 Wanjun Bai1†
Wanjun Bai1† Shuo Shi1,2†
Shuo Shi1,2† Deliang Yin3†Caihui Guo1Haojing Song1Yiting Hu1Jin Gao1
Deliang Yin3†Caihui Guo1Haojing Song1Yiting Hu1Jin Gao1 Bo Qiu1Xueyuan Zhang4Lusha Bi5
Bo Qiu1Xueyuan Zhang4Lusha Bi5 Huizhen Wu1*
Huizhen Wu1* Zhanjun Dong1*
Zhanjun Dong1*- 1Department of Pharmacy, Hebei General Hospital, Hebei Key Laboratory of Clinical Pharmacy, Shijiazhuang, Hebei, China
- 2College of Pharmacy, Hebei Medical University, Shijiazhuang, China
- 3Beijing Kangchuanglian Biopharmaceutical Technology Research Co., Ltd., Beijing, China
- 4Shanghai Innovstone Therapeutics Limited, Shanghai, China
- 5CSPC Ouyi Pharmaceutical Co., Ltd., Shijiazhuang, China
Objectives: This study assessed the pharmacokinetics, safety, and bioequivalence of generic and original palbociclib tablets in healthy Chinese subjects under fasting conditions with rabeprazole pre-treatment.
Methods: This was a single-dose, randomized, open-label, two-period crossover bioequivalence study conducted under fasting conditions with rabeprazole pre-treatment. In each trial, healthy Chinese subjects received 40 mg oral rabeprazole enteric-coated tablets once daily before breakfast for 6 days. Following an overnight fast of at least 10 h, they took the seventh dose of rabeprazole and maintained fasting. They then received a single 125 mg oral dose of either the test or reference palbociclib tablet, followed by a 14-day washout interval between periods. Blood samples were collected from 0 to 96 h post-dose in each period, and palbociclib plasma concentrations were determined using a validated method. The primary pharmacokinetic parameters were calculated using the non-compartmental method. The geometric mean ratios of the two formulations and the corresponding 90% confidence intervals were acquired for bioequivalence analysis. The safety of both formulations was also evaluated.
Results: The 90% confidence intervals for the primary pharmacokinetic parameters of Cmax (84.53%–91.72%), AUC0-t (87.81%–92.49%), and
Conclusion: The trial confirmed that the pharmacokinetic parameters of the generic and original palbociclib tablets were bioequivalent in healthy Chinese subjects under fasting conditions with rabeprazole pre-treatment. Both formulations were safe and well tolerated.
Clinical Trial Registration: http://www.chinadrugtrials.org.cn, identifier CTR20232617; https://www.chictr.org.cn, identifier ChiCTR2400084355.
1 Introduction
Breast cancer is the most common malignant tumor in women. According to GLOBOCAN data, there were approximately 2.3 million new breast cancer cases worldwide in 2022, ranking second in cancer incidence. In the same year, it caused about 666,000 deaths, making it the fourth leading cause of cancer-related death. The high incidence and mortality rates have made breast cancer a major global health challenge (Wilkinson and Gathani, 2022). From the perspective of molecular classification, the hormone receptor-positive/human epidermal growth factor receptor 2-negative (HR+/HER2-) subtype is the most common. Its growth relies on the estrogen signaling pathway, accounting for approximately 70% of all breast cancer cases. Traditional treatment for this subtype primarily involves monotherapy with endocrine drugs, including selective estrogen receptor modulators and degraders. However, the frequent occurrence of drug resistance in clinical practice has severely limited patients’ long-term survival benefits (Burstein et al., 2021; Xiong et al., 2025).
Palbociclib, as the world’s first approved cyclin-dependent kinase (CDK) 4/6 inhibitor, specifically binds to the CDK4/6-cyclin D complex, blocks the phosphorylation of retinoblastoma protein, inhibits the transition of the cell cycle from the G1 phase to the S phase, and thus suppresses DNA synthesis and proliferation of tumor cells (Dhillon, 2015). Multiple key clinical trials have confirmed that palbociclib combined with letrozole or fulvestrant can significantly prolong the survival of HR+/HER2-breast cancer patients with good tolerability, and has become a first-line treatment regime (Brufsky et al., 2021; Cristofanilli et al., 2022; Rugo et al., 2022).
In clinical practice, the use of acid-reducing agents is common among cancer patients, with prevalence proportions of 20% in one major US healthcare database and 33% in another (Smelick et al., 2013). Proton pump inhibitors (PPIs) are the first choice due to their potent acid-suppressing effects and cost-effectiveness (Raoul et al., 2021). However, as a weakly basic compound, the solubility of palbociclib is pH-dependent (Chang et al., 2023; Kuminek et al., 2023). When PPIs inhibit gastric acid secretion and raise the intragastric pH above 4.5, the solubility of palbociclib drops sharply to <0.5 mg/mL, thereby significantly affecting its bioavailability (Eser et al., 2022). Studies have shown that when the palbociclib capsule formulation is co-administered with rabeprazole, under postprandial conditions, palbociclib’s Cmax decreases by 41% and Area Under the Curve (AUC) decreases by 13%; under fasting conditions, its Cmax decreases by 80% and AUC decreases by 62%. When taken with food, PPIs have no clinically significant impact on palbociclib exposure (Sun et al., 2017). Therefore, the capsule formulation requires administration with food (FDA, 2017). However, this is impractical for many cancer patients with poor appetite, leading to poor medication compliance and highlighting the clinical limitations of this dosage form. To address the insufficient exposure of palbociclib capsules under fasting conditions, a palbociclib tablet formulation was developed, with one of the key excipients being succinic acid. The addition of succinic acid effectively improves the environmental adaptability of the tablets, weakens the impact of intragastric pH fluctuations on drug dissolution, and ensures stable drug dissolution and good bioavailability under different intragastric environments (Zhang et al., 2019). Research results show that the exposure of the tablet is unaffected by the intragastric environment, and the prescribing information for the tablet states it can be taken with or without food (FDA, 2019a; FDA,2019b). Due to the pronounced pH-dependent pharmacokinetic (PK) properties of palbociclib (Chang et al., 2023; Kuminek et al., 2023), conventional bioequivalence (BE) studies alone may be inadequate to fully assess the equivalence between generic and original palbociclib tablets across all conditions. This is particularly critical for patients on long-term acid-reducing therapy, as such variations could directly result in significant fluctuations in clinical efficacy. To address this, the FDA mandates—in addition to standard fasting and fed BE studies—an additional BE study under fasting conditions with acid-reducing agents pre-treatment (FDA,2022). This ensures that generic versions maintain therapeutic equivalence to the original drug even in specific clinical scenarios involving acid suppression. However, due to the more intricate study design and execution required, along with higher associated costs, available data on BE studies under fasting conditions with acid-reducing agents pre-treatment remain limited. This research gap may lead to suboptimal drug exposure in clinical practice, potentially compromising efficacy and increasing the risk of treatment failure.
Recently, a generic palbociclib tablet, produced by the CSPC Ouyi Pharmaceutical Co., Ltd. (Hebei, China), is the first generic drug of its type in China. According to the National Medical Products Administration (NMPA) guidelines, this study compared the PK parameters of the new test, palbociclib tablet, with those of the reference product (Ibrance®). To support the marketing approval of the newly developed generic formulation in China, a pharmacokinetics and bioequivalence study was performed in healthy Chinese subjects under fasting conditions with rabeprazole pre-treatment.
2 Materials and methods
2.1 Study drugs
The acid-reducing agent was a rabeprazole enteric-coated tablet (20 mg/tablet, batch number: 2,111,129; expiry date: October 2024), which was produced by Eisai (China) Pharmaceutical Co., Ltd. under the trade name Pariet® and provided by CSPC Ouyi Pharmaceutical Co., Ltd., Hebei, China. The test formulation was a palbociclib tablet, which was produced and provided by CSPC Ouyi Pharmaceutical Co., Ltd., Hebei, China (125 mg/tablet, batch number: R46220203; expiry date: 9 February 2025).The reference formulation (125 mg/tablet, batch number GT2029; expiry date: June 2025), which is marketed under the brand name Ibrance® and produced by Pfizer Manufacturing Deutschland GmbH, was also provided by CSPC Ouyi Pharmaceutical Co., Ltd.
2.2 Ethics approval and study population
The study was registered on the Drug Clinical Trial Registration and Information Disclosure Platform [http://www.chinadrugtrials.org.cn] (number: CTR20232617, date: 23 August 2023) and retrospectively registered on the Chinese Clinical Trial Registry [https://www.chictr.org.cn] (number: ChiCTR2400084355, date: 15 May 2024). The study protocol and informed consent form were reviewed and informed consent forms (ICFs) were reviewed and approved by the independent ethics committee of Hebei General Hospital [Ethics Number: (2023) (21-01)]. The implementation of this study adhered to the ethical principles of the Declaration of Helsinki and the Guidelines for Good Clinical Practice recommended by NMPA.
This study enrolled healthy adult participants aged 18 years or older with a BMI of 19.0–26.0 kg/m2 (male≥50 kg, female≥45 kg) through open recruitment. The health status of all participants was confirmed through medical history review, physical examination, vital signs monitoring, 12-lead electrocardiogram (ECG), chest X-ray, and laboratory tests including complete blood count, blood biochemistry, coagulation function, urinalysis, virology screening, alcohol breath test, and drug abuse screening. Participants were excluded if they had any clinically relevant acute or chronic diseases, a history of smoking or substance abuse, an allergic constitution (particularly hypersensitivity to any component of palbociclib tablets), had donated ≥400 mL of blood or received a blood transfusion within the past 3 months, had taken investigational drugs within 3 months prior to screening, were lactating or pregnant (positive pregnancy test), or had any other factors deemed by the investigator as potentially affecting study outcomes. All participants and their partners were required to use effective contraception during the study and for 6 months after its completion. Written informed consent was obtained from all participants following a detailed explanation of the study’s purpose, content, procedures, and potential risks.
All participants signed the written informed consent form after having sufficient time to review the document and discuss any questions with the study staff. They were informed that they were free to withdraw from the study at any time. The generation of the random allocation sequence was solely managed and kept strictly confidential by the Principal Investigator. Throughout the study period, internists were responsible for the continuous medical supervision of the participants, monitoring medication safety, and managing adverse events (AEs). All AEs were documented in detail and promptly addressed, with monitoring and follow-up continuing throughout the entire study period until resolution, return to baseline, or stabilization to a level deemed acceptable by the investigators. Furthermore, the study would be terminated prematurely in the event of any of the following circumstances, including but not limited to: serious adverse events (SAEs) related to the investigational drug, major protocol deviations that significantly affected the study outcomes, or a termination order issued by the NMPA or the ethics committee.
2.3 Study design
This was a single-center, randomized, open-label, single-dose, two-period crossover bioequivalence study conducted under fasting conditions with rabeprazole pre-treatment. It was carried out at the Phase I Clinical Research Center of Hebei General Hospital in Shijiazhuang, Hebei Province, China, from 12 September 2023, to 27 October 2023. Using SAS statistical software (version 9.4), subjects were randomly allocated in a 1:1 ratio to either the T-R or R-T sequence (where T represents the test formulation and R denotes the reference formulation), with a 14-day washout period justified by palbociclib’s half-life of 29 ± 5 h (FDA.,2019), and exceeding seven elimination half-lives between the two treatment periods. As shown in Figure 1, all subjects received 40 mg oral rabeprazole enteric-coated tablets (2 × 20 mg) with 240 mL water once daily approximately 30 min before breakfast from Day −6 to Day −1. Water intake was prohibited from 1 h before to 1 h after each dose (except for breakfast and water used for medication administration). On Day 1, under fasted conditions (at least 10 h), subjects received their seventh dose of rabeprazole and maintained fasting for an additional 4 h. They then took a single 125 mg dose (1 tablet) of either the test or reference formulation orally, according to the randomized sequence, with 240 mL of water. Water intake was prohibited from 1 h before to 1 h after administration. Standardized meals were provided at 4 and 10 h post-dose.
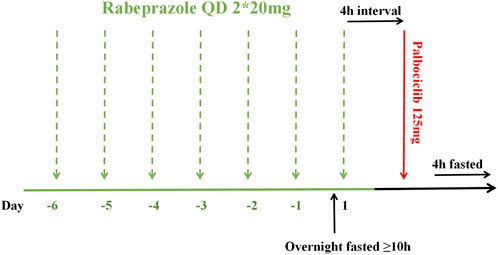
Figure 1. Dosing schedule timeline.
Previous studies have demonstrated that palbociclib exhibits an intra-individual coefficient of variation (Intra-CV) of approximately 26% (FDA.,2019). This study utilized AUC and Cmax as primary pharmacokinetic parameters. Assuming a one-sided test with α = 0.05, power of 0.8 (β = 0.2), and Intra-CV = 25%. The anticipated geometric mean ratio between test and reference formulations was 0.95–1.05, with bioequivalence acceptance criteria set at 80.00%–125.00%. Based on these parameters, the calculated sample size requirement was 28 evaluable subjects. To account for potential dropouts, the study planned to enroll 36 participants.
2.4 Blood sampling and bioanalytical assay
In this study, serial blood samples for palbociclib pharmacokinetic analysis were collected at baseline (1 h before dosing) and at 1.0, 2.0, 3.0, 4.0, 5.0, 6.0, 7.0, 8.0, 9.0, 10.0, 11.0, 12.0, 24.0, 48.0, 72.0, and 96.0 h after dosing under fasting conditions with rabeprazole pre-treatment. Blood samples were centrifuged at 1700 g for 10 min at 2 °C–8 °C within 60 min of collection to obtain plasma supernatant, which was then stored at −60 °C until analysis. Plasma concentrations of palbociclib were quantified using a validated high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) method at a specialized analytical laboratory (Nanjing Kelitai Pharmaceutical Technology Co., Ltd., Nanjing, China), in compliance with both China NMPA and FDA guidelines.
2.5 Pharmacokinetic analysis
The primary pharmacokinetic endpoints for palbociclib were Cmax, AUC0-t and
2.6 Bioequivalence and statistical analysis
After log-transforming Cmax, AUC0−t, and
2.7 Safety assessment
Safety assessments included AEs, SAEs, concomitant medications, laboratory tests, clinical symptoms, vital sign measurements, 12-lead ECG and physical examination results. Seated vital signs of subjects (axillary temperature, pulse and blood pressure) were measured within 1 h before and within 1 h after taking rabeprazole each time, as well as within 1 h before and at 2.0 ± 0.5, 6.0 ± 0.5, 24.0 ± 1.0, 48.0 ± 1.0, 72.0 ± 1.0and 96.0 ± 1.0 h after taking palbociclib. Laboratory tests, physical examinations, and 12-lead ECG were performed at screening and during the follow-up period. All AEs were monitored throughout the study by the research doctors and spontaneously reported by the subjects. AEs were coded using MedDRA (Medical Dictionary for Regulatory Activities: Version 26.1) and summarized according to System Organ Class (SOC) and Preferred Term (PT), including the number and percentage of subjects experiencing AEs and the frequency of AEs. The severity of AEs was graded according to the Common Terminology Criteria for Adverse Events (CTCAE, Version 5.0) issued by the U.S. National Cancer Institute. Missing safety data were labeled as “Missing” in the datasets and were not imputed in the subsequent analyses.
3 Results
3.1 Study population
The flow of participant screening, enrollment, and study completion is summarized in Figure 2. A total of 121 potential Chinese adults were screened. Of these, 33 healthy subjects met the eligibility criteria for the protocol and were enrolled and randomized, with 17 subjects assigned to the T-R group and 16 subjects to the R-T group. Table 1 summarizes the demographic characteristics of all subjects. The age, sex, weight, height, BMI, and race of the subjects were similar between the two parts of the study. In the T-R group, one subject (Y004) withdrew for personal reasons after the end of the first cycle, and the remaining 32 subjects completed both periods.
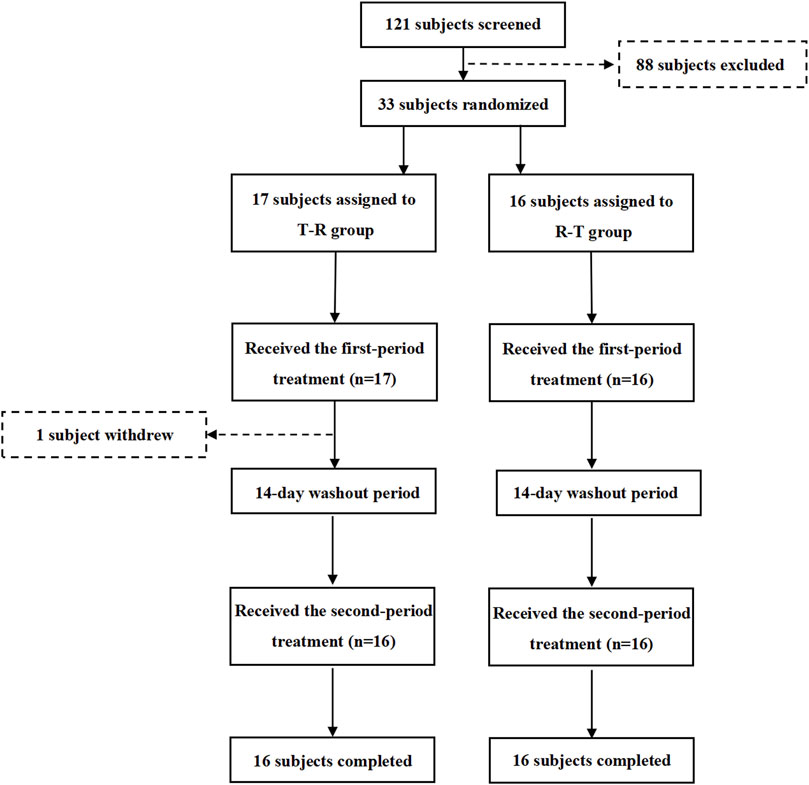
Figure 2. Study design and disposition of subjects.
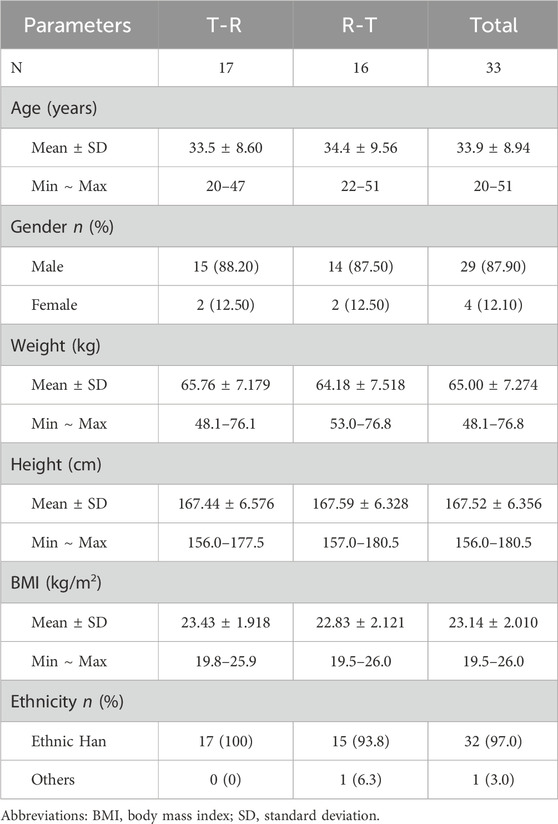
Table 1. Demographic characteristics of the healthy subjects.
3.2 Pharmacokinetics results
All PK parameters were analyzed based on the pharmacokinetics concentration set (PKCS) and pharmacokinetics parameter set (PKPS). In the T-R group, one subject withdrew during the second cycle, and only the data from the first cycle of this subject were included in PKCS and PKPS. Therefore, in the PK analysis, 33 datasets were included for the test formulation, and 32 datasets were included for the reference formulation. The major PK parameters of palbociclib were calculated using a non-compartmental model and are summarized in Table 2.The results showed that a Cmax value of 51.68 ± 12.96 ng/mL (CV% 25.07) of palbociclib was achieved within 5.0–7.0 h after oral administration of the test palbociclib tablet; the mean AUC0-t and
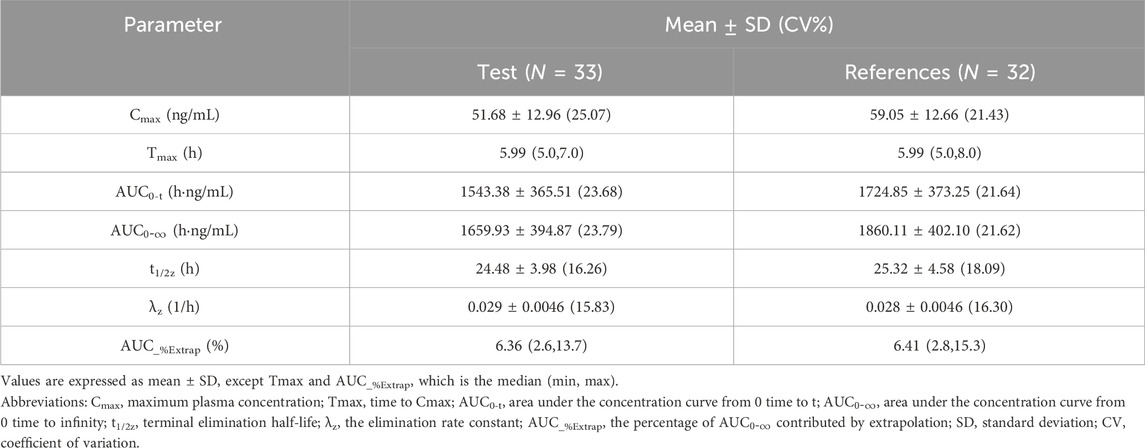
Table 2. Pharmacokinetic parameters.
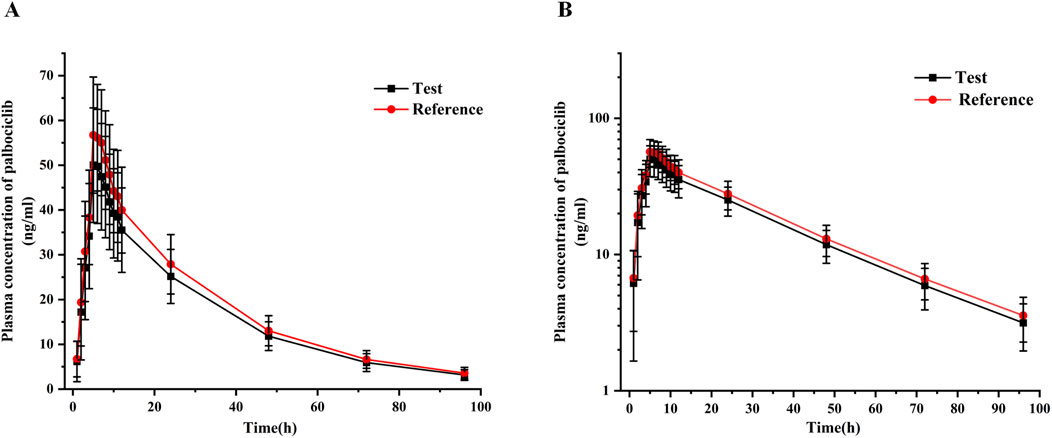
Figure 3. Mean plasma concentration-time curves of test and reference palbociclib (125 mg) under fasting conditions with rabeprazole pre-treatment. (A) Linear-scale profile; (B) Semi-logarithmic-scale profile. Data are presented as mean ± SD.
3.3 Bioequivalence results
For PK bioequivalence evaluation, the subjects were enrolled in the bioequivalence analysis set (BES) as with PKPS under fasting conditions with rabeprazole pre-treatment. As shown in Table 3, the coefficient of intra-subject variation (CV) of Cmax, AUC0-t and
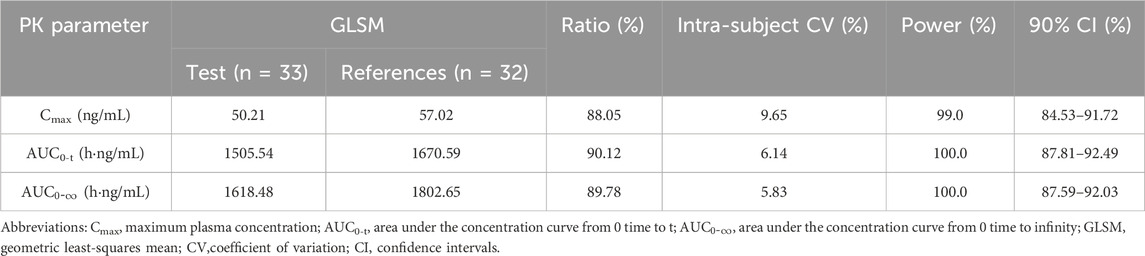
Table 3. Bioequivalence assessment of the primary pharmacokinetic parameters.
To assess whether period, sequence, and formulation factors had an impact on the current study, a multivariate analysis of variance was performed using a linear mixed-effects model for the natural logarithmic of Cmax, AUC0-t and
3.4 Safety assessment
The safety and tolerability of palbociclib were assessed based on the safety analysis set (SS).In this study, 33 subjects received at least one dose of the study drug and were included in SS. Twenty-eight AEs for 17 subjects were reported, and the incidence of AEs was 51.5% (17/33) (Table 4). Of these, 15 AEs were reported for 11 subjects in the test product; the incidence of AEs was 33.3% (11/33) (Table 4), whereas 13 AEs were reported for nine subjects and an incidence of 28.1% (9/32) in the reference product (Table 4). Among the AEs related to the test product, two subjects experienced serum thyroid-stimulating hormone (TSH) increased and nausea, respectively, which were defined as adverse drug reactions (ADRs). Two AEs for two subjects related to the reference product were defined as ADRs, including hemoglobin decreased and urinary tract infection, respectively. All AEs were mild and reported as grade 1. No SAEs occurred during the study. All AEs or ADRs were spontaneously resolved without any specific treatment. The results indicated that palbociclib had good safety and was well tolerated in healthy subjects under fasting conditions with rabeprazole pre-treatment.
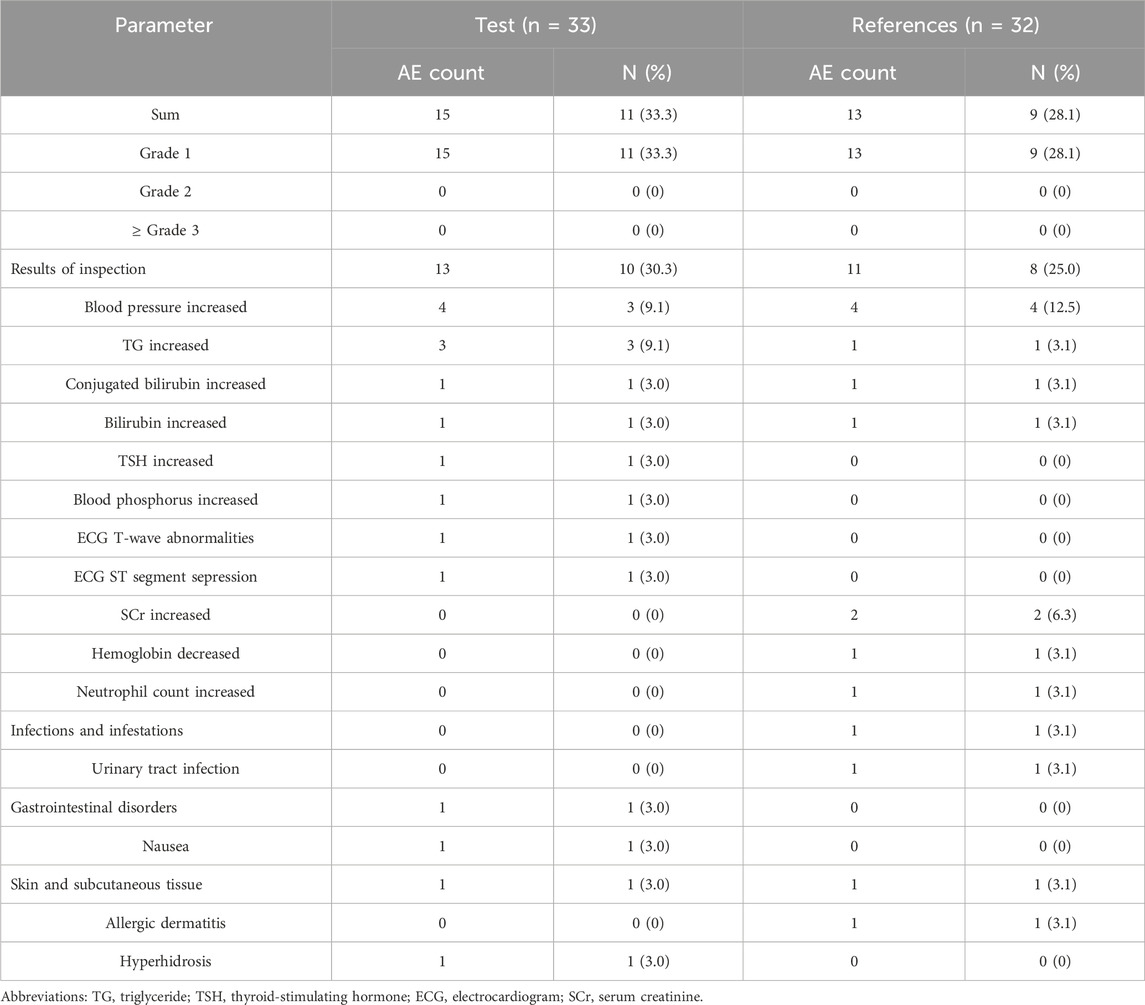
Table 4. Summary of treatment-emergent AEs.
4 Discussion
This study adheres to the guidelines issued by the NMPA, providing evidence of bioequivalence between generic and original palbociclib tablets in healthy Chinese subjects under fasting conditions with rabeprazole pre-treatment. Both formulations met pharmacokinetic bioequivalence criteria and were well-tolerated, offering substantial basis for understanding the drug’s pharmacokinetic characteristics in Chinese populations and supporting abbreviated new drug application approvals.
Palbociclib is categorized as a Biopharmaceutics Classification System (BCS) Class II drug, characterized by low solubility and high intestinal permeability. Drugs within this class often exhibit a greater likelihood of demonstrating food-dependent pharmacokinetic variations (Chu et al., 2022; Kuminek et al., 2023). The FDA mandates fasting, postprandial, and acid-reducing agent pre-treatment bioequivalence studies (FDA, 2022). This trial employed a single-dose, two-period crossover design under fasting conditions with rabeprazole pre-treatment, with a 14-day washout period to ensure drug elimination given palbociclib’s 29 ± 5-h half-life (FDA, 2019a). For pharmacokinetic research, a scientifically validated sampling design holds critical importance, as it necessitates a sample size capable of fully capturing the plasma concentration-time profile to ensure accurate exposure assessment. This benchmark is fulfilled when the AUC0-t constitutes over 80% of the
To evaluate the most extreme impact of PPIs on palbociclib PK, rabeprazole was chosen as the PPI due to its superior 24-h median intragastric pH, quicker onset of action, and prolonged acid suppression duration compared with other PPIs (EMA, 2010; Smelick et al., 2013). Additionally, rabeprazole is almost completely metabolized via non-enzymatic metabolic pathways, with metabolites excreted through the kidneys. Minor enzyme-mediated metabolism involves CYP3A4 and CYP2C1922 (Pace et al., 2007; Swan et al., 1999). Its metabolic pathways, primarily via CYP3A and SULT2A1 for palbociclib (FDA, 2019a), show no significant overlap, thus avoiding drug-drug interactions.
The pharmacokinetic characteristics as we showed in this study, were compared with capsules in healthy Chinese subjects under fed conditions (Chu et al., 2022). The results showed that the AUC0-t of the tablets was 1543.38 h·ng/mL, representing a 4% decrease compared with the capsule’s 1608.10 h·ng/mL. Meanwhile, the Cmax (51.68 ng/mL vs. 57.6 ng/mL) and
The safety analysis showed that a total of 17 subjects reported 28 adverse events, among which 4 were confirmed as adverse reactions to palbociclib. Neutropenia represents the most frequent adverse drug reaction associated with CDK4/6 inhibitor class agents (FDA, 2019a; Finn et al., 2015). The underlying mechanism involves blockade of the CDK4/6-Rb pathway, which induces cell cycle arrest (G1 phase) in early bone marrow progenitor cells, consequently impairing neutrophil differentiation and maturation (FDA, 2019a; Dhillon, 2015; Johnson et al., 2010). Although no typical neutropenic events were observed in this study, the reported hemoglobin reduction may be attributed to analogous inhibitory effects on erythroid progenitors, a finding consistent with palbociclib’s established myelosuppressive profile.The increase in serum thyroid-stimulating hormone (TSH) may be attributed to palbociclib interfering with the proliferation cycle of thyroid follicular cells, causing G1-S phase arrest, which indirectly affects the synthesis and feedback regulation of thyroid hormones, leading to TSH elevation (FDA, 2019b; Dhillon, 2015). Nausea is associated with direct stimulation of the gastric mucosa by drug dissolution after fasting administration and activation of intestinal 5-HT3 receptors. The mechanism of urinary tract infection has not been fully elucidated. Although succinic acid was added to optimize the tablet formulation used in this study, the types and incidence of drug adverse reactions observed were basically consistent with those of the capsule formulation (FDA, 2017), and all events were mild, indicating that the palbociclib tablet has good overall tolerance in healthy humans.
5 Conclusion
According to the results of the current study, the generic palbociclib tablets showed bioequivalence with the reference preparation (Ibrance®) in terms of the extent and rate of absorption under fasting conditions with rabeprazole pre-treatment, and the bioavailability of the test preparation was similar to that of the reference preparation. Both formulations are generally well-tolerated in healthy Chinese populations and can be used interchangeably in clinical practice.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by The Ethics Committee of Hebei General Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
WB: Conceptualization, Writing – review and editing, Methodology, Funding acquisition. SS: Writing – original draft, Visualization. DY: Conceptualization, Writing – review and editing, Investigation. CG: Investigation, Writing – review and editing, Methodology. HS: Methodology, Investigation, Writing – review and editing. YH: Writing – review and editing, Methodology, Investigation. JG: Methodology, Investigation, Writing – review and editing. BQ: Methodology, Writing – review and editing, Investigation. XZ: Writing – review and editing, Investigation, Resources. LB: Software, Investigation, Writing – review and editing. HW: Supervision, Writing – review and editing, Resources. ZD: Supervision, Resources, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was sponsored and funded by CSPC Ouyi Pharmaceutical Co., Ltd. (Hebei, China) and the Hebei Provincial Government-funded Clinical Medicine Outstanding Talents.
Acknowledgments
AcknowledgementsThe authors thank the subjects and staff who participated in the study. During the preparation of this work, the author used DeepSeek solely for the purpose of language translation and polishing of the manuscript content. After using this service, the author reviewed and edited the content as needed and takes full responsibility for the content of the published manuscript.
Conflict of interest
The authors declare that this study received funding from CSPC Ouyi Pharmaceutical Co., Ltd. The funder had the following involvement in the study: study design and data analysis.
Author DY was employed by Beijing Kangchuanglian Biopharmaceutical Technology Research Co., Ltd.
Author XZ was employed by Shanghai Innovstone Therapeutics Limited.
Author LB was employed by CSPC Ouyi Pharmaceutical Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. During the preparation of this work, the author used DeepSeek solely for the purpose of language translation and polishing of the manuscript content.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Brufsky, A., Liu, X., Li, B., McRoy, L., and Layman, R. M. (2021). Real-world tumor response of palbociclib plus letrozole versus letrozole for metastatic breast cancer in US clinical practice. Target Oncol. 16 (5), 601–611. doi:10.1007/s11523-021-00826-1
Burstein, H. J., Curigliano, G., Thürlimann, B., Weber, W. P., Poortmans, P., Regan, M. M., et al. (2021). Customizing local and systemic therapies for women with early breast cancer: the St. Gallen International consensus Guidelines for treatment of early breast cancer 2021. Ann. Oncol. 32 (10), 1216–1235. doi:10.1016/j.annonc.2021.06.023
Chang, Y. C., Song, J., Chang, Y., Huang, C. H., Sudan, A., Chen, P. C., et al. (2023). The Association between proton pump inhibitors and the effectiveness of CDK inhibitors in HR+/HER- advanced breast cancer patients: a systematic review and meta-analysis. Cancers (Basel) 15 (21), 5133. doi:10.3390/cancers15215133
Chu, N. N., Zhang, L., Wang, J., Gu, X., Ding, Y., Huang, K., et al. (2022). Bioequivalence Study of Palbociclib capsules in healthy Chinese subjects under fasting and Fed conditions. Clin. Drug Investig. 42 (1), 53–63. doi:10.1007/s40261-021-01103-9
Cristofanilli, M., Rugo, H. S., Im, S. A., Slamon, D. J., Harbeck, N., Bondarenko, I., et al. (2022). Overall survival with palbociclib and fulvestrant in women with HR+/HER2- ABC: updated exploratory analyses of PALOMA-3, a double-blind, phase III randomized Study. Clin. Cancer Res. 28 (16), 3433–3442. doi:10.1158/1078-0432.Ccr-22-0305
Dhillon, S. (2015). Palbociclib: first global approval. Drugs 75 (5), 543–551. doi:10.1007/s40265-015-0379-9
EMA (2010). Guideline on the investigation of bioequivalence. Available online at: https://www.gmp-compliance.org/files/guidemgr/2016_EMEA_Bioequivalence.pdf (Accessed May 16, 2025).
Eser, K., Önder, A. H., Sezer, E., Çil, T., İnal, A., Öztürk, B., et al. (2022). Proton pump inhibitors may reduce the efficacy of ribociclib and palbociclib in metastatic breast cancer patients based on an observational study. BMC Cancer 22 (1), 516. doi:10.1186/s12885-022-09624-y
FDA (2017). Highlights of Prescribing Information for IBRANCE® (palbociclib) capsules, for oral use. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/207103s004lbl.pdf (Accessed May 15.
FDA (2019a). Clinical review for application 212436Orig1s000. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212436Orig1s000ClinPharmR.pdf (Accessed May 15, 2025).
FDA (2019b). Highlights of Prescribing Information for IBRANCE® (palbociclib) tablets, for oral use. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212436lbl.pdf (Accessed May 15, 2025).
FDA (2022). Draft guidance on palbociclib. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/psg/PSG_212436.pdf (Accessed May 16, 2025).
Finn, R. S., Crown, J. P., Lang, I., Boer, K., Bondarenko, I. M., Kulyk, S. O., et al. (2015). The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 16 (1), 25–35. doi:10.1016/s1470-2045(14)71159-3
Johnson, S. M., Torrice, C. D., Bell, J. F., Monahan, K. B., Jiang, Q., Wang, Y., et al. (2010). Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J. Clin. Invest 120 (7), 2528–2536. doi:10.1172/jci41402
Kuminek, G., Salehi, N., Waltz, N. M., Sperry, D. C., Greenwood, D. E., Hate, S. S., et al. (2023). Use of gastrointestinal simulator, mass transport analysis, and absorption simulation to investigate the impact of pH modifiers in mitigating weakly basic drugs' performance issues related to gastric pH: palbociclib case Study. Mol. Pharm. 20 (1), 147–158. doi:10.1021/acs.molpharmaceut.2c00545
Pace, F., Pallotta, S., Casalini, S., and Porro, G. B. (2007). A review of rabeprazole in the treatment of acid-related diseases. Ther. Clin. Risk Manag. 3 (3), 363–379.
Raoul, J. L., Guérin-Charbonnel, C., Edeline, J., Simmet, V., Gilabert, M., and Frenel, J. S. (2021). Prevalence of proton pump inhibitor use among patients with cancer. JAMA Netw. Open 4 (6), e2113739. doi:10.1001/jamanetworkopen.2021.13739
Rugo, H. S., Brufsky, A., Liu, X., Li, B., McRoy, L., Chen, C., et al. (2022). Real-world study of overall survival with palbociclib plus aromatase inhibitor in HR+/HER2-metastatic breast cancer. NPJ Breast Cancer 8 (1), 114. doi:10.1038/s41523-022-00479-x
Smelick, G. S., Heffron, T. P., Chu, L., Dean, B., West, D. A., Duvall, S. L., et al. (2013). Prevalence of acid-reducing agents (ARA) in cancer populations and ARA drug-drug interaction potential for molecular targeted agents in clinical development. Mol. Pharm. 10 (11), 4055–4062. doi:10.1021/mp400403s
Sun, W., Klamerus, K. J., Yuhas, L. M., Pawlak, S., Plotka, A., O'Gorman, M., et al. (2017). Impact of acid-reducing agents on the pharmacokinetics of Palbociclib, a weak base with pH-Dependent solubility, with different food intake conditions. Clin. Pharmacol. Drug Dev. 6 (6), 614–626. doi:10.1002/cpdd.356
Swan, S. K., Hoyumpa, A. M., and Merritt, G. J. (1999). Review article: the pharmacokinetics of rabeprazole in health and disease. Aliment. Pharmacol. Ther. 13 (Suppl. 3), 11–17. doi:10.1046/j.1365-2036.1999.00020.x
Wilkinson, L., and Gathani, T. (2022). Understanding breast cancer as a global health concern. Br. J. Radiol. 95 (1130), 20211033. doi:10.1259/bjr.20211033
Xiong, X., Zheng, L. W., Ding, Y., Chen, Y. F., Cai, Y. W., Wang, L. P., et al. (2025). Breast cancer: pathogenesis and treatments. Signal Transduct. Target Ther. 10 (1), 49. doi:10.1038/s41392-024-02108-4
Keywords: palbociclib, acid-reducing agent, pharmacokinetics, bioequivalence, safety
Citation: Bai W, Shi S, Yin D, Guo C, Song H, Hu Y, Gao J, Qiu B, Zhang X, Bi L, Wu H and Dong Z (2025) Bioequivalence of a single dose of two palbociclib formulations in healthy Chinese subjects under fasting conditions: a two-period crossover study with rabeprazole pre-treatment. Front. Pharmacol. 16:1722151. doi: 10.3389/fphar.2025.1722151
Received: 10 October 2025; Accepted: 31 October 2025;
Published: 24 November 2025.
Edited by:
Fenglei Huang, Boehringer Ingelheim, GermanyReviewed by:
Muhammad Talha Saleem, University of Karachi, PakistanAmira Soliman, University of Florida, United States
Ahmet Inal, Erciyes Üniversitesi Tıp Fakültesi Hastaneleri, Türkiye
Ramanjaneyulu Seemaladinne, Cosette Pharmaceuticals, Inc, United States
Copyright © 2025 Bai, Shi, Yin, Guo, Song, Hu, Gao, Qiu, Zhang, Bi, Wu and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huizhen Wu, MTM1ODIwMDU5ODJAMTYzLmNvbQ==; Zhanjun Dong, ZHpqaGJnaEAxMjYuY29t
†These authors share first authorship