 Fanghua Chen
Fanghua Chen Yuandong Fu1,2†
Yuandong Fu1,2† Keqin Hua
Keqin Hua- 1Obstetrics and Gynecology Hospital of Fudan University, Shanghai Key Lab of Reproduction and Development, Shanghai Key Lab of Female Reproductive Endocrine Related Diseases, Shanghai, China
- 2Shanghai Key Laboratory of Female Reproductive Endocrine Related Diseases, Obstetrics and Gynecology Hospital of Fudan University, Shanghai, China
Tumor drug resistance represents a major challenge in contemporary cancer therapeutics, significantly compromising the clinical efficacy of chemotherapy, targeted therapy, and immunotherapy. While existing research has elucidated the critical role of tumor cell-intrinsic mechanisms in drug resistance—including genomic instability, persistent activation of signaling pathways and aberrant epigenetic modifications—emerging evidence highlights the crucial involvement of dynamic remodeling within the tumor microenvironment (TME) in driving therapeutic resistance. The TME fosters drug resistance through dynamic remodeling, creating hypoxic conditions, immunosuppressive networks, and metabolic stress, which collectively impair treatment response and promote therapeutic escape. Advances in multi-omics technologies now enable a comprehensive, multi-dimensional analysis of these interactions, integrating genomic, epigenomic, transcriptomic, proteomic, and metabolomic data to uncover critical molecular networks and vulnerabilities. In this review, we explore the key mechanisms by which the TME influences drug resistance, discuss how multi-omics approaches enhance our understanding of these processes and evaluate emerging therapeutic strategies aimed at reprogramming the TME to overcome resistance.
1 Introduction
The tumor microenvironment (TME) dynamically orchestrates drug resistance through integrated cellular and molecular networks (Vennin et al., 2018; Albini and Sporn, 2007). Beyond cancer cell-intrinsic mechanisms, stromal components—including cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), and other immunosuppressive cells—co-opt physiological processes such as metabolic symbiosis, extracellular matrix (ECM) remodeling and immune evasion to sustain tumor survival under therapy (Jain, 2013; Casey et al., 2015). This adaptive crosstalk results in spatially heterogeneous resistance niches, posing a fundamental challenge to current treatments.
While traditional approaches have identified individual resistance pathways, they fail to capture the TME’s systems-level complexity (Junttila and de Sauvage, 2013). The advent of multi-omics technologies now offers an unprecedented opportunity to dissect these complexities (He et al., 2023; Sun et al., 2023; Yan et al., 2025). By integrating genomic, transcriptomic, proteomic, and metabolomic data—complemented by spatial and single-cell resolution—researchers can map the multi-dimensional interactions between tumor cells and their microenvironment, revealing how these relationships drive resistance. This review synthesized current knowledge on TME-mediated drug resistance through a multi-omics lens, emphasizing key interactive networks (e.g., immune-stromal-metabolic axes) that sustain resistance, influencing factors (e.g., genetic change and epigenetic remodeling) uncovered via multi-omics, and emerging interventions targeting TME-omics vulnerabilities, including stromal reprogramming, immune-metabolic modulation, and combinatorial strategies.
2 Mechanisms of drug resistance mediated by TME
The TME orchestrates drug resistance through an intricate interplay of immunosuppression (Cappellesso et al., 2022; Pitt et al., 2016), physical drug delivery barriers (Yang et al., 2021; Stylianopoulos et al., 2018), metabolic reprogramming (Yang et al., 2020; Li et al., 2022; Zhang et al., 2025a) and aberrant intercellular communication (Lu et al., 2023; Rebelo et al., 2023; Liu et al., 2024). These mechanisms do not operate in isolation but rather form a coordinated defense network that enables tumors to evade therapeutic pressure. Understanding these processes is critical for developing strategies to overcome treatment resistance.
2.1 Cellular components of TME and drug resistance
2.1.1 CAFs
At the cellular level, CAFs promote therapy resistance through ECM remodeling, generating dense fibrotic barriers that impede drug penetration. In pancreatic ductal adenocarcinoma (PDAC), for instance, TGFβ-high PDACs drive fibrosis, resulting in elevated collagen fiber density and tissue stiffness, which further restricts chemotherapeutic delivery (Hosein et al., 2020). Beyond physical obstruction, CAFs serve as a major source of cytokines, chemokines, and growth factors within the TME. By recruiting Tregs via CXCL12 and activating stromal stiffening pathways such as VAV2, CAFs reinforce immunosuppression—a mechanism linked to trastuzumab resistance in HER2+ breast cancer (Yang et al., 2016; Kieffer et al., 2020). CAFs could also secrete extracellular vesicles (EVs) in autocrine and paracrine signaling, enhancing cancer cell aggressiveness and therapeutic resistance. For instance, CAF-derived exosomal miR-423-5p promotes taxane resistance in prostate cancer by targeting GREM2 and amplifying TGF-β signaling, leading to increased proliferation and reduced apoptosis upon taxane exposure (Shan et al., 2020). Similarly, in colorectal cancer (CRC), CAFs-derived exosomes confer radioresistance by suppressing DNA damage and inhibiting apoptosis in CRC cells (Chen et al., 2021). Additionally, metabolic reprogramming of CAFs fosters clinical drug tolerance. For example, EGFR- or MET-expressing cancer cells enhance glycolytic activity and lactate production, stimulating CAFs to secrete HGF via NF-κB, thereby activating MET signaling and driving TKI resistance (Apicella et al., 2018).
2.1.2 TAMs
Ideally, TAMs within the TME could theoretically eliminate tumor cells through phagocytosis and activate T-cell-mediated antitumor immunity, but their inherent plasticity and the complex TME often limit this therapeutic potential (Wang et al., 2025). For example, in advanced lymphoma, CD47-targeted therapy fails due to compromised macrophage phagocytic function within the TME (Cao et al., 2022).
M2-polarized TAMs exacerbate therapeutic resistance by secreting IL-10 and TGF-β, expressing PD-L1, and sequestering drugs, which collectively suppress cytotoxic T-cell activity and immunotherapy efficacy (Kanlikilicer et al., 2020; Shi et al., 2022; Croci et al., 2014). These macrophages also promote tumor vascularization through angiogenesis induction, basement membrane degradation, and secretion of pro-angiogenic factors such as VEGF and MMPs (MMP7, MMP9, MMP12) (Giraudo et al., 2004; Chryplewicz et al., 2022). VEGF reinforces M2-like polarization while collaborating with TAMs to disrupt vascular function, reducing drug perfusion and increasing treatment tolerance. In glioblastoma, bevacizumab-induced VEGF depletion unexpectedly elevates macrophage migration inhibitory factor (MIF) at the tumor periphery, expanding TAM infiltration (Castro et al., 2017). Lung adenocarcinoma TAMs similarly suppress p53 and PTEN while overexpressing VEGF-C/VEGFR3, inhibiting apoptosis and conferring doxorubicin resistance (Li et al., 2017). Macrophage-derived EVs mediate intercellular communication by transporting proteins, metabolites, and nucleic acids across the TME. The multidrug resistance protein P-glycoprotein (P-gp), encoded by MDR1, actively exports chemotherapeutics; in ovarian cancer, exosomal miR-1246 amplifies P-gp function through the Cav1/P-gP/PRPS2 axis, reducing paclitaxel uptake and accelerating chemoresistance (Kanlikilicer et al., 2020). Additionally, TAMs elevate glucose metabolism to stimulate OGT-dependent O-GlcNAcylation of Cathepsin B, enhancing its secretion into the TME and facilitating therapeutic evasion (Shi et al., 2022).
2.1.3 Endothelial cells and immune cells
Tumor endothelial cells form abnormal, leaky blood vessels with heterogeneous permeability but inadequate perfusion, creating hypoxic niches that enhance cancer cell survival and therapy resistance. Hypoxia triggered by anti-VEGF therapy increases galectin-1 expression in tumor cells, where it binds glycosylated VEGFR2 on endothelial cells to sustain angiogenesis through VEGFA-mimetic signaling (Croci et al., 2014). Endothelial cells also develop acquired resistance to antiangiogenic tyrosine kinase inhibitors (Wu, S. et al., 2020) and chemotherapeutic agents (Huang et al., 2013a), employing mechanisms similar to tumor cells, such as P-gp upregulation (Akiyama et al., 2012). Functional reprogramming, phenotypic switching, and altered secretory profiles further enable endothelial evasion of antiangiogenic therapy.
Tregs accumulate in the TME through chemokine-mediated recruitment (e.g., CCL17/CCL22-CCR4, CCL28–CCR10, CCL5–CCR5) (Faget et al., 2011; Facciabene et al., 2011; Halvorsen et al., 2016) and peripheral conversion via TGF-β and IL-10 (Park et al., 2022). They suppress cytotoxic T cells by downregulating effector molecules such as granzyme B and IFN-γ (Yu et al., 2021; Huang et al., 2020), further compromising therapeutic efficacy. MDSCs suppress various immune cells, primarily targeting T cells, through the production of ARG1, iNOS, TGF-β, IL-10 and COX2, and also promote immunosuppression by inhibiting CD8+ T cell activity and inducing the differentiation of Tregs (Gabrilovich, 2017; Huang et al., 2006). In cisplatin-resistant bladder cancer, MDSCs are recruited via chemokine upregulation in response to cisplatin treatment and suppress CD8+ T cell responses through enhanced ARG1 and iNOS expression. Their accumulation not only promotes resistance to cisplatin but also impairs the efficacy of PD-L1 blockade, highlighting their role in mediating resistance to both chemotherapy and immune checkpoint inhibitors (ICIs) (Takeyama et al., 2020). These cellular interactions create a self-reinforcing cycle of immune suppression that undermines both conventional therapies and immunotherapies.
2.1.4 Cancer stem cells (CSCs)
CSCs demonstrate dynamic plasticity by alternating between proliferative and quiescent states, with dormancy facilitating long-term survival through metabolic suppression while retaining cell cycle re-entry capacity under specific conditions (Vira et al., 2012). The biological adaptability makes quiescent CSCs especially refractory to cycle-dependent chemotherapeutics such as taxanes. Furthermore, their characteristic slow-cycling behavior, frequently involving extended G1 or S phase arrest, additionally confers resistance to diverse agents including cisplatin, taxol, and doxorubicin (Gao et al., 2010). A notable example is the Zinc Finger E-Box-Binding Homeobox 2 (ZEB2) in CRC, where its overexpression increases the proportion of CSCs in G0/G1 phase, directly contributing to platinum resistance (Francescangeli et al., 2020). Paradoxically, radiation therapy enriches CSC populations due to their inherent radioresistance relative to differentiated tumor cells. Radiation-resistant non-small cell lung cancer (NSCLC) cell lines exhibit marked upregulation of CSC markers like SOX2, CD133, and ALDH, with SOX2 critically enhancing DNA repair mechanisms and radioresistance (Qi et al., 2017). Glioblastoma studies similarly demonstrate that OXM1 induces SOX2 expression, amplifying radioresistance in this aggressive malignancy.
CSCs reside in specialized protective niches within the TME, particularly in perivascular and hypoxic zones, where stromal components, including CAFs and TAMs, sustain their survival and therapy resistance through diverse pathways. CSCs also maintain intricate bidirectional communication with immune cell populations. M2-polarized TAMs preserve CSC populations via chemokine secretion (e.g., IL-6, CCL2), activation of stemness pathways such as Wnt/β-catenin, and evasion of phagocytosis through CD47-SIRPα interactions (Liu et al., 2017; Ma et al., 2009). MDSCs reinforce immunosuppression by inhibiting T cell function while simultaneously enhancing CSC properties through STAT3 and Notch pathway activation (Ouzounova et al., 2017). CSCs systematically evade T cell recognition by downregulating antigen presentation components, losing MHC-I expression, inducing T cell tolerance, and secreting immunosuppressive cytokines like TGF-β and IL-10 (Rothstein and Sayegh, 2003). Although natural killer (NK) cells can target CSCs through cytotoxic mechanisms and differentiation induction, CSCs develop resistance strategies including ligand shedding, Treg recruitment, and modulation of activating receptor expression. While B cells and tertiary lymphoid structures (TLS) within the TME influence overall tumor prognosis and immune responses (Petitprez et al., 2020; Rodriguez et al., 2020), their specific interplay with CSCs requires further elucidation.
2.2 Biochemical and metabolic barriers in TME
2.2.1 Hypoxia and acidosis
Hypoxia and acidosis represent two fundamental and interconnected features of the TME that significantly influence therapeutic outcomes. The landmark discovery of hypoxia-induced angiogenesis and subsequent elucidation of HIF-mediated transcriptional reprogramming revealed not only fundamental drivers of tumor progression but also major obstacles to successful cancer therapy. Notably, these hypoxia-driven adaptations promote chemoresistance through multiple mechanisms, including the upregulation of ATP-binding cassette (ABC) transporters such as ABCB1/P-gp, ABCC1, and ABCG2, which actively efflux diverse chemotherapeutic agents such as vinca alkaloids, anthracyclines, and platinum compounds (Dong et al., 2020; Li et al., 2016; He et al., 2016). Hypoxia simultaneously activates pro-survival pathways (e.g., Ras-MAPK, PI3K-Akt-mTOR) and stemness programs (e.g., Wnt, Notch), while suppressing apoptotic machinery through stabilization of anti-apoptotic proteins (e.g., IAP3, Bcl-2) and TP53 destabilization (Chen et al., 2023a; Kaloni et al., 2023; Cao et al., 2020). Tumor acidosis, another defining TME characteristic, mediates significant immunosuppression. Kreutz et al. established that proton and lactate exposure progressively compromises immune cell function, suppressing T cell secretion of IL-2, IFN-γ, granzyme B, and perforin while reducing monocyte-derived TNF (Sennino and McDonald, 2012). These observations demonstrate how the acidic TME fosters an immune-privileged environment conducive to tumor immune evasion.
2.2.2 Nutrient deprivation and metabolic reprogramming
Metabolic reprogramming constitutes a fundamental cancer hallmark, allowing malignant cells to thrive in nutrient-scarce environments while developing therapeutic resistance through dysregulated glucose, lipid, and amino acid metabolism. The Warburg effect, marked by preferential aerobic glycolysis, depends on key regulatory enzymes such as hexokinase-2 (HK2), phosphofructokinase-1 (PFK1), and pyruvate kinase (PK). HK2 overexpression is associated with aggressive breast cancer phenotypes, and its pharmacological inhibition using 2-deoxy-D-glucose (2-DG) enhances the efficacy of conventional chemotherapeutics like doxorubicin and paclitaxel (Patra et al., 2013), with clinical studies confirming 2-DG’s chemosensitizing potential (Raez et al., 2013). Glutaminolysis also functions as a crucial anaplerotic pathway, sustaining both glycolytic and oxidative phosphorylation fluxes in cancer cells while maintaining redox balance and driving resistance to targeted therapies, as observed in HER2-positive cancers with reactivated mTOR signaling following lapatinib treatment (Deblois et al., 2016; Yang et al., 2017). Cancer cells display significant lipid metabolism alterations, including increased fatty acid oxidation regulated by carnitine palmitoyltransferase 1B (CPT1B) and activation of the JAK/STAT3-CPT1B-FAO axis, which reinforces stemness and chemoresistance—a metabolic shift particularly pronounced in cisplatin-resistant ovarian cancer (Wang et al., 2018; Yoshida, 2015). Parallel metabolic adaptations, including pyrimidine metabolism and IDO-mediated tryptophan catabolism, provide tumors with metabolic flexibility while creating an immunosuppressive niche (Balkwill et al., 2012; Brandacher et al., 2006). Notably, dysregulated pyrimidine metabolism, a recognized cancer hallmark, has emerged as a key modulator of immunotherapy response. Preclinical evidence demonstrates that inhibition of pyrimidine synthesis reduces the frequency of CTLA4+ T cells within the TME, suggesting a metabolic checkpoint that shapes immune evasion (Siddiqui and Ceppi, 2020). Further supporting this link, pyrimidine metabolism-related genes correlate with immunotherapy outcomes, highlighting their potential as a therapeutic target (Luo et al., 2022). The complex crosstalk among these metabolic pathways highlights the intricacies of tumor metabolism while revealing new therapeutic avenues for treatment-resistant malignancies.
2.3 Adaptive evolution of TME under therapy
The TME exhibits substantial plasticity in response to conventional and targeted therapies, developing complex resistance mechanisms through dynamic cellular and molecular adaptations. Chemotherapy often induces EMT, which upregulates drug-resistance genes and recruits immunosuppressive cell populations, creating a therapy-resistant niche (Glaviano et al., 2025). Radiation therapy similarly alters the immune landscape by enriching MDSCs and Tregs, fostering an immunosuppressive environment (Jarosz-Biej et al., 2019). Although reactive oxygen species (ROS) generation mediates radiation-induced cytotoxicity, hypoxia-driven ROS elevation activates a compensatory antioxidant response that induces cytoprotective autophagy, ultimately conferring radioresistance (Eales et al., 2016).
Immunotherapy encounters distinct challenges from the TME’s adaptive responses, particularly through compensatory upregulation of alternative immune checkpoints. In anti-PD-1-treated lung cancer models, T cells show significant TIM-3 overexpression following PD-1 blockade (Koyama et al., 2016). Similarly, prostate tumors responding to CTLA-4 inhibition exhibit elevated PD-L1 and VISTA expression in immune subsets and malignant cells, potentially explaining the limited efficacy of ipilimumab in this context (Gao et al., 2017; Le et al., 2014). Metabolic crosstalk between tumor and stromal cells further drives microenvironmental adaptation. Enhanced glycolysis and mitophagy alter drug pharmacokinetics and sustain energy homeostasis, reinforcing therapeutic resistance (Okoye et al., 2023; Wu, H. et al., 2020). Tumor cells dynamically rewire their metabolic networks to support proliferation while shaping an immune-evasive TME, a reprogramming that not only promotes survival but also compromises immunotherapy through diverse resistance mechanisms (Wu et al., 2021a). Consequently, the evolving interplay between tumor cells and their microenvironment during treatment necessitates longitudinal monitoring to optimize therapeutic strategies.
2.4 Genetic and epigenetic determinants of resistance
High tumor mutation burden (TMB) enhances tumor immunogenicity by generating neoantigens that facilitate T-cell recognition and tumor clearance, correlating with improved responses to ICIs. However, the predictive power of TMB is context-dependent, influenced by factors such as the mutational landscape and the efficiency of neoantigen presentation. For instance, in melanoma, the variable clinical outcomes following PD-1/PD-L1 blockade can be attributed, in part, to the differential kinetics between genomic mutation acquisition and the final steps of MHC-mediated antigen presentation (Goodman et al., 2017). Beyond genetic alterations, epigenetic dysregulation—including aberrant DNA methylation, histone modifications, and chromatin remodeling—plays a pivotal role in shaping immune cell function and tumor immune escape (Yoo and Jones, 2006). Key epigenetic regulators such as EZH2 and DNMT1 suppress antitumor immunity by limiting CD8+ T-cell infiltration, amplifying Treg activity, and downregulating MHC-I expression, thereby fostering an immune-privileged niche (Khodayari et al., 2023; Kundu et al., 2024).
The complexity of these interactions underscores why conventional single-target approaches often fail against TME-mediated resistance. The interconnected nature of these mechanisms - where physical barriers influence drug distribution, metabolic changes alter the therapeutic landscape, and immune suppression protects surviving tumor cells - demands integrated therapeutic strategies. Recent advances in multi-omics approaches are beginning to unravel these complex networks, revealing novel vulnerabilities that could be targeted to overcome the TME’s formidable defenses. As our understanding of these resistance mechanisms deepens, so too does the potential for developing more effective combination therapies that simultaneously target multiple aspects of the resistant TME.
3 Multi-omics technologies deciphering TME-mediated drug resistance
Multi-omics technologies (genomics, epigenomics, transcriptomics, proteomics and metabolomics) now enable comprehensive mapping of these resistance networks across biological scales. As we explore specific omics applications in subsequent sections, this systems biology framework will highlight how multi-omics uncovers resistant mechanisms and latent therapeutic vulnerabilities (Figure 1), revealing the convergent mechanisms by which localized perturbations propagate into system-wide treatment failure.
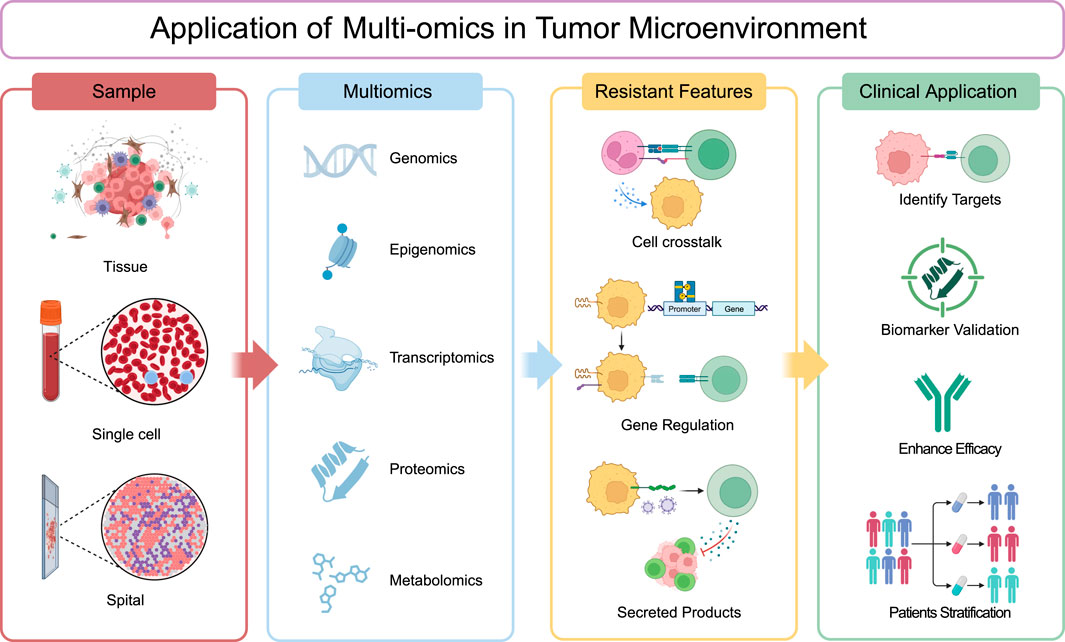
Figure 1. Application of multi-omics in tumor microenvironment. Tumor tissue samples are subjected to comprehensive analysis via single-cell and spatial profiling, as well as through various omics approaches including genomics, epigenomics, transcriptomics, proteomics, and metabolomics. These analyses can be conducted independently or integrated to provide a holistic view. The data obtained are then meticulously evaluated to uncover resistant features within the tumor microenvironment, such as cell-to-cell communication, gene regulatory mechanisms, and the secretion of bioactive molecules. The insights gleaned from these analyses are subsequently translated into clinical applications aimed at identifying therapeutic targets, validating biomarkers, enhancing treatment efficacy, and facilitating patient stratification for tailored therapeutic interventions.
3.1 Genomic alterations shape TME adaptation and reveal therapeutic vulnerabilities
Genomics has enabled the precise detection of single nucleotide variants, copy number variations and structural variations at single-cell resolution through whole-genome sequencing (WGS) and whole-exome sequencing (WES) technologies, revealing how genomic heterogeneity shapes TME evolution (Sherman and Salzberg, 2020; Gawad et al., 2016). Tumor-intrinsic alterations create dual therapeutic barriers by concurrently activating oncogenic pathways and sculpting an immunosuppressive niche. In NSCLC, epidermal growth factor receptor (EGFR) mutations identified by genomics not only sustain tumor cell survival but also correlate with an immune-cold TME phenotype characterized by low TMB and elevated PD-L1 expression (Madeddu et al., 2022), explaining the limited efficacy of PD-1 blockade in this subset. This genomic-immune axis was clinically validated in KEYNOTE-789 (NCT03515837), where pembrolizumab-chemotherapy failed to improve outcomes in EGFR-mutant NSCLC (Yang et al., 2024a). Similarly, WES identified KRAS/MEK1 compensatory alterations driving resistance to RAF/EGFR inhibition (Ahronian et al., 2015), prompting ongoing evaluation of triplet therapy (BRAF+EGFR+MEK) in NCT05217446. Beyond tumor cells, stromal genomic alterations also contribute substantially to therapeutic resistance. In PDAC, SMAD4-deficient CAFs activate a TGF-β/IL-11 signaling axis that drives stromal fibrosis and chemoresistance (Matsumura et al., 2022). These findings have spurred clinical exploration of stromal-targeting strategies, including the TGF-β receptor inhibitor galunisertib, which suppresses SMAD phosphorylation and disrupts canonical TGF-β signaling. A phase Ib study (NCT02734160) evaluating galunisertib plus the anti-PD-L1 antibody durvalumab in refractory metastatic PDAC suggests that TGF-β pathway inhibition may potentiate immunotherapy efficacy in this setting.
Collectively, these insights underscore the necessity of integrating tumor and stromal genomic profiling to fully decipher resistance mechanisms and optimize therapeutic strategies. Serial monitoring of clonal evolution—such as tracking trunk mutations (reflecting TMB) and resistance-associated variants (e.g., RAS/BRAF) in circulating tumor DNA (ctDNA)—further enables dynamic assessment of therapeutic vulnerability, as demonstrated in metastatic colorectal cancer (Vidal et al., 2023).
3.2 Epigenomic regulation of therapy resistance in the TME
Recent advances in epigenomic profiling, including chromatin immunoprecipitation sequencing (ChIP-seq) and transposase-accessible chromatin using sequencing (ATAC-seq), have elucidated how DNA methylation, histone modifications, and chromatin remodeling dynamically regulate tumor evolution and therapy resistance independent of genetic alterations (Weichenhan et al., 2022). Single-cell epigenomic analyses reveal direct links between epigenetic silencing and treatment failure - for instance, chemoresistant stem-like populations in relapsed pediatric AML exhibit reduced chromatin accessibility (Lambo et al., 2023). Emerging technologies like EPIC-seq further demonstrate the translational potential of epigenomics, enabling noninvasive cancer subtyping and prediction of PD-(L)1 inhibitor response through cfDNA-based promoter fragmentation entropy analysis (Esfahani et al., 2022). Epigenomics has found that hypomethylation promotes tumor cell resistance to treatment by affecting gene expression, signaling pathway activity, and epigenetic regulation. In gliomas, CAV1 demethylation confers resistance to oxidative phosphorylation inhibitors (Liu et al., 2023), while clinically, hypomethylating agents show promise for overcoming resistance in hematologic malignancies, as evidenced by the encouraging activity of oral decitabine/cedazuridine plus venetoclax in a recent phase 1/2 trial (NCT04655755). Super-enhancer remodeling represents another conserved resistance mechanism. Integrative analysis of H3K27ac ChIP-seq and RNA-seq data in microsatellite-stable CRC identified KLF3 as a critical regulator of chemoresistance via ABCB1/MDR1 activation (Li et al., 2021), suggesting potential for epigenetic-targeted combination strategies. These findings highlight epigenetic plasticity as a critical determinant of TME-driven resistance, highlighting the need for integrated approaches combining functional epigenomics, CRISPR-based screens, and clinical epigenome editing to develop next-generation therapeutic strategies.
3.3 Transcriptomic insights into the multilayered resistance mechanisms in the TME
Transcriptomic technologies have revolutionized our understanding of therapy resistance by decoding the dynamic interplay between tumor cells and their microenvironment across multiple resolution scales. Bulk RNA sequencing remains a cornerstone for clinical biomarker discovery, offering cost-effectiveness and compatibility with archival specimens. Computational deconvolution algorithms now enable precise reconstruction of TME composition from bulk data, identifying PD-1+ CD8+ T cells as predictive biomarkers of immunotherapy response—potentially refining clinical decision-making.
High-resolution single-cell sequencing (scRNA-seq) has further uncovered tumor-intrinsic and stromal mechanisms of resistance. In NRAS-mutant melanoma, scRNA-seq revealed rapid upregulation of the purinergic receptor P2RX7 within 72 h of MEK/CDK4/6 inhibition, promoting survival via calcium influx (Randic et al., 2023). This aligns with preclinical evidence that P2RX7 blockade enhances anti-PD-1 efficacy through IL-18-mediated NK and CD4+ T cell activation (Douguet et al., 2021). scRNA-seq has also exposed critical stromal contributions. In soft tissue sarcomas, a glycolytic CAF subset drives immunosuppression through GLUT1-mediated metabolic competition and CXCL16-dependent CD8+ T cell exclusion, spurring the development of GLUT1 inhibitors (Broz et al., 2024). In PDAC, LRRC15+CAFs form tumor-peripheral barriers absent in normal tissue, correlate with anti-PD-L1 resistance in 600+ patients, and represent a TGFβ-dependent therapeutic target (Dominguez et al., 2020).
Spatial transcriptomics has added an architectural dimension to resistance mechanisms. In triple-negative breast cancer (TNBC), three distinct immune microenvironments dictate therapeutic outcomes: (1) immunoreactive (intratumoral CD8+/IDO+), (2) immune-cold (B7-H4+/fibrotic stroma), and (3) immunomodulatory (stromal PD-L1+/cholesterol-rich) (Shiao et al., 2024). Immune-cold tumors exhibit pronounced checkpoint inhibitor resistance while macrophage-targeting therapies like bexmarilimab show context-dependent efficacy—activating “cold” TMEs but suppressing IFN-rich niches, as demonstrated in patient-derived explant cultures (Rannikko et al., 2025). A pan-cancer stromal atlas further delineated 39 functionally distinct subsets across 16 malignancies, revealing conserved resistance mechanisms: PGF+ endothelial tip cells mediate immune exclusion, while boundary CAFs drive LGALS9/TIM-3-dependent immune evasion (Du et al., 2024). Integrated single-cell and spatial transcriptomics in rectal cancer, CAF polarization dictates chemotherapy response, with tumor-suppressive CAFs organizing protective immune networks and FAP+ CAFs driving EMT (Qin et al., 2023). This multi-scale transcriptomic framework - from bulk biomarkers to single-cell dynamics and spatial architectures - provides an integrated roadmap for overcoming TME-mediated resistance through precision target identification, patient stratification, and rational combination therapies.
3.4 Metabolic reprogramming in the TME: a key driver of drug resistance
Recent advances in metabolomics have revolutionized our understanding of the TME, revealing how metabolic rewiring drives therapy resistance. Cutting-edge technologies—including liquid chromatography-tandem mass spectrometry (LC-MS/MS), matrix-assisted laser desorption/ionization mass spectrometry imaging (MALDI-MSI), and nuclear magnetic resonance (NMR) spectroscopy—now enable spatially resolved metabolic profiling, uncovering dynamic adaptations that sustain tumor survival under therapeutic pressure (Wishart, 2019). Metabolomic signatures have emerged as powerful predictors of drug response. Chemoresistance has been linked to specific metabolic alterations, including elevated levels of hypotaurine, uridine, dodecanoylcarnitine, choline, dimethylglycine, niacinamide, and L-palmitoylcarnitine (Tian et al., 2018). Similarly, metabolic profiling of patient-derived xenografts has identified resistance biomarkers for multiple anticancer agents (Kaoutari et al., 2021). In TNBC, glutamine accumulation sustains glutathione synthesis while NAD+ depletion impairs DNA repair, driving chemotherapy resistance (Carneiro et al., 2023). Then, it also led to the preclinical trial of glutaminase inhibitor CB-839 with platinum-based chemotherapy (Shen et al., 2020). In diffuse large B-cell lymphoma, MALDI-MSI identified resistant niches characterized by elevated AMP/ATP ratios and phosphatidylinositol accumulation (Raez et al., 2013), which guided the development of the PI3Kδ inhibitor, which normalized CD4/CD8 ratios and maximized the number of CD8+ T-stem cell memory, naive, and central memory T-cells with dose-dependent decreases in expression of the TIM-3 exhaustion marker (Funk et al., 2022). Similarly, in bladder cancer, LC-MS/MS revealed that HK2-mediated glycolytic flux drives cisplatin resistance, prompting the development of the HK2 inhibitor, which synergized with sorafenib to inhibit tumor growth (Gong et al., 2025; DeWaal et al., 2018).
Spatially resolved metabolomics has been instrumental in mapping tumor-stroma interactions and drug distribution. In glioblastoma, the oncometabolite D-2-hydroxyglutarate (D-2-HG), produced by IDH-mutant tumor cells, is taken up by CD8+ T cells, altering their metabolism and impairing cytotoxicity (Notarangelo et al., 2022). Multimodal imaging confirmed that D-2-HG-rich tumor regions exhibit cytotoxic T-cell exclusion, suggesting differential immunotherapy responses based on D-2-HG levels (Altea-Manzano et al., 2023). By elucidating the metabolic basis of resistance—metabolomics has become indispensable for developing precision therapies. Its integration into multi-omics frameworks promises to overcome TME-mediated treatment failure, offering new avenues for patient stratification and rational combination strategies.
3.5 Proteomic profiling of TME-driven resistance: networks and post-translational modifications
Proteomic technologies have emerged as indispensable tools for dissecting the molecular mechanisms underlying therapeutic resistance in cancer. By characterizing protein abundance, post-translational modifications (PTMs), and protein-protein interactions, proteomics provides a functional readout of cellular states that complements genomic and transcriptomic analyses. Recent advances in MS-based proteomics and spatial proteomics have revealed intricate networks of resistance mechanisms operating through tumor-intrinsic pathways and microenvironmental crosstalk. In PTEN-null prostate cancer, proteomic analyses have uncovered multifaceted resistance mechanisms involving MEK-dependent immunosuppressive TAMs and Wnt/β-catenin-driven metabolic adaptation. These findings directly informed the development of combination therapies targeting MEK/Wnt with PI3K inhibitors, achieving complete and durable responses in preclinical models (Chaudagar et al., 2023). Integrated lipidomics-proteomics approaches further identified a FABP4/SCD1-mediated redox maintenance circuit sustained by lipid droplet formation, offering both prognostic biomarkers and therapeutic targets to prevent recurrence (Luis et al., 2021).
While early studies primarily cataloged protein abundance changes—such as EphA2 enrichment in gemcitabine-resistant PDAC (Fan et al., 2018), or mitochondrial Complex I upregulation in trastuzumab-resistant HER2+ breast cancer (Tapia et al., 2024)—contemporary proteomics has shifted focus to PTMs as direct mediators of resistance. First, direct modulation of drug-target interactions occurs, such as hyper-N-glycosylation of PD-L1 in melanoma, which masks checkpoint inhibitor binding sites. This has motivated clinical trials combining PD-1 inhibitors with glycosylation modulators (Loison et al., 2024). Second, by rewiring the function of the protein, hypoxic conditions in ovarian cancer induce PIAS4-mediated SUMOylation of KDM5B, preventing its ubiquitin-dependent degradation and maintaining chemoresistance (Gu et al., 2023), which promotes the development of novel agents, such as the SUMOylation inhibitor TAK-981. In addition, USP5-mediated MDH2 deubiquitination drives ripretinib resistance in gastrointestinal stromal tumors (Wang et al., 2023), while the OTUD6A-CDC6 deubiquitination axis promotes bladder cancer chemoresistance (Cui et al., 2024). Third, the facilitation of intercellular crosstalk is another mechanism. Phosphoproteomic analyses have revealed resistance mechanisms, including EGFR/HMAG1 hyperphosphorylation and SRC-PRKCD cascade activation in EGFR inhibitor-resistant NSCLC. IL-6-induced STAT3 phosphorylation upregulates BCL-2 in breast cancer, conferring radioresistance and justifying JAK2 inhibitor trials (ruxolitinib, NCT04418154). Lactylation has emerged as a critical PTM in immune evasion. In CD8+ T cells, IL-11 activates JAK2/STAT3 to drive immune checkpoint expression via lactylation, promoting exhaustion (Zhou et al., 2024). KRAS-mutant tumors exhibit elevated histone lactylation (H3K18la) in tumor-specific CD8+ T cells, while p300-catalyzed APOC2 lactylation in NSCLC induces extracellular lipolysis and Treg accumulation, driving immune checkpoint blockade (ICB) resistance (Chen et al., 2024). Proteomic technologies have transformed our understanding of therapy resistance by revealing the dynamic post-translational landscape of cancer cells and their microenvironment. The integration of proteomic data with other omics layers and clinical outcomes promises to accelerate the development of effective combination therapies to overcome treatment resistance.
3.6 Integrative multi-omics approaches to decode TME-Mediated resistance
The multifaceted nature of tumor drug resistance necessitates a paradigm shift from single-omics to integrative multi-omics approaches. While genomics identifies driver mutations, transcriptomics reveals dynamic gene expression patterns, proteomics characterizes functional protein networks, and metabolomics uncovers metabolic adaptations, none of these layers alone can fully capture the complexity of treatment resistance. Integrative multi-omics strategies bridge this gap by systematically connecting molecular alterations across biological scales, offering unprecedented insights into the TME’s adaptive responses.
In esophageal cancer, combining genomic, transcriptomic, proteomic, and metabolomic data with machine learning uncovered three clinically relevant findings: DNA repair deficiencies predicting chemoradiotherapy response, glutaminolysis-driven metabolic rewiring underlying resistance, and immune-evasive phenotypes detectable only through integrated protein/RNA profiling (Yang et al., 2024b). Integrated scRNA-seq and single-cell ATAC-seq in tamoxifen-resistant breast cancer delineated tumor heterogeneity, revealing distinct cancer cell states, a core resistance gene signature, and BMP7’s role in oncogenic pathway modulation (Fang et al., 2024). In PDAC, combined scRNA-seq, metabolomics, and flow cytometry exposed chemotherapy-resistant TAMs that evade treatment via altered deoxycytidine metabolism, suggesting new therapeutic targets (Zhang et al., 2023a). Multi-omics profiling in HR+/HER2− metastatic breast cancer further identified APOBEC mutation patterns and homologous recombination deficiency-high clusters linked to CDK4/6 inhibitor resistance, refining patient stratification (Park et al., 2023).
Spatial multi-omics has been particularly transformative. In hepatocellular carcinoma (HCC), combined spatial transcriptomics and proteomics uncovered CXCL12+ tumor-associated endothelial cells that mediate immunosuppression by recruiting MDSCs and inhibiting CD8+ T cells - a resistance mechanism missed in bulk analyses (Lu et al., 2025). NSCLC studies merging scRNA-seq with spatial transcriptomics distinguished tertiary lymphoid structure states and tumor-stroma interactions predictive of chemoimmunotherapy response (Yan et al., 2025). Such spatial insights are vital for designing targeted combination therapies. Prostate cancer research further highlights multi-omics integration: single-cell analyses identified EpCAM-negative circulating tumor cells with unique androgen receptor (AR)-driven biology (Lambros et al., 2018), while spatial approaches revealed KLK3-mediated micrometastatic niches (Heidegger et al., 2022) and CXCL12-driven angiogenesis through combined scRNA-seq and functional assays. These discoveries are already informing clinical applications, including liquid biopsy platforms for AR-variant detection, KLK3-targeted adjuvant strategies, and trials evaluating CXCL12 inhibition. The clinical impact of multi-omics integration is becoming evident across multiple cancers. The gastric cancer study employed WES, whole-transcriptome sequencing and scRNA-seq to show that higher pre-treatment CD3+ T cell infiltration predicts poor response to neoadjuvant PD-1 inhibitors combined with chemotherapy (Ji et al., 2024). Bladder cancer studies identified BCAT2 as a key regulator of noninflamed TME, enabling novel ICB combinations (Cai et al., 2023). The cholangiocarcinoma study integrated analysis of 85 PANoptosis-related genes to uncover POSTN+ CAFs as key resistance orchestrators spatially linked to immunosuppressive TAMs and PD-L1/2, while developing the clinically actionable PANRS model (POSTN/SFN/MYOF/HOGA1/PECR) (Yu et al., 2025). TNBC analyses of 465 samples defined three metabolic subtypes with distinct therapeutic vulnerabilities, guiding subtype-specific strategies from lipid metabolism inhibitors to glycolytic blockade combined with anti-PD-1 immunotherapy (Gong et al., 2021). Glioblastoma studies leveraging multi-omics implicated PKCδ and DNA-PK as master kinases driving molecular subtypes, prompting an RNA-based classifier for clinical stratification and kinase inhibitor trials (Migliozzi et al., 2023). Comprehensive multi-omic profiling of hyperthermia-treated ovarian cancer cells uncovered rapid CDK1 hyperactivation, prompting the rational combination of WEE1 inhibitors with hyperthermic intraperitoneal chemotherapy to improve outcomes (Yang et al., 2024c). Similarly, a landmark study combining single-cell and spatial transcriptomics with bulk sequencing identified CXCL12+ CAFs as key architects of chemoresistance in ovarian cancer, leading to a clinically actionable 24-gene predictive signature (Wang et al., 2024a). These findings exemplify how integration provides insights unattainable through single-omics approaches.
4 Therapeutic strategies targeting the TME to overcome drug resistance
The intricate interplay between cellular and non-cellular components within the TME has necessitated the development of multifaceted therapeutic approaches to combat drug resistance. Recent advances in our understanding of TME biology, coupled with insights from multi-omics analyses, have enabled the design of precision strategies targeting key resistance mechanisms while preserving normal tissue function (Table 1).
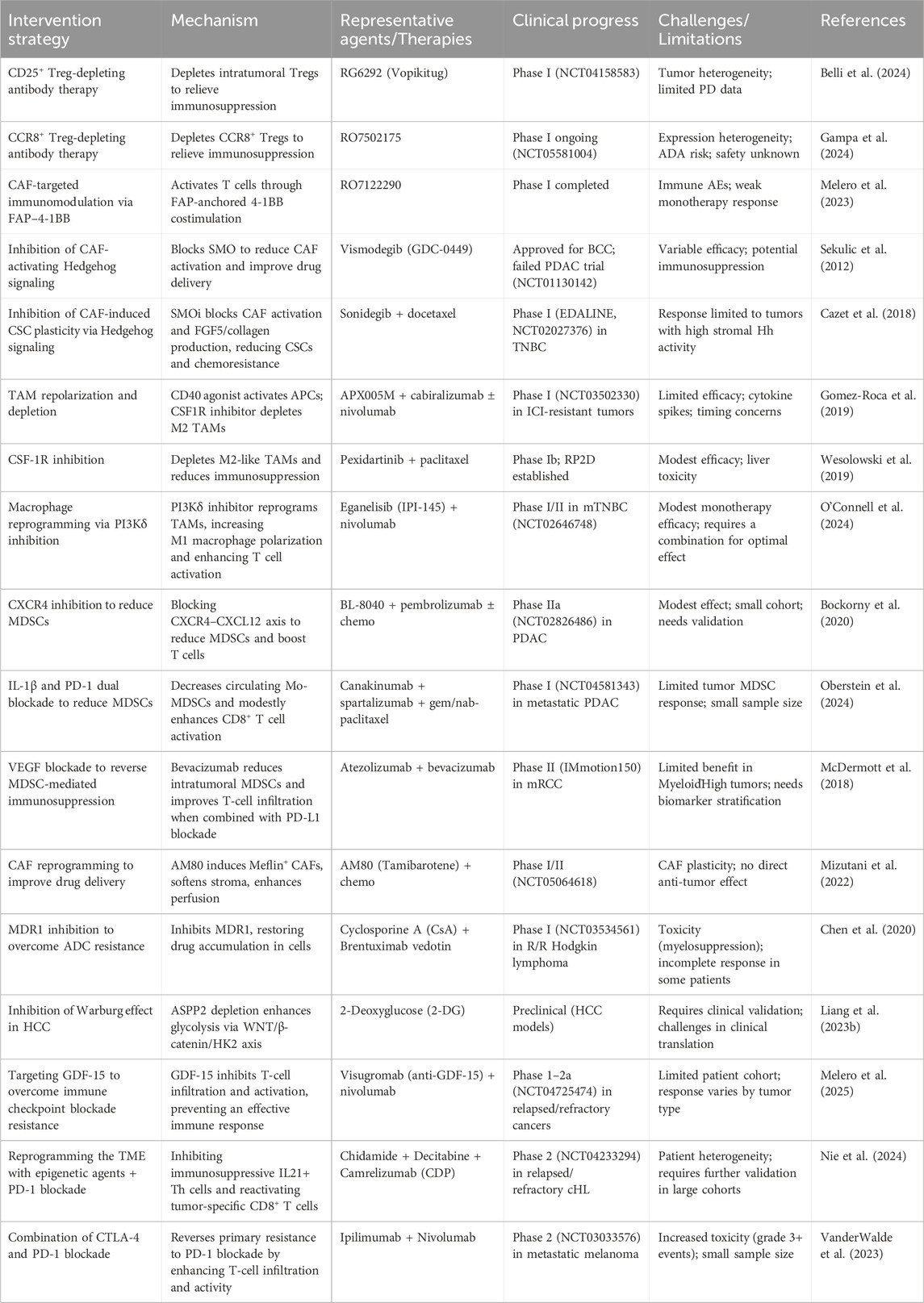
Table 1. Therapeutic strategies targeting the TME to overcome drug resistance: Mechanisms, agents, and clinical progress.
4.1 Targeting cellular components of the TME
4.1.1 CAFs
The immunosuppressive cellular network in the TME provides several potential therapeutic targets. Multi-omics studies have characterized CAF heterogeneity and their dynamic crosstalk with immune cells, informing two refined intervention approaches: direct CAF depletion and disruption of CAF-mediated immunosuppressive signaling. Early CAF depletion efforts focused on broad markers like FAP, α-SMA, and PDGFR (Qin et al., 2023; Grout et al., 2022). While FAP-directed CAR T cells and vaccines effectively eliminated FAP+ CAFs, increased CD8+ T-cell infiltration, and decreased fibrotic stroma (Kieffer et al., 2020; Wehrli et al., 2024; Pure and Blomberg, 2018; Wang et al., 2014), their clinical application was limited by on-target/off-tumor toxicity due to FAP expression in healthy tissues (Tran et al., 2013). Multi-omics data demonstrate that specific CAF subpopulations engage in reciprocal interactions with immune cells; for example, Cluster 0 FAP+ CAFs elevate PD-1 and CTLA-4 expression in Tregs, which subsequently drive the proliferation of immunosuppressive Cluster 3 CAFs associated with immunotherapy resistance (Kieffer et al., 2020). Spatial transcriptomics revealed that PDGFRβ+ CAF clusters recruit PMN-MDSCs through CXCL secretion, and disrupting these niches enhanced anti-PD-1 responses (Akiyama et al., 2023). Recent multi-omic profiling has uncovered CAF-specific surface markers including CD10, GPR77, POSTN, and LRR15, enabling more precise depletion strategies (Su et al., 2018; Purcell et al., 2018; Wang et al., 2024b; Krishnamurty et al., 2022). These findings underscore the need for more precise CAF-targeting strategies that account for functional heterogeneity and microenvironmental crosstalk, prompting recent efforts to develop subtype-specific interventions by multi-omic analysis.
An alternative strategy focuses on blocking CAF-derived immunosuppressive pathways, with multi-omics studies implicating TGF-β signaling, the IL-6/JAK/STAT3 axis, and chemokine networks. Clinical trials have demonstrated benefits with TGF-β inhibitors and bispecific anti-PD-L1/TGF-β antibodies (Kalluri, 2016; Faivre et al., 2019; Melisi et al., 2018; Ravi et al., 2018). IL-6R blockade enhanced CD8+ T-cell activation and synergized with checkpoint inhibitors (Heichler et al., 2020; Dijkgraaf et al., 2015), while CCR2 and CXCR4 inhibition improved therapeutic outcomes when combined with other agents (Xiang et al., 2020; Zhang et al., 2023b). Combined targeting of TGF-β signaling to prevent fibroblast activation and the CXCL12-CXCR4 axis to mitigate immune exclusion has proven particularly effective in restoring T-cell infiltration and potentiating checkpoint inhibitors (Rivas et al., 2022; Mariathasan et al., 2018; Feig et al., 2013). Emerging evidence also supports NOX4 inhibition as a viable approach, as it reverses CAF differentiation, promotes fibroblast normalization, and alleviates CD8+ T-cell exclusion to enhance anti-PD-1 responses (Ford et al., 2020).
4.1.2 MDSCs
MDSCs represent a critical therapeutic target in cancer immunotherapy. Multi-omics profiling has identified four key intervention strategies: disrupting MDSC recruitment, depleting existing populations, inducing differentiation, and neutralizing their immunosuppressive mechanisms. Chemokine networks play a central role in MDSC trafficking, as demonstrated by single-cell RNA sequencing and immune profiling showing that PPARγ-mediated VEGF-A upregulation drives MDSC expansion through the PPARγ/VEGF-A axis (Xiong et al., 2023). Clinical evidence confirms bevacizumab reduces MDSC frequencies in both NSCLC and glioblastoma patients (Koinis et al., 2016; Peereboom et al., 2019). The CCL2-CCR2 axis further contributes to immune suppression, with CCR2 inhibitors like CCX872 showing promise in combination with PD-L1 blockade for HCC (Liang et al., 2023a; Xie et al., 2023). Metabolic vulnerabilities in MDSCs have emerged through multi-omics studies, including gemcitabine’s selective depletion of MDSCs while enhancing NK cell activity via NKG2D ligand upregulation. Cryo-thermal therapy similarly activates NK cells to eliminate MDSCs through the NKG2D-NKG2DL axis while IFN-γ drives their maturation, converting immunosuppressive MDSCs into immunostimulatory phenotypes (Peng et al., 2022). Multi-omics approaches have also identified hypoxic tumor-associated MDSCs that could be activated by the IL6/STAT3 pathway. Tyrosine kinase inhibitors such as sunitinib and cabozantinib have been shown to suppress MDSCs and sensitize tumors to CAR-NK cells (Zhang et al., 2017). Ongoing multi-omics investigations continue to reveal novel targets, including recent metabolomic findings that GLUT1-mediated glucose accumulation sustains MDSC function in murine models, suggesting metabolic disruption as a viable therapeutic approach (Kim et al., 2023).
4.1.3 TAMs
Emerging scRNA-seq and spatial multi-omics technologies have revealed specialized TAM subpopulations that drive immune evasion, angiogenesis, and therapy resistance within the TME. Recent studies have revealed spatially restricted distributions of pro-tumoral (M1-like) and anti-tumoral (M2-like) TAM subsets, providing new opportunities for targeted intervention. Current interventions primarily focus on three mechanistic strategies. The first approach disrupts TAM recruitment by targeting chemokine axes, such as the CSF1-CSF1R pathway, where inhibitors like pexidartinib have demonstrated preclinical efficacy and are undergoing clinical assessment (Ries et al., 2014). CCL2-CCR2 axis blockade similarly reduces TAM infiltration while enhancing CD8+ T cell recruitment, improving antitumor immunity (Pittet et al., 2022). A second strategy reprograms TAM polarization, exemplified by CD40 agonists that convert M2-like TAMs to tumoricidal M1-like phenotypes—an approach showing synergistic effects with PD-1 blockade in clinical trials(NCT02376699) (Coveler et al., 2023). Additionally, inhibition of TREM2, a marker of lipid-metabolizing TAMs identified through omic analyses, has demonstrated the potential to enhance anti-tumor immunity (Kirschenbaum et al., 2024; Molgora et al., 2020). The third approach selectively depletes pro-tumoral subsets, including angiogenesis-associated SPP1+ TAMs and phagocytosis-impaired C1QC+ TAMs identified via multi-omics profiling (Cheng et al., 2021). Targeted therapies against these subsets may overcome resistance while preserving beneficial macrophage functions. Besides, the interaction between TAMs and blood vessels significantly promotes tumor growth, proliferation, and migration. Strategies such as co-targeting EGFR and VEGFR signaling pathways and inhibiting Notch signaling to block carcinogenic reprogramming may prove beneficial (Sharma et al., 2020).
4.2 Targeting non-cellular components: remodeling the ECM in the TME
Among ECM components, collagen forms fundamental structural networks that significantly influence drug distribution. Proteomic and imaging studies reveal that disseminated tumor cells remodel their microenvironment by forming type III collagen-enriched niches, sustaining dormancy via the DDR1-STAT1 signaling axis. Disrupting this pathway induces tumor reactivation, implying that ECM modulation may prolong dormancy and inhibit metastasis (Di Martino et al., 2022). Stromal profiling in pancreatic cancer datasets identifies LOXL2-rich niches as protective barriers in primary tumors, whereas LOXL2 depletion in liver metastases correlates with accelerated progression, warranting reassessment of anti-stromal therapies targeting collagen remodeling (Jiang et al., 2020).
Multi-omics integration has advanced the understanding of ECM-mediated resistance. Neuroendocrine prostate cancer adapts to therapy through tumor-specific ECM-driven epigenetic reprogramming, a vulnerability addressed by sequentially targeting epigenetic modifiers and DRD2 in synthetic organoids and xenografts (Mosquera et al., 2022). Furthermore, proteomics-defined metastasis-associated ECM signatures containing tenascin-C have enabled the development of tumor-agnostic nanobodies for targeted delivery of imaging and therapeutic agents, demonstrating the potential of ECM-guided precision targeting as evidenced by immuno-PET/CT validation in triple-negative breast and colorectal cancer metastases (Jailkhani et al., 2023). Single-cell multiomics in renal cell carcinoma implicates SERPINE2 as a metastatic regulator orchestrating EMT and tumor-TME crosstalk, suggesting its blockade could disrupt progression in advanced disease (Chen et al., 2023b).
Paradoxically, ECM softening promotes chemoresistance through DRP1/MIEF1/2-dependent mitochondrial fission and NRF2 activation, posing a therapeutic dilemma requiring nuanced targeting (Jain, 2014; Qin et al., 2020; Romani et al., 2022). Future investigations must delineate ECM-tumor interactions, refine ECM-directed strategies, and evaluate combinatorial approaches.
4.3 Targeting non-cellular components: remodeling the vascular reprogramming in the TME
Recent multi-omics studies have provided unprecedented insights into the mechanisms of vascular reprogramming in drug-resistant tumors and have identified novel therapeutic interventions for targeted vascular therapy. ScRNA-seq of brain metastasis vasculature uncovered CD276-enriched endothelial subtypes with immunoregulatory properties, and CD276 blockade improved survival in preclinical models, suggesting vascular immune checkpoint inhibition as a potential therapeutic strategy across multiple primary tumor types (Bejarano et al., 2024). Metabolomic profiling identified the sphingosine-1-phosphate receptor 1 (S1PR1)-STAT3-CerS3 signaling axis in endothelial cells as a driver of HCC angiogenesis through ceramide metabolism modulation, with S1PR1 inhibition showing synergistic effects when combined with Lenvatinib (Wang et al., 2022). Further omics analysis demonstrated that hypoxia-induced DGKG overexpression in HCC vascular endothelial cells, mediated by the USP16-ZEB2-TGF-β1 axis, facilitates immune evasion, while pathway inhibition enhances the efficacy of anti-PD-1/VEGFR2 therapy (Zhang et al., 2024).
These omics-derived insights are now informing novel clinical strategies. Dual-targeting agents, such as the bispecific anti-Ang2/VEGF-A antibody A2V, have shown promise in normalizing tumor vasculature while simultaneously reprogramming the TME (Kloepper et al., 2016). Multi-omics investigations of vascular-mediated drug resistance have yielded innovative approaches, including the FAP1V2 intracellular nanobody that concurrently targets PD-L1/PD-1 immune checkpoints and VEGFR2-driven metastasis, inducing durable tumor control through TCRβhi T-cell activation while preventing PD-1hi T-cell exhaustion (Zhang et al., 2025b). Hence, future research should focus on further elucidating the complex interplay between vascular reprogramming and the TME by using multi-omics approaches, developing more precise dual-targeting agents, and exploring the potential of rational combination therapies to enhance clinical outcomes.
4.4 Metabolic targeting in the TME
Multi-omics integration has revolutionized the design and optimization of metabolic therapies by systematically identifying actionable targets, predicting resistance mechanisms, and enabling precise patient stratification. Transcriptomic and metabolomic profiling of therapy-resistant tumors revealed consistent overexpression of glycolytic enzymes (GLUT1, HK2, PKM2), thereby nominating them as therapeutic targets (Liu et al., 2012; Feng et al., 2020). Single-cell multi-omics further validated HK2 as a biomarker for noninvasive bladder cancer detection, demonstrating 90% sensitivity and 98% specificity in urine-based liquid biopsies (Wang et al., 2020). This not only highlights the clinical utility of multi-omics in biomarker discovery but also underscores its potential for noninvasive cancer detection. Integrated genomics and metabolomics in lung cancer revealed BCKDK as an upstream regulator of MYC-dependent HK2 transcription, supporting dual BCKDK/HK2 inhibition to counter trametinib resistance (Wu et al., 2025).
Multi-omics approaches have clarified compensatory metabolic responses to targeted therapies, facilitating rational combination strategies. In castration-resistant prostate cancer, integrated transcriptomic and metabolomic data connected TP53 loss to ASNS-mediated asparagine dependence, informing the use of glutaminase inhibitors (e.g., CB-839) with L-asparaginase to induce synthetic lethality (Yoo et al., 2024). Proteomic flux analyses revealed that GLS inhibition triggers FAO/glycolysis upregulation (Choi and Park, 2018; Cheng et al., 2011), prompting clinical trials pairing GLS inhibitors (e.g., telaglenastat) with FAO blockers (e.g., etomoxir). Phosphoproteomic and lipidomic profiling in glioblastoma exposed an SREBP-1-ASCT2-glutamine resistance axis following lysosomal inhibition, guiding dual-targeting approaches (Zhong et al., 2024). Multi-omics has also been instrumental in connecting metabolic dysregulation to immunotherapy resistance. For instance, ARID1A-mutated ovarian cancers showed GLS1 dependency via metabolomics, motivating CB-839 + anti-PD-L1 trials (Wu et al., 2021b). Spatial metabolomics and transcriptomics identified BCAA metabolism (DLD/IL4I1-OXCT1 axis) and glutamine reprogramming as features of PD-1-high, therapy-resistant nasopharyngeal carcinoma, suggesting metabolic-immune co-targeting to address treatment resistance (Ji et al., 2025). Additionally, combining HK2 inhibitors with anti-PD-1 to reverse lactate-driven immunosuppression (Guo et al., 2022).
5 Discussion
Single-omics technologies have significantly advanced our understanding of tumor biology but remain insufficient for comprehensively characterizing the complex TME. Genomics fails to detect low-frequency variants, struggles with tumor purity, and cannot functionally annotate non-coding regions. Epigenomics encounters difficulties in establishing causal relationships due to low signal-to-noise ratios and sparse data coverage. Transcriptomics is constrained by the imperfect correlation between mRNA and protein levels, compounded by technical artifacts in single-cell RNA-seq. Proteomics currently captures only half of the human proteome, exhibits quantification variability, and misses transient molecular interactions. Metabolomics grapples with compound instability, methodological inconsistencies, and microbial contamination. These constraints underscore the imperative for multi-omics integration in TME research, as single-layer analyses cannot adequately capture cross-network communication or niche-specific signals obscured by bulk measurement limitations. Integrated multi-omics approaches overcome individual method weaknesses by uncovering higher-order biological relationships, distinguishing causal mechanisms from correlations, identifying compensatory pathways, and revealing non-cell-autonomous resistance patterns.
Multi-omics integration has revolutionized our comprehension of TME-mediated drug resistance, overturning conventional oncology paradigms. Where single-omics studies traditionally identified static biomarkers like PD-L1 expression for immune checkpoint blockade response, multi-omics uncovers context-dependent resistance networks—such as the requirement for CXCL13+ T cell niches to validate PD-L1 predictive value (Bassez et al., 2021)—and detects hybrid cellular states (e.g., EMT-metabolic intermediates in PDAC) invisible to single-layer analyses (Lin et al., 2020). However, key debates persist, including whether metabolic symbiosis mechanisms like the CAF-tumor lactate shuttle represent universal targets or context-dependent vulnerabilities (Ippolito et al., 2022). Tissue-level analyses obscure signals by averaging across heterogeneous cell populations, diminishing discovery potential and reproducibility. Advanced deconvolution algorithms using cell-type-specific reference signatures may address this limitation (Avila Cobos et al., 2020). The escalating volume of multi-omics data also necessitates optimized computational frameworks for multimodal integration. Although machine learning models show potential for improving clinical utility such as treatment response prediction (Sammut et al., 2022), obstacles remain in developing robust analytical pipelines and extracting biologically meaningful interpretations (Kang et al., 2022; Miao et al., 2021).
Three testable hypotheses may bridge these findings to clinical practice: First, spatial multi-omics can resolve discordance between tumor cell genotypes and TME phenotypes through predictable spatiotemporal phases, such as from fibrotic to immunosuppressive to metabolic states—detectable via integrated spatial transcriptomics, proteomics, and metabolomics, enabling novel target discovery (Peran et al., 2021). Second, cross-omics network analysis will reveal context-dependent resistance hubs—such as POSTN+ CAF/SPP1+ macrophage crosstalk via IL-6/STAT3—where combinatorial targeting (e.g., stromal-immune co-targeting) outperforms single biomarkers (Chen et al., 2023c). Third, Longitudinal integration of noninvasive dynamics (e.g., 2HG-MRS in gliomas, FDG-PET in breast cancer) with genomic clonal tracking can distinguish adaptive resistance(metabolic plasticity) from selection(subclone expansion), guiding mechanism-specific interventions (Suh et al., 2018; Zhou et al., 2018). Collectively, this framework links multi-omics layers to actionable clinical strategies while accounting for TME plasticity and evolutionary dynamics, thereby facilitating preoperative diagnosis, treatment monitoring, and recurrence surveillance.
Clinical translation of TME-targeting strategies faces substantial yet addressable barriers that require multi-omics-informed solutions. For instance, single-cell RNA-seq reveals niche-specific VEGF expression patterns that demand spatially precise delivery systems, with emerging solutions including MMP2-responsive nanoparticles and spatial multi-omics-guided combinations (Huang et al., 2013b). Adaptive resistance mechanisms, such as therapy-induced kynurenine accumulation, require real-time metabolite tracking coupled with preemptive pathway inhibition. Historical failures emphasize the critical need for proteomic validation in target prioritization. Emerging solutions include modular clinical trials testing stromal disruptors combined with immune modulators and “digital twin” approaches pairing patient-derived organoids with multi-omics simulations. To overcome these hurdles, innovative trial designs are emerging, including modular platform trials testing stromal disruptors with ICIs and “digital twin” approaches that integrate patient-derived organoids with longitudinal multi-omics profiling and AI-based treatment response prediction. These solutions directly address the translational challenges while leveraging multi-omics integration to develop clinically actionable strategies.
6 Conclusion
Therapeutic resistance represents a complex and evolving challenge in cancer treatment, with the TME serving as a central and dynamic contributor. As our understanding of TME-mediated resistance continues to deepen, the integration of multi-omics technologies offers a powerful lens through which to uncover hidden regulatory networks and guide precision intervention. Shortly, leveraging multi-omics insights to develop context-specific, combinatorial strategies that modulate the TME in real-time will be critical. Continued efforts toward translating these findings into clinically actionable approaches hold promise for overcoming resistance and improving durable patient outcomes.
Author contributions
FC: Data curation, Writing – original draft, Writing – review and editing. YF: Data curation, Methodology, Writing – original draft. GB: Investigation, Methodology, Writing – review and editing. JQ: Funding acquisition, Writing – review and editing. KH: Funding acquisition, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (Grant Nos. 82173188 and 82472993), Medical Innovation Research Special Project under the Science and Technology Innovation Action Plan of the Shanghai Municipal Science and Technology Commission (22Y31900501), Medical Innovation Research of Shanghai Science and Technology (21Y11906900).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahronian, L. G., Sennott, E. M., Van Allen, E. M., Wagle, N., Kwak, E. L., Faris, J. E., et al. (2015). Clinical acquired resistance to RAF inhibitor combinations in BRAF-mutant colorectal cancer through MAPK pathway alterations. Cancer Discov. 5 (4), 358–367. doi:10.1158/2159-8290.CD-14-1518
Akiyama, K., Ohga, N., Hida, Y., Kawamoto, T., Sadamoto, Y., Ishikawa, S., et al. (2012). Tumor endothelial cells acquire drug resistance by MDR1 up-regulation via VEGF signaling in tumor microenvironment. Am. J. Pathol. 180 (3), 1283–1293. doi:10.1016/j.ajpath.2011.11.029
Akiyama, T., Yasuda, T., Uchihara, T., Yasuda-Yoshihara, N., Tan, B. J. Y., Yonemura, A., et al. (2023). Stromal reprogramming through dual PDGFRα/β blockade boosts the efficacy of Anti-PD-1 immunotherapy in fibrotic tumors. Cancer Res. 83 (5), 753–770. doi:10.1158/0008-5472.CAN-22-1890
Albini, A., and Sporn, M. B. (2007). The tumour microenvironment as a target for chemoprevention. Nat. Rev. Cancer 7 (2), 139–147. doi:10.1038/nrc2067
Altea-Manzano, P., Doglioni, G., Liu, Y., Cuadros, A. M., Nolan, E., Fernandez-Garcia, J., et al. (2023). A palmitate-rich metastatic niche enables metastasis growth via p65 acetylation resulting in pro-metastatic NF-κB signaling. Nat. Cancer 4 (3), 344–364. doi:10.1038/s43018-023-00513-2
Apicella, M., Giannoni, E., Fiore, S., Ferrari, K. J., Fernandez-Perez, D., Isella, C., et al. (2018). Increased lactate secretion by cancer cells sustains non-cell-autonomous adaptive resistance to MET and EGFR targeted therapies. Cell Metab. 28 (6), 848–865. doi:10.1016/j.cmet.2018.08.006
Avila Cobos, F., Alquicira-Hernandez, J., Powell, J. E., Mestdagh, P., and De Preter, K. (2020). Benchmarking of cell type deconvolution pipelines for transcriptomics data. Nat. Commun. 11 (1), 5650. doi:10.1038/s41467-020-19015-1
Balkwill, F. R., Capasso, M., and Hagemann, T. (2012). The tumor microenvironment at a glance. J. Cell Sci. 125 (Pt 23), 5591–5596. doi:10.1242/jcs.116392
Bassez, A., Vos, H., Van Dyck, L., Floris, G., Arijs, I., Desmedt, C., et al. (2021). A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat. Med. 27 (5), 820–832. doi:10.1038/s41591-021-01323-8
Bejarano, L., Kauzlaric, A., Lamprou, E., Lourenco, J., Fournier, N., Ballabio, M., et al. (2024). Interrogation of endothelial and mural cells in brain metastasis reveals key immune-regulatory mechanisms. Cancer Cell 42 (3), 378–395.e10. doi:10.1016/j.ccell.2023.12.018
Belli, S., Amann, M., Hutchinson, L., Pousse, L., Abdolzade-Bavil, A., Justies, N., et al. (2024). Optimizing early clinical investigations in cancer immunotherapy: the translational journey of RG6292, a novel, selective treg-depleting antibody. Clin. Pharmacol. Ther. 116 (3), 834–846. doi:10.1002/cpt.3303
Bockorny, B., Semenisty, V., Macarulla, T., Borazanci, E., Wolpin, B. M., Stemmer, S. M., et al. (2020). BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat. Med. 26 (6), 878–885. doi:10.1038/s41591-020-0880-x
Brandacher, G., Perathoner, A., Ladurner, R., Schneeberger, S., Obrist, P., Winkler, C., et al. (2006). Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin. Cancer Res. 12 (4), 1144–1151. doi:10.1158/1078-0432.CCR-05-1966
Broz, M. T., Ko, E. Y., Ishaya, K., Xiao, J., De Simone, M., Hoi, X. P., et al. (2024). Metabolic targeting of cancer associated fibroblasts overcomes T-cell exclusion and chemoresistance in soft-tissue sarcomas. Nat. Commun. 15 (1), 2498. doi:10.1038/s41467-024-46504-4
Cai, Z., Chen, J., Yu, Z., Li, H., Liu, Z., Deng, D., et al. (2023). BCAT2 shapes a noninflamed tumor microenvironment and induces resistance to Anti-PD-1/PD-L1 immunotherapy by negatively regulating proinflammatory chemokines and anticancer immunity. Adv. Sci. (Weinh) 10 (8), e2207155. doi:10.1002/advs.202207155
Cao, X., Hou, J., An, Q., Assaraf, Y. G., and Wang, X. (2020). Towards the overcoming of anticancer drug resistance mediated by p53 mutations. Drug Resist Updat 49, 100671. doi:10.1016/j.drup.2019.100671
Cao, X., Wang, Y., Zhang, W., Zhong, X., Gunes, E. G., Dang, J., et al. (2022). Targeting macrophages for enhancing CD47 blockade-elicited lymphoma clearance and overcoming tumor-induced immunosuppression. Blood 139 (22), 3290–3302. doi:10.1182/blood.2021013901
Cappellesso, F., Orban, M. P., Shirgaonkar, N., Berardi, E., Serneels, J., Neveu, M. A., et al. (2022). Targeting the bicarbonate transporter SLC4A4 overcomes immunosuppression and immunotherapy resistance in pancreatic cancer. Nat. Cancer 3 (12), 1464–1483. doi:10.1038/s43018-022-00470-2
Carneiro, T. J., Carvalho, ALMB, Vojtek, M., Carmo, I. F., Marques, M. P. M., Diniz, C., et al. (2023). Disclosing a metabolic signature of cisplatin resistance in MDA-MB-231 triple-negative breast cancer cells by NMR metabolomics. Cancer Cell Int. 23 (1), 310. doi:10.1186/s12935-023-03124-0
Casey, S. C., Amedei, A., Aquilano, K., Azmi, A. S., Benencia, F., Bhakta, D., et al. (2015). Cancer prevention and therapy through the modulation of the tumor microenvironment. Seminars Cancer Biol. 35 (Suppl. l), S199–S223. doi:10.1016/j.semcancer.2015.02.007
Castro, B. A., Flanigan, P., Jahangiri, A., Hoffman, D., Chen, W., Kuang, R., et al. (2017). Macrophage migration inhibitory factor downregulation: a novel mechanism of resistance to anti-angiogenic therapy. Oncogene 36 (26), 3749–3759. doi:10.1038/onc.2017.1
Cazet, A. S., Hui, M. N., Elsworth, B. L., Wu, S. Z., Roden, D., Chan, C.-L., et al. (2018). Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat. Commun. 9 (1), 2897. doi:10.1038/s41467-018-05220-6
Chaudagar, K., Hieromnimon, H. M., Kelley, A., Labadie, B., Shafran, J., Rameshbabu, S., et al. (2023). Suppression of tumor cell lactate-generating signaling pathways eradicates murine PTEN/p53-deficient aggressive-variant prostate cancer via macrophage phagocytosis. Clin. Cancer Res. 29 (23), 4930–4940. doi:10.1158/1078-0432.CCR-23-1441
Chen, C., Guo, Q., Liu, Y., Hou, Q., Liao, M., Guo, Y., et al. (2023c). Single-cell and spatial transcriptomics reveal POSTN(+) cancer-associated fibroblasts correlated with immune suppression and tumour progression in non-small cell lung cancer. Clin. Transl. Med. 13 (12), e1515. doi:10.1002/ctm2.1515
Chen, J., Zhao, D., Wang, Y., Liu, M., Zhang, Y., Feng, T., et al. (2024). Lactylated apolipoprotein C-II induces immunotherapy resistance by promoting extracellular lipolysis. Adv. Sci. (Weinh) 11 (38), e2406333. doi:10.1002/advs.202406333
Chen, R., Herrera, A. F., Hou, J., Chen, L., Wu, J., Guo, Y., et al. (2020). Inhibition of MDR1 overcomes resistance to Brentuximab vedotin in Hodgkin Lymphoma. Clin. Cancer Res. 26 (5), 1034–1044. doi:10.1158/1078-0432.CCR-19-1768
Chen, W. J., Dong, K. Q., Pan, X. W., Gan, S. S., Xu, D., Chen, J. X., et al. (2023b). Single-cell RNA-seq integrated with multi-omics reveals SERPINE2 as a target for metastasis in advanced renal cell carcinoma. Cell Death Dis. 14 (1), 30. doi:10.1038/s41419-023-05566-w
Chen, X., Liu, Y., Zhang, Q., Liu, B., Cheng, Y., Zhang, Y., et al. (2021). Exosomal miR-590-3p derived from cancer-associated fibroblasts confers radioresistance in colorectal cancer. Mol. Ther. Nucleic Acids 24, 113–126. doi:10.1016/j.omtn.2020.11.003
Chen, Y., Xu, X., Wang, Y., Zhang, Y., Zhou, T., Jiang, W., et al. (2023a). Hypoxia-induced SKA3 promoted cholangiocarcinoma progression and chemoresistance by enhancing fatty acid synthesis via the regulation of PAR-dependent HIF-1a deubiquitylation. J. Exp. Clin. Cancer Res. 42 (1), 265. doi:10.1186/s13046-023-02842-7
Cheng, S., Li, Z., Gao, R., Xing, B., Gao, Y., Yang, Y., et al. (2021). A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184 (3), 792–809.e23. doi:10.1016/j.cell.2021.01.010
Cheng, T., Sudderth, J., Yang, C., Mullen, A. R., Jin, E. S., Matés, J. M., et al. (2011). Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl. Acad. Sci. U. S. A. 108 (21), 8674–8679. doi:10.1073/pnas.1016627108
Choi, Y.-K., and Park, K.-G. (2018). Targeting glutamine metabolism for cancer treatment. Biomol. Ther. 26 (1), 19–28. doi:10.4062/biomolther.2017.178
Chryplewicz, A., Scotton, J., Tichet, M., Zomer, A., Shchors, K., Joyce, J. A., et al. (2022). Cancer cell autophagy, reprogrammed macrophages, and remodeled vasculature in glioblastoma triggers tumor immunity. Cancer Cell 40 (10), 1111–1127.e9. doi:10.1016/j.ccell.2022.08.014
Coveler, A. L., Smith, D. C., Phillips, T., Curti, B. D., Goel, S., Mehta, A. N., et al. (2023). Phase 1 dose-escalation study of SEA-CD40: a non-fucosylated CD40 agonist, in advanced solid tumors and lymphomas. J. Immunother. Cancer 11 (6), e005584. doi:10.1136/jitc-2022-005584
Croci, D. O., Cerliani, J. P., Dalotto-Moreno, T., Mendez-Huergo, S. P., Mascanfroni, I. D., Dergan-Dylon, S., et al. (2014). Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell 156 (4), 744–758. doi:10.1016/j.cell.2014.01.043
Cui, J., Liu, X., Shang, Q., Sun, S., Chen, S., Dong, J., et al. (2024). Deubiquitination of CDC6 by OTUD6A promotes tumour progression and chemoresistance. Mol. Cancer 23 (1), 86. doi:10.1186/s12943-024-01996-y
Deblois, G., Smith, H. W., Tam, I. S., Gravel, S. P., Caron, M., Savage, P., et al. (2016). ERRα mediates metabolic adaptations driving lapatinib resistance in breast cancer. Nat. Commun. 7, 12156. doi:10.1038/ncomms12156
DeWaal, D., Nogueira, V., Terry, A. R., Patra, K. C., Jeon, S. M., Guzman, G., et al. (2018). Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat. Commun. 9 (1), 446. doi:10.1038/s41467-017-02733-4
Dijkgraaf, E. M., Santegoets, S. J., Reyners, A. K., Goedemans, R., Wouters, M. C., Kenter, G. G., et al. (2015). A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferon-α2b in patients with recurrent epithelial ovarian cancer. Ann. Oncol. 26 (10), 2141–2149. doi:10.1093/annonc/mdv309
Di Martino, J. S., Nobre, A. R., Mondal, C., Taha, I., Farias, E. F., Fertig, E. J., et al. (2022). A tumor-derived type III collagen-rich ECM niche regulates tumor cell dormancy. Nat. Cancer 3 (1), 90–107. doi:10.1038/s43018-021-00291-9
Dominguez, C. X., Muller, S., Keerthivasan, S., Koeppen, H., Hung, J., Gierke, S., et al. (2020). Single-cell RNA sequencing reveals stromal evolution into LRRC15(+) myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. 10 (2), 232–253. doi:10.1158/2159-8290.CD-19-0644
Dong, J., Qin, Z., Zhang, W. D., Cheng, G., Yehuda, A. G., Ashby, C. R., et al. (2020). Medicinal chemistry strategies to discover P-glycoprotein inhibitors: an update. Drug Resist Updat 49, 100681. doi:10.1016/j.drup.2020.100681
Douguet, L., Janho Dit Hreich, S., Benzaquen, J., Seguin, L., Juhel, T., Dezitter, X., et al. (2021). A small-molecule P2RX7 activator promotes anti-tumor immune responses and sensitizes lung tumor to immunotherapy. Nat. Commun. 12 (1), 653. doi:10.1038/s41467-021-20912-2
Du, Y., Shi, J., Wang, J., Xun, Z., Yu, Z., Sun, H., et al. (2024). Integration of pan-cancer single-cell and spatial transcriptomics reveals stromal cell features and therapeutic targets in tumor microenvironment. Cancer Res. 84 (2), 192–210. doi:10.1158/0008-5472.CAN-23-1418
Eales, K. L., Hollinshead, K. E., and Tennant, D. A. (2016). Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 5 (1), e190. doi:10.1038/oncsis.2015.50
Esfahani, M. S., Hamilton, E. G., Mehrmohamadi, M., Nabet, B. Y., Alig, S. K., King, D. A., et al. (2022). Inferring gene expression from cell-free DNA fragmentation profiles. Nat. Biotechnol. 40 (4), 585–597. doi:10.1038/s41587-022-01222-4
Facciabene, A., Peng, X., Hagemann, I. S., Balint, K., Barchetti, A., Wang, L.-P., et al. (2011). Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 475 (7355), 226–230. doi:10.1038/nature10169
Faget, J., Biota, C., Bachelot, T., Gobert, M., Treilleux, I., Goutagny, N., et al. (2011). Early detection of tumor cells by innate immune cells leads to T(reg) recruitment through CCL22 production by tumor cells. Cancer Res. 71 (19), 6143–6152. doi:10.1158/0008-5472.CAN-11-0573
Faivre, S., Santoro, A., Kelley, R. K., Gane, E., Costentin, C. E., Gueorguieva, I., et al. (2019). Novel transforming growth factor beta receptor I kinase inhibitor galunisertib (LY2157299) in advanced hepatocellular carcinoma. Liver Int. 39 (8), 1468–1477. doi:10.1111/liv.14113
Fan, J., Wei, Q., Koay, E. J., Liu, Y., Ning, B., Bernard, P. W., et al. (2018). Chemoresistance transmission via exosome-mediated EphA2 transfer in pancreatic cancer. Theranostics 8 (21), 5986–5994. doi:10.7150/thno.26650
Fang, K., Ohihoin, A. G., Liu, T., Choppavarapu, L., Nosirov, B., Wang, Q., et al. (2024). Integrated single-cell analysis reveals distinct epigenetic-regulated cancer cell states and a heterogeneity-guided core signature in tamoxifen-resistant breast cancer. Genome Med. 16 (1), 134. doi:10.1186/s13073-024-01407-3
Feig, C., Jones, J. O., Kraman, M., Wells, R. J. B., Deonarine, A., Chan, D. S., et al. (2013). Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. U. S. A. 110 (50), 20212–20217. doi:10.1073/pnas.1320318110
Feng, J., Li, J., Wu, L., Yu, Q., Ji, J., Wu, J., et al. (2020). Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 39 (1), 126. doi:10.1186/s13046-020-01629-4
Ford, K., Hanley, C. J., Mellone, M., Szyndralewiez, C., Heitz, F., Wiesel, P., et al. (2020). NOX4 inhibition potentiates immunotherapy by overcoming cancer-associated fibroblast-mediated CD8 T-cell exclusion from tumors. Cancer Res. 80 (9), 1846–1860. doi:10.1158/0008-5472.CAN-19-3158
Francescangeli, F., Contavalli, P., De Angelis, M. L., Careccia, S., Signore, M., Haas, T. L., et al. (2020). A pre-existing population of ZEB2(+) quiescent cells with stemness and mesenchymal features dictate chemoresistance in colorectal cancer. J. Exp. Clin. Cancer Res. 39 (1), 2. doi:10.1186/s13046-019-1505-4
Funk, C. R., Wang, S., Chen, K. Z., Waller, A., Sharma, A., Edgar, C. L., et al. (2022). PI3Kδ/γ inhibition promotes human CART cell epigenetic and metabolic reprogramming to enhance antitumor cytotoxicity. Blood 139 (4), 523–537. doi:10.1182/blood.2021011597
Gabrilovich, D. I. (2017). Myeloid-derived suppressor cells. Cancer Immunol. Res. 5 (1), 3–8. doi:10.1158/2326-6066.CIR-16-0297
Gampa, G., Spinosa, P., Getz, J., Zhong, Y., Halpern, W., Esen, E., et al. (2024). Preclinical and translational pharmacology of afucosylated anti-CCR8 antibody for depletion of tumour-infiltrating regulatory T cells. Br. J. Pharmacol. 181 (13), 2033–2052. doi:10.1111/bph.16326
Gao, J., Ward, J. F., Pettaway, C. A., Shi, L. Z., Subudhi, S. K., Vence, L. M., et al. (2017). VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 23 (5), 551–555. doi:10.1038/nm.4308
Gao, M. Q., Choi, Y. P., Kang, S., Youn, J. H., and Cho, N. H. (2010). CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene 29 (18), 2672–2680. doi:10.1038/onc.2010.35
Gawad, C., Koh, W., and Quake, S. R. (2016). Single-cell genome sequencing: current state of the science. Nat. Rev. Genet. 17 (3), 175–188. doi:10.1038/nrg.2015.16
Giraudo, E., Inoue, M., and Hanahan, D. (2004). An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Invest 114 (5), 623–633. doi:10.1172/JCI22087
Glaviano, A., Lau, H. S., Carter, L. M., Lee, E. H. C., Lam, H. Y., Okina, E., et al. (2025). Harnessing the tumor microenvironment: targeted cancer therapies through modulation of epithelial-mesenchymal transition. J. Hematol. Oncol. 18 (1), 6. doi:10.1186/s13045-024-01634-6
Gomez-Roca, C. A., Italiano, A., Le Tourneau, C., Cassier, P. A., Toulmonde, M., D'Angelo, S. P., et al. (2019). Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2-like macrophages. Ann. Oncol. 30 (8), 1381–1392. doi:10.1093/annonc/mdz163
Gong, Y., Gao, D., Shi, Y., Fan, G., Yu, X., Yang, E., et al. (2025). SRC enhanced cisplatin resistance in bladder cancer by reprogramming glycolysis and pentose phosphate pathway. Commun. Biol. 8 (1), 36. doi:10.1038/s42003-024-07284-1
Gong, Y., Ji, P., Yang, Y. S., Xie, S., Yu, T. J., Xiao, Y., et al. (2021). Metabolic-pathway-based subtyping of triple-negative breast cancer reveals potential therapeutic targets. Cell Metab. 33 (1), 51–64.e9. doi:10.1016/j.cmet.2020.10.012
Goodman, A. M., Kato, S., Bazhenova, L., Patel, S. P., Frampton, G. M., Miller, V., et al. (2017). Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 16 (11), 2598–2608. doi:10.1158/1535-7163.MCT-17-0386
Grout, J. A., Sirven, P., Leader, A. M., Maskey, S., Hector, E., Puisieux, I., et al. (2022). Spatial positioning and matrix programs of cancer-associated fibroblasts promote T-cell exclusion in human lung tumors. Cancer Discov. 12 (11), 2606–2625. doi:10.1158/2159-8290.CD-21-1714
Gu, Y., Fang, Y., Wu, X., Xu, T., Hu, T., Xu, Y., et al. (2023). The emerging roles of SUMOylation in the tumor microenvironment and therapeutic implications. Exp. Hematol. Oncol. 12 (1), 58. doi:10.1186/s40164-023-00420-3
Guo, D., Tong, Y., Jiang, X., Meng, Y., Jiang, H., Du, L., et al. (2022). Aerobic glycolysis promotes tumor immune evasion by hexokinase2-mediated phosphorylation of IκBα. Cell Metab. 34 (9), 1312–1324.e6. doi:10.1016/j.cmet.2022.08.002
Halvorsen, E. C., Hamilton, M. J., Young, A., Wadsworth, B. J., LePard, N. E., Lee, H. N., et al. (2016). Maraviroc decreases CCL8-mediated migration of CCR5(+) regulatory T cells and reduces metastatic tumor growth in the lungs. Oncoimmunology 5 (6), e1150398. doi:10.1080/2162402X.2016.1150398
He, X., Liu, X., Zuo, F., Shi, H., and Jing, J. (2023). Artificial intelligence-based multi-omics analysis fuels cancer precision medicine. Semin. Cancer Biol. 88, 187–200. doi:10.1016/j.semcancer.2022.12.009
He, X., Wang, J., Wei, W., Shi, M., Xin, B., Zhang, T., et al. (2016). Hypoxia regulates ABCG2 activity through the activivation of ERK1/2/HIF-1α and contributes to chemoresistance in pancreatic cancer cells. Cancer Biol. Ther. 17 (2), 188–198. doi:10.1080/15384047.2016.1139228
Heichler, C., Scheibe, K., Schmied, A., Geppert, C. I., Schmid, B., Wirtz, S., et al. (2020). STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut 69 (7), 1269–1282. doi:10.1136/gutjnl-2019-319200
Heidegger, I., Fotakis, G., Offermann, A., Goveia, J., Daum, S., Salcher, S., et al. (2022). Comprehensive characterization of the prostate tumor microenvironment identifies CXCR4/CXCL12 crosstalk as a novel antiangiogenic therapeutic target in prostate cancer. Mol. Cancer 21 (1), 132. doi:10.1186/s12943-022-01597-7
Hosein, A. N., Brekken, R. A., and Maitra, A. (2020). Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 17 (8), 487–505. doi:10.1038/s41575-020-0300-1
Huang, B., Pan, P.-Y., Li, Q., Sato, A. I., Levy, D. E., Bromberg, J., et al. (2006). Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 66 (2), 1123–1131. doi:10.1158/0008-5472.CAN-05-1299
Huang, K.-W., Hsu, F.-F., Qiu, J. T., Chern, G.-J., Lee, Y.-A., Chang, C.-C., et al. (2020). Highly efficient and tumor-selective nanoparticles for dual-targeted immunogene therapy against cancer. Sci. Adv. 6 (3), eaax5032. doi:10.1126/sciadv.aax5032
Huang, L., Perrault, C., Coelho-Martins, J., Hu, C., Dulong, C., Varna, M., et al. (2013a). Induction of acquired drug resistance in endothelial cells and its involvement in anticancer therapy. J. Hematol. Oncol. 6, 49. doi:10.1186/1756-8722-6-49
Huang, S., Shao, K., Liu, Y., Kuang, Y., Li, J., An, S., et al. (2013b). Tumor-targeting and microenvironment-responsive smart nanoparticles for combination therapy of antiangiogenesis and apoptosis. ACS Nano 7 (3), 2860–2871. doi:10.1021/nn400548g
Ippolito, L., Comito, G., Parri, M., Iozzo, M., Duatti, A., Virgilio, F., et al. (2022). Lactate rewires lipid metabolism and sustains a metabolic-epigenetic axis in prostate cancer. Cancer Res. 82 (7), 1267–1282. doi:10.1158/0008-5472.CAN-21-0914
Jailkhani, N., Clauser, K. R., Mak, H. H., Rickelt, S., Tian, C., Whittaker, C. A., et al. (2023). Proteomic profiling of extracellular matrix components from patient metastases identifies consistently elevated proteins for developing nanobodies that target primary tumors and metastases. Cancer Res. 83 (12), 2052–2065. doi:10.1158/0008-5472.CAN-22-1532
Jain, R. K. (2013). Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J. Clin. Oncol. 31 (17), 2205–2218. doi:10.1200/JCO.2012.46.3653
Jain, R. K. (2014). Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 26 (5), 605–622. doi:10.1016/j.ccell.2014.10.006
Jarosz-Biej, M., Smolarczyk, R., Cichon, T., and Kulach, N. (2019). Tumor microenvironment as A “Game Changer” in cancer radiotherapy. Int. J. Mol. Sci. 20 (13), 3212. doi:10.3390/ijms20133212
Ji, L., Wang, D., Zhuo, G., Chen, Z., Wang, L., Zhang, Q., et al. (2025). Spatial metabolomics and transcriptomics reveal metabolic reprogramming and cellular interactions in nasopharyngeal carcinoma with high PD-1 expression and therapeutic response. Theranostics 15 (7), 3035–3054. doi:10.7150/thno.102822
Ji, Z., Wang, X., Xin, J., Ma, L., Zuo, D., Li, H., et al. (2024). Multiomics reveals tumor microenvironment remodeling in locally advanced gastric and gastroesophageal junction cancer following neoadjuvant immunotherapy and chemotherapy. J. Immunother. Cancer 12 (12), e010041. doi:10.1136/jitc-2024-010041
Jiang, H., Torphy, R. J., Steiger, K., Hongo, H., Ritchie, A. J., Kriegsmann, M., et al. (2020). Pancreatic ductal adenocarcinoma progression is restrained by stromal matrix. J. Clin. Invest 130 (9), 4704–4709. doi:10.1172/JCI136760
Junttila, M. R., and de Sauvage, F. J. (2013). Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 501 (7467), 346–354. doi:10.1038/nature12626
Kalluri, R. (2016). The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16 (9), 582–598. doi:10.1038/nrc.2016.73
Kaloni, D., Diepstraten, S. T., Strasser, A., and Kelly, G. L. (2023). BCL-2 protein family: attractive targets for cancer therapy. Apoptosis 28 (1-2), 20–38. doi:10.1007/s10495-022-01780-7
Kang, M., Ko, E., and Mersha, T. B. (2022). A roadmap for multi-omics data integration using deep learning. Brief. Bioinform 23 (1), bbab454. doi:10.1093/bib/bbab454
Kanlikilicer, P., Bayraktar, R., Denizli, M., Rashed, M. H., Ivan, C., Aslan, B., et al. (2020). Exosomal miRNA confers chemo resistance via targeting Cav1/p-gp/M2-type macrophage axis in ovarian cancer. EBioMedicine 38, 100–112. doi:10.1016/j.ebiom.2018.11.004
Kaoutari, A. E., Fraunhoffer, N. A., Hoare, O., Teyssedou, C., Soubeyran, P., Gayet, O., et al. (2021). Metabolomic profiling of pancreatic adenocarcinoma reveals key features driving clinical outcome and drug resistance. EBioMedicine 66, 103332. doi:10.1016/j.ebiom.2021.103332
Khodayari, S., Khodayari, H., Saeedi, E., Mahmoodzadeh, H., Sadrkhah, A., and Nayernia, K. (2023). Single-cell transcriptomics for unlocking personalized cancer immunotherapy: toward targeting the origin of tumor development immunogenicity. Cancers (Basel) 15 (14), 3615. doi:10.3390/cancers15143615
Kieffer, Y., Hocine, H. R., Gentric, G., Pelon, F., Bernard, C., Bourachot, B., et al. (2020). Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov. 10 (9), 1330–1351. doi:10.1158/2159-8290.CD-19-1384
Kim, J., Lee, H., Choi, H. K., and Min, H. (2023). Discovery of myeloid-derived suppressor cell-specific metabolism by metabolomic and lipidomic profiling. Metabolites 13 (4), 477. doi:10.3390/metabo13040477
Kirschenbaum, D., Xie, K., Ingelfinger, F., Katzenelenbogen, Y., Abadie, K., Look, T., et al. (2024). Time-resolved single-cell transcriptomics defines immune trajectories in glioblastoma. Cell 187 (1), 149–165.e23. doi:10.1016/j.cell.2023.11.032
Kloepper, J., Riedemann, L., Amoozgar, Z., Seano, G., Susek, K., Yu, V., et al. (2016). Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. U. S. A. 113 (16), 4476–4481. doi:10.1073/pnas.1525360113
Koinis, F., Vetsika, E. K., Aggouraki, D., Skalidaki, E., Koutoulaki, A., Gkioulmpasani, M., et al. (2016). Effect of first-line treatment on myeloid-derived suppressor cells’ subpopulations in the peripheral blood of patients with non-small cell lung cancer. J. Thorac. Oncol. 11 (8), 1263–1272. doi:10.1016/j.jtho.2016.04.026
Koyama, S., Akbay, E. A., Li, Y. Y., Herter-Sprie, G. S., Buczkowski, K. A., Richards, W. G., et al. (2016). Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 7, 10501. doi:10.1038/ncomms10501
Krishnamurty, A. T., Shyer, J. A., Thai, M., Gandham, V., Buechler, M. B., Yang, Y. A., et al. (2022). LRRC15(+) myofibroblasts dictate the stromal setpoint to suppress tumour immunity. Nature 611 (7934), 148–154. doi:10.1038/s41586-022-05272-1
Kundu, M., Butti, R., Panda, V. K., Malhotra, D., Das, S., Mitra, T., et al. (2024). Modulation of the tumor microenvironment and mechanism of immunotherapy-based drug resistance in breast cancer. Mol. Cancer 23 (1), 92. doi:10.1186/s12943-024-01990-4
Lambo, S., Trinh, D. L., Ries, R. E., Jin, D., Setiadi, A., Ng, M., et al. (2023). A longitudinal single-cell atlas of treatment response in pediatric AML. Cancer Cell 41 (12), 2117–35.e12. doi:10.1016/j.ccell.2023.10.008
Lambros, M. B., Seed, G., Sumanasuriya, S., Gil, V., Crespo, M., Fontes, M., et al. (2018). Single-cell analyses of prostate cancer liquid biopsies acquired by apheresis. Clin. Cancer Res. 24 (22), 5635–5644. doi:10.1158/1078-0432.CCR-18-0862
Le, M. I., Chen, W., Lines, J. L., Day, M., Li, J., Sergent, P., et al. (2014). VISTA regulates the development of protective antitumor immunity. Cancer Res. 74 (7), 1933–1944. doi:10.1158/0008-5472.CAN-13-1506
Li, Q.-L., Lin, X., Yu, Y.-L., Chen, L., Hu, Q.-X., Chen, M., et al. (2021). Genome-wide profiling in colorectal cancer identifies PHF19 and TBC1D16 as oncogenic super enhancers. Nat. Commun. 12 (1), 6407. doi:10.1038/s41467-021-26600-5
Li, W., Zhang, H., Assaraf, Y. G., Zhao, K., Xu, X., Xie, J., et al. (2016). Overcoming ABC transporter-mediated multidrug resistance: molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Updat. 27, 14–29. doi:10.1016/j.drup.2016.05.001
Li, Y., Weng, Y., Zhong, L., Chong, H., Chen, S., Sun, Y., et al. (2017). VEGFR3 inhibition chemosensitizes lung adenocarcinoma A549 cells in the tumor-associated macrophage microenvironment through upregulation of p53 and PTEN. Oncol. Rep. 38 (5), 2761–2773. doi:10.3892/or.2017.5969
Li, Z., Low, V., Luga, V., Sun, J., Earlie, E., Parang, B., et al. (2022). Tumor-produced and aging-associated oncometabolite methylmalonic acid promotes cancer-associated fibroblast activation to drive metastatic progression. Nat. Commun. 13 (1), 6239. doi:10.1038/s41467-022-33862-0
Liang, B., Jiang, Y., Song, S., Jing, W., Yang, H., Zhao, L., et al. (2023b). ASPP2 suppresses tumour growth and stemness characteristics in HCC by inhibiting Warburg effect via WNT/β-catenin/HK2 axis. J. Cell. Mol. Med. 27 (5), 659–671. doi:10.1111/jcmm.17687
Liang, T., Tao, T., Wu, K., Liu, L., Xu, W., Zhou, D., et al. (2023a). Cancer-associated fibroblast-induced remodeling of tumor microenvironment in recurrent bladder cancer. Adv. Sci. (Weinh) 10 (31), e2303230. doi:10.1002/advs.202303230
Lin, W., Noel, P., Borazanci, E. H., Lee, J., Amini, A., Han, I. W., et al. (2020). Single-cell transcriptome analysis of tumor and stromal compartments of pancreatic ductal adenocarcinoma primary tumors and metastatic lesions. Genome Med. 12 (1), 80. doi:10.1186/s13073-020-00776-9
Liu, H., Tang, L., Li, Y., Xie, W., Zhang, L., Tang, H., et al. (2024). Nasopharyngeal carcinoma: current views on the tumor microenvironment's impact on drug resistance and clinical outcomes. Mol. Cancer 23 (1), 20. doi:10.1186/s12943-023-01928-2
Liu, L., Zhang, L., Yang, L., Li, H., Li, R., Yu, J., et al. (2017). Anti-CD47 antibody as a targeted therapeutic agent for human lung cancer and cancer stem cells. Front. Immunol. 8, 404. doi:10.3389/fimmu.2017.00404
Liu, Y., Cao, Y., Zhang, W., Bergmeier, S., Qian, Y., Akbar, H., et al. (2012). A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 11 (8), 1672–1682. doi:10.1158/1535-7163.MCT-12-0131
Liu, Y., Chen, Y., Wang, F., Lin, J., Tan, X., Chen, C., et al. (2023). Caveolin-1 promotes glioma progression and maintains its mitochondrial inhibition resistance. Discov. Oncol. 14 (1), 161. doi:10.1007/s12672-023-00765-5
Loison, I., Pioger, A., Paget, S., Metatla, I., Orga, R., Vincent, A., et al. (2024). O-GlcNAcylation inhibition redirects the response of colon cancer cells to chemotherapy from senescence to apoptosis. Cell Death Dis. 15 (10), 762. doi:10.1038/s41419-024-07131-5
Lu, J., Li, J., Lin, Z., Li, H., Lou, L., Ding, W., et al. (2023). Reprogramming of TAMs via the STAT3/CD47-SIRPα axis promotes acquired resistance to EGFR-TKIs in lung cancer. Cancer Lett. 564, 216205. doi:10.1016/j.canlet.2023.216205
Lu, Y., Liu, Y., Zuo, X., Li, G., Wang, J., Liu, J., et al. (2025). CXCL12(+) tumor-associated endothelial cells promote immune resistance in hepatocellular carcinoma. J. Hepatol. 82 (4), 634–648. doi:10.1016/j.jhep.2024.09.044
Luis, G., Godfroid, A., Nishiumi, S., Cimino, J., Blacher, S., Maquoi, E., et al. (2021). Tumor resistance to ferroptosis driven by Stearoyl-CoA Desaturase-1 (SCD1) in cancer cells and Fatty Acid Biding Protein-4 (FABP4) in tumor microenvironment promote tumor recurrence. Redox Biol. 43, 102006. doi:10.1016/j.redox.2021.102006
Luo, Y., Tian, W., Lu, X., Zhang, C., Xie, J., Deng, X., et al. (2022). Prognosis stratification in breast cancer and characterization of immunosuppressive microenvironment through a pyrimidine metabolism-related signature. Front. Immunol. 13, 1056680. doi:10.3389/fimmu.2022.1056680
Madeddu, C., Donisi, C., Liscia, N., Lai, E., Scartozzi, M., and Macciò, A. (2022). EGFR-mutated non-small cell lung cancer and resistance to immunotherapy: role of the tumor microenvironment. Int. J. Mol. Sci. 23 (12), 6489. doi:10.3390/ijms23126489
Majeti, R., Chao, M. P., Alizadeh, A. A., Pang, W. W., Jaiswal, S., Gibbs, K. D., et al. (2009). CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 138 (2), 286–299. doi:10.1016/j.cell.2009.05.045
Mariathasan, S., Turley, S. J., Nickles, D., Castiglioni, A., Yuen, K., Wang, Y., et al. (2018). TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554 (7693), 544–548. doi:10.1038/nature25501
Matsumura, K., Hayashi, H., Uemura, N., Ogata, Y., Zhao, L., Sato, H., et al. (2022). Thrombospondin-1 overexpression stimulates loss of Smad4 and accelerates malignant behavior via TGF-β signal activation in pancreatic ductal adenocarcinoma. Transl. Oncol. 26, 101533. doi:10.1016/j.tranon.2022.101533
McDermott, D. F., Huseni, M. A., Atkins, M. B., Motzer, R. J., Rini, B. I., Escudier, B., et al. (2018). Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 24 (6), 749–757. doi:10.1038/s41591-018-0053-3
Melero, I., de Miguel Luken, M., de Velasco, G., Garralda, E., Martín-Liberal, J., Joerger, M., et al. (2025). Neutralizing GDF-15 can overcome anti-PD-1 and anti-PD-L1 resistance in solid tumours. Nature 637 (8048), 1218–1227. doi:10.1038/s41586-024-08305-z
Melero, I., Tanos, T., Bustamante, M., Sanmamed, M. F., Calvo, E., Moreno, I., et al. (2023). A first-in-human study of the fibroblast activation protein-targeted, 4-1BB agonist RO7122290 in patients with advanced solid tumors. Sci. Transl. Med. 15 (695), eabp9229. doi:10.1126/scitranslmed.abp9229
Melisi, D., Garcia-Carbonero, R., Macarulla, T., Pezet, D., Deplanque, G., Fuchs, M., et al. (2018). Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 119 (10), 1208–1214. doi:10.1038/s41416-018-0246-z
Miao, Z., Humphreys, B. D., McMahon, A. P., and Kim, J. (2021). Multi-omics integration in the age of million single-cell data. Nat. Rev. Nephrol. 17 (11), 710–724. doi:10.1038/s41581-021-00463-x
Migliozzi, S., Oh, Y. T., Hasanain, M., Garofano, L., D'Angelo, F., Najac, R. D., et al. (2023). Integrative multi-omics networks identify PKCδ and DNA-PK as master kinases of glioblastoma subtypes and guide targeted cancer therapy. Nat. Cancer 4 (2), 181–202. doi:10.1038/s43018-022-00510-x
Mizutani, Y., Iida, T., Ohno, E., Ishikawa, T., Kinoshita, F., Kuwatsuka, Y., et al. (2022). Safety and efficacy of MIKE-1 in patients with advanced pancreatic cancer: a study protocol for an open-label phase I/II investigator-initiated clinical trial based on a drug repositioning approach that reprograms the tumour stroma. BMC cancer 22 (1), 205. doi:10.1186/s12885-022-09272-2
Molgora, M., Esaulova, E., Vermi, W., Hou, J., Chen, Y., Luo, J., et al. (2020). TREM2 modulation remodels the tumor myeloid landscape enhancing Anti-PD-1 immunotherapy. Cell 182 (4), 886–900. doi:10.1016/j.cell.2020.07.013
Mosquera, M. J., Kim, S., Bareja, R., Fang, Z., Cai, S., Pan, H., et al. (2022). Extracellular matrix in synthetic hydrogel-based prostate cancer organoids regulate therapeutic response to EZH2 and DRD2 inhibitors. Adv. Mater 34 (2), e2100096. doi:10.1002/adma.202100096
Nie, J., Wang, C., Zheng, L., Liu, Y., Wang, C., Chang, Y., et al. (2024). Epigenetic agents plus anti-PD-1 reprogram the tumor microenvironment and restore antitumor efficacy in Hodgkin lymphoma. Blood 144 (18), 1936–1950. doi:10.1182/blood.2024024487
Notarangelo, G., Spinelli, J. B., Perez, E. M., Baker, G. J., Kurmi, K., Elia, I., et al. (2022). Oncometabolite d-2HG alters T cell metabolism to impair CD8(+) T cell function. Science 377 (6614), 1519–1529. doi:10.1126/science.abj5104
Oberstein, P. E., Dias Costa, A., Kawaler, E. A., Cardot-Ruffino, V., Rahma, O. E., Beri, N., et al. (2024). Blockade of IL1β and PD1 with combination chemotherapy reduces systemic myeloid suppression in metastatic pancreatic cancer with heterogeneous effects in the tumor. Cancer Immunol. Res. 12 (9), 1221–1235. doi:10.1158/2326-6066.CIR-23-1073
O’Connell, B. C., Hubbard, C., Zizlsperger, N., Fitzgerald, D., Kutok, J. L., Varner, J., et al. (2024). Eganelisib combined with immune checkpoint inhibitor therapy and chemotherapy in frontline metastatic triple-negative breast cancer triggers macrophage reprogramming, immune activation and extracellular matrix reorganization in the tumor microenvironment. J. Immunother. Cancer 12 (8), e009160. doi:10.1136/jitc-2024-009160
Okoye, C. N., Koren, S. A., and Wojtovich, A. P. (2023). Mitochondrial complex I ROS production and redox signaling in hypoxia. Redox Biol. 67, 102926. doi:10.1016/j.redox.2023.102926
Ouzounova, M., Lee, E., Piranlioglu, R., El Andaloussi, A., Kolhe, R., Demirci, M. F., et al. (2017). Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat. Commun. 8, 14979. doi:10.1038/ncomms14979
Park, K., Veena, M. S., and Shin, D. S. (2022). Key players of the immunosuppressive tumor microenvironment and emerging therapeutic strategies. Front. Cell Dev. Biol. 10, 830208. doi:10.3389/fcell.2022.830208
Park, Y. H., Im, S. A., Park, K., Wen, J., Lee, K. H., Choi, Y. L., et al. (2023). Longitudinal multi-omics study of palbociclib resistance in HR-positive/HER2-negative metastatic breast cancer. Genome Med. 15 (1), 55. doi:10.1186/s13073-023-01201-7
Patra, K. C., Wang, Q., Bhaskar, P. T., Miller, L., Wang, Z., Wheaton, W., et al. (2013). Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 24 (2), 213–228. doi:10.1016/j.ccr.2013.06.014
Peereboom, D. M., Alban, T. J., Grabowski, M. M., Alvarado, A. G., Otvos, B., Bayik, D., et al. (2019). Metronomic capecitabine as an immune modulator in glioblastoma patients reduces myeloid-derived suppressor cells. JCI Insight 4 (22), e130748. doi:10.1172/jci.insight.130748
Peng, P., Lou, Y., Wang, S., Wang, J., Zhang, Z., Du, P., et al. (2022). Activated NK cells reprogram MDSCs via NKG2D-NKG2DL and IFN-gamma to modulate antitumor T-cell response after cryo-thermal therapy. J. Immunother. Cancer 10 (12), e005769. doi:10.1136/jitc-2022-005769
Peran, I., Dakshanamurthy, S., McCoy, M. D., Mavropoulos, A., Allo, B., Sebastian, A., et al. (2021). Cadherin 11 promotes immunosuppression and extracellular matrix deposition to support growth of pancreatic tumors and resistance to gemcitabine in mice. Gastroenterology 160 (4), 1359–1372.e13. doi:10.1053/j.gastro.2020.11.044
Petitprez, F., de Reynies, A., Keung, E. Z., Chen, T. W., Sun, C. M., Calderaro, J., et al. (2020). B cells are associated with survival and immunotherapy response in sarcoma. Nature 577 (7791), 556–560. doi:10.1038/s41586-019-1906-8
Pitt, J. M., Marabelle, A., Eggermont, A., Soria, J. C., Kroemer, G., and Zitvogel, L. (2016). Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 27 (8), 1482–1492. doi:10.1093/annonc/mdw168
Pittet, M. J., Michielin, O., and Migliorini, D. (2022). Clinical relevance of tumour-associated macrophages. Nat. Rev. Clin. Oncol. 19 (6), 402–421. doi:10.1038/s41571-022-00620-6
Purcell, J. W., Tanlimco, S. G., Hickson, J., Fox, M., Sho, M., Durkin, L., et al. (2018). LRRC15 is a novel mesenchymal protein and stromal target for antibody-drug conjugates. Cancer Res. 78 (14), 4059–4072. doi:10.1158/0008-5472.CAN-18-0327
Pure, E., and Blomberg, R. (2018). Pro-tumorigenic roles of fibroblast activation protein in cancer: back to the basics. Oncogene 37 (32), 4343–4357. doi:10.1038/s41388-018-0275-3
Qi, X. S., Pajonk, F., McCloskey, S., Low, D. A., Kupelian, P., Steinberg, M., et al. (2017). Radioresistance of the breast tumor is highly correlated to its level of cancer stem cell and its clinical implication for breast irradiation. Radiother. Oncol. 124 (3), 455–461. doi:10.1016/j.radonc.2017.08.019
Qin, P., Chen, H., Wang, Y., Huang, L., Huang, K., Xiao, G., et al. (2023). Cancer-associated fibroblasts undergoing neoadjuvant chemotherapy suppress rectal cancer revealed by single-cell and spatial transcriptomics. Cell Rep. Med. 4 (10), 101231. doi:10.1016/j.xcrm.2023.101231
Qin, X., Lv, X., Li, P., Yang, R., Xia, Q., Chen, Y., et al. (2020). Matrix stiffness modulates ILK-mediated YAP activation to control the drug resistance of breast cancer cells. Biochimica Biophysica Acta Mol. Basis Dis. 1866 (3), 165625. doi:10.1016/j.bbadis.2019.165625
Raez, L. E., Papadopoulos, K., Ricart, A. D., Chiorean, E. G., Dipaola, R. S., Stein, M. N., et al. (2013). A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 71 (2), 523–530. doi:10.1007/s00280-012-2045-1
Randic, T., Magni, S., Philippidou, D., Margue, C., Grzyb, K., Preis, J. R., et al. (2023). Single-cell transcriptomics of NRAS-mutated melanoma transitioning to drug resistance reveals P2RX7 as an indicator of early drug response. Cell Rep. 42 (7), 112696. doi:10.1016/j.celrep.2023.112696
Rannikko, J. H., Turpin, R., Bostrom, P., Virtakoivu, R., Harth, C., Takeda, A., et al. (2025). Macrophage sensitivity to bexmarilimab-induced reprogramming is shaped by the tumor microenvironment. J. Immunother. Cancer 13 (5), e011292. doi:10.1136/jitc-2024-011292
Ravi, R., Noonan, K. A., Pham, V., Bedi, R., Zhavoronkov, A., Ozerov, I. V., et al. (2018). Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFβ enhance the efficacy of cancer immunotherapy. Nat. Commun. 9 (1), 741. doi:10.1038/s41467-017-02696-6
Rebelo, R., Xavier, C. P. R., Giovannetti, E., and Vasconcelos, M. H. (2023). Fibroblasts in pancreatic cancer: molecular and clinical perspectives. Trends Mol. Med. 29 (6), 439–453. doi:10.1016/j.molmed.2023.03.002
Ries, C. H., Cannarile, M. A., Hoves, S., Benz, J., Wartha, K., Runza, V., et al. (2014). Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 25 (6), 846–859. doi:10.1016/j.ccr.2014.05.016
Rivas, E. I., Linares, J., Zwick, M., Gómez-Llonin, A., Guiu, M., Labernadie, A., et al. (2022). Targeted immunotherapy against distinct cancer-associated fibroblasts overcomes treatment resistance in refractory HER2+ breast tumors. Nat. Commun. 13 (1), 5310. doi:10.1038/s41467-022-32782-3
Rodriguez, A. B., and Engelhard, V. H. (2020). Insights into tumor-associated tertiary lymphoid structures: novel targets for antitumor immunity and cancer immunotherapy. Cancer Immunol. Res. 8 (11), 1338–1345. doi:10.1158/2326-6066.CIR-20-0432
Romani, P., Nirchio, N., Arboit, M., Barbieri, V., Tosi, A., Michielin, F., et al. (2022). Mitochondrial fission links ECM mechanotransduction to metabolic redox homeostasis and metastatic chemotherapy resistance. Nat. Cell Biol. 24 (2), 168–180. doi:10.1038/s41556-022-00843-w
Rothstein, D. M., and Sayegh, M. H. (2003). T-cell costimulatory pathways in allograft rejection and tolerance. Immunol. Rev. 196, 85–108. doi:10.1046/j.1600-065x.2003.00088.x
Sammut, S. J., Crispin-Ortuzar, M., Chin, S. F., Provenzano, E., Bardwell, H. A., Ma, W., et al. (2022). Multi-omic machine learning predictor of breast cancer therapy response. Nature 601 (7894), 623–629. doi:10.1038/s41586-021-04278-5
Sekulic, A., Migden, M. R., Oro, A. E., Dirix, L., Lewis, K. D., Hainsworth, J. D., et al. (2012). Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 366 (23), 2171–2179. doi:10.1056/NEJMoa1113713
Sennino, B., and McDonald, D. M. (2012). Controlling escape from angiogenesis inhibitors. Nat. Rev. Cancer 12 (10), 699–709. doi:10.1038/nrc3366
Shan, G., Gu, J., Zhou, D., Li, L., Cheng, W., Wang, Y., et al. (2020). Cancer-associated fibroblast-secreted exosomal miR-423-5p promotes chemotherapy resistance in prostate cancer by targeting GREM2 through the TGF-beta signaling pathway. Exp. Mol. Med. 52 (11), 1809–1822. doi:10.1038/s12276-020-0431-z
Sharma, A., Seow, J. J. W., Dutertre, C. A., Pai, R., Bleriot, C., Mishra, A., et al. (2020). Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell 183 (2), 377–394. doi:10.1016/j.cell.2020.08.040
Shen, Y. A., Hong, J., Asaka, R., Asaka, S., Hsu, F. C., Suryo Rahmanto, Y., et al. (2020). Inhibition of the MYC-regulated glutaminase metabolic axis is an effective synthetic lethal approach for treating chemoresistant ovarian cancers. Cancer Res. 80 (20), 4514–4526. doi:10.1158/0008-5472.CAN-19-3971
Sherman, R. M., and Salzberg, S. L. (2020). Pan-genomics in the human genome era. Nat. Rev. Genet. 21 (4), 243–254. doi:10.1038/s41576-020-0210-7
Shi, Q., Shen, Q., Liu, Y., Shi, Y., Huang, W., Wang, X., et al. (2022). Increased glucose metabolism in TAMs fuels O-GlcNAcylation of lysosomal Cathepsin B to promote cancer metastasis and chemoresistance. Cancer Cell 40 (10), 1207–1222.e10. doi:10.1016/j.ccell.2022.08.012
Shiao, S. L., Gouin, K. H., Ing, N., Ho, A., Basho, R., Shah, A., et al. (2024). Single-cell and spatial profiling identify three response trajectories to pembrolizumab and radiation therapy in triple negative breast cancer. Cancer Cell 42 (1), 70–84.e8. doi:10.1016/j.ccell.2023.12.012
Siddiqui, A., and Ceppi, P. (2020). A non-proliferative role of pyrimidine metabolism in cancer. Mol. Metab. 35, 100962. doi:10.1016/j.molmet.2020.02.005
Stylianopoulos, T., Munn, L. L., and Jain, R. K. (2018). Reengineering the physical microenvironment of tumors to improve drug delivery and efficacy: from mathematical modeling to bench to bedside. Trends Cancer 4 (4), 292–319. doi:10.1016/j.trecan.2018.02.005
Su, S., Chen, J., Yao, H., Liu, J., Yu, S., Lao, L., et al. (2018). CD10(+)GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 172 (4), 841–856. doi:10.1016/j.cell.2018.01.009
Suh, C. H., Kim, H. S., Jung, S. C., Choi, C. G., and Kim, S. J. (2018). 2-Hydroxyglutarate MR spectroscopy for prediction of isocitrate dehydrogenase mutant glioma: a systemic review and meta-analysis using individual patient data. Neuro Oncol. 20 (12), 1573–1583. doi:10.1093/neuonc/noy113
Sun, C., Wang, A., Zhou, Y., Chen, P., Wang, X., Huang, J., et al. (2023). Spatially resolved multi-omics highlights cell-specific metabolic remodeling and interactions in gastric cancer. Nat. Commun. 14 (1), 2692. doi:10.1038/s41467-023-38360-5
Takeyama, Y., Kato, M., Tamada, S., Azuma, Y., Shimizu, Y., Iguchi, T., et al. (2020). Myeloid-derived suppressor cells are essential partners for immune checkpoint inhibitors in the treatment of cisplatin-resistant bladder cancer. Cancer Lett. 479, 89–99. doi:10.1016/j.canlet.2020.03.013
Tapia, I. J., Perico, D., Wolos, V. J., Villaverde, M. S., Abrigo, M., Di Silvestre, D., et al. (2024). Proteomic characterization of a 3D HER2+ breast cancer model reveals the role of mitochondrial complex I in acquired resistance to trastuzumab. Int. J. Mol. Sci. 25 (13), 7397. doi:10.3390/ijms25137397
Tian, Y., Wang, Z., Liu, X., Duan, J., Feng, G., Yin, Y., et al. (2018). Prediction of chemotherapeutic efficacy in non-small cell lung cancer by serum metabolomic profiling. Clin. Cancer Res. 24 (9), 2100–2109. doi:10.1158/1078-0432.CCR-17-2855
Tran, E., Chinnasamy, D., Yu, Z., Morgan, R. A., Lee, C. C., Restifo, N. P., et al. (2013). Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J. Exp. Med. 210 (6), 1125–1135. doi:10.1084/jem.20130110
VanderWalde, A., Bellasea, S. L., Kendra, K. L., Khushalani, N. I., Campbell, K. M., Scumpia, P. O., et al. (2023). Ipilimumab with or without nivolumab in PD-1 or PD-L1 blockade refractory metastatic melanoma: a randomized phase 2 trial. Nat. Med. 29 (9), 2278–2285. doi:10.1038/s41591-023-02498-y
Vennin, C., Murphy, K. J., Morton, J. P., Cox, T. R., Pajic, M., and Timpson, P. (2018). Reshaping the tumor stroma for treatment of pancreatic cancer. Gastroenterology 154 (4), 820–838. doi:10.1053/j.gastro.2017.11.280
Vidal, J., Fernandez-Rodriguez, M. C., Casadevall, D., Garcia-Alfonso, P., Paez, D., Guix, M., et al. (2023). Liquid biopsy detects early molecular response and predicts benefit to first-line chemotherapy plus cetuximab in metastatic colorectal cancer: PLATFORM-B study. Clin. Cancer Res. 29 (2), 379–388. doi:10.1158/1078-0432.CCR-22-1696
Vira, D., Basak, S. K., Veena, M. S., Wang, M. B., Batra, R. K., and Srivatsan, E. S. (2012). Cancer stem cells, microRNAs, and therapeutic strategies including natural products. Cancer Metastasis Rev. 31 (3-4), 733–751. doi:10.1007/s10555-012-9382-8
Wang, H., Liang, Y., Liu, Z., Zhang, R., Chao, J., Wang, M., et al. (2024b). POSTN(+) cancer-associated fibroblasts determine the efficacy of immunotherapy in hepatocellular carcinoma. J. Immunother. Cancer 12 (7), e008721. doi:10.1136/jitc-2023-008721
Wang, L., Hu, Z., Zhang, W., Wang, Z., Cao, M., and Cao, X. (2025). Promoting macrophage phagocytosis of cancer cells for effective cancer immunotherapy. Biochem. Pharmacol. 232, 116712. doi:10.1016/j.bcp.2024.116712
Wang, L. C., Lo, A., Scholler, J., Sun, J., Majumdar, R. S., Kapoor, V., et al. (2014). Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2 (2), 154–166. doi:10.1158/2326-6066.CIR-13-0027
Wang, T., Fahrmann, J. F., Lee, H., Li, Y. J., Tripathi, S. C., Yue, C., et al. (2018). JAK/STAT3-Regulated fatty acid beta-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 27 (1), 1357–50 e5. doi:10.1016/j.cmet.2018.04.018
Wang, T., Tian, L., Wei, B., Li, J., Zhang, C., Long, R., et al. (2024a). Effect of fibroblast heterogeneity on prognosis and drug resistance in high-grade serous ovarian cancer. Sci. Rep. 14 (1), 26617. doi:10.1038/s41598-024-77630-0
Wang, X., Qiu, Z., Dong, W., Yang, Z., Wang, J., Xu, H., et al. (2022). S1PR1 induces metabolic reprogramming of ceramide in vascular endothelial cells, affecting hepatocellular carcinoma angiogenesis and progression. Cell Death Dis. 13 (9), 768. doi:10.1038/s41419-022-05210-z
Wang, X., Zhou, T., Yang, X., Cao, X., Jin, G., Zhang, P., et al. (2023). DDRGK1 enhances osteosarcoma chemoresistance via inhibiting KEAP1-Mediated NRF2 ubiquitination. Adv. Sci. (Weinh) 10 (14), e2204438. doi:10.1002/advs.202204438
Wang, Z., Chen, J., Yang, L., Cao, M., Yu, Y., Zhang, R., et al. (2020). Single-cell sequencing-enabled hexokinase 2 assay for noninvasive bladder cancer diagnosis and screening by detecting rare malignant cells in urine. Anal. Chem. 92 (24), 16284–16292. doi:10.1021/acs.analchem.0c04282
Wehrli, M., Guinn, S., Birocchi, F., Kuo, A., Sun, Y., Larson, R. C., et al. (2024). Mesothelin CAR T cells secreting Anti-FAP/Anti-CD3 molecules efficiently target pancreatic adenocarcinoma and its stroma. Clin. Cancer Res. 30 (9), 1859–1877. doi:10.1158/1078-0432.CCR-23-3841
Weichenhan, D., Lipka, D. B., Lutsik, P., Goyal, A., and Plass, C. (2022). Epigenomic technologies for precision oncology. Semin. Cancer Biol. 84, 60–68. doi:10.1016/j.semcancer.2020.08.004
Wesolowski, R., Sharma, N., Reebel, L., Rodal, M. B., Peck, A., West, B. L., et al. (2019). Phase Ib study of the combination of pexidartinib (PLX3397), a CSF-1R inhibitor, and paclitaxel in patients with advanced solid tumors. Ther. Adv. Med. Oncol. 11, 1758835919854238. doi:10.1177/1758835919854238
Wishart, D. S. (2019). Metabolomics for investigating physiological and pathophysiological processes. Physiol. Rev. 99 (4), 1819–1875. doi:10.1152/physrev.00035.2018
Wu, H., Wang, T., Liu, Y., Li, X., Xu, S., Wu, C., et al. (2020). Mitophagy promotes sorafenib resistance through hypoxia-inducible ATAD3A dependent Axis. J. Exp. Clin. Cancer Res. 39 (1), 274. doi:10.1186/s13046-020-01768-8
Wu, H., Yang, J., Yang, Z., Xiao, Y., Liu, R., Jia, J., et al. (2025). Targeting the BCKDK/BCLAF1/MYC/HK2 axis to alter aerobic glycolysis and overcome Trametinib resistance in lung cancer. Cell Death Differ. doi:10.1038/s41418-025-01531-6
Wu, P., Gao, W., Su, M., Nice, E. C., Zhang, W., Lin, J., et al. (2021a). Adaptive mechanisms of tumor therapy resistance driven by tumor microenvironment. Front. Cell Dev. Biol. 9, 641469. doi:10.3389/fcell.2021.641469
Wu, S., Fukumoto, T., Lin, J., Nacarelli, T., Wang, Y., Ong, D., et al. (2021b). Targeting glutamine dependence through GLS1 inhibition suppresses ARID1A-inactivated clear cell ovarian carcinoma. Nat. Cancer 2 (2), 189–200. doi:10.1038/s43018-020-00160-x
Wu, S., Huang, L., Shen, R., Bernard-Cacciarella, M., Zhou, P., Hu, C., et al. (2020). Drug resistance-related sunitinib sequestration in autophagolysosomes of endothelial cells. Int. J. Oncol. 56 (1), 113–122. doi:10.3892/ijo.2019.4924
Xiang, H., Ramil, C. P., Hai, J., Zhang, C., Wang, H., Watkins, A. A., et al. (2020). Cancer-associated fibroblasts promote immunosuppression by inducing ROS-generating monocytic MDSCs in lung squamous cell carcinoma. Cancer Immunol. Res. 8 (4), 436–450. doi:10.1158/2326-6066.CIR-19-0507
Xie, M., Lin, Z., Ji, X., Luo, X., Zhang, Z., Sun, M., et al. (2023). FGF19/FGFR4-mediated elevation of ETV4 facilitates hepatocellular carcinoma metastasis by upregulating PD-L1 and CCL2. J. Hepatol. 79 (1), 109–125. doi:10.1016/j.jhep.2023.02.036
Xiong, Z., Chan, S. L., Zhou, J., Vong, J. S. L., Kwong, T. T., Zeng, X., et al. (2023). Targeting PPAR-gamma counteracts tumour adaptation to immune-checkpoint blockade in hepatocellular carcinoma. Gut 72 (9), 1758–1773. doi:10.1136/gutjnl-2022-328364
Yan, Y., Sun, D., Hu, J., Chen, Y., Sun, L., Yu, H., et al. (2025). Multi-omic profiling highlights factors associated with resistance to immuno-chemotherapy in non-small-cell lung cancer. Nat. Genet. 57 (1), 126–139. doi:10.1038/s41588-024-01998-y
Yang, E., Wang, X., Gong, Z., Yu, M., Wu, H., and Zhang, D. (2020). Exosome-mediated metabolic reprogramming: the emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct. Target Ther. 5 (1), 242. doi:10.1038/s41392-020-00359-5
Yang, J. C., Lee, D. H., Lee, J. S., Fan, Y., de Marinis, F., Iwama, E., et al. (2024a). Phase III KEYNOTE-789 study of pemetrexed and platinum with or without pembrolizumab for tyrosine kinase inhibitor‒Resistant, EGFR-Mutant, metastatic nonsquamous non-small cell lung cancer. J. Clin. Oncol. 42 (34), 4029–4039. doi:10.1200/JCO.23.02747
Yang, L., Venneti, S., and Nagrath, D. (2017). Glutaminolysis: a hallmark of cancer metabolism. Annu. Rev. Biomed. Eng. 19, 163–194. doi:10.1146/annurev-bioeng-071516-044546
Yang, M., Li, J., Gu, P., and Fan, X. (2021). The application of nanoparticles in cancer immunotherapy: targeting tumor microenvironment. Bioact. Mater 6 (7), 1973–1987. doi:10.1016/j.bioactmat.2020.12.010
Yang, X., Hu, X., Yin, J., Li, W., Fu, Y., Yang, B., et al. (2024c). Comprehensive multi-omics analysis reveals WEE1 as a synergistic lethal target with hyperthermia through CDK1 super-activation. Nat. Commun. 15 (1), 2089. doi:10.1038/s41467-024-46358-w
Yang, X., Lin, Y., Shi, Y., Li, B., Liu, W., Yin, W., et al. (2016). FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res. 76 (14), 4124–4135. doi:10.1158/0008-5472.CAN-15-2973
Yang, Z., Guan, F., Bronk, L., and Zhao, L. (2024b). Multi-omics approaches for biomarker discovery in predicting the response of esophageal cancer to neoadjuvant therapy: a multidimensional perspective. Pharmacol. Ther. 254, 108591. doi:10.1016/j.pharmthera.2024.108591
Yoo, C. B., and Jones, P. A. (2006). Epigenetic therapy of cancer: past, present and future. Nat. Rev. Drug Discov. 5 (1), 37–50. doi:10.1038/nrd1930
Yoo, Y. A., Quan, S., Yang, W., Guo, Q., Rodriguez, Y., Chalmers, Z. R., et al. (2024). Asparagine dependency is a targetable metabolic vulnerability in TP53-Altered castration-resistant prostate cancer. Cancer Res. 84 (18), 3004–3022. doi:10.1158/0008-5472.CAN-23-2910
Yoshida, G. J. (2015). Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 34, 111. doi:10.1186/s13046-015-0221-y
Yu, J., Green, M. D., Li, S., Sun, Y., Journey, S. N., Choi, J. E., et al. (2021). Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat. Med. 27 (1), 152–164. doi:10.1038/s41591-020-1131-x
Yu, Y., You, Y., Duan, Y., Kang, M., Zhou, B., Yang, J., et al. (2025). Multiomics approaches for identifying the PANoptosis signature and prognostic model via a multimachine-learning computational framework for intrahepatic cholangiocarcinoma. Hepatology. doi:10.1097/hep.0000000000001352
Zhang, C., Oberoi, P., Oelsner, S., Waldmann, A., Lindner, A., Tonn, T., et al. (2017). Chimeric antigen receptor-engineered NK-92 cells: an off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front. Immunol. 8, 533. doi:10.3389/fimmu.2017.00533
Zhang, J., Song, J., Tang, S., Zhao, Y., Wang, L., Luo, Y., et al. (2023a). Multi-omics analysis reveals the chemoresistance mechanism of proliferating tissue-resident macrophages in PDAC via metabolic adaptation. Cell Rep. 42 (6), 112620. doi:10.1016/j.celrep.2023.112620
Zhang, L., Lin, Y., Hu, L., Wang, Y., Hu, C., Shangguan, X., et al. (2025b). Transient intracellular expression of PD-L1 and VEGFR2 bispecific nanobody in cancer cells inspires long-term T cell activation and infiltration to combat tumor and inhibit cancer metastasis. Mol. Cancer 24 (1), 119. doi:10.1186/s12943-025-02253-6
Zhang, L., Xu, J., Zhou, S., Yao, F., Zhang, R., You, W., et al. (2024). Endothelial DGKG promotes tumor angiogenesis and immune evasion in hepatocellular carcinoma. J. Hepatol. 80 (1), 82–98. doi:10.1016/j.jhep.2023.10.006
Zhang, L., Zhao, J., Su, C., Wu, J., Jiang, L., Chi, H., et al. (2025a). Organoid models of ovarian cancer: resolving immune mechanisms of metabolic reprogramming and drug resistance. Front. Immunol. 16, 1573686. doi:10.3389/fimmu.2025.1573686
Zhang, Z., Yu, Y., Zhang, Z., Li, D., Liang, Z., Wang, L., et al. (2023b). Cancer-associated fibroblasts-derived CXCL12 enhances immune escape of bladder cancer through inhibiting P62-mediated autophagic degradation of PDL1. J. Exp. Clin. Cancer Res. 42 (1), 316. doi:10.1186/s13046-023-02900-0
Zhong, Y., Geng, F., Mazik, L., Yin, X., Becker, A. P., Mohammed, S., et al. (2024). Combinatorial targeting of glutamine metabolism and lysosomal-based lipid metabolism effectively suppresses glioblastoma. Cell Rep. Med. 5 (9), 101706. doi:10.1016/j.xcrm.2024.101706
Zhou, C., Li, W., Liang, Z., Wu, X., Cheng, S., Peng, J., et al. (2024). Mutant KRAS-activated circATXN7 fosters tumor immunoescape by sensitizing tumor-specific T cells to activation-induced cell death. Nat. Commun. 15 (1), 499. doi:10.1038/s41467-024-44779-1
Zhou, M., Zhou, Y., Liao, H., Rowland, B. C., Kong, X., Arvold, N. D., et al. (2018). Diagnostic accuracy of 2-hydroxyglutarate magnetic resonance spectroscopy in newly diagnosed brain mass and suspected recurrent gliomas. Neuro Oncol. 20 (9), 1262–1271. doi:10.1093/neuonc/noy022
Glossary
ABC ATP-binding cassette
AR androgen receptor
ATAC-seq transposase-accessible chromatin using sequencing
CAF Cancer-associated fibroblast
ChIP-seq chromatin immunoprecipitation sequencing
CSCs cancer stem sells
CTL cytotoxic T lymphocyte
CTLA-4 Cytotoxic T-lymphocyte–associated protein 4
CPT1B carnitine palmitoyltransferase 1B
D-2-HG D-2-hydroxyglutarate
ECM Extracellular matrix
EGFR Epidermal growth factor receptor
EMT epithelial-mesenchymal transition
EVs extracellular vesicles
FAO Fatty acid oxidation
GM-CSF Granulocyte-macrophage colony-stimulating factor
HCC hepatocellular carcinoma
HK2 hexokinase-2
ICIs Immune checkpoint inhibitors
LC-MS/MS liquid chromatography-tandem mass spectrometry
MIF migration inhibitory factor
MALDI-MSI matrix-assisted laser desorption/ionization mass spectrometry imaging
MDSC Myeloid-derived suppressor cell
NMR Nuclear magnetic resonance
NK cell Natural killer cell
NMR nuclear magnetic resonance
NSCLC Non-small cell lung cancer
PD-1 Programmed cell death protein 1
PD-L1 Programmed death-ligand 1
PDGF Platelet-derived growth factor
PDAC pancreatic ductal adenocarcinoma
PTMs post-translational modifications
P-gp P-glycoprotein
PFK1 phosphofructokinase-1
PK pyruvate kinase
scRNA-seq Single-cell RNA sequencing
TAM Tumor-associated macrophage
TCA Tricarboxylic acid cycle
TGF-β Transforming growth factor-beta
TLS tertiary lymphoid structures
TMB High tumor mutation burden
TNBC triple-negative breast cancer
TME tumor microenvironment
Treg Regulatory T cell
VEGF Vascular endothelial growth factor
WES whole-exome sequencing
WGS whole-genome sequencing
ZEB2 Zinc Finger E-Box-Binding Homeobox 2
2-DG 2-deoxy-D-glucose
Keywords: tumor microenvironment, drug resistance, multi-omics, therapeutic strategies, drug delivery, immune evasion
Citation: Chen F, Fu Y, Bai G, Qiu J and Hua K (2025) Multi-omics dissection of tumor microenvironment-mediated drug resistance: mechanisms and therapeutic reprogramming. Front. Pharmacol. 16:1634413. doi: 10.3389/fphar.2025.1634413
Received: 24 May 2025; Accepted: 24 June 2025;
Published: 07 July 2025.
Edited by:
Hailin Tang, Sun Yat-sen University Cancer Center (SYSUCC), ChinaReviewed by:
Wei Du, The First People’s Hospital of Changde City, ChinaXinjun Lu, Sun Yat-sen University, China
Huimig Chen, University of South China, China
Copyright © 2025 Chen, Fu, Bai, Qiu and Hua. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junjun Qiu, cWl1X2p1bmp1bkBmdWRhbi5lZHUuY24=; Keqin Hua, aHVha2VxaW5AZnVkYW4uZWR1LmNu
†These authors have contributed equally to this work