 Tian Yue
Tian Yue Dezhi Zheng
Dezhi Zheng Jian He
Jian He Shiqiang Xiong
Shiqiang Xiong Junbo Xu1
Junbo Xu1 Jun Hou
Jun Hou- 1Department of Cardiology, The Third People’s Hospital of Chengdu/Affiliated Hospital of Southwest Jiaotong University, Chengdu Institute of Cardiovascular Disease, Chengdu, Sichuan, China
- 2Department of Cardiovascular Surgery, The 960th Hospital of the PLA Joint Logistic Support Force, Jinan, Shandong, China
Heart failure (HF) with preserved ejection fraction (HFpEF) accounts for approximately 50% of all HF cases, and its incidence continues to rise with population aging and the surge in metabolic diseases. Unlike heart failure with reduced ejection fraction (HFrEF), HFpEF lacks effective therapeutic regimens to improve prognosis, with a 5-year mortality rate as high as 50%. Mitochondrial dysfunction, as a key link connecting metabolic disorders and abnormal myocardial systolic and diastolic function, has become a critical mechanism in the pathophysiology of HFpEF and a potential therapeutic target. This review systematically elaborates on the molecular mechanisms in HFpEF, such as mitochondrial energy metabolism disorders, dynamic imbalance, oxidative stress injury, and calcium signal dysregulation, comprehensively reviews the evidence for the effects of marketed drugs and drugs in clinical trials that improve mitochondrial function, and simultaneously explores emerging therapeutic strategies targeting mitochondria. This review aims to provide a theoretical reference for mechanistic research and drug development of HFpEF and promote the application of precision therapy targeting mitochondrial dysfunction in clinical practice.
1 Introduction
Heart failure with preserved ejection fraction (HFpEF) is a complex clinical syndrome characterized primarily by elevated left ventricular filling pressure and impaired diastolic function. In contrast, the left ventricular ejection fraction (LVEF) is normal or near normal (≥50%) (Rao et al., 2025; Hamo et al., 2024). Epidemiological studies have shown that HFpEF accounts for approximately 50% of all heart failure hospitalizations (Hamatani et al., 2025), and its prevalence is increasing significantly, which is closely associated with global population aging, obesity, and the epidemic of metabolic diseases such as type 2 diabetes (Rao et al., 2025; Rambarat et al., 2025). Patients commonly present with comorbidities such as hypertension (Redfield and Borlaug, 2023), obesity (Kosiborod et al., 2024a), and chronic kidney disease (Kirkman et al., 2021), as well as drug-drug interactions resulting from polypharmacy. These two factors exacerbate diagnostic confusion (Cheng and Nayar, 2009). Compared with heart failure with reduced ejection fraction (HFrEF), HFpEF patients have more limited treatment options. Most neurohormonal antagonists, such as Sacubitril/Valsartan (Jackson et al., 2021), effective for HFrEF have failed to show significant benefits in HFpEF treatment, resulting in a 5-year mortality rate as high as 50%, posing a severe public health challenge (Hamatani et al., 2025).
The pathophysiological mechanisms of HFpEF are highly heterogeneous, involving multiple factors such as systemic inflammation (Panico et al., 2024; Thorp and Filipp, 2025), microvascular dysfunction (Grootaert et al., 2025), myocardial fibrosis (Yamada et al., 2025), and metabolic disorders (Kumar et al., 2019). Recent studies have revealed that cardiomyocyte mitochondrial dysfunction is a core link throughout the occurrence and development of HFpEF, especially occupying a central position in HFpEF subtypes related to metabolic abnormalities (Givvimani et al., 2012; Madreiter-Sokolowski et al., 2024). As the energy powerhouse of cells, mitochondrial function directly affects the energy supply of the heart. Abnormal mitochondrial metabolism impairs fatty acid oxidation (FAO) processes (Fang and Gustafsson Å, 2024; Fonseka et al., 2025), reduces glucose metabolism efficiency, promotes increased glycolysis (Sun et al., 2024), and causes the accumulation of abnormal FAO metabolites and glycolytic products (Lim and Khunti, 2024). Such abnormal metabolic processes lead to dysfunction of the mitochondrial electron transport chain and electron leakage, during which reactive oxygen species (ROS) are generated. The accumulation of ROS triggers cellular oxidative stress activation (Gibb et al., 2021; Liang et al., 2025; Lozhkin et al., 2022; Gallo et al., 2024).
The accumulation of FAO metabolites and glycolytic products induces inflammatory activation, promotes the accumulation of inflammasomes such as NLRP3, and upregulates the secretion of inflammatory factors (Duewell et al., 2010). Over the long term, this leads to ventricular remodeling and fibrosis, further reducing the heart’s pumping capacity in HF (Pepin et al., 2025). In HFpEF, imbalances in mitochondrial dynamics and metabolic abnormalities cause intracellular calcium homeostasis dysfunction, severely impair myocardial diastolic function, and exacerbate HF (Hohendanner and Bode, 2020; McKenzie and Duchen, 2016).
This review elaborates in detail on the multi-dimensional roles of mitochondria in HFpEF, the current application and development of clinical and preclinical drugs, as well as the research status of active components of traditional Chinese medicine in HFpEF. It aims to provide new insights into a comprehensive understanding of the critical role of mitochondria in HFpEF and the development of mitochondria-targeted drugs.
2 Mitochondrial function and clinical treatment in HFpEF
2.1 Mitochondrial dysfunction and energy metabolism dysregulation
As a high-energy-consuming organ in the human body, more than 90% of the energy supply of cardiac muscle depends on mitochondrial oxidative phosphorylation (Figure 1) (Gallo et al., 2024; Shehadeh et al., 2025). In patients with HFpEF and animal models, cardiomyocytes exhibit a significant reduction in ATP synthesis, and the underlying cause lies in impaired utilization of energy substrates and impaired respiratory chain function (Shehadeh et al., 2025). FAO is a key pathway for cardiomyocytes to obtain energy (Mericskay et al., 2025). Under physiological conditions, the heart primarily utilizes fatty acids as an energy source, with more than 80% of ATP derived from FAO. FAO dysregulation is one of the core changes in mitochondrial energy metabolism in HFpEF, and it leads to further loss of cardiac function in HFpEF by affecting myocardial energy supply, stimulating the occurrence of oxidative stress, and aggravating ventricular remodeling (Zhang S. et al., 2025). AO dysregulation is typically associated with certain underlying metabolic diseases. For example, in HFpEF associated with obesity or diabetes, plasma levels of long-chain acylcarnitines are elevated, while short-chain and medium-chain acylcarnitines are increased in diabetic patients. Both indicate incomplete oxidation of fatty acids (FA), which largely limits mitochondrial ATP production (Hahn et al., 2023). Moreover, due to the abnormal accumulation of FA, increasing FA oxidation and the clearance rate of abnormally stored FA from tissues are therapeutically beneficial (Ying et al., 2021). Relevant studies have also confirmed this. In the HFpEF model induced by a high-fat diet in ApoE knockout (KO) mice, the levels of long-chain acylcarnitines in the myocardium are significantly elevated, and the proportion of saturated fatty acids increases abnormally, which directly promotes macrophage inflammatory response and cardiomyocyte dysfunction (Travers and Robinson, 2024). I addition, downregulation of peroxisome proliferator-activated receptor α (PPARα) signaling and decreased expression of carnitine palmitoyltransferase 1 (CPT1) further impair the efficiency of fatty acid β-oxidation. A recent study has shown that by modulating PPARα and preventing phosphorylation of nuclear factor κB (NF-κB), oxidative stress and inflammatory responses have been reduced (Zhou et al., 2024).
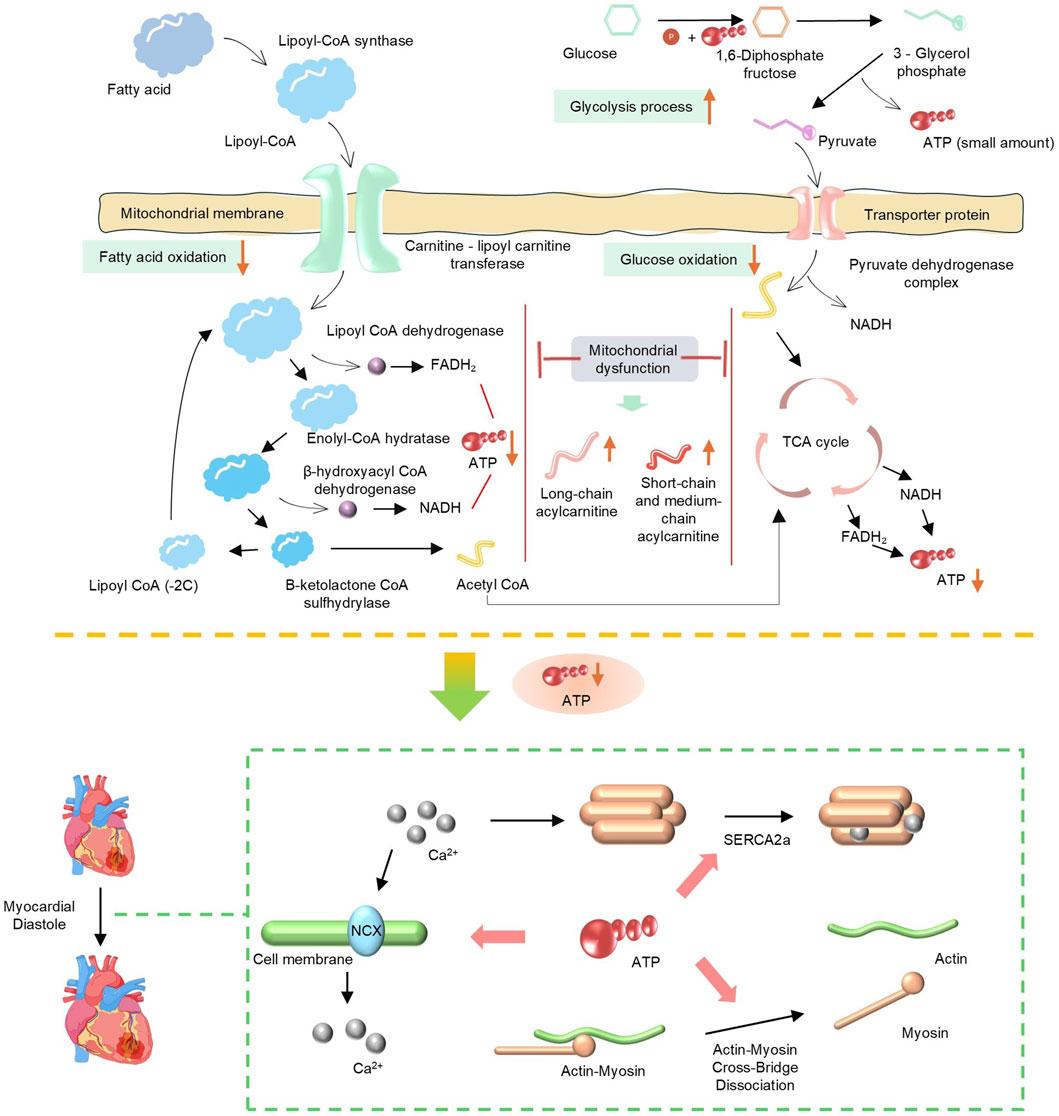
Figure 1. Schematic of mitochondrial energy metabolism pathways (fatty acid and glucose oxidation) and mitochondrial dysfunction, showing key enzymes, processes (TCA cycle, glycolysis), and impact on ATP production.
As another important energy source, abnormal glucose metabolism also significantly affects the progression of HFpEF. Against the backdrop of dominant fatty acid oxidation, the myocardium in HFpEF exhibits a “pseudohypoxic state”; even with normal coronary blood flow, the myocardium still tends to compensate for energy production through glycolysis (Mossmann et al., 2018; Watson et al., 2023). However, the ATP generated by glycolysis is far from sufficient to meet the energy demands of the myocardium, and HFpEF patients often have insulin resistance, resulting in reduced myocardial uptake and utilization of glucose (Hahn et al., 2023). yperglycemia and insulin resistance can stimulate the proliferation of cardiac fibroblasts, increase collagen synthesis, and lead to myocardial fibrosis. Meanwhile, abnormal glucose metabolism can also affect vascular endothelial function, reduce nitric oxide (NO) release, exacerbate coronary microcirculatory dysfunction and myocardial hypoperfusion, and further aggravate abnormalities in myocardial structure and function (Clyne, 2021). In addition, upregulated expression of PDH kinase 4 (PDK4) exacerbates phosphorylation and inactivation of pyruvate dehydrogenase (PDH), blocks the conversion of pyruvate to acetyl-CoA, and further impairs glucose oxidation capacity. A 2025 study revealed that HFpEF mice deficient in FGF21 exhibited significantly increased cardiac PDK4 expression, whereas FGF21 supplementation inhibited PDK4 by activating the PI3K/AKT pathway, restored PDH activity, and increased ATP synthesis (Piristine et al., 2024). Regardless of changes in FFA metabolism, HF is characterized by systemic neurohumoral activation and increased lipolysis, resulting in excess FFA supply that exceeds mitochondrial β-oxidation capacity, promotes the accumulation of toxic metabolic intermediates, and thereby leads to further mitochondrial dysfunction (Del Campo et al., 2021).
The mitochondrial electron transport chain drives transmembrane proton translocation through electron transport, forming a transmembrane proton gradient. Ultimately, Complex Ⅴ utilizes the gradient energy to synthesize ATP. Therefore, dysfunction of respiratory chain complexes is the direct cause of the energy crisis in HFpEF (Schauer et al., 2024). Respiratory chain damage leads to decreased efficiency of oxidative phosphorylation and reduced ATP production. When cardiomyocytes have insufficient energy reserves, the function of ATP-dependent diastolic calcium pumps, such as sarcoplasmic reticulum Ca2+-ATPase, is impaired, resulting in delayed clearance of cytoplasmic Ca2+ and impaired ventricular diastolic relaxation, which is one of the core mechanisms of diastolic dysfunction (Torre et al., 2022). For example, the ZSF1 rat HFpEF model shows that the activity of myocardial mitochondrial Complex I and III is significantly decreased, leading to impairment of electron transport and reduced ATP synthesis (Fopiano et al., 2024). Proteomic analysis further reveals that the stability of respiratory chain supercomplexes is decreased, particularly with reduced content of Cardiolipin, which impairs the assembly and function of the complexes. As a key phospholipid in the inner mitochondrial membrane, Cardiolipin is crucial for maintaining the spatial conformation of respiratory chain complexes and the efficiency of electron transport (Dolinsky et al., 2016).
Under normal physiological conditions, only 4%–15% of ATP in the myocardium is derived from ketone bodies, which is much lower than the contributions from fatty acids (40%–70%) and glucose (20%–30%) (Manolis et al., 2023). Ketone bodies originate from acetyl-CoA generated by fatty acid β-oxidation in hepatic mitochondria, and are converted into β-hydroxybutyrate, acetoacetate, and other ketone bodies through a series of enzymatic reactions (Manolis et al., 2023; Xu et al., 2023).
In terms of metabolic crosstalk, ketone bodies reduce glucose utilization by inhibiting phosphofructokinase (PFK), a key enzyme in glycolysis. Meanwhile, they indirectly regulate FAO through modulating carnitine CPT1, thereby preventing excessive lipid accumulation (Brahma et al., 2022). In heart failure with HFpEF, myocardial metabolic remodeling is accompanied by increased ketone body utilization, which serves as a compensatory mechanism. However, in the diabetic state, decreased activity of 3-oxoacid CoA transferase (SCOT) results in reduced ketone body uptake, exacerbating energy deficiency (Anker et al., 2021). In patients with chronic kidney disease (CKD), the renal capacity for ketone body clearance is impaired (renal clearance accounts for 28% under normal pH conditions), which may induce ketone body accumulation and oxidative stress. These abnormalities exacerbate HFpEF through mitochondrial dysfunction and reduced metabolic flexibility, whereas SGLT2 inhibitors may exert a protective effect by improving ketone body metabolism (Garcia-Ropero et al., 2019; Yurista et al., 2019; Solomon et al., 2022).
2.2 Imbalance in mitochondrial dynamics and dysregulation of quality control
Mitochondria maintain the dynamic balance of their network through continuous fission and fusion, and clear damaged components via mitophagy (Figure 2A). These processes are regulated by specific mitochondrial fusion and fission proteins, ensuring the integrity of mtDNA by eliminating mitochondria with damaged DNA and promoting functional units (Moral et al., 2020). In HFpEF, this dynamic balance is disrupted, characterized by excessive fission and impaired fusion. For example, a recent study has shown that abnormal mitochondrial content and decreased levels of MFN2 are observed in myocardial tissue of elderly HFpEF patients (Molina et al., 2016). In addition, stress factors such as a high-fat diet and hypertension can also induce the translocation of mitochondrial fission protein Drp1 to mitochondria, leading to mitochondrial fragmentation. Preclinical studies have confirmed that Drp1-mediated excessive fission not only reduces mitochondrial oxidative capacity but also promotes cardiomyocyte apoptosis. Conversely, downregulated expression of fusion proteins such as optic atrophy 1 (OPA1) and mitofusin 2 (MFN2) impairs mitochondrial network connectivity and compromises energy transfer efficiency. Some studies have shown that inhibiting Drp1 protein can alleviate left ventricular (LV) dysfunction in HFpEF associated with decreased LC3 and P62 (Givvimani et al., 2012). Furthermore, triggering PKA activation and inhibitory phosphorylation of Drp1 via miR-129-3p has been shown to exert cardioprotective effects (Sotomayor-Flores et al., 2020).
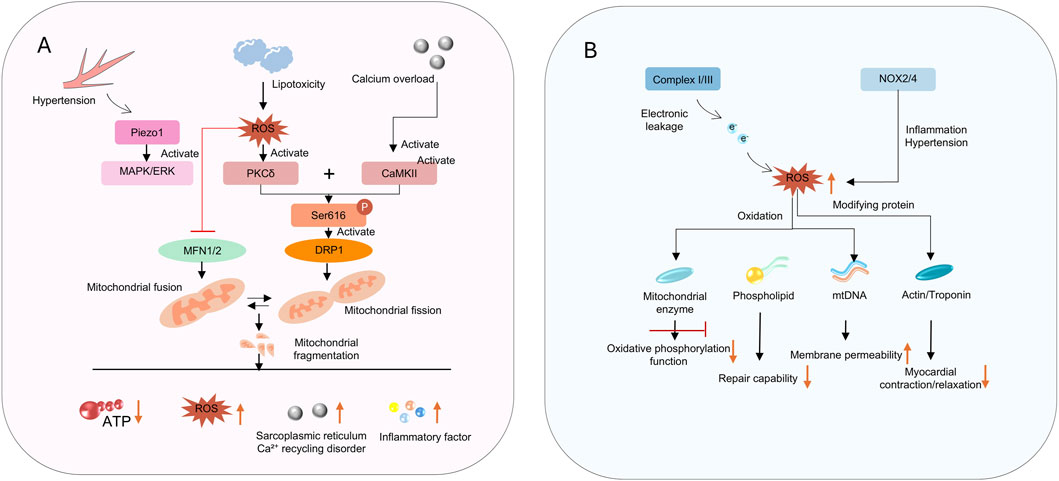
Figure 2. (A) shows signaling pathways disrupting mitochondrial dynamics; (B) depicts ROS - induced impairments in function and myocardial contraction.
Mitophagy is an important process for the clearance of damaged mitochondria and mitochondrial renewal. Patients with HFpEF are usually accompanied by a decline in mitophagy function, failing to clear dysfunctional mitochondria and their accumulation in cardiomyocytes (Abdellatif et al., 2025a; Abdellatif et al., 2025b). On one hand, dysfunctional mitochondria cannot normally perform energy supply. On the other hand, they inhibit the function of normal mitochondria, further reducing energy supply in HFpEF, decreasing myocardial ATP reserves, affecting diastolic calcium ion homeostasis, and exacerbating myocardial diastolic dysfunction (Krammer et al., 2025). Accumulated dysfunctional mitochondria are one of the main sources of ROS release, which leads to cellular oxidative stress and inflammatory activation, and this part will be discussed in depth later (Yoshii et al., 2024). The PINK1/Parkin pathway is the main regulatory mechanism of mitophagy. In the context of HFpEF, chronic inflammation and oxidative stress disrupt the stability of PINK1, leading to insufficient clearance of damaged mitochondria and persistent accumulation of dysfunctional mitochondria. Studies have found that in obesity-related HFpEF models, mitophagy flux in cardiomyocytes is significantly reduced, which is negatively correlated with the degree of insulin resistance (Li et al., 2023; Qiu et al., 2019).
The Unfolded Protein Response (UPR) pathway serves as one of the core defense mechanisms for mitochondria to cope with protein misfolding stress and maintain their own homeostasis. By regulating the efficiency of protein folding in the mitochondrial matrix and promoting the degradation of abnormal proteins, it directly affects mitochondrial respiratory chain function and oxidative stress levels, and is crucial for maintaining the integrity of mitochondria in myocardial cells. Given the key role of the UPR pathway in regulating mitochondrial function, it has become an important therapeutic target for mitochondrial-related diseases. Currently, small-molecule modulators targeting this pathway (such as UPR activators) have entered the early-phase clinical trial stage, and preliminary effects of some drugs—including improving mitochondrial energy metabolism and alleviating myocardial cell damage—have been observed in animal models.
2.3 ROS injury and oxidative stress
The accumulation of ROS in HFpEF is caused by two factors (Figure 2B). First, there is increased ROS production (Nohara et al., 2019). Mitochondria are the main source of intracellular ROS, particularly when the electron transport chain is impaired. Increased electron leakage leads to excessive generation of superoxide anions (O2•−) (Delalat et al., 2025; Teuber et al., 2022). In addition to electron leakage from the respiratory chain, the expression of NADPH oxidase 4 (NOX4) is upregulated in cardiomyocyte membranes and mitochondria in HFpEF, directly promoting O2•− production (Teuber et al., 2022). Furthermore, lipid peroxidation products such as malondialdehyde (MDA) accumulate in myocardial tissue, reflecting the degree of oxidative damage (Costantino et al., 2023). Second, the function of the ROS scavenging system is reduced. The activity of mitochondrial antioxidant enzymes such as superoxide dismutase 2 (SOD2) and glutathione peroxidase 1 (GPX1) decreases, impairing ROS clearance capacity (Russomanno et al., 2017; Su et al., 2023).
The accumulation of ROS exacerbates cardiac dysfunction in HFpEF through multiple mechanisms (Jankauskas et al., 2021). ROS can directly act on the cytoskeleton of cardiomyocytes and affect the function of contractile proteins such as cardiac actin and myosin, impairing normal myocardial contraction and relaxation (Zuo et al., 2015). When acting on fibroblasts, ROS activate the transformation of fibroblasts into myofibroblasts, increase collagen deposition, and exacerbate myocardial fibrosis. Meanwhile, ROS can inhibit the synthesis and release of NO in vascular endothelial cells, impair vascular diastolic function, and increase cardiac load (Zuo et al., 2015). The accumulation of ROS and the state of oxidative stress in cardiomyocytes further damage mitochondrial function, leading to energy metabolism disorders and more ROS release, creating a continuous vicious cycle (Lozhkin et al., 2022).
2.4 Inflammation-metabolism crosstalk and microenvironmental dysregulation
The cardiac immune microenvironment and mitochondrial function form a tightly interconnected network, which is particularly prominent in metabolic HFpEF (Figure 3A) (Kosiborod et al., 2024b; Verma et al., 2024). Dysregulation of macrophage-cardiomyocyte crosstalk, macrophages are highly plastic and can differentiate into various phenotypes in response to microenvironmental signals (Panico et al., 2024; Deng et al., 2021). Studies have shown that in HFpEF hearts, the pro-inflammatory macrophage (M1) phenotype is significantly increased, while the proportion of TIM4+ macrophages, which possess anti-inflammatory and tissue repair functions, is decreased (Kopec et al., 2024; Zhang N. et al., 2022). Activated macrophages release pro-inflammatory factors such as interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6), which impair mitochondrial biogenesis by inhibiting the expression of PPARγ coactivator-1α (PGC-1α) (Agrawal et al., 2023; Gorica et al., 2025). Although anti-inflammatory phenotype macrophages (M2) can alleviate inflammation-induced damage to a certain extent, transforming growth factor-β (TGF-β) secreted by M2 macrophages is one of the potent profibrotic factors, which can activate the transformation of cardiac fibroblasts into myofibroblasts and promote the synthesis and deposition of collagen (types Ⅰ and Ⅲ) (Frangogiannis, 2024). Therefore, finding a balance in the phenotypic changes of different macrophages is crucial for HFpEF.
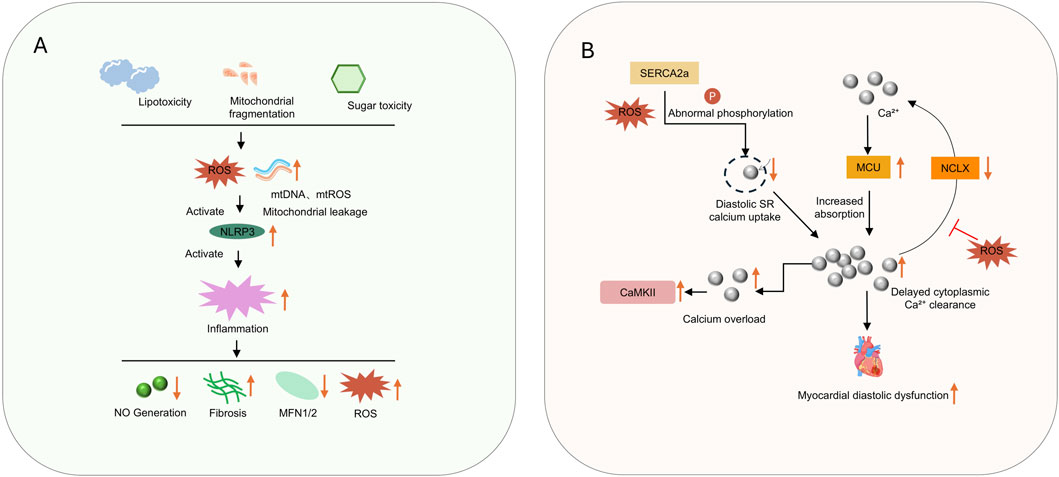
Figure 3. (A) shows lipotoxicity, mitochondrial fragmentation, and sugar toxicity triggering ROS mediated inflammation; (B) illustrates calcium dysregulation causing myocardial diastolic dysfunction.
Furthermore, macrophages can inversely regulate the cardiac immune microenvironment. Matrix metalloproteinases (MMPs) released by macrophages can degrade the extracellular matrix (ECM) (Tourki et al., 2020). However, in HFpEF, the imbalance between MMPs and tissue inhibitors of metalloproteinases (TIMPs) leads to abnormal ECM remodeling and exacerbates fibrosis (Tourki et al., 2020; Krebber et al., 2020). Meanwhile, macrophages can further recruit more inflammatory cells and fibrosis-related cells by phagocytosing signaling molecules (such as ATP) released by apoptotic cardiomyocytes (Smart et al., 2023).
Accumulation of FA is caused by abnormal FAO, i.e., lipotoxicity, which also activates inflammation and exacerbates FAO abnormalities. Excess lipids accumulate as lipid droplets in cardiomyocytes, compressing organelles such as mitochondria and sarcoplasmic reticulum, disrupting the normal structure of cardiomyocytes, and reducing contractile-diastolic coordination (Janssen-Telders et al., 2025; Leggat et al., 2021).
Lipotoxicity is typically associated with obesity, metabolic syndrome, or type 2 diabetes (Fonseka et al., 2025). Incompletely oxidized lipid intermediates (e.g., ceramides, long-chain acyl-CoA, and lipopolysaccharides, LPS) can directly impair the integrity of cardiomyocyte membranes, inducing apoptosis (via activation of the caspase pathway) and necrosis (De Geest and Mishra, 2022; Ketema and Lopaschuk, 2025). For instance, in obesity-related HFpEF, elevated levels of cardiac local free FA (such as lauric acid and myristic acid) directly stimulate pro-inflammatory polarization of macrophages. In vitro experiments have confirmed that bone marrow-derived macrophages from ApoE knockout (KO) mice, when exposed to saturated fatty acids, show significantly upregulated expression of TNF-α and IL-6, forming a “metabolism-inflammation” vicious cycle (Phu et al., 2023; Korbecki and Bajdak-Rusinek, 2019; Vendrov et al., 2024; Riera-Borrull et al., 2017). Lipotoxicity also affects mitochondria: accumulated lipids (especially free fatty acids) in cardiomyocytes can enter mitochondria and undergo excessive oxidation, further impairing the function of the electron transport chain (Da Silva Rosa et al., 2021; Zhang J. et al., 2022).
2.5 Calcium homeostasis and mitochondria-sarcoplasmic reticulum coupling dysfunction
Under physiological conditions, calcium homeostasis in cardiomyocytes relies on the dynamic balance between calcium release during systole and calcium reuptake during diastole (Figure 3B) (Sutanto et al., 2020; Kho, 2023). Imbalance of calcium homeostasis directly impairs ventricular diastolic function and increases fibrosis, mainly manifested as diastolic calcium overload leading to increased diastolic tension in cardiomyocytes, slowed relaxation rate, elevated ventricular filling resistance, increased left ventricular end-diastolic pressure (LVEDP), and reduced compliance—classic hemodynamic features of HFpEF (Adeniran et al., 2015; Tu et al., 2025).
Sustained calcium overload can activate calcium-dependent proteases (e.g., calpains) and transcription factors (e.g., NFAT), promote cardiac fibroblast proliferation and collagen synthesis, exacerbate myocardial interstitial fibrosis, and further increase ventricular stiffness (Tu et al., 2025). By uptake of cytoplasmic calcium ions, mitochondria not only regulate calcium transient kinetics but also provide activation signals for calcium-dependent dehydrogenases (Mira Hernandez et al., 2025).
First, there is abnormal sarcoplasmic reticulum calcium handling: in HFpEF, the activity of sarcoplasmic reticulum Ca2+-ATPase (SERCA2a) is reduced, and phospholamban (PLB) is insufficiently phosphorylated, resulting in delayed diastolic calcium reuptake into the sarcoplasmic reticulum (Grote Beverborg et al., 2021). Excessive phosphorylation of ryanodine receptor 2 (RyR2) causes calcium leakage, reducing sarcoplasmic reticulum calcium storage (Dridi et al., 2023).
Second, there is dysregulated mitochondrial calcium uptake: dysfunction of the mitochondrial calcium uniporter (MCU) complex impairs mitochondrial calcium buffering capacity, leading to delayed cytoplasmic calcium clearance and prolonged myocardial diastolic duration (Weiser et al., 2023). Disrupted calcium signaling simultaneously reduces mitochondrial dehydrogenase activity, further inhibiting ATP production and forming a positive feedback loop between energy deficiency and abnormal calcium handling (Alves-Figueiredo et al., 2024).
2.6 Mitochondrial subtypes
In the myocardium of heart failure with preserved ejection fraction (HFpEF), there are functionally specialized mitochondrial subtypes, and their heterogeneous damage is a key link in disease progression (Glancy et al., 2017). Based on differences in spatial distribution and function, myocardial mitochondria can be divided into interfibrillar mitochondria (adjacent to myofibrils, responsible for immediate ATP supply) and perivascular mitochondria (near capillaries, involved in metabolic substrate exchange). Preclinical studies have shown that in the ZSF1-obese HFpEF rat model, the crista structure disruption, which directly leads to insufficient energy supply for the sarcoplasmic reticulum calcium pump (SERCA2a) and exacerbates diastolic dysfunction (Kumar et al., 2019).
Studies on human HFpEF samples further confirm that the proportion of aging-related mitochondrial subtypes (such as heteroplasmic mitochondria harboring mutant mtDNA) increases, which damages cardiomyocytes via oxidative stress (Tschöpe et al., 2017). These subtype-specific abnormalities suggest that targeted interventions for different mitochondrial subtypes (e.g., improving the crista structure of interfibrillar mitochondria, clearing abnormal perivascular mitochondria) may become a new direction for HFpEF treatment, addressing the previous lack of attention to functional differences among mitochondrial subtypes (Shou and Huo, 2022).
2.7 Clinical treatment of HFpEF
Currently, there is no consensus on the existence of curative drugs for HFpEF. Treatment is usually implemented from aspects including lifestyle modifications, management of underlying diseases, and symptomatic treatment (Heidenreich et al., 2022). Lifestyle is one of the key factors influencing the progression of HFpEF, mainly manifested in salt intake, weight changes, smoking, alcohol consumption, and so on. For HFpEF patients, daily salt intake should be strictly controlled below 5 g, while excessive fluid intake should be avoided to prevent edema caused by it McDonagh et al. (2021), Mullens et al. (2024). For patients with a body mass index (BMI) ≥30 kg/m2, it is necessary to lose weight, restrict the intake of high-calorie foods, and perform 150 min of moderate-intensity aerobic exercise per week. Additionally, tobacco and alcohol intake should be strictly restricted (Mullens et al., 2024).
Patients with HFpEF commonly have comorbid underlying diseases, such as hypertension, type 2 diabetes mellitus, and/or obesity and obstructive sleep apnea (Campbell et al., 2024; Gao et al., 2024). Therefore, strictly controlling these comorbid underlying conditions is one of the core priorities in the treatment of HFpEF. This includes the use of drugs that improve diastolic function, such as angiotensin-converting enzyme inhibitors (ACEIs)/angiotensin II receptor blockers (ARBs) or long-acting calcium channel blockers, to lower blood pressure (Wang et al., 2022). The use of SGLT2 inhibitors to control blood glucose and reduce the risk of heart failure hospitalization, and for patients with moderate to severe obstructive sleep apnea (OSA), the need for nocturnal continuous positive airway pressure (CPAP) therapy (Savarese et al., 2022). When patients present with increased volume overload, loop diuretics should be administered to reduce volume overload.
2.8 Inter-organ interaction and crosstalk
The development of HF with HFpEF is not only affected by cardiac function but also a consequence of the interaction of multiple organ dysfunctions. Pathologies of organs such as the lungs, liver, and kidneys can induce or exacerbate HFpEF through hemodynamic changes, neuroendocrine activation, and inflammatory responses (Shah et al., 2016; Borlaug et al., 2023). The heart and lungs are closely associated; certain pulmonary diseases, such as chronic obstructive pulmonary disease (COPD) (Abdo et al., 2025) and pulmonary embolism (PE) (Reddy et al., 2025), can increase pulmonary vascular resistance, thereby elevating cardiac afterload. Chronic hypoxia caused by COPD and PE activates the renin-angiotensin-aldosterone system (RAAS), leading to myocardial hypertrophy and further exacerbation of cardiac diastolic dysfunction (Mascolo et al., 2022). The kidney is one of the body’s main fluid balance-regulating organs, renal pathologies usually cause fluid retention and electrolyte disturbances, which directly induce or exacerbate HFpEF (Rovin et al., 2023). Electrolyte disturbances, such as changes in potassium and calcium ion levels, can also directly affect the systolic/diastolic function of the heart (Ranasinghe et al., 2024). The liver is one of the important organs regulating coagulation function and neuroendocrine hormones. Hypoalbuminemia resulting from reduced hepatic synthesis of albumin and coagulation factors directly promotes tissue edema and increases cardiac preload (Huynh, 2022). Therefore, HFpEF is a “vicious cycle network” formed by organs including the heart, lungs, kidneys, and liver through hemodynamic abnormalities, neuroendocrine activation (e.g., RAAS, sympathetic nervous system), chronic inflammatory responses, and metabolic disturbances.
3 Mitochondria-targeting therapeutic drugs
Protocols for improving HFpEF by regulating mitochondrial function mainly include direct targeting of mitochondria, regulation of mitochondrial energy metabolism, and acting on mitochondrial quality control (Table 1).

Table 1. Part of drugs acting on mitochondria for treating HFpEF and their mechanisms of action.
3.1 SGLT2 inhibitors: remodeling mitochondrial metabolism and inflammatory balance
3.1.1 SGLT2 inhibitors: mechanisms of action in the treatment of HFpEF
Sodium-glucose cotransporter 2 inhibitors (SGLT2i), initially developed as hypoglycemic agents, have been confirmed in recent years to exert a significant improvement effect on cardiac function and clinical outcomes in patients with HFpEF (Figure 4) (Frampton, 2022). Recent studies have demonstrated that SGLT2i exhibit great potential in restoring mitochondrial respiratory chain to improve energy metabolism, inhibiting inflammation, ameliorating oxidative stress, and alleviating myocardial fibrosis (Withaar et al., 2021; Zafeiropoulos et al., 2024).
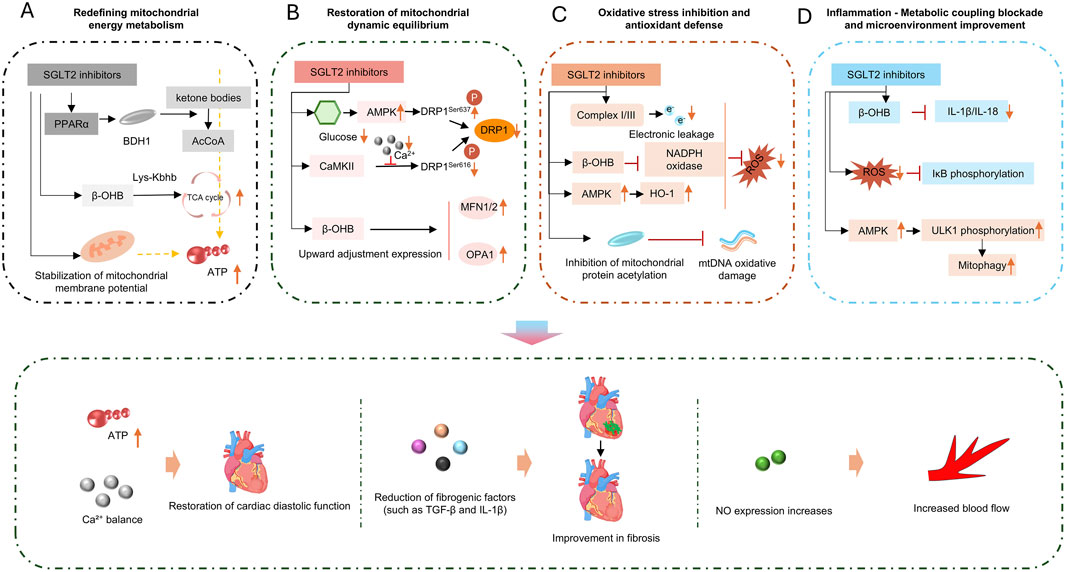
Figure 4. SGLT2 inhibitors act via four pathways: (A) energy metabolism; (B) dynamic equilibrium; (C) oxidative stress; (D) inflammation, to improve cardiac function, including restoring diastolic function and enhancing blood flow.
The improvement of myocardial energy metabolism by SGLT2i is mainly reflected in its ability to reduce myocardial dependence on glucose while promoting ketone body metabolism (Goedeke et al., 2024). SGLT2i increase circulating ketone bodies, including β-hydroxybutyrate and acetoacetate, etc., providing cardiomyocytes with efficiently utilizable ketone bodies as an alternative energy source for ATP production. Unlike glucose, ketone body metabolism produces less NADH, which also reduces the pressure on the mitochondrial electron transport chain, decreases ROS production, and reduces cardiomyocyte dependence on glucose (Uchihashi et al., 2017).
In addition, SGLT2i also affects mitochondrial energy metabolism-related pathways. They may indirectly activate AMP-activated protein kinase (AMPK) by mildly reducing intracellular ATP levels, thereby upregulating peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α). As a core regulator of mitochondrial biogenesis, PGC-1α can promote the gene expression and protein synthesis of respiratory chain complexes (e.g., Complex Ⅳ) and enhance oxidative phosphorylation capacity (Hoong and Chua, 2021). For example, a recent study has shown that in the ZSF1 rat HFpEF model, Empagliflozin treatment for 8 weeks significantly improved the activity of myocardial mitochondrial Complex I and III and increased ATP production (Schauer et al., 2024). Mechanistic studies have indicated that this drug promotes cardiolipin remodeling by upregulating cardiolipin synthases (such as CRLS1 and TAZ), stabilizes the assembly of respiratory chain supercomplexes, and thus restores oxidative phosphorylation efficiency (Jhuo et al., 2023; Santos-Gallego et al., 2019). Proteomic analysis further revealed that Empagliflozin reversed 44% of HFpEF-related abnormal expression of mitochondrial proteins, among which the changes in lipid metabolism- and respiratory chain-related proteins were the most significant (Schauer et al., 2024).
In the pathological process of HFpEF associated with diabetic cardiomyopathy, activation of the mammalian target of rapamycin (mTOR) pathway and inhibition of mitophagy are also observed. Some studies have found that SGLT2i can mildly inhibit mTOR, promote mitophagy, clear dysfunctional mitochondria, and preserve the respiratory chain function of healthy mitochondria (Saha et al., 2023; Park et al., 2020).
SGLT2 inhibitors can inhibit inflammation and oxidative stress through multiple mechanisms, exerting positive effects on HF. They can improve myocardial injury and cardiac function, reduce the risk of hospitalization and cardiovascular death in HF patients, mainly reflected in the inhibition of inflammatory responses and alleviation of oxidative stress (Ijaz et al., 2024).
For example, dapagliflozin can reduce the expression of NLRP3 inflammasome in doxorubicin-induced HFpEF, thereby significantly decreasing the systemic expression levels of IL-1β, IL-6, TNF-α, and granulocyte colony-stimulating factor (G-CSF) in cardiomyocytes (Quagliariello et al., 2024). In a high-fat diet-induced HFpEF model, dapagliflozin significantly reduces cardiac macrophage infiltration and inhibits the expression of pro-inflammatory cytokines (IL-1β, TNF-α, CXCL10) (Crespo-Masip et al., 2025).
In terms of improving oxidative stress, SGLT2 inhibitors can potently block oxidative stress responses, inhibit cardiomyocyte hypertrophy and fibrosis, and reverse ventricular remodeling (Li et al., 2024b). Taking empagliflozin as an example, results from a randomized, double-blind, placebo-controlled study showed that empagliflozin significantly enhances antioxidant capacity by increasing the activities of serum SOD and glutathione peroxidase (GPx) (Eshraghi et al., 2025). Similarly, dapagliflozin can also reduce NADPH oxidase activity in myocardial tissue, decrease ROS production, and improve glutathione antioxidant reserves (Evans et al., 2022).
SGLT2 inhibitors (SGLT2i) also show great potential in improving myocardial fibrosis. Several indirect factors, such as ameliorating inflammation, alleviating cellular oxidative stress, and regulating calcium homeostasis in cardiomyocytes, can effectively reduce myocardial fibrosis. In addition, SGLT2i can directly act on fibroblasts. Studies have demonstrated that SGLT2i can significantly attenuate fibroblast activation induced by the TGF-β1/Smad3 pathway and reduce extracellular collagen deposition (Daud et al., 2021). Taking dapagliflozin as an example, after 8 weeks of intervention with dapagliflozin, glucose metabolism parameters (including blood glucose, glucose tolerance, and insulin sensitivity) in ZDF rats were significantly improved. Dapagliflozin also significantly reduced ventricular hypertrophy and restored diastolic function in ZDF rats (Feng et al., 2023).
3.1.2 Current status of SGLT2 inhibitors in clinical treatment of HFpEF
SGLT2 inhibitors have been widely recognized in the clinical treatment of heart failure with preserved ejection fraction (HFpEF) and are an important therapeutic agent recommended by guidelines, with clear efficacy in reducing the risk of cardiovascular death or heart failure hospitalization (Anker et al., 2021).
The 2024 Chinese Guidelines for the Diagnosis and Treatment of Heart Failure recommend SGLT2 inhibitors for HFpEF patients to reduce the risk of the composite endpoint of heart failure hospitalization or cardiovascular death, as a Class I recommendation (Zhang, 2025). The 2023 Update of the ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure also includes SGLT2 inhibitors for HFpEF treatment as a Class IA recommendation (SEC Working Group for the 2023 update of the 2021 ESC guidelines for the diagnosis and treatment of ac ute and chronic heart failure and SEC Guidelines Committee, 2024).
Pooled analyses show that SGLT2 inhibitors reduce the risk of cardiovascular death or heart failure hospitalization by 21% in HFpEF patients, with a 29% reduction in heart failure hospitalization. In terms of hemodynamic improvement, there is a significant decrease in the E/e' ratio, which is positively correlated with the recovery of mitochondrial function (Anker et al., 2021; Böhm et al., 2022). It indicates that SGLT2 inhibitors have shown certain potential for the treatment of HFpEF in clinical practice.
Furthermore, SGLT2 inhibitors have advantages in the co-management of diabetes, heart, and kidney. For HFpEF patients with comorbid type 2 diabetes mellitus (T2DM) or chronic kidney disease (CKD), they can reduce the risk of cardiovascular death or heart failure hospitalization while improving blood glucose and renal function. No titration is required, and tolerability is good, providing consistent benefits to patients (Jhund et al., 2022; Damman et al., 2014).
3.2 GLP-1 receptor agonists
Glucagon-like peptide-1 receptor agonists (GLP-1RAs) were initially used for the treatment of type 2 diabetes mellitus. As of 2024, GLP-1RAs have not yet been applied as therapeutic drugs for HFpEF in clinical practice (Ying et al., 2015). However, studies (Karlström et al., 2025) have shown that GLP-1RAs can directly activate glucagon-like peptide-1 receptors (GLP-1R) (Hullon et al., 2025) on mitochondria, and regulate mitochondrial biogenesis, optimize mitochondrial function, and counteract mitochondrial oxidative stress, thereby demonstrating potential therapeutic application value for HFpEF. For example, in a recent study (Shigeno et al., 2016), researchers found that the GLP-1 analog Exendin-4 upregulates AMPK activity, activates sirtuin 1 (SIRT1) deacetylase, promotes the expression of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), and reduces the production of mitochondrial ROS. GLP-1RAs also exert a positive effect on mitochondrial structure, researchers observed that in a HFpEF mouse model induced by a high-fat diet, liraglutide activates the Erbb4 receptor, inhibits the phosphorylation of protein kinase C alpha (PKCα) and extracellular signal-regulated kinase 1/2 (ERK1/2), reduces myocardial mitochondrial swelling and cristae fragmentation, and enhances adenosine ATP production simultaneously (Ni et al., 2024). As therapeutic drugs for diabetes, GLP-1RAs also have certain application potential in improving mitochondrial energy metabolism and optimizing the utilization of energy substrates. For instance, semaglutide promotes myocardial glucose oxidation via the Creb5/nuclear receptor subfamily 4 group A member 1 (NR4a1) axis. In HFpEF mice induced by transverse aortic constriction (TAC), semaglutide (Ma et al., 2024) downregulates the nuclear receptor NR4a1 and its translocation to mitochondria, promotes the entry of pyruvate into the tricarboxylic acid cycle, restores myocardial ATP levels to 85% of those in the normal group, and reduces mitochondrial lipid accumulation at the same time.
In addition to preclinical animal studies, findings from some clinical investigations have also indicated the potential application value of GLP-1RAs in HFpEF (Waqas et al., 2025). A real-world analysis of 1.67 million patients with HFpEF (Lee and Elmore, 2025) demonstrated that treatment with semaglutide and tirzepatide reduced the risk of heart failure hospitalization or all-cause death by 40%–58%, and this benefit was positively correlated with the degree of improvement in baseline mitochondrial function markers (such as serum mitochondrial DNA copy number). In patients with obesity-related HFpEF (BMI ≥30 kg/m2), after 48 weeks of treatment with semaglutide (2.4 mg/week), skeletal muscle mitochondrial density measured by magnetic resonance spectroscopy (MRS) increased by 18%, which was significantly associated with improvements in 6-min walk distance and left ventricular E/e' ratio (Butler et al., 2023).
3.3 FGF21 Analogues and signal regulation
Fibroblast growth factor 21 (FGF21) is a metabolic regulatory hormone, and recent studies have revealed its protective effect on mitochondrial function in HFpEF (Figure 5A). In uremic heart failure rat models, obese HF rat models, and hypertensive HF mouse models, FGF21 has been shown to improve mitochondrial function, thereby regulating cardiac remodeling and ameliorating HF (Li et al., 2019; Rupérez et al., 2018; Zeng et al., 2020).
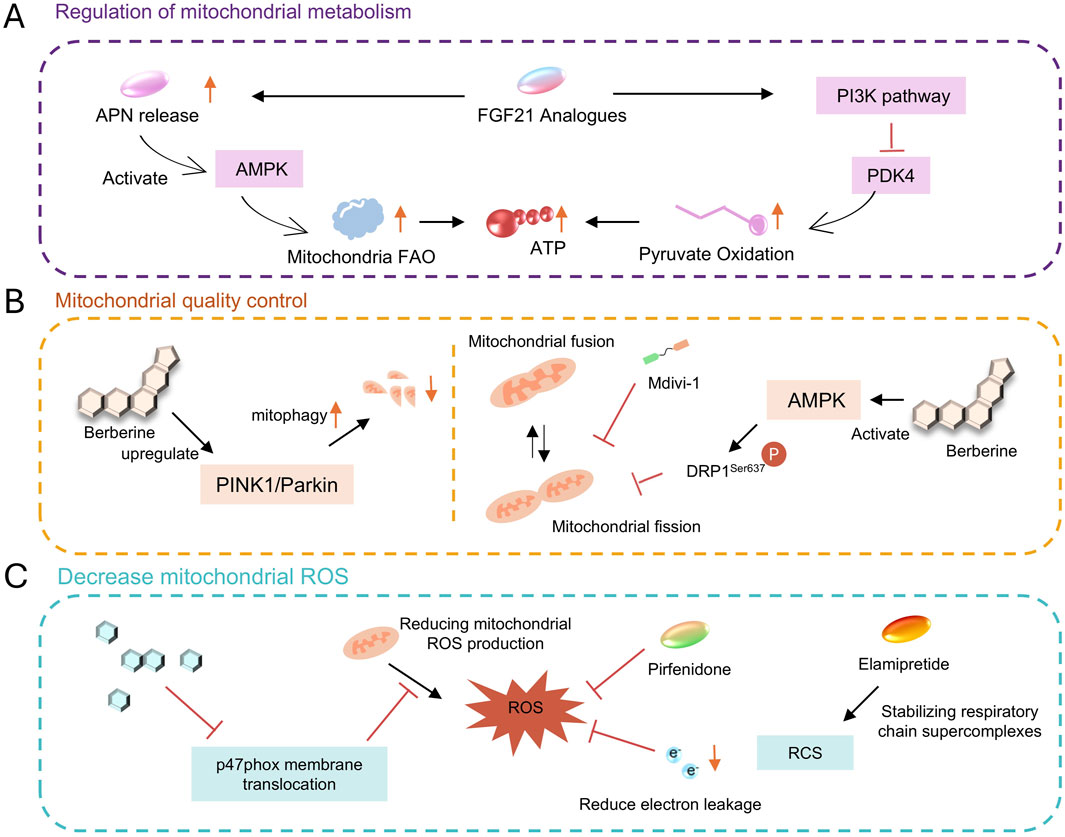
Figure 5. (A) metabolism regulation via AMPK, FGF21, etc. (B) quality control with mitophagy, fusion/fission modulation. (C) ROS reduction using various compounds.
A study demonstrated that FGF21 deficiency significantly exacerbates cardiac diastolic dysfunction in mice after HFpEF, while adipose-specific overexpression of FGF21 can significantly improve this condition. After injecting FGF21 adeno-associated virus (AAV-FGF21) into HFpEF mice, indices of cardiac diastolic function such as the E/A ratio increased, the E/E′ ratio decreased, pulmonary congestion and exercise intolerance were alleviated, and cardiac hypertrophy, oxidative stress, and fibrosis were also attenuated (Zhang K. et al., 2025).The cardioprotective effect of FGF21 against HFpEF is closely associated with its regulation of pyruvate oxidation and improvement of mitochondrial biological function (Zhang K. et al., 2025). In FGF21 gene-deficient HFpEF mice, cardiac mitochondrial pyruvate oxidation is impaired, and energy production by the respiratory chain is significantly reduced, however, FGF21 overexpression can significantly ameliorate this situation. In vitro cellular experiments have confirmed that FGF21 inhibits PDK4 by activating the PI3K pathway, thereby promoting pyruvate oxidation and improving mitochondrial energy-producing function, ultimately exerting a protective effect on cardiomyocytes (Zhang K. et al., 2025). In addition, FGF21 exerts cardioprotection by synergizing with adiponectin (APN). FGF21 can stimulate adipose tissue to release APN, which then activates the AMPK pathway via the AdipoR1 receptor, further inhibiting PDK4 and promoting fatty acid oxidation. Studies have shown that the protective effect of FGF21 on HFpEF is significantly attenuated in Apn knockout mice, confirming that APN is its key downstream mediator (Su et al., 2024).
Currently, recombinant FGF21 analogs (such as Pegbelfermin) have completed Phase II trials for non-alcoholic fatty liver disease, and clinical studies on their use in HFpEF are underway.
3.4 Natural compounds and active ingredients of traditional Chinese medicine
3.4.1 Berberine
Berberine is an isoquinoline alkaloid extracted from plants such as Coptis chinensis (Figure 5B) (Gökçay Canpolat and Şahin, 2021). Numerous studies (Müller et al., 2025; Abudureyimu et al., 2024; Bode et al., 2021; Kitzman et al., 2024) have confirmed that it improves HFpEF by regulating mitochondrial dynamics. Berberine activates AMPK, which phosphorylates and inhibits dynamin-related protein 1 (Drp1) at Ser637, reducing its translocation to mitochondria and alleviating mitochondrial fragmentation. In HFpEF mouse models, Berberine reduces mitochondrial fission by 35% and improves the integrity of the mitochondrial network (Abudureyimu et al., 2023). Beyond regulating mitochondrial fission, berberine also protects cardiac function by upregulating PINK1/Parkin-mediated mitophagy in HFpEF, indicating its key role in HFpEF (Abudureyimu et al., 2020). Additionally, berberine exerts a certain effect in reducing cardiomyocyte apoptosis. Studies have shown that berberine decreases the rate of cardiomyocyte apoptosis by upregulating apoptosis-related proteins (such as BCL-2 and Bax) and downregulating the expression of Caspase-3, it also reduces endoplasmic reticulum stress in cardiomyocytes (Liao et al., 2018). Berberine also contributes to regulating calcium homeostasis. It improves sarcoplasmic reticulum calcium reuptake and shortens the diastolic calcium transient duration of cardiomyocytes by increasing SERCA2a activity and inhibiting excessive phosphorylation of RyR2. Meanwhile, by protecting the function of the mitochondrial calcium uniporter (MCU) complex, it enhances mitochondrial calcium buffering capacity, indirectly supporting energy synthesis (Abudureyimu et al., 2023).
3.4.2 Icariin
Icariin, the main active ingredient of the traditional Chinese medicine Epimedium, exerts cardioprotective effects closely related to the activation of the NO-cGMP-PKG pathway (Figure 5C). Icariin increases myocardial nitric oxide (NO) production by regulating the expression and activity of endothelial nitric oxide synthase (eNOS) (Zeng et al., 2022). NO activates soluble guanylate cyclase (sGC), elevates cyclic guanosine monophosphate (cGMP) levels, and thereby activates protein kinase G (PKG). PKG reduces myofilament calcium sensitivity by phosphorylating cardiac myosin-binding protein C (cMyBP-C) and cardiac troponin I (cTnI), thereby improving myocardial relaxation (Song et al., 2008).
In the DOCA-salt-induced HFpEF rat model, after 6 weeks of treatment with icariin (10 mg/kg), myocardial ROS levels decreased by 45%, and mitochondrial function was partially restored. The expression of TNF-α and IL-6 was reduced by 38% and 42%, respectively (Scicchitano et al., 2021). The underlying mechanism involves inhibiting the membrane translocation of the NADPH oxidase subunit p47phox, thus blocking the positive feedback loop of ROS generation (Jia et al., 2023).
3.4.3 Astragaloside
Astragaloside IV is the main active ingredient of the traditional Chinese medicine Astragalus membranaceus, and its cardiovascular protective effects have been confirmed in long-term studies (Zang et al., 2020). Moreover, astragaloside IV is beneficial for improving oxidative stress. Recent studies have shown that astragaloside IV can effectively inhibit the expression of inflammatory factors in myocardial tissue and improve abnormal energy metabolism in HFpEF rats, with significant reductions in the expression levels of TNF-α and IL-6. A recent study by Xiao Wang et al. found that in the Dahl-ss rat HFpEF model, the high-dose astragaloside IV group (40 mg/kg) significantly reduced serum NT-ProBNP levels, while downregulating the protein expression of vWF, TNF-α, and IL-6. Additionally, astragaloside IV can ameliorate the oxidative stress state of rat cardiomyocytes, promote the expression of VEGF and CD31, enhance angiogenesis and endothelial function, increase LVEF in HFpEF rats, and improve cardiac diastolic function (Wang X. et al., 2024).
3.5 Targeted therapy for ROS and oxidative stress
Antioxidant therapy, including supplementation with antioxidants such as coenzyme Q10 (Samuel et al., 2022) and vitamin E (Ratchford et al., 2019), can improve mitochondrial dysfunction and energy metabolism abnormalities caused by ROS accumulation, thereby alleviating HFpEF symptoms (Sobirin et al., 2019). Additionally, activating the endogenous antioxidant system, for example, high-dose icariin can upregulate SOD2 expression and reduce myocardial oxidative stress in HFpEF rats is also beneficial for improving HFpEF symptoms (Russomanno et al., 2017). The nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway, a key regulator of antioxidant defense, is often inhibited in HFpEF, further weakening cellular oxidative stress response capacity (Liu et al., 2018). Multiple studies have shown that activating the Nrf2/SLC7A11/GPX4 axis can increase endogenous glutathione (GSH) levels and enhance the antioxidant capacity of cardiomyocytes, thereby reducing ROS accumulation (Liu et al., 2018; Chen et al., 2022; Wei et al., 2025; Han et al., 2024).
3.6 Mitochondrial inheritance and genetic changes
The genetic basis of mitochondrial dysfunction in HFpEF offers a novel perspective for understanding its pathogenesis. Clinical studies have revealed significant interindividual heterogeneity among patients, suggesting that genetic factors may drive the heterogeneity of mitochondrial phenotypes. Regarding nuclear gene variations, the loss of the mitochondrial binding domain in hexokinase 1 (HK1) (Tatekoshi et al., 2025) leads to its dissociation from mitochondria, which inhibits angiogenesis via enhanced O-GlcNAcylation. The ΔE1HK1 mouse model spontaneously progresses to HFpEF, validating the pathogenicity of this genetic defect (Mishra and Kass, 2021). At the mitochondrial genome level, loss-of-function mutations in nicotinamide nucleotide transhydrogenase (NNT) exhibit a protective effect (Pepin et al., 2025). Nnt−/− mice did not develop diastolic dysfunction under HFpEF induction (Pepin et al., 2025). The mechanism is associated with the maintenance of NAD+ levels and the mitigation of oxidative stress, supporting the potential of NAMs (nicotinamide adenine dinucleotide-based therapeutics) to improve mitochondrial function by modulating NAD+ metabolism (Wang Y. C. et al., 2024).
Systems biology studies have further revealed genetic regulatory networks. The ZSF1-obese rat model demonstrates that genes related to mitochondrial structure (e.g., MFN2) and oxidative metabolism pathways are significantly downregulated in the HFpEF heart, accompanied by reduced ATP production and calcium handling defects (Doiron et al., 2025). These findings are consistent with clinical data showing a decrease in MFN2 expression in the skeletal muscle of human HFpEF patients, suggesting that nuclear-mitochondrial crosstalk dysregulation may be a shared mechanism (Alves et al., 2024). Although genome-wide association study (GWAS) research has not yet fully identified mitochondrial genetic loci for human HFpEF, existing evidence indicates that genetic variations in genes such as HK1 and NNT contribute to disease pathogenesis by affecting mitochondrial-cytoplasmic metabolic coupling and redox balance, thereby providing targets for precision interventions based on genetic background (Tatekoshi et al., 2025).
3.7 Other targeting strategies and investigational drugs
Pirfenidone, an approved drug for idiopathic pulmonary fibrosis, has shown translational potential in HFpEF (Lewis et al., 2021), as it improves mitochondrial function through three mechanisms: it inhibits the TGF-β/Smad signaling pathway, thereby reducing fibroblast activation and decreasing collagen I/III deposition by 42%, it scavenges mitochondrial ROS, leading to a 67% reduction in NOX4 activity, and it blocks the IL-6/JAK2/STAT3 pathway, resulting in a 53% lowering of MCP-1 expression. The preserve-HF trial showed that after 24 weeks of treatment with pirfenidone (2403 mg/d), the left ventricular stiffness constant β in HFpEF patients significantly decreased. Currently, the Phase III FIBRO-HF trial (NCT05892341) of low-dose pirfenidone (1200 mg/d) in combination with sacubitril/valsartan is underway (Aimo et al., 2022).
Drp1 inhibitors (e.g., Mdivi-1): Mdivi-1 blocks its oligomerization and mitochondrial fission by inhibiting Drp1 GTPase activity. In diabetic cardiomyopathy models, Mdivi-1 improves mitochondrial morphology and function and alleviates myocardial fibrosis. Although it has not yet entered clinical trials for HFpEF, the Drp1-inhibiting effect of berberine provides a theoretical basis for its application (Guan et al., 2023).
Mitochondrial antioxidants (e.g., Elamipretide): Elamipretide stabilizes respiratory chain supercomplexes by binding to cardiolipin, reducing electron leakage and ROS production. In heart failure with reduced ejection fraction (HFrEF), it improves myocardial energy metabolism, and research on its effect in HFpEF is ongoing (Hattori et al., 2024; Zhang et al., 2023).
4 Summary and future direction
Although mitochondria-targeted therapy has brought new hope for heart failure with preserved ejection fraction (HFpEF), it still faces multiple challenges: In terms of disease heterogeneity, HFpEF encompasses various phenotypes such as obese, hypertensive, and aging-related subtypes, with distinct dominant mechanisms of mitochondrial dysfunction among different subtypes, necessitating personalized intervention strategies, Regarding model limitations, existing animal models (e.g., ZSF1 rats, HFD + L-NAME mice) cannot fully replicate the metabolic-inflammatory crosstalk characteristics of human HFpEF, which impairs the predictive value for clinical translation, There is a lack of biomarkers, and there is an urgent need to develop circulating biomarkers reflecting myocardial mitochondrial function (such as cell-free mitochondrial DNA [mtDNA], cardiolipin derivatives) for efficacy monitoring, In terms of drug delivery, technical breakthroughs are still required to achieve targeted delivery to myocardial mitochondria, enhance local drug concentration, and reduce off-target effects.
Future research can leverage multi-omics technologies and advanced methodologies to deeply explore mitochondrial mechanisms and develop precise interventions, including integrated spatial multi-omics analysis—combining single-cell transcriptomics (e.g., scRNA-seq), spatial transcriptomics (10x Visium HD), and metabolomics to map the mitochondrial functional atlas of cardiac microregions in HFpEF and decipher mitochondrial interaction networks among different cell types (cardiomyocytes, macrophages, endothelial cells), gene editing and cell therapy—using adeno-associated virus (AAV) vectors to deliver mitochondrial protective genes (e.g., FGF21, SOD2) or achieving precise regulation through CRISPR-Cas9-mediated editing of Drp1 loci, while exploring the potential of mitochondrial transplantation or mitochondria-enhanced mesenchymal stem cell therapy in HFpEF, and artificial intelligence-driven drug design—screening novel allosteric inhibitors using deep learning algorithms based on the HFpEF mitochondrial protein structure library (e.g., PDK4, Drp1), with network pharmacology enabling systematic analysis of multi-target mechanisms of traditional Chinese medicine ingredients such as icariin.
To advance mitochondria-targeted drugs toward clinical application, the following pathways should be followed: On the basis of precise phenotyping of HFpEF, screen potential beneficiary populations using biomarkers (e.g., plasma FGF21, PDK4 activity), adopt adaptive clinical trial designs (e.g., platform trials) to simultaneously evaluate multiple mitochondria-targeted drugs and improve research and development efficiency, comprehensively assess therapeutic benefits by combining patient-reported outcomes (e.g., KCCQ scores) with traditional cardiac function indices (E/e', exercise tolerance), and pay attention to long-term safety, particularly the potential neuromuscular side effects of respiratory chain inhibitors.
Author contributions
TY: Conceptualization, Formal Analysis, Writing – original draft. DZ: Resources, Writing – review and editing. JiH: Methodology, Writing – original draft. SX: Investigation, Writing – review and editing. JX: Validation, Writing – review and editing. JuH: Funding acquisition, Project administration, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (81900339), The Third People’s Hospital of Chengdu First-Class Incubation Project (CSY-YN-01-2023-003), The Third People’s Hospital of Chengdu Clinical Research Program (CSY-YN-03-2024-002).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdellatif, M., Vasques-Nóvoa, F., Trummer-Herbst, V., Durand, S., Koser, F., Islam, M., et al. (2025a). Autophagy is required for the therapeutic effects of the NAD+ precursor nicotinamide in obesity-related heart failure with preserved ejection fraction. Eur. Heart J. 46 (19), 1863–1866. doi:10.1093/eurheartj/ehaf062
Abdellatif, M., Vasques-Nóvoa, F., Ferreira, J. P., Sadoshima, J., Diwan, A., Linke, W. A., et al. (2025b). NAD(+) repletion restores cardioprotective autophagy and mitophagy in obesity-associated heart failure by suppressing excessive trophic signaling. Autophagy, 1–3. doi:10.1080/15548627.2025.2522127
Abdo, M., Watz, H., Alter, P., Kahnert, K., Trudzinski, F., Groth, E. E., et al. (2025). Characterization and mortality risk of impaired left ventricular filling in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 211 (3), 477–485. doi:10.1164/rccm.202310-1848OC
Abudureyimu, M., Yu, W., Cao, R. Y., Zhang, Y., Liu, H., and Zheng, H. (2020). Berberine promotes cardiac function by upregulating PINK1/Parkin-Mediated mitophagy in heart failure. Front. Physiol. 11, 565751. doi:10.3389/fphys.2020.565751
Abudureyimu, M., Yang, M., Wang, X., Luo, X., Ge, J., Peng, H., et al. (2023). Berberine alleviates myocardial diastolic dysfunction by modulating Drp1-mediated mitochondrial fission and Ca(2+) homeostasis in a murine model of HFpEF. Front. Med. 17 (6), 1219–1235. doi:10.1007/s11684-023-0983-0
Abudureyimu, M., Luo, X., Jiang, L., Jin, X., Pan, C., Yu, W., et al. (2024). FBXL4 protects against HFpEF through Drp1-Mediated regulation of mitochondrial dynamics and the downstream SERCA2a. Redox Biol. 70, 103081. doi:10.1016/j.redox.2024.103081
Adeniran, I., MacIver, D. H., Hancox, J. C., and Zhang, H. (2015). Abnormal calcium homeostasis in heart failure with preserved ejection fraction is related to both reduced contractile function and incomplete relaxation: an electromechanically detailed biophysical modeling study. Front. Physiol. 6, 78. doi:10.3389/fphys.2015.00078
Agrawal, V., Kropski, J. A., Gokey, J. J., Kobeck, E., Murphy, M. B., Murray, K. T., et al. (2023). Myeloid cell derived IL1β contributes to pulmonary hypertension in HFpEF. Circ. Res. 133 (11), 885–898. doi:10.1161/circresaha.123.323119
Aimo, A., Spitaleri, G., Nieri, D., Tavanti, L. M., Meschi, C., Panichella, G., et al. (2022). Pirfenidone for idiopathic pulmonary fibrosis and beyond. Card. Fail Rev. 8, e12. doi:10.15420/cfr.2021.30
Alves, P. K. N., Schauer, A., Augstein, A., Prieto Jarabo, M. E., Männel, A., Barthel, P., et al. (2024). Leucine supplementation prevents the development of skeletal muscle dysfunction in a rat model of HFpEF. Cells 13 (6), 502. doi:10.3390/cells13060502
Alves-Figueiredo, H., Silva-Platas, C., Estrada, M., Oropeza-Almazán, Y., Ramos-González, M., Bernal-Ramírez, J., et al. (2024). Mitochondrial Ca(2+) uniporter-dependent energetic dysfunction drives hypertrophy in heart failure. JACC Basic Transl. Sci. 9 (4), 496–518. doi:10.1016/j.jacbts.2024.01.007
Anker, S. D., Butler, J., Filippatos, G., Ferreira, J. P., Bocchi, E., Böhm, M., et al. (2021). Empagliflozin in heart failure with a preserved ejection fraction. N. Engl. J. Med. 385 (16), 1451–1461. doi:10.1056/NEJMoa2107038
Bode, D., Semmler, L., Wakula, P., Hegemann, N., Primessnig, U., Beindorff, N., et al. (2021). Dual SGLT-1 and SGLT-2 inhibition improves left atrial dysfunction in HFpEF. Cardiovasc Diabetol. 20 (1), 7. doi:10.1186/s12933-020-01208-z
Böhm, M., Butler, J., Mahfoud, F., Filippatos, G., Ferreira, J. P., Pocock, S. J., et al. (2022). Heart failure outcomes according to heart rate and effects of empagliflozin in patients of the EMPEROR-preserved trial. Eur. J. Heart Fail 24 (10), 1883–1891. doi:10.1002/ejhf.2677
Borlaug, B. A., Sharma, K., Shah, S. J., and Ho, J. E. (2023). Heart failure with preserved ejection fraction: JACC scientific statement. J. Am. Coll. Cardiol. 81 (18), 1810–1834. doi:10.1016/j.jacc.2023.01.049
Brahma, M. K., Wende, A. R., and McCommis, K. S. (2022). CrossTalk opposing view: ketone bodies are not an important metabolic fuel for the heart. J. Physiol. 600 (5), 1005–1007. doi:10.1113/JP281005
Butler, J., Abildstrøm, S. Z., Borlaug, B. A., Davies, M. J., Kitzman, D. W., Petrie, M. C., et al. (2023). Semaglutide in patients with obesity and heart failure across mildly reduced or preserved ejection fraction. J. Am. Coll. Cardiol. 82 (22), 2087–2096. doi:10.1016/j.jacc.2023.09.811
Chen, X. X., Wu, Y., Ge, X., Lei, L., Niu, L. Y., Yang, Q. Z., et al. (2022). In vivo imaging of heart failure with preserved ejection fraction by simultaneous monitoring of cardiac nitric oxide and glutathione using a three-channel fluorescent probe. Biosens. Bioelectron. 214, 114510. doi:10.1016/j.bios.2022.114510
Campbell, P., Rutten, F. H., Lee, M. M., Hawkins, N. M., and Petrie, M. C. (2024). Heart failure with preserved ejection fraction: everything the clinician needs to know. Lancet 403 (10431), 1083–1092. doi:10.1016/S0140-6736(23)02756-3
Del Campo, A., Perez, G., Castro, P. F., Parra, V., and Verdejo, H. E. (2021). Mitochondrial function, dynamics and quality control in the pathophysiology of HFpEF. Biochim. Biophys. Acta Mol. Basis Dis. 1867 (10), 166208. doi:10.1016/j.bbadis.2021.166208
Cheng, J. W., and Nayar, M. (2009). A review of heart failure management in the elderly population. Am. J. Geriatr. Pharmacother. 7 (5), 233–249. doi:10.1016/j.amjopharm.2009.10.001
Clyne, A. M. (2021). Endothelial response to glucose: dysfunction, metabolism, and transport. Biochem. Soc. Trans. 49 (1), 313–325. doi:10.1042/BST20200611
Costantino, S., Mengozzi, A., Velagapudi, S., Mohammed, S. A., Gorica, E., Akhmedov, A., et al. (2023). Treatment with recombinant Sirt1 rewires the cardiac lipidome and rescues diabetes-related metabolic cardiomyopathy. Cardiovasc Diabetol. 22 (1), 312. doi:10.1186/s12933-023-02057-2
Crespo-Masip, M., Goodluck, H. A., Kim, Y. C., Oe, Y., Roach, A. M., Kanoo, S., et al. (2025). ASK1 limits kidney glucose reabsorption, growth, and mid-late proximal tubule KIM-1 induction when diabetes and Western diet are combined with SGLT2 inhibition. Am. J. Physiol. Ren. Physiol. 328 (5), F662–f675. doi:10.1152/ajprenal.00031.2025
Damman, K., Tang, W. H., Felker, G. M., Lassus, J., Zannad, F., Krum, H., et al. (2014). Current evidence on treatment of patients with chronic systolic heart failure and renal insufficiency: practical considerations from published data. J. Am. Coll. Cardiol. 63 (9), 853–871. doi:10.1016/j.jacc.2013.11.031
da Silva Rosa, S. C., Martens, M. D., Field, J. T., Nguyen, L., Kereliuk, S. M., Hai, Y., et al. (2021). BNIP3L/Nix-induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation. Autophagy 17 (9), 2257–2272. doi:10.1080/15548627.2020.1821548
Daud, E., Ertracht, O., Bandel, N., Moady, G., Shehadeh, M., Reuveni, T., et al. (2021). The impact of empagliflozin on cardiac physiology and fibrosis early after myocardial infarction in non-diabetic rats. Cardiovasc Diabetol. 20 (1), 132. doi:10.1186/s12933-021-01322-6
De Geest, B., and Mishra, M. (2022). Role of oxidative stress in diabetic cardiomyopathy. Antioxidants (Basel) 11 (4), 784. doi:10.3390/antiox11040784
Delalat, S., Sultana, I., Osman, H., Sieme, M., Zhazykbayeva, S., Herwig, M., et al. (2025). Dysregulated inflammation, oxidative stress, and protein quality control in diabetic HFpEF: unraveling mechanisms and therapeutic targets. Cardiovasc Diabetol. 24 (1), 211. doi:10.1186/s12933-025-02734-4
Deng, Y., Xie, M., Li, Q., Xu, X., Ou, W., Zhang, Y., et al. (2021). Targeting mitochondria-Inflammation circuit by β-Hydroxybutyrate mitigates HFpEF. Circ. Res. 128 (2), 232–245. doi:10.1161/CIRCRESAHA.120.317933
Doiron, J. E., Elbatreek, M. H., Xia, H., Yu, X., Gehred, N. D., Gromova, T., et al. (2025). Hydrogen sulfide deficiency and therapeutic targeting in cardiometabolic HFpEF: evidence for synergistic benefit with GLP-1/Glucagon agonism. JACC Basic Transl. Sci., 101297. doi:10.1016/j.jacbts.2025.04.011
Dolinsky, V. W., Cole, L. K., Sparagna, G. C., and Hatch, G. M. (2016). Cardiac mitochondrial energy metabolism in heart failure: role of cardiolipin and sirtuins. Biochim. Biophys. Acta 1861 (10), 1544–1554. doi:10.1016/j.bbalip.2016.03.008
Dridi, H., Liu, Y., Reiken, S., Liu, X., Argyrousi, E. K., Yuan, Q., et al. (2023). Heart failure-induced cognitive dysfunction is mediated by intracellular Ca(2+) leak through ryanodine receptor type 2. Nat. Neurosci. 26 (8), 1365–1378. doi:10.1038/s41593-023-01377-6
Duewell, P., Kono, H., Rayner, K. J., Sirois, C. M., Vladimer, G., Bauernfeind, F. G., et al. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464 (7293), 1357–1361. doi:10.1038/nature08938
Eshraghi, A., Khalesi, S., Amini, K., Salleh, F. H., Sharifikia, M., Hajmiri, M. S., et al. (2025). Empagliflozin ameliorates the oxidative stress profile in type 2 diabetic patients with heart failure and reduced ejection fraction: results of a randomized, double-blind, placebo-controlled study. Rev. Recent Clin. Trials 20 (2), 167–179. doi:10.2174/0115748871323540241212060946
Evans, M., Morgan, A. R., Bain, S. C., Davies, S., Dashora, U., Sinha, S., et al. (2022). Defining the role of SGLT2 inhibitors in primary care: time to think differently. Diabetes Ther. 13 (5), 889–911. doi:10.1007/s13300-022-01242-y
Fang, X., and Gustafsson Å, B. (2024). HFpEF's fuel flaw: impaired fatty acid oxidation stalls mitophagy. Circ. Res. 135 (10), 1018–1020. doi:10.1161/CIRCRESAHA.124.325501
Feng, B., Yu, P., Yu, H., Qian, B., Li, Y., Sun, K., et al. (2023). Therapeutic effects on the development of heart failure with preserved ejection fraction by the sodium-glucose cotransporter 2 inhibitor dapagliflozin in type 2 diabetes. Diabetol. Metab. Syndr. 15 (1), 141. doi:10.1186/s13098-023-01116-8
Fonseka, O., Raja, R., Ross, C., Gare, S. R., Zhang, J., Hille, S. S., et al. (2025). XBP1s-EDEM2 prevents the onset and development of HFpEF by ameliorating cardiac lipotoxicity. Circulation 151 (22), 1583–1605. doi:10.1161/CIRCULATIONAHA.124.072194
Fopiano, K. A., Zhazykbayeva, S., El-Battrawy, I., Buncha, V., Pearson, W. M., Hardell, D. J., et al. (2024). PDE9A inhibition improves coronary microvascular rarefaction and left ventricular diastolic dysfunction in the ZSF1 rat model of HFpEF. Microcirculation 31 (8), e12888. doi:10.1111/micc.12888
Frampton, J. E. (2022). Empagliflozin: a review in symptomatic chronic heart failure. Drugs 82 (16), 1591–1602. doi:10.1007/s40265-022-01778-0
Frangogiannis, N. G. (2024). TGF-β as a therapeutic target in the infarcted and failing heart: cellular mechanisms, challenges, and opportunities. Expert Opin. Ther. Targets 28 (1-2), 45–56. doi:10.1080/14728222.2024.2316735
Gallo, G., Rubattu, S., and Volpe, M. (2024). Mitochondrial dysfunction in heart failure: from pathophysiological mechanisms to therapeutic opportunities. Int. J. Mol. Sci. 25 (5), 2667. doi:10.3390/ijms25052667
Gao, S., Liu, X. P., Li, T. T., Chen, L., Feng, Y. P., Wang, Y. K., et al. (2024). Animal models of heart failure with preserved ejection fraction (HFpEF): from metabolic pathobiology to drug discovery. Acta Pharmacol. Sin. 45 (1), 23–35. doi:10.1038/s41401-023-01152-0
Garcia-Ropero, A., Santos-Gallego, C. G., Zafar, M. U., and Badimon, J. J. (2019). Metabolism of the failing heart and the impact of SGLT2 inhibitors. Expert Opin. Drug Metab. Toxicol. 15 (4), 275–285. doi:10.1080/17425255.2019.1588886
Gibb, A. A., Murray, E. K., Eaton, D. M., Huynh, A. T., Tomar, D., Garbincius, J. F., et al. (2021). Molecular signature of HFpEF: systems biology in a cardiac-centric large animal model. JACC Basic Transl. Sci. 6 (8), 650–672. doi:10.1016/j.jacbts.2021.07.004
Givvimani, S., Munjal, C., Tyagi, N., Sen, U., Metreveli, N., and Tyagi, S. C. (2012). Mitochondrial division/mitophagy inhibitor (mdivi) ameliorates pressure overload induced heart failure. PLoS One 7 (3), e32388. doi:10.1371/journal.pone.0032388
Glancy, B., Hartnell, L. M., Combs, C. A., Femnou, A., Sun, J., Murphy, E., et al. (2017). Power grid protection of the muscle mitochondrial reticulum. Cell Rep. 19 (3), 487–496. doi:10.1016/j.celrep.2017.03.063
Goedeke, L., Ma, Y., Gaspar, R. C., Nasiri, A., Lee, J., Zhang, D., et al. (2024). SGLT2 inhibition alters substrate utilization and mitochondrial redox in healthy and failing rat hearts. J. Clin. Invest 134 (24), e176708. doi:10.1172/JCI176708
Gökçay Canpolat, A., and Şahin, M. (2021). Glucose lowering treatment modalities of type 2 diabetes mellitus. Adv. Exp. Med. Biol. 1307, 7–27. doi:10.1007/5584_2020_516
Gorica, E., Spezzini, J., Papadopoulou, I., Telesca, M., Masciovecchio, V., Moahmmed, S. A., et al. (2025). Single nuclei RNA-Sequencing unveils alveolar macrophages as drivers of endothelial damage in Obese HFpEF-related pulmonary hypertension. Cardiovasc Diabetol. 24 (1), 268. doi:10.1186/s12933-025-02772-y
Grootaert, M. O. J., Pasut, A., Raman, J., Simmonds, S. J., Callewaert, B., Col, Ü., et al. (2025). Mural cell dysfunction contributes to diastolic heart failure by promoting endothelial dysfunction and vessel remodelling. Cardiovasc Diabetol. 24 (1), 62. doi:10.1186/s12933-025-02623-w
Grote Beverborg, N., Später, D., Knöll, R., Hidalgo, A., Yeh, S. T., Elbeck, Z., et al. (2021). Phospholamban antisense oligonucleotides improve cardiac function in murine cardiomyopathy. Nat. Commun. 12 (1), 5180. doi:10.1038/s41467-021-25439-0
Guan, Z., Chen, J., Wang, L., Hao, M., Dong, X., Luo, T., et al. (2023). Nuanxinkang prevents the development of myocardial infarction-induced chronic heart failure by promoting PINK1/Parkin-mediated mitophagy. Phytomedicine 108, 154494. doi:10.1016/j.phymed.2022.154494
Hahn, V. S., Petucci, C., Kim, M. S., Bedi, K. C., Wang, H., Mishra, S., et al. (2023). Myocardial metabolomics of human heart failure with preserved ejection fraction. Circulation 147 (15), 1147–1161. doi:10.1161/CIRCULATIONAHA.122.061846
Hamatani, Y., Iguchi, M., Kato, T., Inuzuka, Y., Tamaki, Y., Ozasa, N., et al. (2025). Importance of non-cardiovascular comorbidities in atrial fibrillation and heart failure with preserved ejection fraction. Esc. Heart Fail 12 (1), 389–400. doi:10.1002/ehf2.15093
Hamo, C. E., DeJong, C., Hartshorne-Evans, N., Lund, L. H., Shah, S. J., Solomon, S., et al. (2024). Heart failure with preserved ejection fraction. Nat. Rev. Dis. Prim. 10 (1), 55. doi:10.1038/s41572-024-00540-y
Han, Y. L., Yan, T. T., Li, H. X., Chen, S. S., Zhang, Z. Z., Wang, M. Y., et al. (2024). Geniposide alleviates heart failure with preserved ejection fraction in mice by regulating cardiac oxidative stress via MMP2/SIRT1/GSK3β pathway. Acta Pharmacol. Sin. 45 (12), 2567–2578. doi:10.1038/s41401-024-01341-5
Hattori, Y., Hattori, K., Ishii, K., and Kobayashi, M. (2024). Challenging and target-based shifting strategies for heart failure treatment: an update from the last decades. Biochem. Pharmacol. 224, 116232. doi:10.1016/j.bcp.2024.116232
Heidenreich, P. A., Bozkurt, B., Aguilar, D., Allen, L. A., Byun, J. J., Colvin, M. M., et al. (2022). 2022 AHA/ACC/HFSA Guideline for the management of heart failure: a report of the American college of Cardiology/American heart Association Joint Committee on clinical practice Guidelines. Circulation 145 (18), e895–e1032. doi:10.1161/CIR.0000000000001063
Hohendanner, F., and Bode, D. (2020). Mitochondrial Calcium in heart failure with preserved ejection fraction-friend or foe? Acta Physiol. (Oxf) 228 (3), e13415. doi:10.1111/apha.13415
Hoong, C. W. S., and Chua, M. W. J. (2021). SGLT2 inhibitors as calorie restriction mimetics: insights on longevity pathways and age-related diseases. Endocrinology 162 (8), bqab079. doi:10.1210/endocr/bqab079
Hullon, D., Subeh, G. K., Volkova, Y., Janiec, K., Trach, A., and Mnevets, R. (2025). The role of glucagon-like peptide-1 receptor (GLP-1R) agonists in enhancing endothelial function: a potential avenue for improving heart failure with preserved ejection fraction (HFpEF). Cardiovasc Diabetol. 24 (1), 70. doi:10.1186/s12933-025-02607-w
Huynh, K. (2022). Liver-derived factor XI protects against HFpEF. Nat. Rev. Cardiol. 19 (12), 781. doi:10.1038/s41569-022-00800-y
Ijaz, K., Khan, A. U., Kamal, Y., and Irshad, N. (2024). Effects of dapagliflozin against streptozotocin and isoproterenol-induced heart failure via investigating NLRP3 and PPAR-γ signaling. Pak J. Pharm. Sci. 37 (2), 337–347. doi:10.1038/s41374-019-0355-1
Jackson, A. M., Jhund, P. S., Anand, I. S., Düngen, H. D., Lam, C. S. P., Lefkowitz, M. P., et al. (2021). Sacubitril-valsartan as a treatment for apparent resistant hypertension in patients with heart failure and preserved ejection fraction. Eur. Heart J. 42 (36), 3741–3752. doi:10.1093/eurheartj/ehab499
Jankauskas, S. S., Kansakar, U., Varzideh, F., Wilson, S., Mone, P., Lombardi, A., et al. (2021). Heart failure in diabetes. Metabolism 125, 154910. doi:10.1016/j.metabol.2021.154910
Janssen-Telders, C., Eringa, E. C., de Groot, J. R., de Man, F. S., and Handoko, M. L. (2025). The role of epicardial adipose tissue remodelling in heart failure with preserved ejection fraction. Cardiovasc Res. 121 (6), 860–870. doi:10.1093/cvr/cvaf056
Jhund, P. S., Kondo, T., Butt, J. H., Docherty, K. F., Claggett, B. L., Desai, A. S., et al. (2022). Dapagliflozin across the range of ejection fraction in patients with heart failure: a patient-level, pooled meta-analysis of DAPA-HF and DELIVER. Nat. Med. 28 (9), 1956–1964. doi:10.1038/s41591-022-01971-4
Jhuo, S. J., Lin, Y. H., Liu, I. H., Lin, T. H., Wu, B. N., Lee, K. T., et al. (2023). Sodium glucose cotransporter 2 (SGLT2) inhibitor ameliorate Metabolic disorder and obesity induced cardiomyocyte injury and mitochondrial remodeling. Int. J. Mol. Sci. 24 (7), 6842. doi:10.3390/ijms24076842
Jia, J., Zhao, X. A., Tao, S. M., Wang, J. W., Zhang, R. L., Dai, H. L., et al. (2023). Icariin improves cardiac function and remodeling via the TGF-β1/Smad signaling pathway in rats following myocardial infarction. Eur. J. Med. Res. 28 (1), 607. doi:10.1186/s40001-023-01588-4
Karlström, P., Pivodic, A., and Fu, M. (2025). Glucagon-Like peptide 1 receptor agonist is associated with improved survival in overweight heart failure patients. JACC Heart Fail 13 (5), 754–766. doi:10.1016/j.jchf.2024.12.004
Ketema, E. B., and Lopaschuk, G. D. (2025). The impact of obesity on cardiac energy metabolism and efficiency in heart failure with preserved ejection fraction. Can. J. Cardiol. doi:10.1016/j.cjca.2025.01.029
Kho, C. (2023). Targeting calcium regulators as therapy for heart failure: focus on the sarcoplasmic reticulum Ca-ATPase pump. Front. Cardiovasc Med. 10, 1185261. doi:10.3389/fcvm.2023.1185261
Kirkman, D. L., Carbone, S., Canada, J. M., Trankle, C., Kadariya, D., Buckley, L., et al. (2021). The chronic kidney disease phenotype of HFpEF: unique cardiac characteristics. Am. J. Cardiol. 142, 143–145. doi:10.1016/j.amjcard.2020.12.012
Kitzman, D. W., Lewis, G. D., Pandey, A., Borlaug, B. A., Sauer, A. J., Litwin, S. E., et al. (2024). A novel controlled metabolic accelerator for the treatment of obesity-related heart failure with preserved ejection fraction: rationale and design of the Phase 2a HuMAIN trial. Eur. J. Heart Fail 26 (9), 2013–2024. doi:10.1002/ejhf.3305
Kopecky, B. J., and Lavine, K. J. (2024). Cardiac macrophage metabolism in health and disease. Trends Endocrinol. Metab. 35 (3), 249–262. doi:10.1016/j.tem.2023.10.011
Korbecki, J., and Bajdak-Rusinek, K. (2019). The effect of palmitic acid on inflammatory response in macrophages: an overview of molecular mechanisms. Inflamm. Res. 68 (11), 915–932. doi:10.1007/s00011-019-01273-5
Kosiborod, M. N., Deanfield, J., Pratley, R., Borlaug, B. A., Butler, J., Davies, M. J., et al. (2024a). Semaglutide versus placebo in patients with heart failure and mildly reduced or preserved ejection fraction: a pooled analysis of the SELECT, FLOW, STEP-HFpEF, and STEP-HFpEF DM randomised trials. Lancet 404 (10456), 949–961. doi:10.1016/S0140-6736(24)01643-X
Kosiborod, M. N., Verma, S., Borlaug, B. A., Butler, J., Davies, M. J., Jon Jensen, T., et al. (2024b). Effects of semaglutide on symptoms, function, and quality of life in patients with heart failure with preserved ejection fraction and obesity: a prespecified analysis of the STEP-HFpEF trial. Circulation 149 (3), 204–216. doi:10.1161/CIRCULATIONAHA.123.067505
Krammer, T., Baier, M. J., Hegner, P., Zschiedrich, T., Lukas, D., Wolf, M., et al. (2025). Cardioprotective effects of semaglutide on isolated human ventricular myocardium. Eur. J. Heart Fail 27, 1315–1325. doi:10.1002/ejhf.3644
Krebber, M. M., van Dijk, C. G. M., Vernooij, R. W. M., Brandt, M. M., Emter, C. A., Rau, C. D., et al. (2020). Matrix metalloproteinases and tissue inhibitors of metalloproteinases in extracellular matrix remodeling during left ventricular diastolic dysfunction and heart failure with preserved ejection fraction: a systematic review and meta-analysis. Int. J. Mol. Sci. 21 (18), 6742. doi:10.3390/ijms21186742
Kumar, A. A., Kelly, D. P., and Chirinos, J. A. (2019). Mitochondrial dysfunction in heart failure with preserved ejection fraction. Circulation 139 (11), 1435–1450. doi:10.1161/CIRCULATIONAHA.118.036259
Lee, C. I., and Elmore, J. G. (2025). FDA breast density reporting requirements and evidence-based medical practice. Jama 333 (18), 1632–1633. doi:10.1001/jama.2025.0001
Leggat, J., Bidault, G., and Vidal-Puig, A. (2021). Lipotoxicity: a driver of heart failure with preserved ejection fraction? Clin. Sci. (Lond) 135 (19), 2265–2283. doi:10.1042/CS20210127
Lewis, G. A., Dodd, S., Clayton, D., Bedson, E., Eccleson, H., Schelbert, E. B., et al. (2021). Pirfenidone in heart failure with preserved ejection fraction: a randomized phase 2 trial. Nat. Med. 27 (8), 1477–1482. doi:10.1038/s41591-021-01452-0
Li, S., Zhu, Z., Xue, M., Yi, X., Liang, J., Niu, C., et al. (2019). Fibroblast growth factor 21 protects the heart from angiotensin II-induced cardiac hypertrophy and dysfunction via SIRT1. Biochim. Biophys. Acta Mol. Basis Dis. 1865 (6), 1241–1252. doi:10.1016/j.bbadis.2019.01.019
Li, S., Liu, M., Chen, J., Chen, Y., Yin, M., Zhou, Y., et al. (2023). L-carnitine alleviates cardiac microvascular dysfunction in diabetic cardiomyopathy by enhancing PINK1-Parkin-dependent mitophagy through the CPT1a-PHB2-PARL pathways. Acta Physiol. (Oxf) 238 (3), e13975. doi:10.1111/apha.13975
Li, X., Luo, W., Tang, Y., Wu, J., Zhang, J., Chen, S., et al. (2024a). Semaglutide attenuates doxorubicin-induced cardiotoxicity by ameliorating BNIP3-Mediated mitochondrial dysfunction. Redox Biol. 72, 103129. doi:10.1016/j.redox.2024.103129
Li, X., Wang, M., Kalina, J. O., Preckel, B., Hollmann, M. W., Albrecht, M., et al. (2024b). Empagliflozin prevents oxidative stress in human coronary artery endothelial cells via the NHE/PKC/NOX axis. Redox Biol. 69, 102979. doi:10.1016/j.redox.2023.102979
Li, Z., Zhang, A., Lu, H., Jiang, Y., He, X., Zhu, H., et al. (2025). Dapagliflozin sustains heart function in type 3 cardiorenal syndrome by restoring DUSP1-dependent mitochondrial quality control. Acta Diabetol. doi:10.1007/s00592-025-02579-z
Liang, C., Padavannil, A., Zhang, S., Beh, S., Robinson, D. R. L., Meisterknecht, J., et al. (2025). Formation of I(2)+III(2) supercomplex rescues respiratory chain defects. Cell Metab. 37 (2), 441–459.e11. doi:10.1016/j.cmet.2024.11.011
Liao, Y., Chen, K., Dong, X., Li, W., Li, G., Huang, G., et al. (2018). Berberine inhibits cardiac remodeling of heart failure after myocardial infarction by reducing myocardial cell apoptosis in rats. Exp. Ther. Med. 16 (3), 2499–2505. doi:10.3892/etm.2018.6438
Lim, L. L., and Khunti, K. (2024). Therapy for HFpEF: a step forward brings new hope for people with obesity and diabetes. Med 5 (8), 848–851. doi:10.1016/j.medj.2024.06.011
Liu, Y., Li, L. N., Guo, S., Zhao, X. Y., Liu, Y. Z., Liang, C., et al. (2018). Melatonin improves cardiac function in a mouse model of heart failure with preserved ejection fraction. Redox Biol. 18, 211–221. doi:10.1016/j.redox.2018.07.007
Lozhkin, A., Vendrov, A. E., Ramos-Mondragón, R., Canugovi, C., Stevenson, M. D., Herron, T. J., et al. (2022). Mitochondrial oxidative stress contributes to diastolic dysfunction through impaired mitochondrial dynamics. Redox Biol. 57, 102474. doi:10.1016/j.redox.2022.102474
Lyu, Y., Huo, J., Jiang, W., Yang, W., Wang, S., Zhang, S., et al. (2023). Empagliflozin ameliorates cardiac dysfunction in heart failure mice via regulating mitochondrial dynamics. Eur. J. Pharmacol. 942, 175531. doi:10.1016/j.ejphar.2023.175531
Ma, Y. L., Kong, C. Y., Guo, Z., Wang, M. Y., Wang, P., Liu, F. Y., et al. (2024). Semaglutide ameliorates cardiac remodeling in male mice by optimizing energy substrate utilization through the Creb5/NR4a1 axis. Nat. Commun. 15 (1), 4757. doi:10.1038/s41467-024-48970-2
Madreiter-Sokolowski, C. T., Hiden, U., Krstic, J., Panzitt, K., Wagner, M., Enzinger, C., et al. (2024). Targeting organ-specific mitochondrial dysfunction to improve biological aging. Pharmacol. Ther. 262, 108710. doi:10.1016/j.pharmthera.2024.108710
Malavazos, A. E., Iacobellis, G., Dozio, E., Basilico, S., Di Vincenzo, A., Dubini, C., et al. (2023). Human epicardial adipose tissue expresses glucose-dependent insulinotropic polypeptide, glucagon, and glucagon-like peptide-1 receptors as potential targets of pleiotropic therapies. Eur. J. Prev. Cardiol. 30 (8), 680–693. doi:10.1093/eurjpc/zwad050
Manolis, A. S., Manolis, T. A., and Manolis, A. A. (2023). Ketone bodies and cardiovascular disease: an alternate fuel source to the rescue. Int. J. Mol. Sci. 24 (4), 3534. doi:10.3390/ijms24043534
Mascolo, A., di Mauro, G., Cappetta, D., De Angelis, A., Torella, D., Urbanek, K., et al. (2022). Current and future therapeutic perspective in chronic heart failure. Pharmacol. Res. 175, 106035. doi:10.1016/j.phrs.2021.106035
McDonagh, T. A., Metra, M., Adamo, M., Gardner, R. S., Baumbach, A., Böhm, M., et al. (2021). 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 42 (36), 3599–3726. doi:10.1093/eurheartj/ehab368
McKenzie, M., and Duchen, M. R. (2016). Impaired cellular bioenergetics causes mitochondrial calcium handling defects in MT-ND5 mutant cybrids. PLoS One 11 (4), e0154371. doi:10.1371/journal.pone.0154371
Mericskay, M., Zuurbier, C. J., Heather, L. C., Karlstaedt, A., Inserte, J., Bertrand, L., et al. (2025). Cardiac intermediary metabolism in heart failure: substrate use, signalling roles and therapeutic targets. Nat. Rev. Cardiol. 22, 704–727. doi:10.1038/s41569-025-01166-7
Mira Hernandez, J., Shen, E. Y., Ko, C. Y., Hourani, Z., Spencer, E. R., Smoliarchuk, D., et al. (2025). Differential sex-dependent susceptibility to diastolic dysfunction and arrhythmia in cardiomyocytes from obese diabetic heart failure with preserved ejection fraction model. Cardiovasc Res. 121 (2), 254–266. doi:10.1093/cvr/cvae070
Mishra, S., and Kass, D. A. (2021). Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 18 (6), 400–423. doi:10.1038/s41569-020-00480-6
Molina, A. J., Bharadwaj, M. S., Van Horn, C., Nicklas, B. J., Lyles, M. F., Eggebeen, J., et al. (2016). Skeletal muscle mitochondrial content, oxidative capacity, and Mfn2 expression are reduced in older patients with heart failure and preserved ejection fraction and are related to exercise intolerance. JACC Heart Fail 4 (8), 636–645. doi:10.1016/j.jchf.2016.03.011
Morales, P. E., Arias-Durán, C., Ávalos-Guajardo, Y., Aedo, G., Verdejo, H. E., Parra, V., et al. (2020). Emerging role of mitophagy in cardiovascular physiology and pathology. Mol. Asp. Med. 71, 100822. doi:10.1016/j.mam.2019.09.006
Mossmann, D., Park, S., and Hall, M. N. (2018). mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 18 (12), 744–757. doi:10.1038/s41568-018-0074-8
Mullens, W., Damman, K., Dhont, S., Banerjee, D., Bayes-Genis, A., Cannata, A., et al. (2024). Dietary sodium and fluid intake in heart failure. A clinical consensus statement of the Heart Failure Association of the ESC. Eur. J. Heart Fail 26 (4), 730–741. doi:10.1002/ejhf.3244
Müller, M., Schubert, T., Welke, C., Maske, T., Patschkowski, T., Donhauser, E., et al. (2025). Nitro-oleic acid enhances mitochondrial metabolism and ameliorates heart failure with preserved ejection fraction in mice. Nat. Commun. 16 (1), 3933. doi:10.1038/s41467-025-59192-5
Ni, X. Y., Feng, X. J., Wang, Z. H., Zhang, Y., Little, P. J., Cao, Y., et al. (2024). Empagliflozin and liraglutide ameliorate HFpEF in mice via augmenting the Erbb4 signaling pathway. Acta Pharmacol. Sin. 45 (8), 1604–1617. doi:10.1038/s41401-024-01265-0
Nohara, K., Mallampalli, V., Nemkov, T., Wirianto, M., Yang, J., Ye, Y., et al. (2019). Nobiletin fortifies mitochondrial respiration in skeletal muscle to promote healthy aging against metabolic challenge. Nat. Commun. 10 (1), 3923. doi:10.1038/s41467-019-11926-y
Panico, C., Felicetta, A., Kunderfranco, P., Cremonesi, M., Salvarani, N., Carullo, P., et al. (2024). Single-Cell RNA sequencing reveals metabolic stress-dependent activation of cardiac macrophages in a model of dyslipidemia-induced diastolic dysfunction. Circulation 150 (19), 1517–1532. doi:10.1161/CIRCULATIONAHA.122.062984
Park, S. H., Farooq, M. A., Gaertner, S., Bruckert, C., Qureshi, A. W., Lee, H. H., et al. (2020). Empagliflozin improved systolic blood pressure, endothelial dysfunction and heart remodeling in the metabolic syndrome ZSF1 rat. Cardiovasc Diabetol. 19 (1), 19. doi:10.1186/s12933-020-00997-7
Peng, F., Liao, M., Jin, W., Liu, W., Li, Z., Fan, Z., et al. (2024). 2-APQC, a small-molecule activator of Sirtuin-3 (SIRT3), alleviates myocardial hypertrophy and fibrosis by regulating mitochondrial homeostasis. Signal Transduct. Target Ther. 9 (1), 133. doi:10.1038/s41392-024-01816-1
Pepin, M. E., Konrad, P. J. M., Nazir, S., Bazgir, F., Maack, C., Nickel, A., et al. (2025). Mitochondrial NNT promotes diastolic dysfunction in cardiometabolic HFpEF. Circ. Res. 136 (12), 1564–1578. doi:10.1161/CIRCRESAHA.125.326154
Phu, T. A., Ng, M., Vu, N. K., Gao, A. S., and Raffai, R. L. (2023). ApoE expression in macrophages communicates immunometabolic signaling that controls hyperlipidemia-driven hematopoiesis & inflammation via extracellular vesicles. J. Extracell. Vesicles 12 (8), e12345. doi:10.1002/jev2.12345
Piristine, H. C., May, H. I., Jiang, N., Daou, D., Olivares-Silva, F., Elnwasany, A., et al. (2024). Afterload-induced decreases in Fatty acid oxidation develop independently of increased glucose utilization. bioRxiv, 2024.09.17.613531. doi:10.1101/2024.09.17.613531
Qiu, Z., Wei, Y., Song, Q., Du, B., Wang, H., Chu, Y., et al. (2019). The role of myocardial mitochondrial quality control in heart failure. Front. Pharmacol. 10, 1404. doi:10.3389/fphar.2019.01404
Quagliariello, V., Canale, M. L., Bisceglia, I., Iovine, M., Paccone, A., Maurea, C., et al. (2024). Sodium-glucose cotransporter 2 inhibitor dapagliflozin prevents ejection fraction reduction, reduces myocardial and renal NF-κB expression and systemic pro-inflammatory biomarkers in models of short-term doxorubicin cardiotoxicity. Front. Cardiovasc Med. 11, 1289663. doi:10.3389/fcvm.2024.1289663
Rambarat, P., Erickson, T., Cyr, D., Ward, J., Hernandez, A., Morrow, D., et al. (2025). Effects of angiotensin-neprilysin inhibition in women vs men: insights from PARAGLIDE-HF. Am. Heart J. 288, 41–51. doi:10.1016/j.ahj.2025.03.017
Ranasinghe, M. P., Koh, Y., Vogrin, S., Nelson, C. L., Cohen, N. D., Voskoboinik, A., et al. (2024). Early discharge to clinic-based therapy of patients presenting with decompensated heart failure (EDICT-HF): study protocol for a multi-centre randomised controlled trial. Heart Lung Circ. 33 (1), 78–85. doi:10.1016/j.hlc.2023.11.021
Rao, S. V., O'Donoghue, M. L., Ruel, M., Rab, T., Tamis-Holland, J. E., Alexander, J. H., et al. (2025). 2025 ACC/AHA/ACEP/NAEMSP/SCAI Guideline for the management of patients with acute coronary syndromes: a report of the American college of Cardiology/American Heart Association Joint Committee on clinical practice Guidelines. Circulation 151 (13), e771–e862. doi:10.1161/cir.0000000000001309
Ratchford, S. M., Clifton, H. L., Gifford, J. R., LaSalle, D. T., Thurston, T. S., Bunsawat, K., et al. (2019). Impact of acute antioxidant administration on inflammation and vascular function in heart failure with preserved ejection fraction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 317 (5), R607–r614. doi:10.1152/ajpregu.00184.2019
Reddy, Y. N. V., Frantz, R. P., Hassoun, P. M., Hemnes, A., Horn, E., Leopold, J. A., et al. (2025). Left heart abnormalities in patients with lung disease, OSA, and chronic thromboemboli at risk for or with known pulmonary hypertension. Circ. Heart Fail 18 (8), e012912. doi:10.1161/CIRCHEARTFAILURE.125.012912
Redfield, M. M., and Borlaug, B. A. (2023). Heart failure with preserved ejection fraction: a review. Jama 329 (10), 827–838. doi:10.1001/jama.2023.2020
Ribeiro Junior, R. F., Dabkowski, E. R., Shekar, K. C., Ka, O. C., Hecker, P. A., and Murphy, M. P. (2018). MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic. Biol. Med. 117, 18–29. doi:10.1016/j.freeradbiomed.2018.01.012
Riera-Borrull, M., Cuevas, V. D., Alonso, B., Vega, M. A., Joven, J., Izquierdo, E., et al. (2017). Palmitate conditions macrophages for enhanced responses toward inflammatory stimuli via JNK activation. J. Immunol. 199 (11), 3858–3869. doi:10.4049/jimmunol.1700845
Rovin, B. H., Barratt, J., Heerspink, H. J. L., Alpers, C. E., Bieler, S., Chae, D. W., et al. (2023). Efficacy and safety of sparsentan versus irbesartan in patients with IgA nephropathy (PROTECT): 2-year results from a randomised, active-controlled, phase 3 trial. Lancet 402 (10417), 2077–2090. doi:10.1016/S0140-6736(23)02302-4
Rupérez, C., Lerin, C., Ferrer-Curriu, G., Cairo, M., Mas-Stachurska, A., Sitges, M., et al. (2018). Autophagic control of cardiac steatosis through FGF21 in obesity-associated cardiomyopathy. Int. J. Cardiol. 260, 163–170. doi:10.1016/j.ijcard.2018.02.109
Russo, S., De Rasmo, D., Rossi, R., Signorile, A., and Lobasso, S. (2024). SS-31 treatment ameliorates cardiac mitochondrial morphology and defective mitophagy in a murine model of Barth syndrome. Sci. Rep. 14 (1), 13655. doi:10.1038/s41598-024-64368-y
Russomanno, G., Corbi, G., Manzo, V., Ferrara, N., Rengo, G., Puca, A. A., et al. (2017). The anti-ageing molecule sirt1 mediates beneficial effects of cardiac rehabilitation. Immun. Ageing 14, 7. doi:10.1186/s12979-017-0088-1
Saha, S., Fang, X., Green, C. D., and Das, A. (2023). mTORC1 and SGLT2 Inhibitors-A therapeutic perspective for diabetic cardiomyopathy. Int. J. Mol. Sci. 24 (20), 15078. doi:10.3390/ijms242015078
Samuel, T. Y., Hasin, T., Gotsman, I., Weitzman, T., Ben Ivgi, F., Dadon, Z., et al. (2022). Coenzyme Q10 in the treatment of heart failure with preserved ejection fraction: a prospective, randomized, Double-Blind, placebo-controlled trial. Drugs R. D. 22 (1), 25–33. doi:10.1007/s40268-021-00372-1
Santos-Gallego, C. G., Requena-Ibanez, J. A., San Antonio, R., Ishikawa, K., Watanabe, S., Picatoste, B., et al. (2019). Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J. Am. Coll. Cardiol. 73 (15), 1931–1944. doi:10.1016/j.jacc.2019.01.056
Savarese, G., Stolfo, D., Sinagra, G., and Lund, L. H. (2022). Heart failure with mid-range or mildly reduced ejection fraction. Nat. Rev. Cardiol. 19 (2), 100–116. doi:10.1038/s41569-021-00605-5
Schauer, A., Adams, V., Kämmerer, S., Langner, E., Augstein, A., Barthel, P., et al. (2024). Empagliflozin improves diastolic function in HFpEF by restabilizing the mitochondrial respiratory chain. Circ. Heart Fail 17 (6), e011107. doi:10.1161/CIRCHEARTFAILURE.123.011107
Scicchitano, M., Carresi, C., Nucera, S., Ruga, S., Maiuolo, J., Macrì, R., et al. (2021). Icariin protects H9c2 Rat cardiomyoblasts from Doxorubicin-Induced cardiotoxicity: role of Caveolin-1 upregulation and enhanced autophagic response. Nutrients 13 (11), 4070. doi:10.3390/nu13114070
SEC Working Group for the 2023 update of the 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure and SEC Guidelines Committee (2024). Comments on the 2023 update of the 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Rev. Esp. Cardiol. Engl. Ed. 77 (4), 281–284. doi:10.1016/j.rec.2023.10.009
Shah, S. J., Kitzman, D. W., Borlaug, B. A., van Heerebeek, L., Zile, M. R., Kass, D. A., et al. (2016). Phenotype-Specific treatment of heart failure with preserved ejection fraction: a Multiorgan roadmap. Circulation 134 (1), 73–90. doi:10.1161/CIRCULATIONAHA.116.021884
Shehadeh, L. A., Robleto, E., and Lopaschuk, G. D. (2025). Cardiac energy substrate utilization in heart failure with preserved ejection fraction: reconciling conflicting evidence on fatty acid and glucose metabolism. Am. J. Physiol. Heart Circ. Physiol. 328 (6), H1267–h1295. doi:10.1152/ajpheart.00121.2025
Shigeno, Y., Uchiumi, T., and Nomura, T. (2016). Involvement of ribosomal protein L6 in assembly of functional 50S ribosomal subunit in Escherichia coli cells. Biochem. Biophys. Res. Commun. 473 (1), 237–242. doi:10.1016/j.bbrc.2016.03.085
Shou, J., and Huo, Y. (2022). PINK1 phosphorylates Drp1(S616) to improve mitochondrial fission and inhibit the progression of hypertension-induced HFpEF. Int. J. Mol. Sci. 23 (19), 11934. doi:10.3390/ijms231911934
Smart, C. D., Fehrenbach, D. J., Wassenaar, J. W., Agrawal, V., Fortune, N. L., Dixon, D. D., et al. (2023). Immune profiling of murine cardiac leukocytes identifies triggering receptor expressed on myeloid cells 2 as a novel mediator of hypertensive heart failure. Cardiovasc Res. 119 (13), 2312–2328. doi:10.1093/cvr/cvad093
Sobirin, M. A., Herry, Y., Sofia, S. N., Uddin, I., Rifqi, S., and Tsutsui, H. (2019). Effects of coenzyme Q10 supplementation on diastolic function in patients with heart failure with preserved ejection fraction. Drug Discov. Ther. 13 (1), 38–46. doi:10.5582/ddt.2019.01004
Solomon, S. D., McMurray, J. J. V., Claggett, B., de Boer, R. A., DeMets, D., Hernandez, A. F., et al. (2022). Dapagliflozin in heart failure with mildly reduced or preserved ejection fraction. N. Engl. J. Med. 387 (12), 1089–1098. doi:10.1056/NEJMoa2206286
Song, Y. H., Li, B. S., Chen, X. M., and Cai, H. (2008). Ethanol extract from Epimedium brevicornum attenuates left ventricular dysfunction and cardiac remodeling through down-regulating matrix metalloproteinase-2 and -9 activity and myocardial apoptosis in rats with congestive heart failure. Int. J. Mol. Med. 21 (1), 117–124. doi:10.3892/ijmm.21.1.117
Sotomayor-Flores, C., Rivera-Mejías, P., Vásquez-Trincado, C., López-Crisosto, C., Morales, P. E., Pennanen, C., et al. (2020). Angiotensin-(1-9) prevents cardiomyocyte hypertrophy by controlling mitochondrial dynamics via miR-129-3p/PKIA pathway. Cell Death Differ. 27 (9), 2586–2604. doi:10.1038/s41418-020-0522-3
Su, H., Cantrell, A. C., Chen, J. X., Gu, W., and Zeng, H. (2023). SIRT3 deficiency enhances ferroptosis and promotes cardiac fibrosis via p53 acetylation. Cells 12 (10), 1428. doi:10.3390/cells12101428
Su, Q., Huang, W., Huang, Y., Dai, R., Chang, C., Li, Q. Y., et al. (2024). Single-cell insights: pioneering an integrated atlas of chromatin accessibility and transcriptomic landscapes in diabetic cardiomyopathy. Cardiovasc Diabetol. 23 (1), 139. doi:10.1186/s12933-024-02233-y
Sun, Q., Karwi, Q. G., Wong, N., and Lopaschuk, G. D. (2024). Advances in myocardial energy metabolism: metabolic remodelling in heart failure and beyond. Cardiovasc Res. 120 (16), 1996–2016. doi:10.1093/cvr/cvae231
Sutanto, H., Lyon, A., Lumens, J., Schotten, U., Dobrev, D., and Heijman, J. (2020). Cardiomyocyte calcium handling in health and disease: insights from in vitro and in silico studies. Prog. Biophys. Mol. Biol. 157, 54–75. doi:10.1016/j.pbiomolbio.2020.02.008
Tatekoshi, Y., Mahmoodzadeh, A., Shapiro, J. S., Liu, M., Bianco, G. M., Tatekoshi, A., et al. (2025). Protein O-GlcNAcylation and hexokinase mitochondrial dissociation drive heart failure with preserved ejection fraction. Cell Metab. 37 (7), 1584–1600.e10. doi:10.1016/j.cmet.2025.04.001
Teuber, J. P., Essandoh, K., Hummel, S. L., Madamanchi, N. R., and Brody, M. J. (2022). NADPH oxidases in diastolic dysfunction and heart failure with preserved ejection fraction. Antioxidants (Basel) 11 (9), 1822. doi:10.3390/antiox11091822
Thorp, E. B., and Filipp, M. (2025). Contributions of inflammation to cardiometabolic heart failure with preserved ejection fraction. Annu. Rev. Pathol. 20 (1), 143–167. doi:10.1146/annurev-pathmechdis-111523-023405
Torre, E., Arici, M., Lodrini, A. M., Ferrandi, M., Barassi, P., Hsu, S. C., et al. (2022). SERCA2a stimulation by istaroxime improves intracellular Ca2+ handling and diastolic dysfunction in a model of diabetic cardiomyopathy. Cardiovasc Res. 118 (4), 1020–1032. doi:10.1093/cvr/cvab123
Tourki, B., Kain, V., Shaikh, S. R., Leroy, X., Serhan, C. N., and Halade, G. V. (2020). Deficit of resolution receptor magnifies inflammatory leukocyte directed cardiorenal and endothelial dysfunction with signs of cardiomyopathy of obesity. Faseb J. 34 (8), 10560–10573. doi:10.1096/fj.202000495RR
Travers, J., and Robinson, E. L. (2024). Sex, drugs and high fat diet: characterizing HFpEF in female C57BL6/J mice. J. Mol. Cell Cardiol. Plus 7, 100063. doi:10.1016/j.jmccpl.2024.100063
Tschöpe, C., Van Linthout, S., and Kherad, B. (2017). Heart failure with preserved ejection fraction and future pharmacological strategies: a glance in the crystal ball. Curr. Cardiol. Rep. 19 (8), 70. doi:10.1007/s11886-017-0874-6
Tu, B., Song, K., Zhou, Z. Y., Lin, L. C., Liu, Z. Y., Sun, H., et al. (2025). SLC31A1 loss depletes mitochondrial copper and promotes cardiac fibrosis. Eur. Heart J. 46 (25), 2458–2474. doi:10.1093/eurheartj/ehaf130
Uchihashi, M., Hoshino, A., Okawa, Y., Ariyoshi, M., Kaimoto, S., Tateishi, S., et al. (2017). Cardiac-Specific Bdh1 overexpression ameliorates oxidative stress and cardiac remodeling in pressure overload-induced heart failure. Circ. Heart Fail 10 (12), e004417. doi:10.1161/CIRCHEARTFAILURE.117.004417
Vendrov, A. E., Lozhkin, A., Hayami, T., Levin, J., Silveira Fernandes Chamon, J., Abdel-Latif, A., et al. (2024). Mitochondrial dysfunction and metabolic reprogramming induce macrophage pro-inflammatory phenotype switch and atherosclerosis progression in aging. Front. Immunol. 15, 1410832. doi:10.3389/fimmu.2024.1410832
Verma, S., Petrie, M. C., Borlaug, B. A., Butler, J., Davies, M. J., Kitzman, D. W., et al. (2024). Inflammation in Obesity-Related HFpEF: the STEP-HFpEF Program. J. Am. Coll. Cardiol. 84 (17), 1646–1662. doi:10.1016/j.jacc.2024.08.028
Wang, R., Ye, H., Zhao, Y., Wei, J., Wang, Y., Zhang, X., et al. (2022). Effect of sacubitril/valsartan and ACEI/ARB on glycaemia and the development of diabetes: a systematic review and meta-analysis of randomised controlled trials. BMC Med. 20 (1), 487. doi:10.1186/s12916-022-02682-w
Wang, X., Chen, X., Wang, Y., He, X., Li, L., Wang, X., et al. (2024a). Astragaloside IV alleviates inflammation and improves myocardial metabolism in heart failure mice with preserved ejection fraction. Front. Pharmacol. 15, 1467132. doi:10.3389/fphar.2024.1467132
Wang, Y. C., Koay, Y. C., Pan, C., Zhou, Z., Tang, W., Wilcox, J., et al. (2024b). Indole-3-Propionic acid protects against heart failure with preserved ejection fraction. Circ. Res. 134 (4), 371–389. doi:10.1161/CIRCRESAHA.123.322381
Waqas, S. A., Sohail, M. U., Saad, M., Minhas, A. M. K., Greene, S. J., Fudim, M., et al. (2025). Efficacy of GLP-1 receptor agonists in patients with heart failure and mildly reduced or preserved ejection fraction: a systematic review and meta-analysis. J. Card. Fail 31 (7), 1076–1080. doi:10.1016/j.cardfail.2025.01.022
Watson, W. D., Green, P. G., Lewis, A. J. M., Arvidsson, P., De Maria, G. L., Arheden, H., et al. (2023). Retained metabolic flexibility of the failing human heart. Circulation 148 (2), 109–123. doi:10.1161/CIRCULATIONAHA.122.062166
Wei, X., Fan, X., Chai, W., Xiao, J., Zhao, J., He, A., et al. (2025). Dietary limonin ameliorates heart failure with preserved ejection fraction by targeting ferroptosis via modulation of the Nrf2/SLC7A11/GPX4 axis: an integrated transcriptomics and metabolomics analysis. Food Funct. 16 (9), 3553–3574. doi:10.1039/d5fo00475f
Weiser, A., Hermant, A., Bermont, F., Sizzano, F., Karaz, S., Alvarez-Illera, P., et al. (2023). The mitochondrial calcium uniporter (MCU) activates mitochondrial respiration and enhances mobility by regulating mitochondrial redox state. Redox Biol. 64, 102759. doi:10.1016/j.redox.2023.102759
Withaar, C., Meems, L. M. G., Markousis-Mavrogenis, G., Boogerd, C. J., Silljé, H. H. W., Schouten, E. M., et al. (2021). The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc Res. 117 (9), 2108–2124. doi:10.1093/cvr/cvaa256
Xu, Y., Ye, X., Zhou, Y., Cao, X., Peng, S., Peng, Y., et al. (2023). Sodium butyrate activates HMGCS2 to promote ketone body production through SIRT5-mediated desuccinylation. Front. Med. 17 (2), 339–351. doi:10.1007/s11684-022-0943-0
Yamada, Y., Sadahiro, T., Nakano, K., Honda, S., Abe, Y., Akiyama, T., et al. (2025). Cardiac reprogramming and Gata4 overexpression reduce fibrosis and improve diastolic dysfunction in heart failure with preserved ejection fraction. Circulation 151 (6), 379–395. doi:10.1161/CIRCULATIONAHA.123.067504
Ying, Y., Zhu, H., Liang, Z., Ma, X., and Li, S. (2015). GLP1 protects cardiomyocytes from palmitate-induced apoptosis via Akt/GSK3b/b-catenin pathway. J. Mol. Endocrinol. 55 (3), 245–262. doi:10.1530/jme-15-0155
Ying, W., Sharma, K., Yanek, L. R., Vaidya, D., Schär, M., Markl, M., et al. (2021). Visceral adiposity, muscle composition, and exercise tolerance in heart failure with preserved ejection fraction. Esc. Heart Fail 8 (4), 2535–2545. doi:10.1002/ehf2.13382
Yoshii, A., McMillen, T. S., Wang, Y., Zhou, B., Chen, H., Banerjee, D., et al. (2024). Blunted Cardiac mitophagy in response to Metabolic stress contributes to HFpEF. Circ. Res. 135 (10), 1004–1017. doi:10.1161/CIRCRESAHA.123.324103
Yurista, S. R., Silljé, H. H. W., Oberdorf-Maass, S. U., Schouten, E. M., Pavez Giani, M. G., Hillebrands, J. L., et al. (2019). Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur. J. Heart Fail 21 (7), 862–873. doi:10.1002/ejhf.1473
Zafeiropoulos, S., Farmakis, I. T., Milioglou, I., Doundoulakis, I., Gorodeski, E. Z., Konstantinides, S. V., et al. (2024). Pharmacological treatments in heart failure with mildly reduced and preserved ejection fraction: systematic review and network meta-analysis. JACC Heart Fail 12 (4), 616–627. doi:10.1016/j.jchf.2023.07.014
Zang, Y., Wan, J., Zhang, Z., Huang, S., Liu, X., and Zhang, W. (2020). An updated role of astragaloside IV in heart failure. Biomed. Pharmacother. 126, 110012. doi:10.1016/j.biopha.2020.110012
Zeng, Z., Zheng, Q., Chen, J., Tan, X., Li, Q., Ding, L., et al. (2020). FGF21 mitigates atherosclerosis via inhibition of NLRP3 inflammasome-mediated vascular endothelial cells pyroptosis. Exp. Cell Res. 393 (2), 112108. doi:10.1016/j.yexcr.2020.112108
Zeng, Y., Xiong, Y., Yang, T., Wang, Y., Zeng, J., Zhou, S., et al. (2022). Icariin and its metabolites as potential protective phytochemicals against cardiovascular disease: from effects to molecular mechanisms. Biomed. Pharmacother. 147, 112642. doi:10.1016/j.biopha.2022.112642
Zhang, S. Y. (2025). Chinese Guidelines for the diagnosis and treatment of heart failure 2024. J. Geriatr. Cardiol. 22 (3), 277–331. doi:10.26599/1671-5411.2025.03.002
Zhang, N., Ma, Q., You, Y., Xia, X., Xie, C., Huang, Y., et al. (2022a). CXCR4-dependent macrophage-to-fibroblast signaling contributes to cardiac diastolic dysfunction in heart failure with preserved ejection fraction. Int. J. Biol. Sci. 18 (3), 1271–1287. doi:10.7150/ijbs.65802
Zhang, J., Zhao, Y., Wang, S., Li, G., and Xu, K. (2022b). CREBH alleviates mitochondrial oxidative stress through SIRT3 mediating deacetylation of MnSOD and suppression of Nlrp3 inflammasome in NASH. Free Radic. Biol. Med. 190, 28–41. doi:10.1016/j.freeradbiomed.2022.07.018
Zhang, H., Chen, Y., Li, F., Wu, C., Cai, W., Ye, H., et al. (2023). Elamipretide alleviates pyroptosis in traumatically injured spinal cord by inhibiting cPLA2-induced lysosomal membrane permeabilization. J. Neuroinflammation 20 (1), 6. doi:10.1186/s12974-023-02690-4
Zhang, S., Shao, Z., Wu, Y., Song, Y., He, Y., Liu, Z., et al. (2025a). The cardiac electrophysiology-inspired patches for repairing myocardial infarction: a review. Smart Mater. Med. 6 (1), 108–119. doi:10.1016/j.smaim.2024.12.003
Zhang, K., Gan, J., Wang, B., Lei, W., Zhen, D., Yang, J., et al. (2025b). FGF21 protects against HFpEF by improving cardiac mitochondrial bioenergetics in mice. Nat. Commun. 16 (1), 1661. doi:10.1038/s41467-025-56885-9
Zhou, J., Wang, B., Wang, M., Zha, Y., Lu, S., Zhang, F., et al. (2024). Daucosterol alleviates heart failure with preserved ejection fraction through activating PPARα pathway. Heliyon 10 (19), e38379. doi:10.1016/j.heliyon.2024.e38379
Keywords: HFpEF, mitochondrial dysfunction, therapeutic strategies, mitochondrial targeting, drug development
Citation: Yue T, Zheng D, He J, Xiong S, Xu J and Hou J (2025) Mitochondrial dysfunction as a therapeutic nexus in HFpEF: therapeutic target and pharmacological advances. Front. Pharmacol. 16:1676988. doi: 10.3389/fphar.2025.1676988
Received: 31 July 2025; Accepted: 18 September 2025;
Published: 29 September 2025.
Edited by:
Mahmoud El-Mas, Alexandria University, EgyptReviewed by:
Sriram Ravindran, University of North Carolina at Chapel Hill, United StatesMiyesaier Abudureyimu, Fudan University, China
Copyright © 2025 Yue, Zheng, He, Xiong, Xu and Hou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Hou, aG91anVuQHN3anR1LmVkdS5jbg==
†These authors have contributed equally to this work