 Caroline Madeira1
Caroline Madeira1 Charles Vargas-Lopes1
Charles Vargas-Lopes1 Carlos Otávio Brandão2
Carlos Otávio Brandão2 Taylor Reis2
Taylor Reis2 Jerson Laks2
Jerson Laks2 Rogerio Panizzutti1,2*
Rogerio Panizzutti1,2* Sergio T. Ferreira3,4*
Sergio T. Ferreira3,4*- 1Institute of Biomedical Sciences, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 2Institute of Psychiatry, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 3Institute of Medical Biochemistry Leopoldo de Meis, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 4Institute of Biophysics Carlos Chagas Filho, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
Recent evidence suggests that Alzheimer's disease (AD) and depression share common mechanisms of pathogenesis. In particular, deregulation of glutamate-mediated excitatory signaling may play a role in brain dysfunction in both AD and depression. We have investigated levels of glutamate and its precursor glutamine in the cerebrospinal fluid (CSF) of patients with a diagnosis of probable AD or major depression compared to healthy controls and patients with hydrocephalus. Patients with probable AD or major depression showed significantly increased CSF levels of glutamate and glutamine compared to healthy controls or hydrocephalus patients. Furthermore, CSF glutamate and glutamine levels were inversely correlated to the amyloid tau index, a biomarker for AD. Results suggest that glutamate and glutamine should be further explored as potential CSF biomarkers for AD and depression.
Introduction
Epidemiological and clinical studies suggest an association between Alzheimer's disease (AD) and depression (1, 2), the latter being a risk factor for development of AD and other forms of dementia (3–5). About half of the patients with major depression show cognitive impairment that can be persistent and last even after remission of the acute phase of symptoms (6). This could mean cognitive impairment precedes or predisposes to depression or, alternatively, that depression produces persistent cognitive deficits (6, 7). Recent studies have further suggested that similar pathogenic mechanisms may underlie cognitive changes in depression and in AD. Indeed, we have demonstrated that soluble oligomers of the amyloid-β peptide (AβOs), neurotoxins that accumulate in the AD brain and are thought to cause synapse failure and memory loss in AD (8, 9), induce depressive-like behavior in mice (10, 11), providing insight into molecular/cellular mechanisms potentially connecting both disorders.
Glutamate is the major excitatory neurotransmitter in the mammalian brain. However, glutamate at high concentrations in the synaptic cleft is toxic and may result in neuronal death, a phenomenon generally termed excitotoxicity (12). Excitotoxicity mediated by aberrant activation of glutamate receptors, notably of N-methyl-D-aspartate (NMDA) receptors, has been related to the neuropathology of AD (13–16). Consistent with a role of abnormal NMDA receptor function in AD, memantine, an NMDA receptor blocker, is one of the few drugs in clinical use for treatment of moderate to severe AD (17, 18). Interestingly, NMDA receptor antagonists, in particular ketamine, also produce rapid and consistent antidepressant action in subjects with major depression (19, 20).
To prevent excitotoxicity after physiological neurotransmission, glutamate is rapidly removed from the synaptic cleft and converted into glutamine by glutamine synthetase in glial cells (21). Glutamine is then transported back to the presynaptic neuron, where it is converted to glutamate by glutaminase (22). Dysfunction in the glutamate-glutamine cycle could contribute to excitotoxicity mediated by glutamate (22).
Changes in glutamate and glutamine may occur in AD and depression, previous studies of glutamate and glutamine levels in the cerebrospinal fluid (CSF) present controversial findings. Some studies have reported that CSF glutamate levels in AD patients were higher than in the controls (23, 24) or MCI (25), whereas others studies have found decreased (26, 27) or unchanged glutamate levels in AD patients compared to healthy controls (28, 29). Studies that measured CSF glutamine levels have also reported controversial results, with studies reporting increased (30), decreased (28, 31) or no change in glutamine levels in AD patients compared with controls (23, 24, 26).
Controversial findings have also been observed in CSF glutamate and glutamine levels of patients with depression. One study found an increase in CSF glutamine levels (32), whereas another study showed a decrease in CSF glutamate levels in patients with depression compared to controls (33). Moreover, recent studies reported no differences in CSF glutamate and glutamine levels in patients with depression compared to controls (34, 35).
Here, we investigated whether changes in brain levels of glutamate and glutamine are present in major depression and AD. We have studied glutamate and glutamine levels in the CSF of age-matched patients with probable AD or major depression compared to two control groups: healthy subjects and patients with an unrelated neurological condition (normal pressure hydrocephalus). We further investigated the correlation between glutamate and glutamine levels, Mini-Mental State Examination (MMSE) scores, and the Innotest amyloid tau index, a biomarker for AD (36, 37).
Materials and Methods
Human Subjects
The current study was approved by the Committee of Research Ethics involving human subjects of the Institute of Psychiatry of Federal University of Rio de Janeiro (protocol # 32liv2/07). Subjects provided written informed consent before study admission.
All subjects underwent an evaluation comprising full medical history, physical and neurological examination, laboratory tests and neuropsychological assessments. The complete work up is detailed elsewhere (38). The severity of dementia was classified according to the Clinical Dementia Rating (CDR) (39). Mini-Mental State Examination (MMSE) (40) was additionally used to evaluate cognitive state. Exclusion criteria included more than 10 packs/year of cigarette smoking, alcohol abuse or other current or previous psychiatric or clinical disorder. The probable AD group included 21 subjects recruited from the AD Center of the Institute of Psychiatry of the Federal University of Rio de Janeiro. Subjects were diagnosed according to National Institute of Neurological and Communicative Disorders and Stroke (NINCDS), Alzheimer's Disease and Related Disorders Association (ADRDA) and Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) criteria (41). The depression group included 9 subjects with major depression, recruited from the Institute of Psychiatry of the Federal University of Rio de Janeiro and diagnosed according to DSM-IV criteria. The severity of depression was measured by the Brazilian version of the Hamilton Depression Scale (HAM-D) (42–44). Only patients in the first episode of major depression were included in the study. The healthy control group included 10 subjects without any clinical disease or neuropsychiatric disorder. We also studied an additional control group including 9 patients with hydrocephalus, diagnosed according to International Classification of Diseases (ICD-10) (45). Both healthy and hydrocephalus subjects were recruited at Neurolife Laboratory, a private clinic specialized in CSF analysis in the city of Rio de Janeiro. Selected characteristics of each studied group are presented in Table 1. Detailed demographics of individual subjects are presented in Table S1.

Table 1. Characteristics of study subjects.
Psychotropic medications used by probable AD patients were: rivastigmine (47.6%; n = 10), risperidone (38.1%; n = 8), memantine (28.6%; n = 6), donepezil (23.8%; n = 5), clonazepam (19.0%; n = 4), citalopram (4.8%; n = 1), trazodone (4.8%; n = 1), biperiden (4.8%; n = 1), escitalopram (4.8%; n = 1), and mirtazapine (4.8%; n = 1). Two (9.5%) patients with probable AD were not taking any medication at the time of the study. Medications used by major depression patients were: citalopram (11.1%; n = 1), clonazepam (33.3%; n = 3), fluoxetine (22.2%; n = 2), desvenlafaxine (11.1%; n = 1), paroxetine (11.1%; n = 1), buspirone (11.1%; n = 1), sertraline (11.1%; n = 1), venlafaxine (11.1%; n = 1). One (11.1%) patient with major depression was not taking any medication at the time of the study.
CSF Collection
CSF samples were collected by lumbar puncture in the L3-4 or L4-5 interspace at Neurolife Laboratories (Rio de Janeiro, Brazil) and immediately stored at −80°C. All lumbar punctures were performed between 10 am and noon to limit potential circadian fluctuation in CSF content.
Glutamate and Glutamine Measurements in CSF
Glutamate and glutamine levels in CSF were measured by high performance liquid chromatography (HPLC) as previously described (46–48).
Determination of the Amyloid Tau Index
CSF concentrations of Aβ1-42, total tau (T-tau) and phosphorylated tau (P-tau181) were measured using commercially available enzyme-linked immunosorbent assays (ELISA INNOTEST p-tau-181, INNOTEST htau, INNOTEST β-amyloid (1-42) kits; Innogenetics, Gent, Belgium) according to manufacturer's instructions. The Innotest amyloid tau index (IATI) is a score that combines CSF levels of Aβ1-42 and T-tau (36, 37) and was calculated as:
IATI = Aβ1−42/(240+1.18*T-tau).
Statistical Analysis
Results are presented as means (±S.D.) unless otherwise indicated. Statistical significances between groups were determined by one-way analysis of variance (ANOVA) followed by Bonferroni test for multiple comparisons. Sex distribution was studied using Chi-Square. Interactions between years of education, age, disease duration and levels of glutamate or glutamine were studied using correlation analysis and were demonstrated using Pearson's correlation coefficient (r).
Results
Patient groups did not differ in age, but the sex distribution was significantly different between groups (Table 1). However, logistic regression analysis did not reveal significant interactions between sex and levels of glutamate or glutamine (χ2 = 0.21, p = 0.64 for glutamate; χ2 = 0.93, p = 0.33 for glutamine). Groups also differed in terms of numbers of years of education (Table 1). However, no significant correlations were found between years of education and levels of glutamate or glutamine (r = −0.19, p = 0.18 for glutamate; r = −0.02, p = 0.87 for glutamine). Moreover, glutamate and glutamine levels were not significantly correlated to age (r = 0.04, p = 0.78 for glutamate; r = 0.22, p = 0.12 for glutamine) or disease duration (r = 0.22, p = 0.23 for glutamate; r = 0.03, p = 0.86 for glutamine). None of the medications in use by the patients showed a significant effect on glutamate and glutamine levels (Table S2).
Mean CSF glutamate levels were significantly higher in patients with probable AD compared to healthy controls and hydrocephalus patients (F = 50.8, p < 0.0001) (Table 1; Figure 1A). Interestingly, CSF glutamate levels in the major depression group were similar to the levels found in the probable AD group (Table 1, Figure 1A). The sensitivity and specificity were 95.2 and 100%, respectively (using a cutoff of 13.63 μmol glutamate/l) for the diagnosis of probable AD compared to healthy controls (AUC = 0.99, p < 0.0001). The corresponding ROC curve is presented in Figure S1.
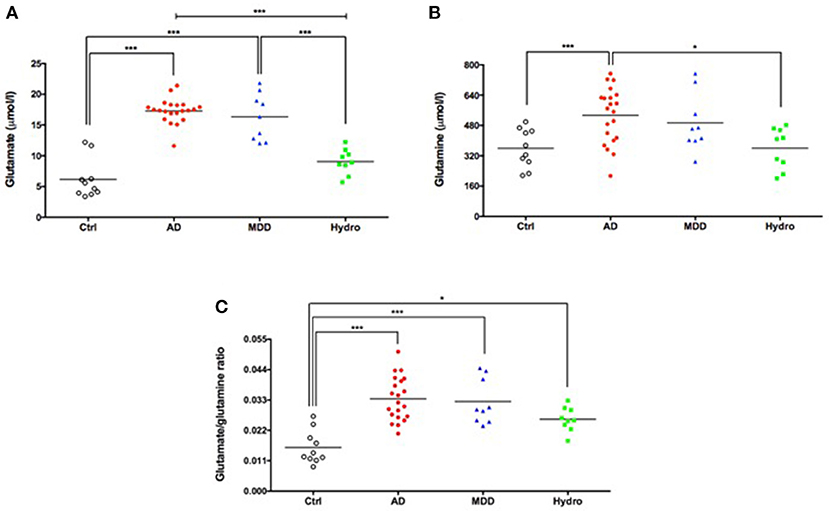
Figure 1. Increased CSF levels of glutamate (A), glutamine (B) and glutamate/glutamine ratio (C) in patients with probable AD and major depression. Symbols correspond to individual subjects. Horizontal lines represent mean values for each group. Statistical significances assessed by one-way ANOVA followed by Bonferroni adjustment for multiple comparisons. *P < 0.05; ***P < 0.001. AD, probable Alzheimer's disease; Ctrl, healthy controls; MDD, major depressive disorder; Hydro, hydrocephalus.
Mean glutamine levels were significantly higher in patients with probable AD than in healthy controls and in the hydrocephalus group (F = 5.92, p = 0.002). Moreover, the mean CSF glutamine level in the major depression group was similar to the mean level found in the group of patients with probable AD (Table 1; Figure 1B).
The glutamate/glutamine ratio, an index of glutamine-glutamate cycle in the brain (49), was significantly higher in all three patient groups (probable AD, major depression and hydrocephalus) compared to healthy controls (Table 1; Figure 1C). Thus, while measurements of CSF glutamate alone robustly separated probable AD and depressive patients from controls and hydrocephalus patients, the glutamate/glutamine ratio was elevated in all three disorders compared to controls, suggesting it was a less specific biomarker for AD and depression.
As expected, the mean MMSE score was significantly lower in patients with probable AD than in healthy controls, major depression and hydrocephalus (Table 1). Interestingly, in subjects without dementia (MMSE above 23) (50), lower MMSE scores were significantly associated with higher CSF glutamate (r = −0.51, p = 0.006) (Figure 2A) and glutamine levels (r = −0.47, p = 0.013) (Figure 2B).
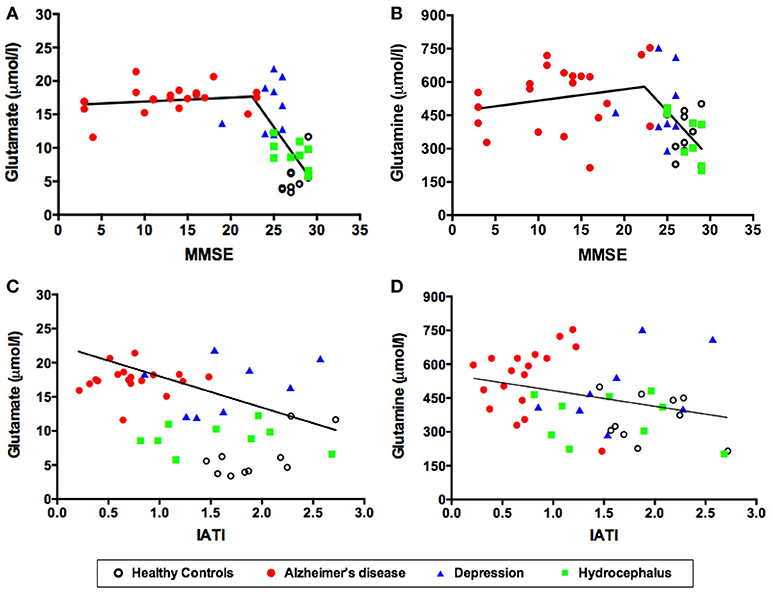
Figure 2. CSF levels of glutamate (A) and glutamine (B) as a function of MMSE scores. CSF levels of glutamate (C) and glutamine (D) as a function of the IATI index. Symbols correspond to individual subjects. Statistical significances assessed by Pearson correlation. MMSE, Mini Mental State Examination; IATI, INNOTEST amyloid/tau index.
As also expected, the mean CSF Innotest amyloid tau index (IATI) was significantly lower in the AD group compared to the other three groups (Table 1). Remarkably, CSF glutamate levels were significantly and inversely correlated to the IATI across the four subject groups (r = −0.45, p = 0.002) (Figure 2C). Glutamine levels were also inversely correlated to the IATI, although the correlation was less robust and significant (r = −0.31, p = 0.04) (Figure 2D).
Finally, CSF glutamate and glutamine levels were analyzed in subject groups separated by their clinical dementia rating (CDR) scores. Individuals with CDR 0.5, 1, 2, and 3 showed significantly elevated mean glutamate levels compared to non-cognitively impaired individuals (CDR 0) (Figure 3A). On the other hand, only the group with CDR 2 exhibited significantly higher glutamine than the CDR 0 group (Figure 3B).
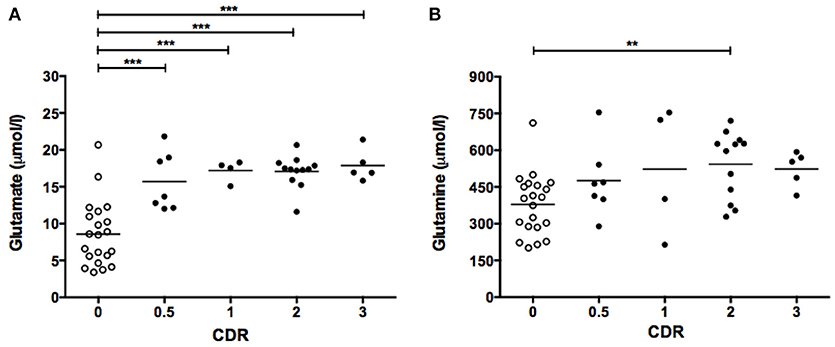
Figure 3. CSF levels of glutamate (A) and glutamine (B) as a function of CDR score. Symbols correspond to individual subjects. Horizontal lines represent mean values for each group. Statistical significances assessed by one-way ANOVA followed by Bonferroni adjustment for selected groups: CDR 0.5, 1, 2, and 3 vs. CDR 0. **P < 0.01; ***P < 0.001. CDR, Clinical Dementia Ratio.
Discussion
We report increased CSF levels of glutamate and glutamine in AD and major depression compared to healthy controls and to patients with normal pressure hydrocephalus. Significantly, lower MMSE and IATI scores were correlated with higher glutamate and glutamine levels in our study cohort.
The current findings are consistent with previous studies reporting increased glutamate in the CSF of patients with probable AD (23, 24). In addition, CSF glutamate levels were found to be significantly elevated in patients with AD in comparison to patients with mild cognitive impairment (25). However, other groups have reported decreased (26, 27) or unchanged glutamate levels in patients with probable AD compared to healthy controls (28, 29). Moreover, our finding of increased CSF glutamine in AD patients is in accord with a previous study (30) but differs from other studies that reported a decrease (28, 31) or no change in glutamine levels in patients with probable AD compared to healthy controls (23, 24, 26). It is noteworthy that the criteria for diagnosis of probable AD in early studies included only clinical features (51), which could lead to greater heterogeneity amongst individuals included in the studies and, hence, to greater variability in results from CSF analysis. More recent revisions in diagnostic and research criteria have advocated the use of biomarkers as closely as possible to the pathological criteria for AD so as to increase the level of certainty in the diagnosis for research purposes (52, 53). All subjects with probable AD studied here were positive for the biomarker amyloid tau index (IATI), thus showing clear evidence of both amyloid-β and tau neuropathology. Interestingly, glutamate and glutamine levels were inversely correlated with individual IATI values across all groups of subjects. We further note that we have previously reported measurements of CSF levels of D-serine, L-serine and glycine in the same patient cohort investigated in the current study (48). We found that D-serine levels in patients with probable AD were significantly higher than in healthy controls, but there were no differences in L-serine and glycine levels.
We further found elevated CSF glutamate and glutamine in major depression patients, similar to levels found in the AD group and significantly higher than the levels found in both healthy control individuals and hydrocephalus patients. Previous studies have measured glutamate and glutamine in the CSF of patients with major depression, and they report controversial findings. Levine et al. (32) reported increased glutamine levels in the CSF of depressed patients compared to controls, whereas Frye et al. (33) showed decreased glutamate levels in depression. More recent studies reported no differences in glutamate or glutamine levels in the CSF of depressed patients compared to controls (34, 35). Of note, Levine et al. (32), Frye et al. (33) and Garakani et al. (34) evaluated middle-aged adult patients, while here we studied older patients. On the other hand, Hashimoto et al. (35) also studied older patients, but the MMSE scores average of their study was slightly higher than in our study. More studies are thus warranted to clarify whether glutamate and glutamine are altered in individuals affected by depression and belonging to different age groups. Increased CSF glutamate may be related to the neurodegenerative process thought to occur in major depression. Evidence indicates that inflammation leading to neurodegeneration plays an important role in depression (54–56). Glutamate excitotoxicity may be involved in this cascade as inflammatory mediators increase glutamate release and decrease glutamate uptake in the central nervous system (57–59).
CSF glutamate and glutamine levels were similar in age-matched patients with probable AD or depression, suggesting another shared mechanism of pathogenesis in these two disorders. Accordingly, a prospective study in a large cohort suggested that depression in the older adult is a prodrome rather than a risk factor for AD (60). Given the central role of glutamatergic neurotransmission in synaptic plasticity, learning and memory (61), as well as in regulation of mood (62), the impact of altered glutamate and glutamine levels on neuropathological mechanisms connecting dementia and depression warrants further investigation. Notably, evidence from animals models indicates that AD and depression involves shared mechanisms, such as hyperactivation of NMDA receptors through increased D-serine (48), inflammatory process (10, 11) and serotonergic signaling (11).
What is the significance of increased glutamate and glutamine levels in the CSF of AD patients? The increase in CSF glutamine may be due to increased activity of aspartate aminotransferase, an enzyme that forms glutamine, in the AD brain (30). A possible mechanism to explain the increase in CSF glutamate involves the build-up of Aβ oligomers (AβOs) in the AD brain. AβOs are increasingly recognized as proximal neurotoxins in AD (8, 9, 63, 64) and oligomer levels increase in AD brains. Of note, we have previously demonstrated that AβOs cause an increase in extracellular levels of glutamate in hippocampal neurons (65), and intracerebroventricular injection of AβOs induces both depressive-like behavior and cognitive deficits in mice (10, 11, 66, 67). It may thus be that AβO-instigated increases in brain glutamate levels underlie, at least in part, cognitive and mood alterations in AD.
Interestingly, we found that glutamate and (albeit somewhat less strongly) glutamine levels are significantly and inversely associated with the MMSE score in subjects without dementia (with MMSE above 23). This implies that alterations in CSF glutamate levels sensitively correlate with sub-clinical deterioration in cognitive performance. Thus, CSF measurements of glutamate/glutamine may serve as a biomarker of subtle cognitive changes that, while still within the range of normality, may reveal underlying mechanisms of pathogenesis potentially leading to future dementia. This could be of major clinical utility in terms of detecting pre-clinical dementia, with clear implications for inclusion of subjects in clinical trials and initiation of preventive/treatment strategies prior to overt cognitive deterioration.
A limitation of the present study was the absence of males in the group of patients with major depression. We note, however, that no sex differences in glutamate and glutamine levels were found in the other three patient groups studied. Additionally, the groups of patients with probable AD and major depression showed a clear trend (albeit not statistically significant) toward lower education than the control and hydrocephalus groups. However, analysis of differences in glutamate levels using education as a covariate confirmed that differences between patient groups remained significant (data not shown). Moreover, the cohort we have studied was of modest size, and it may not be representative of the population as a whole. However, the highly significant differences we have found between groups strongly suggest that the differences may hold in larger patient groups. Nevertheless, despite the current statistically robust results, we acknowledge that this is an initial investigation and, thus, a larger study is warranted to confirm and extend the validity of our findings.
In conclusion, glutamate and glutamine levels are increased in the CSF of patients with probable AD. Significantly increased glutamate levels were detected in CDR 0.5 patients, raising the possibility that determination of CSF glutamate levels might constitute an additional biomarker for pre-clinical cognitive deterioration or early stages of dementia. Finally, our finding that glutamate and glutamine levels were also increased in the CSF of older patients with major depression suggests that these amino acids may be involved in shared mechanisms of pathogenesis between AD and major depression.
Author Contributions
All authors certify that they have participated sufficiently in the work to take public responsibility for the content. RP and SF participated in the conception and design of study. CM, RP, and SF wrote the manuscript. CM and CV-L performed the analysis and interpretation of the data. CB, TR, and JL conduced analysis of patients.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank A. C. Rangel and A. Fantinatti for administrative and technical support. We also thank Dr. L. Scoriels for review of the manuscript. This research was supported by grants from DECIT/SCTIE/MS, SESDC, Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) to RP and SF; Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) to RP, SF, and JL; Instituto Nacional de Ciência e Tecnologia de Biologia Estrutural e Bioimagem (INBEB) to RP; and National Institute for Translational Neuroscience to SF. RP was recipient of a Long-Term Fellowship from the Human Frontier Science Program. CM and CV-L were supported by pre-doctoral fellowships from FAPERJ and CNPq.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2018.00561/full#supplementary-material
Abbreviations
AD, Alzheimer's disease; ADRDA, Alzheimer's Disease and Related Disorders Association; ANOVA, Analysis of variance; Aβ, amyloid-β peptide; AβO, amyloid-β oligomer; CDR, Clinical Dementia Rating; CSF, cerebrospinal fluid; DSM-IV, Diagnostic and Statistical Manual of Mental Disorders; GLX, glutamate plus glutamine concentrations; HPLC, high performance liquid chromatography; IATI, Innotest amyloid tau index; ICD-10, International Classification of Diseases; MMSE, Mini-Mental State Examination; MRS, magnetic resonance spectroscopy; NINCDS, National Institute of Neurological and Communicative Disorders and Stroke; NMDA, N-methyl-D-aspartate; P-tau181, phosphorylated tau; T-tau, total tau.
References
1. Zubenko GS, Zubenko WN, McPherson S, Spoor E, Marin DB, Farlow MR, et al. A collaborative study of the emergence and clinical features of the major depressive syndrome of Alzheimer's disease. Am J Psychiatry (2003) 160:857–66. doi: 10.1176/appi.ajp.160.5.857
2. Gualtieri CT, Johnson LG. Age-related cognitive decline in patients with mood disorders. Prog Neuropsychopharmacol Biol Psychiatry (2008) 32:962–7. doi: 10.1016/j.pnpbp.2007.12.030
3. Green RC, Cupples LA, Kurz A, Auerbach S, Go R, Sadovnick D, et al. Depression as a risk factor for Alzheimer disease: the MIRAGE Study. Arch Neurol. (2003) 60:753–759. doi: 10.1001/archneur.60.5.753
4. Saczynski JS, Beiser A, Seshadri S, Auerbach S, Wolf PA, Au R. Depressive symptoms and risk of dementia: the Framingham Heart Study. Neurology (2010) 75:35–41. doi: 10.1212/WNL.0b013e3181e62138
5. Diniz BS, Butters MA, Albert SM, Dew MA, Reynolds CF 3rd. Late-life depression and risk of vascular dementia and Alzheimer's disease: systematic review and meta-analysis of community-based cohort studies. Br J Psychiatry (2013) 202:329–35. doi: 10.1192/bjp.bp.112.118307
6. Reppermund S, Ising M, Lucae S, Zihl J. Cognitive impairment in unipolar depression is persistent and non-specific: further evidence for the final common pathway disorder hypothesis. Psychol Med. (2009) 39:603–14. doi: 10.1017/S003329170800411X
7. Reppermund S, Zihl J, Lucae S, Horstmann S, Kloiber S, Holsboer F, et al. Persistent cognitive impairment in depression: the role of psychopathology and altered hypothalamic-pituitary-adrenocortical (HPA) system regulation. Biol Psychiatry (2007) 62:400–6. doi: 10.1016/j.biopsych.2006.09.027
8. Ferreira ST, Klein WL. The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer's disease. Neurobiol Learn Mem. (2011) 96:529–543. doi: 10.1016/j.nlm.2011.08.003
9. Mucke L, Selkoe DJ. Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. (2012) 2:a006338. doi: 10.1101/cshperspect.a006338
10. Ledo JH, Azevedo EP, Clarke JR, Ribeiro FC, Figueiredo CP, Foguel D, et al. Amyloid-beta oligomers link depressive-like behavior and cognitive deficits in mice. Mol Psychiatry (2013) 18:1053–4. doi: 10.1038/mp.2012.168
11. Ledo JH, Azevedo EP, Beckman D, Ribeiro FC, Santos LE, Razolli DS, et al. Cross talk between brain innate immunity and serotonin signaling underlies depressive-like behavior induced by Alzheimer's amyloid-β oligomers in mice. J Neurosci. (2016) 36:12106–16 doi: 10.1523/JNEUROSCI.1269-16.2016
12. Greene JG, Greenamyre JT. Bioenergetics and glutamate excitotoxicity. Prog Neurobiol. (1996) 48:613–34.
13. Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer's disease. Neurochem Int. (2004) 45:583–95. doi: 10.1016/j.neuint.2004.03.007
14. Dong XX, Wang Y, Qin ZH. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin. (2009) 30:379–87. doi: 10.1038/aps.2009.24
15. Paula-Lima AC, Brito-Moreira J, Ferreira ST. Deregulation of excitatory neurotransmission underlying synapse failure in Alzheimer's disease. J Neurochem. (2013) 126:191–202. doi: 10.1111/jnc.12304
16. Wang R, Reddy PH. Role of glutamate and NMDA receptors in Alzheimer's disease. J Alzheimers Dis. (2017) 57:1041–8. doi: 10.3233/JAD-160763
17. Lipton SA. The molecular basis of memantine action in Alzheimer's disease and other neurologic disorders: low-affinity, uncompetitive antagonism. Curr Alzheimer Res. (2005) 2:155–65. doi: 10.2174/1567205053585846
18. McKeage K. Memantine: a review of its use in moderate to severe Alzheimer's disease. CNS Drugs (2009) 23:881–97. doi: 10.2165/11201020-000000000-00000
19. Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry (2000) 47:351–4. doi: 10.1016/S0006-3223(99)00230-9
20. Zarate CA Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry (2006) 63:856–64. doi: 10.1001/archpsyc.63.8.856
21. Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem. (2006) 98:641–53. doi: 10.1111/j.1471-4159.2006.03913.x
22. Walton HS, Dodd PR. Glutamate-glutamine cycling in Alzheimer's disease. Neurochem Int. (2007) 50:1052–66. doi: 10.1016/j.neuint.2006.10.007
23. Pomara N, Singh R, Deptula D, Chou JC, Schwartz MB, LeWitt PA. Glutamate and other CSF amino acids in Alzheimer's disease. Am J Psychiatry (1992) 149:251–4. doi: 10.1176/ajp.149.2.251
24. Jimenez-Jimenez FJ, Molina JA, Gomez P, Vargas C, de Bustos F, Benito-Leon J, et al. Neurotransmitter amino acids in cerebrospinal fluid of patients with Alzheimer's disease. J Neural Transm. (1998) 105:269–77. doi: 10.1007/s007020050073
25. Kaiser E, Schoenknecht P, Kassner S, Hildebrandt W, Kinscherf R, Schroeder J. Cerebrospinal fluid concentrations of functionally important amino acids and metabolic compounds in patients with mild cognitive impairment and Alzheimer's disease. Neurodegener Dis. (2010) 7:251–9. doi: 10.1159/000287953
26. Tohgi H, Abe T, Takahashi S, Kimura M. A selective reduction of excitatory amino acids in cerebrospinal fluid of patients with Alzheimer type dementia compared with vascular dementia of the Binswanger type. Neurosci Lett. (1992) 141:5–8. doi: 10.1016/0304-3940(92)90321-W
27. Martinez M, Frank A, Diez-Tejedor E, Hernanz A. Amino acid concentrations in cerebrospinal fluid and serum in Alzheimer's disease and vascular dementia. J Neural Transm Park Dis Dement Sect. (1993) 6:1–9. doi: 10.1007/BF02252617
28. Smith CC, Bowen DM, Francis PT, Snowden JS, Neary D. Putative amino acid transmitters in lumbar cerebrospinal fluid of patients with histologically verified Alzheimer's dementia. J Neurol Neurosurg Psychiatry (1985) 48:469–471. doi: 10.1136/jnnp.48.5.469
29. Degrell I, Hellsing K, Nagy E, Niklasson F. Amino acid concentrations in cerebrospinal fluid in presenile and senile dementia of Alzheimer type and multi-infarct dementia. Arch Gerontol Geriatr. (1989) 9:123–35. doi: 10.1016/0167-4943(89)90033-2
30. D'Aniello A, Fisher G, Migliaccio N, Cammisa G, D'Aniello E, Spinelli P. Amino acids and transaminases activity in ventricular CSF and in brain of normal and Alzheimer patients. Neurosci Lett. (2005) 388:49–53. doi: 10.1016/j.neulet.2005.06.030
31. Procter AW, Palmer AM, Francis PT, Lowe SL, Neary D, Murphy E, et al. Evidence of glutamatergic denervation and possible abnormal metabolism in Alzheimer's disease. J Neurochem. (1988) 50:790–802. doi: 10.1111/j.1471-4159.1988.tb02983.x
32. Levine J, Panchalingam K, Rapoport A, Gershon S, McClure RJ, Pettegrew JW. Increased cerebrospinal fluid glutamine levels in depressed patients. Biol Psychiatry (2000) 47:586–593. doi: 10.1016/S0006-3223(99)00284-X
33. Frye MA, Tsai GE, Huggins T, Coyle JT, Post RM. Low cerebrospinal fluid glutamate and glycine in refractory affective disorder. Biol Psychiatry (2007) 61:162–166. doi: 10.1016/j.biopsych.2006.01.024
34. Garakani A, Martinez JM, Yehuda R, Gorman JM. Cerebrospinal fluid levels of glutamate and corticotropin releasing hormone in major depression before and after treatment. J Affect Disord. (2013) 146:262–265. doi: 10.1016/j.jad.2012.06.037
35. Hashimoto K, Bruno D, Nierenberg J, Marmar CR, Zetterberg H, Blennow K, et al. Abnormality in glutamine-glutamate cycle in the cerebrospinal fluid of cognitively intact elderly individuals with major depressive disorder: a 3-year follow-up study. Transl Psychiatry (2016) 6:e744. doi: 10.1038/tp.2016.8
36. Hulstaert F, Blennow K, Ivanoiu A, Schoonderwaldt HC, Riemenschneider M, De Deyn PP, et al. Improved discrimination of AD patients using beta-amyloid(1-42) and tau levels in CSF. Neurology (1999) 52:1555–62. doi: 10.1212/WNL.52.8.1555
37. Tabaraud F, Leman JP, Milor AM, Roussie JM, Barriere G, Tartary M, et al. Alzheimer CSF biomarkers in routine clinical setting. Acta Neurol Scand. (2012) 125:416–23. doi: 10.1111/j.1600-0404.2011.01592.x
38. Reis T, Brandao CO, Freire Coutinho ES, Engelhardt E, Laks J. Cerebrospinal fluid biomarkers in Alzheimer's disease and geriatric depression: preliminary findings from Brazil. CNS Neurosci Ther. (2012) 18:524–9. doi: 10.1111/j.1755-5949.2012.00311.x
39. Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology (1993) 43:2412–4. doi: 10.1212/WNL.43.11.2412-a
40. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. (1975) 12:189–98. doi: 10.1016/0022-3956(75)90026-6
41. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorder, 4th ed. Washington, DC: American Psychiatric Association (1994).
42. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry (1960) 23:56–62. doi: 10.1136/jnnp.23.1.56
43. Moreno DH, Moreno RA, Calil HM. A Brazilian experience of treatment-resistant depression. In Clin Psychopharmacol. (1994) 2:11–6. doi: 10.1097/00004850-199406002-00003
44. Maier W, Philipp M. Comparative analysis of observer depression scales. Acta Psychiatr Scand. (1985) 72:239–45. doi: 10.1111/j.1600-0447.1985.tb02601.x
45. WHO. Mental Behavioral and Developmental Disorders. In International Statistical Classification of Diseases. Geneva: WHO (1992).
46. Hashimoto A, Nishikawa T, Oka T, Takahashi K, Hayashi T. Determination of free amino acid enantiomers in rat brain and serum by high-performance liquid chromatography after derivatization with N-tert-butyloxycarbonyl-L-cysteine and o-phthaldialdehyde. J Chromatogr. (1992) 582:41–8. doi: 10.1016/0378-4347(92)80300-F
47. Calcia MA, Madeira C, Alheira FV, Silva TC, Tannos FM, Vargas-Lopes C, et al. Plasma levels of D-serine in Brazilian individuals with schizophrenia. Schizophr Res. (2012) 142:83–7. doi: 10.1016/j.schres.2012.09.014
48. Madeira C, Lourenco MV, Vargas-Lopes C, Suemoto CK, Brandão CO, Reis T, et al. D-serine levels in Alzheimer's disease: implications for novel biomarker development. Transl Psychiatry (2015) 5:e561. doi: 10.1038/tp.2015.52
49. Hashimoto K, Malchow B, Falkai P, Schmitt A. Glutamate modulators as potential therapeutic drugs in schizophrenia and affective disorders. Eur Arch Psychiatry Clin Neurosci. (2013) 263:367. doi: 10.1007/s00406-013-0399-y
50. Kochhann R, Varela JS, Lisboa CS de M, Chaves MLF. The Mini Mental State Examination: Review of cutoff points adjusted for schooling in a large Southern Brazilian sample. Dement Neuropsychol. (2010) 4:35–41. doi: 10.1590/S1980-57642010DN40100006
51. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology (1984) 34:939–44. doi: 10.1212/WNL.34.7.939
52. Jack CR Jr, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. (2011) 7:257–62. doi: 10.1016/j.jalz.2011.03.004
53. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr., Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. (2011) 7:263–9. doi: 10.1016/j.jalz.2011.03.005
54. Hurley LL, Tizabi Y. Neuroinflammation, neurodegeneration, and depression. Neurotox Res. (2013) 23:131–44. doi: 10.1007/s12640-012-9348-1
55. Leonard BE. The concept of depression as a dysfunction of the immune system. Curr Immunol Rev. (2010) 6:205–12. doi: 10.2174/157339510791823835
56. Hermida AP, McDonald WM, Steenland K, Levey A. The association between late-life depression, mild cognitive impairment and dementia: is inflammation the missing link? Expert Rev Neurother. (2012) 12:1339–50. doi: 10.1586/ern.12.127
57. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry (2009) 65:732–41. doi: 10.1016/j.biopsych.2008.11.029
58. Niciu MJ, Ionescu DF, Richards EM, Zarate CA. Glutamate and its receptors in the pathophysiology and treatment of major depressive disorder. J Neural Transm. (2014) 121:907–24. doi: 10.1007/s00702-013-1130-x
59. Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. (2016) 16:22–34. doi: 10.1038/nri.2015.5
60. Heser K, Tebarth F, Wiese B, Eisele M, Bickel H, Kohler M, et al. Age of major depression onset, depressive symptoms, and risk for subsequent dementia: results of the German Study on Ageing, Cognition, and Dementia in Primary Care Patients (AgeCoDe). Psychol Med. (2012) 43:1597–610. doi: 10.1017/S0033291712002449
61. Riedel G, Platt B, Micheau J. Glutamate receptor function in learning and memory. Behav Brain Res. (2003) 140:1–47. doi: 10.1016/S0166-4328(02)00272-3
62. Machado-Vieira R, Salvadore G, Ibrahim LA, Diaz-Granados N, Zarate CA Jr. Targeting glutamatergic signaling for the development of novel therapeutics for mood disorders. Curr Pharm Des. (2009) 15:1595–611. doi: 10.2174/138161209788168010
64. Ferreira ST, Lourenco MV, Oliveira MM, De Felice FG. Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer's disease. Front Cell Neurosci. (2015) 9:191. doi: 10.3389/fncel.2015.00191
65. Brito-Moreira J, Paula-Lima AC, Bomfim TR, Oliveira FB, Sepulveda FJ, De Mello FG, et al. Abeta oligomers induce glutamate release from hippocampal neurons. Curr Alzheimer Res. (2011) 8:552–62. doi: 10.2174/156720511796391917
66. Figueiredo CP, Clarke JR, Ledo JH, Ribeiro FC, Costa CV, Melo HM, et al. Memantine rescues transient cognitive impairment caused by high-molecular-weight aβ oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J Neurosci. (2013) 33:9626–34. doi: 10.1523/JNEUROSCI.0482-13.2013
Keywords: Alzheimer's disease, depression, glutamate, glutamine, cerebrospinal fluid, innotest amyloid tau index
Citation: Madeira C, Vargas-Lopes C, Brandão CO, Reis T, Laks J, Panizzutti R and Ferreira ST (2018) Elevated Glutamate and Glutamine Levels in the Cerebrospinal Fluid of Patients With Probable Alzheimer's Disease and Depression. Front. Psychiatry 9:561. doi: 10.3389/fpsyt.2018.00561
Received: 19 June 2018; Accepted: 17 October 2018;
Published: 06 November 2018.
Edited by:
Hsien-Yuan Lane, China Medical University, TaiwanReviewed by:
Kenji Hashimoto, Chiba University, JapanChih-Chiang Chiu, Taipei City Psychiatric Center, Taiwan
Cheng-Sheng Chen, Kaohsiung Medical University Hospital, Taiwan
Copyright © 2018 Madeira, Vargas-Lopes, Brandão, Reis, Laks, Panizzutti and Ferreira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rogerio Panizzutti, cm9nZXJpb0BpY2IudWZyai5icg==
Sergio T. Ferreira, ZmVycmVpcmFAYmlvcW1lZC51ZnJqLmJy