 Ouissam Al Jarroudi
Ouissam Al Jarroudi Khalid El Bairi
Khalid El Bairi Sami Aziz Brahmi1,2
Sami Aziz Brahmi1,2- 1Department of Medical Oncology, Mohammed VI University Hospital, Oujda, Morocco
- 2Faculty of Medicine and Pharmacy, Mohamed I University, Oujda, Morocco
- 3Faculty of Medical Sciences, UM6P Hospitals, Mohammed VI Polytechnic University, Benguerir, Morocco
- 4National Community Oncology Dispensing Association, Cazenovia, NY, United States
Non-Gastrointestinal Stromal Tumors (Non-GIST) Soft Tissue Sarcomas (STS) are highly aggressive and challenging diseases with poor prognosis and limited therapeutic options. Molecular profiling is urgently required to gain a deeper understanding of STS pathogenesis and to identify a comprehensive landscape of genomic alterations in order to develop effective targeted therapies. The mitogen-activated protein kinase (MAPK) signaling pathway is a key molecular mechanism involved in sarcoma development. This study aims to conduct a literature review on the involvement of the MAPK cascade in non-GIST STS, with a focus on the role of MAPK inhibitors in the current treatment paradigm for STS. Furthermore, recent data have provided promising preliminary findings regarding the use of new molecular agents targeting the MAPK pathway, either as single therapies or in combination with other drugs. Numerous clinical trials are currently ongoing, and their outcomes are eagerly awaited. Further research is required in both translational and clinical settings to molecularly characterize STS, identify novel causal alterations, accelerate target discovery, and identify potential biomarkers. Moreover, the development of novel nanomaterials provides a promising perspective that may lead to significant advancements in clinical practice.
Introduction
Non-Gastrointestinal Stromal Tumor Soft Tissue Sarcomas (non-GIST STS) constitute a broadly heterogeneous group of rare malignant mesenchymal tumors that originate from different tissues, including muscle, adipose, bone, and fibrous tissues (1). These aggressive neoplasms present a significant challenge owing to limited therapeutic options (2). There are approximately 100 distinct histological subtypes, each with unique biological behavior and treatment response (3, 4). Common subtypes include liposarcoma (LPS), leiomyosarcoma (LMS), and undifferentiated pleomorphic sarcoma (UPS). Less prevalent subtypes such as angiosarcoma (AS) and malignant peripheral nerve sheath tumor (MPNST) are also recognized (5). Treatment effectiveness varies among sarcoma subtypes owing to distinct oncogenesis mechanisms. Recent advances in molecular diagnostics have enhanced the understanding of sarcoma genetics, enabling the development of more tailored therapies. Currently, driver mutations have been identified in nearly one-third of sarcoma subtypes. For instance, well-differentiated liposarcoma (WDLPS) and dedifferentiated liposarcoma (DDLPS) are characterized by gene amplification of murine double minute-2 (MDM2) and cyclin dependent kinase 4 (CDK4) (6). Overexpression of hepatocyte growth factor receptor (MET) is observed in clear cell sarcoma (CCS). Anaplastic lymphoma kinase (ALK) gene rearrangement occurs in half of inflammatory myofibroblastic tumor (IMT) cases (7), while disruptions in the mammalian target of rapamycin (mTOR) signaling pathway due to tuberous sclerosis complex 1 and 2 (TSC1 and TSC2) gene mutations are common in perivascular epithelioid cell tumors (PEComas) (8). Notably, angiogenesis, which involves the activation of vascular endothelial growth factor receptors (VEGFR-1 to VEGFR-3), platelet-derived growth factor receptors (PDGFRA and PDGFRB), and other targets, is a common pathway for disease progression in certain sarcoma subtypes. Antiangiogenic drugs (such as pazopanib and regorafenib) have shown effectiveness in common subtypes, such as LMS and synovial sarcoma (SS), but not in lipomatous tumors. These drugs exhibit activity in several rare sarcoma subtypes that are resistant to chemotherapy, such as alveolar soft-part sarcoma (ASPS) (9) or solitary fibrous tumors (SFTs) (10).
A comprehensive, interdisciplinary management strategy is essential for addressing non-GIST STS, as conventional treatments such as surgery, radiation, and chemotherapy have not improved overall survival rates (11, 12). The past two decades have witnessed a significant transformation in GIST management. Extensive research has substantially advanced the understanding of GIST’s pathogenesis and biology. The discovery of the c-KIT mutation was a crucial development, enabling enhanced characterization and identification of GIST through molecular studies. The introduction of imatinib, which selectively inhibits KIT protein tyrosine kinase, has markedly impacted GIST treatment approaches (13). For advanced GIST cases, newly developed TKIs have considerably improved the PFS and OS rates. However, the treatment options for STS remain largely unchanged (14).
The poor prognosis and limited effective therapies for non-GIST STS, particularly in the advanced or metastatic stages, underscore the urgent need to identify targetable molecular alterations and develop novel therapies (15). The integration of advanced molecular techniques into clinical practice has significantly enhanced STS subtyping and treatment options (16). Molecular profiling aims to shift away from a one-treatment-fits-all approach towards more efficacious treatments specific to each non-GIST STS subgroup. Given the high heterogeneity of non-GIST STS, a comprehensive analysis of genetic and molecular profiles utilizing genome- and RNA-sequencing is essential for suggesting molecule-based personalized therapy and improving prognosis (17, 18). To date, the most promising treatment for non-GIST STS involves a molecularly targeted approach that requires elucidation of key molecular mechanisms associated with sarcomagenesis as potential therapeutic targets (19). Furthermore, patients with advanced STS should be encouraged to participate in clinical trials, when available.
The mitogen-activated protein kinase (MAPK) signaling pathway is crucial for the proliferation, migration, and metastasis of STS tumor cells (20). The MAPK cascades facilitate signal transduction through the sequential activation of three to five layers of protein kinases, designated as MAPK kinase kinase kinase (MAPKKKK), MAPK kinase kinase (MAPKKK), MAPK kinase (MAPKK), MAPK, and MAPK-activated protein kinases (MAPKAPK). The initial three central layers constitute a fundamental core unit essential for cell differentiation, proliferation, survival, and angiogenesis. Notably, this pathway is frequently overactive in several malignant tumors, including STS (21). Therefore, inhibition of the MAPK pathway is an important approach for managing non-GIST STS. In this narrative review, we conducted a comprehensive study on the involvement of the MAPK signaling pathway in non-GIST STS and outlined the current state of applying MAPK inhibitors as a potential therapeutic strategy in non-GIST STS.
MAPK signaling pathway: overview and regulatory mechanisms
The MAPK signaling pathway is a complex network of cellular signal transduction pathways that has been extensively studied in eukaryotic biology, particularly in budding yeast. Early research in this area revealed mechanisms that activate the MAPK pathway and regulate downstream processes (22). The MAPK signaling pathway regulates cellular processes including proliferation, immune responses, and apoptosis. MAPK activation occurs through phosphorylation of substrates in the cytosol and nucleus, modifying protein function and gene expression (23). Activation of the MAPK signaling pathway can be triggered by various factors, such as changes in Ca2+ levels, RAS activation, and PKC-mediated or G protein-coupled receptors, in a complex, multistep process involving protein kinase cascades (24) (Figure 1). Over a dozen MAPK enzymes regulate cell growth, survival, and differentiation (25). Researchers have identified two types of MAPK enzymes: conventional MAPKs and atypical MAPKs (26, 27). Conventional MAPKs include three families of sequentially activated kinases: classical MAPK or ERK (extracellular signal-related kinase), C-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK), and p38 MAPK (28). Each group of conventional MAPKs consists of three kinases (MAPK, MAPKK, and MAPKKK) acting sequentially. MAPKKK activation stimulates MAPKK phosphorylation and activation, ultimately triggering MAPK activation. Mammalian cells contain approximately 14 MAPKKKs, 7 MAPKKs, and 12 MAPKs, including ERK1, ERK2, p38α, p38β, p38γ, p38δ, JNK1, JNK2, JNK3, ERK3, ERK4, and ERK5 (24). Conversely, atypical MAPKs form a single group with glycine or glutamine acids, replacing the tyrosine residues in ERK3/ERK4 and NLR (29). The regulatory mechanisms and physiological functions of atypical MAPKs are poorly understood (30).
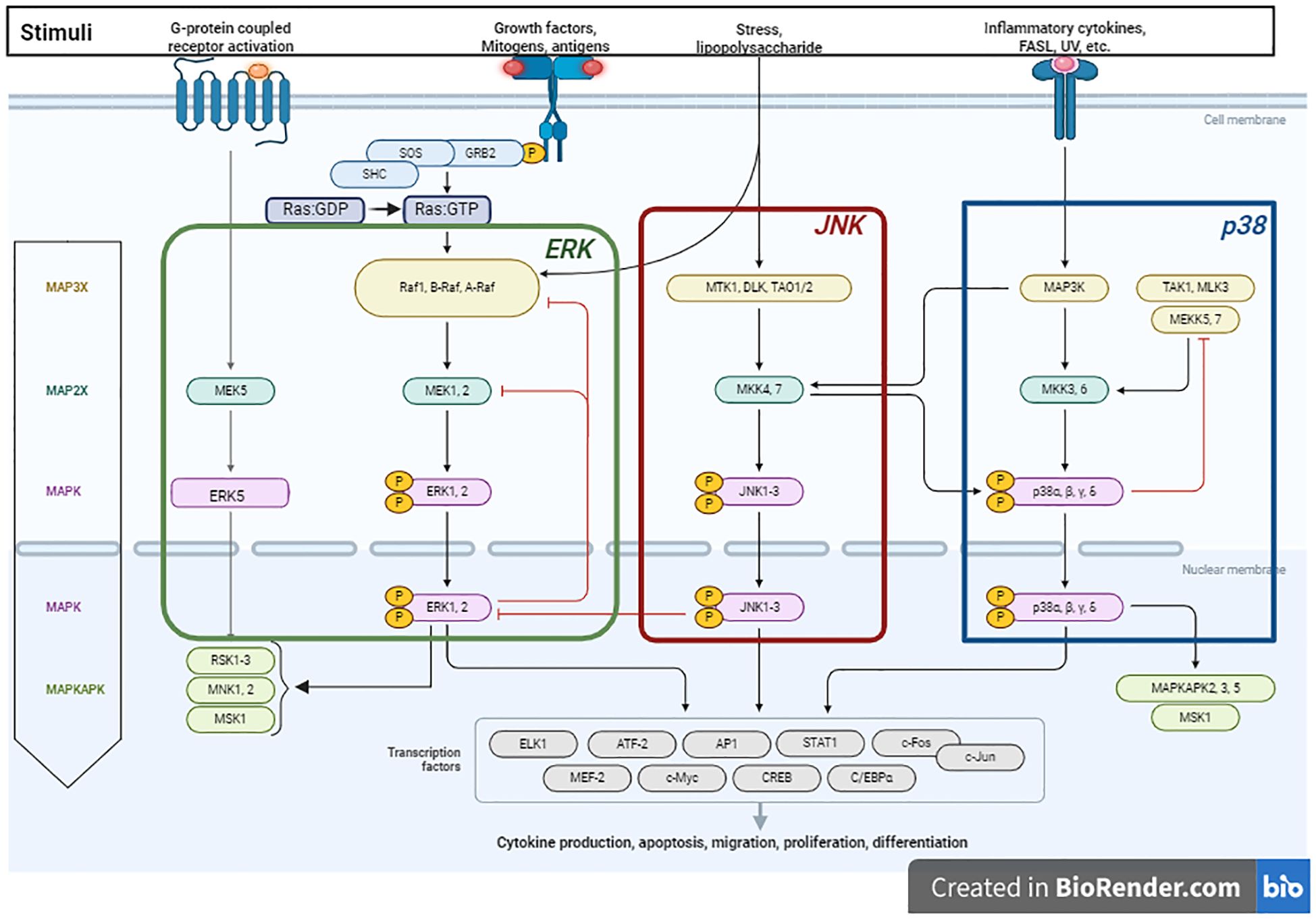
Figure 1. Schematic illustration of the MAPK pathway and its role in promoting tumorigenesis, proliferation and migration. The process is initiated upon the activation of receptor tyrosine kinases (RTKs) by stimuli such as growth factors, cytokines, or stress. The activation of RTKs subsequently leads to the recruitment of the adapter protein Grb2 and the guanine-nucleotide exchange factor Son of Sevenless (SOS), which convert Ras: GDP to Ras: GTP. In mammals, the MAPK pathway comprises three major modules: Extracellular signal-regulated kinase (ERK), C-Jun N-terminal kinase (JNK), and p38 MAPK. The ERK1/2 cascade is activated through the sequential phosphorylation and activation of a series of protein kinases (Raf – MEK1/MEK2 - ERK1/ERK2). RAF kinases, which serve as the MAPKKKK in the ERK1/2 pathway, consist of three members (A-Raf, B-Raf and C-Raf or Raf1). Once activated, ERK translocates to the nucleus, where it activates transcription factors, promoting tumor growth and differentiation by modulating gene expression. The JNK cascade is activated via the phosphorylation of MKK4 and MKK7, which are in turn activated by a variety of MAPKKKs (DLK, MTK1, and TAO1/2). The p38-MAPK module is primarily activated by the protein kinases MKK3 and MKK6. p38 activation extends MAPK cascades by phosphorylating MAPKAPK family members. Figure is adapted from “MAPK Signaling Pathway”, by BioRender.com (2024). Retrieved from https://app.biorender.com/biorender-templates.
Insights into the role of the MAPK signaling pathway in non-GIST STS
The multidisciplinary management of STS, including surgery, radiation therapy, and chemotherapy, has shown limited antitumor efficacy and short survival rates (31). Current research focuses on analyzing diverse signaling pathways in STS to determine their influence on patient outcomes and to identify potential therapeutic targets (20). The MAPK signaling pathway has emerged as a promising target for specific molecular-directed therapy for non-GIST STS (19). Previous preclinical studies have established a strong correlation between the MAPK signaling pathway and cell proliferation in sarcomas, emphasizing the potential therapeutic advantages of selective MAPK inhibitors for treating bone and STS (5). An additional study analyzed the prognostic relevance of MAPK pathway hyperactivation in STS. High expression of phospho-ERK1/2 is associated with aggressive behavior in UPS (32). In vitro studies of bone sarcomas have demonstrated the involvement of the MAPK pathway in modulating their behavior by enhancing aggressive properties, such as proliferation, angiogenesis, and inflammation (33). A recent study showed high intrinsic heterogeneity among STS subtypes; however, the MAPK signaling pathway consistently mediates stimuli in STS, regardless of the specific subtype (34). These findings have led to the emergence of MAPK targeting as a potential treatment modality for sarcomas.
Inhibition of the MAPK pathway: implications for current and future therapy
Recent studies have highlighted the role of MAPK pathway in STS pathophysiology and its potential as a therapeutic target. Several MAPK-targeted inhibitors are currently available, and additional compounds are being investigated in preclinical and clinical trials (35). Furthermore, crosstalk exists between the AKT/mTOR and MAPK pathways, with RAS activating the RAS/MEK/ERK and PI3K/AKT/mTOR pathways. Mutations in several STS subtypes activate pro-survival and growth factor signaling cascades, thereby promoting sarcomagenesis through downstream pathways (36). Growth factors such as IGF, c-MET, VEGF, and PDGF contribute to STS pathogenesis via the RAS/MEK/ERK and/or PI3K/AKT/mTOR pathways (19). This summary provides an overview of relevant treatments targeting the MAPK signaling pathway in non-GIST STS management (Figure 2).
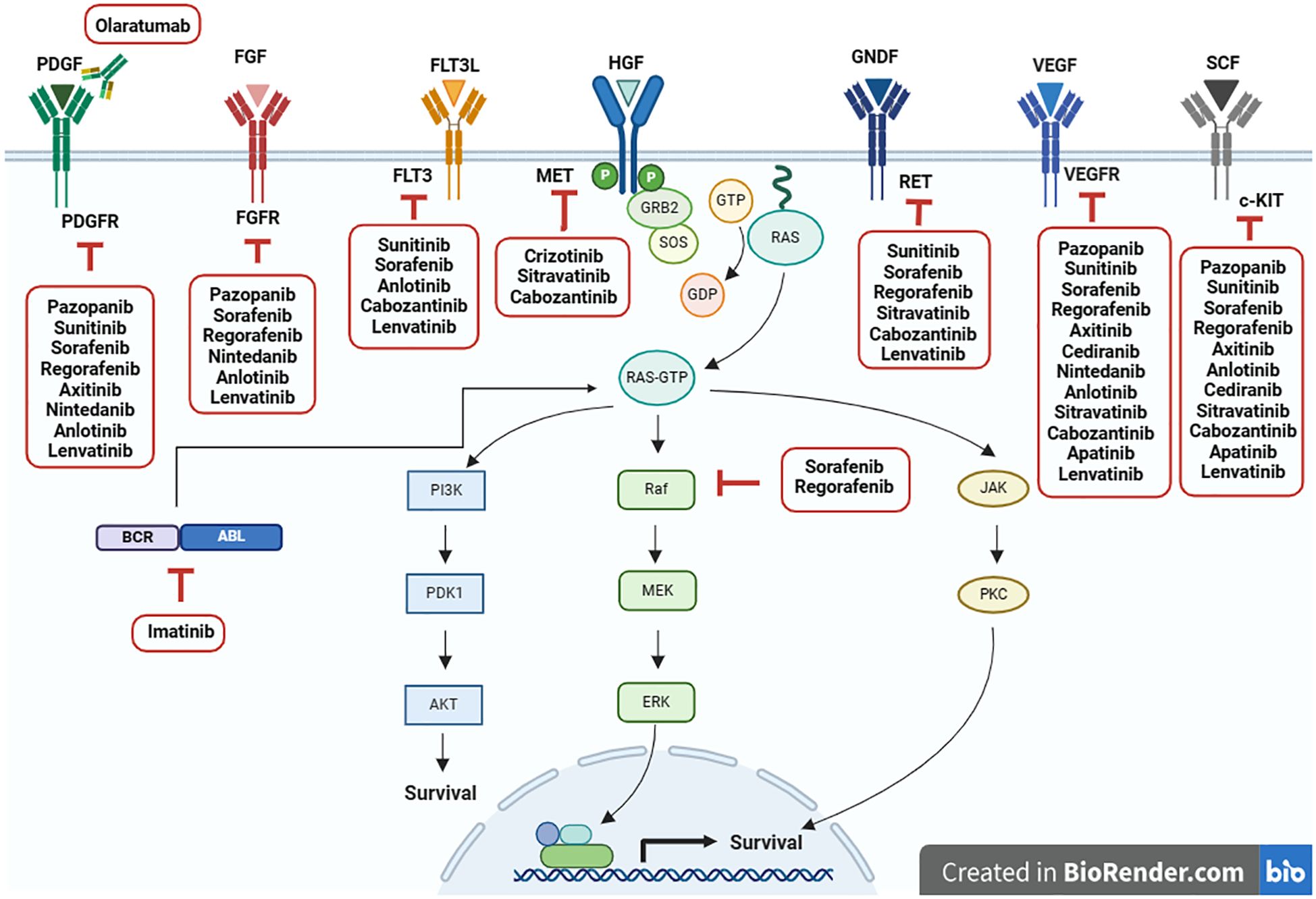
Figure 2. Current targeted therapies for inhibiting MAPK signaling pathway in non-GIST STS. Adapted from “Ras Pathway”, by BioRender.com (2024). Retrieved from https://app.biorender.com/biorender-templates.
Pazopanib
Pazopanib is a targeted therapy that specifically inhibits several growth factor receptors, including VEGFR, FGFR, PDGFR, and c-KIT (37). Preclinical studies show its antitumor effects are attributed to inhibiting angiogenesis and inducing apoptosis. Pazopanib also functions as a pan-RAF inhibitor, blocking the MAPK pathway in cancer cells (38). In a phase II clinical trial (EORTC 62043, NCT00297258), pazopanib exhibited safety and potential antitumor activity in advanced STS, except for the adipocytic subtype (39). However, a phase III trial (PALETTE, NCT00753688) found that pazopanib prolonged progression-free survival (PFS) in previously treated non-adipocytic STS patients, without significant improvement in overall survival (OS) (40). This trial resulted in the approval of pazopanib for STS treatment, excluding LPS (41, 42). Given the overall modest activity of pazopanib as a single agent in STS with varying efficacy across histotypes, further research is needed to identify biomarkers for patient selection and to elucidate resistance mechanisms (43). Several combinations of pazopanib with other drugs, such as chemotherapy (NCT01593748) (44) or a PDL1 inhibitor (NCT03798106) (45), have been investigated in phase II clinical trials to improve the incremental activity of pazopanib as monotherapy (46, 47). Two ongoing clinical trials have assessed pazopanib-based combinations. The first trial was a randomized, multi-center phase II study (NCT05679921) that explored the combination of pazopanib with a PD-1 inhibitor (pembrolizumab) in metastatic STS. The rationale for this combination is based on the potentiating antitumor effect of combining antiangiogenic tyrosine kinase inhibitors (TKIs) with immune checkpoint inhibitors (ICI) (48). The second trial was a phase I/II study (AMPHISARC, NCT05180695) with two steps (dose-escalation and dose-extension) that analyzes the potential of combining pazopanib with an MDM2 inhibitor (HDM201) in advanced/metastatic STS with p53-wild type. The aim of this combination is to enhance the anti-growth effect by blocking angiogenesis and inhibiting p53-MDM2 interaction, thereby increasing p53 tumor suppressive activity (49).
Imatinib
Imatinib is a specific BCR-ABL inhibitor that blocks BCR-ABL-dependent signaling pathways such as the p38 MAPK cascade (50). Following the significant breakthrough of imatinib as the first TKI approved for GIST management, it was subsequently assessed in bone and STS (51). Preclinical data demonstrated the promising efficacy of imatinib in MPNST (52), malignant rhabdoid tumor (MRT) (53), LMS (54), giant cell fibroblastoma and dermatofibrosarcoma protuberans (DFSP) cell lines (55). Its antitumor effect was attributed to reduced cell proliferation and induced apoptosis in both in vitro and in vivo models (56). In an open-label, single-arm, phase II trial (NCT00031915), imatinib was evaluated in pretreated patients with metastatic and locally advanced sarcomas. This study analyzed 10 subtypes of bone and STS (LMS, LPS, SS, MPNST, fibrosarcoma, rhabdomyosarcoma (RMS), malignant fibrous histiocytoma, AS, osteosarcoma, and Ewing’s sarcoma) (51). The primary endpoint was a clinical benefit rate (CBR) of > 30% for each histotype. However, this objective was not met, indicating a lack of imatinib activity in these selected sarcoma subtypes. Subsequently, an unplanned cohort of desmoid tumors (DT) was embedded in this trial, with promising outcomes: a 58% PFS rate and 84% of patients experiencing stable disease (57). A single-arm phase II study (NCT00287846) dedicated to DT involved 40 patients with progressive disease who received imatinib. The one-year PFS rate was 67%, and 10% of patients discontinued treatment due to toxicities (58). Another phase II trial (NCT01137916) enrolled 38 patients with progressive DT, achieving a one-year PFS rate of 59% (59). Imatinib was also evaluated in DFSP. Two open-label, single-arm, single-agent phase II trials (EORTC-62027, NCT00085475) and (SWOG-S0245, NCT00084630) were conducted. The latter failed to reach its target enrollment of 40 patients due to slow accrual, resulting in termination. Pooled data analysis demonstrated that imatinib is a promising therapeutic option for inoperable DFSP (60). In conclusion, imatinib has shown antitumor efficacy in progressive DT and unresectable DFSP (61).
Sunitinib
Sunitinib is a multi-target TKI that blocks several growth factor receptors such as VEGFRs, PDGFR, FLT3, RET, CSFR, and c-KIT (62). Mechanistically, its activity is explained by its ability to arrest cell growth resulting from the blockade of ERK, JNK, and p38 MAPK cascades (63). Preclinical studies have shown potential efficacy in selected sarcoma subtypes such as MPNST, MRT, and LMS (64). Various clinical trials have evaluated the effects of sunitinib in bone and STS. In a phase II trial, 53 adult patients with advanced non-GIST STS received sunitinib, which resulted in a median PFS of 1.8 months. After 24 weeks of treatment, 14 patients had disease stability, mirroring the outcomes observed in the placebo arm of the PALETTE phase II trial. This underscores the need to further define the role of sunitinib in STS. In a subsequent phase II trial involving 19 patients with unresectable DT, sunitinib demonstrated antitumor activity. Five patients experienced partial disease, while eight patients had stable disease. However, serious toxicities were observed, likely because of the high prevalence of mesenteric DT (63.2%) (65). Furthermore, retrospective case series have suggested the utility of sunitinib in specific subtypes of non-GIST STS, including ASPS (66), extraskeletal myxoid chondrosarcoma (67), and SFT (68). Although the small number of patients and non-randomized design in these studies limit the ability to draw firm conclusions about sunitinib’s effectiveness in non-GIST STS, it may be considered as a salvage therapy for specific challenging subtypes, which are known for their indolent nature and chemoresistance (61). Combination strategies represent an interesting path for STS management. Sunitinib was explored in combination with nivolumab in a single-arm, phase Ib/II trial (IMMUNOSARC, NCT03277924), which appears to be an active and safe regimen for patients with advanced STS (69).
Sorafenib
Sorafenib is a specific multitarget inhibitor that selectively inhibits Raf-1, B-Raf, PDGFRβ, VEGFR2, FLT3, RET, and c-KIT (70). In preclinical models, sorafenib has demonstrated antiproliferative activity in DT (71), MPNST (72), and RMS cell lines (73) by blocking ERK, MEK, and AKT signaling cascades (74, 75). The efficacy of sorafenib has been evaluated in clinical trials for various STS, primarily in vascular STS. A phase II single-arm trial (NCT00874874) tested sorafenib in AS, with disappointing results. The median PFS was 1.8 months in the superficial AS group and 3.8 months in the visceral AS cohort (76). A subgroup analysis of patients with progressive SFT (N = 5) showed that two out of five patients achieved 6-month disease control. However, the small sample size limited conclusive findings regarding the antitumor efficacy of sorafenib in STS. Further studies are needed to evaluate the role of sorafenib in this STS subgroup (77). In the same trial, 15 patients with metastatic or inoperable locally advanced epithelioid hemangio-endothelioma (EHE) were evaluated with a response rate of approximately 30.7%. Three patients required a dose reduction, and five patients discontinued the drug (78). A retrospective study of 26 patients with aggressive DT evaluated the sorafenib’s efficacy, showing that 25% of patients experienced partial responses and 70% had disease stability (79). In a randomized, double-blind, placebo-controlled phase III trial (ALLIANCE A091105, NCT02066181), sorafenib was compared to placebo in DT or aggressive fibromatosis (AF). The primary endpoint was PFS rate, and 2-year PFS rate was at 81% in the sorafenib arm versus 36% in the placebo arm. However, OS outcomes were not reported (80). To our knowledge, the ALLIANCE A091105 trial is the only phase III study evaluating TKI in DT, highlighting the promising role of sorafenib in this specific sarcoma histotype.
Regorafenib
Regorafenib is a multi-kinase inhibitor that effectively targets VEGFR, PDGFR, c-KIT, RET, FGFR, and RAF (81). To achieve its antitumor effect, regorafenib induces the activation of MAPK pathways, including the JNK and p38 MAPK signaling cascades (82, 83). Preclinical studies with different STS cell models demonstrated promising activity in LMS (56), MRT (84), and SFT (61). Subsequently, the efficacy of regorafenib was evaluated in a clinical setting. In a phase II, randomized, double-blind trial (REGOSARC, NCT01900743), regorafenib was compared with placebo in adult pretreated advanced STS patients. The results were stratified according to histological subtype (LPS, SS, LMS, and other STS). Regorafenib showed improved PFS in the LMS, SS, and other STS subtypes but not in the LPS cohort (85). An updated analysis of the REGOSARC trial in 2018 confirmed PFS improvement in the regorafenib arm for patients with non-adipocytic STS after a median follow-up of 32.4 months. However, no OS benefit was observed owing to crossover to the regorafenib group once progression was confirmed in the placebo group (86, 87). Another phase II, prospective, non-randomized, single-center trial (NCT02307500) assessed the clinical activity and safety of regorafenib in adult pretreated advanced STS patients. 21 patients were enrolled, with the primary endpoint being the PFS rate at 8 weeks. After a median follow-up of 33.5 months, the PFS rate was 62% (13 out of 21 patients), confirming the incremental clinical activity of regorafenib in advanced non-adipocytic STS (88). Another phase II, randomized, double-blind, multicenter trial (NCT01900743) was designed to compare regorafenib with placebo in adult patients with unresectable advanced or metastatic STS previously treated with chemotherapy and pazopanib. A significant improvement in PFS was observed in the regorafenib group compared to the placebo group, with a median PFS of 2.1 vs. 1.1 months (p = 0.0007), highlighting its promising role as salvage therapy for heavily pretreated non-adipocytic STS (89).
Regorafenib showed promise in bone sarcomas, as demonstrated in the REGOBONE trial (NCT02389244), a randomized, controlled phase II study assessing its efficacy and safety in metastatic bone sarcomas (chondromas, chondrosarcomas, osteosarcomas, and Ewing sarcomas). In the osteosarcoma cohort, regorafenib offered a meaningful benefit over placebo in PFS and OS outcomes, with median PFS of 16.4 weeks and median OS of 11.3 months in the regorafenib group, compared to 4.1 weeks and 5.9 months in the placebo group (90). Therefore, regorafenib can be considered an effective treatment option for osteosarcomas and non-adipocytic STS. It is a promising addition to the armamentarium of targeted therapies for STS treatment. It also represents an interesting option for maintenance therapy of bone sarcomas. Two-phase II clinical trials (NCT04698785 and NCT04055220) are currently recruiting patients with bone sarcomas to study the role of regorafenib as a maintenance treatment. To improve STS outcomes, combinations targeting the VEGFR and PD1/PDL1 pathways have been explored. A recent phase II basket trial (NCT03475953) evaluated the regorafenib-avelumab combination in different solid tumors, including STS (91). Based on these data, further larger studies are required to investigate the best partner to combine with regorafenib and identify potential biomarkers for better patient selection (92). Another phase Ib trial (NCT03475953) is ongoing to further assess the regorafenib-avelumab combination in STS with an immune signature (Tertiary Lymphoid Structure Signature +) (Table 1).
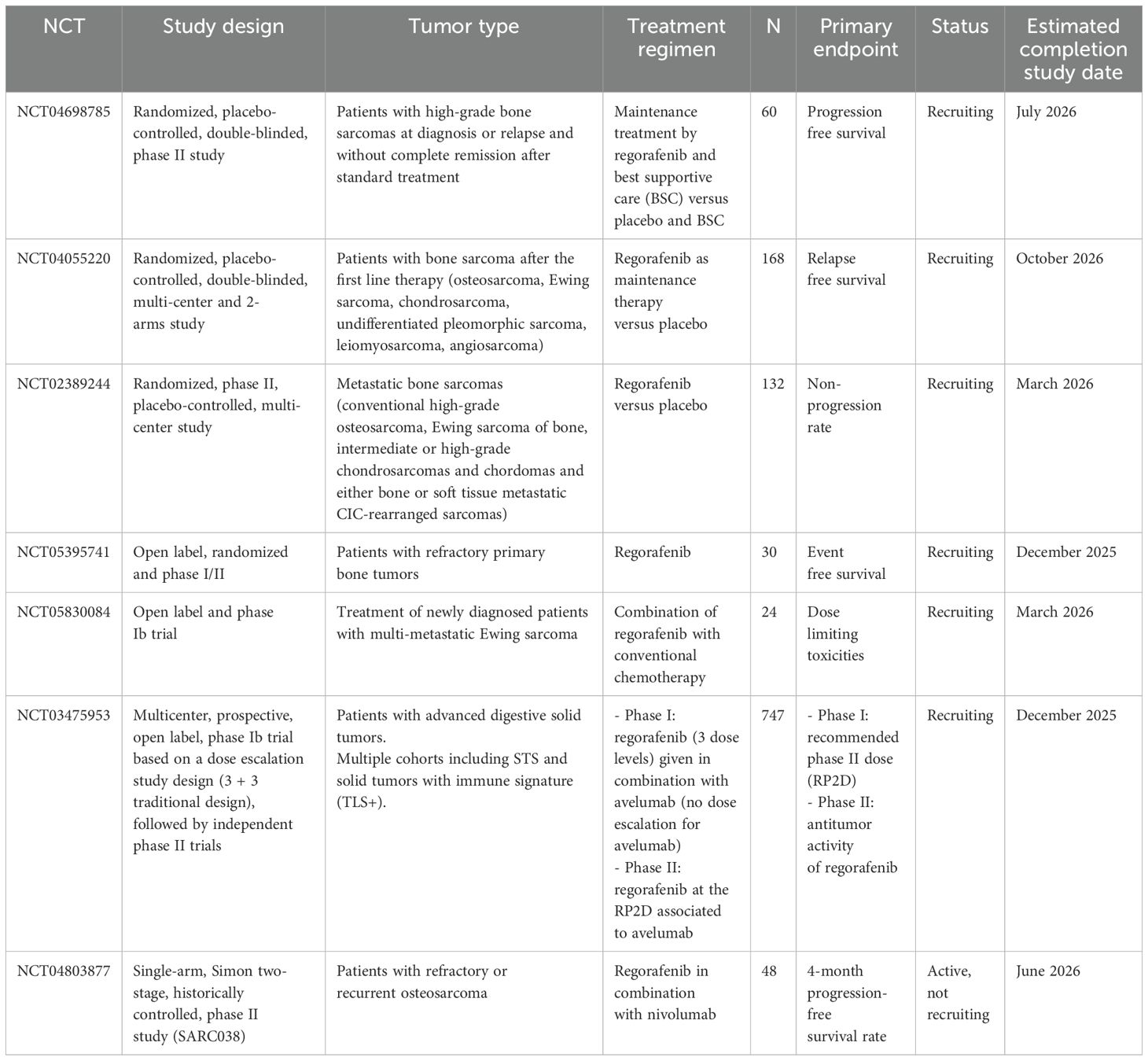
Table 1. Ongoing clinical trials assessing regorafenib in non-GIST STS.
Axitinib
Axitinib is a small-molecule TKI that selectively targets VEGFR, PDGFR, and c-KIT (93). It inhibits tumor growth by blocking VEGFR2, AKT, and ERK signaling pathways (94). Preclinical studies have shown that axitinib is effective in myxoid LPS cell lines and xenografts (95), MRT, SS, and LMS models (56). In a prospective, open-label, non-randomized phase II trial (NCT02261207) involving 17 adult patients with progressive advanced SFT, a partial response was observed in seven patients (7/17, 41%) and stable disease in six patients, while four patients experienced progression. Among the four patients with high-grade or dedifferentiated SFT, no response was reported with axitinib. Furthermore, seven of the 17 patients included in this trial had previously received pazopanib, with half of them responding to axitinib (96). Based on these findings, axitinib may be considered an interesting therapeutic option for advanced SFT after progression on pazopanib. A recent multicenter, open-label, non-randomized, histologically stratified phase II study (Axi-STS, ISRCTN 60791336) of 145 patients with advanced STS found that axitinib demonstrated clinical activity in four histological strata (AS, LMS, SS, and other non-adipocytic STS), with further confirmation needed in phase III trials (97). The combination of VEGFR and ICI is being explored due to the role of angiogenesis in sarcoma proliferation and immunosuppression. In a phase II trial (NCT02636725), the axitinib-pembrolizumab combination was assessed in 36 patients with advanced or metastatic STS, showing meaningful clinical activity, with a 3-month PFS rate of 65.6% in all patients and 72.7% in the ASPS cohort (98).
Cediranib
Cediranib is a TKI that inhibits VEGFR and c-KIT (99), causing antiproliferative effects by suppressing the VEGFR, AKT/mTOR, and ERK/MAPK signaling cascades (100). Tumor regression has been observed in specific rhabdoid tumor xenograft models (101) and MRT, SS, and LMS cell lines (56). Cediranib has demonstrated preliminary activity in ASPS patients (35). In a single-arm phase II study (NCT00942877), cediranib was investigated in 46 adult patients with unresectable or metastatic ASPS. Radiological response was assessed using the RECIST criteria. Partial response and stable disease were reported in 15 (35%) and 26 (60%) patients, respectively, providing evidence of cediranib activity in ASPS (102). In a phase II trial (CASPS, NCT01337401), cediranib was compared with placebo in metastatic ASPS patients. An improvement in response rate was reported in the cediranib arm versus the placebo group (19% vs. 0%, p = 0.072). However, no significant benefit was observed between the two cohorts (p = 0.28) (103). The data suggest a preliminary activity of cediranib in ASPS (104), but larger cohorts are needed to confirm these results and understand the resistance mechanisms.
Nintedanib
Nintedanib is a TKI targeting VEGFR, PDGFR, and FGFR (105), with antitumor effects mainly attributed to the dual inhibition of the ERK and AKT signaling pathways (106). It showed preclinical activity in MPNST (107) and SS models (108), leading to further clinical development. However, a phase II trial (EORTC1506, NCT02808247) comparing nintedanib with ifosfamide in second-line STS treatment was halted due to futility. The trial failed to provide evidence supporting the clinical use of nintedanib in advanced, unselected STS (109).
Anlotinib
Anlotinib is a selective TKI that inhibits VEGFR, PDGFR, FGFR, FLT3, and c-KIT (110). Its antitumor effect is attributed to blockade of the ERK/MAPK and PI3K/AKT signaling pathways (111, 112). In preclinical settings, anlotinib has shown activity in SS models (113). In the phase II single-arm trial (NCT01878448), anlotinib was evaluated as a second-line treatment after progression on anthracycline therapy for 166 patients with advanced STS. They achieved a 74% disease control rate with median PFS and median OS of 5.6 and 10.7 months, respectively (114). This trial suggests a meaningful activity of anlotinib in STS (115). In a phase II/III clinical study comparing the efficacy and safety of anlotinib with a placebo in 233 patients with STS (ALTN-02-IIB, NCT02449343), anlotinib was identified as a novel treatment option for patients with advanced STS after the failure of standard chemotherapy (116). A subsequent phase III trial (APROMISS, NCT03016819) confirmed the acceptable benefit-risk profile of anlotinib in patients with advanced SS, demonstrating improved disease control and superior PFS compared with dacarbazine in advanced SS (117).
Anlotinib has also been utilized as a first-line treatment for locally advanced or metastatic STS in patients unfit for chemotherapy, exhibiting encouraging anti-tumor activity and favorable tolerability in a Chinese phase II clinical trial (NCT03792542) (118). Further studies are necessary to identify the most beneficial STS subgroup. Based on these preliminary promising results, anlotinib is currently being investigated in advanced ASPS, LMS, and SS in an ongoing phase III trial (APROMISS, NCT03016819) (117). Additionally, anlotinib was combined with irinotecan for advanced Ewing sarcoma after standard therapy failure in a multicenter, single-arm phase Ib/II trial (NCT03416517), demonstrating promising clinical efficacy (119). An open-label and single-arm phase II trial (ALTER-S006, NCT03890068) assessed its efficacy as a maintenance therapy in 49 patients with advanced STS who achieved stability after first-line chemotherapy (anthracycline-based treatment). It showed meaningful activity as a post-chemotherapy maintenance treatment in advanced STS, with a median PFS of 9.1 months (120). In a double-blind phase II clinical trial (NCT03951571), the role of anlotinib as an adjuvant therapy for completely resected high-grade STS was evaluated, suggesting its impact in reducing disease recurrence risk with an acceptable toxicity profile. Anlotinib is currently being studied in combination with other therapies in retrospective real-world studies. The anlotinib-liposomal doxorubicin combination was evaluated in an observational study that analyzed the efficacy and safety of this combination in 27 patients with metastatic STS. This Chinese study highlighted the efficacy of anlotinib-liposomal doxorubicin followed by anlotinib monotherapy in advanced STS (121). Another study found that anlotinib was associated with PD-L1 inhibitors in a retrospective cohort of 32 patients with pretreated metastatic STS, achieving an ORR of 34.4% and median PFS of 7.6 months. This study provides real-world evidence of the efficacy of anlotinib-based combinations in advanced STS (122). However, further prospective clinical trials are needed to identify the optimal partner of anlotinib in STS management. Several anlotinib-based combinations are ongoing, including chemotherapy (NCT05121350, NCT05747521), anti-CD137 (NCT05301764), and ICI (NCT04172805, NCT05481645, NCT05193188, and NCT05926700) (Table 2).
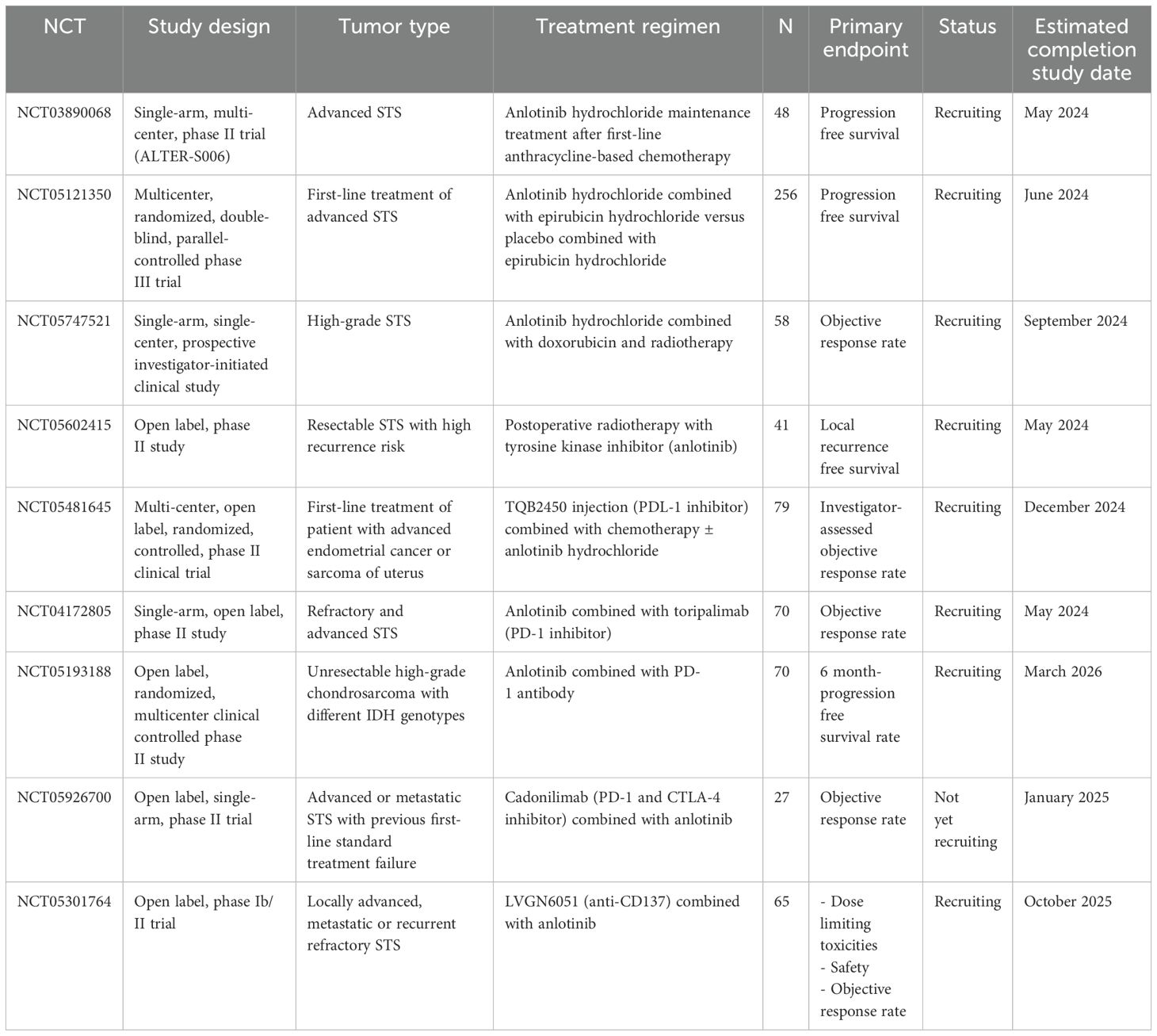
Table 2. Ongoing clinical trials investigating anlotinib in non-GIST STS.
Overall, anlotinib demonstrates potential as a treatment for advanced sarcomas, functioning as an anti-angiogenesis TKI with significant effects, manageable adverse reactions, and enhanced efficacy in combination therapies. However, several challenges persist, including drug resistance, determination of optimal dosage, combining with conventional anti-cancer medications, sequencing, and assessing of treatment effectiveness. To achieve optimal outcomes utilizing anlotinib as targeted therapy for advanced sarcoma patients, these issues warrant investigation for individual sarcoma types (115).
Sitravatinib
Sitravatinib inhibits VEGFR, c-KIT, RET, and MET (123). In preclinical models, sitravatinib induced growth inhibition in DDLPS and MPNST cell models by deactivating the PI3K/AKT and RAS/MAPK pathways (124). This preclinical activity has translated into clinical settings, leading to an ongoing single-arm phase II trial (NCT02978859) in advanced LPS and other STS (125). The trial evaluated patients with advanced LPS (WDLPS and DDLPS) who received at least one systemic therapy in the metastatic setting. The primary endpoint was PFS rate at 12 weeks. 12 of the 29 patients included had free progression at 12 weeks, suggesting the potent efficacy of sitravatinib in advanced LPS (126).
Crizotinib
Crizotinib is a small-molecule TKI that targets the ALK and MET signaling cascades (127), demonstrating antitumor effects in small round-cell tumors and SS models (128). It inhibits cell proliferation by blocking the ERK, AKT, and STAT3 pathways (129). The CREATE trial (EORTC90101, NCT01524926) is a non-randomized, single-arm phase II study that investigated the efficacy of crizotinib in IMT, ASPS, CCS, and aRMS (130, 131). This evaluation is based on ALK and/or MET activation in the pathogenesis of these subgroups (132, 133). In this trial, a cohort of 48 patients with unresectable advanced or metastatic ASPS was analyzed, with a partial response in two patients and stable disease in 39 patients. Concerning crizotinib safety, grade 3 and 4 adverse events were reported in six out of the 48 patents (12.5%) included in the ASPS cohort. In the IMT cohort, crizotinib showed a 50% ORR in patients with ALK gene rearrangement. The updated results in 2021 showed a 66.7% ORR in ALK-positive IMT and a 14.3% ORR for ALK-negative IMT. These findings confirmed that crizotinib is an effective therapy for advanced ALK-positive IMT (134).
CCS are rare tumors characterized by a pathognomonic translocation of t (12,22)(q13; q12), leading to an EWSR1/ATF1 gene fusion (135, 136). The ESWR1/ATF1 transcriptional activator is regulated by the upstream p38 MAPK signaling pathway (137, 138). In the CCS cohort of the CREATE trial, 34 patients were enrolled. Only one patient showed a partial response, while 17 patients had stable disease. The median PFS reported with crizotinib (4.4 months) was higher than that observed in CCS patients treated with cytotoxic chemotherapy in the metastatic setting (131, 139). CREATE is the first genotype-driven trial to evaluate TKI activity in non-GIST STS patients. These findings underscore the importance of screening for ALK status as a biomarker for crizotinib’s efficacy in ALK-rearranged IMT.
Dasatinib
Dasatinib is another TKI that targets SFK members and some TK receptors (such as EGFR and ephrin receptors), inhibiting their corresponding downstream signaling pathways (RAS/ERK, STAT3, FAK, and PI3K/AKT) (140). It demonstrated meaningful preclinical activity in various STS subgroup models (aRMS, LPS, SS, ASPS, and MRT) (56, 141–143). However, it failed to show clinical activity in a phase II trial (SARC009, NCT00464620) for advanced STS patients with SFT, ASPS, epithelioid sarcomas, chondrosarcomas, and chondromas (144).
Cabozantinib
Cabozantinib is an interesting multi-target TKI that inhibits umpteen TK receptors (VEGFR, FLT3, AXL, RET, c-KIT, and MET) (145–147). These targets are involved in the pathogenesis of sarcomas. In preclinical studies, cabozantinib showed significant activity in ASPS and RMS cell lines (143, 148). Additionally, it has been demonstrated to inhibit cell growth in osteosarcomas and Ewing sarcomas in preclinical models (149, 150). In the clinical setting, after demonstrating potent efficacy in GIST, cabozantinib was further investigated in bone and STS in a phase II clinical trial (NCT02216578) (151). Subsequently, it was investigated in a phase I trial (NCT01709435) for refractory solid tumors, including six patients with STS (ASPS, CCS, RMS, and SS) and six patients with bone sarcomas (osteosarcomas and Ewing sarcomas), leading to further research on cabozantinib in bone and STS (152). A phase II study (NCT01755195) was designed to assess the efficacy and safety of cabozantinib in 54 patients with advanced and previously treated STS. The study reported an ORR of six patients (11.1%) and a 6-month PFS rate of 49.3%. Cabozantinib shows promise as a therapy for selected STS subtypes (ASPS, LMS, UPS, and extra-skeletal myxoid chondrosarcoma).
The CABONE trial (NCT02243605) was a phase II study that enrolled 90 patients with heavily pretreated osteosarcomas (N = 45) and Ewing sarcomas (N = 45). The primary endpoints were ORR and 6-month PFS. In the osteosarcoma cohort, the ORR was 12% with a median PFS and median OS of 6.7 and 10.6 months, respectively. In the Ewing sarcoma cohort, the ORR was 26%, with median PFS of 4.4 months and median OS of 10.2 months. Cabozantinib caused serious adverse events in 68% of patients, but no grade 5 toxicity was reported (153, 154). In a recent phase II trial (NCT02867592), the activity of cabozantinib was analyzed in children and young adults with refractory solid tumors, including osteosarcomas, Ewing sarcomas, RMS, and non-RMS STS. This trial found promising activity of cabozantinib in osteosarcoma patients, with an ORR of 34% (155). The combination of cabozantinib with other therapies was also evaluated in heavily pretreated LMS patients, showing an increase in CBR of 33% (156). The cabozantinib-nivolumab combination was assessed in patients with advanced or metastatic AS who had previously received taxane chemotherapy in a phase II trial (Alliance A091902, NCT04339738). The study found a preliminary antitumor effect in patients with advanced taxane-pretreated AS. Exploratory analyses are still awaited (157). Cabozantinib-based combination with dual ICI (PD-1 and CTLA-4 inhibitors) was investigated in a phase II trial (NCT04551430) in metastatic STS. The study aimed to compare two arms: cabozantinib as monotherapy and cabozantinib in association with ipilimumab and nivolumab for 4 cycles, followed by nivolumab as maintenance. The triplet arm had a significantly higher disease control rate (DCR) than the single-agent arm (80% vs. 42%, p = 0.0004). PFS was also improved in the triplet arm compared to the single arm with a median PFS of 5.4 vs. 3.8 months, respectively (p = 0.016) (158). Cabozantinib is a potent and effective therapeutic option for bone and STS management. The drug is still being explored in various ongoing clinical trials for patients with STS in different settings. In the neoadjuvant setting, cabozantinib is being analyzed in combination with radiation therapy for extremities sarcomas in a phase I/II trial (NCT04220229). Cabozantinib is being investigated as a maintenance therapy in an ongoing phase II trial (NCT01979393) for patients with high-grade uterine sarcomas (159). Moreover, cabozantinib has been assessed in combination with ICI in three ongoing phase II clinical studies in patients with bone and STS: with PD-1 inhibitor (pembrolizumab, NCT05182164), PDL-1 inhibitor (atezolizumab, NCT05019703), and PD-1/CTLA-4 inhibitors (nivolumab plus ipilimumab, NCT04551430) (Table 3).
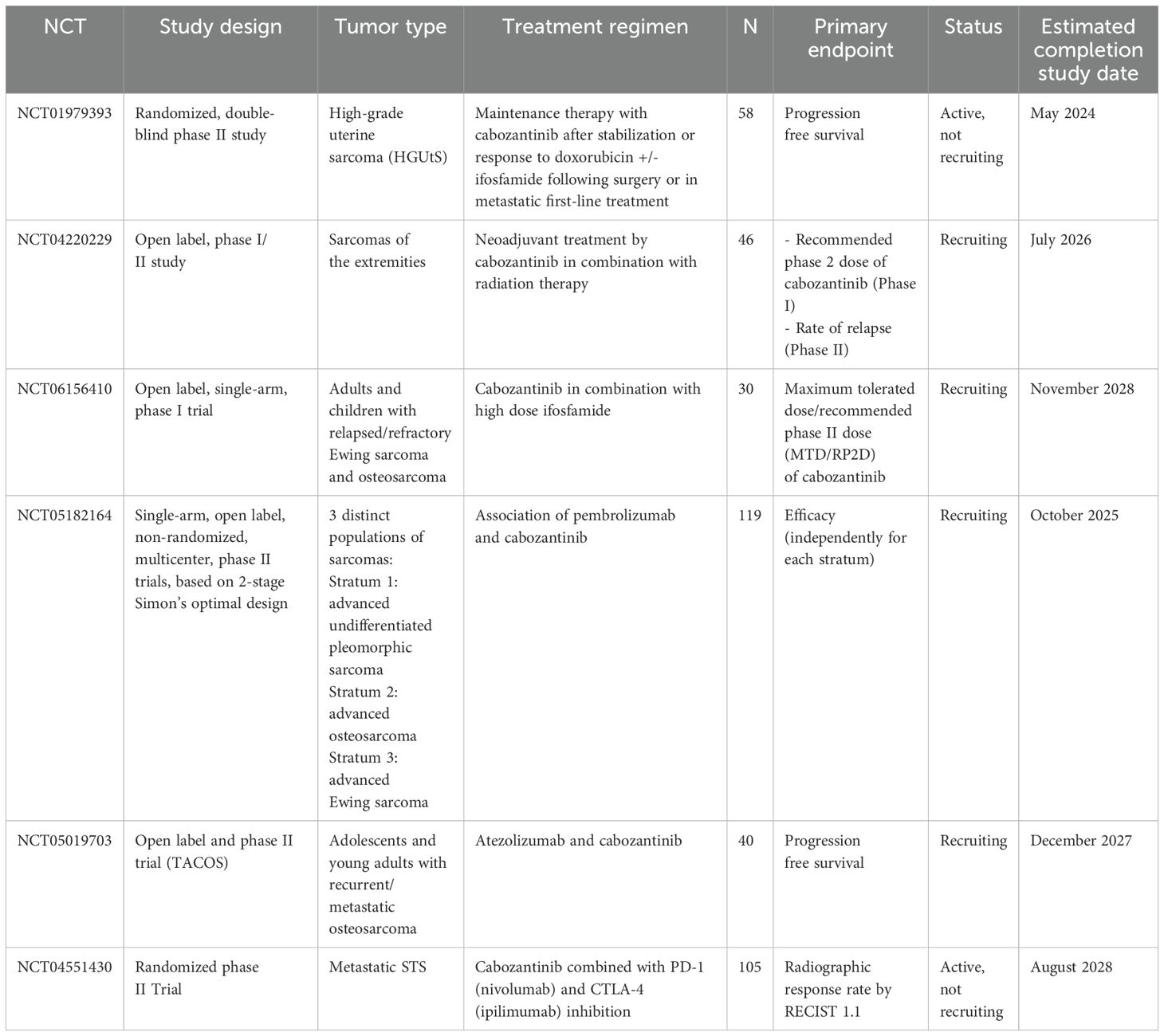
Table 3. Ongoing clinical trials assessing cabozantinib in non-GIST STS.
Apatinib
Apatinib is a small-molecule TKI that inhibits VEGFR, c-Src, and c-KIT tyrosine kinases (160), resulting in an antitumor effect due to the inhibition of ERK/MAPK and PI3K/AKT signaling cascades (161, 162). Apatinib was explored in a phase II trial (NCT03121846) in metastatic STS patients. The PFS rate at 12 weeks was 70%, with an ORR of 26.3% (163). However, it did not show a significant improvement in survival in patients with LMS compared to other STS subtypes (164). In a phase II clinical trial, the efficacy and safety of apatinib were analyzed in patients with advanced previously treated STS, which showed moderate antitumor activity of apatinib (165). To improve its effectiveness, apatinib-based combinations were explored. A meaningful antitumor effect was reported in patients with refractory osteosarcomas treated with apatinib combined with cytotoxic chemotherapy (ifosfamide and etoposide) in a retrospective study (NCT04690231) (166). Another combination of apatinib was evaluated in a phase II trial (NCT03359018), showing a PFS benefit in patients with advanced osteosarcomas receiving camrelizumab (a PD1 inhibitor) and apatinib compared to apatinib alone (167). There are three ongoing clinical trials assessing apatinib-based combinations, including two with cytotoxics (NCT04012827 and NCT04824352) and one with ICI (NCT04074564) (Table 4).
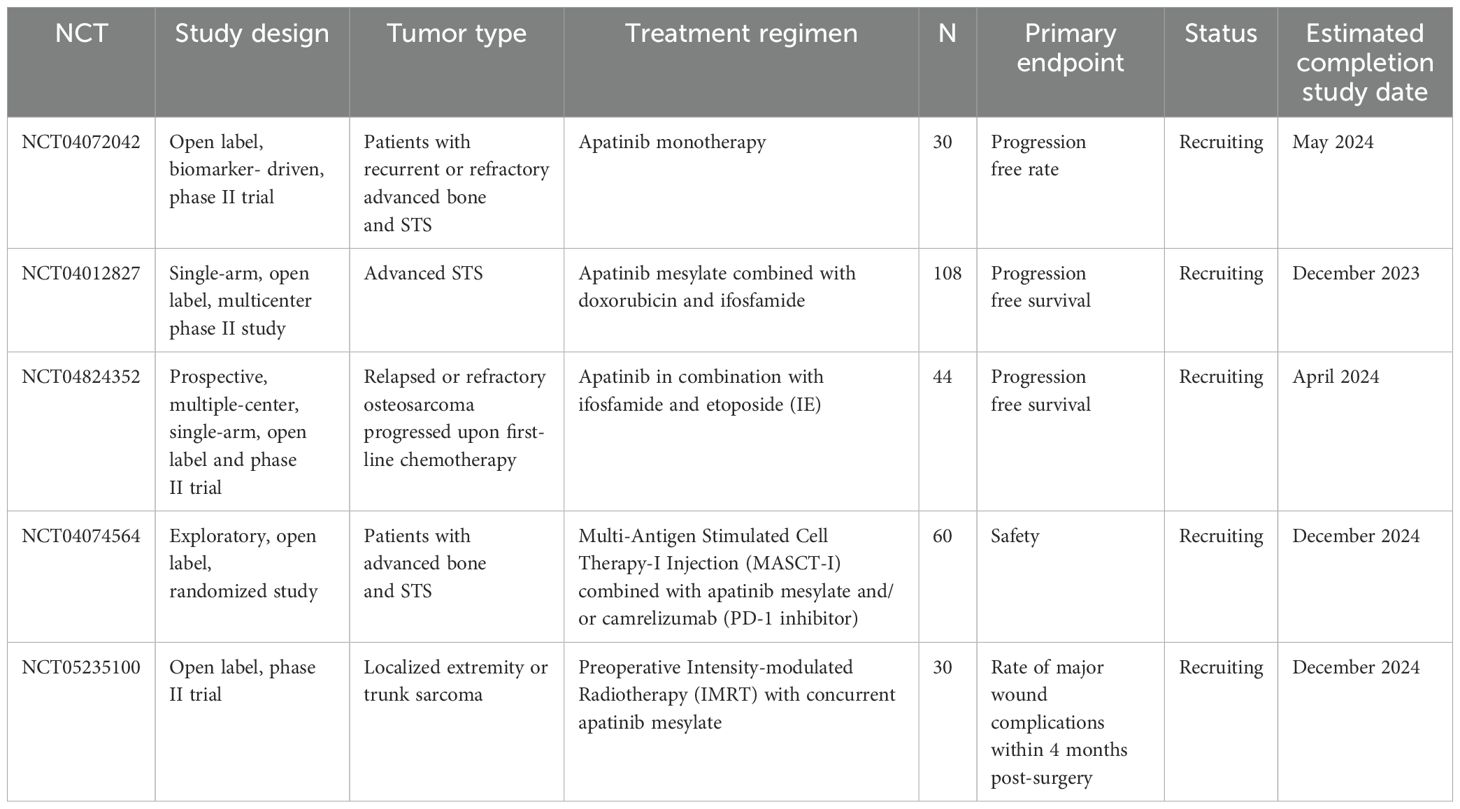
Table 4. Ongoing clinical trials evaluating apatinib in non-GIST STS.
Lenvatinib
Lenvatinib is a potent inhibitor of VEGFR, PDGFR, FGFR, RET, FLT3, and c-KIT. It induces an antitumor effect by blocking the MAPK signaling pathway (168). In a phase Ib/II trial (LEADER, NCT03526679), lenvatinib was combined with eribulin in 20 patients with advanced LPS and LMS, achieving an ORR of 27% and a median PFS of 56 weeks, highlighting the potential efficacy of this combination in LMS and LPS (169). In an open-label, single-arm phase II trial (NCT04784247), lenvatinib was combined with ICI (pembrolizumab) in five cohorts of selected metastatic and unresectable bone and solid tumors (A: LMS; B: high-grade UPS; C: vascular sarcomas (including AS and EHE); D: other STS (including SS and MPNST); and E: bone sarcomas (including osteosarcoma and chondrosarcoma)). Preliminary results indicated the potential antitumor activity of SS, MPNST, AS, and osteosarcoma (170). The trial is ongoing and definitive results are awaited.
Olaratumab
Olaratumab is a monoclonal antibody that inhibits PDGFR alpha, blocking downstream signaling cascades such as the MAPK pathway (171). Inhibition of proliferation was observed with olaratumab in sarcoma cell lines (172). It was first evaluated in two earlier phase I dose-escalation studies in advanced solid tumors, showing preliminary antitumor effects (173, 174). Olaratumab was then investigated in clinical trials as monotherapy and in combination with other agents. In a randomized phase Ib/II trial (NCT01185964), the efficacy and safety of olaratumab and doxorubicin combination compared to doxorubicin alone in 133 patients with advanced STS were evaluated. The median PFS was 6.6 months in the combination arm versus 4.1 months in the doxorubicin monotherapy group. A statistically significant improvement in OS was reported with the olaratumab–doxorubicin combination compared to doxorubicin alone (26.5 vs. 14.7 months, respectively; p = 0.0003) (175).
Olaratumab was the first monoclonal antibody approved by the US Food and Drug Administration (FDA) in October 2016 for treating adult patients with advanced STS (176). However, the combination failed to confirm its efficacy and OS improvement in a subsequent phase III trial (ANNOUNCE, NCT02451943), leading to its withdrawal from the market (177, 178). Recently, a randomized, multicenter phase Ib/II study (ANNOUNCE 2, NCT02659020) was designed to investigate the pharmacokinetics, safety, and efficacy of olaratumab combined with gemcitabine and docetaxel in locally advanced or metastatic STS. This trial failed to demonstrate a significant benefit of OS in the combination arm (179). The addition of olaratumab to PD1 or PDL1 inhibitors appeared to have synergistic activity by increasing the immune response. In a phase Ia/Ib clinical trial (NCT03126591), olaratumab-pembrolizumab combination showed a safe profile, but further studies are required to assess its efficacy (180).
Mechanisms of resistance to MAPK pathway inhibition in non-GIST STS
Resistance to targeted therapies is a prevalent issue in solid tumors, with several overarching mechanisms identified across various anticancer treatments. By examining these resistance mechanisms, the objective is to develop combination therapies that can preemptively address and counteract resistance at the initiation of treatment, thereby enhancing both the extent and duration of clinical efficacy (181). Although distinct resistance mechanisms are reported in STS for each therapy, the most prevalent include drug inactivation or modification, mutation of the target protein, reduced drug accumulation, and bypass of target inhibition (182).
The reactivation of downstream signaling pathways through the development of secondary mutations in the oncogenic target or mechanisms independent of the original drug target leads to the development of acquired resistance. The reactivation of key downstream effectors through parallel signaling pathways of other receptor tyrosine kinases (RTKs) provides an alternate route around the inhibited target to activate downstream signaling and allows the tumor to bypass the inhibition of the driver gene by the TKI drug and continue to survive and grow. This mechanism of resistance is known as bypass activation (183).
Recent data indicate that the tumor microenvironment (TME) remains not sufficiently understood, particularly regarding its role in drug resistance. Consequently, a thorough evaluation of the TME, along with well-designed histotype-specific preclinical studies, may enhance our understanding of the potential effects of combinatorial treatment strategies in STS. This understanding could facilitate the development of therapeutic interventions tailored to patients with STS by inhibiting alternative pathways (184).
Moving forward, there will be an increasing reliance on computational modeling methods capable of analyzing and testing all reasonable drug combinations. These methods should consider and exploit the complex interactions within the MAPK pathways using in silico computational models. Additionally, employing an ex vivo drug sensitivity platform, such as the quadratic phenotypic optimization platform (QPOP), may prove promising in predicting treatment responses and guiding the development of novel therapeutic strategies for STS patients (185).
Perspectives and emerging therapeutic options targeting the MAPK pathway in non-GIST STS
A landscape of targeted therapies has been developed for STS treatment, with numerous molecularly targeted agents under investigation to enhance efficacy and overcome potential resistance mechanisms. While significant challenges persist, including the heterogeneity of non-GIST STS subtypes and the potential for adaptive resistance, the ongoing development of MAPK pathway-targeted therapies represents a promising avenue for improving outcomes in patients with these challenging malignancies (186) (Table 5).
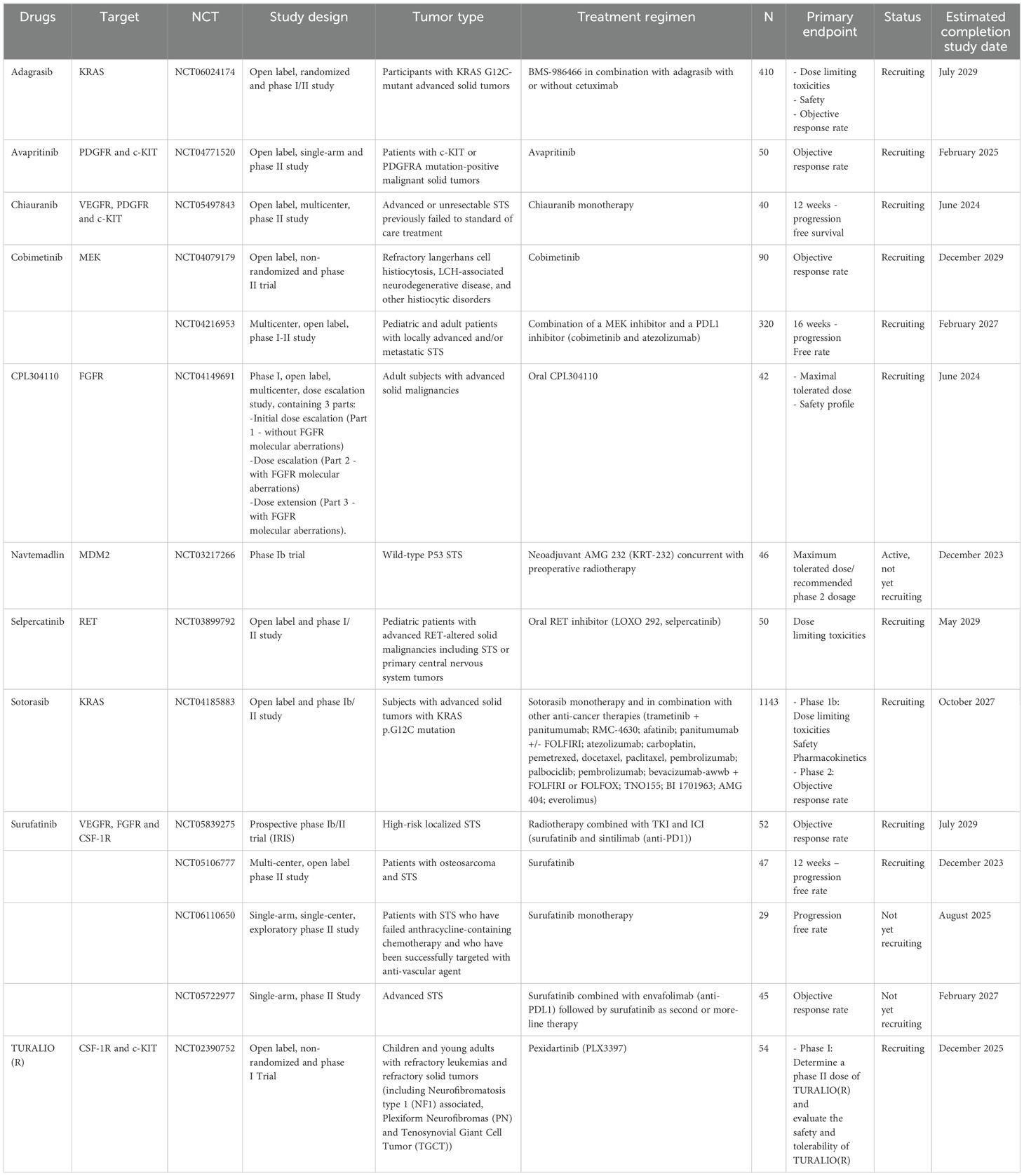
Table 5. Ongoing clinical trials investigating the inhibition of MAPK cascade in non-GIST STS.
Other VEGFR-associated multi-targeted TKIs
Angiogenesis is crucial in sarcomagenesis, and VEGFR overexpression is linked to poor prognosis and resistance to cytotoxic drugs in STS (187). Multi-targeted TKIs that inhibit VEGFR are being explored in clinical trials for their potential in treating sarcomas. Tivozanib, a VEGFR-targeting TKI, showed promising results in a phase II trial with 58 patients with advanced unresectable or metastatic STS, with a PFS at 16 weeks of 36.4%. Based on these promising findings, further investigation of tivozanib in combination with other agents is warranted to improve the outcomes in patients with refractory advanced non-GIST STS. Fruquintinib, another antiangiogenic TKI, demonstrated significant improvement in PFS in an exploratory, retrospective study of 122 patients with advanced bone and STS. The median PFS was 4.8 months in the fruquintinib group versus 1.4 months in the control group (p < 0.001) in an exploratory, retrospective study (NCT06202599). This study concludes the place of fruquintinib, a potentially effective and safe drug, in third- or further-line therapy for advanced bone and STS, and sheds light on the need to continue the exploration of this TKI in STS. Additionally, two new anti-VEGFR TKIs, chiauranib and sufuratinib, are currently under investigation in ongoing clinical trials. Chiauranib is being assessed in a multicenter, single-arm phase II trial (NCT05497843) for patients with advanced STS who have previously failed standard therapy or have no standard of care. Sufuratinib is being explored in multiple studies as a single agent (NCT05106777 and NCT06110650) or in combination with PD1/PDL1 inhibitors (NCT05839275 and NCT05722977).
MEK inhibitors
Inhibition of MEK resulted in the prevention of ERK phosphorylation, which halted tumor growth (188). MEK inhibitors are currently being developed and investigated as monotherapies or in combination with other targeted and cytotoxic agents for STS treatment (189). A recent approach involves combining MEK inhibitors with ICI to enhance immune recognition and augment T cell activity against neoplastic cells. The COTESARC trial (NCT04216953), a phase I/II study, is currently evaluating the efficacy of cobimetinib-atezolizumab combination in both adult and pediatric patients with advanced STS (190).
Furthermore, simultaneous inhibition of MEK and RAF kinase offers advantages in terms of increased efficacy and reduced adverse effects, potentially serving as a promising strategy for targeting the MARK pathway (191). Few cases have reported BRAF mutations and outcomes of BRAF-targeted therapy in various sarcoma types (16). A documented case of BRAF V600E-mutated undifferentiated sarcoma demonstrated successful treatment with a combination of BRAF and MEK inhibitors, suggesting that the BRAF V600E mutation could be a viable therapeutic target when addressed with dual BRAF and MEK inhibition (192).
Considering the high frequency of MAPK pathway abnormalities, particularly in MPNST, MEK inhibitor treatment may prove effective based on preclinical data, warranting further evaluation through clinical trials (193). A comprehensive study of multiple FDA-approved and promising MPNST therapies revealed that low-dose MEK inhibitors demonstrate the strongest synergy and efficacy when combined with other agents. This led to the hypothesis that MEK inhibitors may sensitize MPNST cells to other treatments, potentially rendering combination therapy more effective than mono-therapeutic approaches (194).
Recent research on NF1-deficient MPNST indicated that this subtype develops resistance to MEK inhibitor treatment partly by increasing PDGFRβ transcription and RAF dimer formation. A combination of MEK inhibitors and PDGFR/RAF-dimer inhibitors may overcome this resistance, offering a novel targeted therapeutic approach for NF1-deficient MPNST patients (195). In conclusion, MEK-targeting strategies are particularly relevant for developing effective combination therapies in MPNST treatment (196).
Targeting insulin growth factor receptor in non-GIST STS
The IGF/IGFR pathway is mainly implicated in the pathogenesis of some subtypes of bone and STS, such as osteosarcoma, RMS, and SS. An inhibitor of IGFR type-1 (IGF-1R) was assessed in a phase II trial and showed limited activity in refractory bone and STS. However, patients with RMS and osteosarcoma seem to have a clinical benefit from this drug (197). Given the disappointing findings with IGFR inhibitors as single agents, combinations should be considered to increase the antitumor effects. Translational data highlight the potential synergistic effect of dual targeting of IGFR and CDK4/6 in Ewing sarcoma (198). Exploration of this combination in the clinical setting is required. Similarly, preclinical data on Ewing sarcoma indicate the ability of combinatorial approaches with anti-IGF1R to promote antitumor activity when associated with mTOR inhibitors or chemotherapy (trabectedin) (199).
Kirsten rat sarcoma viral oncogene homolog inhibitors
The major trigger for the development of most cancer types is ERK cascade activation due to pathogenic mutations, particularly in RAS (200). KRAS is one of the most frequently mutated oncogenes in solid tumors, including STS. For many years, KRAS was considered an undruggable target; however, recent advancements have led to the development of selective inhibitors for KRAS G12C mutations, making it possible to target mutant KRAS cancers (201). Adargrasib (MRTX849) and sotorasib (AMG510) are two potent and highly selective KRAS G12C inhibitors (202). These two agents are currently under investigation in ongoing phase I/II clinical trials as single agents or in combination with other therapies for patients with KRAS G12C-mutant advanced solid tumors (NCT06024174 and NCT04185883). However, it is important to note that the KRAS G12C mutation was detected in only a small number of patients with KRAS mutations. Furthermore, data from both preclinical and clinical studies have demonstrated that several mechanisms of acquired resistance to anti-KRAS G12C monotherapies have been described, leading to the activation of alternative RAS-dependent pathways (203). This suggests that combination therapy is an important and prioritized strategy to increase the efficacy and delay the acquisition of drug resistance (204). Additionally, combination approaches and novel alternative targeting methods for RAS-driven cancers are still under clinical investigation (205, 206). Two main strategies for overcoming resistance mechanisms have been explored: vertical inhibition of multiple nodes in the RAS pathway and horizontal inhibition of parallel pathways (207).
MET inhibitors
The downstream response to c-MET activation directly promotes the activation of the RAS and MAPK cascades (208). Interestingly, it was recently shown that the HGF/c-MET signaling axis is implicated in tumor proliferation and metastatic spreading of cancer cells (209). One of the main downstream targets of the c-MET signaling axis is the MAPK: p38 and ERK1/2 pathways (210). A high expression of c-MET was observed in microphthalmia transcription factor-associated (MiT) tumors, including some STS subtypes (CCS and ASPS) (211). Targeting the MET axis is an attractive therapeutic option for these challenging histotypes. Preclinical findings suggest potential synergistic efficacy of a combination of HGF-targeted neutralizing antibodies with CAR-T cell treatments in Ewing sarcoma models (212). Other than multi-targeted TKIs with an activity on HGF/MET interactions, such as cabozantinib and crizotinib, new MET inhibitors are currently explored alone or in combination with EGFR and/or VEGFR inhibitors, particularly in MiT tumors (213).
Other TKIs
Molecular biology studies have improved our understanding of bone and STS pathogenesis, leading to the successful use of novel targeted drugs (214). Various growth factor signaling pathways, such as FGFR, RET, and CSF1R, have been identified in bone and STS (215, 216). Targeting of these oncogenic factors is an interesting therapeutic approach. Based on these data, several clinical trials are ongoing to investigate novel targeted agents, such as CPL304110 (FGFR inhibitor), selpercatinib (RET inhibitor), and pexidartinib (CSF-1R inhibitor) (Table 5).
Era of precision medicine
Recent advances in precision medicine and machine learning-based methods may therefore evolve the management of STS and respond to the urgent need to identify therapeutically targetable genomic and transcriptomic alterations to guide treatment and improve the clinical outcomes of these inherently challenging neoplasms (217, 218). To date, a multicenter, retrospective/prospective, translational study (PROGEN_SARC, NCT06076070) is ongoing to assess the feasibility of genomic profiling for therapeutic purposes in advanced or metastatic sarcomas after evaluation by the molecular tumor board. The results of this study are expected in May 2024. Another interesting randomized, multicenter, phase III trial (MULTISARC, NCT03784014) is ongoing to investigate the feasibility of using molecular profiling by NGS-exome in clinical practice for patients with advanced STS. In parallel, this trial evaluates the efficacy of targeted therapies recommended based on NGS analysis. The treatment strategy guided by NGS should be part of a list of 10 predefined therapeutic target inhibitors (nilotinib, ceritinib, capmatinib, lapatinib, trametinib, trametinib and dabrafenib, olaparib and durvalumab, palbociclib, glasdegib, TAS-120). The study is expected to be completed by October 2025.
Emerging nanotechnology strategies in sarcomas
Nanomedicine is a novel multidisciplinary field that is garnering significant interest and investigation. It is an emerging area experiencing rapid development and is considered a promising strategy for the diagnosis and treatment of cancer (219). Advancements in nanotechnology are enabling responses to tumor microenvironment (TME) changes or external stimuli, thereby improving precise drug release. This innovation significantly enhances targeting specificity and reduces adverse effects of cancer treatment (220).
Nanoparticles: a rising star for therapeutics and drug delivery in sarcomas
Despite significant advancements in tumor treatment, drug resistance and severe toxic side effects remain major challenges for clinicians in clinical practice. Nanoparticles (NPs) have revolutionized the diagnosis and treatment of cancers, addressing the limitations of conventional therapies by enhancing drug retention and permeability at tumor sites (221). NPs are categorized into two main classes: organic and inorganic. NPs are extensively applied in tissue engineering, tissue regeneration, drug-controlled release, and tumor immunotherapy due to their excellent biodegradability, large surface area, low cytotoxicity, and ease of modification (222). They provide advanced drug delivery capabilities, improving overall treatment efficacy through loading, targeting, and controlled release mechanisms, and overcoming the constraints of traditional methods.
Recently, NPs have been developed and tested for osteosarcoma, demonstrating potential applications in the diagnosis and treatment. Despite substantial progress in laboratory research, the clinical translation of nanomedicine still necessitates additional clinical trials and safety assessments (223).
Extracellular vesicles as a next-generation drug delivery platform
Extracellular vesicles (EVs) contain proteins, RNA, genomic DNA, non-coding RNAs, lipids, and metabolites. They are categorized into three types: exosomes, microvesicles, and apoptotic vesicles (224). EVs facilitate information transfer between cancer cells, immune cells, and the TME, eliciting functional responses in receptor cells, promoting phenotypic changes, and influencing their physiological state. They are implicated in processes such as antigen presentation, cell proliferation, metastasis, angiogenesis, inflammation, and apoptosis (225).
Noncoding RNAs (ncRNAs) in EVs from tumor cells can enhance tumor angiogenesis, immune evasion, metastasis, and drug resistance (226). Circular RNAs (circRNAs), a type of ncRNA with covalently closed-loop structures, regulate tumor cell proliferation, invasion, and apoptosis by modulating various genes and signaling pathways. CircRNAs, enriched in exosomes, play roles in tumorigenesis and chemoresistance, including the regulation of cisplatin sensitivity in osteosarcoma cells (227).
In summary, EV-based drug delivery demonstrates significant potential as an advanced nanomaterials for drug delivery and treatment. Their capacity to deliver biologically active substances to target cells renders EVs as promising natural nanocarriers for treating bone sarcomas (228).
Conclusion and future directions
The MAPK signaling pathway has a pivotal role in STS oncogenesis, acting as the foremost orchestrator of tumor cell responses to a diverse array of stimuli. This further opens the door to various components within this complex pathway to be identified as promising therapeutic targets for drug development. The growing focus on molecular therapies that inhibit the MAPK cascade signifies a substantial advancement in STS management. However, most current pharmaceuticals are not specific to MAPK nodes but rather kinase inhibitors or monoclonal antibodies that indirectly affect ERK/p38 activity. With numerous trials still in progress, it remains challenging to draw definitive conclusions regarding the role of MAPK in STS.
KRAS and MEK inhibitors offer more selective MAPK inhibition. Targeting KRAS is particularly promising due to the high frequency of KRAS mutations in non-GIST STS. Although substantial progress is anticipated in treating KRAS-mutant tumors, further research is necessary to elucidate resistance mechanisms and develop potential combination therapies. MEK inhibitors demonstrate potential in combination approaches for MPNST treatment, requiring clinical validation. These findings underscore the critical need to identify genetic and clinical indicators of response, resistance, toxicity, and optimal combination strategies for MAPK-targeted therapies in STS. Given the heterogeneous nature of STS pathology and clinical progression, single-agent targeted therapy has not yet demonstrated efficacy due to drug resistance. Vertical strategies targeting multiple MAPK pathway nodes may achieve more profound suppression. However, translational analysis is particularly challenging due to the high heterogeneity and rarity of non-GIST STS.
Recent advancements in genomic and epigenomic analyses of STS have facilitated the identification of novel alterations causally linked to the disease’s development. These alterations may be co-targeted with the MAPK axis in combination strategies to enhance therapeutic efficacy. Insights emphasize the next level of combined treatment in the era of precision medicine. The multi-targeted approach is used to prevent the emergence of resistance due to the activation of compensatory pathways associated with MAPK. This approach may become the cornerstone of new treatment combinations. To increase the likelihood of success, it is imperative to continue exploring diverse methodologies to further characterize STS at the molecular level, accelerate target discovery, and identify potential biomarkers. Additionally, the development of novel nanomaterials presents a promising avenue that may lead to breakthroughs in clinical practice.
Author contributions
OA: Conceptualization, Writing – original draft, Writing – review & editing. KE: Writing – review & editing. SAB: Writing – review & editing, Supervision. SA: Writing – review & editing, Supervision.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Conflict of interest
KE reports leadership roles and honoraria from NCODA, as well as expert testimony collaborations through Techspert.io (none in relation to this work).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tang F, Tie Y, Wei YQ, Tu CQ, and Wei XW. Targeted and immuno-based therapies in sarcoma: mechanisms and advances in clinical trials. Biochim Biophys Acta BBA - Rev Cancer. (2021) 1876:188606. doi: 10.1016/j.bbcan.2021.188606
2. Tan CJ, Yang JL, Crowe P, and Goldstein D. Targeted therapy in soft tissue sarcoma—a novel direction in therapeutics. Chin Clin Oncol. (2013) 2:22–2. doi: 10.3978/j.issn.2304-3865.2013.08.01
3. Fletcher CDM. The evolving classification of soft tissue tumours – an update based on the new 2013 WHO classification. Histopathology. (2014) 64:2–11. doi: 10.1111/his.2013.64.issue-1
4. Ramu EM, Houdek MT, Isaac CE, Dickie CI, Ferguson PC, and Wunder JS. Management of soft-tissue sarcomas; treatment strategies, staging, and outcomes. SICOT-J. (2017) 3:20. doi: 10.1051/sicotj/2017010
5. Tang S, Wang Y, Luo R, Fang R, Liu Y, Xiang H, et al. Proteomic characterization identifies clinically relevant subgroups of soft tissue sarcoma. Nat Commun. (2024) 15:1381. doi: 10.1038/s41467-024-45306-y
6. Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. (2012) 13:1133–40. doi: 10.1016/S1470-2045(12)70474-6
7. Gros L, Dei Tos AP, Jones RL, and Digklia A. Inflammatory myofibroblastic tumour: state of the art. Cancers. (2022) 14:3662. doi: 10.3390/cancers14153662
8. Kenerson H, Folpe AL, Takayama TK, and Yeung RS. Activation of the mTOR pathway in sporadic angiomyolipomas and other perivascular epithelioid cell neoplasms. Hum Pathol. (2007) 38:1361–71. doi: 10.1016/j.humpath.2007.01.028
9. Fujiwara T, Kunisada T, Nakata E, Nishida K, Yanai H, Nakamura T, et al. Advances in treatment of alveolar soft part sarcoma: an updated review. Jpn J Clin Oncol. (2023) 53:1009–18. doi: 10.1093/jjco/hyad102
10. Katz D, Palmerini E, and Pollack SM. More than 50 subtypes of soft tissue sarcoma: paving the path for histology-driven treatments. Am Soc Clin Oncol Educ Book. (2018) 38):925–38. doi: 10.1200/EDBK_205423
11. de Juan Ferré A, Álvarez Álvarez R, Casado Herráez A, Cruz Jurado J, Estival González A, Martín-Broto J, et al. SEOM Clinical Guideline of management of soft-tissue sarcoma (2020). Clin Transl Oncol. (2021) 23:922–30. doi: 10.1007/s12094-020-02534-0
12. Gamboa AC, Gronchi A, and Cardona K. Soft-tissue sarcoma in adults: An update on the current state of histiotype-specific management in an era of personalized medicine. CA Cancer J Clin. (2020) 70:200–29. doi: 10.3322/caac.21605
13. Alfagih A, AlJassim A, Alshamsan B, Alqahtani N, and Asmis T. Gastrointestinal stromal tumors: 10-year experience in cancer center—The ottawa hospital (TOH). Curr Oncol. (2022) 29:7148–57. doi: 10.3390/curroncol29100562
14. Blay JY, Hindi N, Bollard J, Aguiar S, Angel M, Araya B, et al. SELNET clinical practice guidelines for soft tissue sarcoma and GIST. Cancer Treat Rev. (2022) 102:102312. doi: 10.1016/j.ctrv.2021.102312
15. Tirumani SH, Jagannathan JP, O’Regan K, Kim KW, Shinagare AB, Krajewski KM, et al. Molecular targeted therapies in non-GIST soft tissue sarcomas: what the radiologist needs to know. Cancer Imaging. (2013) 13:197–211. doi: 10.1102/1470-7330.2013.0022
16. Jacobs C and Lapeire L. Translating molecular profiling of soft tissue sarcomas into daily clinical practice. Diagnostics. (2021) 11:512. doi: 10.3390/diagnostics11030512
17. Nacev BA, Sanchez-Vega F, Smith SA, Antonescu CR, Rosenbaum E, Shi H, et al. Clinical sequencing of soft tissue and bone sarcomas delineates diverse genomic landscapes and potential therapeutic targets. Nat Commun. (2022) 13:3405. doi: 10.1038/s41467-022-30453-x
18. Schipper LJ, Monkhorst K, Samsom KG, Bosch LJW, Snaebjornsson P, van Boven H, et al. Clinical impact of prospective whole genome sequencing in sarcoma patients. Cancers. (2022) 14:436. doi: 10.3390/cancers14020436
19. Damerell V, Pepper MS, and Prince S. Molecular mechanisms underpinning sarcomas and implications for current and future therapy. Signal Transduct Target Ther. (2021) 6:1–19. doi: 10.1038/s41392-021-00647-8
20. Sasaki K, Hitora T, Nakamura O, Kono R, and Yamamoto T. The role of MAPK pathway in bone and soft tissue tumors. Anticancer Res. (2011) 31:549–53.
21. Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, and Hu LL. ERK/MAPK signalling pathway and tumorigenesis (Review). Exp Ther Med. (2020) 19:1997–2007. doi: 10.3892/etm.2020.8454
22. Chen RE and Thorner J. Function and regulation in MAPK signaling pathways. Biochim Biophys Acta. (2007) 1773:1311–40. doi: 10.1016/j.bbamcr.2007.05.003
23. Morrison DK. MAP kinase pathways. Cold Spring Harb Perspect Biol. (2012) 4:a011254. doi: 10.1101/cshperspect.a011254
24. Zhang W and Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. (2002) 12:9–18. doi: 10.1038/sj.cr.7290105
25. Cargnello M and Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev MMBR. (2011) 75:50–83. doi: 10.1128/MMBR.00031-10
26. Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. (2001) 22:153–83. doi: 10.1210/edrv.22.2.0428
27. Coulombe P and Meloche S. Atypical mitogen-activated protein kinases: structure, regulation and functions. Biochim Biophys Acta. (2007) 1773:1376–87. doi: 10.1016/j.bbamcr.2006.11.001
28. Xia D, Tian S, Chen Z, Qin W, and Liu Q. miR302a inhibits the proliferation of esophageal cancer cells through the MAPK and PI3K/Akt signaling pathways. Oncol Lett. (2018) 15:3937–43. doi: 10.3892/ol.2018.7782
29. Deane AR, Potemkin N, and Ward RD. Mitogen-activated protein kinase (MAPK) signalling corresponds with distinct behavioural profiles in a rat model of maternal immune activation. Behav Brain Res. (2021) 396:112876. doi: 10.1016/j.bbr.2020.112876
30. Kassouf T and Sumara G. Impact of conventional and atypical MAPKs on the development of metabolic diseases. Biomolecules. (2020) 10:1256. doi: 10.3390/biom10091256
31. Spira AI and Ettinger DS. The use of chemotherapy in soft-tissue sarcomas. Oncologist. (2002) 7:348–59. doi: 10.1634/theoncologist.7-4-348
32. Serrano C, Romagosa C, Hernández-Losa J, Simonetti S, Valverde C, Moliné T, et al. RAS/MAPK pathway hyperactivation determines poor prognosis in undifferentiated pleomorphic sarcomas. Cancer. (2016) 122:99–107. doi: 10.1002/cncr.v122.1
33. Chandhanayingyong C, Kim Y, Staples JR, Hahn C, and Lee FY. MAPK/ERK signaling in osteosarcomas, ewing sarcomas and chondrosarcomas: therapeutic implications and future directions. Sarcoma. (2012) 2012:404810. doi: 10.1155/2012/404810
34. Dodd RD, Mito JK, Eward WC, Chitalia R, Sachdeva M, Ma Y, et al. NF1 deletion generates multiple subtypes of soft-tissue sarcoma that respond to MEK inhibition. Mol Cancer Ther. (2013) 12:1906–17. doi: 10.1158/1535-7163.MCT-13-0189
35. Skafida E, Kokkali S, Nikolaou M, and Digklia A. Metastatic soft tissue sarcoma: current treatment landscape and future perspectives. Expert Rev Anticancer Ther. (2017) 17:537–43. doi: 10.1080/14737140.2017.1321989
36. Yamnik RL and Holz MK. mTOR/S6K1 and MAPK/RSK signaling pathways coordinately regulate estrogen receptor alpha serine 167 phosphorylation. FEBS Lett. (2010) 584:124–8. doi: 10.1016/j.febslet.2009.11.041
37. Li H, Wozniak A, Sciot R, Cornillie J, Wellens J, Van Looy T, et al. Pazopanib, a receptor tyrosine kinase inhibitor, suppresses tumor growth through angiogenesis in dedifferentiated liposarcoma xenograft models. Transl Oncol. (2014) 7:665–71. doi: 10.1016/j.tranon.2014.09.007
38. Gril B, Palmieri D, Qian Y, Smart D, Ileva L, Liewehr DJ, et al. Pazopanib reveals a role for tumor cell B-Raf in the prevention of HER2+ breast cancer brain metastasis. Clin Cancer Res Off J Am Assoc Cancer Res. (2011) 17:142–53. doi: 10.1158/1078-0432.CCR-10-1603
39. Sleijfer S, Ray-Coquard I, Papai Z, Le Cesne A, Scurr M, Schöffski P, et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: A phase II study from the european organisation for research and treatment of cancer–soft tissue and bone sarcoma group (EORTC study 62043). J Clin Oncol. (2009) 27:3126–32. doi: 10.1200/JCO.2008.21.3223
40. van der Graaf WTA, Blay JY, Chawla SP, Kim DW, Bui-Nguyen B, Casali PG, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Lond Engl. (2012) 379:1879–86. doi: 10.1016/S0140-6736(12)60651-5
41. Cesne AL, Bauer S, Demetri GD, Han G, Dezzani L, Ahmad Q, et al. Safety and efficacy of Pazopanib in advanced soft tissue sarcoma: PALETTE (EORTC 62072) subgroup analyses. BMC Cancer. (2019) 19:794. doi: 10.1186/s12885-019-5988-3
42. Kasper B, Sleijfer S, Litière S, Marreaud S, Verweij J, Hodge RA, et al. Long-term responders and survivors on pazopanib for advanced soft tissue sarcomas: subanalysis of two European Organisation for Research and Treatment of Cancer (EORTC) clinical trials 62043 and 62072. Ann Oncol. (2014) 25:719–24. doi: 10.1093/annonc/mdt586
43. Lee ATJ, Jones RL, and Huang PH. Pazopanib in advanced soft tissue sarcomas. Signal Transduct Target Ther. (2019) 4:16. doi: 10.1038/s41392-019-0049-6
44. Somaiah N, Van Tine BA, Wahlquist AE, Milhem MM, Hill EG, Garrett-Mayer E, et al. A randomized, open-label, phase 2, multicenter trial of gemcitabine with pazopanib or gemcitabine with docetaxel in patients with advanced soft-tissue sarcoma. Cancer. (2021) 127:894–904. doi: 10.1002/cncr.v127.6
45. Cho HJ, Yun KH, Shin SJ, Lee YH, Kim SH, Baek W, et al. Durvalumab plus pazopanib combination in patients with advanced soft tissue sarcomas: a phase II trial. Nat Commun. (2024) 15:685. doi: 10.1038/s41467-024-44875-2
46. Subbiah V, Meyer C, Zinner R, Meric-Bernstam F, Zahurak M, O’Connor A, et al. Phase Ib/II study of the safety and efficacy of combination therapy with multikinase vascular endothelial growth factor inhibitor Pazopanib and MEK inhibitor Trametinib in advanced soft tissue sarcoma. Clin Cancer Res Off J Am Assoc Cancer Res. (2017) 23:4027–34. doi: 10.1158/1078-0432.CCR-17-0272
47. Dembla V, Groisberg R, Hess K, Fu S, Wheler J, Hong DS, et al. Outcomes of patients with sarcoma enrolled in clinical trials of pazopanib combined with histone deacetylase, mTOR, Her2, or MEK inhibitors. Sci Rep. (2017) 7:15963. doi: 10.1038/s41598-017-13114-8
48. Yi M, Jiao D, Qin S, Chu Q, Wu K, and Li A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol Cancer. (2019) 18:60. doi: 10.1186/s12943-019-0974-6
49. Stein EM, DeAngelo DJ, Chromik J, Chatterjee M, Bauer S, Lin CC, et al. Results from a first-in-human phase I study of siremadlin (HDM201) in patients with advanced wild-type TP53 solid tumors and acute leukemia. Clin Cancer Res. (2022) 28:870–81. doi: 10.1158/1078-0432.CCR-21-1295
50. Haertel N, Klag T, Hanfstein B, Mueller MC, Hehlmann R, Hochhaus A, et al. Imatinib treatment induces growth and survival promoting MAPK1/2 in hematopoietic cell lines expressing BCR-ABLT315I. Blood. (2007) 110:3460. doi: 10.1182/blood.V110.11.3460.3460
51. Chugh R, Wathen JK, Maki RG, Benjamin RS, Patel SR, Meyers PA, et al. Phase II multicenter trial of imatinib in 10 histologic subtypes of sarcoma using a bayesian hierarchical statistical model. J Clin Oncol Off J Am Soc Clin Oncol. (2009) 27:3148–53. doi: 10.1200/JCO.2008.20.5054
52. Ohishi J, Aoki M, Nabeshima K, Suzumiya J, Takeuchi T, Ogose A, et al. Imatinib mesylate inhibits cell growth of Malignant peripheral nerve sheath tumors in vitro and in vivothrough suppression of PDGFR-β. BMC Cancer. (2013) 13:224. doi: 10.1186/1471-2407-13-224
53. Frühwald MC, Biegel JA, Bourdeaut F, Roberts CWM, and Chi SN. Atypical teratoid/rhabdoid tumors-current concepts, advances in biology, and potential future therapies. Neuro-Oncol. (2016) 18:764–78. doi: 10.1093/neuonc/nov264
54. Chapagain U, Krigman HR, Hagemann IS, Weiss MC, and Sun L. COL1A1::PDGFB fusion-associated uterine sarcoma and response to Imatinib: A case report. Gynecol Oncol Rep. (2023) 49:101270. doi: 10.1016/j.gore.2023.101270
55. Sjöblom T, Shimizu A, O’Brien KP, Pietras K, Dal Cin P, Buchdunger E, et al. Growth inhibition of dermatofibrosarcoma protuberans tumors by the platelet-derived growth factor receptor antagonist STI571 through induction of apoptosis. Cancer Res. (2001) 61:5778–83.
56. Teicher BA, Polley E, Kunkel M, Evans D, Silvers T, Delosh R, et al. Sarcoma cell line screen of oncology drugs and investigational agents identifies patterns associated with gene and microRNA expression. Mol Cancer Ther. (2015) 14:2452–62. doi: 10.1158/1535-7163.MCT-15-0074
57. Chugh R, Wathen JK, Patel SR, Maki RG, Meyers PA, Schuetze SM, et al. Efficacy of imatinib in aggressive fibromatosis: Results of a phase II multicenter Sarcoma Alliance for Research through Collaboration (SARC) trial. Clin Cancer Res Off J Am Assoc Cancer Res. (2010) 16:4884–91. doi: 10.1158/1078-0432.CCR-10-1177
58. Penel N, Le Cesne A, Bui BN, Perol D, Brain EG, Ray-Coquard I, et al. Imatinib for progressive and recurrent aggressive fibromatosis (desmoid tumors): an FNCLCC/French Sarcoma Group phase II trial with a long-term follow-up. Ann Oncol Off J Eur Soc Med Oncol. (2011) 22:452–7. doi: 10.1093/annonc/mdq341
59. Kasper B, Gruenwald V, Reichardt P, Bauer S, Rauch G, Limprecht R, et al. Imatinib induces sustained progression arrest in RECIST progressive desmoid tumours: Final results of a phase II study of the German Interdisciplinary Sarcoma Group (GISG). Eur J Cancer Oxf Engl 1990. (2017) 76:60–7. doi: 10.1016/j.ejca.2017.02.001
60. Fields RC, Hameed M, Qin LX, Moraco N, Jia X, Maki RG, et al. Dermatofibrosarcoma protuberans (DFSP): predictors of recurrence and the use of systemic therapy. Ann Surg Oncol. (2011) 18:328–36. doi: 10.1245/s10434-010-1316-5
61. Wilding CP, Elms ML, Judson I, Tan AC, Jones RL, and Huang PH. The landscape of tyrosine kinase inhibitors in sarcomas: looking beyond pazopanib. Expert Rev Anticancer Ther. (2019) 19:971–91. doi: 10.1080/14737140.2019.1686979
62. Papaetis GS and Syrigos KN. Sunitinib. BioDrugs. (2009) 23:377–89. doi: 10.2165/11318860-000000000-00000
63. Fenton MS, Marion KM, Salem AK, Hogen R, Naeim F, and Hershman JM. Sunitinib inhibits MEK/ERK and SAPK/JNK pathways and increases sodium/iodide symporter expression in papillary thyroid cancer. Thyroid Off J Am Thyroid Assoc. (2010) 20:965–74. doi: 10.1089/thy.2010.0008
64. Wong JP, Todd JR, Finetti MA, McCarthy F, Broncel M, Vyse S, et al. Dual targeting of PDGFRα and FGFR1 displays synergistic efficacy in Malignant rhabdoid tumors. Cell Rep. (2016) 17:1265–75. doi: 10.1016/j.celrep.2016.10.005
65. Jo JC, Hong YS, Kim KP, Lee JL, Lee J, Park YS, et al. A prospective multicenter phase II study of sunitinib in patients with advanced aggressive fibromatosis. Invest New Drugs. (2014) 32:369–76. doi: 10.1007/s10637-013-0059-0
66. Jagodzińska-Mucha P, Świtaj T, Kozak K, Koseła-Paterczyk H, Klimczak A, Ługowska I, et al. Long-term results of therapy with sunitinib in metastatic alveolar soft part sarcoma. Tumori. (2017) 103:231–5. doi: 10.5301/tj.5000617
67. Stacchiotti S, Pantaleo MA, Astolfi A, Dagrada GP, Negri T, Dei Tos AP, et al. Activity of sunitinib in extraskeletal myxoid chondrosarcoma. Eur J Cancer Oxf Engl 1990. (2014) 50:1657–64. doi: 10.1016/j.ejca.2014.03.013
68. Stacchiotti S, Negri T, Libertini M, Palassini E, Marrari A, De Troia B, et al. Sunitinib malate in solitary fibrous tumor (SFT). Ann Oncol Off J Eur Soc Med Oncol. (2012) 23:3171–9. doi: 10.1093/annonc/mds143
69. Martin-Broto J, Hindi N, Grignani G, Martinez-Trufero J, Redondo A, Valverde C, et al. Nivolumab and sunitinib combination in advanced soft tissue sarcomas: a multicenter, single-arm, phase Ib/II trial. J Immunother Cancer. (2020) 8:e001561. doi: 10.1136/jitc-2020-001561
70. Tang W, Chen Z, Zhang W, Cheng Y, Zhang B, Wu F, et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects. Signal Transduct Target Ther. (2020) 5:87. doi: 10.1038/s41392-020-0187-x
71. Schut ARW, Vriends ALM, Sacchetti A, Timbergen MJM, Alman BA, Al-Jazrawe M, et al. In desmoid-type fibromatosis cells sorafenib induces ferroptosis and apoptosis, which are enhanced by autophagy inhibition. Eur J Surg Oncol J Eur Soc Surg Oncol Br Assoc Surg Oncol. (2022) 48:1527–35. doi: 10.1016/j.ejso.2022.02.020
72. Ambrosini G, Cheema HS, Seelman S, Teed A, Sambol EB, Singer S, et al. Sorafenib inhibits growth and mitogen-activated protein kinase signaling in Malignant peripheral nerve sheath cells. Mol Cancer Ther. (2008) 7:890–6. doi: 10.1158/1535-7163.MCT-07-0518
73. Abraham J, Chua YX, Glover JM, Tyner JW, Loriaux MM, Kilcoyne A, et al. An adaptive src-PDGFRA-raf axis in rhabdomyosarcoma. Biochem Biophys Res Commun. (2012) 426:363–8. doi: 10.1016/j.bbrc.2012.08.092
74. Ortega-Muelas M, Roche O, Fernández-Aroca DM, Encinar JA, Albandea-Rodríguez D, Arconada-Luque E, et al. ERK5 signalling pathway is a novel target of sorafenib: Implication in EGF biology. J Cell Mol Med. (2021) 25:10591–603. doi: 10.1111/jcmm.v25.22
75. Zhu YJ, Zheng B, Wang HY, and Chen L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin. (2017) 38:614–22. doi: 10.1038/aps.2017.5
76. Ray-Coquard I, Italiano A, Bompas E, Le Cesne A, Robin YM, Chevreau C, et al. Sorafenib for patients with advanced angiosarcoma: A phase II trial from the french sarcoma group (GSF/GETO). Oncologist. (2012) 17:260–6. doi: 10.1634/theoncologist.2011-0237
77. Valentin T, Fournier C, Penel N, Bompas E, Chaigneau L, Isambert N, et al. Sorafenib in patients with progressive Malignant solitary fibrous tumors: a subgroup analysis from a phase II study of the French Sarcoma Group (GSF/GETO). Invest New Drugs. (2013) 31:1626–7. doi: 10.1007/s10637-013-0023-z
78. Chevreau C, Le Cesne A, Ray-Coquard I, Italiano A, Cioffi A, Isambert N, et al. Sorafenib in patients with progressive epithelioid hemangioendothelioma: a phase 2 study by the French Sarcoma Group (GSF/GETO). Cancer. (2013) 119:2639–44. doi: 10.1002/cncr.v119.14
79. Gounder MM, Lefkowitz RA, Keohan ML, D’Adamo DR, Hameed M, Antonescu CR, et al. Activity of Sorafenib against desmoid tumor/deep fibromatosis. Clin Cancer Res Off J Am Assoc Cancer Res. (2011) 17:4082–90. doi: 10.1158/1078-0432.CCR-10-3322
80. Gounder MM, Mahoney MR, Van Tine BA, Ravi V, Attia S, Deshpande HA, et al. Sorafenib for advanced and refractory desmoid tumors. N Engl J Med. (2018) 379:2417–28. doi: 10.1056/NEJMoa1805052
81. Ettrich TJ and Seufferlein T. Regorafenib. Recent Results Cancer Res Fortschr Krebsforsch Progres Dans Rech Sur Cancer. (2018) 211:45–56. doi: 10.1007/978-3-319-91442-8_3
82. Subramonian D, Phanhthilath N, Rinehardt H, Flynn S, Huo Y, Zhang J, et al. Regorafenib is effective against neuroblastoma in vitro and in vivo and inhibits the RAS/MAPK, PI3K/Akt/mTOR and Fos/Jun pathways. Br J Cancer. (2020) 123:568–79. doi: 10.1038/s41416-020-0905-8
83. Sui H, Xiao S, Jiang S, Wu S, Lin H, Cheng L, et al. Regorafenib induces NOX5-mediated endoplasmic reticulum stress and potentiates the anti-tumor activity of cisplatin in non-small cell lung cancer cells. Neoplasia N Y N. (2023) 39:100897. doi: 10.1016/j.neo.2023.100897
84. Daudigeos-Dubus E, Le Dret L, Lanvers-Kaminsky C, Bawa O, Opolon P, Vievard A, et al. Regorafenib: antitumor activity upon mono and combination therapy in preclinical pediatric Malignancy models. PloS One. (2015) 10:e0142612. doi: 10.1371/journal.pone.0142612
85. Mir O, Brodowicz T, Italiano A, Wallet J, Blay JY, Bertucci F, et al. Safety and efficacy of regorafenib in patients with advanced soft tissue sarcoma (REGOSARC): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. (2016) 17:1732–42. doi: 10.1016/S1470-2045(16)30507-1
86. Grothey A, Blay JY, Pavlakis N, Yoshino T, and Bruix J. Evolving role of regorafenib for the treatment of advanced cancers. Cancer Treat Rev. (2020) 86:101993. doi: 10.1016/j.ctrv.2020.101993
87. Brodowicz T, Mir O, Wallet J, Italiano A, Blay JY, Bertucci F, et al. Efficacy and safety of regorafenib compared to placebo and to post-cross-over regorafenib in advanced non-adipocytic soft tissue sarcoma. Eur J Cancer Oxf Engl 1990. (2018) 99:28–36. doi: 10.1016/j.ejca.2018.05.008
88. Marrari A, Bertuzzi A, Bozzarelli S, Gennaro N, Giordano L, Quagliuolo V, et al. Activity of regorafenib in advanced pretreated soft tissue sarcoma. Med (Baltimore). (2020) 99:e20719. doi: 10.1097/MD.0000000000020719
89. Penel N, Mir O, Wallet J, Ray-Coquard I, Le Cesne A, Italiano A, et al. A double-blind placebo-controlled randomized phase II trial assessing the activity and safety of regorafenib in non-adipocytic sarcoma patients previously treated with both chemotherapy and pazopanib. Eur J Cancer Oxf Engl 1990. (2020) 126:45–55. doi: 10.1016/j.ejca.2019.12.001
90. Duffaud F, Mir O, Boudou-Rouquette P, Piperno-Neumann S, Penel N, Bompas E, et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: a non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. (2019) 20:120–33. doi: 10.1016/S1470-2045(18)30742-3
91. Cousin S, Bellera C, Guegan JP, Valentin T, Bahleda R, Metges JP, et al. 1494P Regomune - a phase II study of regorafenib + avelumab in solid tumors: Results of the soft tissue sarcoma (STS) cohort. Ann Oncol. (2022) 33:S1230. doi: 10.1016/j.annonc.2022.07.1597
92. Agulnik M and Attia S. Growing role of regorafenib in the treatment of patients with sarcoma. Target Oncol. (2018) 13:417–22. doi: 10.1007/s11523-018-0575-0
93. Chen Y, Tortorici MA, Garrett M, Hee B, Klamerus KJ, and Pithavala YK. Clinical pharmacology of axitinib. Clin Pharmacokinet. (2013) 52:713–25. doi: 10.1007/s40262-013-0068-3
94. Paik ES, Kim TH, Cho YJ, Ryu J, Choi JJ, Lee YY, et al. Preclinical assessment of the VEGFR inhibitor axitinib as a therapeutic agent for epithelial ovarian cancer. Sci Rep. (2020) 10:4904. doi: 10.1038/s41598-020-61871-w
95. Kerr LT, Donoghue JF, Wilding AL, and Johns TG. Axitinib has antiangiogenic and antitumorigenic activity in myxoid liposarcoma. Sarcoma. (2016) 2016:3484673. doi: 10.1155/2016/3484673
96. Stacchiotti S, Simeone N, Lo Vullo S, Morosi C, Greco FG, Gronchi A, et al. Activity of axitinib in progressive advanced solitary fibrous tumour: Results from an exploratory, investigator-driven phase 2 clinical study. Eur J Cancer Oxf Engl 1990. (2019) 106:225–33. doi: 10.1016/j.ejca.2018.10.024
97. Woll PJ, Gaunt P, Gaskell C, Young R, Benson C, Judson IR, et al. Axitinib in patients with advanced/metastatic soft tissue sarcoma (Axi-STS): an open-label, multicentre, phase II trial in four histological strata. Br J Cancer. (2023) 129:1490–9. doi: 10.1038/s41416-023-02416-6
98. Wilky BA, Trucco MM, Subhawong TK, Florou V, Park W, Kwon D, et al. Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: a single-centre, single-arm, phase 2 trial. Lancet Oncol. (2019) 20:837–48. doi: 10.1016/S1470-2045(19)30153-6
99. Heckman CA, Holopainen T, Wirzenius M, Keskitalo S, Jeltsch M, Ylä-Herttuala S, et al. The tyrosine kinase inhibitor cediranib blocks ligand-induced vascular endothelial growth factor receptor-3 activity and lymphangiogenesis. Cancer Res. (2008) 68:4754–62. doi: 10.1158/0008-5472.CAN-07-5809
100. Guo M, Liu Z, Si J, Zhang J, Zhao J, Guo Z, et al. Cediranib induces apoptosis, G1 phase cell cycle arrest, and autophagy in non-small-cell lung cancer cell A549 in vitro. BioMed Res Int. (2021) 2021:5582648. doi: 10.1155/2021/5582648
101. Morton CL, Maris JM, Keir ST, Gorlick R, Kolb EA, Billups CA, et al. Combination testing of cediranib (AZD2171) against childhood cancer models by the pediatric preclinical testing program. Pediatr Blood Cancer. (2012) 58:566–71. doi: 10.1002/pbc.23159
102. Kummar S, Allen D, Monks A, Polley EC, Hose CD, Ivy SP, et al. Cediranib for metastatic alveolar soft part sarcoma. J Clin Oncol. (2013) 31:2296–302. doi: 10.1200/JCO.2012.47.4288
103. Judson I, Morden JP, Kilburn L, Leahy M, Benson C, Bhadri V, et al. Cediranib in patients with alveolar soft-part sarcoma (CASPS): a double-blind, placebo-controlled, randomised, phase 2 trial. Lancet Oncol. (2019) 20:1023–34. doi: 10.1016/S1470-2045(19)30215-3
104. Schöffski P. SY20-2 - Exploring novel therapies in advanced soft tissue sarcoma: the path from multi-sarcoma to highly subtype-specific trials. Ann Oncol. (2019) 30:vi44. doi: 10.1093/annonc/mdz350
105. Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. (2008) 68:4774–82. doi: 10.1158/0008-5472.CAN-07-6307
106. Landi C, Carleo A, Vantaggiato L, Bergantini L, d'Alessandro M, Cameli P, et al. Common molecular pathways targeted by nintedanib in cancer and IPF: A bioinformatic study. Pulm Pharmacol Ther. (2020) 64:101941. C L, A C, L V, L BP C. doi: 10.1016/j.pupt.2020.101941
107. Hilberg F, Tontsch-Grunt U, Baum A, Le AT, Doebele RC, Lieb S, et al. Triple angiokinase inhibitor nintedanib directly inhibits tumor cell growth and induces tumor shrinkage via blocking oncogenic receptor tyrosine kinases. J Pharmacol Exp Ther. (2018) 364:494–503. doi: 10.1124/jpet.117.244129
108. Patwardhan PP, Musi E, and Schwartz GK. Preclinical evaluation of nintedanib, a triple angiokinase inhibitor, in soft-tissue sarcoma: potential therapeutic implication for synovial sarcoma. Mol Cancer Ther. (2018) 17:2329–40. doi: 10.1158/1535-7163.MCT-18-0319
109. Schöffski P, Toulmonde M, Estival A, Marquina G, Dudzisz-Śledź M, Brahmi M, et al. Randomised phase 2 study comparing the efficacy and safety of the oral tyrosine kinase inhibitor nintedanib with single agent ifosfamide in patients with advanced, inoperable, metastatic soft tissue sarcoma after failure of first-line chemotherapy: EORTC-1506-STBSG “ANITA. Eur J Cancer. (2021) 152:26–40. doi: 10.1016/j.ejca.2021.04.015
110. Shen G, Zheng F, Ren D, Du F, Dong Q, Wang Z, et al. Anlotinib: a novel multi-targeting tyrosine kinase inhibitor in clinical development. J Hematol OncolJ Hematol Oncol. (2018) 11:120. doi: 10.1186/s13045-018-0664-7
111. Ji Y and Qu P. Anlotinib suppresses growth and metastasis through MAPK signaling pathway in cisplatin-resistant ovarian cancer cells. J Clin Oncol. (2022) 40(16_suppl). doi: 10.1200/JCO.2022.40.16_suppl.e17542
112. Chen S, Gao D, Sun R, Bao J, Lu C, Zhang Z, et al. Anlotinib prove to be a potential therapy for the treatment of pulmonary fibrosis complicated with lung adenocarcinoma. Pulm Pharmacol Ther. (2023) 80:102202. doi: 10.1016/j.pupt.2023.102202
113. Tang L, Yu W, Wang Y, Li H, and Shen Z. Anlotinib inhibits synovial sarcoma by targeting GINS1: a novel downstream target oncogene in progression of synovial sarcoma. Clin Transl Oncol Off Publ Fed Span Oncol Soc Natl Cancer Inst Mex. (2019) 21:1624–33. doi: 10.1007/s12094-019-02090-2
114. Chi Y, Fang Z, Hong X, Yao Y, Sun P, Wang G, et al. Safety and efficacy of anlotinib, a multikinase angiogenesis inhibitor, in patients with refractory metastatic soft-tissue sarcoma. Clin Cancer Res Off J Am Assoc Cancer Res. (2018) 24:5233–8. doi: 10.1158/1078-0432.CCR-17-3766
115. Li S. Anlotinib: A novel targeted drug for bone and soft tissue sarcoma. Front Oncol. (2021) 11:664853. doi: 10.3389/fonc.2021.664853
116. Chi Y, Yao Y, Wang S, Huang G, Cai Q, Shang G, et al. Anlotinib for metastasis soft tissue sarcoma: A randomized, double-blind, placebo-controlled and multi-centered clinical trial. J Clin Oncol. (2018) 36:11503–3. doi: 10.1200/JCO.2018.36.15_suppl.11503
117. Van Tine BA, Chawla SP, Trent JC, Wilky BA, Chugh R, Chmielowski B, et al. A phase III study (APROMISS) of AL3818 (Catequentinib, Anlotinib) hydrochloride monotherapy in subjects with metastatic or advanced synovial sarcoma. J Clin Oncol. (2021) 39:11505–5. doi: 10.1200/JCO.2021.39.15_suppl.11505
118. Li T, Ye Z, Wei Y, Wang S, Liu Y, and Chen J. A phase II study of anlotinib in the first-line treatment of locally advanced or metastatic soft-tissue sarcoma: Updated results. J Clin Oncol. (2022) 40:e23559–9. doi: 10.1200/JCO.2022.40.16_suppl.e23559
119. Xu J, Xie L, Guo W, Tang X, Yang R, Yan T, et al. Anlotinib and irinotecan for advanced Ewing sarcoma after failure of standard therapy: A multicenter, two-cohort, single-arm, open label, phase Ib/II trial (NCT03416517). J Clin Oncol. (2019) 37:11012–2. doi: 10.1200/JCO.2019.37.15_suppl.11012
120. Xu B, Pan Q, Pan H, Li H, Li X, Chen J, et al. Anlotinib as a maintenance treatment for advanced soft tissue sarcoma after first-line chemotherapy (ALTER-S006): a multicentre, open-label, single-arm, phase 2 trial. eClinicalMedicine. (2023) 64:102240. doi: 10.1016/j.eclinm.2023.102240
121. Liu Z, Yao W, Zhao Y, Liu O, Zhang P, and Ge H. Efficacy and safety of anlotinib combined with liposomal doxorubicin followed by anlotinib maintenance in metastatic soft tissue sarcomas. Cancer Manag Res. (2021) 13:1009–16. doi: 10.2147/CMAR.S286322
122. Sun X, Xu J, Xie L, and Guo W. Effectiveness and tolerability of anlotinib plus PD-1 inhibitors for patients with previously treated metastatic soft-tissue sarcoma. Int J Gen Med. (2022) 15:7581–91. doi: 10.2147/IJGM.S379269
123. Bauer T, Cho BC, Heist R, Bazhenova L, Werner T, Goel S, et al. First-in-human phase 1/1b study to evaluate sitravatinib in patients with advanced solid tumors. Invest New Drugs. (2022) 40:990–1000. doi: 10.1007/s10637-022-01274-y
124. Patwardhan PP, Ivy KS, Musi E, de StanChina E, and Schwartz GK. Significant blockade of multiple receptor tyrosine kinases by MGCD516 (Sitravatinib), a novel small molecule inhibitor, shows potent anti-tumor activity in preclinical models of sarcoma. Oncotarget. (2016) 7:4093–109. doi: 10.18632/oncotarget.6547
125. Oza J, Doshi S, Lee SM, Van Tine BA, Choy E, Oppelt PJ, et al. A phase II trial of sitravatinib, a multireceptor tyrosine kinase inhibitor, in patients with advanced well-differentiated/dedifferentiated liposarcoma. J Clin Oncol. (2021) 39:11513–3. doi: 10.1200/JCO.2021.39.15_suppl.11513
126. Ingham M, Lee S, Van Tine BA, Choy E, Oza J, Doshi S, et al. A single-arm phase II trial of sitravatinib in advanced well-differentiated/dedifferentiated liposarcoma. Clin Cancer Res. (2023) 29:1031–9. doi: 10.1158/1078-0432.CCR-22-3351
127. Sahu A, Prabhash K, Noronha V, Joshi A, and Desai S. Crizotinib: A comprehensive review. South Asian J Cancer. (2013) 2:91–7. doi: 10.4103/2278-330X.110506
128. Oyama R, Takahashi M, Yoshida A, Sakumoto M, Takai Y, Kito F, et al. Generation of novel patient-derived CIC- DUX4 sarcoma xenografts and cell lines. Sci Rep. (2017) 7:4712. doi: 10.1038/s41598-017-04967-0
129. Megiorni F, McDowell HP, Camero S, Mannarino O, Ceccarelli S, Paiano M, et al. Crizotinib-induced antitumour activity in human alveolar rhabdomyosarcoma cells is not solely dependent on ALK and MET inhibition. J Exp Clin Cancer Res. (2015) 34:112. doi: 10.1186/s13046-015-0228-4
130. Schöffski P, Wozniak A, Kasper B, Aamdal S, Leahy MG, Rutkowski P, et al. Activity and safety of crizotinib in patients with alveolar soft part sarcoma with rearrangement of TFE3: European Organization for Research and Treatment of Cancer (EORTC) phase II trial 90101 “CREATE. Ann Oncol Off J Eur Soc Med Oncol. (2018) 29:758–65. doi: 10.1093/annonc/mdx774
131. Schöffski P, Wozniak A, Stacchiotti S, Rutkowski P, Blay JY, Lindner LH, et al. Activity and safety of crizotinib in patients with advanced clear-cell sarcoma with MET alterations: European Organization for Research and Treatment of Cancer phase II trial 90101 “CREATE. Ann Oncol Off J Eur Soc Med Oncol. (2017) 28:3000–8. doi: 10.1093/annonc/mdx527
132. Antonescu CR, Suurmeijer AJH, Zhang L, Sung YS, Jungbluth AA, Travis WD, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 gene fusions and rare novel RET rearrangement. Am J Surg Pathol. (2015) 39:957–67. doi: 10.1097/PAS.0000000000000404
133. van der Graaf WTA and Gelderblom H. New systemic therapy options for advanced sarcomas. Curr Treat Options Oncol. (2012) 13:306–17. doi: 10.1007/s11864-012-0196-2
134. Schöffski P, Kubickova M, Wozniak A, Blay JY, Strauss SJ, Stacchiotti S, et al. Long-term efficacy update of crizotinib in patients with advanced, inoperable inflammatory myofibroblastic tumour from EORTC trial 90101 CREATE. Eur J Cancer Oxf Engl 1990. (2021) 156:12–23. doi: 10.1016/j.ejca.2021.07.016
135. Hisaoka M, Ishida T, Kuo TT, Matsuyama A, Imamura T, Nishida K, et al. Clear cell sarcoma of soft tissue: a clinicopathologic, immunohistochemical, and molecular analysis of 33 cases. Am J Surg Pathol. (2008) 32:452–60. doi: 10.1097/PAS.0b013e31814b18fb
136. Davis IJ, McFadden AW, Zhang Y, Coxon A, Burgess TL, Wagner AJ, et al. Identification of the receptor tyrosine kinase c-Met and its ligand, Hepatocyte Growth Factor, as therapeutic targets in clear cell sarcoma. Cancer Res. (2010) 70:639–45. doi: 10.1158/0008-5472.CAN-09-1121
137. Yamada K, Ohno T, Aoki H, Semi K, Watanabe A, Moritake H, et al. EWS/ATF1 expression induces sarcomas from neural crest-derived cells in mice. J Clin Invest. (2013) 123:600–10. doi: 10.1172/JCI63572
138. Mae H, Outani H, Imura Y, Chijimatsu R, Inoue A, Kotani Y, et al. Targeting the clear cell sarcoma oncogenic driver fusion gene EWSR1::ATF1 by HDAC inhibition. Cancer Res Commun. (2023) 3:1152–65. doi: 10.1158/2767-9764.CRC-22-0518
139. Schöffski P, Wozniak A, Stacchiotti S, Rutkowski P, Blay JY, Lindner LH, et al. Activity and safety of crizotinib in patients with advanced clear-cell sarcoma with MET alterations: European Organization for Research and Treatment of Cancer phase II trial 90101 ‘CREATE.’. Ann Oncol. (2019) 30:344. doi: 10.1093/annonc/mdx823
140. Choi KM, Cho E, Bang G, Lee SJ, Kim B, Kim JH, et al. Activity-based protein profiling reveals potential dasatinib targets in gastric cancer. Int J Mol Sci. (2020) 21:9276. doi: 10.3390/ijms21239276
141. Aslam MI, Abraham J, Mansoor A, Druker BJ, Tyner JW, and Keller C. PDGFRβ reverses EphB4 signaling in alveolar rhabdomyosarcoma. Proc Natl Acad Sci U S A. (2014) 111:6383–8. doi: 10.1073/pnas.1403608111
142. Michels S, Trautmann M, Sievers E, Kindler D, Huss S, Renner M, et al. SRC signaling is crucial in the growth of synovial sarcoma cells. Cancer Res. (2013) 73:2518–28. doi: 10.1158/0008-5472.CAN-12-3023
143. Mukaihara K, Tanabe Y, Kubota D, Akaike K, Hayashi T, Mogushi K, et al. Cabozantinib and dastinib exert anti-tumor activity in alveolar soft part sarcoma. PloS One. (2017) 12:e0185321. doi: 10.1371/journal.pone.0185321
144. Schuetze SM, Bolejack V, Choy E, Ganjoo KN, Staddon AP, Chow WA, et al. Phase 2 study of dasatinib in patients with alveolar soft part sarcoma, chondrosarcoma, chordoma, epithelioid sarcoma, or solitary fibrous tumor. Cancer. (2017) 123:90–7. doi: 10.1002/cncr.v123.1
145. Xiao X, Garbutt CC, Hornicek F, Guo Z, and Duan Z. Advances in chromosomal translocations and fusion genes in sarcomas and potential therapeutic applications. Cancer Treat Rev. (2018) 63:61–70. doi: 10.1016/j.ctrv.2017.12.001
146. DuBois S and Demetri G. Markers of angiogenesis and clinical features in patients with sarcoma. Cancer. (2007) 109:813–9. doi: 10.1002/cncr.v109:5
147. Dantas-Barbosa C, Lesluyes T, Loarer FL, Chibon F, Treilleux I, Coindre JM, et al. Expression and role of TYRO3 and AXL as potential therapeutical targets in leiomyosarcoma. Br J Cancer. (2017) 117:1787–97. doi: 10.1038/bjc.2017.354
148. Kahen E, Yu D, Harrison DJ, Clark J, Hingorani P, Cubitt CL, et al. Identification of clinically achievable combination therapies in childhood rhabdomyosarcoma. Cancer Chemother Pharmacol. (2016) 78:313–23. doi: 10.1007/s00280-016-3077-8
149. Fioramonti M, Fausti V, Pantano F, Iuliani M, Ribelli G, Lotti F, et al. Cabozantinib affects osteosarcoma growth through A direct effect on tumor cells and modifications in bone microenvironment. Sci Rep. (2018) 8:4177. doi: 10.1038/s41598-018-22469-5
150. Assi A, Farhat M, Hachem MCR, Zalaquett Z, Aoun M, Daher M, et al. Tyrosine kinase inhibitors in osteosarcoma: Adapting treatment strategiesa. J Bone Oncol. (2023) 43:100511. doi: 10.1016/j.jbo.2023.100511
151. Schöffski P, Blay JY, and Ray-Coquard I. Cabozantinib as an emerging treatment for sarcoma. Curr Opin Oncol. (2020) 32:321–31. doi: 10.1097/CCO.0000000000000644
152. Chuk MK, Widemann BC, Minard CG, Liu X, Kim A, Bernhardt MB, et al. A phase 1 study of cabozantinib in children and adolescents with recurrent or refractory solid tumors, including CNS tumors: trial ADVL1211, A report from the children’s oncology group. Pediatr Blood Cancer. (2018) 65:e27077. doi: 10.1002/pbc.27077
153. Italiano A, Mir O, Mathoulin-Pelissier S, Penel N, Piperno-Neumann S, Bompas E, et al. Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): a multicentre, single-arm, phase 2 trial. Lancet Oncol. (2020) 21:446–55. doi: 10.1016/S1470-2045(19)30825-3
154. Maroto P, Porta C, Capdevila J, Apolo AB, Viteri S, Rodriguez-Antona C, et al. Cabozantinib for the treatment of solid tumors: a systematic review. Ther Adv Med Oncol. (2022) 14:17588359221107112. doi: 10.1177/17588359221107112
155. Akshintala S, Widemann BC, Barkauskas DA, Hall D, Reid JM, Voss SD, et al. Phase 2 trial of cabozantinib in children and young adults with refractory sarcomas, Wilms tumor, and rare tumors: Children’s Oncology Group Study (ADVL1622). J Clin Oncol. (2021) 39:10010–0. doi: 10.1200/JCO.2021.39.15_suppl.10010
156. Ikeda S, Kudoh K, Sasaki N, Takano M, Goto T, Kikuchi R, et al. Synergistic effects of cabozantinib to temozolomide and bevacizumab in patients with heavily pretreated relapsed uterine leiomyosarcoma. J Clin Oncol. (2015) 33:5590–0. doi: 10.1200/jco.2015.33.15_suppl.5590
157. Grilley-Olson JE, Allred JB, Schuetze S, Davis EJ, Wagner MJ, Poklepovic AS, et al. A multicenter phase II study of cabozantinib + nivolumab for patients (pts) with advanced angiosarcoma (AS) previously treated with a taxane (Alliance A091902). J Clin Oncol. (2023) 41:11503–3. doi: 10.1200/JCO.2023.41.16_suppl.11503
158. Eulo VA, Wilky BA, Luo J, Hirbe AC, Weiss MC, Oppelt PJ, et al. A randomized phase II trial of cabozantinib combined with PD-1 and CTLA-4 inhibition in metastatic soft tissue sarcoma. J Clin Oncol. (2021) 39:TPS11583–TPS11583. doi: 10.1200/JCO.2021.39.15_suppl.TPS11583
159. Ray-Coquard I, Hatcher H, Bompas E, Casado A, Westermann A, Isambert N, et al. A randomized double-blind phase II study evaluating the role of maintenance therapy with cabozantinib in high-grade uterine sarcoma after stabilization or response to doxorubicin ± ifosfamide following surgery or in metastatic first line treatment (EORTC62113). Int J Gynecol Cancer Off J Int Gynecol Cancer Soc. (2020) 30:1633–7. doi: 10.1136/ijgc-2020-001519
160. Zhang H. Apatinib for molecular targeted therapy in tumor. Drug Des Devel Ther. (2015) 9:6075–81. doi: 10.2147/DDDT.S97235
161. Yu X, Fan H, Jiang X, Zheng W, Yang Y, Jin M, et al. Apatinib induces apoptosis and autophagy via the PI3K/AKT/mTOR and MAPK/ERK signaling pathways in neuroblastoma. Oncol Lett. (2020) 20:52. doi: 10.3892/ol.2020.11913
162. Hu Y, Jing J, Shi Y, Zhang P, Dong D, Wu Y, et al. Apatinib inhibits pancreatic cancer growth, migration and invasion through the PI3K/AKT and ERK1/2/MAPK pathways. Transl Cancer Res. (2021) 10:3306–16. doi: 10.21037/tcr-21-207
163. Liu X, Xu J, Li F, Liao Z, Ren Z, Zhu L, et al. Efficacy and safety of the VEGFR2 inhibitor Apatinib for metastatic soft tissue sarcoma: Chinese cohort data from NCT03121846. BioMed Pharmacother Biomedecine Pharmacother. (2020) 122:109587. doi: 10.1016/j.biopha.2019.109587
164. Zhu B, Li J, Xie Q, Diao L, Gai L, and Yang W. Efficacy and safety of apatinib monotherapy in advanced bone and soft tissue sarcoma: An observational study. Cancer Biol Ther. (2018) 19:198–204. doi: 10.1080/15384047.2017.1416275
165. Xie L, Xu J, Sun X, Tang X, Yan T, Yang R, et al. Apatinib for advanced osteosarcoma after failure of standard multimodal therapy: an open label phase II clinical trial. Oncologist. (2019) 24:e542–50. doi: 10.1634/theoncologist.2018-0542
166. Xie L, Xu J, Sun X, Li X, Liu K, Liang X, et al. Apatinib plus ifosfamide and etoposide for relapsed or refractory osteosarcoma: A retrospective study in two centres. Oncol Lett. (2021) 22:552. doi: 10.3892/ol.2021.12813
167. Xie L, Xu J, Sun X, Guo W, Gu J, Liu K, et al. Apatinib plus camrelizumab (anti-PD1 therapy, SHR-1210) for advanced osteosarcoma (APFAO) progressing after chemotherapy: a single-arm, open-label, phase 2 trial. J Immunother Cancer. (2020) 8:e000798. doi: 10.1136/jitc-2020-000798
168. Wu CH, Hsu FT, Chao TL, Lee YH, and Kuo YC. Revealing the suppressive role of protein kinase C delta and p38 mitogen-activated protein kinase (MAPK)/NF-κB axis associates with lenvatinib-inhibited progression in hepatocellular carcinoma in vitro and in vivo. BioMed Pharmacother Biomedecine Pharmacother. (2022) 145:112437. doi: 10.1016/j.biopha.2021.112437
169. Chen TWW, Hsu CL, Hong RL, Lee JC, Chang K, Yu CW, et al. A single-arm phase ib/II study of lenvatinib plus eribulin in advanced liposarcoma and leiomyosarcoma. Clin Cancer Res Off J Am Assoc Cancer Res. (2022) 28:5058–65. doi: 10.1158/1078-0432.CCR-22-2092
170. Movva S, Avutu V, Chi P, Dickson MA, Gounder MM, Kelly CM, et al. A pilot study of lenvatinib plus pembrolizumab in patients with advanced sarcoma. J Clin Oncol. (2023) 41:11517–7. doi: 10.1200/JCO.2023.41.16_suppl.11517
171. Davis EJ and Chugh R. Spotlight on olaratumab in the treatment of soft-tissue sarcoma: design, development, and place in therapy. Drug Des Devel Ther. (2017) 11:3579–87. doi: 10.2147/DDDT.S121298
172. Higuchi T, Sugisawa N, Miyake K, Oshiro H, Yamamoto N, Hayashi K, et al. The combination of olaratumab with doxorubicin and cisplatinum regresses a chemotherapy-resistant osteosarcoma in a patient-derived orthotopic xenograft mouse model. Transl Oncol. (2019) 12:1257–63. doi: 10.1016/j.tranon.2019.06.002
173. Chiorean EG, Sweeney C, Youssoufian H, Qin A, Dontabhaktuni A, Loizos N, et al. A phase I study of olaratumab, an anti-platelet-derived growth factor receptor alpha (PDGFRα) monoclonal antibody, in patients with advanced solid tumors. Cancer Chemother Pharmacol. (2014) 73:595–604. doi: 10.1007/s00280-014-2389-9
174. Doi T, Ma Y, Dontabhaktuni A, Nippgen C, Nippgen J, and Ohtsu A. Phase I study of olaratumab in Japanese patients with advanced solid tumors. Cancer Sci. (2014) 105:862–9. doi: 10.1111/cas.2014.105.issue-7
175. Tap WD, Jones RL, Van Tine BA, Chmielowski B, Elias AD, Adkins D, et al. Olaratumab and doxorubicin versus doxorubicin alone in soft tissue sarcoma. Lancet Lond Engl. (2016) 388:488–97. doi: 10.1016/S0140-6736(16)30587-6
176. Moore DC and Lavery LA. Olaratumab: A new strategy in the treatment of advanced soft-tissue sarcoma. J Adv Pract Oncol. (2018) 9:235–40.
177. Tap WD, Wagner AJ, Schöffski P, Martin-Broto J, Krarup-Hansen A, Ganjoo KN, et al. Effect of doxorubicin plus olaratumab vs doxorubicin plus placebo on survival in patients with advanced soft tissue sarcomas: the ANNOUNCE randomized clinical trial. JAMA. (2020) 323:1266–76. doi: 10.1001/jama.2020.1707
178. Zerdan MB, Bidikian AH, Alameh I, Nakib CE, and Assi HI. Olaratumab’s failure in soft tissue sarcoma. Rare Tumors. (2021) 13:20363613211034115. doi: 10.1177/20363613211034115
179. Attia S, Villalobos V, Hindi N, Wagner AJ, Chmielowski B, Oakley GJ, et al. Randomized phase 2 clinical trial of olaratumab in combination with gemcitabine and docetaxel in advanced soft tissue sarcomas. Cancers. (2023) 15:4871. doi: 10.3390/cancers15194871
180. Schöffski P, Bahleda R, Wagner AJ, Burgess MA, Junker N, Chisamore M, et al. Results of an open-label, phase ia/b study of pembrolizumab plus olaratumab in patients with unresectable, locally advanced, or metastatic soft-tissue sarcoma. Clin Cancer Res. (2023) 29:3320–8. doi: 10.1158/1078-0432.CCR-23-0742
181. Goetz EM and Garraway LA. Mechanisms of resistance to mitogen-activated protein kinase pathway inhibition in BRAF-mutant melanoma. Am Soc Clin Oncol Educ Book. (2012) 32):680–4. doi: 10.14694/EdBook_AM.2012.32.189
182. Lee S, Rauch J, and Kolch W. Targeting MAPK signaling in cancer: mechanisms of drug resistance and sensitivity. Int J Mol Sci. (2020) 21:1102. doi: 10.3390/ijms21031102
183. Liu Q, Yu S, Zhao W, Qin S, Chu Q, and Wu K. EGFR-TKIs resistance via EGFR-independent signaling pathways. Mol Cancer. (2018) 17:53. doi: 10.1186/s12943-018-0793-1
184. Strizova Z and Ozaniak A. 100P Combinatorial treatment strategies for overcoming the immunotherapy resistance in soft tissue sarcomas. ESMO Open. (2023) 8. doi: 10.1016/j.esmoop.2023.101137
185. Chan SPY, Rashid MBMA, Lim JJ, Goh JJN, Wong WY, Hooi L, et al. Functional combinatorial precision medicine for predicting and optimizing soft tissue sarcoma treatments. NPJ Precis Oncol. (2025) 9:1–16. doi: 10.1038/s41698-025-00851-7
186. Kyriazoglou A, Gkaralea LE, Kotsantis I, Anastasiou M, Pantazopoulos A, Prevezanou M, et al. Tyrosine kinase inhibitors in sarcoma treatment. Oncol Lett. (2022) 23:183. doi: 10.3892/ol.2022.13303
187. Rocchi L, Caraffi S, Perris R, and Mangieri D. The angiogenic asset of soft tissue sarcomas: a new tool to discover new therapeutic targets. Biosci Rep. (2014) 34:e00147. doi: 10.1042/BSR20140075
188. Mahapatra DK, Asati V, and Bharti SK. MEK inhibitors in oncology: a patent review (2015-Present). Expert Opin Ther Pat. (2017) 27:887–906. doi: 10.1080/13543776.2017.1339688
189. Cheng Y and Tian H. Current development status of MEK inhibitors. Mol Basel Switz. (2017) 22:1551. doi: 10.3390/molecules22101551
190. Dufresne A, Brahmi M, Cropet C, Bahleda R, Watson S, Pautier P, et al. 59MO COTESARC – A multicentre, phase I-II clinical trial evaluating the combination of MEK and PDL-1 inhibitors in patients (pts) with advanced soft tissue sarcoma (STS): Results from complex (CG) and simple genomic (SG) STS cohorts. ESMO Open. (2024) 9. doi: 10.1016/j.esmoop.2024.102449
191. Liu H, Nazmun N, Hassan S, Liu X, and Yang J. BRAF mutation and its inhibitors in sarcoma treatment. Cancer Med. (2020) 9:4881–96. doi: 10.1002/cam4.v9.14
192. Saijo K, Imai H, Katayama H, Fujishima F, Nakamura K, Kasahara Y, et al. BRAF and MEK inhibitor treatment for metastatic undifferentiated sarcoma of the spermatic cord with BRAF V600E mutation. Case Rep Oncol. (2022) 15:762–9. doi: 10.1159/000526018
193. Nagabushan S, Lau LMS, Barahona P, Wong M, Sherstyuk A, Marshall GM, et al. Efficacy of MEK inhibition in a recurrent Malignant peripheral nerve sheath tumor. NPJ Precis Oncol. (2021) 5:9. doi: 10.1038/s41698-021-00145-8
194. González-Muñoz T, Kim A, Ratner N, and Peinado H. The need for new treatments targeting MPNST: the potential of strategies combining MEK inhibitors with antiangiogenic agents. Clin Cancer Res Off J Am Assoc Cancer Res. (2022) 28:3185–95. doi: 10.1158/1078-0432.CCR-21-3760
195. Miranda-Román MA, Lee CJ, Fishinevich E, Ran L, Patel AJ, Yan J, et al. MEK inhibitors lead to PDGFR pathway upregulation and sensitize tumors to RAF dimer inhibitors in NF1-deficient Malignant peripheral nerve sheath tumor. Clin Cancer Res Off J Am Assoc Cancer Res. (2024) 30:5154–65. doi: 10.1158/1078-0432.CCR-24-1750
196. Ortega-Bertran S, Fernández-Rodríguez J, Magallón-Lorenz M, Zhang X, Creus-Bachiller E, Diazgranados AP, et al. Triple combination of MEK, BET, and CDK inhibitors significantly reduces human Malignant peripheral nerve sheath tumors in mouse models. Clin Cancer Res Off J Am Assoc Cancer Res. (2024) 31(5):907–20. doi: 10.1158/1078-0432.c.7700607
197. Pappo AS, Vassal G, Crowley JJ, Bolejack V, Hogendoorn PCW, Chugh R, et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a Sarcoma Alliance for Research Through Collaboration study. Cancer. (2014) 120:2448–56. doi: 10.1002/cncr.v120.16
198. Guenther LM, Dharia NV, Ross L, Conway A, Robichaud AL, Catlett JL II, et al. A combination CDK4/6 and IGF1R inhibitor strategy for Ewing sarcoma. Clin Cancer Res Off J Am Assoc Cancer Res. (2019) 25:1343–57. doi: 10.1158/1078-0432.CCR-18-0372
199. Mancarella C, Morrione A, and Scotlandi K. Unraveling the IGF system interactome in sarcomas exploits novel therapeutic options. Cells. (2021) 10:2075. doi: 10.3390/cells10082075
200. Huang L, Guo Z, Wang F, and Fu L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther. (2021) 6:1–20. doi: 10.1038/s41392-021-00780-4
201. Zhu C, Guan X, Zhang X, Luan X, Song Z, Cheng X, et al. Targeting KRAS mutant cancers: from druggable therapy to drug resistance. Mol Cancer. (2022) 21:159. doi: 10.1186/s12943-022-01629-2
202. Wu X, Song W, Cheng C, Liu Z, Li X, Cui Y, et al. Small molecular inhibitors for KRAS-mutant cancers. Front Immunol. (2023) 14:1223433. doi: 10.3389/fimmu.2023.1223433
203. Cheema PK, Banerji SO, Blais N, Chu QSC, Juergens RA, Leighl NB, et al. Canadian consensus recommendations on the management of KRAS G12C-mutated NSCLC. Curr Oncol Tor Ont. (2023) 30:6473–96. doi: 10.3390/curroncol30070476
204. Perurena N, Situ L, and Cichowski K. Combinatorial strategies to target RAS-driven cancers. Nat Rev Cancer. (2024) 24:316–37. doi: 10.1038/s41568-024-00679-6
205. Singhal A, Li BT, and O’Reilly EM. Targeting KRAS in cancer. Nat Med. (2024) 30:969–83. doi: 10.1038/s41591-024-02903-0
206. Indini A, Rijavec E, Ghidini M, Cortellini A, and Grossi F. Targeting KRAS in solid tumors: current challenges and future opportunities of novel KRAS inhibitors. Pharmaceutics. (2021) 13:653. doi: 10.3390/pharmaceutics13050653
207. Punekar SR, Velcheti V, Neel BG, and Wong KK. The current state of the art and future trends in RAS-targeted cancer therapies. Nat Rev Clin Oncol. (2022) 19:637–55. doi: 10.1038/s41571-022-00671-9
208. Fu J, Su X, Li Z, Deng L, Liu X, Feng X, et al. HGF/c-MET pathway in cancer: from molecular characterization to clinical evidence. Oncogene. (2021) 40:4625–51. doi: 10.1038/s41388-021-01863-w
209. Organ SL and Tsao MS. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. (2011) 3:S7–19. doi: 10.1177/1758834011422556
210. Chen W, Wu S, Huang Y, Zhang T, Dong H, Zheng X, et al. A c-Met Inhibitor Suppresses Osteosarcoma Progression via the ERK1/2 Pathway in Human Osteosarcoma Cells. OncoTargets Ther. (2021) 14:4791–804. doi: 10.2147/OTT.S317122
211. Quesada J and Amato R. The molecular biology of soft-tissue sarcomas and current trends in therapy. Sarcoma. (2012) 2012:e849456. doi: 10.1155/2012/849456
212. Charan M, Dravid P, Cam M, Audino A, Gross AC, Arnold MA, et al. GD2-directed CAR-T cells in combination with HGF-targeted neutralizing antibody (AMG102) prevent primary tumor growth and metastasis in Ewing sarcoma. Int J Cancer. (2020) 146:3184–95. doi: 10.1002/ijc.v146.11
213. Wagner AJ, Goldberg JM, Dubois SG, Choy E, Rosen L, Pappo A, et al. Tivantinib (ARQ 197), a selective inhibitor of MET, in patients with microphthalmia transcription factor-associated tumors: results of a multicenter phase 2 trial. Cancer. (2012) 118:5894–902. doi: 10.1002/cncr.v118.23
214. Zhang Q, Yang Y, You X, Ju Y, Zhang Q, Sun T, et al. Comprehensive genomic analysis of primary bone sarcomas reveals different genetic patterns compared with soft tissue sarcomas. Front Oncol. (2023) 13:1173275. doi: 10.3389/fonc.2023.1173275
215. Napolitano A, Ostler AE, Jones RL, and Huang PH. Fibroblast growth factor receptor (FGFR) signaling in GIST and soft tissue sarcomas. Cells. (2021) 10:1533. doi: 10.3390/cells10061533
216. Wen J, Wang S, Guo R, and Liu D. CSF1R inhibitors are emerging immunotherapeutic drugs for cancer treatment. Eur J Med Chem. (2023) 245:114884. doi: 10.1016/j.ejmech.2022.114884
217. Crombé A, Cousin S, Spalato-Ceruso M, Le Loarer F, Toulmonde M, Michot A, et al. Implementing a machine learning strategy to predict pathologic response in patients with soft tissue sarcomas treated with neoadjuvant chemotherapy. JCO Clin Cancer Inform. (2021) 5):958–72. doi: 10.1200/CCI.21.00062
218. Crombé A, Spinnato P, Italiano A, Brisse HJ, Feydy A, Fadli D, et al. Radiomics and artificial intelligence for soft-tissue sarcomas: Current status and perspectives. Diagn Interv Imaging. (2023) 104:567–83. doi: 10.1016/j.diii.2023.09.005
219. Wang B, Hu S, Teng Y, Chen J, Wang H, Xu Y, et al. Current advance of nanotechnology in diagnosis and treatment for Malignant tumors. Signal Transduct Target Ther. (2024) 9:200. doi: 10.1038/s41392-024-01889-y
220. Moradi Kashkooli F, Jakhmola A, Hornsby TK, Tavakkoli JJ, and Kolios MC. Ultrasound-mediated nano drug delivery for treating cancer: Fundamental physics to future directions. J Control Release Off J Control Release Soc. (2023) 355:552–78. doi: 10.1016/j.jconrel.2023.02.009
221. Zhu X and Li S. Nanomaterials in tumor immunotherapy: new strategies and challenges. Mol Cancer. (2023) 22:94. doi: 10.1186/s12943-023-01797-9
222. Pei Z, Lei H, and Cheng L. Bioactive inorganic nanomaterials for cancer theranostics. Chem Soc Rev. (2023) 52:2031–81. doi: 10.1039/D2CS00352J
223. Guan Y, Zhang W, Mao Y, and Li S. Nanoparticles and bone microenvironment: a comprehensive review for Malignant bone tumor diagnosis and treatment. Mol Cancer. (2024) 23:246. doi: 10.1186/s12943-024-02161-1
224. Kim HI, Park J, Zhu Y, Wang X, Han Y, and Zhang D. Recent advances in extracellular vesicles for therapeutic cargo delivery. Exp Mol Med. (2024) 56:836–49. doi: 10.1038/s12276-024-01201-6
225. Gao X, Gao B, and Li S. Extracellular vesicles: A new diagnostic biomarker and targeted drug in osteosarcoma. Front Immunol. (2022) 13:1002742. doi: 10.3389/fimmu.2022.1002742
226. Guo X, Gao C, Yang DH, and Li S. Exosomal circular RNAs: A chief culprit in cancer chemotherapy resistance. Drug Resist Update Rev Comment Antimicrob Anticancer Chemother. (2023) 67:100937. doi: 10.1016/j.drup.2023.100937
227. Li S, Liu F, Zheng K, Wang W, Qiu E, Pei Y, et al. CircDOCK1 promotes the tumorigenesis and cisplatin resistance of osteogenic sarcoma via the miR-339-3p/IGF1R axis. Mol Cancer. (2021) 20:161. doi: 10.1186/s12943-021-01453-0
Keywords: soft tissue sarcomas, MAPK pathway, targeted therapy, precision medicine, clinical trials
Citation: Al Jarroudi O, El Bairi K, Brahmi SA and Afqir S (2025) MAPK-targeted therapies in non-gastrointestinal stromal tumor soft tissue sarcomas: current landscape and future directions. Front. Oncol. 15:1418537. doi: 10.3389/fonc.2025.1418537
Received: 16 April 2024; Accepted: 17 June 2025;
Published: 20 August 2025.
Edited by:
Nicolas N. Nassar, Cincinnati Children’s Hospital Medical Center, United StatesReviewed by:
Aixi Yu, Wuhan University, ChinaShenglong Li, Dalian University of Technology, China
Angelina Vaseva, The University of Texas Health Science Center at San Antonio, United States
Copyright © 2025 Al Jarroudi, El Bairi, Brahmi and Afqir. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ouissam Al Jarroudi, YWxqYXJyb3VkaS5vdWlzc2FtQGdtYWlsLmNvbQ==
†ORCID: Ouissam Al Jarroudi, orcid.org/0000-0001-9946-921X