 Jerillyn S. Kent
Jerillyn S. Kent Amanda R. Bolbecker
Amanda R. Bolbecker Brian F. O’Donnell
Brian F. O’Donnell William P. Hetrick
William P. Hetrick- 1Department of Psychological and Brain Sciences, Indiana University, Bloomington, IN, USA
- 2Minneapolis Veterans Affairs Health Care System, Minneapolis, MN, USA
- 3Department of Psychiatry, Indiana University School of Medicine, Indianapolis, IN, USA
- 4Larue D. Carter Memorial Hospital, Indianapolis, IN, USA
There is accruing evidence of cerebellar abnormalities in schizophrenia. The theory of cognitive dysmetria considers cerebellar dysfunction a key component of schizophrenia. Delay eyeblink conditioning (EBC), a cerebellar-dependent translational probe, is a behavioral index of cerebellar integrity. The circuitry underlying EBC has been well characterized by non-human animal research, revealing the cerebellum as the essential circuitry for the associative learning instantiated by this task. However, there have been persistent inconsistencies in EBC findings in schizophrenia. This article thoroughly reviews published studies investigating EBC in schizophrenia, with an emphasis on possible effects of antipsychotic medication and stimulus and analysis parameters on reports of EBC performance in schizophrenia. Results indicate a consistent finding of impaired EBC performance in schizophrenia, as measured by decreased rates of conditioning, and that medication or study design confounds do not account for this impairment. Results are discussed within the context of theoretical and neurochemical models of schizophrenia.
Introduction
Growing empirical evidence suggests cerebellar abnormalities in schizophrenia. In terms of cerebellar morphology, imaging studies report reduced cerebellar volume in chronic (1–4), neuroleptic-naïve (5), adolescent (6), first-episode (7–9), and childhood-onset (10) schizophrenia [for exceptions see Ref. (11, 12)]. Postmortem studies have also found reduced size and density of Purkinje cells in schizophrenia (13–15). In addition to structure, cerebellar function has also been reported to be abnormal in schizophrenia. Functional neuroimaging studies report abnormal cerebellar activation at rest (16–18) and during cognitive tasks [Ref. (19–21); see Ref. (22) for critical review] in individuals with schizophrenia.
These structural and functional cerebellar abnormalities appear to have clinical and functional implications in schizophrenia. Specifically, cerebellar abnormalities are associated with clinical symptoms, cognitive deficits, and outcome measures in schizophrenia (3, 23–25). For example, deficits in working memory and mental flexibility correlate with cerebellar volume (26), and fronto-cerebellar metabolic abnormalities are associated with anhedonia and ambivalence (27). Moreover, increased connectivity between frontal–parietal and cerebellar regions predicts better cognitive performance in controls and individuals with schizophrenia, and individuals with schizophrenia with improved connectivity have fewer disorganization symptoms (28).
These empirical findings are often integrated into the cognitive dysmetria theory of schizophrenia, which places the cerebellum prominently in the cortico-cerebellar-thalamic-cortical circuit (CCTCC). The theory of cognitive dysmetria proposes a model of schizophrenia wherein deficits in this circuit are associated with both motor dysfunction and the clinical presentation of schizophrenia, and abnormalities in the CCTCC are believed to mediate the disordered cognition, behavior, and motor function characteristic of individuals with schizophrenia (29). A behavioral measure of cerebellar integrity, such as eyeblink conditioning (EBC), that can be administered to individuals with schizophrenia as an index of how well the cerebellum and interrelated circuits are performing is vital to the investigation of the cerebellum as a critical node in the CCTCC and locus of dysfunction in this influential theory of schizophrenia.
Eyeblink conditioning is a widely used measure of cerebellar-dependent associative learning. In the delay form of this task, a conditioned stimulus (e.g., brief tone) is paired, and co-terminates, with an unconditioned stimulus (e.g., air puff to the eye) that elicits an unconditioned response (e.g., eyeblink). Over the course of repeated paired presentations, a conditioned eyeblink response (CR) occurs in response to the tone and preceding the onset of the unconditioned stimulus. EBC is used in the study of clinical disorders such as schizophrenia and autism as well as aging for several reasons. First, the neural circuit underlying EBC has been well-characterized in non-human animals, with the specific brain stem nuclei associated with both stimulus encoding and motor output remarkably well-understood [see Ref. (30), for review]. Furthermore, the neural plasticity underlying standard delay EBC has been localized to the ipsilateral dorsal lateral anterior interpositus nucleus, and specific areas of the cerebellar cortex involved with timing and gain control of the conditioned response have also been identified [again see Ref. (30), for review]. Second, the conditioned response that develops over the course of delay EBC is well-preserved across species including rodents [e.g., Ref. (31, 32)], rabbits [e.g., Ref. (33)], cats [e.g., Ref. (34)], and humans [e.g., Ref. (35)], making EBC a widely used translational probe of cerebellar function. Finally, the associative learning induced by EBC is a non-declarative form of learning that occurs outside of intention and conscious awareness (35). Because performance on EBC is not dependent on higher-order cognitive function or the ability to follow complex instructions, it can be studied in individuals across a variety of ages and clinical presentations.
Importantly, the robust identification of cerebellar circuitry underlying delay EBC in non-human species is remarkably consistent with human EBC findings. Such evidence has emerged from studies involving patients with cerebellar lesions, dual-task interference, transcranial direct current stimulation (tDCS), and functional brain imaging. Specifically, individuals with cerebellar strokes demonstrate impairments in delay EBC performance (36–38). In addition, studies have demonstrated a significant relationship between performance on delay EBC and cerebellar-dependent timed interval tapping (39) as well as dual-task interference during simultaneous delay EBC and timed interval tapping (40) in non-psychiatric controls. tDCS applied to the cerebellum during acquisition has been shown to modify delay EBC performance (41). Finally, human brain imaging studies investigating the neural substrates of EBC converge with the lesion and dual-task studies described above, as well as further localize the site of EBC learning-related plasticity in humans. Specifically, positron emission tomography (PET) studies have revealed changes in cerebellar activation during EBC (42–46), and functional magnetic resonance imaging (fMRI) BOLD activation changes in the cerebellum are consistently reported during EBC (47–50).
In the first published review of EBC studies and schizophrenia (51), the author concluded that overall the EBC findings were inconclusive and any observed EBC deficits may be accounted for by antipsychotic medication administration. Lubow (51) called for an explicit comparison between medicated and non-medicated individuals with schizophrenia. In addition, concerns were raised about drawing firm conclusions regarding EBC impairment in schizophrenia due to inconsistencies in the analysis of EBC (i.e., whether or not studies accounted for alpha responses and spontaneous blink rate), possible group differences in processing and encoding EBC stimuli, the notorious heterogeneity present in the diagnostic category of schizophrenia, and the small sample sizes and disproportionate number of male individuals with schizophrenia reported in the literature (51).
Two subsequent brief reviews have appeared as subsections in two recently published articles, one reviewing EBC performance across many neurodevelopmental disorders (52) and another reviewing cerebellar-related motor dysfunction in schizophrenia and high-risk populations (53). The authors of both brief reviews largely emphasized the emerging pattern of abnormal EBC performance in schizophrenia, citing the large sample sizes and the persistent deficit in EBC performance in an unmedicated subsample reported in studies published after Lubow’s (51) review (52), as well as even more recent studies of EBC impairment in individuals with schizotypal personality disorder, first-degree relatives of individuals with schizophrenia, and individuals with schizophrenia who are medication-free for a period of several weeks (53). However, both groups also acknowledged the possible role of antipsychotic medication and methodological variability in the inconsistent findings across studies (52, 53).
Importantly, since the publication of Lubow’s (51) initial review of nine articles, six additional studies have been published examining EBC in the schizophrenia spectrum. These six studies account for 48% of all individuals in the schizophrenia spectrum that have participated in delay EBC studies, nearly doubling the number of participants in the schizophrenia spectrum that have been studied since Lubow’s (51) review. However, questions still persist regarding the source of inconsistency in the literature examining EBC in schizophrenia, specifically related to the potential effects of antipsychotic medication and heterogeneity in methodology.
The purpose of the present review was to conduct a thorough and integrative review of published studies of EBC in the schizophrenia spectrum. Given Lubow’s (51) findings and cautions as well as the conclusions of Reeb-Sutherland and Fox (52) and Bernard and Mittal (53), special attention was paid to (1) evidence of antipsychotic medication effects, (2) inconsistencies between studies in and any systematic effects of stimulus and analysis parameters, and (3) differences in sample size and sample characteristics. Finally, the findings of this review are interpreted within the context of existing models of schizophrenia.
Method
Tables 1–5 catalog 15 studies examining EBC in individuals with schizophrenia. These studies were first identified using Lubow’s existing review of EBC in schizophrenia. Studies examining EBC in the schizophrenia spectrum published subsequent to this review were identified using PubMed, a resource of the National Center for Biotechnology Information (NCBI), at the National Institutes of Health’s (NIH) U.S. National Library of Medicine (NLM).
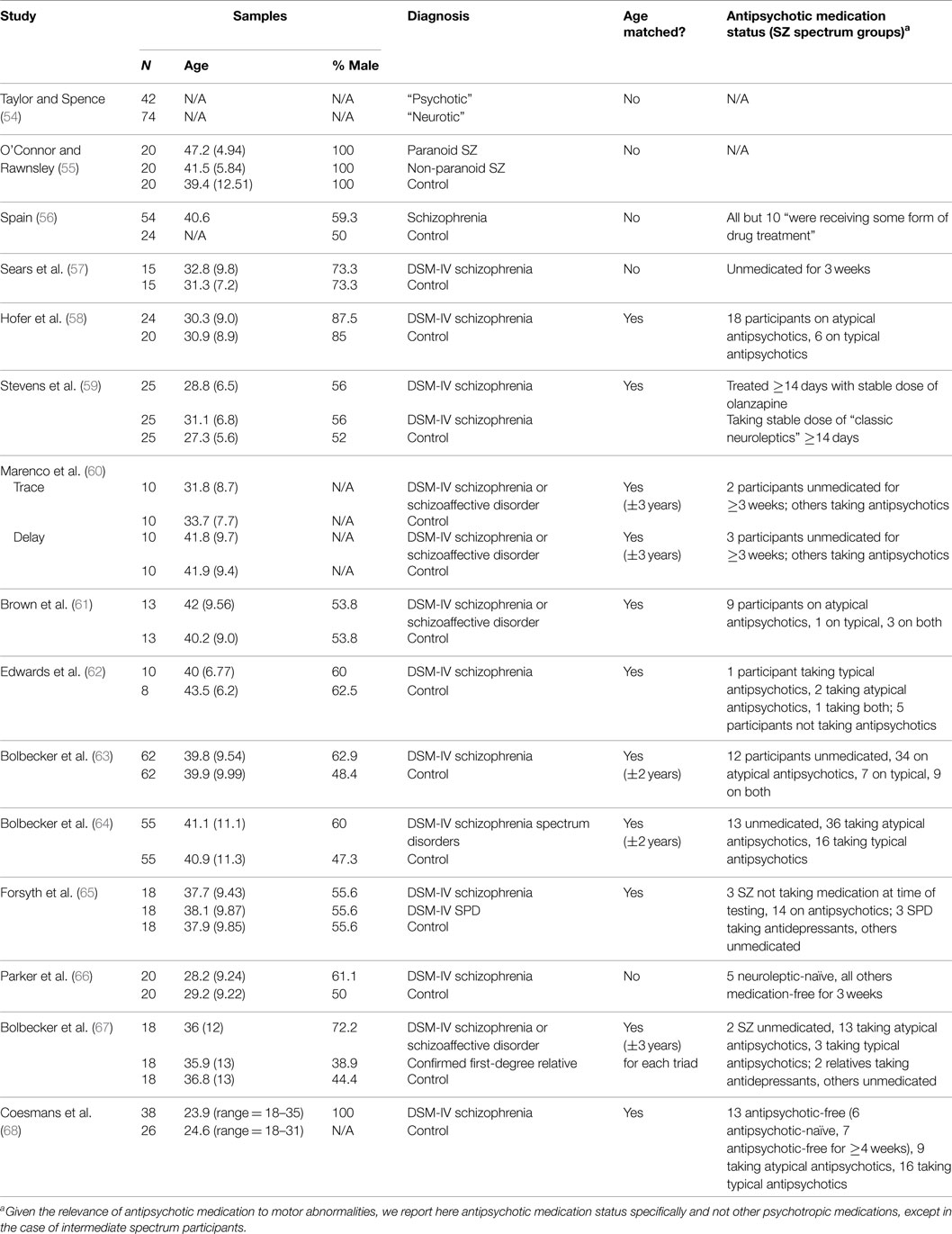
Table 1. Sample characteristics for studies of EBC in schizophrenia.
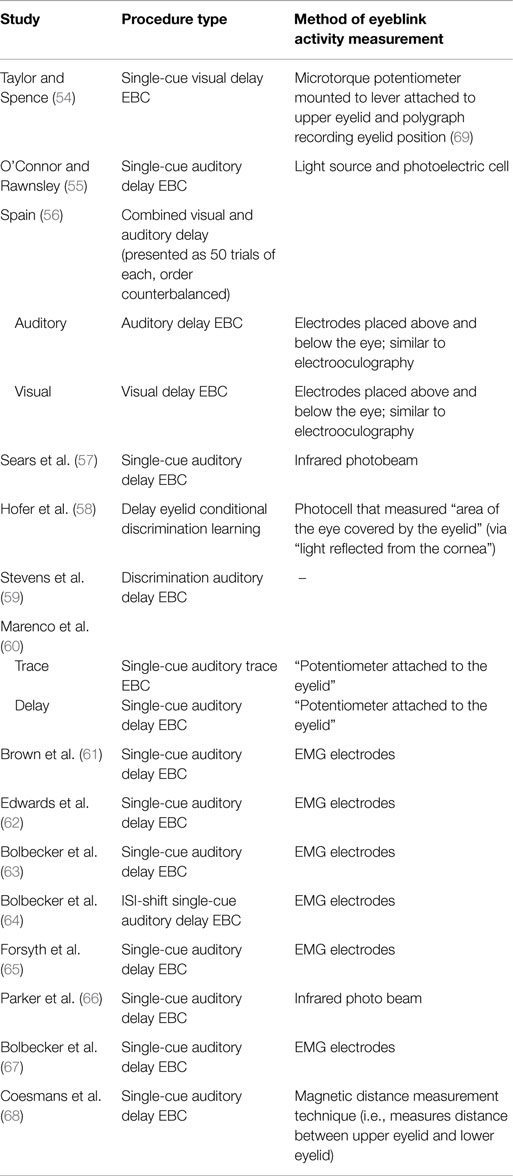
Table 2. EBC paradigms and measurement techniques for studies of EBC in schizophrenia.
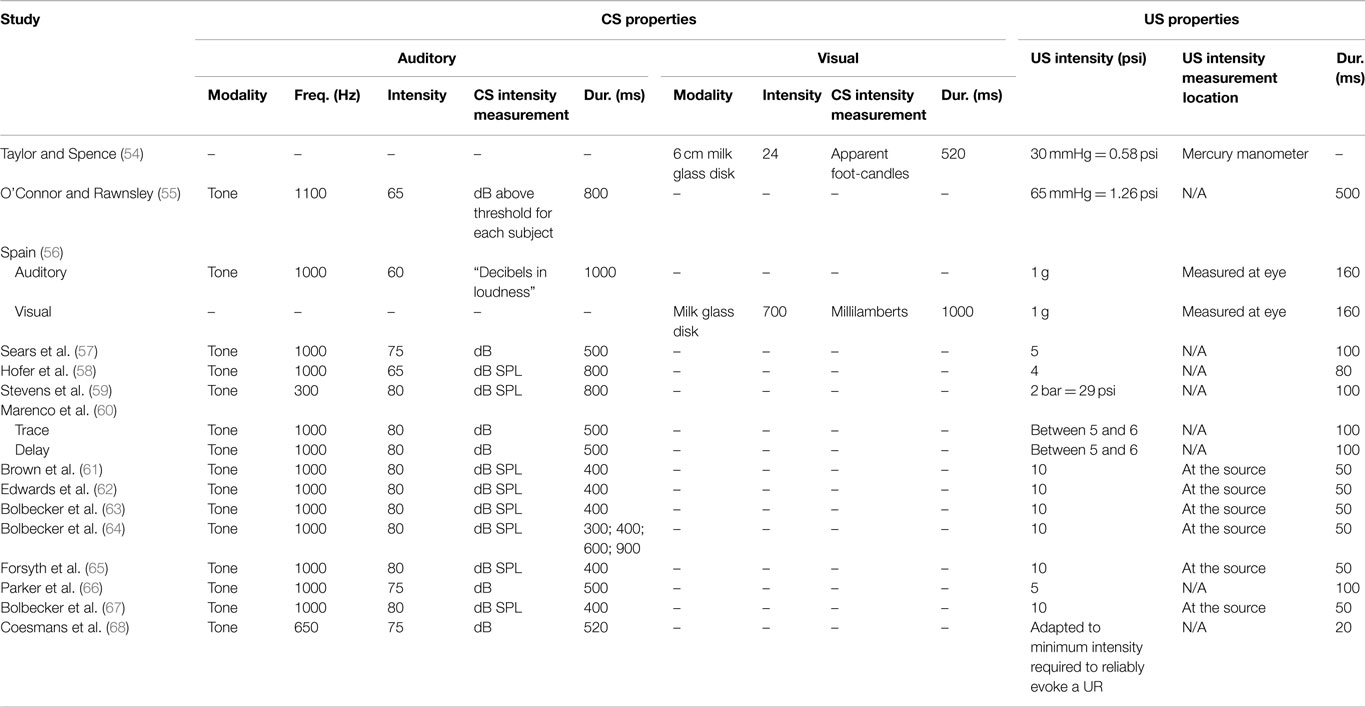
Table 3. EBC stimulus properties for studies of EBC in schizophrenia.
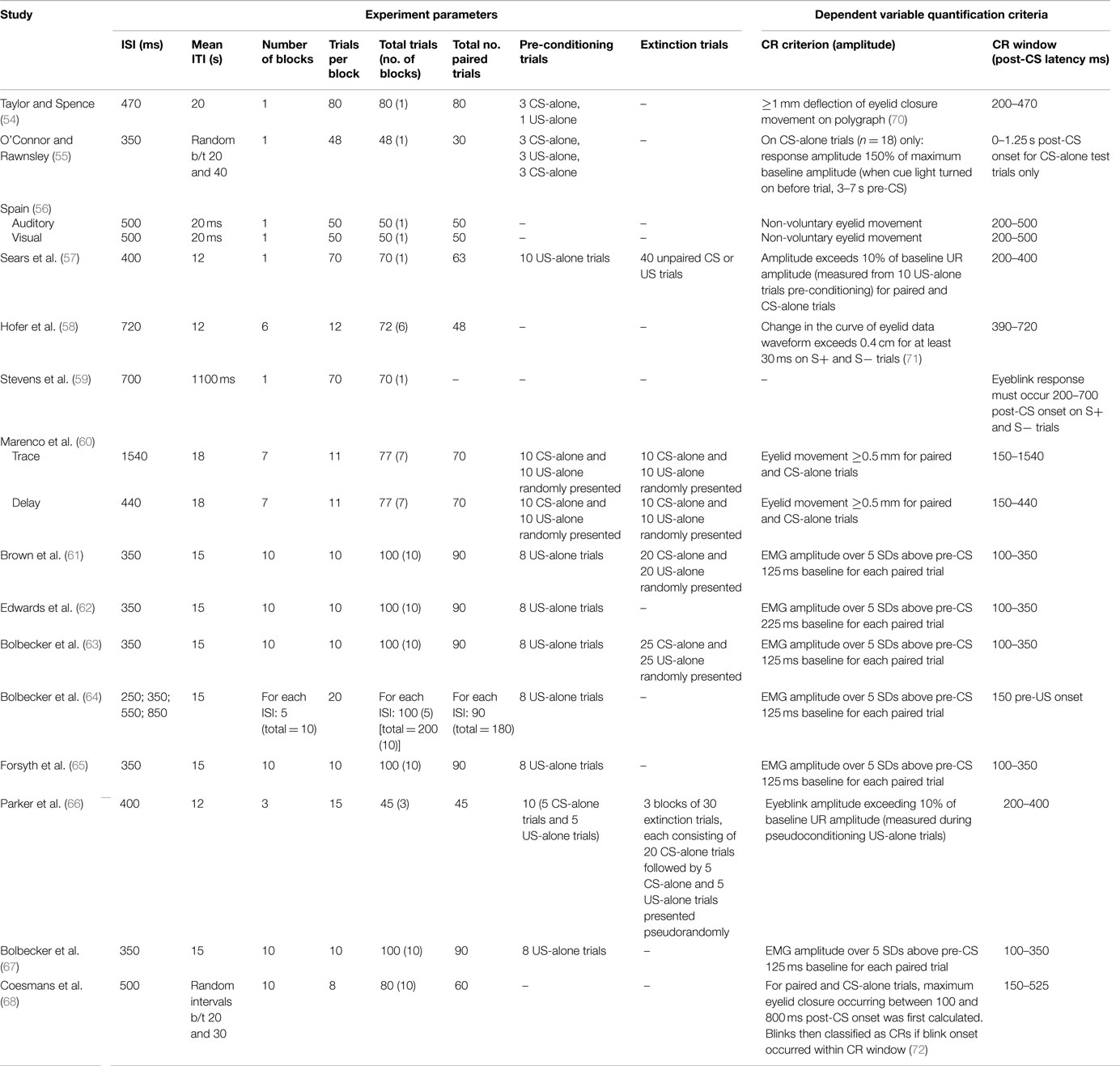
Table 4. EBC experiment and analysis parameters for studies of EBC in schizophrenia.

Table 5. Summary of main findings from studies of EBC in schizophrenia.
Various domains of information from these 15 studies examining EBC in the schizophrenia spectrum were then recorded and organized, including sample characteristics (see Table 1), parametric properties of the EBC tasks and analyses, and major findings (see Tables 2–5). In the review of this literature, careful attention was paid to (1) findings that occur consistently across studies and across research groups, (2) the relationship of medication status to consistent findings, (3) any sample characteristics or parametric variability (in either EBC paradigms or analyses) that may contribute to heterogeneity of findings, (4) correlates of EBC performance in individuals along the schizophrenia spectrum, and (5) the implications of the findings of this review for current systems-level and neurobiological theories of schizophrenia.
Results
Conditioning
Conditioned Responding (e.g., %CRs)
Of the 15 studies of delay EBC in schizophrenia, 9 demonstrated decreased CRs compared to controls (58, 61–68), 4 found no group differences in rates of conditioned responding (54, 55, 59, 60), and 2 reported facilitated conditioning in schizophrenia (56, 57). It should be noted, however, in one study (56) which reported overall increased percent CRs in schizophrenia vs. controls, that when the auditory and visual EBC results are considered separately, schizophrenia patients yielded fewer CRs when the CS was an auditory vs. visual stimulus.
CR Onset Latency
One study reported shorter CR onset latencies in individuals with schizophrenia vs. controls (61). Two studies reported longer CR onset latencies in schizophrenia vs. controls (60, 64). Two studies reported no significant differences between groups (66, 67). One study reported blink onset latency results regardless of CR or UR performance, and therefore cannot be considered with either CR or UR results [see Ref. (57) in Table 5 for these and CS-alone latency findings].
CR Peak Latency
Three studies reported shorter peak latency in individuals with schizophrenia vs. controls (61, 63, 66). One study reported longer CR peak latency in schizophrenia vs. controls (60), and three studies reported no significant differences between groups (62, 64, 65).
CR Amplitude
Five studies reported no significant differences between groups for CR peak amplitude (60, 61, 63, 66, 67). Sears and colleagues (57) reported increased CR amplitude in individuals with schizophrenia vs. controls in CS-alone trials. In post hoc analyses of individual blocks, Forsyth and colleagues (65) found increased CR amplitudes in controls vs. schizophrenia and SPD in later but not earlier blocks of conditioning.
Medication Effects
Of the 15 published studies, 13 reported medication status and all but one of these (56) included information specific to antipsychotic medication status. In 10 of these 12 studies, most participants in the schizophrenia sample were currently taking antipsychotic medication. In terms of conditioning effects, 8 of these 10 studies of medicated individuals reported decreased conditioning (e.g., decreased percent CRs) in individuals with schizophrenia compared to controls (58, 61–65, 67, 68). In the other two studies of medicated individuals, no group differences in conditioning rates were found (59, 60).
In 2 of the 12 studies, the entire schizophrenia group was antipsychotic-free for 3 weeks (57, 66). Sears and colleagues (57) reported facilitated conditioning in these participants, whereas Parker and colleagues (66) reported impaired conditioning. In addition, 3 of the 12 studies analyzed data from antipsychotic-free subsamples of individuals with schizophrenia (63, 64, 68). When Bolbecker and colleagues (63) re-analyzed their data including only the medication-free subset of individuals with schizophrenia and their age-matched controls (with a sample size in each group of n = 13, similar to other stand-alone studies of antipsychotic-free schizophrenia), they found decreased CRs and shorter CR peak latencies in these individuals with schizophrenia – with even larger effect sizes than in the full sample of individuals with schizophrenia. The authors reported no significant correlations between EBC dependent variables and chlorpromazine equivalent dosages (63), as did Brown and colleagues (61). Similarly, in a later study, Bolbecker and colleagues (64) reported no significant differences between schizophrenia participants medicated with antipsychotics vs. those who were medication-free. Finally, Coesmans and colleagues (68) reported no effect of group on percent CRs or “learning index” (change in number of CRs from first to last conditioning block) when comparing the three subgroups of individuals with schizophrenia (those taking atypical antipsychotics, typical antipsychotics, and those who were antipsychotic medication-free), and no significant correlation between learning index and chlorpromazine equivalent dosages.
Finally, both studies including intermediate schizophrenia spectrum participants [individuals with SPD (65) and first-degree relatives (67)] reported that there was no antipsychotic use in either of these populations. In these studies both individuals with SPD and first-degree relatives of individuals with schizophrenia were impaired in EBC.
Unconditioned Responses
UR measures on paired trials are reported less frequently in the literature. With regard to percentage of URs, one study reported decreased percent URs in individuals with schizophrenia vs. controls (60). With regard to UR latency, two studies reported slower UR peak latency in individuals with schizophrenia vs. controls (63, 64), while three other studies reported no significant differences between groups (61, 65, 67). Finally, with regard to UR amplitude, three studies reported increased UR amplitude in schizophrenia vs. controls (63, 65, 67), whereas three studies reported no significant group difference (61, 62, 64). And, one study reported a significant group by block interaction showing consistently diminished UR amplitude in individuals with schizophrenia compared to controls, and larger initial UR amplitude in controls that decreased across blocks (60).
Importantly, several studies explored group differences in URs to unpaired unconditioned stimuli during pre-conditioning trials or pseudoconditioning (prior to paired trial presentation). Such pre-conditioning measures test for pre-existing differences between groups in the ability to generate a blink in the absence of recent associatively salient stimuli and habituation. Marenco and colleagues (60) reported no group differences in baseline UR activity; Edwards and colleagues (62) reported no group difference in baseline UR amplitude. Bolbecker and colleagues reported no group differences in UR peak amplitude or latency in individuals with schizophrenia compared to controls in one article (67) and increased UR amplitude in another (63) – in both cases suggesting that conditioning deficits could not be accounted for by pre-existing group differences in eyeblink responses. However, Sears and colleagues (57) reported longer UR latency in individuals with schizophrenia compared to controls for US-alone trials.
Extinction
Four studies reported no significant differences between extinction rate in individuals with schizophrenia and controls (60, 61, 63, 66). However, interpretation of this finding is complicated by the group differences in percent CRs during the acquisition phase reported by three of the studies (61, 63, 66). Finally, Brown and colleagues (61) reported shorter CR onset and peak latency in individuals with schizophrenia vs. controls during extinction.
Spontaneous Blink Rate
Several studies excluded individual trials in which a blink occurred at a time during a trial that would render CR production impossible (i.e., immediately prior to CS onset) [Ref. (61–65, 67); see Ref. (60) for a more liberal window for trial exclusion]. Most of these studies also reported no significant group differences in this rough estimate of spontaneous blink rate [Ref. (60, 63–65, 67), but see Ref. (61)].
Alpha Responses
Three studies examined group differences in alpha responses, which are reflexive orienting responses to the tone (importantly, alpha responses are non-associative). All three studies reported no group differences in the rate of alpha responses (57, 58, 60). Marenco and colleagues (60) reported earlier onset of the alpha response in controls vs. individuals with schizophrenia.
EBC Correlates
Symptoms and Demographic Variables
Multiple studies have failed to find significant relationships between schizophrenia symptom severity and EBC dependent variables (61, 63, 68). Brown and colleagues (61) and Bolbecker and colleagues (63) also reported null results between symptom severity and extinction dependent variables. Parker and colleagues (66) found no significant correlations between positive or negative symptoms and the three phases of conditioning the authors used to analyze their EBC data (i.e., early, middle, and late); however, negative symptoms were significantly correlated with late-phase extinction of the CR. In the earliest examination of symptom correlates of EBC, O’Connor and Rawnsley (55) reported no significant correlation between EBC and introversion scores [but see Spain (56) for EBC correlates of clinician-rated withdrawal]. Finally, in their investigation of demographic correlates of EBC, Coesmans and colleagues (68) also reported non-significant correlations between learning index and age and years of education.
Neuropsychological Variables
Bolbecker and colleagues (63) reported significant positive correlations between average percent CRs and both WASI IQ estimates and the WASI Vocabulary subscale in controls, but not in individuals with schizophrenia. The Matrix Reasoning subscale was not significantly correlated with average percent CRs in either group. Forsyth and colleagues (65) reported a significant positive correlation between percent CRs and Digit Symbol score (a subscale of the WAIS) for schizophrenia spectrum participants (i.e., individuals with schizophrenia and SPD were combined into one group). This significant correlation held when individuals with schizophrenia were analyzed separately, but not when individuals with SPD were analyzed separately. Additionally, the authors reported no significant correlations between Digit Symbol score and percent CRs in controls, or between percent CRs and the Picture Completion, Similarities, or Digit Span WAIS subscales in either controls or schizophrenia spectrum participants (65). Using aggregate cognitive domain scores from a battery of neuropsychological tests in patients, Parker and colleagues (66) reported a significant positive relationship between both aggregate language and motor scores and CR timing during early conditioning; motor scores were also correlated with middle-phase extinction of the CR. Finally, Coesmans and colleagues (68) reported a significant positive correlation between EBC learning index and saccade adaptation strength in controls, but not individuals with schizophrenia, while no significant correlations were found in either group between EBC learning index and saccade adaptation speed.
Neuroimaging Measures
In a study of cerebellar volumetric correlates of EBC, Edwards and colleagues (62) reported a significant positive correlation between anterior lobe volume and CR onset latency, and a significant negative correlation between anterior lobe volume and UR amplitude (in response to paired trials) in controls, but no significant correlations between cerebellar MRI volume and EBC dependent variables in individuals with schizophrenia. Parker and colleagues (66) analyzed PET data according to phases of conditioning (i.e., early, middle, and late), and reported decreased rCBF in individuals with schizophrenia compared to controls in frontal, thalamic, and cerebellar regions during both acquisition and extinction (among other loci). In summarizing findings of hypofrontality during EBC, the authors highlighted decreased rCBF in individuals with schizophrenia compared to controls in the contralateral medial frontal gyrus during all phases of conditioning, and the contralateral middle frontal gyrus during the early and middle phases of conditioning. The authors also highlighted decreased rCBF in contralateral cerebellar lobules IV and V in individuals with schizophrenia compared to controls during all phases of conditioning, with a group difference in ipsilateral cerebellar lobule VI during late acquisition only. Finally, group differences in rCBF in the thalamus were significant during early and late conditioning. Regarding rCBF during extinction, the authors highlighted decreased rCBF in individuals with schizophrenia compared to controls during all phases of extinction in the medial and middle frontal gyri and in cerebellar lobule IX. Additional loci of decreased cerebellar rCBF in individuals with schizophrenia compared to controls included cerebellar lobules IV and V during middle extinction, and cerebellar lobules IV, V, and VI during late extinction. Finally, the authors highlighted that decreased thalamic rCBF in individuals with schizophrenia was significant during early phase extinction (66).
Discussion
Conditioning (i.e., %CRs)
In reviewing the literature investigating delay EBC in schizophrenia, decreased percent conditioned responses in individuals with schizophrenia compared to non-psychiatric controls emerges as the single consistent, robust, and replicated finding. Diminished conditioning in schizophrenia is highly suggestive of cerebellar dysfunction, given the crucial role of the cerebellum in the circuit underlying delay EBC. Moreover, as discussed in the following paragraphs, there are no extraneous variables (i.e., medication status, sample size, different analytical approaches, parametric variability, non-associative blinking function, and investigative group) that could fully account for these EBC deficits in schizophrenia.
In investigating the possible driving role of medication in the observed EBC deficits (i.e., decreased %CRs) in individuals with schizophrenia, it is crucial to note that both medicated and non-medicated samples demonstrate conditioning deficits in individuals with schizophrenia (see Medication Effects subsection of Section “RESULTS”). Also important to this question of the effect of antipsychotic medication on EBC are the findings of EBC deficits in a non-medicated subsample (63), as well as the failure to find group differences in medicated vs. unmedicated individuals with schizophrenia (64, 68).
However, it is important to note that even the “medication-free” samples and subsamples reported above are not medication-naïve samples. While a small number of participants in the most recent studies [n = 5 in Parker et al. (66), and n = 6 in Coesmans et al. (68)] were naïve to antipsychotics, the small sizes of these groups precluded meaningful analyses investigating the effect of antipsychotic-naïve medication status. Therefore, while it appears unlikely based on the current review that recent use of antipsychotic medication drives EBC deficits, it is impossible to rule-out the long-term effects of antipsychotic use in individuals with schizophrenia in the results of the study of “medication-free” samples and subsamples.
Eyeblink conditioning studies of intermediate genotypes and phenotypes of schizophrenia such as first-degree relatives (67) and SPD (65) that have demonstrated conditioning deficits in these groups are very important, especially given the absence of studies using medication-naïve or first-episode schizophrenia groups. Neither of these study groups were taking antipsychotic medication. This suggests that EBC deficits are related to the genetic/biological pathophysiology of schizophrenia, not the history of or current antipsychotic medication use.
In addition to medication status, examination of Tables 1–5 reveals no systematic sample characteristic, parameter, or analytic approach that could be driving this review’s main finding of EBC deficits in schizophrenia. Indeed, EBC deficits occur across samples of varying ages and gender composition, and in studies using a range of EBC stimulus parameters and experimental design (e.g., CS/US duration, ISI, ITI, and pre-conditioning trials or pseudoconditioning) and analysis (e.g., CR window and criterion) specifications. Furthermore, potentially confounding issues such as spontaneous blink rate and baseline blinking function have been investigated by several groups, with no convincing evidence that these variables bias EBC experimental results.
Furthermore, it appears as though many studies reporting null findings or facilitated conditioning may have parametric or analytic variations that could account for such results. Specifically, Taylor and Spence (54) used a visual delay EBC paradigm, and the diagnostic criteria for the disorder differed substantially from those used in recent decades. Furthermore, the idiosyncratic analytic approaches of other studies may account for the reported null findings. For example, rather than quantifying rate of conditioning, Stevens and colleagues (59) measured the number of trials it took for participants to reach “criterion,” or five consecutive CRs. This style of analysis is not reported in most other studies. Another study appeared to restrict their analysis such that relatively less data are included compared to other studies. Specifically, O’Connor and Rawnsley (55) only used 18 unpaired CS-alone trials to measure conditioning, rather than attempting to detect CRs across all paired trials over the course of conditioning. Finally, Sears and colleagues (57) did not include a measure of spontaneous blink rate; it is therefore possible that group differences in non-associative blinking could have confounded the reported findings of facilitated conditioning in schizophrenia. More research is necessary to determine whether these varied findings are due to these methodological differences or, in fact, reflect inconsistencies in EBC deficits in schizophrenia across studies.
CR Timing
Group differences in timing of the conditioned response (i.e., onset and peak latency) have been reported far less frequently than rate of conditioning (i.e., percent CRs). Among studies reporting these variables, there is inconsistency in how onset latency is calculated and whether the algorithm used to calculate onset latency is reported. Results are also inconsistent, with findings reported in both directions and null results. However, the proportion of findings reporting some group difference (N = 7) in CR timing vs. null results (N = 6) suggests that there may be abnormalities in the timing of the conditioned response in individuals with schizophrenia.
Interpretation of Correlate Findings
Parker and colleagues’ (66) findings of both impaired conditioning and decreased cerebellar blood flow in individuals with schizophrenia compared to controls during delay EBC strongly suggest that cerebellar neural dysfunction underlies the behavioral EBC abnormalities consistently reported in individuals with schizophrenia. This is a crucial piece of evidence, as authors reporting previous findings of impaired delay EBC in individuals with schizophrenia have inferred underlying cerebellar dysfunction given the well-established delay EBC cerebellar circuitry in non-human animals.
In addition, EBC correlates of neuropsychological performance are reported by a few studies (63, 65, 66). This shared variance between cerebellar-dependent EBC performance and cognition indicates that the cerebellum may be a shared neural substrate between these two processes, which is consistent with cerebellar involvement in cognitive as well as motor function.
Limitations and Future Directions
One critical conclusion from this review is that antipsychotic medications do not appear to be driving the EBC deficit observed consistently in schizophrenia. However, this conclusion is primarily based on the study of EBC in unmedicated (rather than never-medicated) individuals with schizophrenia, first-degree relatives and individuals with schizotypal personality disorder. While the robustness of the EBC deficit in these populations is obviously compelling, a logical and important next step is conducting delay EBC in first episode and/or never-medicated individuals. Second, significant variability in methodological and analytic strategies across EBC studies precluded a meta-analytic approach; therefore, as more studies are conducted using consistent methods, statistical analyses, and reporting, this approach should be considered. Third, further replication of the main findings of this review article (i.e., an EBC deficit in schizophrenia) is essential given that one investigative group has accounted for most patients studied (6 of 15 studies).
Finally, further work investigating the neural activity in the cerebellum during delay EBC in schizophrenia is essential to elucidating the specific contribution of the cerebellum in driving impairments in delay EBC. Specifically, the fine-grained spatial resolution of fMRI could prove essential to understanding which regions of the cerebellum underlie delay EBC in humans, and where this circuit is degraded in schizophrenia.
EBC Findings Within the Context of Theories of Schizophrenia
Overall, the reported deficits in cerebellar-dependent EBC in individuals with schizophrenia are consistent with the theory of cognitive dysmetria, in which the cerebellum is one node in a circuit regulating the fluid temporal coordination of motor, cognitive, and affective information, the disruption of which is hypothesized to be a common underlying precursor to the heterogeneous downstream expressions of the phenomenology of schizophrenia (29, 73). The cerebellum is believed to play a unique role in this circuit mediating the coordination (or instantiating the discoordination) of mental activity, which also includes the prefrontal cortex and the thalamus. Specifically, it is the feedback (via the thalamus) between the prefrontal cortex (and the higher-order cognitive processes instantiated therein) and the cerebellum (which is notable for cytoarchitecture conducive to large-scale parallel processing and its role in coordination, sequencing, and timing) that is hypothesized to instantiate the fluid temporal coordination of mental activity (29, 73). As stated above, consistent deficits in performance on one of the most robust and well-understood (with respect to underlying circuitry) cerebellar tasks in individuals with schizophrenia provide evidence consistent with the theory of cognitive dysmetria, and this finding is germane to its arguably most critical node [Andreasen (29) initially identified assays of cerebellar function, specifically citing EBC as a potential example, as the litmus test through which the theory can be falsified]. In addition, the relationship between cerebellar function and cognitive function supports this theory (63, 65, 66).
More specifically, the possible mechanisms of the cerebellum’s contribution to higher-order cognitive function have been hypothesized to parallel that proposed by control theory in the domain of motor control [see Ref. (74, 75), for review]. The cerebellum is hypothesized to contribute to the coordination of movement via internal models (both forward and reverse) (74), which are neural representations that can be trained to simulate the dynamics of motor action (74–76). Forward models are believed to receive input that duplicates the motor command (termed an efference copy) sent by the motor cortex (which controls movement) after the motor cortex receives a higher-order instructor command (i.e., from the premotor cortex), and output a prediction of what the sensory consequences of that command will be (a corollary discharge). The forward model is tuned by a mechanism that compares sensory predictions of the model to actual sensory input, which has been hypothesized to occur in the inferior olive. Once a forward model is adequately trained, it can provide useful feedback (via the thalamus) to the primary motor cortex, which executes motor commands (74, 75). An inverse model, conversely, can eventually conduct feed-forward motor control in response to a higher-order instructor command. While error-related feedback processing in the inferior olive is also hypothesized to tune inverse models, inverse models are trained by comparing motor output to the initial instructor command, and this feedback is mediated through the motor cortex (74).
In generalizing the function of internal models in the cerebellum to a role in cognition, it has been proposed that there are areas in the prefrontal cortex (following a higher-order instructor command, as in the example using motor function) that send commands to areas of the cortex that instantiate psychological processes and manipulate these areas in much the same way the motor cortex manipulates the motor system. In this way, the cerebellum receives an efference copy of this command and can learn and execute forward models that would simulate processing in the target brain area and provide feedback to the prefrontal cortex (74, 75). Using an inverse model, the cerebellum could actually perform feed-forward control of cognitive function (again following an instructor signal) by acting directly on the target brain area (74).
These putative mechanisms of cerebellar contributions to cognition are supported by the frequently cited uniformity of cerebellar cytoarchitecture, which, along with its circuitry suggest that the cerebellum is performing a uniform process across a variety of cortical inputs (74, 75). In addition, the matched increase in both cerebral and cerebellar neurons in humans as well as high connectivity between the cortex and cerebellum also indicate the proposed mechanisms of cerebellar contributions to cognition are physiologically plausible (77, 78). Finally, translational evidence in support of cerebellar contributions to cognition can be found in comparing cortical projections to the cerebellum in humans and macaque monkeys, where the largest proportion of the projections in humans originates in prefrontal cortex vs. motor areas in macaque monkeys [see Ref. (75) for review]. In light of this physiological and translational evidence, the proposed function of internal models as a mechanism of cerebellar contributions to cognition seems both anatomically and evolutionarily sound. Ramnani (75) has further described internal models as ideally suited to rapid, highly accurate, efficient processing of routine, well-practiced cognitive processes, whereas cortical mechanisms are best suited for flexible though less efficient processing, which would be important for processing novel problems or generalizing cognitive processes across different contexts.
In addition to being a robust assay of cerebellar function, EBC is especially germane to the putative mechanisms outlined above in light of the proposed mechanism of error correction of internal models. Specifically, the feedback-related tuning of internal models is believed to be instantiated through error signals sent from climbing fibers (originating in the inferior olive), which results in LTD at the parallel fiber–Purkinje cell synapse when climbing and parallel fibers are simultaneously activated (74). In EBC, US information is transmitted through climbing fibers from the inferior olive, and is often conceptualized as an error signal, and an identical LTD mechanism as that described above is believed to be an integral part of cerebellar cortical plasticity during conditioning [see Ref. (79) for review]. It has previously been suggested that dysfunctional internal models may be the mechanism of cerebellar-mediated cognitive and affective dysfunction in schizophrenia (22, 74). It is therefore notable that the findings of this review indicating deficits in cerebellar function in schizophrenia, and more importantly EBC deficits specifically, may be indicative of dysfunctional cerebellar internal models, which may be mediating the cardinal cognitive and affective symptoms of the disorder.
However, neuropsychological correlates of EBC in individuals with schizophrenia have been rarely investigated. Furthermore, the consistently reported non-significant correlations between delay EBC and symptom severity in individuals with schizophrenia is surprising given the putative role of the cerebellum in the pathophysiology of schizophrenia. It is possible that the contributions of cerebellar deficits to the pathological processes of schizophrenia are more proximal effects on timing and coordination of information, whereas symptoms and impaired neuropsychological function are more distal manifestations of the disorder that are affected by many factors and are not linearly related in magnitude to cerebellar dysfunction. Restricted range in neuropsychological and symptom measures and/or floor effects might also obscure any systematic relationships between these variables and EBC performance. Finally, symptoms are a state-dependent variable; the potentially transient and fluctuating nature of symptom severity might also account for the lack of reported correlates. Alternatively, it is possible that cerebellar dysfunction in areas outside of the delay EBC circuitry is related to symptom severity and neuropsychological function. Still, more research is necessary to understand the relationships between cerebellar-mediated dysfunction and cognitive and clinical variables.
Importantly, EBC performance deficits in schizophrenia may have implications for glutamatergic models of the disorder given that glutamate is the primary excitatory neurotransmitter in the cerebellum [see Ref. (80) for review]. The glutamate model of schizophrenia hypothesizes dysfunction of the NMDA type of glutamate receptor [see Ref. (81) for overview]. Non-human animal research has implicated NMDA receptors in the interpositus nucleus in CR acquisition [Ref. (82); see Ref. (79) for a thorough review of the neural mechanisms of EBC]. Given that the “memory trace” of delay EBC has been localized to the anterior interpositus nucleus, it is therefore possible that impairments in conditioning in schizophrenia (reported most frequently as a decrease in percent CRs, or impaired CR acquisition) are related to NMDA receptor dysfunction in the interpositus nucleus in schizophrenia.
There is also substantial glutamatergic transmission in the cerebellar cortex; therefore, abnormalities in CR timing (largely mediated by the cerebellar cortex) may also be indicative of NMDA receptor dysfunction in schizophrenia. While NMDA receptors have been reported in the cerebellar cortex (80), they were traditionally not believed to play a role in the cellular mechanism (i.e., LTD at the parallel fiber-Purkinje cell synapse following both parallel and climbing fiber input to Purkinje cells) believed to underlie EBC-related learning in the cerebellar cortex [see Ref. (83) for review]. Importantly, however, there is more recent evidence that NMDA receptors at the climbing fiber–Purkinje cell synapse may in fact contribute to LTD at the parallel fiber–Purkinje cell synapse (84). Furthermore, more broad conceptualizations of the substrates of cerebellar learning are emerging that suggest that mechanisms of cerebellar cortical plasticity and neural activity beyond LTD at the parallel fiber-Purkinje cell synapse (some involving NMDA receptors) may be involved in EBC (85, 86). Accordingly, more research is necessary to determine the role of glutamate in reported EBC timing abnormalities in schizophrenia.
In addition to the glutamate hypothesis, abnormalities in the endocannabinoid system in schizophrenia [see Ref. (87) for brief review] are also implicated by the current review findings. Edwards and Skosnik (87) have proposed EBC neural circuitry including endocannabinoids as retrograde signals serving to neuromodulate cerebellar cortical activity, thereby influencing CR timing and morphology. It is therefore possible that CR timing abnormalities in schizophrenia are indicative of abnormalities in the endocannabinoid system [see Ref. (87) for discussion].
Author Contributions
JK, WH, AB, and BO conceptualized the review article. JK conducted the review. JK and WH drafted the paper, and AB and BO provided critical review. All authors approved and agree to be accountable for the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by NIMH Grant 2R01MH074983 to WH and an NSF predoctoral Graduate Research Fellowship to JK.
References
1. Loeber RT, Cintron CM, Yurgelun-Todd DA. Morphometry of individual cerebellar lobules in schizophrenia. Am J Psychiatry (2001) 158:952–4. doi:10.1176/appi.ajp.158.6.952
2. Molina V, Martín C, Ballesteros A, de Herrera AG, Hernández-Tamames JA. Optimized voxel brain morphometry: association between brain volumes and the response to atypical antipsychotics. Eur Arch Psychiatry Clin Neurosci (2011) 261:407–16. doi:10.1007/s00406-010-0182-2
3. Nopoulos PC, Ceilley JW, Gailis EA, Andreasen NC. An MRI study of cerebellar vermis morphology in patients with schizophrenia: evidence in support of the cognitive dysmetria concept. Biol Psychiatry (1999) 46:703–11. doi:10.1016/S0006-3223(99)00093-1
4. Volz H, Gaser C, Sauer H. Supporting evidence for the model of cognitive dysmetria in schizophrenia – a structural magnetic resonance imaging study using deformation-based morphometry. Schizophr Res (2000) 46:45–56. doi:10.1016/S0920-9964(99)00236-4
5. Ichimiya T, Okubo Y, Suhara T, Sudo Y. Reduced volume of the cerebellar vermis in neuroleptic-naive schizophrenia. Biol Psychiatry (2001) 49:20–7. doi:10.1016/S0006-3223(00)01081-7
6. Henze R, Brunner R, Thiemann U, Parzer P, Richterich A, Essig M, et al. Gray matter alterations in first-admission adolescents with schizophrenia. J Neuroimaging (2011) 21:241–6. doi:10.1111/j.1552-6569.2010.00504.x
7. Bottmer C, Bachmann S, Pantel J, Essig M, Amann M, Schad LR, et al. Reduced cerebellar volume and neurological soft signs in first-episode schizophrenia. Psychiatry Res (2005) 140:239–50. doi:10.1016/j.pscychresns.2005.02.011
8. Kašpárek T, Mareček R, Schwarz D, Prikryl R, Vaníček J, Mikl M, et al. Source-based morphometry of gray matter volume in men with first-episode schizophrenia. Hum Brain Mapp (2009) 31:300–10. doi:10.1002/hbm.20865
9. Rasser PE, Schall U, Peck G, Cohen M, Johnston P, Khoo K, et al. Cerebellar grey matter deficits in first-episode schizophrenia mapped using cortical pattern matching. Neuroimage (2010) 53:1175–80. doi:10.1016/j.neuroimage.2010.07.018
10. Jacobsen LK, Giedd JN, Berquin PC, Krain AL, Hamburger SD, Kumra S, et al. Quantitative morphology of the cerebellum and fourth ventricle in childhood-onset schizophrenia. Am J Psychiatry (1997) 154:1663–9. doi:10.1176/ajp.154.12.1663
11. Cahn W, Hulshoff Pol HE, Bongers M, Schnack HG, Mandl RC, Van Haren NE, et al. Brain morphology in antipsychotic-naive schizophrenia: a study of multiple brain structures. Br J Psychiatry (2002) 181:s66–72. doi:10.1192/bjp.181.43.s66
12. Levitt JJ, McCarley RW, Nestor PG, Petrescu C, Donnino R, Hirayasu Y, et al. Quantitative volumetric MRI study of the cerebellum and vermis in schizophrenia: clinical and cognitive correlates. Am J Psychiatry (1999) 156:1105–7.
13. Maloku E, Covelo IR, Hanbauer I, Guidotti A, Kadriu B, Hu Q, et al. Lower number of cerebellar Purkinje neurons in psychosis is associated with reduced reelin expression. Proc Natl Acad Sci U S A (2010) 107:4407–11. doi:10.1073/pnas.0914483107
14. Reyes MG, Gordon A. Cerebellar vermis in schizophrenia. Lancet (1981) 2:700–1. doi:10.1016/S0140-6736(81)91039-4
15. Tran KD, Smutzer GS, Doty RL, Arnold SE. Reduced Purkinje cell size in the cerebellar vermis of elderly patients with schizophrenia. Am J Psychiatry (1998) 155:1288–90. doi:10.1176/ajp.155.9.1288
16. Loeber RT, Sherwood AR, Renshaw PF, Cohen BM, Yurgelun-Todd DA. Differences in cerebellar blood volume in schizophrenia and bipolar disorder. Schizophr Res (1999) 37:81–9. doi:10.1016/S0920-9964(98)00137-6
17. Steinberg JL, Devous MD, Moeller FG, Paulman RG, Raese JD, Gregory RR. Cerebellar blood flow in schizophrenic patients and normal control subjects. Psychiatry Res (1995) 61:15–31. doi:10.1016/0925-4927(95)02574-H
18. Volkow ND, Levy A, Brodie JD, Wolf AP, Cancro R, Van Gelder P, et al. Low cerebellar metabolism in medicated patients with chronic schizophrenia. Am J Psychiatry (1992) 149:686–8. doi:10.1176/ajp.149.5.686
19. Andreasen NC, O’Leary DS, Cizadlo T, Arndt S, Rezai K, Ponto LL, et al. Schizophrenia and cognitive dysmetria: a positron-emission tomography study of dysfunctional prefrontal-thalamic-cerebellar circuitry. Proc Natl Acad Sci U S A (1996) 93:9985–90. doi:10.1073/pnas.93.18.9985
20. Crespo-Facorro B, Paradiso S, Andreasen NC, O’Leary DS, Watkins GL, Boles Ponto LL, et al. Recalling word lists reveals “cognitive dysmetria” in schizophrenia: a positron emission tomography study. Am J Psychiatry (1999) 156:386–92.
21. Kim JJ, Mohamed S, Andreasen NC, O’Leary DS, Watkins GL, Boles Ponto LL, et al. Regional neural dysfunctions in chronic schizophrenia studied with positron emission tomography. Am J Psychiatry (2000) 157:542–8. doi:10.1176/appi.ajp.157.4.542
22. Bernard JA, Mittal VA. Dysfunctional activation of the cerebellum in schizophrenia: a functional neuroimaging meta-analysis. Clin Psychol Sci (2014) 3(4):545–66. doi:10.1177/2167702614542463
23. Ho B-C, Mola C, Andreasen NC. Cerebellar dysfunction in neuroleptic naive schizophrenia patients: clinical, cognitive, and neuroanatomic correlates of cerebellar neurologic signs. Biol Psychiatry (2004) 55:1146–53. doi:10.1016/j.biopsych.2004.02.020
24. Potkin SG, Alva G, Fleming K, Anand R, Keator D, Carreon D, et al. A PET study of the pathophysiology of negative symptoms in schizophrenia. Positron emission tomography. Am J Psychiatry (2002) 159:227–37. doi:10.1176/appi.ajp.159.2.227
25. Wassink TH, Andreasen NC, Nopoulos P, Flaum M. Cerebellar morphology as a predictor of symptom and psychosocial outcome in schizophrenia. Biol Psychiatry (1999) 45:41–8. doi:10.1016/S0006-3223(98)00175-9
26. Segarra N, Bernardo M, Valdes M, Caldu X, Falcón C, Rami L, et al. Cerebellar deficits in schizophrenia are associated with executive dysfunction. Neuroreport (2008) 19:1513–7. doi:10.1097/WNR.0b013e3283108bd8
27. Park K-M, Kim J-J, Seok JH, Chun JW, Park H-J, Lee JD. Anhedonia and ambivalence in schizophrenic patients with fronto-cerebellar metabolic abnormalities: a fluoro-d-glucose positron emission tomography study. Psychiatry Investig (2009) 6:72–7. doi:10.4306/pi.2009.6.2.72
28. Repovs G, Csernansky JG, Barch DM. Brain network connectivity in individuals with schizophrenia and their siblings. Biol Psychiatry (2011) 69:967–73. doi:10.1016/j.biopsych.2010.11.009
29. Andreasen NC. A unitary model of schizophrenia: Bleuler’s “fragmented phrene” as schizencephaly. Arch Gen Psychiatry (1999) 56:781–7. doi:10.1001/archpsyc.56.9.781
30. Thompson RF, Steinmetz JE. The role of the cerebellum in classical conditioning of discrete behavioral responses. Neuroscience (2009) 162:732–55. doi:10.1016/j.neuroscience.2009.01.041
31. Kotani S, Kawahara S, Kirino Y. Classical eyeblink conditioning in decerebrate guinea pigs. Eur J Neurosci (2002) 15:1267–70. doi:10.1046/j.1460-9568.2002.01963.x
32. Rogers RF, Britton GB, Steinmetz JE. Learning-related interpositus activity is conserved across species as studied during eyeblink conditioning in the rat. Brain Res (2001) 905:171–7. doi:10.1016/S0006-8993(01)02532-X
33. McCormick DA, Thompson RF. Neuronal responses of the rabbit cerebellum during acquisition and performance of a classically conditioned nictitating membrane-eyelid response. J Neurosci (1984) 4:2811–22.
34. Norman RJ, Villablanca JR, Brown KA, Schwafel JA, Buchwald JS. Classical eyeblink conditioning in the bilaterally hemispherectomized cat. Exp Neurol (1974) 44:363–80. doi:10.1016/0014-4886(74)90202-7
35. Clark RE, Squire LR. Classical conditioning and brain systems: the role of awareness. Science (1998) 280:77–81. doi:10.1126/science.280.5360.77
36. Gerwig M, Dimitrova A, Kolb FP, Maschke M, Brol B, Kunnel A, et al. Comparison of eyeblink conditioning in patients with superior and posterior inferior cerebellar lesions. Brain J Neurol (2003) 126:71–94. doi:10.1093/brain/awg011
37. Timmann D, Gerwig M, Frings M, Maschke M, Kolb FP. Eyeblink conditioning in patients with hereditary ataxia: a one-year follow-up study. Exp Brain Res (2005) 162:332–45. doi:10.1007/s00221-004-2181-x
38. Woodruff-Pak DS, Papka M, Ivry RB. Cerebellar involvement in eyeblink classical conditioning in humans. Neuropsychology (1996) 10:443–58. doi:10.1037/0894-4105.10.4.443
39. Woodruff-Pak DS, Jaeger ME. Predictors of eyeblink classical conditioning over the adult age span. Psychol Aging (1998) 13:193–205. doi:10.1037/0882-7974.13.2.193
40. Papka M, Ivry RB, Woodruff-Pak DS. Selective disruption of eyeblink classical conditioning by concurrent tapping. Neuroreport (1995) 6:1493–7. doi:10.1097/00001756-199507310-00007
41. Zuchowski ML, Timmann D, Gerwig M. Acquisition of conditioned eyeblink responses is modulated by cerebellar tDCS. Brain Stimul (2014) 7:525–31. doi:10.1016/j.brs.2014.03.010
42. Blaxton TA, Zeffiro TA, Gabrieli JD, Bookheimer SY, Carrillo MC, Theodore WH, et al. Functional mapping of human learning: a positron emission tomography activation study of eyeblink conditioning. J Neurosci (1996) 16:4032–40.
43. Logan CG, Grafton ST. Functional anatomy of human eyeblink conditioning determined with regional cerebral glucose metabolism and positron-emission tomography. Proc Natl Acad Sci U S A (1995) 92:7500–4. doi:10.1073/pnas.92.16.7500
44. Molchan SE, Sunderland T, McIntosh AR, Herscovitch P, Schreurs BG. A functional anatomical study of associative learning in humans. Proc Natl Acad Sci U S A (1994) 91:8122–6. doi:10.1073/pnas.91.17.8122
45. Parker KL, Andreasen NC, Liu D, Freeman JH, Ponto LL, O’Leary DS. Eyeblink conditioning in healthy adults: a positron emission tomography study. Cerebellum (2012) 11:946–56. doi:10.1007/s12311-012-0377-3
46. Schreurs BG, McIntosh AR, Bahro M, Herscovitch P, Sunderland T, Molchan SE. Lateralization and behavioral correlation of changes in regional cerebral blood flow with classical conditioning of the human eyeblink response. J Neurophysiol (1997) 77:2153–63.
47. Cheng DT, Meintjes EM, Stanton ME, Desmond JE, Pienaar M, Dodge NC, et al. Functional MRI of cerebellar activity during eyeblink classical conditioning in children and adults: eyeblink conditioning in children and adults. Hum Brain Mapp (2014) 35:1390–403. doi:10.1002/hbm.22261
48. Cheng DT, Disterhoft JF, Power JM, Ellis DA, Desmond JE. Neural substrates underlying human delay and trace eyeblink conditioning. Proc Natl Acad Sci U S A (2008) 105:8108–13. doi:10.1073/pnas.0800374105
49. Knuttinen M-G, Parrish TB, Weiss C, LaBar KS, Gitelman DR, Power JM, et al. Electromyography as a recording system for eyeblink conditioning with functional magnetic resonance imaging. Neuroimage (2002) 17:977–87. doi:10.1006/nimg.2002.1199
50. Ramnani N, Toni I, Josephs O, Ashburner J, Passingham RE. Learning- and expectation-related changes in the human brain during motor learning. J Neurophysiol (2000) 84:3026–35.
51. Lubow RE. Classical eyeblink conditioning and schizophrenia: a short review. Behav Brain Res (2009) 202:1–4. doi:10.1016/j.bbr.2009.03.006
52. Reeb-Sutherland BC, Fox NA. Eyeblink conditioning: a non-invasive biomarker for neurodevelopmental disorders. J Autism Dev Disord (2015) 45:376–94. doi:10.1007/s10803-013-1905-9
53. Bernard JA, Mittal VA. Cerebellar-motor dysfunction in schizophrenia and psychosis-risk: the importance of regional cerebellar analysis approaches. Front Psychiatry (2014) 5:160. doi:10.3389/fpsyt.2014.00160
54. Taylor JA, Spence KW. Conditioning level in the behavior disorders. J Abnorm Psychol (1954) 49:497–502. doi:10.1037/h0055951
55. O’Connor N, Rawnsley K. Two types of conditioning in psychotics and normals. J Abnorm Psychol (1959) 58:157–61. doi:10.1037/h0043677
56. Spain B. Eyelid conditioning and arousal in schizophrenic and normal subjects. J Abnorm Psychol (1966) 71:260–6. doi:10.1037/h0023596
57. Sears LL, Andreasen NC, O’Leary DS. Cerebellar functional abnormalities in schizophrenia are suggested by classical eyeblink conditioning. Biol Psychiatry (2000) 48:204–9. doi:10.1016/S0006-3223(00)00247-X
58. Hofer E, Doby D, Anderer P, Dantendorfer K. Impaired conditional discrimination learning in schizophrenia. Schizophr Res (2001) 51:127–36. doi:10.1016/S0920-9964(00)00118-3
59. Stevens A, Schwarz J, Schwarz B, Ruf I, Kolter T, Czekalla J. Implicit and explicit learning in schizophrenics treated with olanzapine and with classic neuroleptics. Psychopharmacology (2002) 160:299–306. doi:10.1007/s00213-001-0974-1
60. Marenco S, Weinberger DR, Schreurs BG. Single-cue delay and trace classical conditioning in schizophrenia. Biol Psychiatry (2003) 53:390–402. doi:10.1016/S0006-3223(02)01506-8
61. Brown SM, Kieffaber PD, Carroll CA, Vohs JL, Tracy JA, Shekhar A, et al. Eyeblink conditioning deficits indicate timing and cerebellar abnormalities in schizophrenia. Brain Cogn (2005) 58:94–108. doi:10.1016/j.bandc.2004.09.011
62. Edwards CR, Newman S, Bismark A, Skosnik PD, O’Donnell BF, Shekhar A, et al. Cerebellum volume and eyeblink conditioning in schizophrenia. Psychiatry Res (2008) 162:185–94. doi:10.1016/j.pscychresns.2007.06.001
63. Bolbecker AR, Mehta CS, Edwards CR, Steinmetz JE, O’Donnell BF, Hetrick WP. Eye-blink conditioning deficits indicate temporal processing abnormalities in schizophrenia. Schizophr Res (2009) 111:182–91. doi:10.1016/j.schres.2009.03.016
64. Bolbecker AR, Steinmetz AB, Mehta CS, Forsyth JK, Klaunig MJ, Lazar EK, et al. Exploration of cerebellar-dependent associative learning in schizophrenia: effects of varying and shifting interstimulus interval on eyeblink conditioning. Behav Neurosci (2011) 125:687–98. doi:10.1037/a0025150
65. Forsyth JK, Bolbecker AR, Mehta CS, Klaunig MJ, Steinmetz JE, O’Donnell BF, et al. Cerebellar-dependent eyeblink conditioning deficits in schizophrenia spectrum disorders. Schizophr Bull (2012) 38:751–9. doi:10.1093/schbul/sbq148
66. Parker KL, Andreasen NC, Liu D, Freeman JH, O’Leary DS. Eyeblink conditioning in unmedicated schizophrenia patients: a positron emission tomography study. Psychiatry Res (2013) 214:402–9. doi:10.1016/j.pscychresns.2013.07.006
67. Bolbecker AR, Kent JS, Petersen IT, Klaunig MJ, Forsyth JK, Howell JM, et al. Impaired cerebellar-dependent eyeblink conditioning in first-degree relatives of individuals with schizophrenia. Schizophr Bull (2014) 40:1001–10. doi:10.1093/schbul/sbt112
68. Coesmans M, Röder CH, Smit AE, Koekkoek SK, De Zeeuw CI, Frens MA, et al. Cerebellar motor learning deficits in medicated and medication-free men with recent-onset schizophrenia. J Psychiatry Neurosci (2014) 39:E3–11. doi:10.1503/jpn.120205
69. Spence K. Learning and performance in eyelid conditioning as a function of intensity of the UCS. J Exp Psychol (1953) 45:57–63. doi:10.1037/h0058815
70. Spence K, Taylor JA. The relation of conditioned response strength to anxiety in normal, neurotic, and psychotic subjects. J Exp Psychol (1953) 45:265–72. doi:10.1037/h0056392
71. Daum I, Channon S, Polkey CE, Gray JA. Classical conditioning after temporal lobe lesions in man: impairment in conditional discrimination. Behav Neurosci (1991) 105:396–408. doi:10.1037/0735-7044.105.3.396
72. Smit AE, van der Geest JN, Vellema M, Koekkoek SK, Willemsen R, Govaerts LC, et al. Savings and extinction of conditioned eyeblink responses in fragile X syndrome. Genes Brain Behav (2008) 7:770–7. doi:10.1111/j.1601-183X.2008.00417.x
73. Andreasen NC, Paradiso S, O’Leary DS. “Cognitive dysmetria” as an integrative theory of schizophrenia: a dysfunction of the cortical-subcortical-cerebellar circuitry. Schizophr Bull (1998) 24:203–18. doi:10.1093/oxfordjournals.schbul.a033321
74. Ito M. Control of mental activities by internal models in the cerebellum. Nat Rev Neurosci (2008) 9:304–13. doi:10.1038/nrn2332
75. Ramnani N. The primate cortico-cerebellar system: anatomy and function. Nat Rev Neurosci (2006) 7:511–22. doi:10.1038/nrn1953
76. Wolpert DM, Miall RC. Forward models for physiological motor control. Neural Netw (1996) 9:1265–79. doi:10.1016/S0893-6080(96)00035-4
77. Herculano-Houzel S. Not all brains are made the same: new views on brain scaling in evolution. Brain Behav Evol (2011) 78:22–36. doi:10.1159/000327318
78. Strick PL, Dum RP, Fiez JA. Cerebellum and nonmotor function. Annu Rev Neurosci (2009) 32:413–34. doi:10.1146/annurev.neuro.31.060407.125606
79. Christian KM, Thompson RF. Neural substrates of eyeblink conditioning: acquisition and retention. Learn Mem (2003) 10(6):427–55. doi:10.1101/lm.59603
80. Yeganeh-Doost P, Gruber O, Falkai P, Schmitt A. The role of the cerebellum in schizophrenia: from cognition to molecular pathways. Clinics (São Paulo) (2011) 66:71–7. doi:10.1590/S1807-59322011001300009
81. Javitt DC. Twenty-five years of glutamate in schizophrenia: are we there yet? Schizophr Bull (2012) 38:911–3. doi:10.1093/schbul/sbs100
82. Chen G, Steinmetz JE. Intra-cerebellar infusion of NMDA receptor antagonist AP5 disrupts classical eyeblink conditioning in rabbits. Brain Res (2000) 887:144–56. doi:10.1016/S0006-8993(00)03005-5
83. Linden DJ. From molecules to memory in the cerebellum. Science (2003) 301:1682–5. doi:10.1126/science.1090462
84. Piochon C, Levenes C, Ohtsuki G, Hansel C. Purkinje cell NMDA receptors assume a key role in synaptic gain control in the mature cerebellum. J Neurosci (2010) 30:15330–5. doi:10.1523/JNEUROSCI.4344-10.2010
85. Hansel C, Linden DJ, D’Angelo E. Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat Neurosci (2001) 4:467–75. doi:10.1038/87419
86. Gao Z, van Beugen BJ, De Zeeuw CI. Distributed synergistic plasticity and cerebellar learning. Nat Rev Neurosci (2012) 13:619–35. doi:10.1038/nrn3312
Keywords: schizophrenia, cerebellum, eyeblink conditioning, associative learning, cognitive dysmetria
Citation: Kent JS, Bolbecker AR, O’Donnell BF and Hetrick WP (2015) Eyeblink Conditioning in Schizophrenia: A Critical Review. Front. Psychiatry 6:146. doi: 10.3389/fpsyt.2015.00146
Received: 31 May 2015; Accepted: 22 September 2015;
Published: 18 December 2015
Edited by:
Tracy L. Greer, University of Texas Southwestern Medical Center, USAReviewed by:
Litao Sun, The Scripps Research Institute (TSRI), USAJohn T. Green, University of Vermont, USA
Copyright: © 2015 Kent, Bolbecker, O’Donnell and Hetrick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William P. Hetrick, d2hldHJpY2tAaW5kaWFuYS5lZHU=