Abstract
Background:
Primary Aldosteronism (PA), a form of endocrine hypertension (EH), often manifests as Resistant Hypertension (RHTN). RHTN is an increasingly prevalent clinical condition associated with target organ damage and a poor prognosis. Accurate diagnosis and management of EH and PA are challenging due to their diverse clinical manifestations, complex laboratory findings, and potential drug-drug interactions (DDIs). These DDIs, often overlooked in practice, can complicate the diagnostic and treatment processes.
Case Presentation:
A 56-year-old man with uncontrolled hypertension was admitted to our hospital. He was suspected of having Primary Aldosteronism (PA) and subclinical Cushing’s Syndrome (SCS) based on elevated aldosterone-to-renin ratio (ARR), captopril challenge test results (CCT), and low-dose dexamethasone suppression test (LDDST) results. Adrenal CT showed mild bilateral adrenal hyperplasia. Despite being on six antihypertensive medications, including spironolactone, his blood pressure remained uncontrollable. His medical history revealed prior use of rifampicin for brucellosis. Rifampicin, a CYP450 inducer, caused drug-drug interactions (DDIs), leading to a false-positive dexamethasone suppression test (DST) and reduced efficacy of antihypertensive drugs. After discontinuing rifampicin, his blood pressure was controlled with fewer medications. One month later, repeated ARR and CCT were still positive. Adrenal venous sampling (AVS) indicated bilateral aldosterone secretion without a dominant side, confirming Idiopathic Hyperaldosteronism (IHA). Targeted treatment with MRA led to partial clinical and biochemical remission of PA.
Conclusion:
This case highlights the diagnostic and therapeutic challenges of Endocrine Hypertension (EH) and Primary Aldosteronism complicated by CYP450 enzyme inducers. Specifically, the use of rifampicin, a potent CYP450 inducer, resulted in false-positive diagnostic test results and diminished efficacy of antihypertensive medications, thereby contributing to RHTN. When encountering uncontrolled hypertension, particularly when standard treatments fail, awareness of DDIs is crucial for accurate diagnosis and effective management.
Introduction
Resistant hypertension (RHTN), a severe form of hypertension, is more likely to cause target organ damage and has a poor prognosis. It includes Endocrine hypertension (EH), where elevated blood pressure (BP) is closely linked to hormone secretion, most commonly due to adrenal diseases such as Primary aldosteronism (PA). Additionally, numerous factors influence the diagnosis and treatment of EH, including drug-drug interactions (DDIs). Studies indicate that 10%–20% of hypertensive patients develop resistant hypertension (RHTN), and roughly 5% of these cases are classified as extreme refractory hypertension (Siddiqui and Calhoun, 2017). Prompt identification of the underlying cause can markedly reduce blood pressure or even cure the hypertension. Nevertheless, the differential diagnostic process is not always straightforward in clinical practice. Here, we report a rare case of primary aldosteronism (PA) presenting as RHTN, in which diagnosis and treatment were complicated by DDIs involving rifampicin.
Case presentation
A 56-year-old Chinese man was admitted to the endocrinology department with suspected Primary aldosteronism (PA). He had an 18-year history of hypertension (regularly treated with nifedipine 30 mg QD), spontaneous hypokalemia and a history of cerebral infarction. The blood pressure (BP) had been significantly elevated for 1 month prior to hospitalization, with an admission BP of 174/98 mmHg. Physical examination revealed no Cushingoid features except for being overweight (BMI = 27.6 kg/m2). Laboratory tests revealed a positive aldosterone-to-renin ratio (ARR) due to an elevated plasma aldosterone concentration (PAC = 356.0 pg/mL; reference range: 30–353.0 pg/mL) and a decreased plasma renin concentration (PRC = 1.5µIU/mL; reference range 4.4–46.1μIU/mL). Cortisol levels at 8a.m. and 0a.m. (358.0 nmol/L and 145.0 nmol/L, respectively) suggested an abnormal circadian rhythm. Blood and urine catecholamine levels were within the normal range. Adrenal enhanced CT (Figure 1F1a) showed mild bilateral adrenal hyperplasia. Synthesizing the above situation, the patient was prescribed Doxazosin 4 mg BID and Diltiazem 90 mg BID for drug elution, and was also prescribed Isosorbide mononitrate 40 mg QD due to chest tightness. Two weeks later, he underwent a series of retests.
FIGURE 1
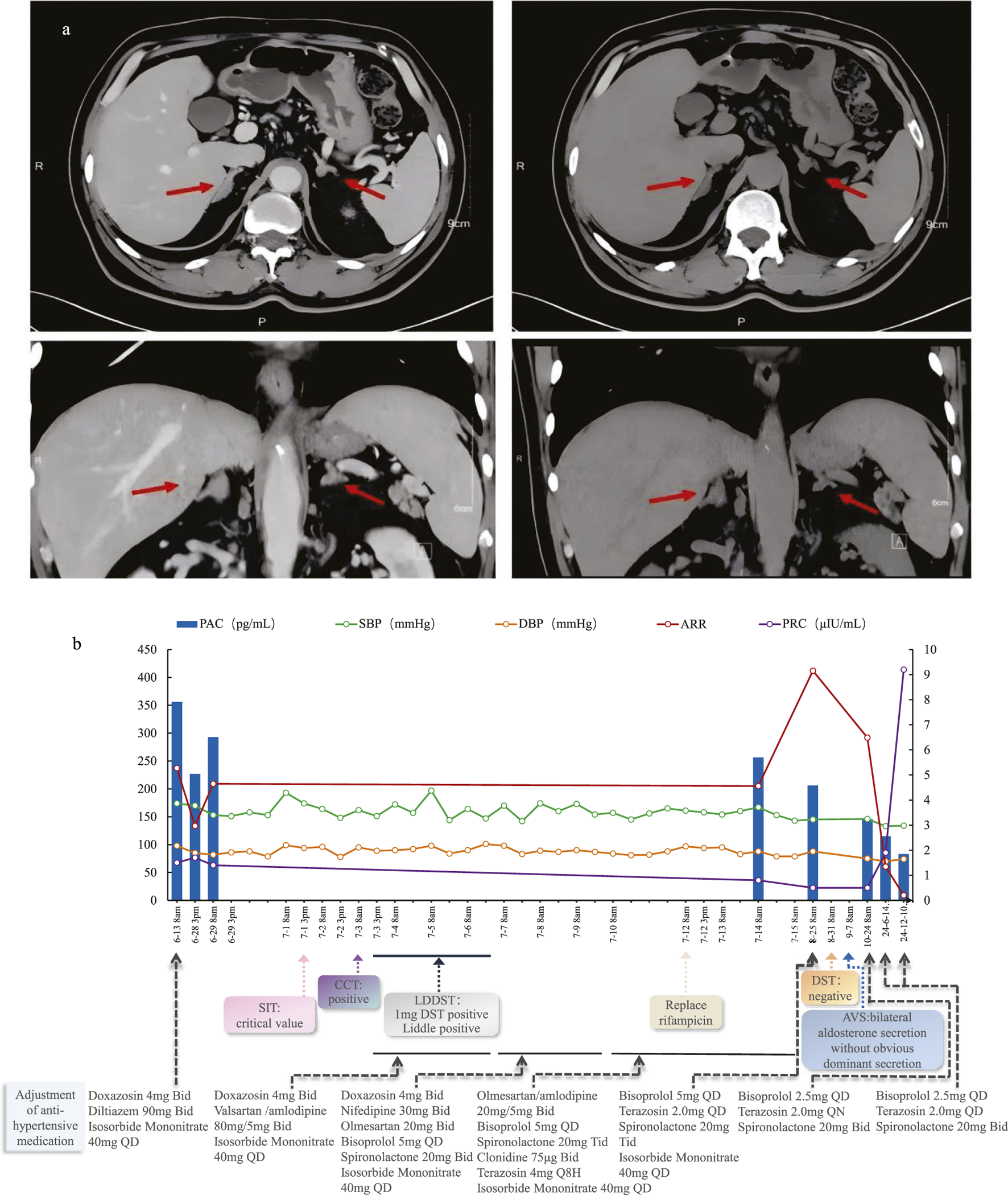
(a) Axial and Coronal adrenal contrast-enhanced CT of this patient; (b) Patient’s diagnostic and treatment process chart.
Routine laboratory tests revealed normal renal function and fasting blood glucose levels, and a negative renal artery ultrasound. An echocardiogram showed an ejection fraction (EF) of 67%, left atrial enlargement, and left ventricular hypertrophy. 24-h ambulatory BP monitoring Before admission indicated the disappearance of the BP circadian rhythm, with a non-dipper curve. His average systolic BP was 177 mmHg, peaking at 211 mmHg, while the average diastolic BP was 97 mmHg, peaking at 120 mmHg. Despite adequate potassium supplement, the serum potassium was only 3.16 mmol/L. A positive result (ARR = 209.29) was obtained again after a 2-week drug washout. Further confirmatory tests revealed that the saline suppression test (SIT) and captopril challenge test (CCT) showed all positive results, confirming PA. We re-evaluated his cortisol levels [(8a.m.-4p.m.-0a.m.) = 187.0-167.6-259.6 nmol/L, respectively; reference range: 166.0–507.0 nmol/L for 8a.m.] and adrenocorticotropic hormone (ACTH) levels [(8a.m.-4p.m.-0a.m.) = 61.3-46.2-66.5 pg/mL, respectively; reference range: 7.2–63.3 pg/mL for 8a.m.], which implied a disrupted ACTH-cortisol rhythm. The overnight 1-mg dexamethasone suppression test (ODST) was performed first, yielding a cortisol level of 113.0 nmol/L. Subsequently, the low-dose 2-day dexamethasone suppression test (LDDST) resulted in a cortisol level of 193 nmol/L. These findings suggested the possibility of endogenous hypercortisolism.
Diagnosis and treatment process
Given the above situation, we considered the following preliminary diagnoses: Endocrine hypertension (EH): Primary aldosteronism (PA), possible Cushing’s syndrome (CS). Although spironolactone (20 mg TID) and amlodipine (5 mg BID) were added for the treatment of PA, there was no significant change in systolic BP. The BP occasionally reached 200/100 mmHg. Subsequent treatment involved bisoprolol (5 mg QD), terazosin (4 mg Q8H), clonidine (75 μg BID), and Olmesartan (20 mg BID) at near-maximum doses, but hypertension remained uncontrolled, indicating the presence of RHTN.
Considering the ACTH levels and positive LDDST results, we initially suspected that Cushing’s Syndrome (CS) was causing the resistant hypertension. However, a pituitary MRI yielded negative findings. Then, after a multidisciplinary team (MDT) discussion and a review of the patient’s medical history, it was revealed that he had been regularly taking rifampicin (600 mg QD) combined with doxycycline (100 mg BID) for brucellosis 1 month before admission. The patient did not disclose this history initially, as he believed that the previous treatment for brucellosis was irrelevant to the current hospitalization. Moreover, upon further reflection, he confirmed that his blood pressure (BP) became uncontrollable after 10 days of brucellosis treatment. Given that rifampicin acts as a hepatic microsomal CYP450 enzyme inducer and can lead to drug-drug interactions (DDIs), we decided to replace it with sulfamethoxazole (SMZ) (Qureshi et al., 2023). Three days after changing from rifampicin to SMZ, the diurnal BP fluctuations improved significantly. This further confirmed that the patient’s uncontrollable BP, despite the use of multiple antihypertensive agents, was associated with the decreased plasma concentrations of some antihypertensive drugs caused by rifampicin, indicating the occurrence of DDIs in this patient’s treatment process. We recommended that the patient undergo follow-up and re-examination in the outpatient clinic 1 month later.
Follow-up and clinical outcome
Two weeks after discharge, the patient’s BP decreased. His antihypertensive regimen had been reduced to spironolactone (20 mg TID), bisoprolol (5 mg QD), terazosin (2.0 mg QD), isosorbide mononitrate (40 mg QD), and potassium supplementation 1.0 g per day. Approximately 1 month after discharge, he underwent an outpatient follow-up, 1 month after completing the brucellosis treatment course. Laboratory tests revealed a plasma aldosterone concentration (PAC) of 206 pg/mL and a positive ARR. The overnight DST was repeated, yielding a negative result (cortisol = 17.6 nmol/L). The previous LDDST results were attributed to rifampicin use and considered falsely positive. The patient was diagnosed with PA but not CS. Adrenal venous sampling (AVS) indicated bilateral aldosterone secretion without a dominant side. The patient was ultimately diagnosed with idiopathic hyperaldosteronism (IHA) and elected to undergo long-term MRA centered medical therapy.
Three months after discharge, his antihypertensive medications had already been reduced to spironolactone (20 mg BID), bisoprolol (2.5 mg QD), and terazosin (2 mg QD). This antihypertensive regimen remained unchanged until the 1.5-year follow-up after discharge when he returned for another visit. During the 1.5-year follow-up, his PAC was 83.0 pg/mL, PRC was 9.2 µU/mL, and ARR was 9.02. Throughout the entire post-discharge follow-up period, both his blood pressure and potassium levels remained normal. The diagnosis and treatment process is shown in Figure 1F1b.
Discussion and Conclusion
Primary aldosteronism (PA) is the most common cause of endocrine hypertension (EH) and can manifest as resistant hypertension (RHTN). However, some cases of PA are actually complicated by the presence of pseudo-RHTN, which can result from CYP450 enzyme inducers that interfere with diagnostic tests and treatment due to drug-drug interactions (DDIs).
Pharmacokinetic-based DDIs are most common and involve various enzymes, with the cytochrome P450 (CYP450) family being the most critical for human metabolism (Michalets, 1998). As a CYP450 inducer, rifampicin increases drug bioconversion rates and lowers serum drug concentrations, which may reduce efficacy or cause treatment failure. Studies show that the peak induction effect of rifampicin occurs at a daily dose of 600 mg, with higher doses failing to significantly enhance DDIs (Gorski et al., 2003). In this patient, PA was confirmed by hypertension, spontaneous hypokalemia, a positive ARR, and confirmatory CCT. However, the differential diagnosis and treatment of endocrine hypertension were complicated by the use of rifampicin.
CS is a clinical syndrome caused by prolonged excessive cortisol secretion from the adrenal cortex. Some patients with PA can also have concurrent CS. This patient lacked typical CS features but had abnormal initial hormone assays of DST, raising suspicion for subclinical Cushing’s syndrome (SCS). Studies show that approximately 21% of PA patients may have concurrent SCS (Hiraishi et al., 2011). Initially, we hypothesized that the patient’s uncontrollable hypertension was due to combined PA and SCS. However, upon identifying rifampicin use, the dramatic BP fluctuations and false-positive LDDST results were clearly attributed to DDIs.
Drug-drug interactions (DDIs) occur when the pharmacokinetics or pharmacodynamics of a drug are altered by the presence of another drug, potentially leading to adverse effects or changes in therapeutic efficacy. These interactions can be influenced by factors such as drug-metabolizing enzymes, like CYP450, and drug transport proteins (Subr et al.). Rifampicin, an inducer of hepatic microsomal CYP450 enzymes including CYP3A4, CYP2C19, CYP2B6, CYP2C8, and CYP2C9 et al., accelerates the metabolism of various drugs like glucocorticoids, warfarin, and oral contraceptives that are metabolized by the CYP450 enzyme family (Baciewicz and Self, 1984). Among these, CYP3A4 is involved in the metabolism of a large proportion (30%–50%) of clinically used drugs (Liu et al., 2005; Haddad et al., 2007). The enzyme activity changes induced by rifampicin peak 1–2 weeks after oral administration (Bettonte et al., 2023). By inducing CYP3A4 activity, rifampicin can increase the clearance of dexamethasone fivefold and decrease its half-life threefold (Kawai, 1985; Evans et al., 1985). This results in reduced dexamethasone concentrations, which fail to suppress endogenous cortisol, leading to the aforementioned false-positive results in the DST (Chinese Pituitar y Adenoma Collaboration Group, 2016).
Additionally, the biosynthesis and catabolic metabolism of cortisol, aldosterone, and sex steroid hormones also involve multiple enzymes within the CYP450 family. Rifampicin enhances cortisol catabolism, thereby reducing plasma cortisol concentrations. This reduction in cortisol levels leads to elevated ACTH levels, which in turn stimulate an increased rate of cortisol synthesis (Edwards et al., 1974; Elias and Gwinup, 1980). The impact of rifampicin on the metabolism of aldosterone and catecholamines remains to be fully elucidated. However, it has been demonstrated that rifampicin can interfere with high-performance liquid chromatography (HPLC) measurements of urinary catecholamines, resulting in falsely elevated results (Kim et al., 2015).
Furthermore, most antihypertensive drugs are substrates of CYP450 enzymes. Since rifampicin can induce these enzymes, it leads to decreased blood concentrations and reduced efficacy of the antihypertensive medications (see Table 1), thereby causing elevated blood pressure. Consequently, more targeted selection of antihypertensive drugs is required for patients receiving concurrent treatment with rifampicin. Among the antihypertensive drugs available at our hospital, we selected olmesartan (which is not affected by CYP450), amlodipine (which is relatively less affected), and terazosin (for which no definitive evidence of interaction has been observed) for this patient’s subsequent antihypertensive treatment. Although the efficacy of bisoprolol was affected by rifampicin’s CYP enzyme induction, it was still retained and continued to be used to alleviate sympathetic excitation. In this patient’s case, during the period when he was using rifampicin, his antihypertensive regimen was gradually increased to six medications (even with concurrent treatment for PA with mineralocorticoid receptor antagonists such as spironolactone). Despite this, all medications were administered at near-maximal doses, yet his hypertension remained poorly controlled.
TABLE 1
| Drug category | Drug name | Antihypertensive efficacy | The effect of rifampicin on the CYP450 enzyme system | |||
|---|---|---|---|---|---|---|
| CYP3A4 | CYP2C19 | CYP2C9 | CYP2D6 | |||
| CCB | Nifedipine (Yoshimoto et al., 1996) | ↓ | activity↑ | - | - | - |
| Amlodipine (Ye et al., 2019) |
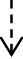
|
activity↑ | - | - | - | |
| Verapamil (Barbarash, 1985) | ↓ | activity↑ | - | - | - | |
| Diltiazem (Adebayo et al., 1989) | ↓ | activity↑ | - | - | - | |
| Beta blocker | Metoprolol (Molden and Spigset, 2011) | ↓ | - | - | - | activity↑ |
| Propranolol (McGinnity et al., 2000) | ↓ | - | - | - | activity↑ | |
| Bisoprolol (Horikiri et al., 1998) | ↓ | activity↑ | - | - | activity↑ | |
| ACEI | Captopril (Gan et al., 2014) | - | - | - | - | - |
| ARB | Losartan (Williamson et al., 1998; Joy et al., 2009) | ↓ | activity↑ | - | activity↑ | - |
| Valsartan (Marino and Vachharajani, 2001) | ↓ | - | - | activity↑ | - | |
| Olmesartan (Laeis et al., 2001) | - | - | - | - | - | |
| Alpha blocker | Prazosin (Akduman and Crawford, 2001) | - | - | - | - | - |
| Terazosin | — | — | — | — | — | |
| Diuretic | Indapamide (Yan et al., 2012; Sun et al., 2009) | ↓ | activity↑ | activity↑ | - | - |
| Furosemide (Gan et al., 2014) | - | - | - | - | - | |
| Hydrochlorothiazide (Gan et al., 2014) | - | - | - | - | - | |
| Active metabolites of Spironolactone (Soliman et al., 2025; Niwa et al., 2020) | ↓ | activity↑ | - | - | - | |
The Mechanism by which Rifampicin Affects the Efficacy of Some Antihypertensive Drugs.
**CCB: calcium channel blockers; ACEI: angiotensin converting enzyme inhibitors; ARB: angiotensin receptor blockers; -: no effect;/: not reported;: The dashed line indicates that amlodipine, which is both metabolized by and acts as an inhibitor of CYP3A4, partially counteracts the CYP3A4 activation induced by rifampicin.
Spironolactone is a prodrug extensively metabolized in the liver to active metabolites, mainly 7α-thiomethyl-spironolactone (7α-TMS) and canrenone, of which 7α-TMS shows greater and more sustained mineralocorticoid receptor antagonism than the parent drug (Soliman et al., 2025; Overdiek and Merkus, 1987; ALDACTONE, 2008). Although spironolactone itself is not primarily metabolized by cytochrome P450 enzymes, 7α-TMS may undergo CYP3A4-mediated hydroxylation to form the less active 6β-hydroxy-7α-TMS (Soliman et al., 2025; Niwa et al., 2020; ALDACTONE, 2008; Flowers et al., 1989). This oxidative pathway regulates the duration of spironolactone’s pharmacologic effect, and rifampicin-induced CYP3A4 upregulation may accelerate 7α-TMS clearance, thereby reducing therapeutic efficacy (Soliman et al., 2025; Niwa et al., 2020).
After replacing rifampicin with sulfamethoxazole for the subsequent brucellosis treatment, blood pressure variability significantly improved (Figure 1b). Additionally, the enzymatic activity changes induced by rifampicin typically disappear about 2 weeks after discontinuation (Niemi et al., 2003; Imai et al., 2011). Although the exact time required for rifampicin elution is not definitively established, some scholars recommend performing DST 15 days after discontinuation (Abdullah and Nowalid, 2010). After rechecking DST and ARR levels 1 month later, a true negative DST outcome was observed, while both the ARR and CTT values remained elevated. These results ultimately led to a diagnosis of idiopathic hyperaldosteronism (IHA) for the patient, with CS ruled out.
During the follow-up phase of treatment, his hypokalemia was corrected and PRC increased to nearly 10µIU/mL. According to the Primary Aldosteronism Medical Treatment Outcome (PAMO) criteria (Yang et al., 2025), the patient achieved a partial clinical and biochemical response. Additionally, using the defined daily dose (DDD) as a standard and following the DDD analysis method recommended by WHO Collaborating Center (WHOCC, 2017), we calculated the antihypertensive drug consumption of the patient on the day of discharge and at the last follow-up visit. Compared to the period when he was using rifampicin, the total accumulated DDD of antihypertensive medications (excluding the targeted medication-spironolactone) decreased by over 50% (Table 2).
TABLE 2
| Drug name | DDD (mg) | Initial antihypertensives (with rifampicin) | Current antihypertensives (without rifampicin) | ||
|---|---|---|---|---|---|
| Dosage and Usage | Accumulated DDD copies | Dosage and Usage | Accumulated DDD copies | ||
| Spironolactone | 37.5 | 20 mg TID | 1.6 | 20 mg BID | 1.07 |
| Terazosin | 5 | 4 mg Q8H | 2.4 | 2 mg QD | 0.4 |
| Bisoprolol | 10 | 5 mg QD | 0.5 | 2.5 mg QD | 0.25 |
| Amlodipine | 5 | 5 mg BID | 2 | ||
| Olmesartan | 20 | 20 mg BID | 2 | ||
| Clonidine | 0.45 | 75 μg BID | 0.33 | ||
| Total accumulated DDD copies (excluding spironolactone) | 7.23 | 0.65 | |||
Total accumulated DDD copies of the patient’s antihypertensive medications.
Through this study, we optimized the diagnostic and therapeutic algorithm for resistant hypertension (RHTN), with particular emphasis on the role of CYP enzyme disruptors (Figure 2). Additionally, after reviewing relevant literature, we have compiled a reference list of common CYP450 enzyme inducers and their effects on CYP enzymes, including those that reduce antihypertensive drug efficacy and cause blood pressure fluctuations through enzyme induction (Table 3).
FIGURE 2
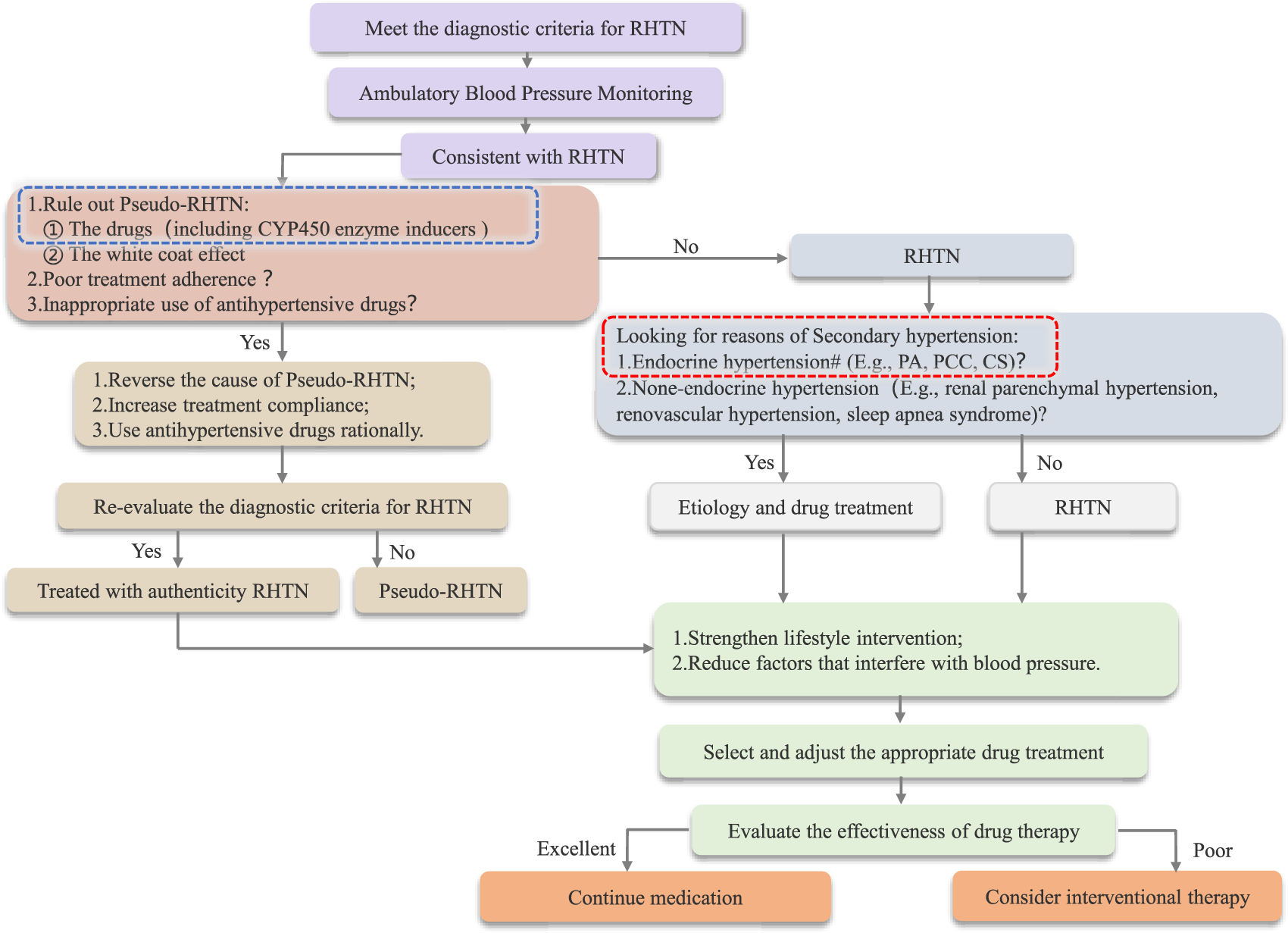
Flowchart Outlining the Diagnosis and Treatment of RHTN *: Drugs that cause DDIs by inducing abnormal metabolic activity of CYP450 enzymes can be categorized into two groups: CYP450 enzyme inducers and inhibitors. #: CYP450 enzyme disruptors can affect endocrine function test results; therefore, it is essential to ensure the proper elution of such disruptors.
TABLE 3
| Enzyme | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Class of inducers | Inducing medication | CYP1A1 | CYP1A2 | CYP2A6 | CYP2B6 | CYP2C8 | CYP2C9 | CYP2C19 | CYP3A4 | CYP3A5 | CYP3A7and CYP3A43 |
| Antibiotic | Rifampicin | W (Hakkola et al., 2020) | M (Hakkola et al., 2020) | M (Stout et al., 2021) | Ma | Ma | S (Hakkola et al., 2020) | Sa | ND | ||
| Dicloxacillin | W (Hakkola et al., 2020) | W (Hakkola et al., 2020) | W (Hakkola et al., 2020) | ||||||||
| Antiepileptics | Carbamazepine | W (Magnusson et al., 2008) | W (Hakkola et al., 2020) | M (Hakkola et al., 2020) | ND | Ma | M (Hakkola et al., 2020) | Sa | ND | ||
| Phenytoin | ND | ND | M (Rodriguez-Vera et al., 2023) | M (Rodriguez-Vera et al., 2023) | M (Hakkola et al., 2020) | ||||||
| Proton pump inhibitors | Omeprazole | ND | Wa | ||||||||
| Glucocorticoids | Dexamethasone | Wa | |||||||||
| Methylprednisolone | W (Hakkola et al., 2020) | ||||||||||
| Prednisolone | W (Hakkola et al., 2020) | ||||||||||
| Prednisone | Min. (Belderbos et al., 2019) | ||||||||||
| Steroidogenesis inhibitors | Mitotane | S (Hakkola et al., 2020) | |||||||||
| Stimulants | Modafinil (and its Renantiomer armodafinil) | Wa | |||||||||
| Nelfinavir | M (Hakkola et al., 2020) | W (Hakkola et al., 2020) | W (Hakkola et al., 2020) | ||||||||
| Antiretrovirals | Ritonavir | M (Hakkola et al., 2020) | W-M (Hakkola et al., 2020) | Min. (Rohr et al., 2024) | M (Rohr et al., 2024) | M (Rohr et al., 2024) | |||||
| Tipranavir | ND | ||||||||||
| Pentobarbital | ND | M (Hakkola et al., 2020) | ND | M (Hakkola et al., 2020) | |||||||
| Barbiturates | Phenobarbital | W (Hakkola et al., 2020) | ND | S (Li et al., 2019) | Wa | W (Hakkola et al., 2020) | Ma | ||||
| Secobarbital | ND | ND | |||||||||
| Antimalarials | Artemisinin | M (Burk et al., 2012) | M (Hakkola et al., 2020) | W (Hakkola et al., 2020) | M (Hakkola et al., 2020) | ||||||
| Estrogens | Ethinyl estradiol (of oral contraceptives) | W (Benowitz et al., 2006) | |||||||||
| Antiandrogens | Apalutamide | S (Hakkola et al., 2020) | W (Hakkola et al., 2020) | M (Hakkola et al., 2020) | |||||||
| Enzalutamide | M (Hakkola et al., 2020) | M (Hakkola et al., 2020) | M (Hakkola et al., 2020) | ||||||||
| Antipyretic analgesic | Metamizole | M (Hakkola et al., 2020) | M (Bachmann et al., 2021) | ||||||||
| Gout medications | Lesinurad | W (G et al., 2017) | |||||||||
| Antiemetics | Aprepitant | W (Hakkola et al., 2020) | W (Shadle et al., 2004) | ||||||||
| Antidiarrheals | Telotristat ethyl | M (FDA, 2017) | |||||||||
| Antineoplastic agents | Vinblastine | W (Smith et al., 2010) | |||||||||
| Bile acid derivatives | Ursodeoxycholic acid | Min. (Dilger et al., 2005) | |||||||||
Main drugs acting as CYP 450 enzyme inducers.
CYP450 induction strength is indicated as follows.
S: strong effect; M: moderate effect; W: weak effect; Min.: minimal effect; ND: Not determined (An inducer, but public AUC/fold-change data are lacking for strength classification).
We thank Dr. David A. flockhart and the division of clinical pharmacology, Indiana University School of Medicine, for maintaining the Cytochrome P450 Drug Interaction Table (https://drug-interactions.medicine.iu.edu), which served as the key reference for the asterisked data in Table 3.
Beyond its potent CYP450 enzymes, rifampicin also upregulates intestinal and hepatic P-glycoprotein (P-gp/ABCB1) expression by approximately 3.5-fold in humans. P-gp is an ATP-dependent efflux transporter that actively pumps absorbed substrates back into the intestinal lumen, thereby reducing their net absorption and systemic bioavailability. Consequently, rifampicin primarily affects the oral exposure of P-gp substrates, including endocrine hormones and certain antihypertensive drugs, through intestinal P-gp induction (Greiner et al., 1999; Fromm, 2004).
Previous research has shown that aldosterone and cortisol are physiological but low-affinity substrates of P-gp (Ueda et al., 1992), whereas renin is not. In Caco-2 cell experiments, Crowe and Tan (2012) reported low efflux ratios (ER ≈ 1.5) for aldosterone and cortisol, indicating weak transport activity with limited clinical significance (Crowe and Tan, 2012). Therefore, rifampicin-induced upregulation of P-gp has minimal impact on plasma aldosterone levels, while renin remains unchanged, leaving the ARR largely unaffected. In contrast, dexamethasone is a well-established P-gp substrate (Caco-2 efflux ratio≈2.1) (Crowe and Tan, 2012). Rifampicin, through the combined induction of CYP450 enzymes and P-gp, markedly reduces oral dexamethasone exposure, which may result in false-positive outcomes during the DST.
On the other hand, across antihypertensive drug classes, the influence of P-gp on systemic exposure and therapeutic efficacy varies considerably. Among CCBs, pharmacogenetic studies indicate that amlodipine plasma levels are affected by ABCB1 polymorphisms, suggesting modest P-gp involvement, though its disposition is largely governed by CYP3A metabolism (Kim et al., 2007). Nifedipine is also primarily metabolized by CYP3A and shows no evidence of being a P-gp substrate (Soldner et al., 1999). Diltiazem demonstrates a mild association with P-gp, as animal studies suggest that intestinal efflux may partially limit its absorption (Athukuri and Neerati, 2017), but it also acts as a P-gp inhibitor (Wessler et al., 2013). For β-adrenoceptor blockers, evidence of P-gp substrate activity is limited and varies by agent. Carvedilol appears to be the clinically relevant substrate and also a moderate inhibitor, whereas bisoprolol shows weak affinity, and other β-blockers such as metoprolol, atenolol, and sotalol exhibit negligible interaction (Wessler et al., 2013; Bachmakov et al., 2006). In the ARB and ACEI classes, olmesartan medoxomil exhibits intestinal absorption partly mediated by OATP2B1 at the prodrug stage. The active metabolite, olmesartan, is predominantly excreted into bile through MRP2, with limited evidence of direct involvement of P-gp (Fukazawa et al., 2024). For α1-adrenergic blockers such as terazosin and doxazosin, there is currently very limited evidence confirming P-gp transport. Among diuretics, spironolactone functions as a P-gp modulator rather than a substrate, primarily inducing P-gp expression through pregnane X receptor (PXR) activation in vitro studies (Rigalli et al., 2011).
Collectively, in this case of endocrine hypertension under rifampicin therapy, secondary induction of P-gp may contribute to clinically relevant effects such as false-positive results in the DST and mild, limited reductions in the efficacy of certain antihypertensive agents.
These combined pharmacokinetic mechanisms underscore the importance of careful diagnostic interpretation and therapeutic monitoring when potent enzyme or transporter inducers are used. Nevertheless, in this case, CYP450 enzyme induction remains the principal mechanism driving rifampicin-related reductions in antihypertensive drug exposure, whereas P-gp modulation serves as a confounding factor in these DDIs.
Summary
This paper reviews a patient with Resistant Hypertension (RHTN) and Primary Aldosteronism (PA), highlighting the challenges in managing Endocrine Hypertension (EH) complicated by drug-drug interactions (DDIs). CYP450 enzyme inducers, particularly rifampicin, can interfere with EH diagnostic tests and alter antihypertensive drug metabolism, leading to treatment failure and persistent hypertension. These DDIs are often overlooked in RHTN. When laboratory results and clinical manifestations are discordant, potential influencing factors, including DDIs, should be carefully investigated. This approach can improve diagnostic accuracy and therapeutic outcomes, reducing the risk of misdiagnosis and inappropriate treatment.
Statements
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by the Second Affiliated Hospital, Zhejiang University School of Medicine. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
XS: Writing – original draft, Writing – review and editing, Data curation, Investigation, Validation, Conceptualization. MJ: Data curation, Writing – review and editing, Investigation, Supervision. HY: Writing – review and editing, Investigation, Formal Analysis. ZD: Data curation, Writing – review and editing, Formal Analysis. KC: Writing – review and editing, Formal Analysis. XP: Writing – original draft, Writing – review and editing, Data curation, Supervision, Validation, Conceptualization, Investigation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Zhejiang Provincial Natural Science Foundation (LMS25H070001), the Zhejiang Provincial Medical and Health Technology Project (2020380946, 2022KY172, and 2024KY1024), and the Zhejiang Province Traditional Chinese Medicine Science and Technology Project (2024ZL565). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
The research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Abdullah H. N. Nowalid W. K. (2010). Abnormal dexamethasone suppression tests in a rifampicin-treated patient with suspected cushing's syndrome. Endokrynol. Pol.61 (6), 706–709. Available online at: https://pubmed.ncbi.nlm.nih.gov/21104646/.
2
Adebayo G. I. Akintonwa A. Mabadeje A. F. (1989). Attenuation of rifampicin-induced theophylline metabolism by diltiazem/rifampicin coadministration in healthy volunteers. Eur. J. Clin. Pharmacol.37 (2), 127–131. 10.1007/BF00558219
3
Akduman B. Crawford E. D. (2001). Terazosin, doxazosin, and prazosin: current clinical experience. Urology58 (6 Suppl. 1), 49–54. 10.1016/s0090-4295(01)01302-4
4
Athukuri B. L. Neerati P. (2017). Enhanced oral bioavailability of diltiazem by the influence of gallic acid and ellagic acid in Male wistar rats: involvement of CYP3A and P-gp inhibition. Phytother. Res.31 (9), 1441–1448. 10.1002/ptr.5873
5
ALDACTONE (2008). Revised Aug 2008. Section Clinical Pharmacology. New York, NY: Pfizer Inc. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/012151s062lbl.pdf.
6
Bachmakov I. Werner U. Endress B. Auge D. Fromm M. F. (2006). Characterization of beta-adrenoceptor antagonists as substrates and inhibitors of the drug transporter P-glycoprotein. Fundam. Clin. Pharmacol.20 (3), 273–282. 10.1111/j.1472-8206.2006.00408.x
7
Bachmann F. Duthaler U. Meyer Zu Schwabedissen H. E. Puchkov M. Huwyler J. Haschke M. et al (2021). Metamizole is a moderate cytochrome P450 inducer via the constitutive androstane receptor and a weak inhibitor of CYP1A2. Clin. Pharmacol. Ther.109 (6), 1505–1516. 10.1002/cpt.2141
8
Baciewicz A. M. Self T. H. (1984). Rifampin drug interactions. Arch. Intern. Med.144 (8), 1667–1671. 10.1001/archinte.144.8.1667
9
Barbarash R. A. (1985). Verapamil-rifampin interaction. Drug Intell. Clin. Pharm.19 (7–8), 559–560. 10.1177/106002808501900712
10
Belderbos B. P. S. Hussaarts KGAM van Harten L. J. Oomen-de Hoop E. de Bruijn P. Hamberg P. et al (2019). Effects of prednisone on docetaxel pharmacokinetics in men with metastatic prostate cancer: a randomized drug-drug interaction study. Br. J. Clin. Pharmacol.85 (5), 986–992. 10.1111/bcp.13889
11
Benowitz N. L. Lessov-Schlaggar C. N. Swan G. E. Jacob P. III (2006). Female sex and oral contraceptive use accelerate nicotine metabolism. Clin. Pharmacol. Ther.79 (5), 480–488. 10.1016/j.clpt.2006.01.008
12
Bettonte S. Berton M. Stader F. Battegay M. Marzolini C. (2023). Management of drug interactions with inducers: onset and disappearance of induction on cytochrome P450 3A4 and uridine diphosphate glucuronosyltransferase 1A1 substrates. Eur. J. Drug Metab. Pharmacokinet48 (04), 353–362. 10.1007/s13318-023-00833-9
13
Burk O. Piedade R. Ghebreghiorghis L. Fait J. T. Nussler A. K. Gil J. P. et al (2012). Differential effects of clinically used derivatives and metabolites of artemisinin in the activation of constitutive androstane receptor isoforms. Br. J. Pharmacol.167 (3), 666–681. 10.1111/j.1476-5381.2012.02033.x
14
Chinese Pituitary Adenoma Collaboration Group (2016). Chinese cushing's disease diagnosis and treatment expert consensus (2015). Chin. Med. J.96 (11), 835–840. 10.3760/cma.j.issn.0376-2491.2016.11.002
15
Crowe A. Tan A. M. (2012). Oral and inhaled corticosteroids: differences in P-glycoprotein (ABCB1) mediated efflux. Toxicol. Appl. Pharmacol.260 (3), 294–302. 10.1016/j.taap.2012.03.008
16
Dilger K. Denk A. Heeg M. H. Beuers U. (2005). No relevant effect of ursodeoxycholic acid on cytochrome P450 3A metabolism in primary biliary cirrhosis. Hepatology41 (3), 595–602. 10.1002/hep.20568
17
Edwards O. M. Courtenay-Evans R. J. Galley J. M. Hunter J. Tait A. D. (1974). Changes in cortisol metabolism following rifampicin therapy. Lancet2 (7880), 548–551. Available online at: https://pubmed.ncbi.nlm.nih.gov/4140269/.
18
Elias A. N. Gwinup G. (1980). Effects of some clinically encountered drugs on steroid synthesis and degradation. Metabolism29 (6), 582–595. 10.1016/0026-0495(80)90086-4
19
Evans M. I. Chrousos G. P. Mann D. W. Larsen J. W. Jr Green I. McCluskey J. et al (1985). Pharmacologic suppression of the fetal adrenal gland in utero. Attempted prevention of abnormal external genital masculinization in suspected congenital adrenal hyperplasia. JAMA253 (7), 1015–1020. 10.1001/jama.1985.03350310097034
20
FDA (2017). Clinical pharmacology and biopharmaceutics review: telotristat ethyl [EB/OL]. Silver Spring, MD, United States: U.S. Food and Drug Administration. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208794Orig1s000ClinPharmR.pdf.
21
Flowers N. L. O’Donnell J. P. Colby H. D. (1989). Spironolactone metabolism in target tissues: characteristics of deacetylation in kidney, liver, adrenal cortex, and testes. Drug Metab. Dispos.17 (2), 186–189. 10.1016/s0090-9556(25)08742-2
22
Fromm M. F. (2004). Importance of P-glycoprotein at blood-tissue barriers. Trends Pharmacol. Sci.25 (8), 423–429. 10.1016/j.tips.2004.06.002
23
Fukazawa N. Nishimura T. Orii K. Noguchi S. Tomi M. (2024). Conversion of olmesartan to Olmesartan Medoxomil, A prodrug that improves intestinal absorption, confers substrate recognition by OATP2B1. Pharm. Res.41 (5), 849–861. 10.1007/s11095-024-03687-1
24
Gillen M. Yang C. Wilson D. Valdez S. Lee C. Kerr B. et al (2017). Evaluation of pharmacokinetic interactions between lesinurad, a new selective urate reabsorption inhibitor, and CYP enzyme substrates Sildenafil, amlodipine, tolbutamide, and repaglinide. Clin. Pharmacol. Drug Dev.6 (4), 363–376. 10.1002/cpdd.324
25
Gan D. J. Peng Q. Su H. (2014). How to select antihypertensive drugs for hypertension patients with tuberculosis. Zhonghua Gao Xue Ya Za Zhi22 (4), 301–303. 10.16439/j.cnki.1673-7245.2014.04.003
26
Gorski J. C. Vannaprasaht S. Hamman M. A. Ambrosius W. T. Bruce M. A. Haehner-Daniels B. et al (2003). The effect of age, sex, and rifampin administration on intestinal and hepatic cytochrome P450 3A activity. Clin. Pharmacol. Ther.74 (3), 275–287. 10.1016/S0009-9236(03)00187-5
27
Greiner B. Eichelbaum M. Fritz P. Kreichgauer H. P. von Richter O. Zundler J. et al (1999). The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J. Clin. Invest104 (2), 147–153. 10.1172/JCI6663
28
Haddad A. Davis M. Lagman R. (2007). The pharmacological importance of cytochrome CYP3A4 in the palliation of symptoms: review and recommendations for avoiding adverse drug interactions. Support Care Cancer15 (3), 251–257. 10.1007/s00520-006-0127-5
29
Hakkola J. Hukkanen J. Turpeinen M. Pelkonen O. (2020). Inhibition and induction of CYP enzymes in humans: an update. Arch. Toxicol.94 (11), 3671–3722. 10.1007/s00204-020-02936-7
30
Hiraishi K. Yoshimoto T. Tsuchiya K. Minami I. Doi M. Izumiyama H. et al (2011). Clinicopathological features of primary aldosteronism associated with subclinical Cushing's syndrome. Endocr. J.58 (7), 543–551. 10.1507/endocrj.k10e-402
31
Horikiri Y. Suzuki T. Mizobe M. (1998). Pharmacokinetics and metabolism of bisoprolol enantiomers in humans. J. Pharm. Sci.87 (3), 289–294. 10.1021/js970316d
32
Imai H. Kotegawa T. Ohashi K. (2011). Duration of drug interactions: putative time courses after mechanism-based inhibition or induction of CYPs. Expert Rev. Clin. Pharmacol.4 (4), 409–411. 10.1586/ecp.11.30
33
Joy M. S. Dornbrook-Lavender K. Blaisdell J. Hilliard T. Boyette T. Hu Y. et al (2009). CYP2C9 genotype and pharmacodynamic responses to losartan in patients with primary and secondary kidney diseases. Eur. J. Clin. Pharmacol.65 (9), 947–953. 10.1007/s00228-009-0707-7
34
Kawai S. (1985). A comparative study of the accelerated metabolism of cortisol, prednisolone and dexamethasone in patients under rifampicin therapy. Nihon Naibunpi Gakkai Zasshi61 (3), 145–161. 10.1507/endocrine1927.61.3_145
35
Kim K. A. Park P. W. Park J. Y. (2007). Effect of ABCB1 (MDR1) haplotypes derived from G2677T/C3435T on the pharmacokinetics of amlodipine in healthy subjects. Br. J. Clin. Pharmacol.63 (1), 53–58. 10.1111/j.1365-2125.2006.02733.x
36
Kim H. K. Ko D. H. Jeong T. D. Lee S. H. Lee W. Lee S. Y. et al (2015). Rifampicin interference in the measurement of urinary catecholamines by high-performance liquid chromatography. Ann. Clin. Lab. Sci.45 (3), 356–359. Available online at: https://pubmed.ncbi.nlm.nih.gov/26116604/.
37
Laeis P. Püchler K. Kirch W. (2001). The pharmacokinetic and metabolic profile of olmesartan medoxomil limits the risk of clinically relevant drug interaction. J. Hypertens. Suppl.19 (1), S21–S32. 10.1097/00004872-200106001-00004
38
Li L. Welch M. A. Li Z. Mackowiak B. Heyward S. Swaan P. W. et al (2019). Mechanistic insights of phenobarbital-mediated activation of human but not mouse Pregnane X receptor. Mol. Pharmacol.96 (3), 345–354. 10.1124/mol.119.116616
39
Liu Z. W. Hu Z. H. Cai Y. Y. (2005). The combined effect of isoniazid and rifampicin on the activities of CYP4501A2 and CYP4503A4 in primary hepatocytes from healthy human adults. Zhonghua Jie He He Hu Xi Za Zhi28 (11), 785–788. Available online at: https://pubmed.ncbi.nlm.nih.gov/16324277/.
40
Magnusson M. O. Dahl M. L. Cederberg J. Karlsson M. O. Sandström R. (2008). Pharmacodynamics of carbamazepine-mediated induction of CYP3A4, CYP1A2, and Pgp as assessed by probe substrates midazolam, caffeine, and digoxin. Clin. Pharmacol. Ther.84 (1), 52–62. 10.1038/sj.clpt.6100431
41
Marino M. R. Vachharajani N. N. (2001). Drug interactions with irbesartan. Clin. Pharmacokinet.40 (8), 605–614. 10.2165/00003088-200140080-00004
42
McGinnity D. F. Parker A. J. Soars M. Riley R. J. (2000). Automated definition of the enzymology of drug oxidation by the major human drug metabolizing cytochrome P450s. Drug Metab. Dispos.28 (11), 1327–1334. 10.1016/s0090-9556(24)15081-7
43
Michalets E. L. (1998). Update: clinically significant cytochrome P-450 drug interactions. Pharmacotherapy18 (1), 84–112. 10.1002/j.1875-9114.1998.tb03830.x
44
Molden E. Spigset O. (2011). Interactions between metoprolol and antidepressants. Tidsskr. Nor. Laegeforen.131 (18), 1777–1779. 10.4045/tidsskr.11.0143
45
Niemi M. Backman J. T. Fromm M. F. Neuvonen P. J. Kivistö K. T. (2003). Pharmacokinetic interactions with rifampicin: clinical relevance. Clin. Pharmacokinet.42 (9), 819–850. 10.2165/00003088-200342090-00003
46
Niwa T. Okamoto A. Narita K. Toyota M. Kato K. Kobayashi K. et al (2020). Comparison of steroid hormone hydroxylation mediated by cytochrome P450 3A subfamilies. Arch. Biochem. Biophys.682, 108283. 10.1016/j.abb.2020.108283
47
Overdiek H. W. Merkus F. W. (1987). The metabolism and biopharmaceutics of spironolactone in man. Rev. Drug Metab. Drug Interact.5 (4), 273–302. 10.1515/dmdi.1987.5.4.273
48
Qureshi K. A. Parvez A. Fahmy N. A. Abdel H. B. H. Kumar S. Ganguly A. et al (2023). Brucellosis: epidemiology, pathogenesis, diagnosis and treatment-a comprehensive review. Ann. Med.55 (2), 2295398. 10.1080/07853890.2023.2295398
49
Rigalli J. P. Ruiz M. L. Perdomo V. G. Villanueva S. S. Mottino A. D. Catania V. A. (2011). Pregnane X receptor mediates the induction of P-glycoprotein by spironolactone in HepG2 cells. Toxicology285 (1-2), 18–24. 10.1016/j.tox.2011.03.015
50
Rodriguez-Vera L. Yin X. Almoslem M. Romahn K. Cicali B. Lukacova V. et al (2023). Comprehensive physiologically based pharmacokinetic model to assess drug-drug interactions of phenytoin. Pharmaceutics15 (10), 2486. 10.3390/pharmaceutics15102486
51
Rohr B. S. Krohmer E. Foerster K. I. Burhenne J. Schulz M. Blank A. et al (2024). Time course of the interaction between oral short-term ritonavir therapy with three factor Xa inhibitors and the activity of CYP2D6, CYP2C19, and CYP3A4 in healthy volunteers. Clin. Pharmacokinet.63 (4), 469–481. 10.1007/s40262-024-01350-x
52
Shadle C. R. Lee Y. Majumdar A. K. Petty K. J. Gargano C. Bradstreet T. E. et al (2004). Evaluation of potential inductive effects of aprepitant on cytochrome P450 3A4 and 2C9 activity. J. Clin. Pharmacol.44 (3), 215–223. 10.1177/0091270003262950
53
Siddiqui M. Calhoun D. A. (2017). Refractory versus resistant hypertension. Curr. Opin. Nephrol. Hypertens.26 (1), 14–19. 10.1097/MNH.0000000000000286
54
Smith N. F. Mani S. Schuetz E. G. Yasuda K. Sissung T. M. Bates S. E. et al (2010). Induction of CYP3A4 by vinblastine: role of the nuclear receptor NR1I2. Ann. Pharmacother.44 (11), 1709–1717. 10.1345/aph.1P354
55
Soldner A. Christians U. Susanto M. Wacher V. J. Silverman J. A. Benet L. Z. (1999). Grapefruit juice activates P-glycoprotein-mediated drug transport. Pharm. Res.16 (4), 478–485. 10.1023/a:1011902625609
56
Soliman A. Pippa L. F. Lass J. Leroux S. Vozmediano V. de Moraes N. V. (2025). Model-Informed dose optimization of spironolactone in neonates and infants. Pharm. (Basel)18 (3), 355. 10.3390/ph18030355
57
Stout S. M. Nemerovski C. W. Streetman D. S. Berg M. Hoffman J. Burke K. et al (2021). Interpretation of cytochrome P-450 inhibition and induction effects from clinical data: current standards and recommendations for implementation. Clin. Pharmacol. Ther.109 (1), 82–86. 10.1002/cpt.1918
58
Subramanian A. Adhimoolam M. Kannan S. (2018). Study of drug-drug interactions among the hypertensive patients in a tertiary care teaching hospital. Perspect. Clin. Res.9 (1), 9–14. 10.4103/picr.PICR_145_16
59
Sun H. Moore C. Dansette P. M. Kumar S. Halpert J. R. Yost G. S. (2009). Dehydrogenation of the indoline-containing drug 4-chloro-N-(2-methyl-1-indolinyl)-3-sulfamoylbenzamide (indapamide) by CYP3A4: correlation with in silico predictions. Drug Metab. Dispos.37 (3), 672–684. 10.1124/dmd.108.022707
60
Ueda K. Okamura N. Hirai M. Tanigawara Y. Saeki T. Kioka N. et al (1992). Human P-glycoprotein transports cortisol, aldosterone, and dexamethasone, but not progesterone. J. Biol. Chem.267 (34), 24248–24252. 10.1016/s0021-9258(18)35757-0
61
Wessler J. D. Grip L. T. Mendell J. Giugliano R. P. (2013). The P-glycoprotein transport system and cardiovascular drugs. J. Am. Coll. Cardiol.61 (25), 2495–2502. 10.1016/j.jacc.2013.02.058
62
WHOCC (2017). WHO collaborating centre for drug statistics methodology [EB/OL]. 2017-06-10. Available online at: http://whocc.no/atc_ddd_alterations_cumulative/add_alterations./.
63
Williamson K. M. Patterson J. H. McQueen R. H. Adams K. F. Jr Pieper J. A. (1998). Effects of erythromycin or rifampin on losartan pharmacokinetics in healthy volunteers. Clin. Pharmacol. Ther.63 (3), 316–323. 10.1016/S0009-9236(98)90163-1
64
Yan F. Hu Y. Di B. He P. L. Sun G. (2012). Effects of some antihypertensive drugs on the metabolism and pharmacokinetics of indapamide in rats. J. Pharm. Pharm. Sci.15 (2), 208–220. 10.18433/j3sk5v
65
Yang J. Burrello J. Goi J. Reincke M. Adolf C. Asbach E. et al (2025). Outcomes after medical treatment for primary aldosteronism: an international consensus and analysis of treatment response in an international cohort. Lancet Diabetes Endocrinol.13 (2), 119–133. 10.1016/S2213-8587(24)00308-5
66
Ye H. Yu X. T. Yang S. F. Su F. F. (2019). A comparative analysis of the antihypertensive effects of rifampicin compared to Nitrendipine and Amlodipine. J. Pract. Med.35 (1), 140–142. 10.3969/j.issn.1006-5725.2019.01.031
67
Yoshimoto H. Takahashi M. Saima S. (1996). Influence of rifampicin on antihypertensive effects of dihydropiridine calcium-channel blockers in four elderly patients. Nihon Ronen Igakkai Zasshi33 (9), 692–696. 10.3143/geriatrics.33.692
Summary
Keywords
primary aldosteronism, resistant hypertension, endocrine hypertension, drug-drug interactions, rifampicin
Citation
Song X, Jia M, Yu H, Dong Z, Cheok K and Pan X (2025) Rifampicin-induced challenges in managing endocrine hypertension and primary aldosteronism: a case report and literature review. Front. Pharmacol. 16:1678430. doi: 10.3389/fphar.2025.1678430
Received
02 August 2025
Revised
23 October 2025
Accepted
27 October 2025
Published
07 November 2025
Volume
16 - 2025
Edited by
Muhammad Fawad Rasool, Bahauddin Zakariya University, Pakistan
Reviewed by
Marios Spanakis, University of Crete, Greece
Hiromitsu Imai, Oita University, Japan
Updates
Copyright
© 2025 Song, Jia, Yu, Dong, Cheok and Pan.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Pan, pxpx1987611@hotmail.com; Xiaoxiao Song, xsong103@zju.edu.cn
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.