Abstract
Diabetes mellitus (DM), a globally prevalent metabolic disorder, poses a significant public health threat due to its systemic complications. Recent studies have increasingly recognized the lung as a target organ in diabetic pathology. However, owing to the respiratory system’s complex physiology, the mechanisms underlying DM-associated lung injury remain poorly understood and require further investigation. This review systematically elucidates the multifaceted effects of DM-induced metabolic disturbances on the lung, with a focus on four key pathophysiological axes triggered by hyperglycemic homeostasis, including chronic inflammation, oxidative stress imbalance, endocrine network disruption, and intestinal dysbiosis. Building upon the “metabolism-microbiota-immune” axis framework, this study demonstrates that: persistent hyperglycemia induces pulmonary tissue damage and immune microenvironment disruption through metabolite accumulation and mitochondrial dysfunction; DM-associated intestinal dysbiosis amplifies pulmonary inflammation via the gut-lung axis, mediated by metabolic reprogramming and immune cell trafficking; and metabolic aberration-driven dysregulation of innate/adaptive immunity serves as the pivotal mediator for progressive lung injury. Building on this mechanistic framework, we discuss emerging therapeutic avenues that target metabolic reprogramming, modulation of the gut microbiota, and restoration of immune homeostasis. Promising strategies include repurposed antidiabetic drugs (e.g., SGLT-2 inhibitors, GLP-1 receptor agonists), microbiome-targeted therapies (e.g., fecal microbiota transplantation), and novel immunomodulatory agents. These therapies are offering a new shift towards multi-target treatments for diabetic pulmonary complications.
1 Introduction
Diabetes mellitus (DM) is a systemic metabolic disorder characterized by chronic hyperglycemia resulting from insulin secretion defects and/or insulin resistance (Sacks et al., 2023). According to data from the International Diabetes Federation, the global prevalence of DM among adults aged 20–79 years reached 537 million in 2021, with projections indicating this number will rise to 693 million by 2045 (Cho et al., 2018). In recent years, advances in DM monitoring systems and in-depth research have led to significant progress in understanding the pathogenesis of DM-related complications. Among these, diabetic pulmonary injury has emerged as a novel target organ damage and is increasingly becoming a research focus (Lange et al., 1989; Ahmad et al., 2011). The clinical significance of respiratory complications in DM cannot be overlooked. Growing clinical evidence indicates that these complications pose substantial risks, with diabetic patients exhibiting a two-fold increase in pneumonia-associated mortality (Singh et al., 2020). Furthermore, chronic airway diseases affect approximately 20% of elderly diabetic individuals (Caughey et al., 2010). Moreover, a bidirectional association exists between DM and chronic obstructive pulmonary disease (COPD), with concomitant presentation of these conditions substantially complicating clinical management (Caughey et al., 2013). Collectively, these findings underscore the significant clinical implications of respiratory complications in diabetic patients.
At the pathological level, DM exerts multifaceted effects on pulmonary function through multiple mechanisms, including inflammatory responses, altered immune regulation, and pulmonary microvascular dysfunction (Tiengo et al., 2008; Erener, 2020; Roberts et al., 2018). Early experimental evidence from animal studies in 1997 demonstrated that DM combined with hyperlipidemia induces structural abnormalities in lung tissue, with these alterations being particularly pronounced in hyperlipidemia-associated DM (Popov and Simionescu, 1997). The metabolic disturbances of hyperglycemia, dyslipidemia, and insulin resistance in DM significantly alter systemic metabolic homeostasis. These pathological metabolic states play a crucial role in the development of diabetic complications (Eid et al., 2019). The lungs, with their extensive vascular network, are particularly vulnerable to circulating metabolic byproducts. This anatomical susceptibility suggests that diabetic pulmonary injury may be mechanistically linked to systemic metabolic dysregulation. Emerging evidence highlights the lung as a metabolically active organ, with recent studies identifying dysregulated metabolic pathways that contribute to both pulmonary injury and repair processes in lung diseases (Liu and Summer 2019; Rajesh et al., 2023). Evidence suggests that hyperglycemia directly impairs the metabolic function of pulmonary dendritic cells (DCs) (Nobs et al., 2023) and promotes systemic oxidative stress along with a pro-inflammatory state (Betteridge, 2000). Collectively, these findings indicate that the dysregulated metabolic milieu damages lung tissue through multiple mechanisms, including immune dysregulation, oxidative stress induction, and chronic inflammation.
Notably, the intestinal flora serves as a crucial metabolic regulatory hub, and its dysbiosis may exacerbate pulmonary immune imbalance through the gut-lung axis. Clinical evidence has revealed altered gut microbiome composition in diabetic patients (de Vos et al., 2022). Furthermore, intestinal dysbiosis may independently contribute to metabolic dysregulation. A growing body of evidence indicates that endocrine-disrupting chemicals, particularly those migrating from plastic products and food packaging additives, can induce gut microbiota dysbiosis through disruption of microbial homeostasis. This metabolic perturbation in intestinal flora has been mechanistically linked to impaired glucose regulation in mammalian hosts (Velmurugan et al., 2017). The pulmonary and intestinal mucosal immune systems exhibit bidirectional crosstalk, and DM-induced intestinal flora alterations may further compromise pulmonary mucosal barrier function (Yang et al., 2021; Özçam and Lynch, 2024). Collectively, these findings reveal that aberrant energy metabolism, dysregulated cell death, intestinal dysbiosis, and immune dysfunction collectively constitute the complex pathogenic network underlying diabetic pulmonary injury.
Conventional DM research has predominantly focused on microvascular complications and neural damage, while the pathological mechanisms involving the lung—a highly metabolically active organ—have long been overlooked. Critical questions remain unresolved: How does metabolic dysregulation drive pulmonary injury through inter-organ crosstalk networks? What roles do the microbiome and immune system play in this process? As depicted in Figure 1, we posit that diabetic pulmonary injury results from a synergistic triad of metabolic dysregulation, gut microbiota alterations, and pulmonary immune defects, forming a self-sustaining vicious cycle. Here, we systematically evaluate this “gut-metabolism-immune” axis and highlight promising pharmacological strategies targeting its critical nodes.
FIGURE 1

Crosstalk between metabolism, immunity and the gut microbiota in diabetic lung injury. Diabetic lung injury arises from the synergistic interplay between metabolic dysregulation, gut microbiota alterations, and immune dysfunction. This cooperative axis drives pulmonary pathology through gut-lung signaling, ultimately leading to endothelial barrier disruption, oxidative stress, and fibrotic remodeling.
2 DM-associated metabolic dysregulation
Diabetic pulmonary injury is conceptualized within a framework of a self-perpetuating vicious cycle, driven by the interplay of metabolic dysregulation, gut microbiota disruption, and immune imbalance. The analysis in this review thus begins by delineating the systemic metabolic disturbances inherent to diabetes. This section systematically examines the core mechanistic alterations in carbohydrate, lipid, and amino acid metabolism, which collectively establish the pathological foundation for the subsequent development of multi-organ complications.
In diabetic patients, impaired insulin secretion or insulin resistance disrupts metabolism and cell signal transduction (Sacks et al., 2023; Shahwa et al., 2022). Accumulation of advanced glycation end products (AGEs) resulting from aberrant glycation reactions plays a pivotal role in both DM progression and its associated complications (Khalid et al., 2022). The receptor for advanced glycation end products (RAGE), a transmembrane receptor for AGEs, is abundantly expressed in pulmonary tissue with highly selective localization to the basement membrane of type I alveolar epithelial cells (Fehrenbach et al., 1998). The RAGE pathway amplifies pulmonary damage induced by infection, direct injury, and inflammatory responses (Guo et al., 2012). Thus, the DM-AGEs-RAGE axis may represent a crucial mediator of diabetic pulmonary injury. Furthermore, in gestational diabetes mellitus (GDM) patients, pathological manifestations correlate with circulating AGEs levels (Li and Yang, 2019). Clinical evidence further links GDM to pulmonary sequelae in offspring: maternal hyperglycemia impairs fetal lung development (He et al., 2023) and is associated with reduced pulmonary function in progeny (Yang et al., 2024). Moreover, maternal-neonatal gut microbiota modifications and associated metabolic reprogramming can significantly impair neonatal pulmonary immune function (Stevens et al., 2022; Fonseca et al., 2021). These findings collectively reveal that maternal metabolic disturbances in DM can remotely impact fetal lung development across the placental barrier, closely associated with gut microbiota metabolism. These findings collectively reveal complex crosstalk among diabetic metabotypes, intestinal flora, and immune regulation.
Diabetic patients also showed characteristic lipid metabolism disorders (Golay et al., 1984). For example, disordered ketone body metabolism in DM impairs the synthesis and function of pulmonary surfactant (Jung et al., 2023). Thus, diabetic lipid metabolic alterations frequently elevate the risk of DM-related lung complications (Conte et al., 2024; Eid et al., 2023). In addition, lipid metabolism is closely related to the intestinal flora, which plays a crucial role in diabetic complications through its metabolites. And we will reveal this phenomenon below. This suggests that therapeutic modulation of microbiota shows clinical potential for mitigating diabetic complications.
Furthermore, amino acid metabolism is significantly dysregulated in patients with DM. Clinical evidence indicates that children with type 1 DM (T1DM) exhibit reduced levels of branched-chain amino acids (BCAAs) and decreased Fischer ratios (BCAA/AAA (aromatic amino acid)) (Hassan et al., 2023). Elevated levels of BCAAs (leucine, isoleucine, and valine) have been consistently observed in patients with type 2 DM (T2DM), potentially linking to both immune dysregulation and insulin resistance (Wang et al., 2011). Intriguingly, reduction of fasting plasma BCAAs levels alone is insufficient to improve insulin sensitivity, suggesting a potentially synergistic role of multiple tissues in modulating BCAAs metabolism to alter insulin responsiveness (Blair et al., 2023). The gut microbiota, however, serves as the master regulator of this metabolic network, where microbial BCAAs biosynthesis and catabolism dynamically control systemic BCAAs availability. Collectively, amino acid metabolic disorders can directly or indirectly affect lung function, and intestinal flora is the core regulator of this process.
3 Metabolic dysregulation: molecular bridges between DM and pulmonary injury
DM creates a pathological environment for multi-organ injury through comprehensive dysregulation of carbohydrate, lipid, and amino acid metabolism. Metabolic dysregulation serves as a molecular bridge linking diabetes to pulmonary injury, where systemic imbalances in lipids, amino acids, and glucose generate pathogenic intermediates that travel to the lungs, triggering endothelial barrier breakdown, impaired repair, inflammation, and immune metabolic alterations (Figure 2). Critically, hyperglycemia-mediated injury exhibits distinct organ-specific patterns: experimental animal models reveal tissue-specific metabolic reprogramming (Sas et al., 2016). Notably, metabolites such as AGEs, dysregulated ketone bodies, and BCAAs have been demonstrated to be directly or indirectly associated with structural abnormalities and functional decline in the lung (e.g., impaired surfactant synthesis, diminished respiratory muscle function). This establishes the metabolic basis for considering the lung a target organ in diabetes. Next, we will explore how these circulating metabolites disrupt intrapulmonary metabolic homeostasis, thereby directly linking systemic metabolic dysregulation to local pulmonary pathology.
FIGURE 2
Metabolic Dysregulation as a Bridge from Diabetes to Lung Injury. Diabetes-induced imbalances in lipids, amino acids, and glucose generate pathogenic intermediates that disseminate systemically to the lung. This metabolic insult triggers pulmonary damage via key pathways: endothelial barrier breakdown, impaired tissue repair, inflammatory activation, and immune-metabolic reprogramming, collectively promoting fibrosis and infection susceptibility.
Within the framework of DM-induced metabolic dysregulation (encompassing lipid/amino acid/carbohydrate metabolism), gut microbiota disruption, and loss of immune homeostasis, the lung emerges as a critical nexus of these systemic interactions. Although primarily regarded as a gas-exchange organ, the lung demonstrates remarkable metabolic activity, continuously enduring high oxidative stress and substantial circulatory demands. Its unique microenvironment—where an extensive capillary network interfaces with both inhaled pathogens and circulating metabolites—renders the pulmonary system particularly vulnerable to DM-induced metabolic perturbations.
3.1 Mechanisms of pulmonary metabolic homeostasis
Research indicates that pulmonary secretion, clearance, and other functions are sustained through metabolic pathways including glucose metabolism, lipid metabolism, and oxidative metabolism (Fisher, 1984). Key features of pulmonary glucose metabolism encompass glycolysis and the pentose phosphate pathway, which generate lactate, carbon dioxide, and intermediates for the biosynthesis of lipids, nucleic acids, and amino acids, along with biologically reduced NADPH, NADH and the high-energy compound ATP. Notably, a major fraction (40%–50%) of glucose is metabolized into lactate and pyruvate (Fisher, 1984), which subsequently contribute to fatty acid synthesis and incorporation into pulmonary surfactant lipids (Burgy et al., 2022). The synthesis of pulmonary surfactant represents a hallmark metabolic activity of the lung, which typically maintains a highly active state of lipid metabolism (Burgy et al., 2022). To ensure surfactant renewal, the lung utilizes circulating fatty acids from the bloodstream, which undergo intracellular remodeling before being incorporated into surfactant synthesis and secretion (Harayama et al., 2014). Emerging evidence indicates that pulmonary surfactant not only facilitates respiration but also contributes to host defense by promoting pathogen clearance and immune modulation (Han and Mallampalli, 2015). Furthermore, the lung enhances the clearance of circulating 5-hydroxytryptamine (5-HT; serotonin), a process potentially linked to the metabolic activity of pulmonary capillary endothelial cells and the platelet activation and recruitment (Yu et al., 2009). The serotonin transporter in pulmonary endothelial cells has been identified as a key regulator in these processes, participating in vascular remodeling and adaptation during neonatal transition (Castro et al., 2017). Collectively, these interconnected metabolic networks maintain pulmonary homeostasis, and their dysregulation may trigger pathological cascades.
Growing evidence has linked pulmonary diseases such as pulmonary hypertension, pulmonary fibrosis, and lung cancer to metabolic dysregulation (Rajesh et al., 2023; Xu et al., 2021; Koukourakis et al., 2017). The cellular diversity of the lung creates a complex microenvironment. Metabolites can relay signals through transcytosis across pulmonary endothelial cells (Jones and Minshall, 2020). For instance, mast cell-derived serotonin acts on 5-HT2A receptors, enhancing methacholine-induced airway hyper-responsiveness in house dust mite-triggered experimental asthma (Mendez-Enriquez et al., 2021). Moreover, serotonin metabolites (5-hydroxyindoleacetic acid) derived from activated platelets and mast cells in the lung can act as ligands to drive eosinophil recruitment via the chemotactic receptor G protein-coupled receptor 35, exacerbating fungal infections (De Giovanni et al., 2023). Additionally, dysregulated lipid metabolism is a critical contributing factor in pulmonary diseases. In pulmonary fibrosis, alveolar type II epithelial cells generate foam cells and release them into the distal lung space in response to injury (Romero et al., 2015). Oxidized phospholipids subsequently accumulate in alveolar macrophages, enhancing the production of TGF-β1 and thereby exacerbating fibrotic progression (Romero et al., 2015). Moreover, elevated lactate metabolism is observed in fibrotic lung tissues, which induces pH-dependent activation of TGF-β, promoting fibroblast differentiation and driving fibrotic development (Kottmann et al., 2012). These findings demonstrate that dysregulated lipid metabolism serves as a metabolic reprogramming hallmark of pulmonary fibrosis. In summary, proper pulmonary metabolic function is crucial for maintaining lung homeostasis, and targeting aberrant metabolic pathways represents a promising therapeutic strategy for pulmonary disorders.
3.2 Energy metabolic dysregulation and diabetic pulmonary injury
The stable tricarboxylic acid (TCA) cycle sustains pulmonary surfactant synthesis through ATP production. However, the dynamic pulmonary microenvironment requires adaptive energy regulation via molecular sensors such as adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) and the mammalian target of rapamycin (mTOR). AMPK activation serves to conserve cellular energy, whereas the mTOR pathway governs energy expenditure behaviors (Stapleton et al., 1996; Brown et al., 1994). In patients with DM, dysregulated AMPK coupled with mTORC1 activation drives lipid metabolic disorders and disease complications (Ruderman et al., 2013; Li et al., 2021; Uno et al., 2015). The mTOR signaling plays a pivotal role in pulmonary vascular remodeling, promoting pathological restructuring of pulmonary arterioles (Goncharova, 2013). Therapeutic targeting of both AMPK and mTOR may ameliorate lipid metabolism dysregulation, hyperglycemia, and DM-associated complications (Entezari et al., 2022; Rohm et al., 2022). Activation of the AMPK signaling pathway by metformin treatment in lipopolysaccharide (LPS)-induced acute pulmonary injury attenuates inflammatory responses (Zhang Y. et al., 2022). Similarly, AMPK activation in diabetic pulmonary injury models suppresses high glucose-driven alveolar epithelial-mesenchymal transition, revealing its dual role in maintaining energy homeostasis and preventing fibrosis (Wang L. et al., 2023). In summary, the AMPK/mTOR pathway plays a central role in maintaining pulmonary homeostasis by orchestrating energy metabolic balance. Its dysregulation is closely linked to DM-associated pulmonary injury, including fibrosis and vascular remodeling. Therapeutic strategies targeting this pathway—such as AMPK activation via metformin—demonstrate dual potential in ameliorating metabolic disturbances and suppressing pathological progression.
3.3 Lipid metabolism reprogramming: immune homeostasis disruption
DM sustains systemic chronic inflammation through lipid-mediated pathways, in which prostaglandins (PGs), particularly the abundant PGE2, serve as both dual mediators and amplifiers of inflammatory cascades. PGE2, an arachidonic acid (AA)-derived eicosanoid subclass, acts as a ubiquitous inflammatory mediator that can be potently suppressed by lipid emulsion therapy to mitigate tissue inflammation (Huang et al., 2020). In patients with DM, inflammatory cells markedly elevate the release of lipid mediator PGE2 in affected tissues, which serves as both a cause and consequence of inflammation (Martín-Vázquez et al., 2023). Concurrently, the LTB4 pathway is activated in DM, which upregulating ACE2/TMPRSS2 and arachidonate 5-lipoxygenase expression to exacerbate COVID-19 progression (Bonyek-Silva et al., 2021). Consistent with these findings, patients with COPD exhibit selective elevation of both PGE2 and LTB4 levels in exhaled breath condensate (Montuschi et al., 2003). These shared proinflammatory lipid signatures point to common pathogenic mechanisms driving multiorgan dysfunction in metabolic-pulmonary comorbidities, offering a mechanistic framework for understanding the shared pathophysiology between DM and COPD (Caughey et al., 2013; Hughes et al., 2020). Notably, elevated levels of 12(S)-hydroxyeicosatetraenoic acid [12(S)-HETE], a subclass of eicosanoids, activate the intracellular cation channel transient receptor potential vanilloid one and mediate endothelial dysfunction in DM (Otto et al., 2020). However, high concentrations of 12-HETE disrupt pulmonary immunity and compromise the pulmonary vascular endothelial barrier (Li et al., 2024; Zhang et al., 2018; Pernet et al., 2023). The DM-associated metabolic-immune axis further manifests in alveolar pathophysiology: obesity and T2DM induce alterations in surfactant synthesis within alveolar type II epithelial cells (Schipke et al., 2021). Notably, surfactant protein A (SP-A) and surfactant protein D (SP-D) serve dual roles as pulmonary innate immune proteins. Patients with DM exhibit reduced circulating SP-D levels that correlate with insulin resistance, whereas serum SP-A concentrations are elevated but show no significant association with systemic inflammation (Fernández-Real et al., 2010; Fernández-Real et al., 2008). Importantly, insulin signaling has been demonstrated to be essential for post-inflammatory SP-D recovery (Fernández-Real et al., 2010). These findings underscore metabolic regulation as a critical determinant of pulmonary immunometabolic homeostasis.
3.4 Abnormal amino acid metabolism: BCAA and tryptophan are double-edged swords
As outlined above, BCAA metabolism is severely dysregulated in DM. Systemic BCAA imbalance exerts direct effects on pulmonary function. The β-aminoisobutyric acid, a catabolic derivative of BCAAs, activates the AMPK/Nrf2 pathway to attenuate pulmonary ischemia-reperfusion injury (Zhang et al., 2023). In DM, impaired BCAAs catabolism may disrupt pulmonary repair mechanisms, potentially exacerbating post-injury complications (Lynch and Adams, 2014). Notably, the diaphragm exhibits robust BCAAs metabolism (Huang et al., 2011). Clinical investigations have established that decreased Fischer ratios are significantly associated with both diminished respiratory muscle performance and declined pulmonary function (Yoneda et al., 1992). Consequently, BCAAs metabolic disturbances in T1DM patients may represent a pathophysiological mechanism underlying the observed pulmonary functional alterations. The clinical correlation between DM and lung cancer remains contentious, with population studies demonstrating discordant survival outcomes (Khateeb et al., 2019; Shieh et al., 2012; Hatlen et al., 2011). Mechanistically, non-small cell lung cancer (NSCLC) cells exhibit enhanced BCAAs uptake (Mayers et al., 2016). The elevated BCAAs catabolism in NSCLC cells suppresses m6A demethylase ALKBH5 expression, thereby promoting epithelial-mesenchymal transition and tumor metastasis (Mao et al., 2024). However, increased plasma BCAAs levels have been observed in some NSCLC patients (Wang Y. et al., 2021), potentially attributable to BCAAs catabolic inhibition by branched-chain keto acid dehydrogenase kinase in NSCLC (Xue et al., 2023). These findings demonstrate that BCAAs metabolism is reprogrammed to support tumor growth in the context of lung cancer. This metabolic plasticity likely underlies the observed clinical heterogeneity, as DM-induced systemic metabolic disturbances may interact with tumor-specific BCAAs flux to modulate disease progression.
Tryptophan metabolic dysregulation plays a pivotal role in DM pathogenesis through three primary pathways, including the indole, kynurenine, and serotonin pathways (Gao et al., 2023). Clinical evidence demonstrates that patients with DM exhibit significantly elevated kynurenine levels and increased kynurenine/tryptophan ratios (Kozieł and Urbanska, 2023), which are strongly associated with exacerbated pulmonary inflammatory responses during COVID-19 infection (Conte et al., 2024; Thomas et al., 2020). Moreover, 5-hydroxytryptamine (5-HT) serves as a potent vasoactive mediator of tissue edema and inflammation. 5-HT induces airway plasma extravasation through transient receptor potential vanilloid 4 (TRPV4) (Retamal et al., 2021), a cation channel critical for epithelial and endothelial barrier integrity. Under diabetic and hypertensive conditions, aberrant TRPV4 signaling disrupts pulmonary epithelial/endothelial homeostasis, establishing a vicious cycle of edema and inflammation (Darby et al., 2016). Therapeutic targeting of this pivotal cation channel emerges as a promising strategy for DM-associated pulmonary complications, underscoring its translational potential.
4 Molecular mechanisms of pulmonary injury in DM
Having established that metabolic dysregulation disrupts pulmonary homeostasis, we now examine how these insults propagate into distinct molecular damage signals. This section delineates the core cellular stress pathways—including endoplasmic reticulum stress, oxidative stress, mitophagy dysfunction, and epigenetic reprogramming—that drive pulmonary injury in diabetes at the molecular level (Figure 3).
FIGURE 3
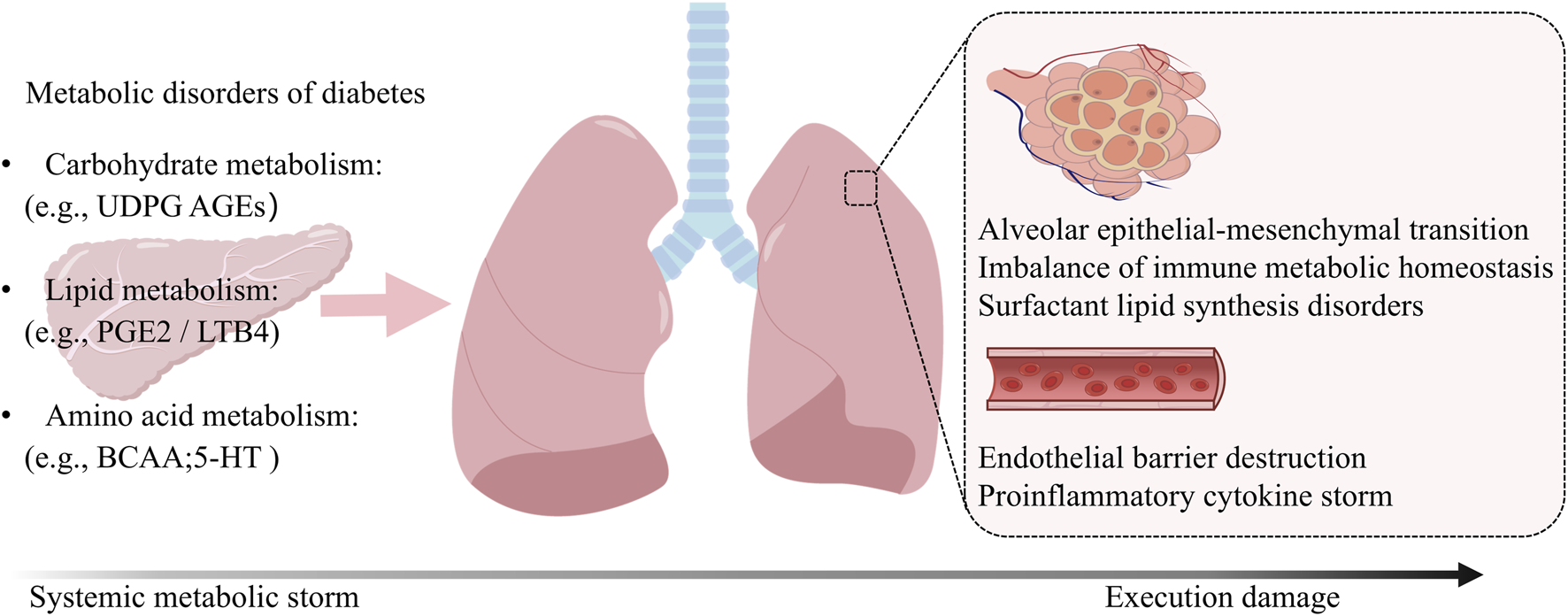
Core Mechanistic Network in Diabetic Pulmonary Injury. Diabetic conditions trigger synchronized immunometabolic disturbances in the lung through multiple signaling pathways. Hyperglycemia disrupts glycogen metabolism, leading to UDPG accumulation and P2Y14R-mediated pro-inflammatory macrophage polarization. Concurrently, elevated BCAAs activate mTOR signaling and generate toxic metabolites, while lipotoxicity and hyperglycemia induce ERS and NLRP3 inflammasome activation. These pathways converge to promote oxidative stress, impaired mitochondrial clearance, and epigenetic modifications, collectively establishing a persistent inflammatory microenvironment that drives progressive lung damage.
4.1 Inflammation-driven pulmonary injury
Circulating inflammatory cells are recruited and secrete inflammatory mediators, which communicate with structural cells (including epithelial cells, endothelial cells and fibroblasts) to trigger tissue inflammation and injury. DM is characterized by disordered glucose metabolism, manifested as decreased glycogen synthesis and increased glycogenolysis. Evidence indicates that loss of UDP-glucose pyrophosphorylase two reduces intracellular glycogen levels and impairs N-glycosylation targets (Wolfe et al., 2021). Notably, we detected upregulated glycogen and N-linked glycans in the fibrotic cores of lung tissue sections from pulmonary fibrosis patients, with lysosomes contributing to glycogen utilization during fibrogenesis (Conroy et al., 2023). Studies show that glycogen metabolism upregulates uridine diphosphate glucose (UDPG) in macrophages (Ma et al., 2020). Binding to the P2Y14R receptor, UDPG synergistically drives macrophage polarization toward a proinflammatory phenotype—characterized by robust TNF-α/IL-6 secretion—via enhancing both STAT1 expression and phosphorylation (Figure 3) (Ma et al., 2020). The pivotal inflammatory role of this pathway is corroborated in P2Y14R-deficient mouse models (Ka et al., 2021). In diabetes, impaired skeletal muscle glycogen synthase activity may disrupt UDPG clearance (Majer et al., 1996), while glutamine metabolism, which possesses UDPG-lowering potential, is itself dysregulated (Petrus et al., 2020; Shi et al., 2024), collectively exacerbating inflammation mediated by the UDPG/P2Y14R axis. We therefore hypothesize that these aberrant glycogen metabolic pathways may collectively contribute to inflammation-driven pulmonary injury in DM.
4.2 ERS-driven pulmonary injury
The endoplasmic reticulum (ER) is a functionally versatile nutrient-sensing platform that plays a critical role in metabolic regulation. Endoplasmic reticulum stress (ERS) has been identified as a major contributor to human diseases, including DM, neurodegenerative disorders, and cancer (Hetz and Papa, 2018). Emerging evidence indicates that ERS disrupts lipid homeostasis and interferes with critical cell signaling pathways in pulmonary disorders, with its role in DM-associated pulmonary injury gaining increasing recognition (Hsu et al., 2017; Chen et al., 2020). ERS not only initiates inflammatory cascades but also determines cellular fate (Hetz and Papa, 2018). Mechanistic investigations reveal that ERS activates the cytosolic pattern recognition receptors NOD1 and NOD2 (nucleotide-binding oligomerization domain-containing proteins one and 2) through bacterial peptidoglycan detection (Zangara et al., 2021), subsequently inducing inflammation and IL-6-dominated cytokine production (Keestra-Gounder et al., 2016). Notably, as a crucial cytosolic pattern recognition receptor, the NLRP3 inflammasome detects diverse stimuli, including microbial pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), while also responding to metabolic disturbances such as hyperglycemia and free fatty acids (Zangara et al., 2021). These findings suggest that ERS-induced inflammatory responses may be mechanistically linked to NLRP3 inflammasome activation (Figure 3). In calcium signaling regulation, the ER serves as the primary intracellular calcium reservoir, with calcium release playing a pivotal role in signal transduction. Research has demonstrated that the DM-associated protein CD36 promotes substantial release of arachidonic acid and PGE2 by modulating ERS-dependent calcium influx, thereby triggering inflammatory cascades (Kuda et al., 2011). Moreover, administration of human islet amyloid polypeptide in diabetic mice was found to induce β-cell dysfunction, concomitant with activation of TRPV4 channels and subsequent ERS induction (Casas et al., 2008). Similar mechanisms were observed in the substantia nigra of Parkinson’s disease mouse models, where TRPV4 signaling was demonstrated to mediate both ERS and inflammatory pathways (Liu et al., 2022). Consistent with these findings, TRPV4 overexpression in human lung adenocarcinoma A549 cells elevated intracellular calcium concentrations and induced ERS (Yoo et al., 2020). Collectively, these results suggest that ERS, functioning as a metabolic stress response system, may transduce diabetic stress into pulmonary tissue damage through circulating mediators.
4.3 Oxidative stress-driven pulmonary injury
Oxidative stress represents a core pathogenic driver in the development and progression of DM. Under pathological conditions including obesity, insulin resistance, hyperglycemia, chronic inflammation and dyslipidemia, excessive reactive oxygen species (ROS) production occurs (Forrester et al., 2018). Notably, the pulmonary system inherently exists in a high redox-pressure environment, where ROS overaccumulation induces both DNA damage and lipid peroxidation - molecular mechanisms now established as central to DM-associated pulmonary injury (Thannickal and Fanburg, 2000). Emerging evidence highlights the pivotal role of BCAAs in modulating oxidative stress responses and their downstream effects (Figure 3). As previously noted, diabetic patients exhibit impaired BCAAs metabolism leading to systemic accumulation. Mechanistically, elevated BCAA slevels exacerbate oxidative stress through two distinct pathways: (i) increased circulating BCAAs enhance ROS production in peripheral blood mononuclear cells (PBMCs) via mTOR signaling hyperactivation, resulting in PBMC overactivation (Zhenyukh et al., 2017); (ii) defective BCAAs catabolism causes accumulation of branched-chain α-keto acids, which induce macrophage oxidative stress and perpetuate chronic inflammatory states (Liu S. et al., 2021). Furthermore, dysfunctional brown adipose tissue (BAT) in diabetic patients contributes to this pathogenic cascade (Okla et al., 2017). Specifically, impaired mitochondrial BCAAs flux and reduced synthesis of BCAA-derived metabolites in BAT exacerbate hepatic oxidative stress while depleting glutathione reserves (Verkerke et al., 2024). This ‘dual-’hit’ mechanism - combining enhanced oxidative stress with compromised redox regulation - not only establishes a pathological link between diabetic metabolic dysregulation and tissue damage, but also suggests the therapeutic potential of modulating BCAAs metabolism to restore redox homeostasis.
4.4 Mitophagy-driven pulmonary injury
In the pathological progression of DM, aberrant accumulation of AGEs disrupts mitophagic homeostasis, thereby exacerbating multi-organ damage (Dewanjee et al., 2022; Xu R. et al., 2023; Zhao et al., 2024). Notably, pulmonary tissue repair processes demonstrate particular dependence on proper autophagic regulation and glucose metabolic function (Li et al., 2019), a characteristic that may fundamentally explain the enhanced pulmonary vulnerability observed in diabetic patients. From the perspective of lipid metabolism, reduced levels of sphingosine-1-phosphate (S1P) in diabetic mouse models lead to significant sphingolipid metabolic defects, which impair both autophagic function and T-cell activity (Jin et al., 2024). More critically, this mechanism is also observed in human lung microvascular endothelial cells, where impaired S1P/S1PR1 signaling directly compromises autophagy, contributing to pulmonary vascular damage (Goel et al., 2021). Collectively, these findings indicate that DM-associated metabolic dysregulation can directly induce pulmonary vascular injury by disrupting autophagy. Notably, autophagy dysfunction and metabolic disturbances form a vicious cycle - impaired autophagic flux exacerbates glucose and lipid metabolic imbalances (Kim and Lee, 2014), which may serve as a key driver for the progressive metabolic deterioration in DM (Figure 3). This recognition provides novel mechanistic insights into the pathogenesis of multi-organ complications in diabetic patients.
4.5 Epigenetic dysregulation-driven pulmonary injury
Previous study has demonstrated complex genetic risk variants during DM pathogenesis (Gao et al., 2019). These variants not only determine disease susceptibility but also reshape tissue-specific epigenomic landscapes (Ling et al., 2022). In T1DM, for instance, genetic variations at risk loci are strongly associated with characteristic inflammatory responses and distinct patterns of immune dysregulation (Gao et al., 2019; Chiou et al., 2021). Persistent hyperglycemia and lipotoxicity synergistically exacerbate metabolic dysregulation through epigenetic modifications such as histone H3 lysine 27 acetylation (H3K27ac). Mechanistic studies reveal that H3K27ac accelerates diabetic cardiomyopathy progression by activating the long non-coding RNA PPARα-seRNA (Ma et al., 2024). Notably, H3K27ac-mediated chromatin remodeling exhibits cell-type-specific effects in pulmonary tissues: in alveolar fibroblasts, it transcriptionally represses autophagy via LINC00941 activation (Zhang J. et al., 2022), whereas in macrophages, serum- and glucocorticoid-inducible kinase 1 (SGK1) disrupts immune homeostasis through H3K27ac-dependent reprogramming (Wu et al., 2024). These findings collectively demonstrate that hyperglycemia-induced chromatin remodeling persistently influences fibroblast activation. Consequently, targeted modulation of dynamic epigenetic marks like H3K27ac may emerge as a novel therapeutic strategy for mitigating metabolic complications and enabling precision risk prediction. Current evidence strongly establishes metabolic dysregulation as a pivotal factor in the pathogenesis of DM and its complications. However, elucidating the precise mechanisms through which diabetic metabolic disorders induce pulmonary tissue injury remains a significant challenge. While abnormal accumulation of metabolic intermediates has been shown to drive pulmonary inflammation and injury (Conroy et al., 2023; Kwon et al., 2019), the mechanistic underpinnings for most metabolites remain undefined, necessitating further investigation to delineate the molecular pathways of metabolite-driven lung damage in DM. Building upon existing findings, glycogen metabolism emerges as a potential therapeutic target, with glycogen synthase modulators and receptor for RAGE antagonists demonstrating promising therapeutic potential.
5 Gut microbiota in DM: metabolic crosstalk with pulmonary pathology
The mechanisms discussed thus far have largely focused on the direct pathogenic effects of metabolic dysregulation in the lung. However, the full picture of diabetic pulmonary injury necessitates a systemic perspective that incorporates the crucial role of the gut microbiota. Diabetes-driven gut dysbiosis acts as a key amplifier, exacerbating systemic metabolic and immune perturbations. The following sections will elucidate how the gut microbiota and its metabolites mediate remote control of pulmonary pathology via the gut-lung axis.
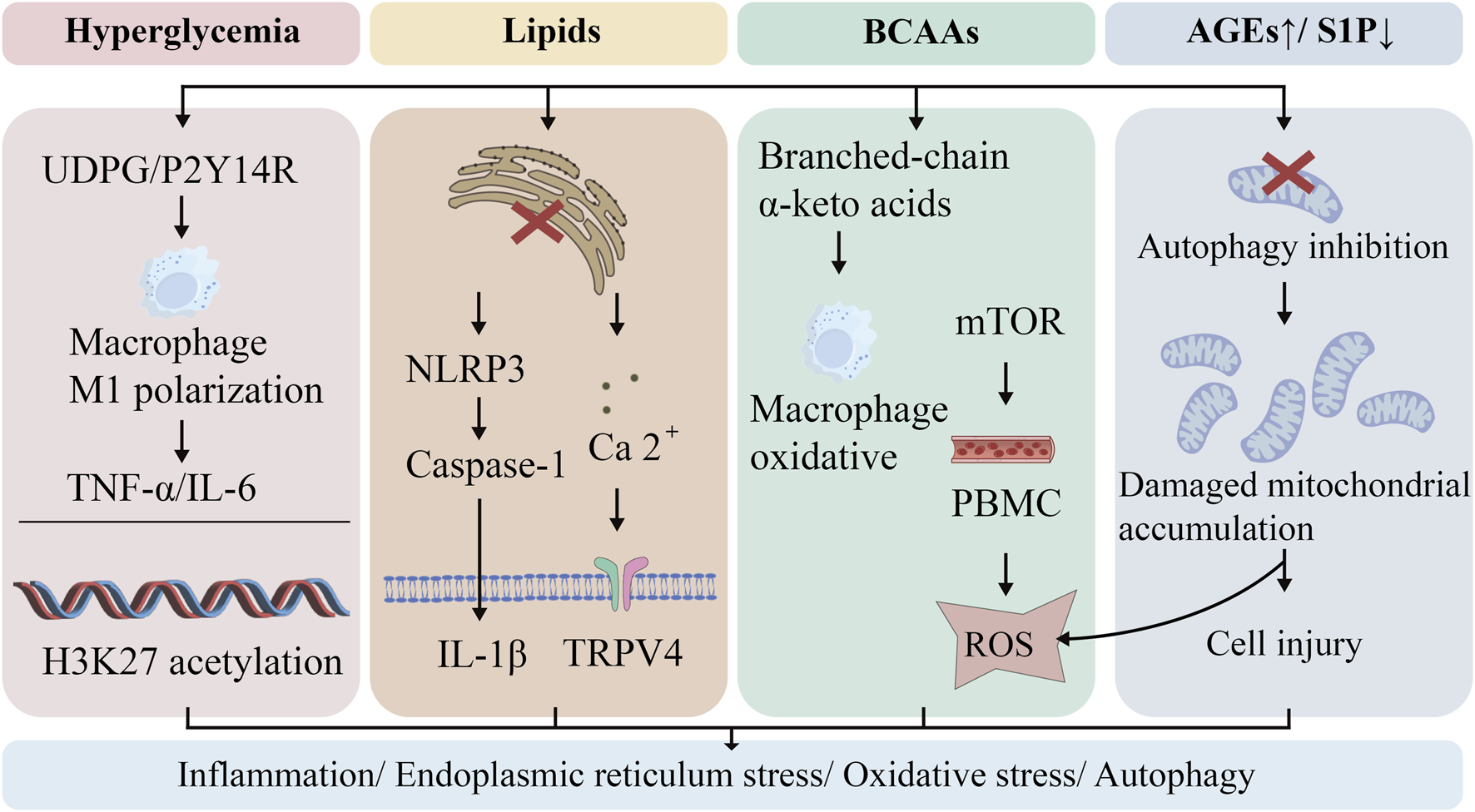
Current evidence has firmly established that intestinal dysbiosis represents a critical pathological component in the development and progression of DM (Yang et al., 2021). The mechanistic interplay between the gut microbiome and host chronic inflammatory states/metabolic disturbances in diabetic patients has garnered increasing research attention (Chávez-Talavera et al., 2017). The small intestine serves as the primary site for lipid reabsorption, where gut microbiota-derived metabolites modulate immune homeostasis (Greer et al., 2013). Emerging evidence suggests that targeted modulation of gut microbial composition and its metabolic output may confer beneficial effects against DM-associated complications (Canfora et al., 2019). Similarly, in T2DM, altered microbial metabolism of histidine leads to elevated imidazole propionate levels (Molinaro et al., 2020), which subsequently exacerbates glucose metabolic dysregulation. Emerging evidence demonstrates significant suppression of ethanolamine-metabolizing microbiota in the gut of individuals with DM and obesity, resulting in ethanolamine accumulation. This metabolic perturbation significantly downregulates the expression of intestinal tight junction proteins, particularly ZO-1 (Mishra et al., 2023), thereby initiating a multistep pathogenic cascade: (i) compromised intestinal barrier integrity promotes bacterial translocation and systemic dissemination of deleterious metabolites; (ii) subsequent activation of systemic inflammatory responses; and (iii) consequent aggravation of insulin resistance, deterioration of glucose homeostasis, and disruption of immunological equilibrium - ultimately precipitating multi-organ dysfunction. As illustrated in Figure 4, the gut microbiota serves as a central hub in diabetic lung injury, where gut-derived metabolites and toxins (e.g., LPS, BCAAs) remotely regulate pulmonary inflammation and immune responses through systemic circulation, ultimately leading to endothelial disruption and oxidative stress.
FIGURE 4
The Gut-Lung Axis in Diabetic Pulmonary Pathogenesis. The gut-lung axis mediates diabetic pulmonary injury through circulating microbial metabolites and bacterial components. Diabetes-induced gut dysbiosis results in reduced SCFA-producing bacteria, impaired bile acid metabolism, and increased gut permeability. This permits translocation of LPS, BCAAs, and other microbial products into systemic circulation. Upon reaching the lung, these gut-derived mediators activate innate immune receptors, triggering endothelial activation, neutrophilic infiltration, and oxidative stress responses that collectively drive pulmonary inflammation and tissue remodeling.
5.1 Metabolite-mediated intestinal-pulmonary axis crosstalk
Gut microbial metabolites, serving as pivotal mediators of host-microbiota interactions, exhibit dynamic composition and functionality regulated by host genetics, dietary patterns, and environmental factors, while reciprocally modulating host homeostasis via the metabolism-immune axis (Schoeler and Caesar, 2019). These bioactive molecules, like short-chain fatty acids (SCFAs) and BCAAs, can enter systemic circulation by crossing the intestinal barrier, thereby modulating the function of distal organs such as the lungs via metabolite-organ crosstalk. During the pathological progression of DM, four classes of critical microbial metabolites (SCFAs, BCAAs, bile acids, bacterial endotoxins) mediate multi-organ damage through the gut-lung axis (Saad et al., 2016). In conclusion, gut microbial metabolites, regulated by host genetics/diet/environment, mediate DM multi-organ damage via gut-lung axis mechanisms.
SCFAs, the principal metabolic end-products of dietary fiber fermentation by obligate anaerobes (e.g., Bacteroides spp.), play a pivotal role in maintaining intestinal redox homeostasis (van Hoek and Merks, 2012). Mechanistic investigations demonstrate that SCFAs markedly enhance insulin sensitivity and optimize energy metabolism in diabetic patients through G protein-coupled receptor activation (Gao et al., 2009). Notably, pulmonary tissues lack essential substrates for SCFA biosynthesis (e.g., dietary fiber), and germ-free mouse models exhibit significantly reduced pulmonary SCFA levels - direct evidence that lung SCFAs are entirely dependent on functional gut microbiota-derived interorgan supply (Liu Q. et al., 2021). In conclusion, SCFAs from gut microbiota enhance insulin sensitivity, maintain intestinal redox, and supply lungs via interorgan transport.
BCAAs (comprising leucine, isoleucine, and valine), as essential amino acids, exhibit metabolic dynamics tightly regulated by the gut microbiota—which participates in both BCAAs synthesis (Prevotella spp.) and catabolism (Roseburia spp.) (Gojda and Cahova, 2021). Mechanistic studies reveal that high-fat diets elevate circulating BCAAs levels, activating the mTORC1 signaling pathway in skeletal muscle, thereby inducing lipid deposition and ultimately promoting obesity-associated insulin resistance (Newgard et al., 2009). Based on the above analysis, gut microbiota-regulated BCAAs drive mTORC1-mediated muscle lipid accumulation and insulin resistance in high-fat diet-induced obesity.
Notably, bile acids form a dynamic regulatory network through enterohepatic circulation. Primary bile acids (e.g., cholic acid) are converted into secondary bile acids (e.g., deoxycholic acid) via microbial 7α-dehydroxylation, which activate FXR and TGR5 receptors to not only improve glucose and lipid metabolism in diabetic patients but also exert potent anti-inflammatory effects (Chávez-Talavera et al., 2017). Of particular significance, ursodeoxycholic acid (UDCA), as a key secondary bile acid, exhibits immunomodulatory functions through a dual mechanism: downregulating CD86 expression on DCs while simultaneously inhibiting Th2 cell differentiation and promoting Treg cell proliferation, thereby effectively ameliorating eosinophilic airway inflammation (Willart et al., 2012). In conclusion, microbial-converted bile acids activate FXR/TGR5 to improve metabolism and inflammation, while UDCA modulates DCs and Tregs to ameliorate eosinophilic airway inflammation.
The pathological association between DM and increased intestinal permeability coupled with gut microbiota dysbiosis has garnered substantial scientific attention. Substantial evidence confirms that gut barrier dysfunction-induced microbial imbalance facilitates bacterial endotoxin (e.g., LPS) translocation across the intestinal epithelium into systemic circulation, precipitating metabolic endotoxemia - a process driving multi-tissue injury through systemic inflammatory responses (Saad et al., 2016). Notably, LPS-CD14 binding via pattern recognition receptors not only mediates innate immune activation but has been mechanistically linked to insulin resistance, obesity, and DM pathogenesis (Cani et al., 2007). To sum up, gut barrier dysfunction in DM drives microbiota-LPS translocation, triggering CD14-mediated systemic inflammation and insulin resistance, exacerbating multi-organ damage.
From a systems biology perspective, gut microbial metabolites serve as critical signaling mediators in gut-lung axis crosstalk, whose homeostatic disruption may reprogram metabolic-immune networks to propagate diabetic multi-organ complications. This trans-organ pathophysiological interconnection provides a novel conceptual framework for understanding DM-associated systemic damage.
5.2 Bacterial endotoxins in DM-associated pulmonary injury
Mounting experimental evidence continues to delineate the crucial involvement of gut-derived endotoxins in pulmonary injury pathogenesis through pattern recognition receptor (PRR)-dependent molecular crosstalk. Mechanistically, microbiota-derived endotoxins orchestrate parallel activation of both caspase-11-mediated non-canonical inflammasome pathways and canonical caspase-1/NLRP3 inflammasome complexes. This dual activation cascade induces catastrophic breakdown of alveolar-capillary barrier integrity, manifesting pathologically as alveolar edema, neutrophilic inflammation, and refractory hypoxemia (Cheng et al., 2017; Jin et al., 2021). In conclusion, gut endotoxins trigger lung injury via PRR-dependent dual inflammasome pathways, disrupting alveolar-capillary integrity.
The Toll-like receptor 4 (TLR4) system emerges as the principal lipopolysaccharide (LPS) recognition apparatus, executing transmembrane signaling through myeloid differentiation primary response 88 (MyD88)-dependent phosphorylation cascades (Poltorak et al., 1998). Of particular pathogenic significance, intestinal epithelial TLR4 activation stimulates systemic release of high-mobility group box 1 (HMGB1), a prototypic damage-associated molecular pattern (DAMP) that mediates remote pulmonary parenchymal injury via hematogenous dissemination (Sodhi et al., 2015). This mechanistic paradigm is substantiated by the complete abrogation of pulmonary injury phenotypes in intestinal epithelium-specific TLR4 knockout murine models (Sodhi et al., 2015). Based on the above analysis, TLR4 recognizes LPS via MyD88, triggering gut HMGB1 release that causes lung injury through hematogenous spread.
This gut-lung interorgan communication mechanism demonstrates significant clinical relevance in chronic respiratory diseases, with large-scale cohort studies establishing a strong inverse correlation between gut microbiota α-diversity reduction and key clinical parameters including asthma exacerbation frequency and annual lung function decline in COPD (Tangedal et al., 2024). In traumatic shock models, gut barrier collapse generates proinflammatory mediators that form a “second hit” through mesenteric lymphatic circulation, exacerbating acute pulmonary injury and precipitating multi-organ dysfunction (Magnotti et al., 1998). Collectively, these findings demonstrate that gut-derived inflammatory cascades mediated by TLRs/NLRs signaling pathways serve not only as key drivers of experimental pulmonary injury, but their persistent activation likely contributes to the pathological progression of advanced diabetic complications (e.g., diabetic lung disease). This paradigm provides novel therapeutic targets for multi-organ protection in metabolic disorders.
5.3 Gut virome dynamics in DM progression
Emerging research has progressively unveiled the dynamic evolution of the gut virome during DM progression and its complications. Contemporary evidence reveals that specific bacteriophages (e.g., Caudovirales members) exert critical regulatory functions in glucolipid metabolic disorders by modulating host-bacterial interaction networks (Fan et al., 2023). Notably, the ecologically integrated system formed by the gut virome and bacteriome generates distinct microbial metabolite signatures across DM subtypes through niche-specific regulatory mechanisms governed by “phage predation-bacterial lysis” dynamics (Nishijima et al., 2022; Hu et al., 2023). These cross-kingdom virus-bacteria interactions exhibit systemic regulatory potential, as circulating bacteriophages can translocate across the intestinal barrier via epithelial transcytosis, contributing to immune homeostasis in distal organs such as pancreatic islets and adipose tissue (Nguyen et al., 2017). Clinical interventions suggest that nutritional modulation (e.g., dietary fiber supplementation) may attenuate DM-associated gut dysbiosis by recalibrating temperate-lytic phage cycle equilibria (Avellaneda-Franco et al., 2024). Nevertheless, population-level virome heterogeneity and its interplay with host genetic determinants demand systematic exploration. Emerging synthetic biology tools enabling precision phage editing may pioneer novel therapeutic paradigms for metabolic reprogramming through “top-down” microbial ecosystem engineering.
In environmental exposure research, a paradigm-shifting discovery has redefined pulmonary microbiology: the lung parenchyma, previously deemed sterile, hosts a unique microbial ecosystem shaped by microbial migration-colonization-clearance dynamics (Natalini et al., 2023). This breakthrough elucidates mechanisms whereby environmental pollutants exacerbate respiratory pathologies via microbiota-immune axis disruption. Significantly, pulmonary microbiome dysbiosis in DM may constitute a novel mediator of gut-lung axis crosstalk, expanding our understanding of systemic complications.
Regarding DM-associated gastrointestinal pathophysiology, studies demonstrate hyperglycemia-driven systemic inflammation through three key mechanisms: (1) Downregulation of intestinal tight junction proteins (e.g., ZO-1) enhances gut permeability, enabling systemic translocation of pathogen-associated molecular patterns (PAMPs, e.g., LPS); (2) Reduced microbiota β-diversity generates individualized metabolic phenotypes, with specific bacterial consortia (e.g., Bacteroidetes/Firmicutes ratios) exhibiting strong correlations with insulin sensitivity; and (3) Gut-derived outer membrane vesicles mediate multi-organ injury via lymphatic trafficking (Yang et al., 2021). While animal models have partially deciphered these mechanisms, human studies face substantial challenges: interindividual microbiota variability and confounding environmental exposures (e.g., antibiotic history) complicate causal inference. From a translational perspective, microbiome signature-guided interventions—including precision probiotics and engineered phage cocktails—show therapeutic promise, though their efficacy necessitates validation through multicenter randomized controlled trials.
6 Dysregulation of the pulmonary immune microenvironment
Gut microbiota, through its metabolites and immunomodulatory functions, constitutes a critical hub linking diabetes to pulmonary pathologies. Signals originating from the gut and circulation ultimately converge within the pulmonary immune microenvironment, determining the balance between inflammation and immune tolerance. Next, we will elucidate how diabetes and its associated metabolic and microbiota dysregulation disrupt the equilibrium of innate and adaptive immune cells in the lung, leading to uncontrolled inflammation and tissue damage (Figure 5).
FIGURE 5
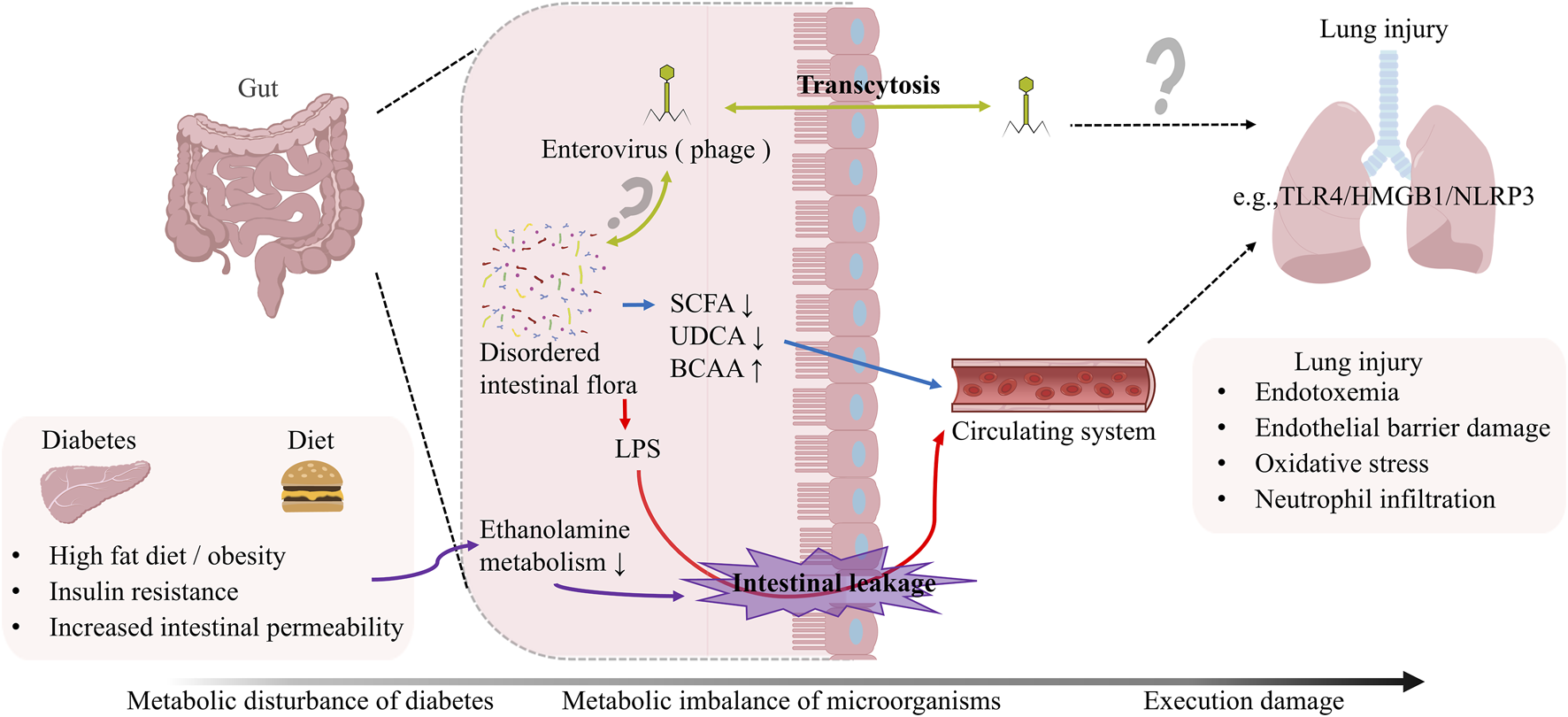
Microbiota-Driven Immune Dysregulation in the Diabetic Lung. Gut microbiota disturbances disrupt pulmonary immune homeostasis through metabolite-mediated mechanisms. First, metabolite imbalances disrupt adaptive immunity: SCFAs depletion impairs Treg differentiation, BCAAs accumulation hyperactivates CD8+ T cell glycolysis, and LCFAs excess promotes Th1/Th17 polarization. Concurrently, innate immunity collapses. This collapse involves S1P deficiency-mediated arrest of Treg autophagy, hyperglycemia-induced DC hyperacetylation, which disrupts antigen presentation, and DMPC/PMPC depletion that cripples surfactant-dependent pathogen clearance. Furthermore, adaptive immunity undergoes metabolic reprogramming; BCAA-PD-1 crosstalk alters the efficacy of NSCLC immunotherapy, and CD8+ memory T cells exhibit failure of the gluconeogenesis-glycogen axis.
The pulmonary immune defense system operates through two interconnected arms: the innate immune network (encompassing neutrophils, macrophages, eosinophils, mast cells, natural killer cells, γδ T cells, innate lymphoid cells, and dendritic cells) and the adaptive immune apparatus (T and B lymphocytes). Under diabetic conditions, synergistic dysregulation of immune and metabolic homeostasis fosters a chronic inflammatory microenvironment, predisposing to pulmonary tissue injury. Established research confirms that diabetes mellitus (DM) and hyperlipidemia compromise pulmonary defense mechanisms by disrupting surfactant biosynthesis. Crucially, DM-associated metabolic dysregulation extends beyond biosynthetic disruption to critically impair surfactant’s antimicrobial functionality. In murine models of diet-induced obesity, pulmonary tissues and bronchoalveolar lavage fluid demonstrate significant depletion of critical phospholipids—dimyristoyl phosphatidylcholine (DMPC) and palmitoyl myristoyl phosphatidylcholine (PMPC). These dysregulated phospholipid profiles not only hinder pathogen clearance but also enhance SARS-CoV-2 infectivity by modulating viral entry mechanisms (Du et al., 2022). Therefore, diabetic metabolic dysregulation critically subverts pulmonary immune resilience by destabilizing surfactant integrity, amplifying infection susceptibility through combined biosynthetic, antimicrobial, and lipidome perturbations.
From a metabolic regulatory perspective, DM-induced hormonal dysregulation impairs pulmonary immunity through two principal pathways: First, hyperactivated glucocorticoid signaling suppresses alveolar macrophage phagocytic capacity; second, systemic insulin resistance reprograms immunometabolic circuits, driving pro-inflammatory polarization of immune cells (Webber et al., 2022). Emerging evidence further identifies gut microbiota dysbiosis in DM as a key driver of pulmonary immunopathology via systemic immune-metabolic axis activation. Gut-derived metabolites interact with pulmonary immune effectors, establishing a self-amplifying inflammatory loop within lung tissue (Burcelin, 2016). This gut-lung axis mechanism has been experimentally validated in COPD and COVID-19 pathobiology, where specific microbial taxa regulate pulmonary immune responses through Th17/Treg balance modulation and associated cytokine networks (Lai et al., 2022; Nagata et al., 2023). Therefore, DM orchestrates multi-organ immunometabolic crosstalk through endocrine-immune circuitry and gut-lung microbial networks, collectively reprogramming pulmonary defense landscapes toward chronic inflammation and pathogenic vulnerability.
Current findings underscore that maintaining metabolic homeostasis is paramount for pulmonary immune equilibrium, with gut microbiota serving as central metabolic regulators. Therapeutic modulation of microbiota-derived metabolites—particularly SCFAs and secondary bile acids—through dietary interventions or pharmacological agents shows significant promise in counteracting DM-associated pulmonary immune dysfunction, potentially via epigenetic modulation of immune cell function.
6.1 Gut microbiota and the immune microenvironment
Gut microbiota metabolism serves as a central immunoregulatory hub, coordinating immune system development and functional maturation through multifactorial mechanisms. During early-life immune programming, vertical microbial transmission from mother to offspring establishes a protective neonatal gut ecosystem. This process not only initiates primary microbial colonization but also activates TLR-dependent signaling to drive immune effector differentiation, establishing the foundation for lifelong immunological tolerance (Koren et al., 2024). Therefore, early-life gut microbiota-host interactions critically shape immunological trajectories by integrating microbial colonization, innate receptor signaling, and tolerance programming, forming an essential developmental axis for pulmonary and systemic immune homeostasis.
Gut microbiota-derived SCFAs are key messengers of the gut-lung axis (Figure 5). At the immune level, metabolic crosstalk critically shapes polarization: persistent long-chain fatty acid (LCFA) exposure depletes intestinal SCFAs while promoting differentiation of naïve T cells into pro-inflammatory Th1/Th17 subsets in the small intestine (Haghikia et al., 2015). In contrast, SCFA enrichment induces dual regulatory mechanisms—JNK1/p38 signaling suppression and Foxp3 transcriptional activation—to promote regulatory T (Treg) cell differentiation, thereby attenuating inflammatory cascades (Haghikia et al., 2015; Smith et al., 2013). Therefore, the metabolic milieu directs T cell lineage commitment through opposing lipid-sensing pathways, wherein SCFA-mediated immunomodulation counterbalances pro-inflammatory fatty acid signaling to maintain mucosal immune equilibrium and restrain pathological inflammation.
This metabolically driven immunomodulation exhibits trans-organ synergy across mucosal systems. The gut-lung axis facilitates dynamic interorgan communication, with gut-derived metabolites modulating distal immunity. Seminal work demonstrates that systemic BCG vaccination triggers intestinal lamina propria-resident innate memory cells via microbiota-dependent spatiotemporal metabolic reprogramming, identifying a paradigm for distal mucosal immune memory establishment (Jeyanathan et al., 2022). Therefore, this trans-mucosal metabolic dialogue establishes a systemic immune memory architecture, wherein gut-orchestrated metabolic reprogramming by microbial and vaccine stimuli epigenetically primes distal barrier sites for enhanced pathogen vigilance through conserved immuno-metabolic circuitry.
Emerging research has further elucidated that two major mucosal interfaces achieve systemic regulation through metabolite-immune crosstalk. On one hand, gut-to-lung axis: Circulating gut-derived metabolites (e.g., tryptophan catabolites) directly regulate pulmonary epithelial immunity; on the other hand, lung-to-gut feedback: Respiratory inflammatory signals reciprocally remodel gut microbial composition, forming a self-reinforcing metabolic-immune network (Özçam and Lynch, 2024). This trans-mucosal metabolic-immune synergy provides a novel theoretical framework for understanding microbiota-mediated systemic immune reprogramming.
6.2 Dysregulation of the innate immune system
T1DM is defined by immunopathology rooted in multifactorial regulatory T cell (Treg) dysfunction, with collapse of this central immune checkpoint driving persistent immune dysregulation (Kukreja et al., 2002). Tregs maintain immune homeostasis through two synergistic pathways: (1) paracrine release of immunosuppressive cytokines (e.g., IL-10, TGF-β) and (2) direct cellular interactions with innate immune populations—including monocyte-derived macrophages, granulocytes, dendritic cells (DCs), and innate lymphoid cells. Mechanistic drivers of Treg impairment converge on autophagy dysregulation. In diabetic murine models, sphingolipid metabolic disruption—marked by sphingosine-1-phosphate (S1P) deficiency—triggers mTOR-dependent autophagosome overproduction, accelerating Foxp3 proteasomal degradation and crippling Treg suppressive function (Jin et al., 2024). Therapeutic S1P restoration rescues Treg fitness via S1PR1-STAT5 signaling, which stabilizes Foxp3 expression and rebalances autophagic flux through lysosomal reactivation (Jin et al., 2024). Therefore, sphingolipid-autophagy crosstalk constitutes a pivotal regulatory nexus governing Treg stability, whose diabetic disruption propagates T1DM immunopathology by crippling immunosuppressive circuits, while pharmacologically restoring this axis emerges as a strategic therapeutic paradigm for re-establishing immune tolerance (Figure 5).
DCs exhibit parallel metabolism-driven dysfunction in T1DM, with subset-specific roles shaping immune polarization: cDC1 skews toward Th1 induction, while pDC preferentially promotes immune tolerance (Lo and Clare-Salzler, 2006). In diabetic pathology, DCs display metabolically programmed functional decay: Impaired efferocytosis in wound-resident DCs correlates with lysosomal acidification defects and dysregulated LC3-II expression (Masch et al., 2022). SLC7A11 inhibition restores efferocytic capacity by rescuing cystine/glutamate antiporter-dependent lysosomal proteolysis, accelerating diabetic wound repair (Masch et al., 2022). Hyperglycemia-induced epigenetic reprogramming in pulmonary DCs suppresses MHC-II and costimulatory molecule expression via histone hyperacetylation, blunting antigen presentation and CD8+ T-cell priming (Figure 5). HDAC inhibitors reverse these defects, reinstating DC immunogenicity (Nobs et al., 2023). Therapeutic implications: T1DM-associated metabolic insults—S1P depletion, glucolipotoxicity—converge on autophagic and epigenetic mechanisms to disable immunoregulatory cells. Targeting sphingolipid metabolism (e.g., S1P analogs), glucose transporters, or HDACs may restore tissue immunometabolic equilibrium, offering novel intervention strategies.
6.3 Pathological remodeling of adaptive immunity
The adaptive immune system orchestrates pathogen defense and tumor immunosurveillance through metabolic reprogramming, with CD8+ T cell remodeling serving as a linchpin. Mechanistic studies identify a tripartite metabolic axis—gluconeogenesis, glycogen metabolism, and the pentose phosphate pathway (PPP)—as essential for memory T cell survival. Cytosolic phosphoenolpyruvate carboxykinase (PCK1) catalyzes oxaloacetate-to-glucose-6-phosphate conversion, diverting metabolic flux toward glycogen synthesis and PPP activation. The resultant NADPH sustains Tm longevity by preserving glutathione redox homeostasis (Ma et al., 2018). This metabolic shunt enables Tm cells to bypass glycolysis-dependent energy production, maintaining functionality in nutrient-poor niches.
In addition to their direct effects on lung tissue, BCAAs also serve as critical immune-related metabolites (Figure 5). At physiological levels, BCAAs enhance CD8+ T cell antitumor activity via FoxO1-mediated glucose transporter 1 (GLUT1) upregulation, facilitating glycolytic-oxidative phosphorylation coupling (Yao et al., 2023). Clinically, gut microbiota-derived BCAAs synergize with PD-1 inhibitors to improve therapeutic responses in non-small cell lung cancer (NSCLC), potentially through microbiota-driven expansion of peripheral Tm cells and natural killer (NK) cell activation (Yao et al., 2023; Zhu et al., 2023; Jin et al., 2019). Strikingly, BCAAs metabolism displays paradoxical roles in diabetic pathophysiology—gut dysbiosis-induced BCAA accumulation confers survival benefits in specific NSCLC-DM cohorts, yet effect directionality depends on individual metabotypes, necessitating longitudinal metabolomics to decode the “DM-lung cancer” risk continuum (Qian et al., 2022). NSCLC patients exhibit aberrant expression of DM-specific microbial dysbiosis markers (CRP, LBP, CD14), implicating microbiota-derived endotoxemia in lung carcinogenesis (Qian et al., 2022). Therefore, BCAA metabolism manifests as a pleiotropic immunological arbitrator, where gut microbiota-driven metabolic reprogramming and host metabotype specificity converge to dictate its paradoxical roles across cancer immunotherapy and diabetic comorbidity, mandating precision metabologenomic approaches to harness its therapeutic duality while mitigating DM-associated oncogenic risk.
Integrating these findings, we propose a tripartite metabolic-immune axis driving DM-associated pulmonary injury. Firstly, pulmonary surfactant lipidome remodeling: DM-induced sphingolipid dysregulation (e.g., sphingosine-1-phosphate depletion) and phospholipid restructuring (reduced DMPC/PMPC ratios) compromise alveolar barrier integrity. Secondly, microbiota metabolite-driven immunometabolic reprogramming: Microbial metabolites (BCAAs, SCFAs) epigenetically modulate immunoregulatory cells (Tregs, DCs) via histone hyperacetylation and JNK1/p38-Foxp3 signaling. Finally, endocrine-immune integration: The gut-lung axis mediates systemic crosstalk through a microbiota-hormone-immunity network, where hyperglycemia-induced metabolic memory persistently impairs DC antigen presentation via HDAC-dependent epigenetic silencing.
Future investigations should prioritize resolving three pivotal scientific questions: (1) Whether microbial antigens drive lung tissue-specific autoimmune responses via molecular mimicry or related mechanisms; (2) How distinct metabolites (e.g., S1P vs BCAAs); cooperatively regulate immune cell fate determination across spatiotemporal dynamics; (3) Strategies to re-establish pulmonary immunometabolic homeostasis through microbiota-targeted metabolic interventions. Therapeutic strategies addressing these mechanisms—such as S1PR agonists or microbiota transplantation combined with immune checkpoint inhibitors—may pioneer a precision therapeutic paradigm for DM-associated pulmonary complications.
7 Therapeutic strategies for DM-associated pulmonary injury
The escalating global prevalence of DM necessitates future advancements in novel biomarker discovery and personalized therapeutic strategies. Clinically, managing pulmonary complications in DM confronts five cardinal challenges: the intricate molecular crosstalk governing metabolic-immune networks; the translational gaps in preclinical animal models recapitulating human-specific immunometabolic pathologies; the absence of standardized efficacy metrics for tissue-specific immunomodulation; the polypharmacy challenges arising from multimorbidity management; and the undercharacterized risks of off-target drug-metabolite interactions.
Having established the “metabolic dysregulation-gut dysbiosis-immune imbalance” axis as the core pathogenic mechanism, we now examine corresponding therapeutic strategies. Interventions targeting this axis—including GLP-1RAs/SGLT-2is, microbiota-directed therapy, and anti-IL-6/NLRP3 agents—hold significant promise (Figure 6). This axis provides the logical framework for the following review of current and emerging treatments, guiding the discussion of their translational potential and challenges.
FIGURE 6
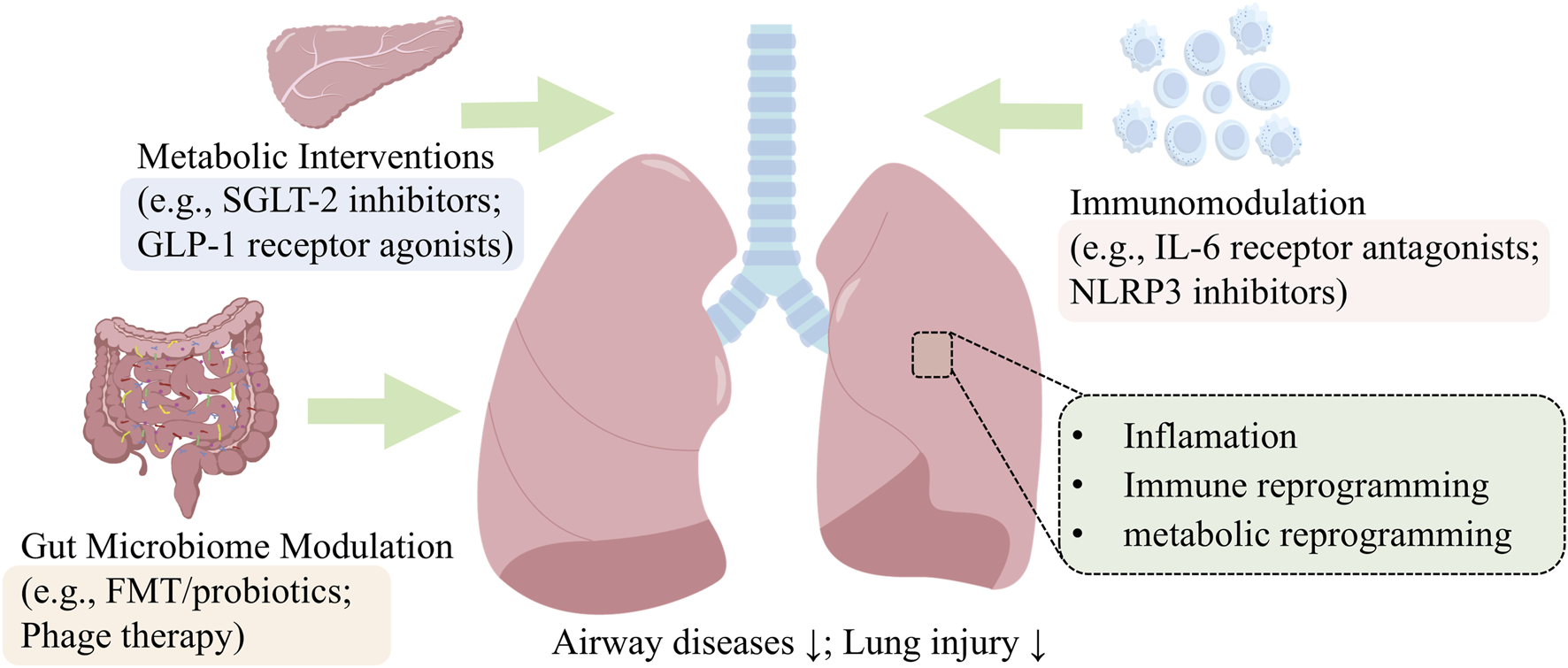
Therapeutic Targeting of the Metabolic-Microbial-Immune Axis. This schematic summarizes promising therapeutic strategies that pharmacologically target the metabolic (GLP-1RAs, SGLT-2is), microbial (FMT, phages), and immune (anti-IL-6, NLRP3) components of diabetic lung injury.
7.1 Targeting shared metabolic-immune pathomechanisms
Emerging therapeutic paradigms in diabetes mellitus (DM) leverage the pleiotropic benefits of sodium-glucose cotransporter-2 inhibitors (SGLT-2Is) and glucagon-like peptide-1 receptor agonists (GLP-1RAs), extending beyond glycemic control to systemic immunomodulation. SGLT-2Is block renal glucose reabsorption to promote urinary excretion, while GLP-1RAs enhance glucose-dependent insulin secretion and suppress glucagon release (Prattichizzo et al., 2021). Both classes exhibit pulmonary protective effects via systemic anti-inflammatory reprogramming, with clinical studies demonstrating reduced exacerbation risk in T2DM patients with comorbid COPD (Foer et al., 2023). Therefore, SGLT-2Is and GLP-1RAs exemplify dual-pathway therapeutics, synergizing antihyperglycemic efficacy with systemic immunometabolic reprogramming to confer multi-organ protection, positioning these agents as cornerstone interventions for mitigating diabetes-associated pulmonary and systemic inflammatory comorbidities.
As previously discussed, metabolic imbalance and mitochondrial dysfunction under diabetic conditions are key mechanisms leading to lung injury. Therefore, restoring metabolic balance represents a promising therapeutic target. Beyond lowering blood glucose by promoting urinary glucose excretion, SGLT-2 inhibitors have been shown to activate the AMPK pathway (El-Horany et al., 2023), thereby improving mitochondrial function, alleviating endoplasmic reticulum stress and oxidative stress, reversing metabolic defects, and consequently protecting lung tissue. The prototypical inhibitor canagliflozin (CANA) attenuates pulmonary injury by skewing alveolar macrophage polarization toward tissue-reparative M2 phenotypes via PPARγ-dependent fatty acid oxidation (Lin et al., 2020). Concurrently, CANA restores mitochondrial homeostasis by activating the PINK1-Parkin mitophagy pathway while suppressing oxidative stress and endoplasmic reticulum stress (ERS), demonstrating efficacy across multiorgan injury models (O'Keefe et al., 2023; Packer, 2020). Although direct clinical evidence for their pulmonary protective effects is still accumulating, meta-analyses of large cardiovascular outcome trials indicate that SGLT-2 inhibitors significantly reduce the risk of acute exacerbations in diabetic patients with comorbid COPD (Foer et al., 2023), suggesting that they provide a pleiotropic organ protection beyond hypoglycemic effects.
GLP-1 receptor activation in sepsis models inhibits Toll-like receptor (TLR)-driven TNF-α production, mitigating systemic inflammation and acute lung injury (Wong et al., 2024). Clinically, GLP-1RAs reduce asthma exacerbations in DM patients, potentially by dampening allergen-induced airway neutrophilia (Lee et al., 2025; Toki et al., 2021). Both drug classes inhibit fibrotic progression and pulmonary inflammation by targeting key pathways—NF-κB signaling and NLRP3 inflammasome activation—thereby disrupting pro-inflammatory cytokine cascades (Shakour et al., 2024; Yang et al., 2022; Wang Y. et al., 2023). Therefore, CANA and GLP-1RAs orchestrate pleiotropic therapeutic benefits by converging on mitochondrial quality control, macrophage polarization, and inflammasome-cytokine axis suppression, thereby resolving immunometabolic crossfire in pulmonary systems and positioning these agents as cornerstone therapies for diabetes-related multiorgan inflammatory sequelae.
Emerging therapeutic strategies emphasize epigenetic precision therapeutics for DM-related tissue injury. Emerging strategies focus on microRNA (miR)-155, a master epigenetic regulator of DM-related multiorgan complications (Huang et al., 2014). The inhibition of miR-155 rescues redox homeostasis and mitochondrial dynamics in diabetic tissues via AMPKα-PGC1β signaling (Prieto et al., 2023; Ying et al., 2017), and suppresses alveolar macrophage M1 polarization in lung injury models by silencing the SOCS1/STAT1 axis, reducing neutrophil extracellular trap (NET) formation and IL-6/IL-1β-driven inflammation (Xu Y. et al., 2023). Antagomirs and CRISPR/Cas9-based editing systems enable tissue-specific miR-155 modulation, offering targeted therapeutic potential.
The dual targeting of immunometabolic pathways (via SGLT-2Is/GLP-1RAs) and epigenetic regulators (e.g., miR-155) represents a paradigm shift in DM therapeutics. These approaches address both systemic inflammation and tissue-specific damage, highlighting opportunities for precision medicine in diabetic complications. However, it must be noted that these pulmonary outcomes are largely derived from exploratory analyses or observational studies, and specifically designed prospective RCTs are warranted to confirm their efficacy against specific pulmonary diseases, such as idiopathic pulmonary fibrosis.
7.2 Metabolic reprogramming
First-line antidiabetic agents targeting AMPK and PPAR-γ exhibit potent immunomodulatory and cytoprotective effects through metabolic pathway modulation. As mentioned earlier, imbalance in the AMPK/mTOR energy-sensing pathway under diabetic conditions is a key mechanism driving metabolic stress and fibrotic transformation in alveolar epithelial cells. Consequently, restoring AMPK activity presents an attractive therapeutic target. Metformin, a canonical AMPK agonist, preserves airway epithelial integrity by stabilizing tight junctions (ZO-1/occludin) and restoring CFTR-mediated ion transport, reducing neutrophilic inflammation (Patkee et al., 2016; Myerburg et al., 2010). Furthermore, it exerts potent antifibrotic effects in pulmonary systems via TGF-β1/Smad3 inhibition, collagen I/III suppression, and PPAR-γ-driven adipogenic reprogramming of lung fibroblasts (evidenced by lipid droplet accumulation and FABP4 upregulation) (Kheirollahi et al., 2019). Pioglitazone, a PPAR-γ agonist, ameliorates pulmonary vascular dysfunction in hypertension by enhancing mitochondrial fatty acid oxidation and transcriptional remodeling (Legchenko et al., 2018). Furthermore, it restores immune equilibrium via PPAR-γ-dependent macrophage polarization and Treg/Th17 balance regulation (Carvalho et al., 2021; Nobs and Kopf, 2018). Therefore, by dual targeting of AMPK and PPAR-γ signaling, these first-line agents orchestrate pulmonary protection through mitochondrial bioenergetic restoration, epithelial barrier stabilization, and immunofibrotic balance, forging a metabolic-immunological nexus that mitigates diabetic pulmonary complications beyond conventional glycemic management. However, their efficacy and optimal dosing in human diabetic pulmonary fibrosis lack support from large-scale clinical data. Furthermore, future research needs to identify which patient subgroups are most likely to benefit from the pulmonary protective effects of these agents.
As noted earlier, aberrant glycogen metabolism in macrophages within the diabetic milieu leads to UDP-glucose (UDPG) accumulation. Binding and activation of the purinergic receptor P2Y14R by UDPG drives STAT1-dependent pro-inflammatory polarization, representing a key initiating mechanism for the inflammatory storm in diabetic lung injury (Ma et al., 2020). Therefore, P2Y14R emerges as a novel and highly potent target for directly interrupting this specific ‘metabolite-immune’ crosstalk for precise anti-inflammatory therapy. The development of new antagonists for this target has made preliminary progress. A recent study reported a highly potent P2Y14R antagonist, compound 25L, based on a 3-sulfonamidobenzoic acid scaffold. This compound demonstrated excellent properties in preclinical studies: its inhibitory activity against P2Y14R (IC50 = 5.6 nM) surpassed that of the lead compound PPTN, with more favorable pharmacokinetic characteristics (Ma et al., 2025). Crucially, in an LPS-induced mouse model of acute lung injury, treatment with compound 25L significantly alleviated pulmonary inflammatory infiltration and levels of pro-inflammatory cytokines (IL-1β, IL-6, TNF-α), providing direct pharmacological evidence supporting the hypothesis that P2Y14R inhibition alleviates lung inflammation (Ma et al., 2025). However, it must be clearly recognized that applying such P2Y14R antagonists to diabetic pulmonary injury faces significant challenges. All reported P2Y14R antagonists remain in the preclinical stage and have not entered clinical development. Common bottlenecks include: first, the need to confirm their long-term efficacy and safety in the context of chronic metabolic diseases such as diabetes, which differs significantly from acute injury models; second, the precise inhibition of the macrophage-specific UDPG-P2Y14R axis in the diabetic state by these compounds requires validation in disease models. Therefore, future research urgently needs to evaluate the efficacy of compound 25L and its analogs in diabetic animal models, focusing on resolving potential off-target effects and long-term toxicological issues to advance this promising target toward clinical translation.
Acetyl-CoA carboxylase (ACC) inhibition demonstrates therapeutic potential for metabolic syndrome by modulating lipid biosynthesis and energy homeostasis (Harwood, 2005). Concurrently, ACC inhibition reduces inflammatory macrophage recruitment through blockade of chemokine receptors CCR2/CCR5 (e.g., via cenicriviroc), thereby attenuating pro-inflammatory cytokine production in pulmonary niches (Tacke and Weiskirchen, 2018). Furthermore, ACC-targeted interventions enhance lipid metabolism in pulmonary macrophages, restricting intracellular lipid droplet availability and thereby reducing Mycobacterium tuberculosis survival and infection likelihood (Brandenburg et al., 2021). BAM15, a mitochondrial uncoupling agent, reverses diet-induced obesity and insulin resistance in murine models while enhancing mitochondrial bioenergetics through cristae remodeling and UCP1-independent thermogenesis (Alexopoulos et al., 2020; Axelrod et al., 2020). Furthermore, in sepsis models, BAM15 polarizes macrophages toward anti-inflammatory phenotypes and reduces pro-inflammatory cytokine production via AMPK-SIRT1-PGC1α axis activation (Dang et al., 2021). These multimodal mechanisms underscore BAM15’s therapeutic potential as a pharmacological candidate for metabolic-inflammatory disorders. However, it is important to note that mitochondrial uncouplers, due to their impact on systemic energy metabolism, may cause difficult-to-control side effects. Achieving lung-specific targeting while avoiding systemic adverse reactions is a prerequisite for developing them into viable drugs. Currently, the research focus in this area should be on developing lung-targeted delivery systems.
7.3 Microbiome-based targeted therapeutics
Metabolites resulting from gut dysbiosis (e.g., decreased SCFAs, increased BCAAs) and endotoxin (LPS) translocation exacerbate pulmonary inflammation via the gut-lung axis. Fecal microbiota transplantation (FMT) from healthy donors to diabetic recipients restores gut microbial diversity. FMT restores SCFA-producing bacteria (e.g., Faecalibacterium), thereby elevating SCFA levels and subsequently activating the GLP-1R axis to improve metabolism (de Groot et al., 2021; Ng et al., 2022; Tanase et al., 2020; Wu et al., 2022). Specific bacterial consortia (e.g., Akkermansia muciniphila + Bifidobacterium longum) further repair intestinal barrier integrity via P9 peptide secretion and AMPK-FOXO1 pathway activation, stabilizing postprandial glycemia (Yoon et al., 2021). Therefore, FMT-mediated restoration of gut microbial diversity activates SCFA-GLP-1 axis-driven metabolic reprogramming and bacterial consortia-enhanced barrier repair, synergistically ameliorating diabetic immunometabolic dyshomeostasis through microbiome-pancreatic crosstalk.
This review highlighted that metabolic endotoxemia resulting from gut dysbiosis (e.g., LPS translocation) is a core event driving pulmonary hyperinflammation via the TLR4/NF-κB pathway. Precision modulation of the gut microbiota, particularly reducing LPS-producing Gram-negative pathogens, represents a potential therapeutic target. Against this backdrop, targeted phage therapy demonstrates unique promise. The gut virome of individuals with DM exhibits characteristic alterations, including an enrichment of Caudovirales phages, which enhance antibiotic resistance in pathogens (e.g., Klebsiella pneumoniae) via horizontal gene transfer. Targeted phage cocktail therapy achieves a reduction in pathogenic bacterial load while preserving commensal microbiota stability (Nishijima et al., 2022; Hu et al., 2023; Shkoporov et al., 2022). Gut-derived phages cross the intestinal barrier via receptor-mediated transcytosis and migrate to pulmonary tissues through the portal venous system, enabling tissue-specific colonization (Ling et al., 2023; Mousavinasab et al., 2023; Naveen Kumar et al., 2025). The pH-responsive microcapsules enable site-specific intestinal release of encapsulated phages, followed by systemic circulation-mediated targeting to inflamed pulmonary niches. Integration of phage pharmacokinetic modeling with real-time host metabolomic profiling enables dynamic assessment of gut-lung axis modulation, allowing iterative refinement of personalized regimens. However, such therapies are currently largely confined to preclinical proof-of-concept. Translating this strategy from the laboratory to the clinic faces numerous severe challenges. As exogenous proteins, the inherent immunogenicity of phages and their colonization stability in humans remain largely unknown. Therefore, future research must prioritize addressing the challenges of precise phage delivery, in vivo kinetics, and strategies to evade host immune clearance.
7.4 Inflammatory network regulation and precision therapy
Although PGE2 is a classic inflammatory mediator, its downstream signaling pathways through different receptors (such as EP4) may be precisely regulated to achieve anti-inflammatory and tissue repair effects. PGE2-EP4 activation elevates cAMP, upregulating 15-lipoxygenase (15-LOX) to shift lipid mediator production from pro-inflammatory leukotriene B4 (LTB4) to anti-inflammatory lipoxin A4 (LXA4). This metabolic switch suppresses neutrophil pulmonary infiltration by downregulating CXCR2 expression and inhibiting NADPH oxidase assembly, reducing bronchoalveolar lavage fluid (BALF) protein extravasation in septic models (Jiao et al., 2023). During pulmonary recovery, PGE2-EP2/EP4 signaling activates CREB, epigenetically enhancing PD-1 expression in lung tissue-resident memory T (TRM) cells. This primes TRM cells for rapid pathogen response while maintaining immune homeostasis (Feehan et al., 2024). These findings collectively demonstrate the critical role of PGE2 in orchestrating long-term immune dynamics and suggest that precision-targeted modulation of its inflammatory pathways—through spatiotemporal regulation of EP receptor signaling—may confer dual therapeutic benefits for both diabetic complications and pulmonary injury.
Emerging study demonstrates that targeted delivery of nanomedicines disrupts inflammatory signaling pathways commonly associated with DM and pulmonary injury, thereby achieving therapeutic efficacy. As described above, the activation of NLRP3 inflammasome leads to the maturation of IL-1β and IL-18, driving lung inflammation. Through structural optimization, the orally administered inhibitor DFV890 achieves colon-specific release, suppressesing ASC oligomerization and IL-1β maturation in diabetic lung injury models. It can enter enterohepatic recirculation to sustain pyroptosis pathway inhibition, offering durable anti-inflammatory effects (Gatlik et al., 2024). Although DFV890, as an oral NLRP3 inhibitor, has shown good tolerability in initial human trials, this represents only the first step in its clinical translation. Key questions remain regarding its ability to effectively inhibit NLRP3 inflammasome activation within the complex milieu of diabetic lung injury, and whether long-term suppression of this core innate immune pathway might lead to the activation of compensatory inflammatory pathways or other unknown off-target effects, all of which require longer-term clinical investigation.
Within the complex inflammatory network of diabetic pulmonary injury described earlier, IL-6 has been identified as a key cytokine driving the acute phase response and immune cell infiltration. Therefore, directly blocking the IL-6 signaling pathway is a rational strategy for controlling aberrant pulmonary inflammation. Tocilizumab, a humanized monoclonal antibody against the IL-6 receptor, is re-evaluated in this context. Tocilizumab is innovatively encapsulated in pH-sensitive nanoparticles, enabling site-specific release at inflammatory loci (pH < 6.5), which enhances pulmonary soluble IL-6 receptor (sIL-6R) neutralization efficiency. In clinical studies of diabetes with comorbid interstitial lung disease, tocilizumab effectively slowed the decline in diffusing capacity for carbon monoxide, potentially by neutralizing IL-6 and breaking the inflammatory cycle (Tanaka et al., 2018; Khanna et al., 2022). Case reports have documented its therapeutic application in managing post-pulmonary infection complications in elderly diabetic patients (Kai et al., 2022). This suggests that combining validated biologics with advanced delivery technologies represents a future direction for precision immunomodulation. However, caution is warranted as the IL-6 pathway plays a crucial role in host defense; thus, long-term use may increase the risk of serious infections. For diabetic patients who are susceptible to themselves, such risks need to be highly valued in future risk-benefit assessments.
In diabetes, Treg cells exhibit a functional impairment that compromises their immunosuppressive capacity. CRISPR/dCas9-mediated precision editing of the FOXP3 enhancer region generates stabilized Treg cells enhanced suppressive activity in vitro. Adoptive transfer of these cells in diabetic pulmonary injury models reduced neutrophil proportion in bronchoalveolar lavage fluid, while maintaining antimicrobial defense capacity (Esensten et al., 2018; Ferreira et al., 2019). A thermosensitive hydrogel loaded with IL-2/JES6-1 complexes enables inflammation-triggered sustained release in pulmonary microenvironments, enhancing Treg recruitment efficiency at lesion sites. This strategy demonstrated greater improvement in PaO2/FiO2 ratio compared to conventional therapies in graft-versus-host disease-associated pulmonary injury (Esensten et al., 2018; Ferreira et al., 2019; Wang C. et al., 2021). Thus, adoptive transfer of engineered Treg cells provides a precise tool for restoring immune tolerance. However, the high technical complexity and cost of this approach severely limit its broad application. More critically, safety remains a paramount hurdle: Are the edited Treg cells stable in vivo? Is there a risk of their conversion into pathogenic effector cells? Before considering any clinical application, stringent safety guidelines must be established, and long-term follow-up studies in larger animal models are imperative.
7.4.1 Summary and prospect
Emerging mechanistic insights into DM-associated pulmonary injury have unveiled a sophisticated multi-axis interplay among metabolic regulatory networks, gut microbial homeostasis, and host immune response systems. This field necessitates comprehensive investigations from a tripartite metabolic-microbial-immune interaction perspective to systematically elucidate the pathogenesis, comorbidity progression patterns, and prognostic determinants of diabetic pulmonary complications. Clinical epidemiological studies have established significant comorbidities between DM and respiratory disorders including lung cancer and COPD. The metabolic regulatory network integrates nutrient-sensing pathways encompassing glucose metabolism, lipid homeostasis, and amino acid flux, forming the pivotal pathological nexus linking DM to its pulmonary complications. Mechanistically, DM-induced perturbations in the AMPK/mTOR signaling axis, impaired branched-chain amino acid catabolism, and tryptophan-kynurenine axis dysregulation collectively drive oxidative stress and immune homeostasis disruption through cascade reactions, culminating in pulmonary parenchymal injury. Crucially, gut microbiota exerts central regulatory control over diabetic pulmonary injury progression via bidirectional gut-lung axis crosstalk. Operative mechanisms include: 1) Immune network remodeling mediated by microbiota-derived metabolites; 2) Synergistic amplification of endogenous metabolic disturbances and exogenous environmental stressors. Under pathological conditions such as endotoxemia and intestinal barrier dysfunction, dysbiosis-driven metabolic reprogramming converts entero-derived metabolic signals into pro-inflammatory cytokine cascades, thereby potentiating pulmonary inflammatory microenvironment formation.
7.4.2 Emerging therapeutic dilemmas in DM-pulmonary comorbidity management
While the causal relationship between DM and pulmonary disorders remains incompletely elucidated, the clinical management of their comorbid status presents emerging challenges. Shared inflammatory pathophysiology and multisystem interactions substantially amplify clinical complexity, particularly evident in diabetic patients with concurrent COPD. Current practice reveals a critical therapeutic paradox: although international guidelines endorse inhaled corticosteroids (ICS) as standard COPD therapy (Agustí et al., 2023), their dose-dependent induction of glycemic variability (Slatore et al., 2009) establishes potentially conflicting treatment paradigms.
Dual Therapeutic Benefits of GLP-1 Receptor Agonists: Glucagon-like peptide-1 (GLP-1), an incretin hormone secreted by intestinal L-cells, activates cAMP/PKA signaling through GLP-1 receptor (GLP-1R) engagement, not only potentiating glucose-dependent insulin secretion for glycemic control (Kim and Egan, 2008) but also exerting novel respiratory effects. Clinical evidence demonstrates that GLP-1 receptor agonists (GLP-1RAs) confer dual benefits in DM-COPD comorbidity: reducing asthma exacerbation risk (Foer et al., 2023), while ameliorating bronchial hyperresponsiveness via PKA activation in airway epithelial cells (Rogliani et al., 2016). Mechanistically, these agents remodel alveolar macrophage metabolic phenotypes and suppress NF-κB-mediated inflammatory cascades.
7.4.3 Future directions in metabo-inflammatory modulation
These breakthroughs underscore the therapeutic potential of targeting metabo-inflammatory crosstalk for multidimensional comorbidity management (Table 1). Advancing this frontier requires establishing multi-omics predictive models to optimize personalized dosing regimens within evidence-based frameworks, thereby achieving synergistic glycemic and pulmonary functional outcomes.
TABLE 1
| Therapeutic target | Mechanism of action | Representative agents/Interventions | Clinical evidence |
|---|---|---|---|
| Metabolic interventions | |||
| SGLT-2 inhibitors | Inhibit glucose reabsorption; reduce oxidative/ER stress; modulate macrophage polarization | Canagliflozin, Dapagliflozin | Reduces COPD exacerbation risk; attenuates sepsis-induced lung injury (Lin et al., 2020; O'Keefe et al., 2023; Packer, 2020) |
| GLP-1 receptor agonists | Stimulate insulin secretion; suppress TLR-mediated TNF-α release; regulate neutrophil recruitment | Liraglutide, Semaglutide | Decreases asthma incidence; inhibits pulmonary fibrosis (Wong et al., 2024; Lee et al., 2025; Toki et al., 2021) |
| AMPK agonists | Maintain airway epithelial integrity; suppress TGF-β1-driven fibrosis | Metformin | Anti-fibrotic effects; enhances infection resistance (Patkee et al., 2016; Myerburg et al., 2010; Kheirollahi et al., 2019) |
| PPAR-γ agonists | Regulate fatty acid oxidation; improve mitochondrial function; suppress pulmonary inflammation | Pioglitazone | Modulates lung immune development; improves hypertensive vascular function (Legchenko et al., 2018; Carvalho et al., 2021; Nobs and Kopf, 2018) |
| P2Y14R antagonists | Suppress pulmonary inflammation | Compound 25L | Reduced the inflammatory response and the levels of proinflammatory cytokines in lung tissues (Ma et al., 2025) |
| ACC inhibitors | Block CCR2/CCR5-dependent inflammatory macrophage recruitment; enhance lipid metabolism | Cenicriviroc | Reduces tuberculosis susceptibility (Harwood, 2005; Tacke and Weiskirchen, 2018; Brandenburg et al., 2021) |
| Mitochondrial uncouplers | Reverse insulin resistance; enhance mitochondrial efficiency; promote anti-inflammatory macrophages | BAM15 | Mitigates sepsis-associated inflammation (Alexopoulos et al., 2020; Axelrod et al., 2020; Dang et al., 2021) |
| Gut microbiome modulation | |||
| FMT/probiotics | Restore microbial diversity; reverse gut hyperpermeability; balance SCFA/BCAA metabolism | Human-derived probiotics (e.g., Bifidobacterium) | Improves insulin sensitivity; reduces systemic inflammation (de Groot et al., 2021; Ng et al., 2022; Tanase et al., 2020; Wu et al., 2022) |
| Phage therapy | Precisely edit gut virome; block gut-lung axis transmission of LPS | CRISPR-engineered phages | Regulates pulmonary immune responses (Ling et al., 2023; Mousavinasab et al., 2023) |
| Immunomodulation | |||
| PGE2 pathway modulators | Shift lipid mediator profile (LTB4→LXA4); inhibit neutrophil recruitment/NET formation | EP4 receptor agonists | Controls sepsis-induced injury; enhances T cell memory (Jiao et al., 2023; Feehan et al., 2024) |
| NLRP3 inhibitors | Block inflammasome activation; reduce IL-1β release | DFV890 | Specifically targets NLRP3 signaling (Gatlik et al., 2024) |
| IL-6 receptor antagonists | Neutralize IL-6 signaling; suppress systemic inflammation | Tocilizumab | Effective in interstitial lung disease; treats diabetes-associated pulmonary infection (Tanaka et al., 2018; Khanna et al., 2022; Kai et al., 2022) |
| Treg cell therapy | Re-establish immune tolerance; suppress hyperinflammation | Exogenous Treg infusion | Demonstrates safety in type 1 diabetes and transplantation (Esensten et al., 2018; Ferreira et al., 2019) |
| Epigenetic modulation | |||
| miRNA regulators | Inhibit pro-inflammatory miRNAs (e.g., miR-155); reduce M1 macrophage polarization | miRNA antagonists (e.g., antagomir-155) | Attenuates lung injury-associated inflammation (Huang et al., 2014; Prieto et al., 2023; Ying et al., 2017; Xu et al., 2023b) |
Multidimensional therapeutic strategies targeting diabetic pulmonary injury.
Persisting knowledge gaps surround the mechanistic contributions of dysregulated metabolic functions to disease pathogenesis. Groundbreaking advances in metabolomics and metagenomics have enabled integrated multi-omics frameworks to systematically deconvolute the multidimensional “metabolism-microbiota-immune” interactome. This field is undergoing a paradigm shift—wherein cross-scale biological network modeling allows precise identification of critical regulatory nodes governing metabolic imbalance and immune dysregulation. Such systems biology perspectives not only drive fundamental understanding of comorbidity mechanisms but also provide a theoretical foundation for developing next-generation trans-organ therapeutic strategies targeting systemic immunometabolic homeostasis.
Statements
Author contributions
JS: Data curation, Formal Analysis, Investigation, Methodology, Writing – original draft. JC: Data curation, Formal Analysis, Investigation, Methodology, Writing – original draft. YS: Data curation, Formal Analysis, Writing – original draft. XY: Formal Analysis, Investigation, Writing – review and editing. HS: Formal Analysis, Investigation, Writing – review and editing. BC: Conceptualization, Funding acquisition, Writing – review and editing. JF: Conceptualization, Investigation, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (No. 82401633), Jiangsu Provincial Research Hospital (YJXYY202204-XKA02), the Natural Science Research Projects in Universities of Jiangsu Province (No. 24KJA310007, No. 24KJB310013), and Suzhou Science and Technology Development Program (No. SYW2024048).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Agustí A. Celli B. R. Criner G. J. Halpin D. Anzueto A. Barnes P. et al (2023). Global initiative for chronic obstructive lung disease 2023 report: GOLD executive summary. Eur. Respir. J.61 (4), 2300239. 10.1183/13993003.00239-2023
2
Ahmad N. Srinivasan K. Moudgil H. (2011). Diabetes and lung function: part of a wider spectrum. Chest139 (1), 235–236. 10.1378/chest.10-2095
3
Alexopoulos S. J. Chen S. Y. Brandon A. E. Salamoun J. M. Byrne F. L. Garcia C. J. et al (2020). Mitochondrial uncoupler BAM15 reverses diet-induced obesity and insulin resistance in mice. Nat. Commun.11 (1), 2397. 10.1038/s41467-020-16298-2
4
Avellaneda-Franco L. Xie L. Nakai M. Barr J. J. Marques F. Z. (2024). Dietary fiber intake impacts gut bacterial and viral populations in a hypertensive mouse model. Gut microbes16 (1), 2407047. 10.1080/19490976.2024.2407047
5
Axelrod C. L. King W. T. Davuluri G. Noland R. C. Hall J. Hull M. et al (2020). BAM15-mediated mitochondrial uncoupling protects against obesity and improves glycemic control. EMBO Mol. Med.12 (7), e12088. 10.15252/emmm.202012088
6
Betteridge D. J. (2000). What is oxidative stress?Metabolism Clin. Exp.49 (2 Suppl. 1), 3–8. 10.1016/s0026-0495(00)80077-3
7
Blair M. C. Neinast M. D. Jang C. Chu Q. Jung J. W. Axsom J. et al (2023). Branched-chain amino acid catabolism in muscle affects systemic BCAA levels but not insulin resistance. Nat. Metab.5 (4), 589–606. 10.1038/s42255-023-00794-y
8
Bonyek-Silva I. Machado A. F. A. Cerqueira-Silva T. Nunes S. Silva Cruz M. R. Silva J. et al (2021). LTB(4)-Driven inflammation and increased expression of ALOX5/ACE2 during severe COVID-19 in individuals with diabetes. Diabetes70 (9), 2120–2130. 10.2337/db20-1260
9
Brandenburg J. Marwitz S. Tazoll S. C. Waldow F. Kalsdorf B. Vierbuchen T. et al (2021). WNT6/ACC2-induced storage of triacylglycerols in macrophages is exploited by Mycobacterium tuberculosis. J. Clin. investigation131 (16), e141833. 10.1172/JCI141833
10
Brown E. J. Albers M. W. Shin T. B. Ichikawa K. Keith C. T. Lane W. S. et al (1994). A Mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature369 (6483), 756–758. 10.1038/369756a0
11
Burcelin R. (2016). Gut microbiota and immune crosstalk in metabolic disease. Mol. Metab.5 (9), 771–781. 10.1016/j.molmet.2016.05.016
12
Burgy O. Loriod S. Beltramo G. Bonniaud P. (2022). Extracellular lipids in the lung and their role in pulmonary fibrosis. Cells11 (7), 1209. 10.3390/cells11071209
13
Canfora E. E. Meex R. C. R. Venema K. Blaak E. E. (2019). Gut microbial metabolites in obesity, NAFLD and T2DM. Nat. Rev. Endocrinol.15 (5), 261–273. 10.1038/s41574-019-0156-z
14
Cani P. D. Amar J. Iglesias M. A. Poggi M. Knauf C. Bastelica D. et al (2007). Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes56 (7), 1761–1772. 10.2337/db06-1491
15
Carvalho M. V. Gonçalves-de-Albuquerque C. F. Silva A. R. (2021). PPAR gamma: from definition to molecular targets and therapy of lung diseases. Int. J. Mol. Sci.22 (2), 805. 10.3390/ijms22020805
16
Casas S. Novials A. Reimann F. Gomis R. Gribble F. M. (2008). Calcium elevation in mouse pancreatic beta cells evoked by extracellular human islet amyloid polypeptide involves activation of the mechanosensitive ion channel TRPV4. Diabetologia51 (12), 2252–2262. 10.1007/s00125-008-1111-z
17
Castro E. C. Sen P. Parks W. T. Langston C. Galambos C. (2017). The role of serotonin transporter in human lung development and in neonatal lung disorders. Can. Respir. J.2017, 9064046. 10.1155/2017/9064046
18
Caughey G. E. Roughead E. E. Vitry A. I. McDermott R. A. Shakib S. Gilbert A. L. (2010). Comorbidity in the elderly with diabetes: identification of areas of potential treatment conflicts. Diabetes Res. Clin. Pract.87 (3), 385–393. 10.1016/j.diabres.2009.10.019
19
Caughey G. E. Preiss A. K. Vitry A. I. Gilbert A. L. Roughead E. E. (2013). Comorbid diabetes and COPD: impact of corticosteroid use on diabetes complications. Diabetes care36 (10), 3009–3014. 10.2337/dc12-2197
20
Chávez-Talavera O. Tailleux A. Lefebvre P. Staels B. (2017). Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic Fatty liver disease. Gastroenterology152 (7), 1679–1694.e1673. 10.1053/j.gastro.2017.01.055
21
Chen Y. Zhang F. Wang D. Li L. Si H. Wang C. et al (2020). Mesenchymal stem cells attenuate diabetic lung fibrosis via adjusting Sirt3-Mediated stress responses in rats. Oxidative Med. Cell. Longev.2020, 8076105. 10.1155/2020/8076105
22
Cheng K. T. Xiong S. Ye Z. Hong Z. Di A. Tsang K. M. et al (2017). Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J. Clin. investigation127 (11), 4124–4135. 10.1172/JCI94495
23
Chiou J. Geusz R. J. Okino M. L. Han J. Y. Miller M. Melton R. et al (2021). Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature594 (7863), 398–402. 10.1038/s41586-021-03552-w
24
Cho N. H. Shaw J. E. Karuranga S. Huang Y. da Rocha Fernandes J. D. Ohlrogge A. W. et al (2018). IDF Diabetes Atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract.138, 271–281. 10.1016/j.diabres.2018.02.023
25
Conroy L. R. Clarke H. A. Allison D. B. Valenca S. S. Sun Q. Hawkinson T. R. et al (2023). Spatial metabolomics reveals glycogen as an actionable target for pulmonary fibrosis. Nat. Commun.14 (1), 2759. 10.1038/s41467-023-38437-1
26
Conte C. Cipponeri E. Roden M. (2024). Diabetes Mellitus, energy metabolism, and COVID-19. Endocr. Rev.45 (2), 281–308. 10.1210/endrev/bnad032
27
Dang C. P. Issara-Amphorn J. Charoensappakit A. Udompornpitak K. Bhunyakarnjanarat T. Saisorn W. et al (2021). BAM15, a mitochondrial uncoupling agent, attenuates inflammation in the LPS Injection Mouse model: an adjunctive anti-inflammation on macrophages and hepatocytes. J. innate Immun.13 (6), 359–375. 10.1159/000516348
28
Darby W. G. Grace M. S. Baratchi S. McIntyre P. (2016). Modulation of TRPV4 by diverse mechanisms. Int. J. Biochem. and Cell. Biol.78, 217–228. 10.1016/j.biocel.2016.07.012
29
De Giovanni M. Dang E. V. Chen K. Y. An J. Madhani H. D. Cyster J. G. (2023). Platelets and mast cells promote pathogenic eosinophil recruitment during invasive fungal infection via the 5-HIAA-GPR35 ligand-receptor system. Immunity56 (7), 1548–1560.e5. 10.1016/j.immuni.2023.05.006
30
de Groot P. Nikolic T. Pellegrini S. Sordi V. Imangaliyev S. Rampanelli E. et al (2021). Faecal microbiota transplantation halts progression of human new-onset type 1 diabetes in a randomised controlled trial. Gut70 (1), 92–105. 10.1136/gutjnl-2020-322630
31
de Vos W. M. Tilg H. Van Hul M. Cani P. D. (2022). Gut microbiome and health: mechanistic insights. Gut71 (5), 1020–1032. 10.1136/gutjnl-2021-326789
32
Dewanjee S. Chakraborty P. Bhattacharya H. Chacko L. Singh B. Chaudhary A. et al (2022). Altered glucose metabolism in Alzheimer's disease: role of mitochondrial dysfunction and oxidative stress. Free Radic. Biol. and Med.193 (Pt 1), 134–157. 10.1016/j.freeradbiomed.2022.09.032
33
Du K. Sun L. Luo Z. Cao Y. Sun Q. Zhang K. et al (2022). Reduced DMPC and PMPC in lung surfactant promote SARS-CoV-2 infection in obesity. Metabolism Clin. Exp.131, 155181. 10.1016/j.metabol.2022.155181
34
Eid S. Sas K. M. Abcouwer S. F. Feldman E. L. Gardner T. W. Pennathur S. et al (2019). New insights into the mechanisms of diabetic complications: role of lipids and lipid metabolism. Diabetologia62 (9), 1539–1549. 10.1007/s00125-019-4959-1
35
Eid S. A. Rumora A. E. Beirowski B. Bennett D. L. Hur J. Savelieff M. G. et al (2023). New perspectives in diabetic neuropathy. Neuron111 (17), 2623–2641. 10.1016/j.neuron.2023.05.003
36
El-Horany H. E. Atef M. M. Abdel Ghafar M. T. Fouda M. H. Nasef N. A. Hegab I. I. et al (2023). Empagliflozin ameliorates bleomycin-induced pulmonary fibrosis in rats by modulating Sesn2/AMPK/Nrf2 signaling and targeting ferroptosis and autophagy. Int. J. Mol. Sci.24 (11), 9481. 10.3390/ijms24119481
37
Entezari M. Hashemi D. Taheriazam A. Zabolian A. Mohammadi S. Fakhri F. et al (2022). AMPK signaling in diabetes mellitus, insulin resistance and diabetic complications: a pre-clinical and clinical investigation. Biomed. and Pharmacother. = Biomedecine and Pharmacother.146, 112563. 10.1016/j.biopha.2021.112563
38
Erener S. (2020). Diabetes, infection risk and COVID-19. Mol. Metab.39, 101044. 10.1016/j.molmet.2020.101044
39
Esensten J. H. Muller Y. D. Bluestone J. A. Tang Q. (2018). Regulatory T-cell therapy for autoimmune and autoinflammatory diseases: the next frontier. J. allergy Clin. Immunol.142 (6), 1710–1718. 10.1016/j.jaci.2018.10.015
40
Fan G. Cao F. Kuang T. Yi H. Zhao C. Wang L. et al (2023). Alterations in the gut virome are associated with type 2 diabetes and diabetic nephropathy. Gut microbes15 (1), 2226925. 10.1080/19490976.2023.2226925
41
Feehan K. T. Bridgewater H. E. Stenkiewicz-Witeska J. De Maeyer R. P. H. Ferguson J. Mack M. et al (2024). Post-resolution macrophages shape long-term tissue immunity and integrity in a mouse model of pneumococcal pneumonia. Nat. Commun.15 (1), 4326. 10.1038/s41467-024-48138-y
42
Fehrenbach H. Kasper M. Tschernig T. Shearman M. S. Schuh D. Müller M. (1998). Receptor for advanced glycation endproducts (RAGE) exhibits highly differential cellular and subcellular localisation in rat and human lung. Cellular and molecular biology (Noisy-le-Grand, France). Cell. Mol. Biol.44 (7), 1147–1157.
43
Fernández-Real J. M. Chico B. Shiratori M. Nara Y. Takahashi H. Ricart W. (2008). Circulating surfactant protein A (SP-A), a marker of lung injury, is associated with insulin resistance. Diabetes care31 (5), 958–963. 10.2337/dc07-2173
44
Fernández-Real J. M. Valdés S. Manco M. Chico B. Botas P. Campo A. et al (2010). Surfactant protein d, a marker of lung innate immunity, is positively associated with insulin sensitivity. Diabetes care33 (4), 847–853. 10.2337/dc09-0542
45
Ferreira L. M. R. Muller Y. D. Bluestone J. A. Tang Q. (2019). Next-generation regulatory T cell therapy. Nat. Rev. Drug Discov.18 (10), 749–769. 10.1038/s41573-019-0041-4
46
Fisher A. B. (1984). Intermediary metabolism of the lung. Environ. health Perspect.55, 149–158. 10.1289/ehp.8455149
47
Foer D. Strasser Z. H. Cui J. Cahill K. N. Boyce J. A. Murphy S. N. et al (2023). Association of GLP-1 receptor agonists with chronic obstructive pulmonary disease exacerbations among patients with type 2 diabetes. Am. J. Respir. Crit. care Med.208 (10), 1088–1100. 10.1164/rccm.202303-0491OC
48
Fonseca W. Malinczak C. A. Fujimura K. Li D. McCauley K. Li J. et al (2021). Maternal gut microbiome regulates immunity to RSV infection in offspring. J. Exp. Med.218 (11), e20210235. 10.1084/jem.20210235
49
Forrester S. J. Kikuchi D. S. Hernandes M. S. Xu Q. Griendling K. K. (2018). Reactive oxygen species in metabolic and inflammatory signaling. Circulation Res.122 (6), 877–902. 10.1161/CIRCRESAHA.117.311401
50
Gao Z. Yin J. Zhang J. Ward R. E. Martin R. J. Lefevre M. et al (2009). Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes58 (7), 1509–1517. 10.2337/db08-1637
51
Gao P. Uzun Y. He B. Salamati S. E. Coffey J. K. M. Tsalikian E. et al (2019). Risk variants disrupting enhancers of T(H)1 and T(REG) cells in type 1 diabetes. Proc. Natl. Acad. Sci. U. S. A.116 (15), 7581–7590. 10.1073/pnas.1815336116
52
Gao J. Yang T. Song B. Ma X. Ma Y. Lin X. et al (2023). Abnormal tryptophan catabolism in diabetes mellitus and its complications: opportunities and challenges. Biomed. and Pharmacother. = Biomedecine and Pharmacother.166, 115395. 10.1016/j.biopha.2023.115395
53
Gatlik E. Mehes B. Voltz E. Sommer U. Tritto E. Lestini G. et al (2024). First-in-human safety, tolerability, and pharmacokinetic results of DFV890, an oral low-molecular-weight NLRP3 inhibitor. Clin. Transl. Sci.17 (5), e13789. 10.1111/cts.13789
54
Goel K. Beatman E. L. Egersdorf N. Scruggs A. Cao D. Berdyshev E. V. et al (2021). Sphingosine 1 phosphate (S1P) receptor 1 is decreased in human lung Microvascular endothelial cells of smokers and mediates S1P effect on autophagy. Cells10 (5), 1200. 10.3390/cells10051200
55
Gojda J. Cahova M. (2021). Gut microbiota as the link between elevated BCAA serum levels and insulin resistance. Biomolecules11 (10), 1414. 10.3390/biom11101414
56
Golay A. Felber J. P. Meyer H. U. Curchod B. Maeder E. Jéquier E. (1984). Study on lipid metabolism in obesity diabetes. Metabolism Clin. Exp.33 (2), 111–116. 10.1016/0026-0495(84)90121-5
57
Goncharova E. A. (2013). mTOR and vascular remodeling in lung diseases: current challenges and therapeutic prospects. FASEB J. official Publ. Fed. Am. Soc. Exp. Biol.27 (5), 1796–1807. 10.1096/fj.12-222224
58
Greer R. L. Morgun A. Shulzhenko N. (2013). Bridging immunity and lipid metabolism by gut microbiota. J. allergy Clin. Immunol.132 (2), 253–262. 10.1016/j.jaci.2013.06.025
59
Guo W. A. Knight P. R. Raghavendran K. (2012). The receptor for advanced glycation end products and acute lung injury/acute respiratory distress syndrome. Intensive care Med.38 (10), 1588–1598. 10.1007/s00134-012-2624-y
60
Haghikia A. Jörg S. Duscha A. Berg J. Manzel A. Waschbisch A. et al (2015). Dietary fatty acids directly impact central nervous System autoimmunity via the small intestine. Immunity43 (4), 817–829. 10.1016/j.immuni.2015.09.007
61
Han S. Mallampalli R. K. (2015). The role of surfactant in lung disease and host defense against pulmonary infections. Ann. Am. Thorac. Soc.12 (5), 765–774. 10.1513/AnnalsATS.201411-507FR
62
Harayama T. Eto M. Shindou H. Kita Y. Otsubo E. Hishikawa D. et al (2014). Lysophospholipid acyltransferases mediate phosphatidylcholine diversification to achieve the physical properties required in vivo. Cell. metab.20 (2), 295–305. 10.1016/j.cmet.2014.05.019
63
Harwood H. J. Jr. (2005). Treating the metabolic syndrome: acetyl-coa carboxylase inhibition. Expert Opin. Ther. targets9 (2), 267–281. 10.1517/14728222.9.2.267
64
Hassan M. H. Galal O. Sakhr H. M. Kamaleldeen E. B. Zekry N. F. Fateen E. et al (2023). Profile of plasma free amino acids, carnitine and acylcarnitines, and JAK2(v617f) mutation as potential metabolic markers in children with type 1 diabetic nephropathy. Biomed. Chromatogr. BMC37 (12), e5747. 10.1002/bmc.5747
65
Hatlen P. Grønberg B. H. Langhammer A. Carlsen S. M. Amundsen T. (2011). Prolonged survival in patients with lung cancer with diabetes mellitus. J. Thorac. Oncol. official Publ. Int. Assoc. Study Lung Cancer6 (11), 1810–1817. 10.1097/JTO.0b013e31822a75be
66
He M. Y. Zhang P. Shi N. Li T. Wang J. He L. et al (2023). Nrf2 is involved in hyperglycemia-induced abnormal lung development through both antioxidation-dependent and antioxidation-independent activation. Am. J. Respir. Cell. Mol. Biol.69 (2), 197–209. 10.1165/rcmb.2022-0345OC
67
Hetz C. Papa F. R. (2018). The unfolded protein response and cell fate control. Mol. Cell.69 (2), 169–181. 10.1016/j.molcel.2017.06.017
68
Hsu H. S. Liu C. C. Lin J. H. Hsu T. W. Hsu J. W. Su K. et al (2017). Involvement of ER stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Sci. Rep.7 (1), 14272. 10.1038/s41598-017-14612-5
69
Hu J. Ding J. Li X. Li J. Zheng T. Xie L. et al (2023). Distinct signatures of gut microbiota and metabolites in different types of diabetes: a population-based cross-sectional study. EClinicalMedicine62, 102132. 10.1016/j.eclinm.2023.102132
70
Huang Y. Zhou M. Sun H. Wang Y. (2011). Branched-chain amino acid metabolism in heart disease: an epiphenomenon or a real culprit?Cardiovasc. Res.90 (2), 220–223. 10.1093/cvr/cvr070
71
Huang Y. Liu Y. Li L. Su B. Yang L. Fan W. et al (2014). Involvement of inflammation-related miR-155 and miR-146a in diabetic nephropathy: implications for glomerular endothelial injury. BMC Nephrol.15, 142. 10.1186/1471-2369-15-142
72
Huang L. M. Hu Q. Huang X. Qian Y. Lai X. H. (2020). Preconditioning rats with three lipid emulsions prior to acute lung injury affects cytokine production and cell apoptosis in the lung and liver. Lipids health Dis.19 (1), 19. 10.1186/s12944-019-1137-x
73
Hughes M. J. McGettrick H. M. Sapey E. (2020). Shared mechanisms of multimorbidity in COPD, atherosclerosis and type-2 diabetes: the neutrophil as a potential inflammatory target. Eur. Respir. Rev. official J. Eur. Respir. Soc.29 (155), 190102. 10.1183/16000617.0102-2019
74
Jeyanathan M. Vaseghi-Shanjani M. Afkhami S. Grondin J. A. Kang A. D'Agostino M. R. et al (2022). Parenteral BCG vaccine induces lung-resident memory macrophages and trained immunity via the gut-lung axis. Nat. Immunol.23 (12), 1687–1702. 10.1038/s41590-022-01354-4
75
Jiao Y. Zhang T. Liu M. Zhou L. Qi M. Xie X. et al (2023). Exosomal PGE2 from M2 macrophages inhibits neutrophil recruitment and NET formation through lipid mediator class switching in sepsis. J. Biomed. Sci.30 (1), 62. 10.1186/s12929-023-00957-9
76
Jin Y. Dong H. Xia L. Yang Y. Zhu Y. Shen Y. et al (2019). The diversity of gut Microbiome is associated with favorable responses to anti-programmed death 1 immunotherapy in Chinese patients with NSCLC. J. Thorac. Oncol. official Publ. Int. Assoc. Study Lung Cancer14 (8), 1378–1389. 10.1016/j.jtho.2019.04.007
77
Jin S. Ding X. Yang C. Li W. Deng M. Liao H. et al (2021). Mechanical ventilation exacerbates poly (I:C) induced acute lung injury: central role for Caspase-11 and gut-lung axis. Front. Immunol.12, 693874. 10.3389/fimmu.2021.693874
78
Jin Y. Wang Y. Feng M. Ni X. Qiang L. Xue J. et al (2024). Sphingosine-1-phosphate alleviates Sjögren's syndrome-like symptoms via inducing autophagy and regulating status of Treg cells in NOD mice. Int. Immunopharmacol.143 (Pt 3), 113514. 10.1016/j.intimp.2024.113514
79
Jones J. H. Minshall R. D. (2020). Lung Endothelial Transcytosis. Compr. Physiol.10 (2), 491–508. 10.1002/cphy.c190012
80
Jung S. J. Hadinoto K. Park J. W. (2023). Mechanical properties of 3-Hydroxybutyric acid-induced vesicles. Mol. Basel, Switz.28 (6), 2742. 10.3390/molecules28062742
81
Karcz T. P. Whitehead G. S. Nakano K. Nakano H. Grimm S. A. Williams J. G. et al (2021). UDP-glucose and P2Y14 receptor amplify allergen-induced airway eosinophilia. J. Clin. investigation131 (7), e140709. 10.1172/JCI140709
82
Kai Y. Matsuda M. Suzuki K. Kasamatsu T. Kajita A. Uno K. et al (2022). Tocilizumab and baricitinib for recovery from acute exacerbation of combined pulmonary fibrosis and emphysema secondary to COVID-19 infection: a case report. Cureus14 (3), e23411. 10.7759/cureus.23411
83
Keestra-Gounder A. M. Byndloss M. X. Seyffert N. Young B. M. Chávez-Arroyo A. Tsai A. Y. et al (2016). NOD1 and NOD2 signalling links ER stress with inflammation. Nature532 (7599), 394–397. 10.1038/nature17631
84
Khalid M. Petroianu G. Adem A. (2022). Advanced glycation end products and diabetes mellitus: mechanisms and perspectives. Biomolecules12 (4), 542. 10.3390/biom12040542
85
Khanna D. Lin C. J. F. Furst D. E. Wagner B. Zucchetto M. Raghu G. et al (2022). Long-Term safety and efficacy of Tocilizumab in early systemic sclerosis-interstitial lung disease: Open-Label extension of a phase 3 randomized controlled trial. Am. J. Respir. Crit. care Med.205 (6), 674–684. 10.1164/rccm.202103-0714OC
86
Khateeb J. Fuchs E. Khamaisi M. (2019). Diabetes and lung disease: a neglected relationship. Rev. Diabet. Stud. RDS15, 1–15. 10.1900/RDS.2019.15.1
87
Kheirollahi V. Wasnick R. M. Biasin V. Vazquez-Armendariz A. I. Chu X. Moiseenko A. et al (2019). Metformin induces lipogenic differentiation in myofibroblasts to reverse lung fibrosis. Nat. Commun.10 (1), 2987. 10.1038/s41467-019-10839-0
88
Kim W. Egan J. M. (2008). The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol. Rev.60 (4), 470–512. 10.1124/pr.108.000604
89
Kim K. H. Lee M. S. (2014). Autophagy--a key player in cellular and body metabolism. Nat. Rev. Endocrinol.10 (6), 322–337. 10.1038/nrendo.2014.35
90
Koren O. Konnikova L. Brodin P. Mysorekar I. U. Collado M. C. (2024). The maternal gut microbiome in pregnancy: implications for the developing immune system. Nat. Rev. Gastroenterology and hepatology21 (1), 35–45. 10.1038/s41575-023-00864-2
91
Kottmann R. M. Kulkarni A. A. Smolnycki K. A. Lyda E. Dahanayake T. Salibi R. et al (2012). Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am. J. Respir. Crit. care Med.186 (8), 740–751. 10.1164/rccm.201201-0084OC
92
Koukourakis M. I. Kalamida D. Mitrakas A. G. Liousia M. Pouliliou S. Sivridis E. et al (2017). Metabolic cooperation between co-cultured lung cancer cells and lung fibroblasts. Laboratory investigation; a J. Tech. methods pathology97 (11), 1321–1331. 10.1038/labinvest.2017.79
93
Kozieł K. Urbanska E. M. (2023). Kynurenine pathway in diabetes mellitus-novel pharmacological target?Cells12 (3), 460. 10.3390/cells12030460
94
Kuda O. Jenkins C. M. Skinner J. R. Moon S. H. Su X. Gross R. W. et al (2011). CD36 protein is involved in store-operated calcium flux, phospholipase A2 activation, and production of prostaglandin E2. J. Biol. Chem.286 (20), 17785–17795. 10.1074/jbc.M111.232975
95
Kukreja A. Cost G. Marker J. Zhang C. Sun Z. Lin-Su K. et al (2002). Multiple immuno-regulatory defects in type-1 diabetes. J. Clin. investigation109 (1), 131–140. 10.1172/JCI13605
96
Kwon S. Crowley G. Caraher E. J. Haider S. H. Lam R. Veerappan A. et al (2019). Validation of predictive metabolic syndrome biomarkers of world Trade Center lung injury: a 16-Year longitudinal Study. Chest156 (3), 486–496. 10.1016/j.chest.2019.02.019
97
Lai H. C. Lin T. L. Chen T. W. Kuo Y. L. Chang C. J. Wu T. R. et al (2022). Gut microbiota modulates COPD pathogenesis: role of anti-inflammatory Parabacteroides goldsteinii lipopolysaccharide. Gut71 (2), 309–321. 10.1136/gutjnl-2020-322599
98
Lange P. Groth S. Kastrup J. Mortensen J. Appleyard M. Nyboe J. et al (1989). Diabetes mellitus, plasma glucose and lung function in a cross-sectional population study. Eur. Respir. J.2 (1), 14–19. 10.1183/09031936.93.02010014
99
Lee B. Man K. K. C. Wong E. Tan T. Sheikh A. Bloom C. I. (2025). Antidiabetic medication and asthma attacks. JAMA Intern. Med.185 (1), 16–25. 10.1001/jamainternmed.2024.5982
100
Legchenko E. Chouvarine P. Borchert P. Fernandez-Gonzalez A. Snay E. Meier M. et al (2018). PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci. Transl. Med.10 (438), eaao0303. 10.1126/scitranslmed.aao0303
101
Li S. Yang H. (2019). Relationship between advanced glycation end products and gestational diabetes mellitus. J. maternal-fetal and neonatal Med. official J. Eur. Assoc. Perinat. Med. Fed. Asia Ocean. Perinat. Soc. Int. Soc. Perinat. Obstet32 (17), 2783–2789. 10.1080/14767058.2018.1449201
102
Li K. Li M. Li W. Yu H. Sun X. Zhang Q. et al (2019). Airway epithelial regeneration requires autophagy and glucose metabolism. Cell. death and Dis.10 (12), 875. 10.1038/s41419-019-2111-2
103
Li M. Zhang C. S. Feng J. W. Wei X. Zhang C. Xie C. et al (2021). Aldolase is a sensor for both low and high glucose, linking to AMPK and mTORC1. Cell. Res.31 (4), 478–481. 10.1038/s41422-020-00456-8
104
Li C. Gao P. Zhuang F. Wang T. Wang Z. Wu G. et al (2024). Inhibition of ALOX12-12-HETE alleviates lung ischemia-reperfusion injury by reducing endothelial ferroptosis-mediated neutrophil extracellular trap Formation, 7:0473. 10.34133/research.0473
105
Lin F. Song C. Zeng Y. Li Y. Li H. Liu B. et al (2020). Canagliflozin alleviates LPS-induced acute lung injury by modulating alveolar macrophage polarization. Int. Immunopharmacol.88, 106969. 10.1016/j.intimp.2020.106969
106
Ling C. Bacos K. Rönn T. (2022). Epigenetics of type 2 diabetes mellitus and weight change - a tool for precision medicine?Nat. Rev. Endocrinol.18 (7), 433–448. 10.1038/s41574-022-00671-w
107
Ling K. M. Stick S. M. Kicic A. (2023). Pulmonary bacteriophage and cystic fibrosis airway mucus: friends or foes?Front. Med.10, 1088494. 10.3389/fmed.2023.1088494
108
Liu G. Summer R. (2019). Cellular metabolism in lung health and disease. Annu. Rev. physiology81, 403–428. 10.1146/annurev-physiol-020518-114640
109
Liu S. Li L. Lou P. Zhao M. Wang Y. Tang M. et al (2021a). Elevated branched-chain α-keto acids exacerbate macrophage oxidative stress and chronic inflammatory damage in type 2 diabetes mellitus. Free Radic. Biol. and Med.175, 141–154. 10.1016/j.freeradbiomed.2021.08.240
110
Liu Q. Tian X. Maruyama D. Arjomandi M. Prakash A. (2021b). Lung immune tone via gut-lung axis: gut-derived LPS and short-chain fatty acids' immunometabolic regulation of lung IL-1β, FFAR2, and FFAR3 expression. Am. J. physiology Lung Cell. Mol. physiology321 (1), L65–l78. 10.1152/ajplung.00421.2020
111
Liu N. Bai L. Lu Z. Gu R. Zhao D. Yan F. et al (2022). TRPV4 contributes to ER stress and inflammation: implications for Parkinson's disease. J. neuroinflammation19 (1), 26. 10.1186/s12974-022-02382-5
112
Lo J. Clare-Salzler M. J. (2006). Dendritic cell subsets and type I diabetes: focus upon DC-based therapy. Autoimmun. Rev.5 (6), 419–423. 10.1016/j.autrev.2005.12.001
113
Lynch C. J. Adams S. H. (2014). Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol.10 (12), 723–736. 10.1038/nrendo.2014.171
114
Ma R. Ji T. Zhang H. Dong W. Chen X. Xu P. et al (2018). A Pck1-directed glycogen metabolic program regulates formation and maintenance of memory CD8(+) T cells. Nat. Cell. Biol.20 (1), 21–27. 10.1038/s41556-017-0002-2
115
Ma J. Wei K. Liu J. Tang K. Zhang H. Zhu L. et al (2020). Glycogen metabolism regulates macrophage-mediated acute inflammatory responses. Nat. Commun.11 (1), 1769. 10.1038/s41467-020-15636-8
116
Ma X. Mei S. Wuyun Q. Zhou L. Cai Z. Ding H. et al (2024). Super-enhancer-driven LncRNA PPARα-seRNA exacerbates glucolipid metabolism and diabetic cardiomyopathy via recruiting KDM4B. Mol. Metab.86, 101978. 10.1016/j.molmet.2024.101978
117
Ma S. Wang M. Wu Y. Meng D. Zhang B. Zhu H. et al (2025). Discovery of a series of novel 3-sulfonamido benzoic acid derivatives as promising P2Y(14)R antagonists for acute lung injury. Eur. J. Med. Chem.290, 117588. 10.1016/j.ejmech.2025.117588
118
Magnotti L. J. Upperman J. S. Xu D. Z. Lu Q. Deitch E. A. (1998). Gut-derived mesenteric lymph but not portal blood increases endothelial cell permeability and promotes lung injury after hemorrhagic shock. Ann. Surg.228 (4), 518–527. 10.1097/00000658-199810000-00008
119
Majer M. Mott D. M. Mochizuki H. Rowles J. C. Pedersen O. Knowler W. C. et al (1996). Association of the glycogen synthase locus on 19q13 with NIDDM in Pima Indians. Diabetologia39 (3), 314–321. 10.1007/BF00418347
120
Mao L. Wang L. Lyu Y. Zhuang Q. Li Z. Zhang J. et al (2024). Branch chain amino acid metabolism promotes brain metastasis of NSCLC through EMT occurrence by regulating ALKBH5 activity. Int. J. Biol. Sci.20 (9), 3285–3301. 10.7150/ijbs.85672
121
Martín-Vázquez E. Cobo-Vuilleumier N. López-Noriega L. Lorenzo P. I. Gauthier B. R. (2023). The PTGS2/COX2-PGE(2) signaling cascade in inflammation: pro or anti? A case study with type 1 diabetes mellitus. Int. J. Biol. Sci.19 (13), 4157–4165. 10.7150/ijbs.86492
122
Maschalidi S. Mehrotra P. Keçeli B. N. De Cleene H. K. L. Lecomte K. Van der Cruyssen R. et al (2022). Targeting SLC7A11 improves efferocytosis by dendritic cells and wound healing in diabetes. Nature606 (7915), 776–784. 10.1038/s41586-022-04754-6
123
Mayers J. R. Torrence M. E. Danai L. V. Papagiannakopoulos T. Davidson S. M. Bauer M. R. et al (2016). Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Sci. (New York, NY)353 (6304), 1161–1165. 10.1126/science.aaf5171
124
Mendez-Enriquez E. Alvarado-Vazquez P. A. Abma W. Simonson O. E. Rodin S. Feyerabend T. B. et al (2021). Mast cell-derived serotonin enhances methacholine-induced airway hyperresponsiveness in house dust mite-induced experimental asthma. Allergy76 (7), 2057–2069. 10.1111/all.14748
125
Mishra S. P. Wang B. Jain S. Ding J. Rejeski J. Furdui C. M. et al (2023). A mechanism by which gut microbiota elevates permeability and inflammation in obese/diabetic mice and human gut. Gut72 (10), 1848–1865. 10.1136/gutjnl-2022-327365
126
Molinaro A. Bel Lassen P. Henricsson M. Wu H. Adriouch S. Belda E. et al (2020). Imidazole propionate is increased in diabetes and associated with dietary patterns and altered microbial ecology. Nat. Commun.11 (1), 5881. 10.1038/s41467-020-19589-w
127
Montuschi P. Kharitonov S. A. Ciabattoni G. Barnes P. J. (2003). Exhaled leukotrienes and prostaglandins in COPD. Thorax58 (7), 585–588. 10.1136/thorax.58.7.585
128
Mousavinasab F. Karimi R. Taheri S. Ahmadvand F. Sanaaee S. Najafi S. et al (2023). Microbiome modulation in inflammatory diseases: progress to microbiome genetic engineering. Cancer Cell. Int.23 (1), 271. 10.1186/s12935-023-03095-2
129
Myerburg M. M. King J. D. Jr. Oyster N. M. Fitch A. C. Magill A. Baty C. J. et al (2010). AMPK agonists ameliorate sodium and fluid transport and inflammation in cystic fibrosis airway epithelial cells. Am. J. Respir. Cell. Mol. Biol.42 (6), 676–684. 10.1165/2009-0147OC
130
Nagata N. Takeuchi T. Masuoka H. Aoki R. Ishikane M. Iwamoto N. et al (2023). Human gut Microbiota and its metabolites impact immune responses in COVID-19 and its complications. Gastroenterology164 (2), 272–288. 10.1053/j.gastro.2022.09.024
131
Natalini J. G. Singh S. Segal L. N. (2023). The dynamic lung microbiome in health and disease. Nat. Rev. Microbiol.21 (4), 222–235. 10.1038/s41579-022-00821-x
132
Naveen Kumar R. Surekha M. V. Gowthami S. D. G. Aditi A. K. Satyavani M. Satyanarayana K. et al (2025). Toxicological evaluation of Salmonella phage NINP13076 in BALB/c mice: histopathological studies. Microb. Pathog.198, 107146. 10.1016/j.micpath.2024.107146
133
Newgard C. B. An J. Bain J. R. Muehlbauer M. J. Stevens R. D. Lien L. F. et al (2009). A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell. metab.9 (4), 311–326. 10.1016/j.cmet.2009.02.002
134
Ng S. C. Xu Z. Mak J. W. Y. Yang K. Liu Q. Zuo T. et al (2022). Microbiota engraftment after faecal microbiota transplantation in obese subjects with type 2 diabetes: a 24-week, double-blind, randomised controlled trial. Gut71 (4), 716–723. 10.1136/gutjnl-2020-323617
135
Nguyen S. Baker K. Padman B. S. Patwa R. Dunstan R. A. Weston T. A. et al (2017). Bacteriophage transcytosis provides a mechanism to cross epithelial cell layers. mBio8 (6), e01874-17. 10.1128/mBio.01874-17
136
Nishijima S. Nagata N. Kiguchi Y. Kojima Y. Miyoshi-Akiyama T. Kimura M. et al (2022). Extensive gut virome variation and its associations with host and environmental factors in a population-level cohort. Nat. Commun.13 (1), 5252. 10.1038/s41467-022-32832-w
137
Nobs S. P. Kopf M. (2018). PPAR-γ in innate and adaptive lung immunity. J. Leukoc. Biol.104 (4), 737–741. 10.1002/JLB.3MR0118-034R
138
Nobs S. P. Kolodziejczyk A. A. Adler L. Horesh N. Botscharnikow C. Herzog E. et al (2023). Lung dendritic-cell metabolism underlies susceptibility to viral infection in diabetes. Nature624 (7992), 645–652. 10.1038/s41586-023-06803-0
139
O'Keefe J. H. Weidling R. O'Keefe E. L. Franco W. G. (2023). SGLT inhibitors for improving Healthspan and lifespan. Prog. Cardiovasc. Dis.81, 2–9. 10.1016/j.pcad.2023.10.003
140
Okla M. Kim J. Koehler K. Chung S. (2017). Dietary factors promoting brown and beige fat development and thermogenesis. Adv. Nutr. (Bethesda, Md)8 (3), 473–483. 10.3945/an.116.014332
141
Otto M. Bucher C. Liu W. Müller M. Schmidt T. Kardell M. et al (2020). 12(S)-HETE mediates diabetes-induced endothelial dysfunction by activating intracellular endothelial cell TRPV1. J. Clin. investigation130 (9), 4999–5010. 10.1172/JCI136621
142
Özçam M. Lynch S. V. (2024). The gut-airway microbiome axis in health and respiratory diseases. Nat. Rev. Microbiol.22 (8), 492–506. 10.1038/s41579-024-01048-8
143
Packer M. (2020). Role of impaired nutrient and oxygen deprivation signaling and deficient autophagic flux in diabetic CKD development: implications for understanding the effects of sodium-glucose cotransporter 2-Inhibitors. J. Am. Soc. Nephrol. JASN31 (5), 907–919. 10.1681/ASN.2020010010
144
Patkee W. R. Carr G. Baker E. H. Baines D. L. Garnett J. P. (2016). Metformin prevents the effects of Pseudomonas aeruginosa on airway epithelial tight junctions and restricts hyperglycaemia-induced bacterial growth. J. Cell. Mol. Med.20 (4), 758–764. 10.1111/jcmm.12784
145
Pernet E. Sun S. Sarden N. Gona S. Nguyen A. Khan N. et al (2023). Neonatal imprinting of alveolar macrophages via neutrophil-derived 12-HETE. Nature614 (7948), 530–538. 10.1038/s41586-022-05660-7
146
Petrus P. Lecoutre S. Dollet L. Wiel C. Sulen A. Gao H. et al (2020). Glutamine links obesity to inflammation in Human white adipose tissue. Cell. metab.31 (2), 375–390. 10.1016/j.cmet.2019.11.019
147
Poltorak A. He X. Smirnova I. Liu M. Y. Van Huffel C. Du X. et al (1998). Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Sci. (New York, NY)282 (5396), 2085–2088. 10.1126/science.282.5396.2085
148
Popov D. Simionescu M. (1997). Alterations of lung structure in experimental diabetes, and diabetes associated with hyperlipidaemia in hamsters. Eur. Respir. J.10 (8), 1850–1858. 10.1183/09031936.97.10081850
149
Prattichizzo F. de Candia P. Ceriello A. (2021). Diabetes and kidney disease: emphasis on treatment with SGLT-2 inhibitors and GLP-1 receptor agonists. Metabolism Clin. Exp.120, 154799. 10.1016/j.metabol.2021.154799
150
Prieto I. Kavanagh M. Jimenez-Castilla L. Pardines M. Lazaro I. Herrero Del Real I. et al (2023). A mutual regulatory loop between miR-155 and SOCS1 influences renal inflammation and diabetic kidney disease. Mol. Ther. Nucleic acids34, 102041. 10.1016/j.omtn.2023.102041
151
Qian X. Zhang H. Y. Li Q. L. Ma G. J. Chen Z. Ji X. M. et al (2022). Integrated microbiome, metabolome, and proteome analysis identifies a novel interplay among commensal bacteria, metabolites and candidate targets in non-small cell lung cancer. Clin. Transl. Med.12 (6), e947. 10.1002/ctm2.947
152
Rajesh R. Atallah R. Bärnthaler T. (2023). Dysregulation of metabolic pathways in pulmonary fibrosis. Pharmacol. and Ther.246, 108436. 10.1016/j.pharmthera.2023.108436
153
Retamal J. S. Grace M. S. Dill L. K. Ramirez-Garcia P. Peng S. Gondin A. B. et al (2021). Serotonin-induced vascular permeability is mediated by transient receptor potential vanilloid 4 in the airways and upper gastrointestinal tract of mice. Laboratory investigation; a J. Tech. methods pathology101 (7), 851–864. 10.1038/s41374-021-00593-7
154
Roberts T. J. Burns A. T. MacIsaac R. J. MacIsaac A. I. Prior D. L. La Gerche A. (2018). Diagnosis and significance of pulmonary microvascular disease in diabetes. Diabetes care41 (4), 854–861. 10.2337/dc17-1904
155
Rogliani P. Calzetta L. Capuani B. Facciolo F. Cazzola M. Lauro D. et al (2016). Glucagon-Like peptide 1 receptor: a novel pharmacological target for treating human bronchial hyperresponsiveness. Am. J. Respir. Cell. Mol. Biol.55 (6), 804–814. 10.1165/rcmb.2015-0311OC
156
Rohm T. V. Keller L. Bosch A. J. T. AlAsfoor S. Baumann Z. Thomas A. et al (2022). Targeting colonic macrophages improves glycemic control in high-fat diet-induced obesity. Commun. Biol.5 (1), 370. 10.1038/s42003-022-03305-z
157
Romero F. Shah D. Duong M. Penn R. B. Fessler M. B. Madenspacher J. et al (2015). A pneumocyte-macrophage paracrine lipid axis drives the lung toward fibrosis. Am. J. Respir. Cell. Mol. Biol.53 (1), 74–86. 10.1165/rcmb.2014-0343OC
158
Ruderman N. B. Carling D. Prentki M. Cacicedo J. M. (2013). AMPK, insulin resistance, and the metabolic syndrome. J. Clin. investigation123 (7), 2764–2772. 10.1172/JCI67227
159
Saad M. J. Santos A. Prada P. O. (2016). Linking gut microbiota and inflammation to obesity and insulin resistance. Physiol. (Bethesda, Md)31 (4), 283–293. 10.1152/physiol.00041.2015
160
Sacks D. B. Arnold M. Bakris G. L. Bruns D. E. Horvath A. R. Lernmark Å. et al (2023). Guidelines and recommendations for laboratory analysis in the diagnosis and management of diabetes mellitus. Diabetes care46 (10), e151–e199. 10.2337/dci23-0036
161
Sas K. M. Kayampilly P. Byun J. Nair V. Hinder L. M. Hur J. et al (2016). Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI insight1 (15), e86976. 10.1172/jci.insight.86976
162
Schipke J. Jütte D. Brandenberger C. Autilio C. Perez-Gil J. Bernhard W. et al (2021). Dietary carbohydrates and fat induce distinct surfactant alterations in mice. Am. J. Respir. Cell. Mol. Biol.64 (3), 379–390. 10.1165/rcmb.2020-0335OC
163
Schoeler M. Caesar R. (2019). Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. and metabolic Disord.20 (4), 461–472. 10.1007/s11154-019-09512-0
164
Shahwan M. Alhumaydhi F. Ashraf G. M. Hasan P. M. Z. Shamsi A. (2022). Role of polyphenols in combating Type 2 Diabetes and insulin resistance. Int. J. Biol. Macromol.206, 567–579. 10.1016/j.ijbiomac.2022.03.004
165
Shakour N. Karami S. Iranshahi M. Butler A. E. Sahebkar A. (2024). Antifibrotic effects of sodium-glucose cotransporter-2 inhibitors: a comprehensive review. Diabetes and metabolic syndrome18 (1), 102934. 10.1016/j.dsx.2023.102934
166
Shi H. Yuan X. Yang X. Huang R. Fan W. Liu G. (2024). A novel diabetic foot ulcer diagnostic model: identification and analysis of genes related to glutamine metabolism and immune infiltration. BMC genomics25 (1), 125. 10.1186/s12864-024-10038-2
167
Shieh S. H. Probst J. C. Sung F. C. Tsai W. C. Li Y. S. Chen C. Y. (2012). Decreased survival among lung cancer patients with co-morbid tuberculosis and diabetes. BMC cancer12, 174. 10.1186/1471-2407-12-174
168
Shkoporov A. N. Stockdale S. R. Lavelle A. Kondova I. Heuston C. Upadrasta A. et al (2022). Viral biogeography of the mammalian gut and parenchymal organs. Nat. Microbiol.7 (8), 1301–1311. 10.1038/s41564-022-01178-w
169
Singh A. K. Gillies C. L. Singh R. Singh A. Chudasama Y. Coles B. et al (2020). Prevalence of co-morbidities and their association with mortality in patients with COVID-19: a systematic review and meta-analysis. Diabetes, Obes. and metabolism22 (10), 1915–1924. 10.1111/dom.14124
170
Slatore C. G. Bryson C. L. Au D. H. (2009). The association of inhaled corticosteroid use with serum glucose concentration in a large cohort. Am. J. Med.122 (5), 472–478. 10.1016/j.amjmed.2008.09.048
171
Smith P. M. Howitt M. R. Panikov N. Michaud M. Gallini C. A. Bohlooly Y. M. et al (2013). The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Sci. (New York, NY)341 (6145), 569–573. 10.1126/science.1241165
172
Sodhi C. P. Jia H. Yamaguchi Y. Lu P. Good M. Egan C. et al (2015). Intestinal epithelial TLR-4 activation is required for the development of Acute lung injury after Trauma/Hemorrhagic shock via the release of HMGB1 from the gut. J. Immunol. Baltim. Md 1950194 (10), 4931–4939. 10.4049/jimmunol.1402490
173
Stapleton D. Mitchelhill K. I. Gao G. Widmer J. Michell B. J. Teh T. et al (1996). Mammalian AMP-activated protein kinase subfamily. J. Biol. Chem.271 (2), 611–614. 10.1074/jbc.271.2.611
174
Stevens J. Steinmeyer S. Bonfield M. Peterson L. Wang T. Gray J. et al (2022). The balance between protective and pathogenic immune responses to pneumonia in the neonatal lung is enforced by gut microbiota. Sci. Transl. Med.14 (649), eabl3981. 10.1126/scitranslmed.abl3981
175
Tacke F. Weiskirchen R. (2018). An update on the recent advances in antifibrotic therapy. Expert Rev. gastroenterology and hepatology12 (11), 1143–1152. 10.1080/17474124.2018.1530110
176
Tanaka T. Narazaki M. Kishimoto T. (2018). Interleukin (IL-6) immunotherapy. Cold Spring Harb. Perspect. Biol.10 (8), a028456. 10.1101/cshperspect.a028456
177
Tanase D. M. Gosav E. M. Neculae E. Costea C. F. Ciocoiu M. Hurjui L. L. et al (2020). Role of Gut Microbiota on onset and progression of microvascular complications of type 2 diabetes (T2DM). Nutrients12 (12), 3719. 10.3390/nu12123719
178
Tangedal S. Nielsen R. Aanerud M. Drengenes C. Husebø G. R. Lehmann S. et al (2024). Lower airway microbiota in COPD and healthy controls. Thorax79, 219–226. 10.1136/thorax-2023-220455
179
Thannickal V. J. Fanburg B. L. (2000). Reactive oxygen species in cell signaling. Am. J. physiology Lung Cell. Mol. physiology279 (6), L1005–L1028. 10.1152/ajplung.2000.279.6.L1005
180
Thomas T. Stefanoni D. Reisz J. A. Nemkov T. Bertolone L. Francis R. O. et al (2020). COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI insight5 (14), e140327. 10.1172/jci.insight.140327
181
Tiengo A. Fadini G. P. Avogaro A. (2008). The metabolic syndrome, diabetes and lung dysfunction. Diabetes and metabolism34 (5), 447–454. 10.1016/j.diabet.2008.08.001
182
Toki S. Newcomb D. C. Printz R. L. Cahill K. N. Boyd K. L. Niswender K. D. et al (2021). Glucagon-like peptide-1 receptor agonist inhibits aeroallergen-induced activation of ILC2 and neutrophilic airway inflammation in obese mice. Allergy76 (11), 3433–3445. 10.1111/all.14879
183
Uno K. Yamada T. Ishigaki Y. Imai J. Hasegawa Y. Sawada S. et al (2015). A hepatic amino acid/mTOR/S6K-dependent signalling pathway modulates systemic lipid metabolism via neuronal signals. Nat. Commun.6, 7940. 10.1038/ncomms8940
184
van Hoek M. J. Merks R. M. (2012). Redox balance is key to explaining full vs. partial switching to low-yield metabolism. BMC Syst. Biol.6, 22. 10.1186/1752-0509-6-22
185
Velmurugan G. Ramprasath T. Gilles M. Swaminathan K. Ramasamy S. (2017). Gut Microbiota, endocrine-disrupting chemicals, and the diabetes epidemic. Trends Endocrinol. metabolism TEM28 (8), 612–625. 10.1016/j.tem.2017.05.001
186
Verkerke A. R. P. Wang D. Yoshida N. Taxin Z. H. Shi X. Zheng S. et al (2024). BCAA-nitrogen flux in brown fat controls metabolic health independent of thermogenesis. Cell.187 (10), 2359–2374.e18. 10.1016/j.cell.2024.03.030
187
Wang T. J. Larson M. G. Vasan R. S. Cheng S. Rhee E. P. McCabe E. et al (2011). Metabolite profiles and the risk of developing diabetes. Nat. Med.17 (4), 448–453. 10.1038/nm.2307
188
Wang Y. Xiao J. Jiang W. Zuo D. Wang X. Jin Y. et al (2021a). BCKDK alters the metabolism of non-small cell lung cancer. Transl. lung cancer Res.10 (12), 4459–4476. 10.21037/tlcr-21-885
189
Wang C. Xie K. Li K. Lin S. Xu F. (2021b). Potential therapeutic effects of interleukin-35 on the differentiation of naïve T cells into Helios+Foxp3+ Tregs in clinical and experimental acute respiratory distress syndrome. Mol. Immunol.132, 236–249. 10.1016/j.molimm.2021.01.009
190
Wang L. Wang J. Ren G. Sun S. Nishikawa K. Yu J. et al (2023a). Ameliorative effects of the Coptis inflorescence extract against lung injury in diabetic mice by regulating AMPK/NEU1 signaling. Phytomedicine Int. J. phytotherapy Phytopharm.118, 154963. 10.1016/j.phymed.2023.154963
191
Wang Y. Deng F. Zhong X. Du Y. Fan X. Su H. et al (2023b). Dulaglutide provides protection against sepsis-induced lung injury in mice by inhibiting inflammation and apoptosis. Eur. J. Pharmacol.949, 175730. 10.1016/j.ejphar.2023.175730
192
Webber T. Ronacher K. Conradie-Smit M. Kleynhans L. (2022). Interplay between the immune and endocrine systems in the lung: implications for TB susceptibility. Front. Immunol.13, 829355. 10.3389/fimmu.2022.829355
193
Willart M. A. van Nimwegen M. Grefhorst A. Hammad H. Moons L. Hoogsteden H. C. et al (2012). Ursodeoxycholic acid suppresses eosinophilic airway inflammation by inhibiting the function of dendritic cells through the nuclear farnesoid X receptor. Allergy67 (12), 1501–1510. 10.1111/all.12019
194
Wolfe A. L. Zhou Q. Toska E. Galeas J. Ku A. A. Koche R. P. et al (2021). UDP-glucose pyrophosphorylase 2, a regulator of glycogen synthesis and glycosylation, is critical for pancreatic cancer growth. Proc. Natl. Acad. Sci. U. S. A.118 (31), e2103592118. 10.1073/pnas.2103592118
195
Wong C. K. McLean B. A. Baggio L. L. Koehler J. A. Hammoud R. Rittig N. et al (2024). Central glucagon-like peptide 1 receptor activation inhibits toll-like receptor agonist-induced inflammation. Cell. metab.36 (1), 130–143.e5. 10.1016/j.cmet.2023.11.009
196
Wu Z. Zhang B. Chen F. Xia R. Zhu D. Chen B. et al (2022). Fecal microbiota transplantation reverses insulin resistance in type 2 diabetes: a randomized, controlled, prospective study. Front. Cell. Infect. Microbiol.12, 1089991. 10.3389/fcimb.2022.1089991
197
Wu J. Gong L. Li Y. Liu T. Sun R. Jia K. et al (2024). SGK1 aggravates idiopathic pulmonary fibrosis by triggering H3k27ac-mediated macrophage reprogramming and disturbing immune homeostasis. Int. J. Biol. Sci.20 (3), 968–986. 10.7150/ijbs.90808
198
Xu W. Janocha A. J. Erzurum S. C. (2021). Metabolism in pulmonary hypertension. Annu. Rev. physiology83, 551–576. 10.1146/annurev-physiol-031620-123956
199
Xu R. Wang F. Zhang Z. Zhang Y. Tang Y. Bi J. et al (2023a). Diabetes-Induced autophagy dysregulation engenders testicular impairment via oxidative stress. Oxidative Med. Cell. Longev.2023, 4365895. 10.1155/2023/4365895
200
Xu Y. Zhang C. Cai D. Zhu R. Cao Y. (2023b). Exosomal miR-155-5p drives widespread macrophage M1 polarization in hypervirulent Klebsiella pneumoniae-induced acute lung injury via the MSK1/p38-MAPK axis. Cell. and Mol. Biol. Lett.28 (1), 92. 10.1186/s11658-023-00505-1
201
Xue M. Xiao J. Jiang W. Wang Y. Zuo D. An H. et al (2023). Loss of BCAA catabolism enhances Rab1A-mTORC1 signaling activity and promotes tumor proliferation in NSCLC. Transl. Oncol.34, 101696. 10.1016/j.tranon.2023.101696
202
Yang G. Wei J. Liu P. Zhang Q. Tian Y. Hou G. et al (2021). Role of the gut microbiota in type 2 diabetes and related diseases. Metabolism Clin. Exp.117, 154712. 10.1016/j.metabol.2021.154712
203
Yang F. Luo X. Li J. Lei Y. Zeng F. Huang X. et al (2022). Application of glucagon-like peptide-1 receptor antagonists in fibrotic diseases. Biomed. and Pharmacother. = Biomedecine and Pharmacother.152, 113236. 10.1016/j.biopha.2022.113236
204
Yang M. Cao Z. Li W. Zhou J. Liu J. Zhong Y. et al (2024). Maternal glycemia during pregnancy and child lung function: a prospective cohort Study. Diabetes care47 (11), 1941–1948. 10.2337/dc24-0865
205
Yao C. C. Sun R. M. Yang Y. Zhou H. Y. Meng Z. W. Chi R. et al (2023). Accumulation of branched-chain amino acids reprograms glucose metabolism in CD8(+) T cells with enhanced effector function and anti-tumor response. Cell. Rep.42 (3), 112186. 10.1016/j.celrep.2023.112186
206
Ying W. Riopel M. Bandyopadhyay G. Dong Y. Birmingham A. Seo J. B. et al (2017). Adipose tissue macrophage-derived exosomal miRNAs can modulate in vivo and in vitro insulin sensitivity. Cell.171 (2), 372–384. 10.1016/j.cell.2017.08.035
207
Yoneda T. Yoshikawa M. Tsukaguchi K. Tokuyama T. Fu A. Tomoda K. et al (1992). Relation of airway obstruction and respiratory muscle weakness to energy metabolism in pulmonary emphysema. Nihon Kyobu Shikkan Gakkai zasshi30 (9), 1667–1672.
208
Yoo Y. M. Jung E. M. Jeon B. H. Tran D. N. Jeung E. B. (2020). Potassium-dependent sodium/calcium exchanger 3 (Nckx3) depletion leads to abnormal motor function and social behavior in mice. J. physiology Pharmacol. official J. Pol. Physiological Soc.71 (5). 10.26402/jpp.2020.4.08
209
Yoon H. S. Cho C. H. Yun M. S. Jang S. J. You H. J. Kim J. H. et al (2021). Akkermansia muciniphila secretes a glucagon-like peptide-1-inducing protein that improves glucose homeostasis and ameliorates metabolic disease in mice. Nat. Microbiol.6 (5), 563–573. 10.1038/s41564-021-00880-5
210
Yu Z. Ohba M. Nakamura M. Sasano T. Ono M. Sugawara S. et al (2009). Dynamics of platelet mobilisation into lungs in response to 5-hydroxytryptamine (serotonin) in mice. Thrombosis haemostasis102 (6), 1251–1258. 10.1160/TH08-06-0406
211
Zangara M. T. Johnston I. Johnson E. E. McDonald C. (2021). Mediators of metabolism: an unconventional role for NOD1 and NOD2. Int. J. Mol. Sci.22 (3), 1156. 10.3390/ijms22031156
212
Zhang C. Ma C. Yao H. Zhang L. Yu X. Liu Y. et al (2018). 12-Lipoxygenase and 12-hydroxyeicosatetraenoic acid regulate hypoxic angiogenesis and survival of pulmonary artery endothelial cells via PI3K/Akt pathway. Am. J. physiology Lung Cell. Mol. physiology314 (4), L606-L616–l616. 10.1152/ajplung.00049.2017
213
Zhang Y. Zhang H. Li S. Huang K. Jiang L. Wang Y. (2022a). Metformin alleviates LPS-Induced acute lung injury by regulating the SIRT1/NF-κB/NLRP3 pathway and inhibiting endothelial cell pyroptosis. Front. Pharmacol.13, 801337. 10.3389/fphar.2022.801337
214
Zhang J. Wang H. Chen H. Li H. Xu P. Liu B. et al (2022b). ATF3 -activated accelerating effect of LINC00941/lncIAPF on fibroblast-to-myofibroblast differentiation by blocking autophagy depending on ELAVL1/HuR in pulmonary fibrosis. Autophagy18 (11), 2636–2655. 10.1080/15548627.2022.2046448
215
Zhang Z. Li X. Guo J. He B. Wu L. Yang R. et al (2023). β-aminoisobutyrics acid, a metabolite of BCAA, activates the AMPK/Nrf-2 pathway to prevent ferroptosis and ameliorates lung ischemia-reperfusion injury. Mol. Med. Camb. Mass29 (1), 164. 10.1186/s10020-023-00729-z
216
Zhao W. Zhao B. Meng X. Li B. Wang Y. Yu F. et al (2024). The regulation of MFG-E8 on the mitophagy in diabetic sarcopenia via the HSPA1L-Parkin pathway and the effect of D-pinitol. J. cachexia, sarcopenia muscle15 (3), 934–948. 10.1002/jcsm.13459
217
Zhenyukh O. Civantos E. Ruiz-Ortega M. Sánchez M. S. Vázquez C. Peiró C. et al (2017). High concentration of branched-chain amino acids promotes oxidative stress, inflammation and migration of human peripheral blood mononuclear cells via mTORC1 activation. Free Radic. Biol. and Med.104, 165–177. 10.1016/j.freeradbiomed.2017.01.009
218
Zhu X. Li K. Liu G. Wu R. Zhang Y. Wang S. et al (2023). Microbial metabolite butyrate promotes anti-PD-1 antitumor efficacy by modulating T cell receptor signaling of cytotoxic CD8 T cell. Gut microbes15 (2), 2249143. 10.1080/19490976.2023.2249143 author reply 236.
Summary
Keywords
diabetes mellitus, pulmonary injury, metabolic dysregulation, gut-lung axis, intestinal dysbiosis
Citation
Sun J, Chen J, Shen Y, Yao X, Sun H, Chen B and Feng J (2025) Diabetes mellitus-driven pulmonary injury: multidimensional mechanisms linking metabolic dysregulation to gut-lung axis and promising therapies. Front. Pharmacol. 16:1689522. doi: 10.3389/fphar.2025.1689522
Received
28 August 2025
Accepted
08 October 2025
Published
23 October 2025
Volume
16 - 2025
Edited by
Venkata Ramireddy Narala, Yogi Vemana University, India
Reviewed by
Yssel Mendoza-Mari, Western University of Health Sciences, United States
Vira Zlatkina, V. N. Karazin Kharkiv National University, Ukraine
Updates
Copyright
© 2025 Sun, Chen, Shen, Yao, Sun, Chen and Feng.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Feng, jfeng68@126.com; Bingqian Chen, cbq0433@suda.edu.cn
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.