 Rocío Rosas-Alonso1,2*
Rocío Rosas-Alonso1,2* Nuria Rodríguez Salas3,4Pablo Perez Wert4Angela Hoyo5
Nuria Rodríguez Salas3,4Pablo Perez Wert4Angela Hoyo5 Susana Martin-López2Daniel Martínez-Pérez4Iciar Ruiz-Gutiérrez4Diego Jiménez-Bou4Jesús Peña4Pedro Arias2Ana Custodio3,4
Susana Martin-López2Daniel Martínez-Pérez4Iciar Ruiz-Gutiérrez4Diego Jiménez-Bou4Jesús Peña4Pedro Arias2Ana Custodio3,4 Itsaso Losantos-García6
Itsaso Losantos-García6 Alberto M. Borobia2,7Jaime Feliu3,4,8Ismael Ghanem3,4*
Alberto M. Borobia2,7Jaime Feliu3,4,8Ismael Ghanem3,4*- 1Biomarkers and Experimental Therapeutics in Cancer, La Paz University Hospital Research Institute (IdiPAZ), Madrid, Spain
- 2Pharmacogenetic Unit, La Paz University Hospital, Madrid, Spain
- 3Translational Oncology, La Paz University Hospital Research Institute (IdiPAZ), Madrid, Spain
- 4Medical Oncology Department, La Paz University Hospital, Madrid, Spain
- 5Pharmacy Department, La Paz University Hospital, Madrid, Spain
- 6Biostatistics Platform. La Paz University Hospital Research Institute (IdiPAZ), Madrid, Spain
- 7Clinical Pharmacology, La Paz University Hospital Research Institute (IdiPAZ), Madrid, Spain
- 8Biomedical Research Networking Center on Oncology-CIBERONC, ISCIII, Madrid, Spain
Introduction: Fluoropyrimidines (FP) are the mainstay of colorectal cancer (CRC) treatment, but can cause severe toxicity in up to 40% of patients. Variants in the DPYD gene are associated with these adverse events. A DPYD-guide dose adjustment is now recommended in Europe. This ambispective study aims to analyze the FP-related severe toxicity and the FP dose adjustment in heterozygous DPYD variant carriers with colorectal cancer, comparing a DPYD-guided FP dose adjustment (DA) approach to the non-DPYD-guided FP dose adjustment (NDA).
Methods: 1.279 DPYD genotyping reports were issued. Sixty patients were identified with DPYD variants. Twenty-five CRC patients (17 in the DA cohort and 8 in the NDA cohort) were included in the analysis. Thirty-five patients were excluded from the analysis because they did not satisfy any of the study’s inclusion criteria. Reasons for exclusion included having a diagnosis other than colorectal cancer, not receiving fluoropyrimidine treatment, participation in a clinical trial, or insufficient follow-up.
Results: Of the twenty-five patients included, sixteen patients (94%) started with a 50% FP dose reduction in the DA cohort while 7 out of 8 patients (87%) in the NDA cohort received 100% of dose in cycle 1. In the DA cohort, 12% of patients experienced severe fluoropyrimidines-related adverse events, compared to 50% in the NDA cohort (OR = 0.13; 95%CI: 0.01–0.93; p = 0.05). FP discontinuation due to severe toxicity occurred in 6% of patients in the DA cohort versus 50% in the NDA cohort (OR = 0.06; 95%CI: 0.003–0.55, p = 0.02).
Discussion: These findings suggest that DPYD-guided dose adjustment significantly reduces both the incidence of severe toxicity and the rate of treatment discontinuation in CRC patients. Initiating treatment with a 50% FP reduction allows for dose escalation in patients who exhibit good tolerance and avoid the discontinuation for those patients intolerant to higher doses thereby improving overall treatment adherence and completion.
1 Introduction
Colorectal cancer (CRC) ranks as the third most common neoplasm globally and stands as the second leading cause of cancer-related mortality (Bray et al., 2024). Chemotherapy based on fluoropyrimidine (FP) is widely utilized for various tumor types, forming a cornerstone in the treatment of gastrointestinal tumors, particularly CRC. FP can be administered as monotherapy or in combination with other agents such as platinum compounds, irinotecan, taxanes, or monoclonal antibodies (Cervantes et al., 2023; Argiles et al., 2020).
Severe toxicity is observed in up to 40% of patients undergoing FP chemotherapy, with mortality rates from adverse events ranging from 0.5% to 1% (Levy et al., 1998; Hoff et al., 2001). Certain clinical characteristics such as an older age, or female sex may be associated with a higher risk of severe toxicity (Knikman et al., 2021). In addition, the critical enzyme in the metabolism and inactivation of FP is dihydropyrimidine dehydrogenase (DPD). Variants in the DPYD gene, which encodes DPD, are key determinants of reduced enzyme activity. A deficiency in DPD function is present in 39%–61% of patients experiencing severe FP toxicity (van Kuilenburg, 2004). Therefore, DPYD variants significantly increase the risk of severe toxicity at standard doses (Rosmarin et al., 2014; Ruzzo et al., 2017; Meulendijks et al., 2015; Henricks et al., 2018). However, a large recent meta-analysis reported that among patients developing grade 3–4-5 hematologic and digestive fluoropyrimidine-related toxicities (no dose adjustment on DPYD), only 14% and 7% carried at least one *2A, *13 or c.2846A>T allele, or c.1236G>A (haplotype B3) allele, respectively (Le et al., 2024).
In a prospective study, Henricks, L. M. et al. Demonstrated that an initial 50% dose reduction of FP may be sufficient for heterozygous carriers of the nonfunctional DPYD*2A and DPYD*13 variants. However, due to the low prevalence of these variants, the study included only 16 patients heterozygous for DPYD*2A and 1 patient heterozygous for DPYD*13. For variants associated with reduced function (c.2846A>T and c.1236G>A), a 25% dose reduction was considered potentially insufficient, necessitating further investigation (Henricks et al., 2018).
Based on the findings of increased risk of severe toxicity with these variants (Rosmarin et al., 2014; Meulendijks et al., 2015; Terrazzino et al., 2013), several clinical guidelines recommend performing DPYD genotyping analysis in all patients prior to starting FP treatment and adjusting the dose if a deficiency is detected (Amstutz et al., 2018; Lunenburg et al., 2020; Garcia-Alfonso et al., 2022). However, these guidelines differ in their recommendations for initial dose adjustments and do not provide detailed guidance on increasing FP doses based on specific DPYD variants.
Furthermore, numerous studies have shown that DPYD variant carriers receiving standard treatment have a higher risk of severe adverse events compared to the wild-type population (Rosmarin et al., 2014; Ruzzo et al., 2017), even after genotype-guided dose adjustments (Henricks et al., 2018; Deenen et al., 2016). However, there are no studies directly comparing the benefits in terms of toxicity of FP treatment in DPYD carries with or without DPYD-guided dose adjustments according to current recommendations, specifically in CRC patients.
Consequently, additional research is imperative to elucidate the clinical management of unresolved questions concerning DPYD-guided FP dose adjustments.
The aim of our study is to analyze the FP toxicity profile in routine clinical practice, as well as the dose FP adjustment dynamics in heterozygous patients carrying any of the four classical DPYD variants, depending on whether they received a DPYD-guided FP dose adjustment or no adjustment.
2 Materials and methods
2.1 Study population
The inclusion criteria of the study were: patients diagnosed with colorectal cancer (CRC) at Hospital Universitario La Paz; identification of a DPYD gene variant by genotyping between 1 June 2020, and 28 June 2024; initiation of first-time treatment with fluoropyrimidines (FP), either before or after DPYD genotyping, administered as monotherapy or in combination with oxaliplatin (with or without targeted therapies); treatment given in routine clinical practice as (neo)adjuvant therapy or first-line therapy for metastatic disease; to have provided informed consent for pharmacogenetic testing.
The exclusion criteria were: participation in a clinical trial; concurrent treatment with irinotecan; diagnosis of a non-colorectal malignancy; and a follow-up duration of less than 3 months.
2.2 Study design
This is a single-center, ambispective study conducted at Hospital Universitario La Paz. The study was conducted with the approval of the Ethics Committee of La Paz University Hospital (PI-5204) and in accordance with the principles outlined in the Declaration of Helsinki. Two cohorts were established: the DPYD-guided FP dose adjustment cohort (DA), consisting of patients who received a FP dose adjustment in cycle 1 for DPYD variant carriers, with subsequent dose escalation based on patient tolerance; and the non-DPYD-guided FP dose adjustment cohort (NDA), comprising patients who received an unadjusted DPYD-guide FP dose, higher than 75%, in the first cycle. Most of the patients in the NDA cohort were treated prior to the implementation of preemptive DPYD genotyping in our hospital (June 2020). These patients were subsequently genotyped due to relapse or second malignancy.
Clinical and genetic variables were collected. Clinical variables were extracted from patient medical records, encompassing a comprehensive array of data. These included sex, age, ECOG performance status, therapeutic intention and treatment regimen administered. Severe (≥ grade 3) FP-related adverse events (SFAEs) were evaluated according to the Common Terminology Criteria for Adverse Events (CTCAE) version 5.0 at cycle 1 and maximum toxicity at any cycle. Maximum FP dose, initial and final dose adjustments, and discontinuation of FP due to toxicity were also collected. To minimize bias from overestimating FP-related toxicity in patients receiving combination regimens, patients treated with irinotecan (associated with overlapping toxicities) were excluded, and only adverse events specifically attributable to FP were considered. FP-related adverse events were defined as follows: diarrhea, fatigue, emesis, hand-foot syndrome, neutropenia, thrombopenia, anemia, febrile neutropenia, mucositis and hyperbilirubinemia.
2.3 DNA extraction and genotyping
The DNA extraction and genotyping were performed during routine clinical practice in the Clinical Pharmacogenetics Unit, a multidisciplinary unit integrating the clinical pharmacology and genetics departments (Stewart et al., 2023; Borobia et al., 2018). For the genetic study, blood samples were collected in Vacutainer tubes containing EDTA anticoagulant (Becton Dickinson, Franklin Lakes, NJ, United States). DNA was then isolated in the preanalytical unit of the genetics department using the Chemagen robot (Perkin-Elmer, Boston, MA, United States). DPYD genotyping was performed in the pharmacogenetics laboratory using a validated OpenArray technology (ThermoFisher, Waltham, MA, United States) (Rosas-Alonso et al., 2021). Specifically, for the pharmacogenetics of FP, the four variants recommended by the Spanish Agency for Medicines and Medical Devices (AEMPS) were analyzed in a clinical setting: DPYD*2A (rs3918290, c.1905 + 1G>A), DPYD*13 (rs55886062, c.1679T>G), c.2846A>T (rs67376798) and [c.1236G>A; c.1129–5923C>G]/HapB3) (rs56038477 and rs75017182). Both HapB3 haplotype variants were analyzed to confirm linkage disequilibrium.
2.4 Treatment recommendations
Based on the genotype of both alleles for these variants, an activity score was assigned with a dose reduction recommendation according to the Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines (Amstutz et al., 2018). Thus, heterozygous c.2846A>T and c.1236G>A carriers (reduced function variants) were assigned an activity score of 1.5 and dose reductions of 25%–50% were recommended, depending on individual patient conditions (and oncologist decision). If the patient showed good clinical tolerance, in subsequent cycles (tipically the second), the dose was usually increased up to 75% at the discretion of the treating physician. For heterozygous DPYD*2A or DPYD*13 carriers (loss of function variants), an activity score of 1.0 was assigned and a dose reduction of 50% was recommended. No dose escalation was commonly performed in subsequent cycles even when clinical tolerance was adequate. However, doses could be adjusted according to individual patient circumstances and medical decisions. For the compound heterozygous carrier with an activity score of 1, the CPIC guideline does not provide specific recommendations; therefore, our recommendations were based on the DPWG guideline (Lunenburg et al., 2020), specifically, an additional phenotyping test measuring uracil levels to determine DPD enzyme activity was performed, as dose adjustments cannot be accurately predicted based on genotype alone according to the DPWG guideline. Relative dose intensity was calculated as the dose administered divided by the standard dose for the regimen administered.
2.5 Statistical analysis
Qualitative variables were presented as absolute frequencies and percentages. Quantitative variables were summarized using means or medians, depending on their distribution. The normality of continuous variables was assessed using the Shapiro-Wilk test. Associations between categorical variables were analyzed using binary logistic regression (severe toxicity and treatment discontinuation), Fisher’s test (qualitative baseline characteristics) and t-Student (age). All statistical tests were two-sided, with p-values ≤0.05 considered statistically significant. Data analysis was performed using R statistical software, version 4.3.3 (de With et al., 2023).
3 Results
3.1 Characteristics of patients
From 1 June 2020 to 28 June 2024 a total of 1,279 DPYD genotyping reports were issued. Among these, 60 patients (4.7%) were identified with DPYD variants (Supplementary Table S1). Specifically, 25 of these DPYD-variant carriers were diagnosed with CRC and received FP based chemotherapy. Thirty-five patients were excluded from the analysis because they did not meet any of the study inclusion criteria. Seventeen patients (68%) were included in the DA cohort while 8 were included in the NDA cohort (Figure 1).
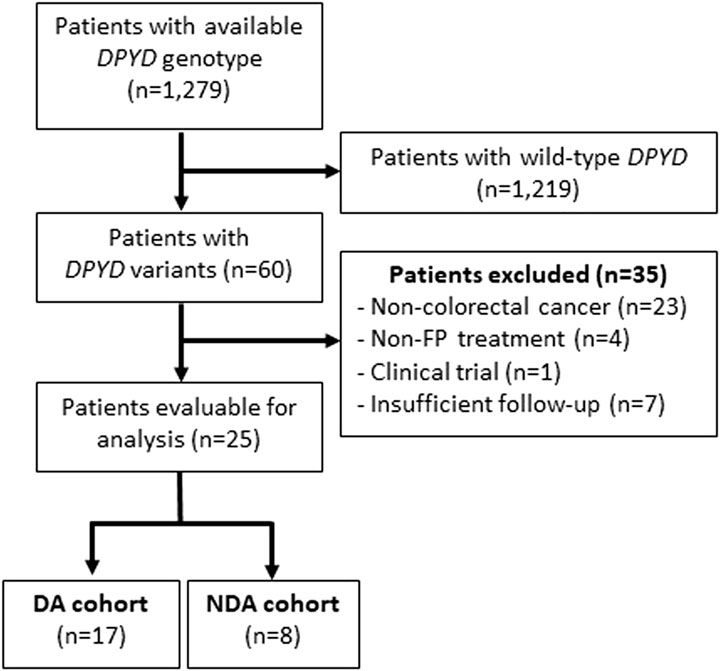
Figure 1. Flow diagram for the inclusion of patients DPYD: dihydropyrimidine dehydrogenase; FP: Fluoropyrimidines; AD: DPYD-guided FP dose adjustment; NDA: Non-DPYD-guided FP dose adjustment.
The baseline characteristics of the patients are presented in Table 1, showing a generally well-balanced distribution between the two cohorts, with the exception of sex (65% female in the DA cohort compared to 12% in the NDA cohort). Most patients exhibited an ECOG performance status of 0–1, with 88% in the DA cohort and 100% in the NDA cohort. All patients in the study received chemotherapy regimens consisting of FP, either as monotherapy or in combination with oxaliplatin. Additionally, six patients (24%) were treated with bevacizumab or anti-EGFR antibodies, and only one patient received concurrent radiotherapy with capecitabine. More than half of the patients carried the c.1236G>A variant, with prevalence rates of 65% and 50% in the DA and NDA cohorts, respectively. There was one patient who was compound heterozygous for both c.1236G>A and c.2846A>T.
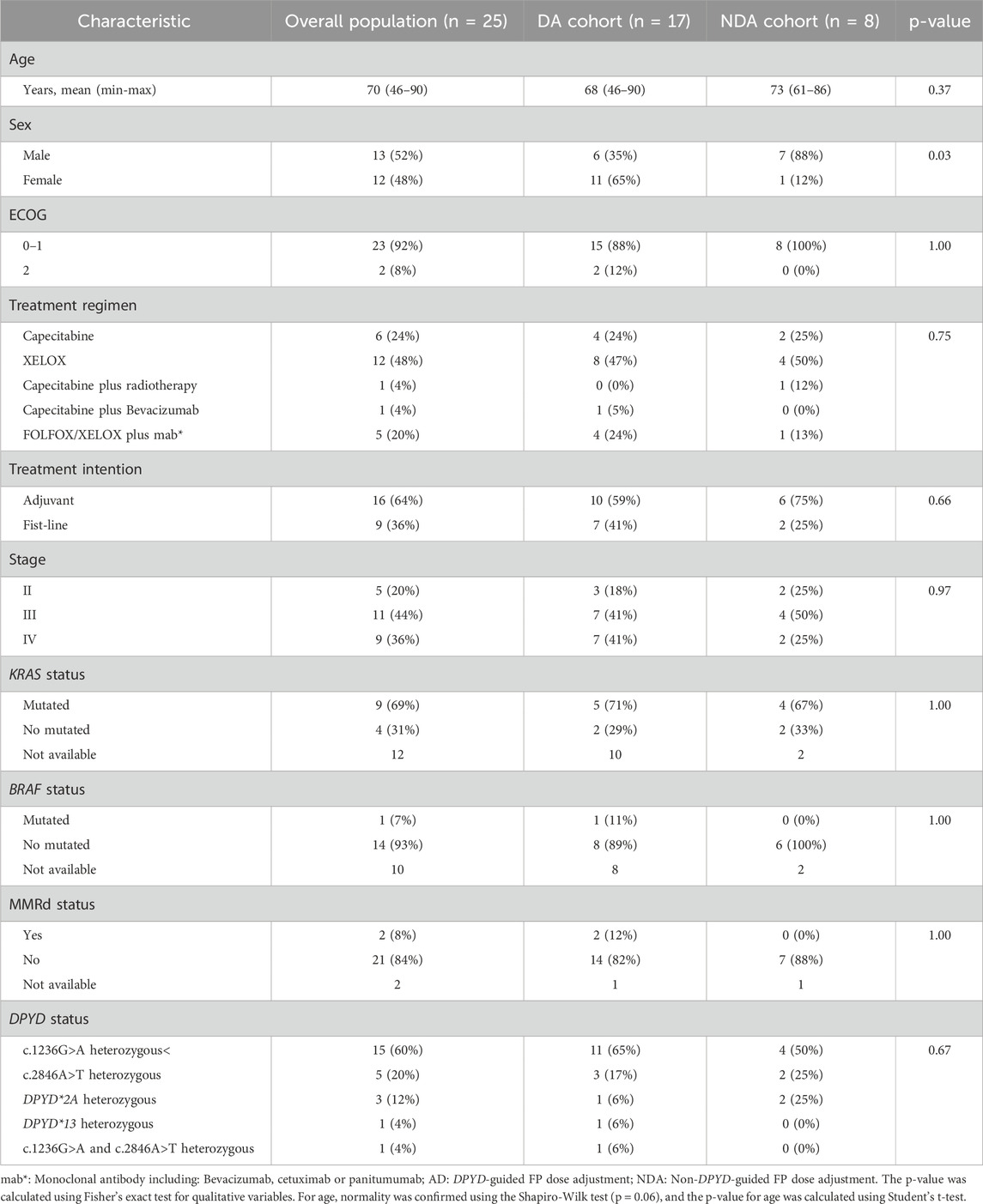
Table 1. Baseline characteristics of patients in the DA and NDA cohorts.
3.2 Severe fluoropyrimidine related toxicity
Overall, 6 patients (24%) experienced SFAEs, 2 patients (12%) from the DA cohort and 4 patients (50%) from the NDA cohort (OR = 0.13; 95%CI: 0.01–0.93; p = 0.05) (Figure 2). Specifically, regarding the most frequent variant (c.1236G>A), there were no SFAEs in patients within the DA cohort, while 50% in the NDA cohort experienced SFAEs (Table 2). In the DA cohort, 6% of the patients were hospitalized due to treatment-related toxicities, whereas in the NDA cohort, the rate was 25%.
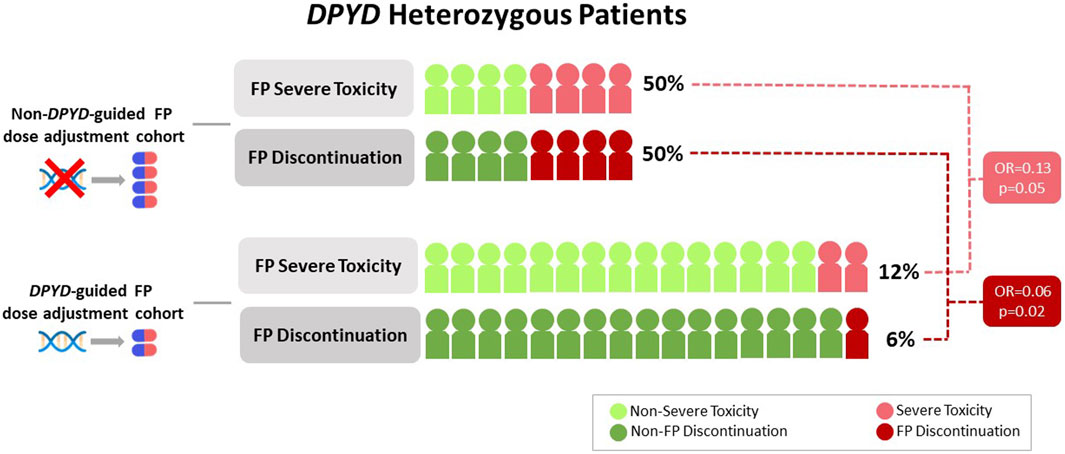
Figure 2. Severe fluoropyrimidine-related toxicity and treatment discontinuation in DA and NDA cohorts.
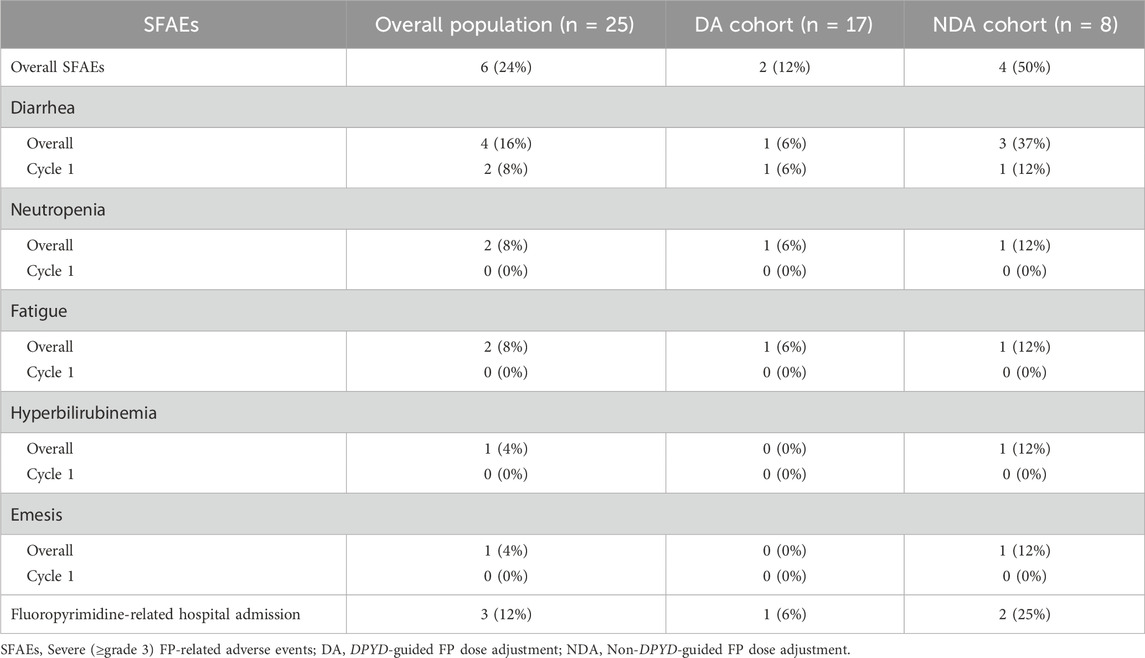
Table 2. Severe (≥ grade 3) FP-related adverse events in DA and NDA cohorts (n ≥ 1).
The most frequent SFAEs was diarrhea in 4 patients (16%). Other SFAEs were neutropenia (8%), fatigue (8%), hyperbilirubinemia (4%) and emesis (4%). All them were most frequent in the NDA cohort (Table 2). There were no deaths related to the FP administration.
The SFAEs according to the type of DPYD variant are presented in Table 3.
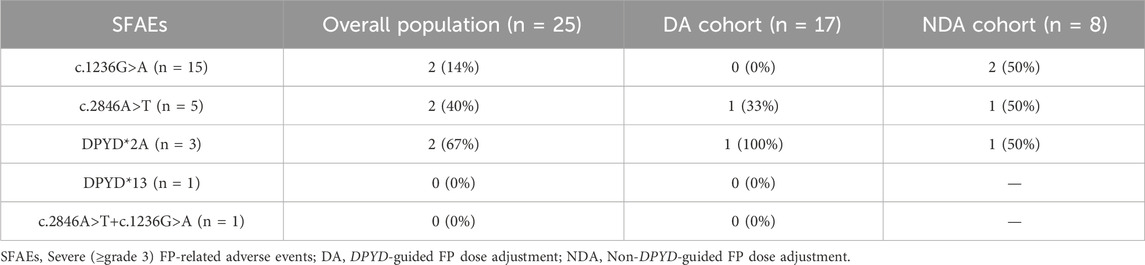
Table 3. Severe (≥ grade 3) FP-related adverse events in DA and NDA cohorts according to DPYD variants.
3.3 Fluoropyrimidine dose adjustment dynamics
Due to SFAEs, FP treatment was adjusted in 7 out of 8 patients (87%) in the NDA cohort. In the DA cohort, dose reduction was performed in only one case (6%), whereas for the remaining patients, the doses were either maintained or increased.
The FP dosing and modifications or dynamics of dose adjustments for the DA and NDA cohorts are summarized in Table 4. A normality test was conducted for the variables initial dose, maximum dose, and final dose using the Shapiro-Wilk test. In all cases, the p-value was <0.05, indicating that the data did not follow a normal distribution. In the DA cohort, the median relative dose intensity for the initial dose was 50%. Notably, 94% of patients received 50% of the standard dose, while one patient received a higher dose (62%). In the NDA cohort 7 out of 8 patients (87%) received 100% of standard dose in cycle 1. Regarding the maximum dose, the DA cohort had a median relative dose intensity of 70%, with 76% of patients receiving doses ranging from 50% to 75%. The NDA cohort had a median relative dose intensity of 100%, with 88% of patients receiving 100%.

Table 4. Fluoropyrimidine dose adjustment dynamics according to the DA and NDA cohorts.
For the final dose, the DA cohort had a median relative dose intensity of 70%, with 70% of patients receiving more than 50%–75%. The NDA cohort had a median relative dose intensity of 25%, with 63% of patients receiving 0%–50%.
FP discontinuation due to toxicity occurred in 6% of patients in the DA cohort and 50% of patients in the NDA cohort (OR = 0.06; 95%CI; 0.003–0.55, p = 0.02) (Figure 2). In the DA cohort, treatment discontinuation due to toxicity occurred after the third cycle. In the NDA cohort, treatment discontinuation occurred after the first, the second, the third and the fourth cycles.
In the subgroup of patients with decreased function DPYD variants, the initial dose in the DA cohort had a median relative dose intensity of 50%, with all patients receiving 50% of the dose. The NDA cohort had a median initial dose intensity of 100%, with all patients receiving doses greater than 75%. At the final dose, the DA cohort had a median intensity of 75%, whereas the NDA cohort had a median relative dose intensity of 37% (Supplementary Table S2). Additionally, 50% of the NDA cohort discontinued FP due to toxicity, compared to none in the DA cohort (p = 0.99).
Only four patients had DPYD variants with loss of function, two in the NDA cohort and two in the DA cohort. In the NDA cohort, both patients were heterozygous for the DPYD*2A variant. One patient discontinued treatment at cycle 2 after experiencing grade 3 diarrhea and grade 3 neutropenia, while the other patient presented with grade 2 hematological toxicity, requiring a 50% dose reduction to complete the treatment. In the DA cohort, one DPYD*2A carrier discontinued FP at the third cycle after experiencing grade 3 diarrhea, while the other patient, a DPYD*13 carrier, completed the treatment without severe adverse effects.
Finally, the compound heterozygous carrier for c.2846A>T and c.1236G>A completed the FP treatment, starting with a 50% dose reduction, increasing the dose to 66%. In this case, both genotyping and phenotyping were performed: while the genotype inferred an activity score of 1 or partial DPD deficiency, the phenotyping result indicated normal DPD activity. Given this discrepancy, we opted for the safest approach, managing the patient as an activity score 1 carrier.
4 Discussion
The implementation of pharmacogenetic testing for fluoropyrimidines has become a reality in Europe following the European Medicines Agency and European Society of Medical Oncology to recommend preemptive DPYD testing (de With et al., 2023; Abad-Santos et al., 2024). However, there remain unresolved issues regarding optimal dose adjustment and dose escalation.
Our study showed a prevalence of 4.7% of carriers, which is lower than the frequencies reported in other studies, which reached 8% (Henricks et al., 2018). However, this discrepancy may be due to a lower frequency of these variants in the Spanish population, with previous reports indicating frequencies below 5% (Miarons et al., 2023).
In our study, patients were treated using either a DPYD-guided FP dose adjustment strategy or a standard dose FP-based treatment. In the DA cohort, patients started with a median FP dose intensity of 50% (n = 17), whereas the NDA cohort received 100% (n = 8). Consequently, patients in the DA cohort experienced fewer SFAEs and were less likely to discontinue treatment.
Remarkably, the rate of severe toxicity in the NDA cohort (50%) was four times higher than that observed in the DA cohort (12%). A recently published study involving 27 patients, but not limited to CRC, also shows that DPYD genotype pretreatment testing reduced severe toxicities in DPYD variant carriers from 64% to 31% (Nguyen et al., 2024). The rate of severe toxicity in the NDA cohort was similar to that reported in previous studies (Rosmarin et al., 2014; Meulendijks et al., 2015; Terrazzino et al., 2013; Nguyen et al., 2024). However, the incidence of SFAEs for the DA cohort was substantially inferior to the 39% reported in the prospective Henricks, L. M. et al. Study, also evaluating the DPYD genotype-guided FP dose adjustment strategy (Henricks et al., 2018). These differences could be explained by the variability secondary to the low incidence of DPYD variants, the partially retrospective data collection that could underreport the toxicity in this work or the homogeneity and less toxic regimens and neoplasms in our study. However, the larger 50% dose reduction performed in the first cycle for decreased function DPYD variants in this study compared to 25% in the Henricks study seems the most consistent explanation. The overall dose escalation rate of 13% in the Dutch study does not justify its higher toxicity (Henricks et al., 2018). Other studies starting on a 50% dose reduction also showed lower severe toxicity rates of 20%–30% (Nguyen et al., 2024; Wang et al., 2022). Although there is a significantly higher proportion of males in the NDA cohort (p = 0.03), this fact does not seem to justify the differences in severe toxicity, since it is the female sex that could potentially be associated with a higher risk of toxicity (Knikman et al., 2021).
Another important question is whether the DPYD-guided reduction of FP doses could affect the prognosis of oncologic diseases, particularly in CRC. The Knikman study suggests that while no apparent loss of efficacy was observed in the overall pool of patients carrying DPYD variants, a loss of efficacy in carriers of the c.1236G>A variant compared to DPYD wild-type patients cannot be ruled out (Knikman et al., 2023). In our study, survival was not evaluated due to a potential selection bias, as the vast majority of patients in the NDA cohort were identified by DPYD analysis after relapse. However, when comparing the dynamics of FP dose administration in both cohorts, 50% of patients carrying DPYD variants treated without dose adjustment discontinued FP in the first cycles, whereas only 6% of patients following DPYD-guided FP dose adjustment had to discontinue FP due to SFAEs. This discontinuation rate is even lower than that reported for the wild-type DPYD population or for DPYD variant carriers treated with a more permissive FP dose reduction strategy, which showed discontinuation rates above 18% (Henricks et al., 2018). Additionally, in the DA cohort, the majority (82%) of patients received a dose greater than 50% in the last cycle, with a median relative dose intensity of 70%.
Therefore, the data from our study which exclusively involved CRC patients support that overall, the DPYD-guided dose adjustment strategy not only reduces the risk of toxicity but also reduces the risk of FP discontinuation, making it unlikely to result in a worse prognosis.
However, other questions remain open: how much to adjust the FP dose in the first cycle, whether dose escalation is indicated, or the rate of dose escalation for each DPYD variant to minimize the risk of toxicity while maintaining the maximum efficacy of FP (Henricks et al., 2018; Amstutz et al., 2018; Lunenburg et al., 2020; Knikman et al., 2023). Focusing on the DPYD variants with reduced function (c.2846A>T and c.1236G>A), our study suggests that initiating treatment with a 50% dose reduction, followed by an escalation up to 75% based on clinical tolerance, results in a marked decrease in the risk of severe toxicity, from 50% to less than 10%. This approach addresses the issue of insufficient decrease in the incidence of severe toxicity with an initial 25% dose reduction shown in the largest prospective study (Henricks et al., 2018). Furthermore, with this approach no patients had to discontinue FP treatment.
This is of particular interest when considering the prognostic uncertainty for patients with the c.1236G>A variant who were treated with an initial 75% dose of FP (Knikman et al., 2023). On the other hand, a less aggressive approach, starting at 50% of the standard dose without dose escalation could not add any benefit in tolerance and potentially lead to under-treatment for most patients (Wang et al., 2022). Our study also suggests the possibility of exploring a dose escalation beyond 75% for carriers of reduced function variants: 93% of patients were able to complete the treatment with a dose escalation at least up to 75%. A small study with 11 patients showed that a tolerance-based capecitabine dose escalation was possible in 4 patients (Kleinjan et al., 2019). However, in the Dutch study, three out of five patients with DPYD variants with reduced function had to reduce the FP dose after a dose escalation beyond 75% (Henricks et al., 2018).
Concerning DPYD variants with loss of function, the evidence is much more limited. The results of our study in only 4 patients (3 for DPYD*2A and 1 for c.1679T>G) showed that one patient in each cohort discontinued the treatment due to severe toxicity, regardless of the 50% dose adjustment. However, the largest prospective study evaluating the dose adjustment in DPYD*2A carriers (n = 22) showed that an initial 50% dose reduction was safe. In 3 patients (17%) a further reduction was necessary because of toxicity (Deenen et al., 2016). Another study with 17 DPYD variant carriers (16 for variant DPYD*2A and 1 for c.1679T>G) suggests that a 50% reduction is safe as well (Henricks et al., 2018).
Therefore, a treatment strategy that begins with lower doses followed by careful dose escalation could potentially identify patients with poor tolerance, preventing severe toxicity and ensuring treatment completion. Additionally, DPD may be an inducible enzyme (Yoshida et al., 2015), which could allow for higher doses to be reached with good tolerance. Hence, for heterozygous patients with loss-of-function DPYD variants an initial dose reduction of ≥50%, with subsequent dose escalation based on patient tolerance up to 50%. For heterozygous patients with reduced-function DPYD variants an initial dose reduction of 50%, followed by dose escalation based on patient tolerance up to 75%. While a higher limit may be tolerable for some patients, we believe that further research is necessary.
These proposed strategies are consistent with international pharmacogenetic recommendations, although some variability exists across clinical guidelines. The Spanish Pharmacogenetics and Pharmacogenomics Society, together with the Spanish Society of Medical Oncology, recommend initiating treatment at 50% of the standard dose, followed by dose titration according to toxicity or pharmacokinetic parameters (Garcia-Alfonso et al., 2022). Likewise, the DPWG advises starting therapy at 50% of the standard dose for patients with an activity score of 1 or 1.5, with subsequent adjustments based on observed toxicity and therapeutic effectiveness (Lunenburg et al., 2020). Both approaches are closely aligned with the strategy we propose. By comparison, the French National Network of Pharmacogenetics and the Italian Association of Medical Oncology propose a somewhat less cautious approach, recommending a 50% dose reduction for heterozygous carriers of *2A, *13, or c.2846A>T variants during the first cycle, while for carriers of c.1236G>A, initiation at 75% of the standard dose is advised (Quaranta and Thomas, 2017). Finally, the CPIC recommends a 50% dose reduction for activity score 1 carriers, whereas for those with an activity score of 1.5 the guidance is less specific, suggesting a reduction of between 25% and 50% depending on clinical circumstances.
This study has several limitations as a single-center ambispective study, potentially leading to underestimated data collection, particularly regarding toxicity. Additionally, the small sample size restricts the ability to draw definitive conclusions. Furthermore, we cannot draw conclusions about efficacy due to potential selection bias. In addition, the fact that the NDA cohort consisted primarily of patients with recurrent or secondary tumors could potentially influence the toxicity results. Nevertheless, the low prevalence of patients with DPYD variants underscores the significance of this study, as it addresses questions that had not been previously explored in the literature. Despite the rarity of these variants, our findings contribute valuable insights and help fill knowledge gaps regarding the clinical management and safety considerations for this specific patient population.
In conclusion, our study suggests that a more conservative DPYD-guided dose adjustment could enhance tolerance and decrease the likelihood of treatment discontinuation. This approach could potentially enable a better selection of patients intolerant to slightly higher doses, allowing them to complete treatment. However, further evidence is needed from larger studies in homogeneous populations.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Ethics Committee of La Paz University Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
RR-A: Writing – original draft, Formal Analysis, Conceptualization, Validation, Methodology, Project administration, Data curation. NS: Data curation, Methodology, Writing – review and editing. PW: Data curation, Writing – review and editing, Methodology. AH: Writing – review and editing, Methodology. SM-L: Writing – review and editing, Methodology. DM-P: Methodology, Writing – review and editing, Data curation. IR-G: Data curation, Writing – review and editing, Methodology. DJ-B: Writing – review and editing, Methodology, Data curation. JP: Methodology, Data curation, Writing – review and editing. PA: Writing – review and editing, Methodology. AC: Methodology, Data curation, Writing – review and editing. IL-G: Methodology, Writing – review and editing, Formal Analysis. AB: Data curation, Methodology, Writing – review and editing. JF: Data curation, Methodology, Writing – review and editing. IG: Writing – original draft, Formal Analysis, Data curation, Methodology, Conceptualization, Validation, Project administration.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We are indebted to the Biostatistics core facility of the Hospital La Paz Institute for Health Research (IdiPAZ) for the technical assistance.
Conflict of interest
IG reports a relationship with Servier, Merck, Amgen, MSD, AstraZeneca that includes: consulting or advisory and travel reimbursement.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2025.1645188/full#supplementary-material
References
Abad-Santos, F., Alino, S. F., Borobia, A. M., Garcia-Martin, E., Gasso, P., Maronas, O., et al. (2024). Developments in pharmacogenetics, pharmacogenomics, and personalized medicine. Pharmacol. Res. 200, 107061. doi:10.1016/j.phrs.2024.107061
Amstutz, U., Henricks, L. M., Offer, S. M., Barbarino, J., Schellens, J. H. M., Swen, J. J., et al. (2018). Clinical pharmacogenetics implementation consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin. Pharmacol. Ther. 103, 210–216. doi:10.1002/cpt.911
Argiles, G., Tabernero, J., Labianca, R., Hochhauser, D., Salazar, R., Iveson, T., et al. (2020). Localised colon cancer: ESMO clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 31, 1291–1305. doi:10.1016/j.annonc.2020.06.022
Borobia, A. M., Dapia, I., Tong, H. Y., Arias, P., Munoz, M., Tenorio, J., et al. (2018). Clinical implementation of pharmacogenetic testing in a Hospital of the Spanish national health System: strategy and experience over 3 years. Clin. Transl. Sci. 11, 189–199. doi:10.1111/cts.12526
Bray, F., Laversanne, M., Sung, H., Ferlay, J., Siegel, R. L., Soerjomataram, I., et al. (2024). Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263. doi:10.3322/caac.21834
Cervantes, A., Adam, R., Rosello, S., Arnold, D., Normanno, N., Taieb, J., et al. (2023). Metastatic colorectal cancer: ESMO clinical practice guideline for diagnosis, treatment and follow-up. Ann. Oncol. 34, 10–32. doi:10.1016/j.annonc.2022.10.003
de With, M., Sadlon, A., Cecchin, E., Haufroid, V., Thomas, F., Joerger, M., et al. (2023). Implementation of dihydropyrimidine dehydrogenase deficiency testing in Europe. ESMO Open 8, 101197. doi:10.1016/j.esmoop.2023.101197
Deenen, M. J., Meulendijks, D., Cats, A., Sechterberger, M. K., Severens, J. L., Boot, H., et al. (2016). Upfront genotyping of DPYD*2A to individualize fluoropyrimidine therapy: a safety and cost analysis. J. Clin. Oncol. 34, 227–234. doi:10.1200/JCO.2015.63.1325
Garcia-Alfonso, P., Saiz-Rodriguez, M., Mondejar, R., Salazar, J., Paez, D., Borobia, A. M., et al. (2022). Consensus of experts from the Spanish pharmacogenetics and pharmacogenomics Society and the Spanish Society of medical oncology for the genotyping of DPYD in cancer patients who are candidates for treatment with fluoropyrimidines. Clin. Transl. Oncol. 24, 483–494. doi:10.1007/s12094-021-02708-4
Henricks, L. M., Lunenburg, C., de Man, F. M., Meulendijks, D., Frederix, G. W. J., Kienhuis, E., et al. (2018). DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. Lancet Oncol. 19, 1459–1467. doi:10.1016/S1470-2045(18)30686-7
Hoff, P. M., Ansari, R., Batist, G., Cox, J., Kocha, W., Kuperminc, M., et al. (2001). Comparison of oral capecitabine versus intravenous fluorouracil plus leucovorin as first-line treatment in 605 patients with metastatic colorectal cancer: results of a randomized phase III study. J. Clin. Oncol. 19, 2282–2292. doi:10.1200/JCO.2001.19.8.2282
Kleinjan, J. P., Brinkman, I., Bakema, R., van Zanden, J. J., and van Rooijen, J. M. (2019). Tolerance-based capecitabine dose escalation after DPYD genotype-guided dosing in heterozygote DPYD variant carriers: a single-center observational study. Anticancer Drugs 30, 410–415. doi:10.1097/CAD.0000000000000748
Knikman, J. E., Gelderblom, H., Beijnen, J. H., Cats, A., Guchelaar, H. J., and Henricks, L. M. (2021). Individualized dosing of fluoropyrimidine-based chemotherapy to prevent severe fluoropyrimidine-related toxicity: what are the options? Clin. Pharmacol. Ther. 109, 591–604. doi:10.1002/cpt.2069
Knikman, J. E., Wilting, T. A., Lopez-Yurda, M., Henricks, L. M., Lunenburg, C., de Man, F. M., et al. (2023). Survival of patients with cancer with DPYD variant alleles and dose-individualized fluoropyrimidine Therapy-A matched-pair analysis. J. Clin. Oncol. 41, 5411–5421. doi:10.1200/JCO.22.02780
Le, T. G., Cozic, N., Boyer, J. C., Boige, V., Diasio, R. B., Taieb, J., et al. (2024). Dihydropyrimidine dehydrogenase gene variants for predicting grade 4-5 fluoropyrimidine-induced toxicity: FUSAFE individual patient data meta-analysis. Br. J. Cancer 130, 808–818. doi:10.1038/s41416-023-02517-2
Levy, E., Piedbois, P., Buyse, M., Pignon, J. P., Rougier, P., Ryan, L., et al. (1998). Toxicity of fluorouracil in patients with advanced colorectal cancer: effect of administration schedule and prognostic factors. J. Clin. Oncol. 16, 3537–3541. doi:10.1200/JCO.1998.16.11.3537
Lunenburg, C., van der Wouden, C. H., Nijenhuis, M., Crommentuijn-van Rhenen, M. H., de Boer-Veger, N. J., Buunk, A. M., et al. (2020). Dutch pharmacogenetics working Group (DPWG) guideline for the gene-drug interaction of DPYD and fluoropyrimidines. Eur. J. Hum. Genet. 28, 508–517. doi:10.1038/s41431-019-0540-0
Meulendijks, D., Henricks, L. M., Sonke, G. S., Deenen, M. J., Froehlich, T. K., Amstutz, U., et al. (2015). Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: a systematic review and meta-analysis of individual patient data. Lancet Oncol. 16, 1639–1650. doi:10.1016/S1470-2045(15)00286-7
Miarons, M., Manzaneque Gordon, A., Riera, P., Gutierrez Nicolas, F., and DrgwtSSoHP, R. (2023). Allelic frequency of DPYD genetic variants in patients with cancer in Spain: the PhotoDPYD Study. Oncologist 28, e304–e308. doi:10.1093/oncolo/oyad077
Nguyen, D. G., Morris, S. A., Hamilton, A., Kwange, S. O., Steuerwald, N., Symanowski, J., et al. (2024). Real-world impact of an In-House dihydropyrimidine dehydrogenase (DPYD) genotype test on fluoropyrimidine dosing, toxicities, and hospitalizations at a multisite cancer center. JCO Precis. Oncol. 8, e2300623. doi:10.1200/PO.23.00623
Quaranta, S., and Thomas, F. (2017). Pharmacogenetics of anti-cancer drugs: state of the art and implementation - recommendations of the French national Network of pharmacogenetics. Therapie 72, 205–215. doi:10.1016/j.therap.2017.01.005
Rosas-Alonso, R., Queiruga, J., Arias, P., Del Monte, A., Yuste, F., Rodriguez-Antolin, C., et al. (2021). Analytical validation of a laboratory-development multigene pharmacogenetic assay. Pharmacogenet Genomics 31, 177–184. doi:10.1097/FPC.0000000000000438
Rosmarin, D., Palles, C., Church, D., Domingo, E., Jones, A., Johnstone, E., et al. (2014). Genetic markers of toxicity from capecitabine and other fluorouracil-based regimens: investigation in the QUASAR2 study, systematic review, and meta-analysis. J. Clin. Oncol. 32, 1031–1039. doi:10.1200/JCO.2013.51.1857
Ruzzo, A., Graziano, F., Galli, F., Galli, F., Rulli, E., Lonardi, S., et al. (2017). Dihydropyrimidine dehydrogenase pharmacogenetics for predicting fluoropyrimidine-related toxicity in the randomised, phase III adjuvant TOSCA trial in high-risk colon cancer patients. Br. J. Cancer 117, 1269–1277. doi:10.1038/bjc.2017.289
Stewart, S., Dodero-Anillo, J. M., Guijarro-Eguinoa, J., Arias, P., Gomez Lopez De Las Huertas, A., Seco-Meseguer, E., et al. (2023). Advancing pharmacogenetic testing in a tertiary hospital: a retrospective analysis after 10 years of activity. Front. Pharmacol. 14, 1292416. doi:10.3389/fphar.2023.1292416
Terrazzino, S., Cargnin, S., Del Re, M., Danesi, R., Canonico, P. L., and Genazzani, A. A. (2013). DPYD IVS14+1G>A and 2846A>T genotyping for the prediction of severe fluoropyrimidine-related toxicity: a meta-analysis. Pharmacogenomics 14, 1255–1272. doi:10.2217/pgs.13.116
van Kuilenburg, A. B. (2004). Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5-fluorouracil. Eur. J. Cancer 40, 939–950. doi:10.1016/j.ejca.2003.12.004
Wang, L., Howlett, S., and Essapen, S. (2022). Treating patients with dihydropyrimidine dehydrogenase (DPD) deficiency with fluoropyrimidine chemotherapy since the onset of routine prospective testing-the experience of a large oncology center in the United Kingdom. Semin. Oncol. 49, 170–177. doi:10.1053/j.seminoncol.2021.11.004
Keywords: fluoropyrimidines, DPYD, colorectal cancer, toxicity, discontinuation
Citation: Rosas-Alonso R, Salas NR, Wert PP, Hoyo A, Martin-López S, Martínez-Pérez D, Ruiz-Gutiérrez I, Jiménez-Bou D, Peña J, Arias P, Custodio A, Losantos-García I, Borobia AM, Feliu J and Ghanem I (2025) DPYD-guided fluoropyrimidine dose adjustment in colorectal cancer DPYD carriers: start slower to finish stronger. Front. Pharmacol. 16:1645188. doi: 10.3389/fphar.2025.1645188
Received: 11 June 2025; Accepted: 02 September 2025;
Published: 18 September 2025.
Edited by:
Baoming Wang, University of Technology Sydney, AustraliaReviewed by:
Carla V. Finkielstein, Virginia Tech, United StatesXiaomeng Sun, Broad Institute, United States
Copyright © 2025 Rosas-Alonso, Salas, Wert, Hoyo, Martin-López, Martínez-Pérez, Ruiz-Gutiérrez, Jiménez-Bou, Peña, Arias, Custodio, Losantos-García, Borobia, Feliu and Ghanem. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rocío Rosas-Alonso, cm9zYXMuYWxvbnNvLnJvY2lvQGdtYWlsLmNvbQ==; Ismael Ghanem, aXNtYV9nX2NAaG90bWFpbC5jb20=