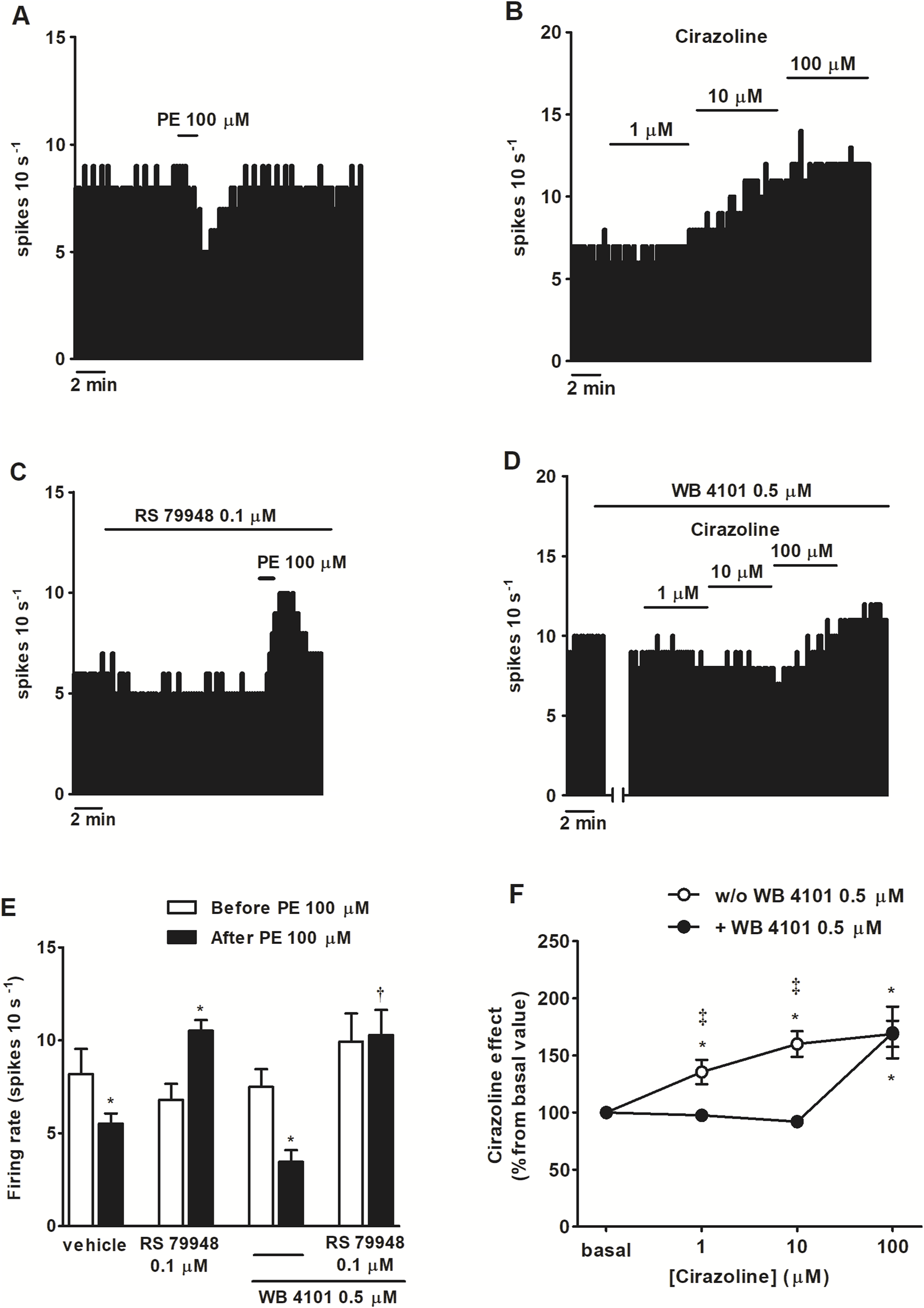
Effect of α1AR agonists PE and cirazoline before and after the administration of α2AR antagonist RS 79948, α1AR antagonist WB 4101 or RS 79948 and WB 4101 on the spontaneous FR of LC neurons. (A–D) Representative examples of recordings of single LC neurons showing the inhibitory effect of PE (100 μM, 1 min) on the basal FR (A), the stimulatory effect of cirazoline (1, 10, and 100 μM; 5–10 min) (B), the stimulatory effect of PE (100 μM, 1 min) in the presence of α2AR antagonist RS 79948 (0.1 μM, 10 min) (C), and the blockade of the stimulatory effect of cirazoline (1, 10 μM, 5–10 min), but not cirazoline (100 μM, 5–10 min), in the presence of α1AR antagonist WB 4101 (0.5 μM, 10 min) (D). Vertical lines represent the integrated firing rates (spikes per 10 s). Drugs were bath-applied at the concentrations and for the durations indicated by horizontal bars. (E) Bar histograms showing the mean ± SEM of LC neurons FR before and after the application of PE (100 μM, 1 min, n = 5), PE (100 μM, 1 min) + RS 79948 (0.1 μM, 10 min, n = 5), PE (100 μM, 1 min) + WB 4101 (0.5 μM, 10 min, n = 7), or PE (100 μM, 1 min) + RS 79948 (0.1 μM, 10 min) + WB 4101 (0.5 μM, 10 min, n = 5). (F) Symbols representing the mean ± SEM of LC neurons FR before and after the application of cirazoline (1, 10, and 100 μM; 5–10 min; n = 12) or cirazoline (1, 10, and 100 μM; 5–10 min) + WB 4101 (0.5 μM, 10 min, n = 5). *P < 0.05, compared with the FR before the application of PE (100 μM, 1 min) using a paired Student’s t-test or with the FR before the application of cirazoline (1, 10, and 100 μM, 5–10 min) using a repeated-measures ANOVA, followed by Bonferroni’s multiple-comparison post hoc test. †P < 0.05, compared with the effect (normalized as the percentage change from the baseline FR) induced by PE (100 μM, 1 min) in the absence of WB 4101 (0.5 μM, 10 min) during RS 79948 (0.1 μM, 10 min) perfusion using an unpaired Student’s t-test. ‡P < 0.05, compared with the effect of cirazoline in the presence of WB 4101 (0.5 μM, 10 min) using an unpaired Student’s t-test.
3.2 Molecular mechanisms involved in the α1AR-mediated effects of NA on the firing rate of LC cells
To characterize which signaling pathways were involved in the stimulatory effect produced by NA after blockade of α2AR, we tested the effect of NA (100 μM, 1 min) in the continuous presence of RS 79948 (0.1 μM, 10 min), before and after the application of inhibitors of several signaling pathways that could be involved in the increase in the FR. Perfusion with the inhibitor of classical type PKC isoenzyme Go 6976 (1 μM, 30 min) (n = 5), PKA inhibitor H-89 (10 μM, 20 min) (n = 5), or GIRK channel blocker BaCl2 (300 μM, 15 min) (n = 5) failed to significantly change NA (100 μM, 1 min)-induced stimulation in the continuous presence of RS 79948 (0.1 μM, 10 min) (Figures 3A–D). To study the role of Gi/o proteins in the stimulatory effect induced by NA (100 μM, 1 min) in the presence of RS 79948 (0.1 μM, 10 min), we treated brain slices containing the LC with the catalyst of ADP-ribosylation of Gi/o proteins PTX (500 ng ml-1, 18 h). To assess whether Gi/o proteins had been correctly inactivated, we tested the inhibitory effect of μ opioid receptor agonist ME (0.8 μM, 1 min) in PTX-treated slices. μ opioid receptors couple to Gi/o proteins, and their activation causes the opening of GIRK channels, which, in turn, activate outward potassium currents that inhibit the FR of LC neurons (Stein, 2016). In PTX-treated slices, the inhibitory effect of ME (0.8 μM, 1 min) was significantly lower than that in control slices. Thus, ME (0.8 μM, 1 min)-induced inhibition in control slices was 96.7% ± 1.3% (n = 17), whereas in PTX-treated slices, it was 30.9% ± 7.6% (n = 5) (P < 0.05). This indicates that Gi/o protein inactivation by bath application of PTX (500 ng ml-1, 18 h) was effective. In slices treated with PTX (500 ng ml-1, 18 h), perfusion with NA (100 μM, 1 min) in the presence of RS 79948 (0.1 μM, 10 min) stimulated the FR by 29.4% ± 5.7% (n = 5, P < 0.05), which was significantly reduced compared to that in the control group (Figures 4A,C). Finally, we tested the effect of the TRPC5/M7 channel blocker 2-APB (3, 10, and 30 μM; 10 min) in the NA-induced stimulatory effect. Perfusion with 2-APB (3 and 10 μM, 10 min) leads to a reduction in the stimulatory effect induced by NA (100 μM, 1 min) in the presence of RS 79948 (0.1 μM, 10 min), which was significant with the highest concentration (30 μM, 10 min, P < 0.05) (Figures 4B,D). Thus, before 2-APB perfusion, the stimulatory effect of NA (100 μM, 1 min) in the presence of RS 79948 (0.1 μM, 10 min) was 449.2% ± 97.7%, whereas after 2-APB perfusion (3 μM, 10 μM, and 30 μM; 10 min), it was 434.8 ± 91.0 (n = 5), 363.6% ± 73.9% (n = 5), and 253.8% ± 49.4% (n = 5, P < 0.05), respectively (Figures 4B,D). These data suggest that both Gi/o proteins and transient receptor potential (TRP) channels are implicated in the signaling pathway that mediates the stimulatory effect induced by NA (100 μM, 1 min) in the presence of RS 79948 (0.1 μM, 10 min) through α1AR.
FIGURE 3
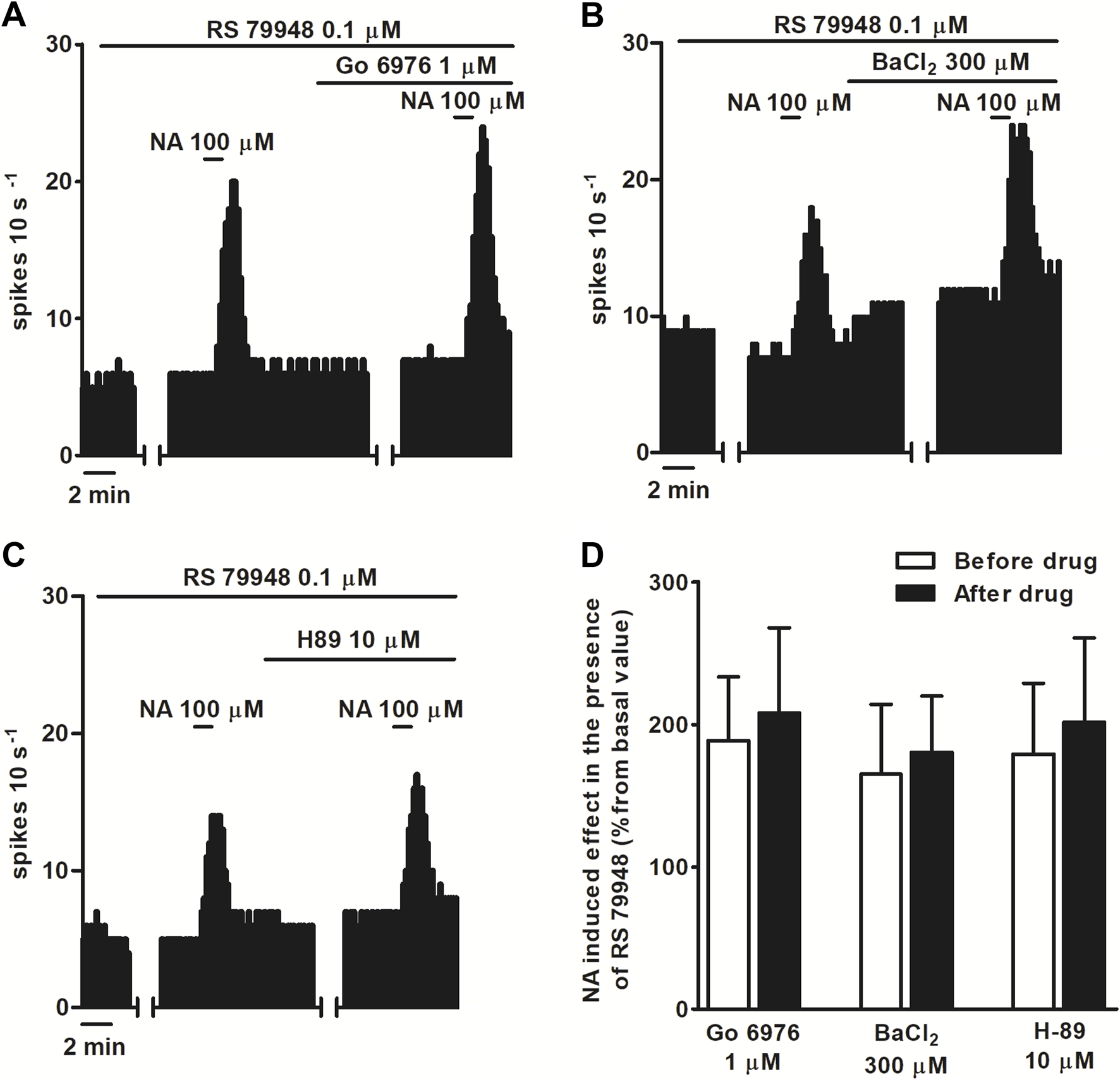
Effect of NA in the presence of RS 79948 before and after the application of PKC inhibitor Go 6976, GIRK blocker BaCl2, or PKA inhibitor H-89. (A–C) Representative examples of recordings of single LC neurons showing the stimulatory effect of NA (100 μM, 1 min) in the presence of RS79948 (0.1 μM, 10 min) before and after the application of Go 6976 (1 μM, 30 min) (A), BaCl2(B), and H89 (C). Vertical lines represent the integrated firing rates (spikes per 10 s). Drugs were bath-applied at the concentrations and for the durations indicated by the horizontal bars. (D) Bar histograms showing the mean ± SEM of the stimulatory effect (normalized as the percentage change from the baseline FR) of NA (100 μM, 1 min) in the presence of RS 79948 (0.1 μM, 10 min), before and after the application of Go 6976 (1 μM, 30 min, n = 5), BaCl2 (300 μM, 15 min, n = 5), or H-89 (10 μM, 20 min, n = 5).
FIGURE 4
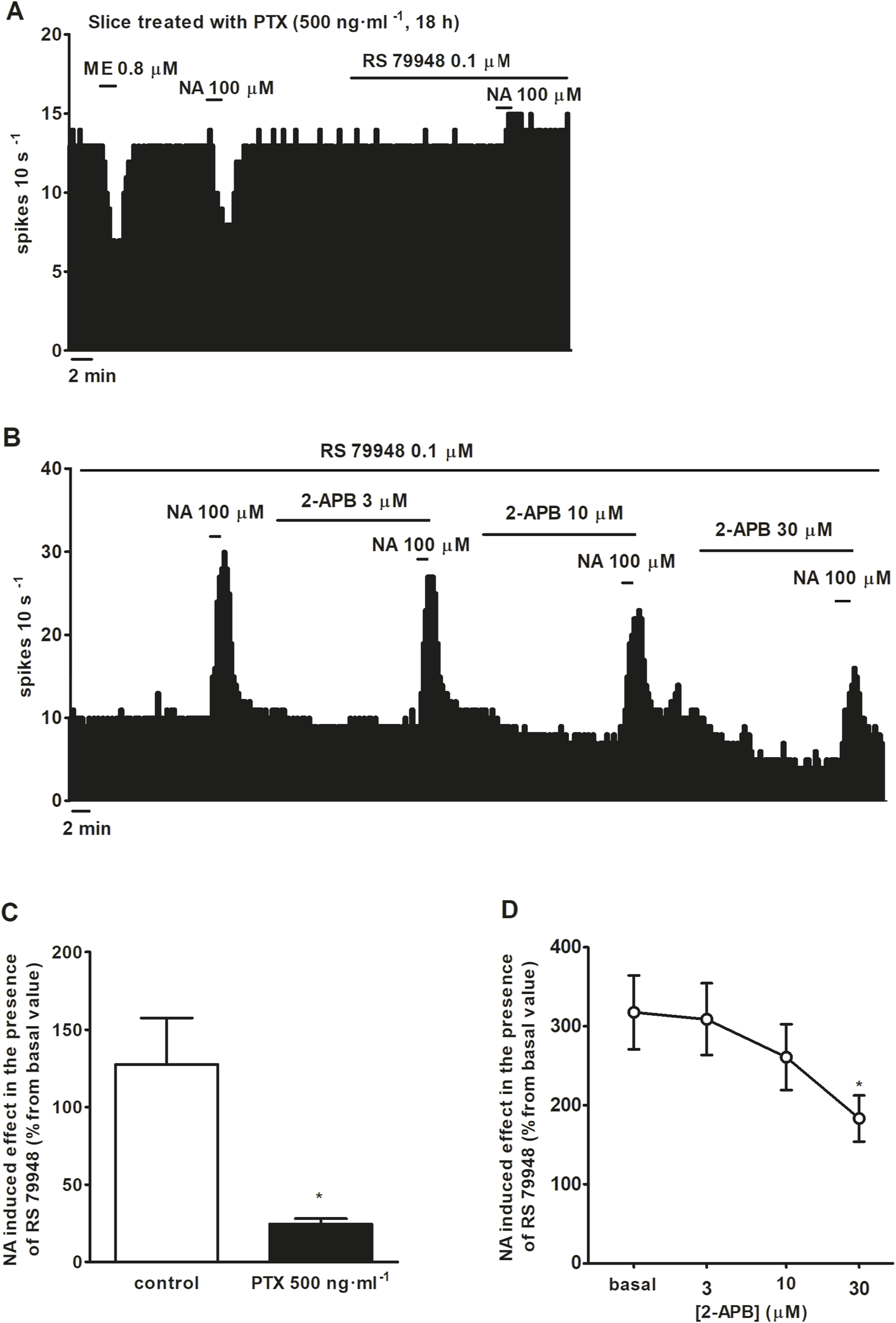
Effect of NA in the presence of RS 79948 before and after the application of the catalyst of ADP-ribosylation of Gi/o proteins PTX or the TRPC5/M7 channel blocker 2-APB. (A,B) Representative examples of the recordings of single LC neurons showing inhibitory effects of ME (0.8 μM, 1 min) and NA (100 μM, 1 min), and effect of NA (100 μM, 1 min) in the presence of RS 79948 (0.1 μM, 10 min), in a slice treated with PTX (500 ng ml-1, 18 h) (A), or the effect of NA (100 μM, 1 min) in the presence of RS 79948 (0.1 μM, 10 min) before and after the application of 2-APB (3, 10, 30 μM, 10 min) (B). Vertical lines represent the integrated firing rates (spikes per 10 s). Drugs (except PTX) were bath-applied at the concentrations and for the durations indicated by horizontal bars. (C) Bar histograms showing the mean ± SEM of the stimulatory effect of NA (100 μM, 1 min) in the presence of RS 79948 (0.1 μM, 10 min) in slices treated with PTX (500 ng ml-1, 18 h, n = 5) or its vehicle (n = 5). (D) Symbols representing the mean ± SEM of the stimulatory effect (normalized as the percentage change from the baseline FR) of NA (100 μM, 1 min) in the presence of RS 79948 (0.1 μM, 10 min), before and after the application of 2-APB (3, 10, and 30 μM; 10 min). *P < 0.05, compared with the stimulatory effect of NA (100 μM, 1 min) during RS 79948 (0.1 μM, 10 min) perfusion in slices that were not treated with PTX using an unpaired Student’s t-test. *P < 0.05, compared with the stimulatory effect of NA (100 μM, 1 min) during RS 79948 (0.1 μM, 10 min) perfusion before the application of 2-APB (3, 10, and 30 μM; 10 min) using a repeated-measures ANOVA, followed by Bonferroni’s multiple-comparison post hoc test.
3.3 Molecular mechanisms involved in the α1AR-mediated effect of cirazoline
To further study the mechanism of the stimulatory effect mediated by α1AR activation, we tested the effect of α1AR agonist cirazoline (10 μM, 5–10 min) after treatment with inhibitors of several signaling pathways. As previously mentioned, the cirazoline (10 μM, 5–10 min)-induced effect was 60.1% ± 11.2% (n = 12, P < 0.05). Unexpectedly, application of the PKC inhibitors Go 6976 (1 μM, 30 min) and chelerythrine (10 μM, 30 min) induced a significant increase in cirazoline (10 μM, 5–10 min) stimulatory effect (cirazoline’s effect after Go 6976 = 209.3 ± 25.6%, n = 5; after chelerythrine = 199.2 ± 11.1%, n = 3; P < 0.05 in both cases) (Figures 5C,D,G). However, slice treatment with the Gi/o protein inactivator PTX (500 ng ml-1, 18 h) (n = 5), the blocker of TRPC5 and TRPM7 subtypes 2-APB (30 μM, 10 min) (n = 5), the PKA inhibitor H-89 (10 μM, 20 min (n = 5), or the PLC inhibitor U73122 (10 μM, 30 min) (n = 6) failed to change cirazoline (10 μM, 5–10 min)-induced stimulation (Figures 5A,B,E–G). These results suggest that the signaling pathways that were studied are not directly involved in the stimulatory effect induced by cirazoline through the α1AR.
FIGURE 5
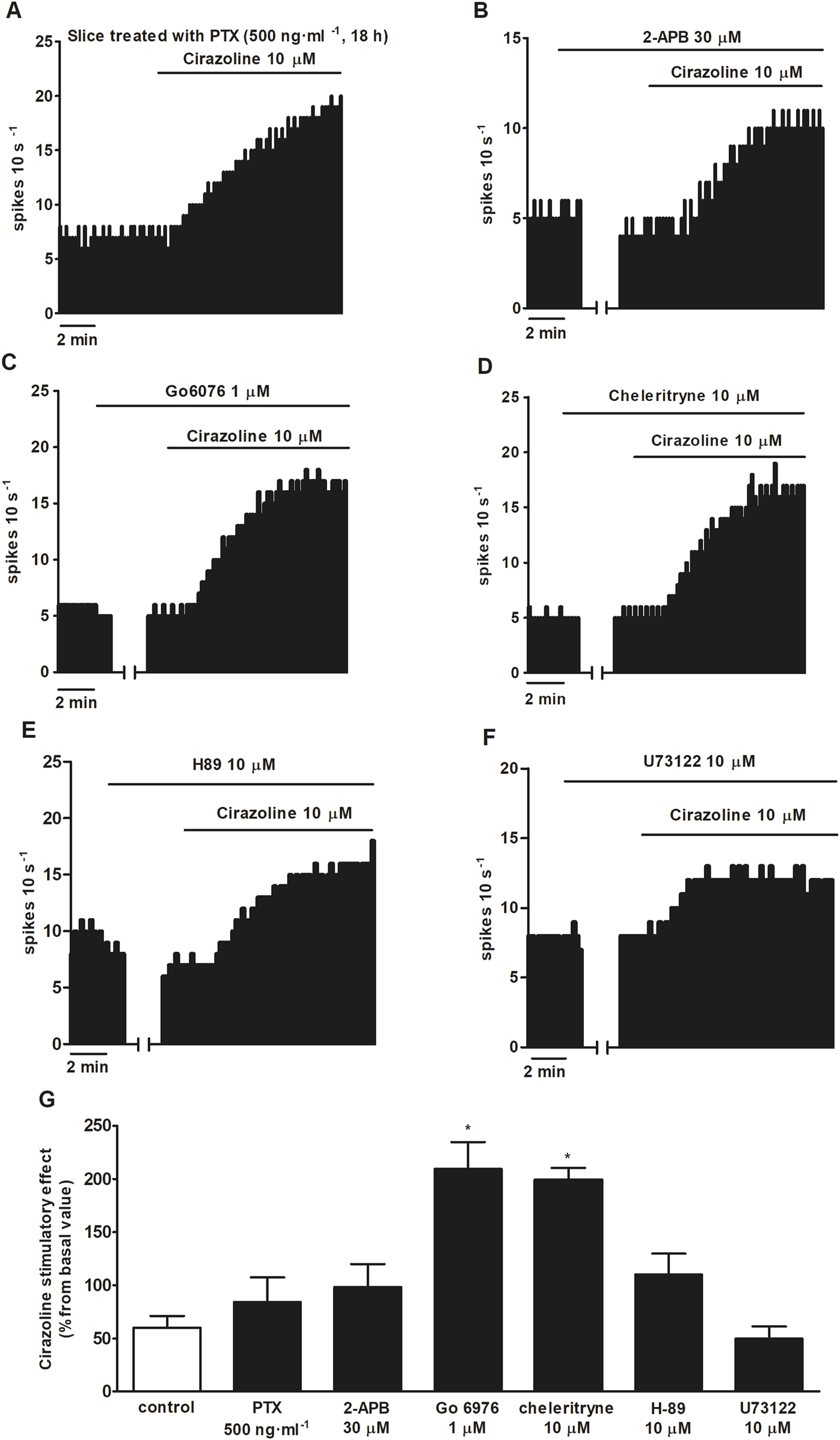
Effect of cirazoline in the absence or presence of PTX, 2-APB, Go 6976, chelerythrine, H-89, and U73122. (A,B) Representative examples of the recordings of single LC neurons showing the stimulatory effect of cirazoline (10 μM, 5–10 min) in a slice treated with PTX (500 ng ml-1, 18 h) (A) or in the presence of 2-APB (30 μM, 10 min) (B), Go 6976 (1 μM, 30 min) (C), chelerythrine (10 μM, 30 min) (D), H-89 (10 μM, 20 min) (E), or U73122 (10 μM, 30 min) (F). Vertical lines represent the integrated firing rates (spikes per 10 s). Drugs (except PTX) were bath-applied at the concentrations and for the durations indicated by horizontal bars. (G) Bar histograms showing the mean ± SEM of the stimulatory effect of cirazoline (normalized as the percentage change from the baseline FR) in the absence (control) and presence of PTX (500 ng ml-1, 18 h), 2-APB (30 μM, 10 min), Go 6976 (1 μM, 30 min), chelerythrine (10 μM, 30 min), H-89 (10 μM, 20 min), and U73122 (10 μM, 30 min). *P < 0.05, compared with the stimulatory effect of cirazoline (10 μM, 5–10 min) in the presence of the vehicle using one-way ANOVA, followed by the Dunnett post hoc test.
4 Discussion
The present work was undertaken to investigate the role of the α1AR in the regulation of the FR of LC neurons and the signaling pathways involved in its effects. Our results reveal that α1AR activation with the adrenergic agonists NA and PE in the presence of an α2AR antagonist or with cirazoline stimulates the FR of LC NA cells in adult male rat brain ex vivo. NA-induced stimulation was reduced by the inhibitor of Gi/o protein PTX and the TRP channel blocker 2-APB. However, none of the inhibitors blocked the stimulatory effect induced by cirazoline.
Perfusion with NA or PE inhibits the FR of LC neurons, whereas in the presence of α2AR antagonist RS 79948, they stimulate the FR. These results show that the inhibitory effects of these adrenergic agonists are mediated by the α2AR, as previously described (Williams et al., 1985; Williams et al., 1991). Even though PE is mainly considered a selective α1AR agonist, some studies performed in vascular tissues have shown that PE can activate α2AR (McGrath et al., 1999; Görnemann et al., 2009; VanLangen et al., 2013). Moreover, PE partially inhibits serotonin release in rat raphe nuclei through α2AAR activation (Hopwood and Stamford, 2001) and also evokes small-membrane hyperpolarizations in LC neurons (Williams et al., 1985). Finally, it has been recently reported that PE stimulates the cytoplasmatic release of NA via the NA transporter (Al-Khrasani et al., 2022), which could explain the inhibitory effect of PE observed in our experiments due to the presence of large reserve of α2AR receptors in LC neurons (Pineda et al., 1997).
In the presence of RS 79948 and α1AR antagonist WB 4101, both the inhibitory and stimulatory effects of NA and PE were blocked, which indicates that the increase in the FR induced by NA or PE after the α2AR blockade is mediated by the α1AR. Our data suggest that the α1AR-mediated excitatory effect of NA in LC neurons in adult rats may be masked by concurrent α2AR-mediated inhibition becoming apparent only when α2ARs are blocked by an antagonist. These results are consistent with those of studies that suggest that the activation of the α1AR may contribute to increase the FR of LC neurons (Ivanov and Aston-Jones, 1995). In contrast, some previous studies have reported that α1AR-mediated effects in LC neurons are restricted to early developmental stages. This discrepancy may be explained, at least in part, through methodological differences. Notably, earlier studies used techniques such as intracellular recordings in organotypic cultures, which may not fully reflect the physiological properties of mature LC neurons in acute brain slices. Moreover, the studies did not examine the effects of cirazoline or assess NA responses in the presence of an α2AR antagonist, both key aspects of our experimental design that may have unmasked α1AR-mediated excitatory effects in adult tissue.
The putative involvement of the β1AR in the stimulatory effects induced by NA and PE could be considered as both compounds bind to this receptor subtype (Ki ≈ 100–300 nM and 13 μM, respectively). However, in the case of NA, the contribution of the β1AR appears unlikely as WB 4101—a selective α1AR antagonist that does not bind to β1AR—completely blocked the stimulatory effect of NA in the presence of α2AR antagonist RS 79948. In contrast, in our slice preparations, WB 4101 significantly, but not completely, inhibited the stimulatory effect of PE under the same conditions. This suggests that β1AR may contribute to the effects of PE, a possibility that cannot be entirely excluded considering their affinity values for different receptors (Ki ≈ 13 µM for β1AR, 6 µM for α1AR, and 0.4 µM for α2AR) and the concentration applied (Chen et al., 1993; Gil and Donello, 2005). Alternatively, the involvement of other receptors (e.g., 5-HT7 receptor) could not be ruled out (European Molecular Biology Laboratory - European Bioinformatics Institute, 2025). It is important to note, however, that the same concentration of PE (100 µM) has previously been administrated in LC slice preparations to investigate α1AR-mediated effects, which aligns with the conditions used in our study (Osborne et al., 2002). Furthermore, the concentrations of all drugs used in this study were selected based on their reported Ki values or data from previous electrophysiological studies in brain slices, taking into account that drug affinities observed in isolated radioligand binding assays can differ significantly—often by 10- to 300-fold depending on the drug hydrosolubility—from those used in functionally active slice preparation. We used antagonists RS 79948 and WB 4101 to block α2AR and α1AR, respectively, because they have been shown to be rather selective for each α-adrenergic receptor type in previous in vivo or binding studies (Drew, 1982; Michel et al., 1995; Milligan et al., 1997; Mateo et al., 2000; Proudman et al., 2022). In other words, WB 4101 has lower affinity for D2 (Ki ≈ 123 nM) and α2AR (Ki ≈ 28–46 nM for α2B–α2AAR) than for α1AR (Ki ≈ 6–8 nM for α1B and Ki ≈ 0.5 nM for α1A). We did not use prazosin or terazosin as antagonists due to their lower selectivity for the α1AAR (Hancock et al., 1995; Yuan et al., 2009). Furthermore, WB 4101 (0.5 µM) blocks the PE-induced effect through the α1AAR in slices from other brain regions (Hancock et al., 1995; Yuan et al., 2009), which could also occur in the LC.
The α1AR agonist cirazoline, which also shows moderate affinity for the 5-HT1A receptor (Ki ≈ 35 nM) and binds to the α2AR (Ki ≈ 59 nM), stimulated the FR of LC cells. The stimulatory effects induced by cirazoline (1 µM and 10 µM) were blocked by perfusion with α1AR antagonist WB 4101, supporting the role of the α1AR in the regulation of the FR of LC neurons. However, at high concentrations (100 µM), cirazoline-induced stimulation was not blocked by WB 4101, which indicates that WB 4101 behaves as a competitive antagonist. The involvement of the non-α1AR receptor-mediated mechanism in the effect produced by the highest concentration of cirazoline could also be considered, including the activation of imidazoline receptors, 5-HT1A receptors, and α2AAR (Angel et al., 1995; Alexander et al., 2023). Thus, in anesthetized rats pretreated with EEDQ (an irreversible α-adrenoceptor antagonist), imidazoline drugs such as clonidine, cirazoline, and rilmenidine stimulate neuronal activity in LC cells through the activation of I1-imidazoline receptors (Pineda et al., 1993), but it seems to be an indirect effect mediated by imidazoline receptors located on paragigantocellularis neurons that project to the LC (Ruiz-Ortega and Ugedo, 1997). Some imidazoline drugs can stimulate the FR of LC neurons by a non-I1/I2 imidazoline receptor located extracellularly (Ugedo et al., 1998). Although this mechanism has not been described for cirazoline, it remains to be studied how cirazoline stimulates the FR of LC neurons after blockade of the α1AR in our system. The putative contribution of the 5-HT1A receptor or α2AR to the observed stimulatory effect could be ruled out as the activation of the 5-HT1A receptor or α2AR would reduce rather than increase the FR of LC cells. To address the study of the signaling pathways involved in the stimulatory effect mediated by α1AR, we used the agonists NA (100 μM; in the presence of the α2AR antagonist RS 79948 0.1 µM) and cirazoline (10 µM) because their effects were fully blocked after α1AR antagonism. Even though α1AR has been considered to be coupled to the Gq/11/PLC/PKC pathway in some areas of the rat brain (Kobayashi et al., 2008), there is evidence showing that this receptor can couple to other G proteins and multiple signaling pathways (Hein and Michel, 2007; Cotecchia, 2010). α1AR activation can also induce cAMP accumulation and PKA activation, which could modulate PKC (García-Sáinz et al., 2000). The enhancement of intracellular levels of cAMP or application of its analogs is known to increase the FR of LC neurons (Wang and Aghajanian, 1987). The activation of the α1AR with PE suppresses currents carried by GIRK channels that are opened by Gi/o protein-coupled receptors such as µ opioid receptor or α2AR (Osborne et al., 2002). In addition, it has been described that TRP channel antagonists suppress inward currents produced after α1AR activation with phenylephrine in LC neurons of SHR juvenile rats (Igata et al., 2014). Moreover, quantitative real-time PCR analysis found high levels of mRNA expression of TRPC5 (the most abundant type), TRPM7, and TRPM2 channels in the LC (Cui et al., 2011). The drugs that were used to inhibit each signaling pathway, such as Go 6976, PTX, BaCl2, 2-APB, H-89, and U73122, had been previously used by other authors in rat brain slices at similar concentrations (Chiu et al., 1995; Chessel et al., 1996; Bailey et al., 2009; Murai et al., 2012; Igata et al., 2014; Jolas et al., 2000).
PKC inhibitor Go 6976, PKA inhibitor H-89, and the blockade of GIRK channels with BaCl2 failed to change the stimulatory effect of NA in the presence of RS 79948. In contrast, Gi/o protein inhibition with PTX and perfusion with the TRPC5 and TRPM7 channel blocker 2-APB significantly reduced the stimulatory effect of NA in the presence of RS 79948, suggesting that the effect induced by NA through α1AR occurs via a pathway that involves Gi/o proteins and TRP channels. Although there is no evidence describing the α1AR/Gi/o protein/TRP channel pathway in neurons, some studies performed in different tissues could support this hypothesis. First, α1AR couples to Gi/o proteins and mediates pertussis toxin-sensitive effects in some vascular systems (Gurdal et al., 1997; Otani et al., 2001; Petitcolin et al., 2001). Second, Gi/o proteins stimulate TRPC5 and TRPM7 channel activities in several cell types (Beech, 2012; Oronowicz et al., 2021). Moreover, TRPC5 channels can be activated by the Gi/o protein-coupled µ opioid receptor (Miller et al., 2011), which is widely expressed in LC neurons, and they also contribute to the development of opioid tolerance in spinal neurons (Chu et al., 2020). Considering the aforementioned studies, we suggest that α1AR activation by adrenergic agonist NA stimulates the FR of LC neurons through its interaction with Gi/o proteins and TRPC5/TRPM7 channels, and we propose two hypotheses that could explain this mechanism. On the one hand, α1AR, Gi/o proteins, and TRP channels could be directly coupled. Then, α1AR activation could regulate Gi/o proteins and lead to the opening of TRP channels, which would induce an inward cationic current to increase the FR. On the other hand, Gi/o proteins could constitutively inhibit TRP channels and α1AR activation could relieve this inhibition, which would result in TRP channel opening and neuron depolarization.
It has been described that α1AR activation by PE in LC neurons reduces GIRK channel conductance induced by α2AR or µ opioid receptors, which are coupled to Gi/o proteins (Osborne et al., 2002). This interaction does not seem to occur in our system as the role of GIRK channels in α1AR activation was discarded. However, a mechanism involving TRP channels could also explain the observed stimulatory effect of PE on the FR of LC cells as in the case of NA. In contrast to NA-induced stimulation, Gi/o protein inhibitor PTX, TRP channel blocker 2-APB, PKC inhibitors Go 6976 and chelerythrine, PKA inhibitor H89, and PLC inhibitor U73122 failed to reduce the cirazoline stimulatory effect.
Furthermore, Go 6976 and chelerythrine enhanced the effect of cirazoline. Although no studies so far have directly linked PKC inhibition to the increased cirazoline effect, it is plausible to hypothesize that PKC activity exerts a constitutive inhibitory influence on the signaling pathway responsible for α1AR-mediated increases in the firing rate of LC neurons. Therefore, inhibition of PKC may relieve this suppression, thereby amplifying the stimulatory effect of cirazoline.
The differences in the results regarding the signaling pathways involved in NA or cirazoline stimulatory effects can be explained because these agonists may display functional selectivity. Functional selectivity or biased signaling refers to the ligand-dependent receptor activation of certain signaling pathways over others (Kolb et al., 2022). This property has been widely characterized in many GPCRs, including α1AR, which has been described by several authors to couple to different G proteins and activate several signaling pathways (Zhong and Minneman, 1999; Alcántara-Hernández et al., 2017; da Silva Junior et al., 2017). Therefore, our results could be explained due to biased signaling as studies performed in CHO-K1 cells expressing α1aAR found that cirazoline displays the signaling bias toward cAMP accumulation relative to Ca2+ release when compared to reference endogenous agonist NA (Evans et al., 2011). Moreover, NA and cirazoline show different affinities for each α1AR receptor subtype (NA: α1d > α1b > α1a; cirazoline: α1a > α1d > α1b) (Horie et al., 1995; Proudman and Baker, 2021).
Our study has some potential limitations. First, the magnitude of the basal stimulatory effect of NA in the presence of RS 79948 (i.e., in the absence of inhibitors or blockers) appeared to differ across some experimental groups (e.g., Figures 1 vs.Figure 4). However, no changes in experimental conditions could account for this discrepancy. Notably, in most groups, the effect of NA in the presence of RS 79948 was compared to the control NA response recorded in the same neuron, minimizing the impact of this variability on the overall conclusions. Second, consistent with previous studies, only male rats were used in this investigation. Future experiments should aim to characterize α1AR-mediated effects in female rats to assess potential sex-dependent differences.
α1AR is involved in several CNS functions and pathologies such as behavioral activity and depression (Stone et al., 2007), pain modulation (Kingery et al., 2002; Nakatsuka et al., 2022), reward processes (Lin et al., 2007), psychostimulant-induced locomotor hyperactivity (Drouin et al., 2002), or neurodegenerative disorders. Some of these have been shown to be related to the LC. In this line, an increase in the neuronal activity induced by the activation of α1-AR has also been reported in other brain regions, such as in the prefrontal cortex (Datta et al., 2019), the paraventricular nucleus of the hypothalamus (Chen et al., 2006), or the interneurons of the layer CA1 of the hippocampus (Hillman et al., 2009). In the latter case, this activation leads to a decrease in the activity of pyramidal neurons and to an antiepileptic effect. Previous studies have revealed a role of α2AR in the regulation of the activity of LC neuron (Aghajanian and VanderMaelen, 1982; Pineda et al., 1997) in the adult rat brain, but the function of α1AR was thought to be reduced during development. Our results reveal a functional importance of α1AR in the adult rat LC. The α1AR can be activated by adrenergic agonists NA and PE (after α2AR blockade) or by α1AR agonist cirazoline, stimulating the FR of LC neurons. The stimulatory effect induced by NA would occur through a signaling pathway that involves Gi/o proteins and TRPC5/TRPM7 channels. More studies will be required to describe in detail the mechanisms involved in the α1AR stimulatory effect and the functional role of this receptor in female rats, but our results suggest that the α1AR receptor in the adult male rat LC could constitute a target in the treatment of several disorders of the CNS.
Statements
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by the Ethical Committee for Research and Teaching of the University of the Basque Country (UPV/EHU, Spain) and the Department of Sustainability and Natural Environment of Provincial Council from Bizkaia. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
IR: data curation, writing – review and editing, investigation, formal analysis, and writing – original draft. AM: writing – review and editing, writing – original draft, supervision, funding acquisition, formal analysis, data curation, and conceptualization. JP: funding acquisition, formal analysis, writing – review and editing, conceptualization, project administration, supervision, and data curation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the University of the Basque Country (UPV/EHU) (GIU 19/076) and by the Ministerio de Sanidad, Consumo y Bienestar Social. Delegación del Gobierno para el Plan Nacional Sobre Drogas, PND 2018I025 (PND18/04). Irati Rodilla was supported by a predoctoral fellowship from the Spanish Ministry of Universities.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
α1AR, α1-adrenoceptor; α2AR, α2-adrenoceptor; aCSF, artificial cerebrospinal fluid; βAR, β-adrenoceptor; CNS, central nervous system; cAMP, cyclic adenosine monophosphate; D2 receptor, dopamine D2 receptor; FR, firing rate; GABA, γ-aminobutyric acid; GIRK, G protein-activated inward rectifier potassium; GPCR, G protein-coupled receptor; 5-HT1A receptor, serotonin 1A receptor; LC, locus coeruleus; ME, [Met]enkephalin; NA, noradrenaline; PAG, periaqueductal gray; PE, phenylephrine; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C; PTX, pertussis toxin; TRP, transient receptor potential channel.
References
1
Aantaa R. Marjamäki A. Scheinin M. (1995). Molecular pharmacology of α 2 -adrenoceptor subtypes. Ann. Med.27, 439–449. 10.3109/07853899709002452
2
Aghajanian G. K. VanderMaelen C. P. (1982). Alpha 2-adrenoceptor-mediated hyperpolarization of locus coeruleus neurons: intracellular studies in vivo. Science215, 1394–1396. 10.1126/science.6278591
3
Al-Khrasani M. Karadi D. A. Galambos A. R. Sperlagh B. Vizi E. S. (2022). The pharmacological effects of phenylephrine are indirect, mediated by noradrenaline release from the cytoplasm. Neurochem. Res.47, 3272–3284. 10.1007/s11064-022-03681-2
4
Alcántara-Hernández R. Hernández-Méndez A. Romero-Ávila M. T. Alfonzo-Méndez M. A. Pupo A. S. García-Sáinz J. A. (2017). Noradrenaline, oxymetazoline and phorbol myristate acetate induce distinct functional actions and phosphorylation patterns of α1A-adrenergic receptors. Biochim. Biophys. Acta - Mol. Cell Res.1864, 2378–2388. 10.1016/j.bbamcr.2017.09.002
5
Alexander S. P. H. Christopoulos A. Davenport A. P. Kelly E. Mathie A. A. Peters J. A. et al (2023). The concise guide to PHARMACOLOGY 2023/24: g protein-coupled receptors. Br. J. Pharmacol.180, S23–S144. 10.1111/bph.16177
6
Andrade R. Aghajanian G. K. (1984). Locus coeruleus activity in vitro: intrinsic regulation by a calcium-dependent potassium conductance but not alpha 2-adrenoceptors. J. Neurosci.4, 161–170. 10.1523/jneurosci.04-01-00161.1984
7
Angel I. Le Rouzic M. Pimoulde C. Graham D. Arbilla S. (1995). [3H]Cirazoline as a tool for the characterization of imidazoline sites, ann. New York acad. Sci.763, 112–124. 10.1111/j.1749-6632.1995.tb32396.x
8
Bailey C. P. Llorente J. Gabra B. H. Smith F. L. Dewey W. L. Kelly E. et al (2009). Role of protein kinase C and µ-opioid receptor (MOPr) desensitization in tolerance to morphine in rat locus coeruleus neurons. Eur. J. Neurosci.29, 307–318. 10.1111/j.1460-9568.2008.06573.x
9
Beech D. J. (2012). Integration of transient receptor potential canonical channels with lipids. Acta Physiol.204, 227–237. 10.1111/j.1748-1716.2011.02311.x
10
Berridge C. W. Waterhouse B. D. (2003). The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res. Rev.42, 33–84. 10.1016/S0165-0173(03)00143-7
11
Chamba G. Weissmann D. Rousset C. Renaud B. Pujol J. F. (1991). Distribution of alpha-1 and alpha-2 binding sites in the rat locus coeruleus. Brain Res. Bull.26, 185–193. 10.1016/0361-9230(91)90225-9
12
Chen G. T. King M. Gusovsky F. Creveling C. R. Daly J. W. Chen B. et al (1993). Syntheses of 2,5- and 2,6-Difluoronorepinephrine, 2,5-Difluoroepinephrine, and 2,6-Difluorophenylephrine: effect of disubstitution with fluorine on adrenergic activity. J. Med. Chem.36, 3947–3955. 10.1021/jm00076a024
13
Chen Q. Li D. P. Pan H. L. (2006). Presynaptic alpha1 adrenergic receptors differentially regulate synaptic glutamate and GABA release to hypothalamic presympathetic neurons. J. Pharmacol. Exp. Ther.316, 733–742. 10.1124/jpet.105.094797
14
Chessel I. Black M. Feniuk W. Humphrey P. (1996). Operational characteristics of somatostatin receptors mediating inhibitory actions on rat locus coeruleus neurones. Br. J. Pharmacol.117, 1673–1678. 10.1111/j.1476-5381.1996.tb15338.x
15
Chiu T. Chen M. Yang Y. Yang J. Tang F. (1995). Action of dexmedetomidine on rat locus coeruleus neurones: intracellular recording in vitro. Eur. J. Pharmacol.285, 261–268. 10.1016/0014-2999(95)00417-j
16
Chu W. G. Wang F. D. Sun Z. C. Bin Ma S. Wang X. Han W. J. et al (2020). TRPC1/4/5 channels contribute to morphine-induced analgesic tolerance and hyperalgesia by enhancing spinal synaptic potentiation and structural plasticity. FASEB J.34, 8526–8543. 10.1096/fj.202000154RR
17
Cotecchia S. (2010). The α1-adrenergic receptors: diversity of signaling networks and regulation. J. Recept. Signal Transduct. Res.30, 410–419. 10.3109/10799893.2010.518152
18
Cui N. Zhang X. Tadepalli J. S. Yu L. Gai H. Petit J. et al (2011). Involvement of TRP channels in the CO₂ chemosensitivity of locus coeruleus neurons. J. Neurophysiol.105, 2791–2801. 10.1152/jn.00759.2010
19
Curtis M. J. Bond R. A. Spina D. Ahluwalia A. Alexander S. P. A. Giembycz M. A. et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br. J. Pharmacol.172, 3461–3471. 10.1111/bph.12856
20
da Silva Junior E. D. Sato M. Merlin J. Broxton N. Hutchinson D. S. Ventura S. et al (2017). Factors influencing biased agonism in recombinant cells expressing the human α1A-adrenoceptor. Br. J. Pharmacol.174, 2318–2333. 10.1111/bph.13837
21
Datta D. Yang S. T. Galvin V. C. Solder J. Luo F. Morozov Y. M. et al (2019). Noradrenergic α1-adrenoceptor actions in the primate dorsolateral prefrontal cortex. J. Neurosci.39, 2722–2734. 10.1523/JNEUROSCI.2472-18.2019
22
Day H. E. W. Campeau S. Watson S. J. Akil H. (1997). Distribution of α1a-α1b- and α1d-adrenergic receptor mRNA in the rat brain and spinal cord. J. Chem. Neuroanat.13, 115–139. 10.1016/S0891-0618(97)00042-2
23
Drew G. M. (1982). Evidence in favour of a selective α1-Adrenoceptor blocking action of WB4101 in vivo. Naunyn Schmied. Arch. Pharmacol.319, 222–225. 10.1007/BF00495869
24
Drouin C. Blanc G. Villgier A. S. Glowinski J. Tassin J. P. (2002). Critical role of α1-adrenergic receptors in acute and sensitized locomotor effects of D-amphetamine, cocaine, and GBR 12783: influence of preexposure conditions and pharmacological characteristics. Synapse43, 51–61. 10.1002/syn.10023
25
Eason M. G. Liggett S. B. (1995). Identification of a Gs coupling domain in the amino terminus of the third intracellular loop of the alpha 2A-adrenergic receptor. Evidence for distinct structural determinants that confer Gs versus Gi coupling. J. Biol. Chem.270, 24753–24760. 10.1074/jbc.270.42.24753
26
European Molecular Biology Laboratory - European Bioinformatics Institute (2025). ChEMBL. Available online at: https://www.ebi.ac.uk/chembl/explore/document/CHEMBL5442175.
27
Evans B. A. Broxton N. Merlin J. Sato M. Hutchinson D. S. Christopoulos A. et al (2011). Quantification of functional selectivity at the human α1A-adrenoceptor. Mol. Pharmacol.79, 298–307. 10.1124/mol.110.067454
28
Finlayson P. G. Marshall K. C. (1984). Hyperpolarizing and age-dependent depolarizing responses of cultured locus coeruleus neurons to noradrenaline, dev. Brain Res.15, 167–175. 10.1016/0165-3806(84)90094-4
29
Foote S. L. Bloom F. E. Aston-Jones G. (1983). Nucleus locus ceruleus: new evidence of anatomical and physiological specificity. Physiol. Rev.63, 844–914. 10.1152/physrev.1983.63.3.844
30
García-Sáinz J. A. Vázquez-Prado J. Villalobos-Molina R. (2000). Alpha 1-adrenoceptors: subtypes, signaling, and roles in health and disease. Arch. Med. Res.30, 449–458. 10.1016/s0188-0128(99)00059-7
31
Ghanemi A. Hu X. (2015). Elements toward novel therapeutic targeting of the adrenergic system. Neuropeptides49, 25–35. 10.1016/j.npep.2014.11.003
32
Gil D. W. Donello J. E. (2005). Novel methods for identifying improved, non-sedating alpha-2 agonists, US 2005/0059664 A1.
33
Görnemann T. Villalón C. M. Centurión D. Pertz H. H. (2009). Phenylephrine contracts porcine pulmonary veins via alpha(1B)-alpha(1D)-and alpha(2)-adrenoceptors. Eur. J. Pharmacol.613, 86–92. 10.1016/j.ejphar.2009.04.011
34
Gurdal H. Seasholtz T. M. Wang H. Y. Brown R. D. Johnson M. D. Friedman E. (1997). Role of Gαq or Gαo proteins in α1-adrenoceptor subtype-mediated responses in fischer 344 rat aorta. Mol. Pharmacol.52, 1064–1070. 10.1124/mol.52.6.1064
35
Hancock A. A. Buckner S. A. Ireland L. M. Knepper S. M. Kerwin J. F. (1995). Actions of terazosin and its enantiomers at subtypes of alpha 1- and alpha 2-adrenoceptors in vitro. J. Recept. Signal Transduct.15, 863–885. 10.3109/10799899509049862
36
Hein P. Michel M. C. (2007). Signal transduction and regulation: are all α1-adrenergic receptor subtypes created equal?Biochem. Pharmacol.73, 1097–1106. 10.1016/j.bcp.2006.11.001
37
Hillman K. L. Lei S. a Doze V. Porter J. E. (2009). Alpha-1A adrenergic receptor activation increases inhibitory tone in CA1 hippocampus. Epilepsy Res.84, 97–109. 10.1016/j.eplepsyres.2008.12.007
38
Hopwood S. E. Stamford J. A. (2001). Noradrenergic modulation of serotonin release in rat dorsal and median raphé nuclei via alpha(1) and alpha(2A) adrenoceptors. Neuropharmacology41, 433–442. 10.1016/S0028-3908(01)00087-9
39
Horie K. Obika K. Foglar R. Tsujimoto G. (1995). Selectivity of the imidazoline α-adrenoceptor agonists (oxymetazoline and cirazoline) for human cloned α1-adrenoceptor subtypes. Br. J. Pharmacol. Pharmacol.116, 1611–1618. 10.1111/j.1476-5381.1995.tb16381.x
40
Igata S. Hayashi T. Itoh M. Akasu T. Takano M. Ishimatsu M. (2014). Persistent α1-adrenergic receptor function in the nucleus locus coeruleus causes hyperexcitability in AD/HD model rats. J. Neurophysiol.111, 777–786. 10.1152/jn.01103.2012
41
Ivanov A. Aston-Jones G. (1995). Extranuclear dendrites of locus coeruleus neurons: activation by glutamate and modulation of activity by alpha adrenoceptors. J. Neurophysiol.74, 2427–2436. 10.1152/jn.1995.74.6.2427
42
Jolas T. Nestler E. J. Aghajanian G. K. (2000). Chronic morphine increases GABA tone on serotonergic neurons of the dorsal raphe nucleus: association with an up-regulation of the cyclic AMP pathway. Neuroscience95, 433–443. 10.1016/S0306-4522(99)00436-4
43
Kingery W. S. Agashe G. S. Guo T. Z. Sawamura S. Frances Davies M. David Clark J. et al (2002). Isoflurane and nociception: spinal alpha2A adrenoceptors mediate antinociception while supraspinal alpha1 adrenoceptors mediate pronociception. Anesthesiology96, 367–374. 10.1097/00000542-200202000-00023
44
Kobayashi M. Sasabe T. Shiohama Y. Koshikawa N. (2008). Activation of alpha1-adrenoceptors increases firing frequency through protein kinase C in pyramidal neurons of rat visual cortex. Neurosci. Lett.430, 175–180. 10.1016/j.neulet.2007.10.047
45
Kolb P. Kenakin T. Alexander S. P. H. Bermudez M. Bohn L. M. Breinholt C. S. et al (2022). Community guidelines for GPCR ligand bias: IUPHAR review 32. Br. J. Pharmacol.179, 3651–3674. 10.1111/bph.15811
46
Lemmens S. Brône B. Dooley D. Hendrix S. Geurts N. (2015). Alpha-AdrenoceptorModulation in central nervous system trauma: pain, spasms, and paralysis – an unlucky triad. Med. Res. Rev.35, 653–677. 10.1002/med.21337
47
Lin Y. Cabeza de Vaca S. Carr K. D. Stone E. A. (2007). Role of alpha(1)-adrenoceptors of the locus coeruleus in self-stimulation of the medial forebrain bundle. Neuropsychopharmacology32, 835–841. 10.1038/sj.npp.1301145
48
Luyo Z. N. M. Lawrence A. B. Stathopoulos T. G. Mitrano D. A. (2023). Localization and neurochemical identity of alpha1-adrenergic receptor-containing elements in the mouse locus coeruleus. J. Chem. Neuroanat.133, 102343–22. 10.1016/j.jchemneu.2023.102343
49
Mateo Y. Ruiz-Ortega J. A. Pineda J. Ugedo L. Meana J. J. (2000). Inhibition of 5-hydroxytryptamine reuptake by the antidepressant citalopram in the locus coeruleus modulates the rat brain noradrenergic transmission in vivo. Neuropharmacology39, 2036–2043. 10.1016/S0028-3908(00)00041-1
50
Matt R. A. Martin R. S. Evans A. K. Gever J. R. Vargas G. A. Shamloo M. et al (2024). Locus coeruleus and noradrenergic pharmacology in neurodegenerative disease. Handb. Exp. Pharmacol.285, 555–616. 10.1007/164_2023_677
51
McGrath J. C. Naghadeh M. A. Pediani J. D. Mackenzie J. F. Daly C. J. (1999). Importance of agonists in alpha-adrenoceptor classification and localisation of alpha1-adrenoceptors in human prostate. Eur. Urol.36, 80–88. 10.1159/000052326
52
Michel M. C. Kenny B. Schwinn D. A. (1995). Classification of α1-adrenoceptor subtypes. Naunyn. Schmiedeb. Arch. Pharmacol.352, 1–10. 10.1007/BF00169183
53
Miller M. Shi J. Zhu Y. Kustov M. Bin Tian J. Stevens A. et al (2011). Identification of ML204, a novel potent antagonist that selectively modulates native TRPC4/C5 ion channels. J. Biol. Chem.286, 33436–33446. 10.1074/jbc.M111.274167
54
Milligan C. M. Linton C. J. Patmore L. Gillard N. Ellis G. J. P T. (1997). [3H]-RS-79948-197, a high affinity radioligand selective for alpha 2-adrenoceptor subtypes. Sci.812, 176–177. 10.1111/j.1749-6632.1997.tb48164.x
55
Murai Y. Okabe Y. Tanaka E. (2012). Activation of protein kinase A and C prevents recovery from persistent depolarization produced by oxygen and glucose deprivation in rat hippocampal neurons. J. Neurophysiol.107, 2517–2525. 10.1152/jn.00537.2011
56
Nakatsuka K. Matsuoka Y. Kurita M. Wang R. Tsuboi C. Sue N. et al (2022). Intrathecal administration of the α1 adrenergic antagonist phentolamine upregulates spinal GLT-1 and improves mirror image pain in SNI model rats. Acta Med. Okayama76, 255–263. 10.18926/AMO/63719
57
Nazabal A. Mendiguren A. Pineda J. (2023). Inhibition of rat locus coeruleus neurons by prostaglandin E2 EP3 receptors: pharmacological characterization ex vivo. Front. Pharmacol.14, 1290605. 10.3389/fphar.2023.1290605
58
Oronowicz J. Reinhard J. Reinach P. S. Ludwiczak S. Luo H. Omar Ba Salem M. H. et al (2021). Ascorbate-induced oxidative stress mediates TRP channel activation and cytotoxicity in human etoposide-sensitive and -resistant retinoblastoma cells. Lab. Investig.101, 70–88. 10.1038/s41374-020-00485-2
59
Osborne P. B. Vidovic M. Chieng B. Hill C. E. Christie M. J. (2002). Expression of mRNA and functional alpha1-adrenoceptors that suppress the GIRK conductance in adult rat locus coeruleus neurons. Br. J. Pharmacol.135, 226–232. 10.1038/sj.bjp.0704453
60
Otani H. Oshiro A. Yagi M. Inagaki C. (2001). Pertussis toxin-sensitive and -insensitive mechanisms of alpha1-adrenoceptor-mediated inotropic responses in rat heart. Eur. J. Pharmacol.419, 249–252. 10.1016/S0014-2999(01)00979-7
61
Perez D. M. (2020). α1-Adrenergic receptors in neurotransmission, synaptic plasticity, and cognition. Front. Pharmacol.11, 581098–22. 10.3389/fphar.2020.581098
62
Petitcolin M. A. Spitzbarth-Régrigny E. Bueb J. L. Capdeville-Atkinson C. Tschirhart E. (2001). Role of Gi-proteins in norepinephrine-mediated vasoconstriction in rat tail artery smooth muscle. Biochem. Pharmacol.61, 1169–1175. 10.1016/S0006-2952(01)00589-5
63
Pineda J. Ugedo L. García-Sevilla J. A. (1993). Stimulatory effects of clonidine, cirazoline and rilmenidine on locus coeruleus noradrenergic neurones: possible involvement of imidazoline-preferring receptors. Naunyn. Schmiedeb. Arch. Pharmacol.348, 134–140. 10.1007/BF00164789
64
Pineda J. a Ruiz-Ortega J. Ugedo L. (1997). Receptor reserve and turnover of alpha-2 adrenoceptors that mediate the clonidine-induced inhibition of rat locus coeruleus neurons in vivo. J. Pharmacol. Exp. Ther.281, 690–698. 10.1016/s0022-3565(24)36670-4
65
Proudman R. G. W. Baker J. G. (2021). The selectivity of α-adrenoceptor agonists for the human α1A, α1B, and α1D-adrenoceptors. Pharmacol. Res. Perspect.9, e00799–23. 10.1002/prp2.799
66
Proudman R. G. W. Akinaga J. Baker J. G. (2022). The affinity and selectivity of α-adrenoceptor antagonists, antidepressants and antipsychotics for the human α2A, α2B, and α2C-adrenoceptors and comparison with human α1 and β-adrenoceptors. Pharmacol. Res. Perspect.10, e00936–18. 10.1002/prp2.936
67
Pudovkina O. L. Westerink B. H. C. (2005). Functional role of alpha1-adrenoceptors in the locus coeruleus: a microdialysis study. Brain Res.1061, 50–56. 10.1016/j.brainres.2005.08.049
68
Pudovkina O. L. Kawahara Y. De Vries J. Westerink B. H. C. (2001). The release of noradrenaline in the locus coeruleus and prefrontal cortex studied with dual-probe microdialysis. Brain Res.906, 38–45. 10.1016/S0006-8993(01)02553-7
69
Ruiz-Ortega J. A. Ugedo L. (1997). The stimulatory effect of clonidine on locus coeruleus neurons of rats with inactivated α2-adrenoceptors: involvement of imidazoline receptors located in the nucleus paragigantocellularis. Naunyn Schmied. Arch. Pharmacol.355, 288–294. 10.1007/pl00004945
70
Schwarz L. A. Luo L. (2015). Organization of the locus Coeruleus-Norepinephrine system. Curr. Biol.25, R1051–R1056. 10.1016/j.cub.2015.09.039
71
Stein C. (2016). Opioid receptors. Annu. Rev. Med.67, 433–451. 10.1146/annurev-med-062613-093100
72
Stone E. A. Quartermain D. Lin Y. Lehmann M. L. (2007). Central alpha1-adrenergic system in behavioral activity and depression. Biochem. Pharmacol.73, 1063–1075. 10.1016/j.bcp.2006.10.001
73
Ugedo L. Pineda J. a Ruiz-Ortega J. Martín-Ruiz R. (1998). Stimulation of locus coeruleus neurons by non-I1/I2-type imidazoline receptors: an in vivo and in vitro electrophysiological study. Br. J. Pharmacol.125, 1685–1694. 10.1038/sj.bjp.0702255
74
VanLangen J. T. H. VanHove C. E. Schrijvers D. M. Martinet W. De Meyer G. R. Fransen P. et al (2013). Contribution of α-adrenoceptor stimulation by phenylephrine to basal nitric oxide production in the isolated mouse aorta. J. Cardiovasc. Pharmacol.61, 318–323. 10.1097/FJC.0b013e318281fa2d
75
Wang Y. Y. Aghajanian G. K. (1987). Excitation of locus coeruleus neurons by an adenosine 3´,5´-Cyclic monophosphate-activated inward current: extracellular and intracellular studies in rat brain slices. Synapse1, 481–487. 10.1002/syn.890010512
76
Williams J. T. Marshall K. C. (1987). Membrane properties and adrenergic responses in locus coeruleus neurons of young rats. J. Neurosci.7, 3687–3694. 10.1523/JNEUROSCI.07-11-03687.1987
77
Williams J. T. Henderson G. North R. A. (1985). Characterization of alpha 2-adrenoceptors which increase potassium conductance in rat locus coeruleus neurones. Neuroscience14, 95–101. 10.1016/0306-4522(85)90166-6
78
Williams J. T. Bobker D. H. Harris G. C. (1991). Synaptic potentials locus coeruleus neurons brain slices11, 167–172. 10.1016/s0079-6123(08)63806-6
79
Yuan W. X. Chen S. R. Chen H. Pan H. L. (2009). Stimulation of alpha(1)-adrenoceptors reduces glutamatergic synaptic input from primary afferents through GABA(A) receptors and T-type Ca(2+) channels. Neuroscience158, 1616–1624. 10.1016/j.neuroscience.2008.11.022
80
Zhong H. Minneman K. P. (1999). Alpha1-adrenoceptor subtypes. Eur. J. Pharmacol.375, 261–276. 10.1016/S0014-2999(99)00222-8
Summary
Keywords
locus coeruleus, α1-adrenoceptor, slice, firing, noradrenaline, rat, cirazoline, phenylephrine
Citation
Rodilla I, Mendiguren A and Pineda J (2025) Functional characterization of the α1-adrenoceptor in adult male rat locus coeruleus neurons ex vivo. Front. Pharmacol. 16:1626019. doi: 10.3389/fphar.2025.1626019
Received
09 May 2025
Accepted
22 July 2025
Published
21 August 2025
Volume
16 - 2025
Edited by
Dirk Feldmeyer, Helmholtz Association of German Research Centres (HZ), Germany
Reviewed by
Jaromir Myslivecek, Charles University, Czechia
Darlene A. Mitrano, Christopher Newport University, United States
Yasutaka Mukai, Nagoya University, Japan
Updates
Copyright
© 2025 Rodilla, Mendiguren and Pineda.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aitziber Mendiguren, aitziber.mendiguren@ehu.eus
ORCID: Aitziber Mendiguren, orcid.org/0000-0002-1990-0493, Irati Rodilla, orcid.org/0000-0003-2236-4158, Joseba Pineda, orcid.org/0000-0002-9421-1081
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.