 Anna Giménez-Palomo1
Anna Giménez-Palomo1 Seetal Dodd2,3
Seetal Dodd2,3 Gerard Anmella1
Gerard Anmella1 Andre F. Carvalho4,5
Andre F. Carvalho4,5 Giselli Scaini6
Giselli Scaini6 Joao Quevedo6,7,8,9
Joao Quevedo6,7,8,9 Isabella Pacchiarotti1Eduard Vieta1Michael Berk10,11,12*
Isabella Pacchiarotti1Eduard Vieta1Michael Berk10,11,12*- 1Bipolar and Depressives Disorders Unit, Hospital Clínic, University of Barcelona, Institut d'Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Mental Health Research Networking Center (CIBERSAM), Madrid, Spain
- 2Deakin University, The Institute for Mental and Physical Health and Clinical Translation, School of Medicine, Barwon Health, Geelong, VIC, Australia
- 3Department of Psychiatry, Centre for Youth Mental Health, The University of Melbourne, Melbourne, VIC, Australia
- 4Centre for Addiction and Mental Health, Toronto, ON, Canada
- 5Department of Psychiatry, University of Toronto, Toronto, ON, Canada
- 6Translational Psychiatry Program, Faillace Department of Psychiatry and Behavioral Sciences, McGovern Medical School, The University of Texas Health Science Center at Houston, Houston, TX, United States
- 7Neuroscience Graduate Program, The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences, Houston, TX, United States
- 8Translational Psychiatry Laboratory, Graduate Program in Health Sciences, University of Southern Santa Catarina, Criciúma, Brazil
- 9Center of Excellence in Mood Disorders, Faillace Department of Psychiatry and Behavioral Sciences, McGovern Medical School, The University of Texas Health Science Center at Houston, Houston, TX, United States
- 10School of Medicine, The Institute for Mental and Physical Health and Clinical Translation, Deakin University, Barwon Health, Geelong, VIC, Australia
- 11Orygen, The National Centre of Excellence in Youth Mental Health, Parkville, VIC, Australia
- 12Centre for Youth Mental Health, Florey Institute for Neuroscience and Mental Health and the Department of Psychiatry, The University of Melbourne, Melbourne, VIC, Australia
Mitochondria are cellular organelles involved in several biological processes, especially in energy production. Several studies have found a relationship between mitochondrial dysfunction and mood disorders, such as major depressive disorder and bipolar disorder. Impairments in energy production are found in these disorders together with higher levels of oxidative stress. Recently, many agents capable of enhancing antioxidant defenses or mitochondrial functioning have been studied for the treatment of mood disorders as adjuvant therapy to current pharmacological treatments. A better knowledge of mitochondrial physiology and pathophysiology might allow the identification of new therapeutic targets and the development and study of novel effective therapies to treat these specific mitochondrial impairments. This could be especially beneficial for treatment-resistant patients. In this article, we provide a focused narrative review of the currently available evidence supporting the involvement of mitochondrial dysfunction in mood disorders, the effects of current therapies on mitochondrial functions, and novel targeted therapies acting on mitochondrial pathways that might be useful for the treatment of mood disorders.
Introduction
Mitochondria are cellular organelles known to be involved in diverse biological processes, such as adenosine triphosphate (ATP) production, metabolism of reactive oxygen species (ROS), calcium (Ca2+) homeostasis, cell death and survival (1), as well as in synaptic plasticity. Mitochondria are abundant in neuronal dendrites and synaptic terminals. In the brain, which uses high amounts of ATP and does not have the ability to store it (2), their activity is crucial for the modulation of neuronal activity, short- and long-term neuronal plasticity, cellular resilience, and behavioral adaptations, mainly through actions on long-term potentiation (3–6). Moreover, several lines of evidence suggest roles for mitochondria in supporting the different bioenergetic requirements of highly proliferative neural stem cells and postmitotic neurons (7). In this respect, the adaptation of the energy supply to the energy demand and mitochondrial health is central to cellular homeostasis, and appropriate neuronal function (8–10).
Mitochondrial dysfunction is considered a multifactorial phenomenon since it may have multiple causes and affects numerous neurobiological processes, altering synapsis and enhancing apoptosis, which could play a role in the potentially progressive long-term course of some psychiatric disorders (1). Several studies have focused on the presence of impaired energy metabolism in patients with mood disorders, which indicates that mitochondrial dysfunction may play an important role in various aspects of these conditions (2). In summary, the “mitochondrial hypothesis” suggests that mood disorders are triggered, in part, by mitochondrial dysfunction, which can be intimately linked to a wide range of processes associated with treatment outcomes, disease progression, and severity (11, 12). Moreover, mitochondrial dysfunction could pre-dispose vulnerable individuals to these disorders and lastly, be an important target for current and novel potential therapies for mood disorders (2).
Every cell depends on energy production from mitochondria, with much higher demand in neurons, especially in gray matter, which has a high number of synapses and mitochondria (13–15). Besides energy production, mitochondria are sources of cellular growth substrates and play crucial roles in oxidative/nitrosative stress, cell resilience, and death pathways (3, 16, 17).
Mitochondria are the only organelles in the cell that contain their own DNA, called mitochondrial DNA (mtDNA), which contains 37 genes that encode 13 proteins, 22 tRNA, and 2 rRNAs. These genes encode 13 protein subunits of the electron transport chain (ETC). Genes from nuclear DNA (nDNA) code the rest of the mitochondrial proteins (15) and play a role in the regulation of mitochondrial function. In contrast to nDNA, mtDNA is vulnerable to DNA damage due to constant exposure to reactive oxygen species (ROS) and at times insufficient DNA repair mechanisms (18). Moreover, a number of proteomic studies have been conducted to decipher the mitochondrial proteome. Several mitochondrial databases that list the number of mitochondrial proteins are available nowadays (19).
Mitochondria contain two membranes, an outer and an inner membrane, an intermembrane space, and an intracellular matrix. The intracellular matrix contains several enzymes, which participate in the tricarboxylic acid (TCA) cycle and are responsible for the generation of NADH and FADH2 (20). These redox cofactors are required for the generation of ATP through oxidative phosphorylation via the ETC, present within the folds on the inner mitochondrial membrane or cristae, as explained in Figure 1 (21–23).
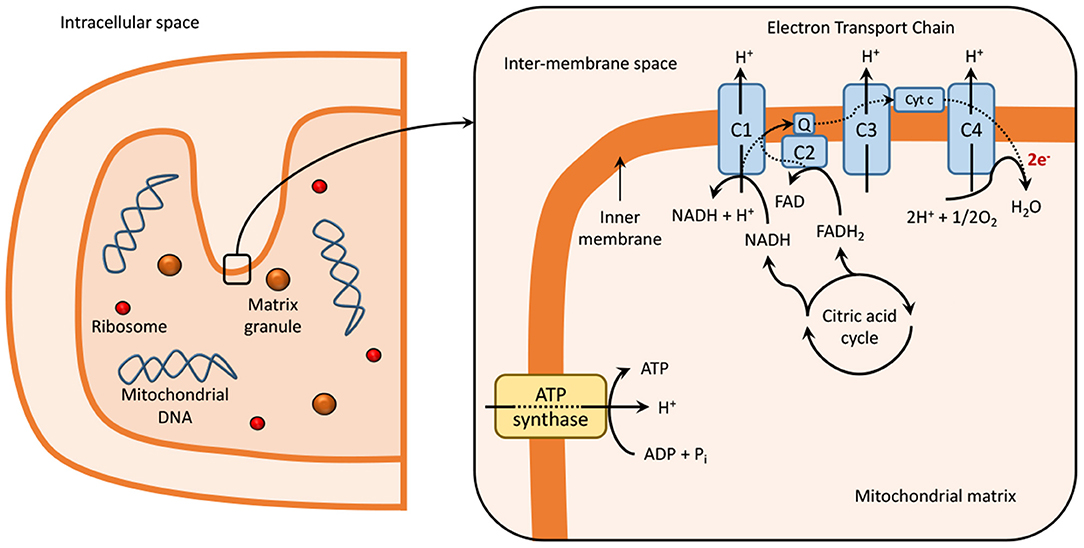
Figure 1. Mitochondria at normal physiological conditions. Dashed lines represent electron transport in the electron transport chain reaction (ETC). The ETC is localized within the inner mitochondrial membrane or cristae of the mitochondria (21–23) and is composed of five multimeric protein complexes (I-IV and ATP-synthase or complex V) that are responsible for ATP production by oxidative phosphorylation. Complex I, or nicotinamide adenine dinucleotide (NAD+), and/or complex II (succinate dehydrogenase), begin the process of oxidative phosphorylation by catalyzing the transfer of electrons from NADH and FADH2, respectively, to coenzyme Q (or ubiquinone). The transfer of electrons is serially conducted through complex III (ubiquinol: cytochrome c oxidoreductase), cytochrome c and complex IV (cytochrome c oxidase), to the terminal acceptor, generating an electrochemical proton gradient that enhances ATP production in complex V via oxidative phosphorylation (1, 24, 25). During this process, single-electrons can escape and produce a single-electron reduction of O2, forming superoxides and other ROS (24, 26, 27). Impaired functioning of ETC can result in excessive ROS production, which leads to the damage of DNA, lipids, proteins, and other molecules in a process known as oxidative damage (1, 24, 27). The generation of ROS is also related to signaling physiological processes, such as synaptic plasticity and memory (28). C, complex; Cyt c, cytocrome c; e-, electron; ECT, electron transport chain; FAD, flavin adenine dinucleotide; Q, coenzyme Q; NAD, nicotinamide adenine dinucleotide.
Given their diverse roles, mitochondria possess several mechanisms to maintain a healthy and functional mitochondrial pool (29), such as neutralizing ROS by antioxidant defenses, the unfolded protein response (UPR), mitochondrial dynamics, biogenesis, and mitophagy (30).
Apart from their involvement in cellular energy production, mitochondria also play an important role in regulating the process of apoptosis through both intrinsic and extrinsic pathways (31). In normal conditions, apoptosis removes those neurons and glia that are functionally compromised or unable to make neuronal connections (15). In the intrinsic mitochondrial-mediated pathway, stimuli such as high levels of intracellular cytoplasmic Ca2+ or ROS, as well as the activation of proapoptotic proteins (i.e., Bcl-2 family members) in the outer mitochondrial membrane (32), trigger a cascade of processes that activate caspases. This results in cleavage of several proteins, DNA fragmentation and cell death (33, 34). In the extrinsic pathway, activation of extracellular death receptors enhances processes that alter membrane permeability, resulting in leakage of proapoptotic factors and apoptosis (34).
Ca2+ homeostasis is another key process in which mitochondria are involved, with Ca2+ a principal secondary messenger that is involved in the regulation of neurotransmission and neuroplasticity in the brain (15). The mitochondrial outer membrane is permeable to Ca2+, and the inner membrane contains Ca2+ uniporters for its inward movement, and Na+/Ca2+ and Ca2+/H+ antiporters for its outward movement (35). Moreover, mitochondria form highly specialized signaling hubs with the ER through the mitochondria-associated membranes (MAMs), allowing the regulation of lipid synthesis and rapid transmission of Ca2+ signals between these organelles (36).
Mitochondrial Ca2+ concentrations increase when cytosolic Ca2+ levels are high and in case of high-energy demand, and decrease when cytosolic levels are low, or the ATP/ADP ratio is high. Ca2+ can modulate oxidative phosphorylation machinery by different mechanisms, including direct binding, enhancing post-transcriptional modification, and also by the activity of a Ca2+-dependent binding protein. It also binds to complex IV and reduces ATP inhibition of this enzyme, enhancing ATP production even in situations of high ATP concentrations (24). ATP synthesis is also enhanced by stimulation of the aspartate-glutamate carriers (AGCs) and the ATP-Mg/Pi (i.e., Ca2+-binding mitochondrial carrier protein, SCaMC-3) transporters on the inner mitochondrial membrane. Ca2+ also leads to increased NADH synthesis and higher production of pyruvate (15).
However, when Ca2+ levels are excessive in the intracellular space or mitochondria they induce stress and excitotoxicity, ATP production is reduced (37, 38), and Ca2+ is extruded through the Na+/Ca2+exchanger and the mitochondrial permeability transition pore (mPTP). Impairment in the control of mitochondrial membrane permeabilization, by mPTP, has been suggested to be responsible for the mitophagy of depolarized mitochondria, induction of apoptosis, and necrosis (15). Ca2+ homeostasis is regulated by different proteins, enzymes, and cellular signaling networks, which may be risk pathways for mood disorders when they are altered (15).
The maintenance of a healthy mitochondrial pool is critically regulated by the dynamics and turnover of the mitochondrial population (29). At the organelle level, mitochondrial quality is sustained through the synthesis of new mitochondria, fusion and fission, and the elimination of damaged mitochondria (30, 39). The balance between fusion and fission events shapes mitochondrial networks to meet metabolic demands (40, 41).
A considerable amount of literature has demonstrated that neuronal activity regulates mitochondria and synapses (42, 43). Neurons depend on oxidative metabolism to meet their high-energy needs (10). Thus, to match the actual local needs in neurons, mitochondria constantly move along microtubes networks, changing mitochondrial trafficking, distribution, anchoring, and membrane dynamics (44).
Mitochondria also regulate synaptic plasticity by transducing some of the effects of glutamate and BDNF. BDNF expression and signaling are promoted by some environmental factors, such as physical activity and cognitive stimulation (2, 45). On the other hand, studies have shown that BDNF enhances ATP synthesis and mitochondrial respiration through several mechanisms, including increases in glucose transport, upregulation of the mitochondrial biogenesis, and respiratory coupling efficiency (46, 47). Moreover, ATP is necessary for the mobilization of synaptic vesicles to the active sites of synapse in neurons. The ATPase complex, by producing cAMP, activates PKA kinase, which allows the mobilization of synaptic vesicles (48). When ATP production is reduced, as in mood disorders, neuronal transmission is consequently impaired (15, 49, 50).
Mitochondria also play a critical role in the neurogenesis, the process of neural stem cell proliferation and differentiation into new neurons. Numerous studies have shown that the mitochondrial genome and mitochondrial proteins are required for neuronal differentiation (51–53). Moreover, accumulating evidence has indicated that the development of a mature mitochondrial network in terms of mitochondrial function and structure is necessary during the differentiation of induced pluripotent stem cells (iPSCs) (54–56).
The aim of this article is to provide a focused narrative review of the currently available evidence supporting (1) the involvement of mitochondrial dysfunction in mood disorders, (2) the effects of current therapies on mitochondria, and (3) novel targeted therapies acting on mitochondrial pathways that might be useful for the treatment of mood disorders. To this end, a literature search was conducted to identify relevant original research articles, reviews including systematic reviews and meta-analyses containing evidence regarding the role of mitochondria in mood disorders, from MEDLINE, SCOPUS, EMBASE, ClinicalTrials, ISI Web of Science and Google Scholar. Based on these reports, we provide a critical overview of the current state of the role of mitochondria in mood disorders, ranging from physiology to pathophysiology, and therapeutic strategies, as well as perspectives on future directions.
Take-Home Message
Mitochondria are cellular organelles involved in a number of biological processes, with a key role in maintaining neuronal homeostasis. They are involved in energy production, metabolism of ROS, calcium homeostasis, apoptosis, synaptic plasticity and neurogenesis, modulating neuronal activity and preventing neuronal damage. In mood disorders, mitochondrial dysfunction leads to the impairment in cellular homeostasis with dysregulation in these mechanisms.
Overview and Discussion
Mitochondrial Dysfunction in Mood Disorders
As mentioned above, mitochondria are the main source for cellular energy but are also responsible for other processes that are crucial for cell functioning and survival, such as apoptosis and Ca2+ homeostasis (57). Impaired mitochondrial functioning may result from a number of causes, including altered expression of mitochondria-related genes, changes in the regulation of mitochondrial biogenesis, mitochondrial structural abnormalities, changes in oxidative phosphorylation and variations in metabolite levels (57). The above-discussed functions make mitochondria indispensable in several network processes, as well as they are associated with aging and a plethora of pathological conditions, such as Alzheimer's, Parkinson's, and Huntington's disease, amyotrophic lateral sclerosis, and psychiatric diseases (6, 8, 57–60). The hypothesis that mitochondrial dysfunction is associated with these conditions is supported by reports that have associated mitochondrial diseases with psychiatric symptoms, especially mood and cognition (1, 61, 62).
Mitochondrial Bioenergetics and Redox in Mood Disorders
The brain is an organ with the highest energy consumption, unique membrane lipid composition, and depends on mitochondrial oxidative phosphorylation, being unable to store glycogen. Since brain mitochondria produce high quantities of ATP but also ROS and RNS, this organ is vulnerable to oxidative damage, which occurs when the oxidative load exceeds antioxidant capacity (2).
Metabolic Changes
A number of studies using neuroimaging and post-mortem brain tissue from patients with BD have shown lower numbers of neuronal and glial cells and lower brain volume in prefrontal and limbic brain regions. Growing evidence suggests mitochondrial dysfunction is implicated in these changes through a reduction in oxidative bioenergetic generation and a shift to anaerobic glycolysis and consequently impaired neuroplasticity, phospholipid metabolism and Ca2+ homeostasis (15, 24). In addition, alterations in various regions of the brain in neurometabolites, including high-energy compounds, have been found in patients with mood disorders. In summary, it has been described that patients with mood disorders have lower levels of phosphocreatine (PCr), N-acetyl-aspartate (NAA), adenosine diphosphate (ADP), and ATP (63, 64). In patients with major depressive disorder (MDD), hypermetabolism could be a consequence of depression severity (65), whereas hypometabolism appears linked to less severe illness (66–69).
Moreover, studies have noted negative correlations between NAA/Creatine + PCr or NAA levels and illness duration in BD (70, 71), with an enzymatic reaction rate abnormality present in BD in the creatine kinase (CK) system, based on the decrease in the forward rate constant of the CK enzyme without alterations on ATP and PCr levels, as well as by downregulation of CK in post-mortem brains of BD patients (72, 73). This hypothesis is consistent with a previous study that showed that individuals with BD could maintain average brain concentrations of high-energy compounds at rest, but there is an underlying abnormality in the mechanism that generates new ATP, which can be uncovered when energy demand is increased (72). Apart from this, studies showing increased lactate and taurine levels and a reduced brain intracellular pH suggest that there is a shift from oxidative phosphorylation to glycolysis as a major source of energy generation in BD (74, 75). Elevated lactate is present, especially in manic phases, in the frontal cortex, caudate, cingulate, and anterior cingulate cortex, which could mean either an overall increase in ATP demand, or defective oxidative metabolism (76).
Mitochondrial Changes in the Electron Transport Chain
By drawing on the hypothesis of mitochondrial impairment on mood disorders, several studies in post-mortem brain, skeletal muscle or blood from patients with mood disorders have shown changes in the enzymatic activities linked to the TCA cycle and ETC, as well as impairment in mitochondrial oxygen consumption. Studies in post-mortem samples of patients with BD and MDD have shown that many mitochondria-related genes are downregulated compared with controls (77). For instance, some studies reported decreased expression of some of the complex I subunits in the cerebellum in bipolar and depressed patients compared with controls (57, 78–81). Not only is decreased expression present, but decreased activity is also reported in MDD and BD patients. A recent study confirmed previous findings showing that the citrate synthase (CS), complex II, and complex IV activities were decreased, while the complex I activity and complex I/citrate synthase ratio were significantly increased in blood platelets of BD patients during a depressive episode. Supporting these findings, Valvassori et al. (82) in isolated mitochondria from peripheral blood mononuclear cells (PBMCs) showed a decrease only in complex II activity in bipolar depressed patients. In contrast, in MDD patients, physiological respiration, the maximal capacity of the electron transport system, and respiratory rate after complex I inhibition are decreased, as well as activity of complex II and CS (83). However, there are studies on mitochondria isolated from PBMCs and blood platelet showing no significant differences in ETC activity in MDD and BD patients (84–87).
Oxidative Damage
Based on the premise that mitochondria are the primary source of ROS, replicated studies documented alterations in multiple aspects of oxidative stress, including an increase in the production of ROS and a reduction of the antioxidant capacity, in MDD and BD patients. Compared to healthy controls, depressed patients show an increase in oxidative stress markers involved in lipid peroxidation (88–90) and a decrease in antioxidant markers (91), as well as lower brain ATP levels (92). Patients with BD have increased lipid peroxidation products in the cingulate cortex (93), and also increased markers of oxidative and nitrosative damage in the prefrontal cortex (1, 94, 95). A meta-analysis that assessed eight oxidative stress markers in patients with BD, including 971 patients with BD and 886 controls, reported an increase of markers of lipid peroxidation, DNA/RNA damage and nitric oxide in the group with BD (96).
Since oxidative damage is the result of the balance between oxidative products and the antioxidant defense, some studies in mood disorders have investigated this system, including superoxide dismutase (SOD), catalase, glutathione S-transferase (GST), and glutathione peroxidase (GPx) (34, 97). Animal studies have shown that chronic stress is associated with lower brain concentrations of GSH, SOD and catalase (98–100). Studies in post-mortem brains of patients with BD have shown lower expression of SOD, microsomal GST, and GPx in frontal areas and lower expression of GPx in the hippocampus (101–103), and reduced activity of SOD and catalase in these patients (104, 105).
However, in some studies SOD activity appears increased in BD during the manic and depressive episodes (104, 106–108), whereas there are studies showing decreased SOD levels in manic patients (1, 109). Savas et al. (106) found increased SOD levels in euthymic bipolar patients (106), whereas others found decreased activity in the euthymic phase (104, 107). A study reported increased activity of GPx in euthymic bipolar patients (107) but not in depressed or manic patients, whereas another showed increased GPx levels in depressed bipolar patients compared to healthy controls (110). Other studies did not find any differences in GPx activity compared to a control group or across different mood states (95, 104). The same uncertain pattern is observed regarding catalase activity. Studies in chronic patients have shown decreased or unaltered catalase activity (105, 107). Contrary to these reports, BD depression at baseline presented a significant increase in catalase levels, with a lower SOD/CAT ratio (110), which was confirmed by previous findings (108). This may be explained by a compensatory mechanism in the early phases of BD, or heterogeneity in other data domains. Compared to controls, reduced GSH and glutathione S-transferase were increased among patients with late-stage BD (95).
Calcium-Dependent Functions
When ATP production is reduced, mitochondrial and cellular functions are impaired due to changes in mitochondrial membrane potential, reducing mitochondrial capability for Ca2+ uptake. Studies in brains from bipolar subjects have shown altered intracellular free Ca2+ levels in blood cells and olfactory neurons (111, 112). Bipolar patients evidence high cellular Ca2+ levels in all states, but especially in mania (113, 114), and also changes in the expression of genes involved in Ca2+ signaling, neuroactive ligand-receptor interaction, and protein kinase PKA/PKC signaling pathways. Moreover, the authors also found changes in the action potential system (115). Indeed, excess Ca2+ affects both neuronal excitability and signaling cascades regulating gene expression, leading to perturbation of multiple neuronal processes, such as dendrite development, synaptic plasticity, and excitatory/inhibitory balance (116).
Calcium/Calmodulin Dependent Protein Kinase Kinase 2 (CaMKK2), is the core component of the Ca2+-calmodulin (Ca2+-CaM) dependent signaling pathway in neurons (117). Through activation of AMP-activated protein kinase (AMPK) and the master mitochondrial regulator, PGC1α, tightly linked to the circadian clock (118), CaMKK2 regulates mitochondrial function and whole-body energy balance. Bipolar disorder is associated with mutations that affect the function or experssion of CaMKK2 (119). Decreased CaMKK2 function leads to decreased BDNF expression, a known biomarker of BD. Lastly, the activity of CAMKK2 is regulated at least in part by the multi-site phosphorylation of the catalytic domain termed the S3-node in a switchable bidirectional manner, a phenomenon critical for understanding the biphasic nature of the disorder (120, 121).
Studies have also reported that DISC1, a protein involved in mitochondrial dynamics and a putative risk factor for BD and MDD (122), interacts with the IP3R1 modulating endoplasmic reticulum-mitochondria Ca2+ transfer (123). One study by Dwivedi et al. (124) showed an increase in IP3R1 binding sites and protein levels in platelets of depressed patients. Moreover, Scaini et al. (125) found that BD patients had higher levels of VDAC and TSPO, suggesting that these could deregulate mitochondrial Ca2+ signaling and increase ROS production.
Mitochondrial Morphology
Other findings that support the hypothesis about the association between mitochondrial dysfunction and mood disorders are changes in mitochondrial morphology, distribution, and degradation. A study undertaken by Cataldo et al. (126) showed that prefrontal neurons from post-mortem brain samples obtained from patients with BD and peripheral cells from patients with BD contain a larger number of smaller-sized mitochondria. The same authors showed an abnormal pattern of clumping and marginalization in the intracellular distribution of mitochondria in peripheral cells, as well as atypically shaped mitochondria (ring- or cup-shaped mitochondrial profiles), suggesting subtle changes in the critical network architecture of mitochondria in the cells (127). Moreover, Mertens et al. (115) showed that iPSC-derived hippocampal dentate gyrus-like neurons of patients with BD had smaller mitochondria than those from healthy controls. As previously described, the balance of fusion and fission modifies the overall morphology of the mitochondrial network (40, 127). Thus, the alterations in these processes observed by downregulation of the mitochondrial fusion-related proteins Mfn-2 and Opa-1 and an upregulation of the fission protein Fis-1 in PBMCs from BD patients (128) might explain the abnormal mitochondrial morphology and distribution findings in patients with BD.
Mitochondrial Degradation and Apoptosis
By drawing on the concept of mitochondrial quality control, Scaini et al. (125) have been able to show that BD patients presented a downregulation of mitophagy-related proteins, Parkin, p62/SQSTM1 and LC3A in PBMCs, followed by NLRP3-inflammasome activation. In summary, the imbalance in mitochondrial fission and fusion toward fission, followed by a decrease in the levels of mitophagy proteins and an increase in the caspase-3 protein levels (125, 128) could suggest that the number of damaged mitochondria exceeds the capacity of mitophagy, and apoptosis becomes the dominant pathway to minimize tissue damage in BD (129, 130). Indeed, evidence has shown that apoptotic genes, such as FAS, BAK and APAF-1 are upregulated in the hippocampus of patients with BD (103). Moreover, Bcl-2, an antiapoptotic protein, is downregulated in BD patients due to different polymorphisms, resulting in Ca2+ homeostasis dysregulation and increased glutamate levels. This is added to the endoplasmic reticulum (ER) stress response seen in all states of BD, mainly in mania (34, 129). Chronic mild stress was shown to reduce the expression of BAG-1, a gene that enhances the anti-apoptotic effects of Bcl-2. This causes the activation of caspases, BCL-2-associated X protein (BAX), and BCL-2 antagonist/killer (BAK) in the mitochondria, which leads to the alteration of membrane permeability and neuronal death (15).
PI3K and Akt are other proteins related to cell survival and proliferation. Their transcription is upregulated in mania, and this pathway is activated by oxidative stress and IL-6, and regulated by AMPK, suggesting that this pathway is active in bipolar mania (103). Akt promotes mitochondrial survival via different routes, such as inhibiting cytochrome c release into the cytosol, which is the final act of mitochondrial apoptosis (131). It also activates the ETC and promotes a shift to glycolytic energy generation in BD. PI3K activates mTOR, which stimulates oxidative phosphorylation (76). Moreover, GSK-3α and GSK-3β are activated in an environment of chronic oxidative stress, such as in BD, with greater activation in mania than in depression. Their inactivation has been correlated with measured clinical improvement (76).
GSK-3 promotes cellular apoptosis by the activation of Fas receptor, also promoted by TNF-α, but which also has a role in neuroprotection. In mania, TNF-α activates GSK-3 to promote neuronal survival, since GSK-3 upregulates NFκβ, and this inhibits TNF-α mediated apoptosis, may inhibit oxidative phosphorylation and promote aerobic glycolysis. TNF-α inhibits mitochondrial biogenesis, which is prevented when SIRT-1 activity is increased (76). Increased levels of NFκβ and SIRT-1 have been found in mania compared to bipolar depression and healthy controls (76). SIRT-1 levels are lower in bipolar depression than in euthymia, and TNF-α levels may be lower in depression than in mania (76). A dysregulated NFκβ system plus genetically influenced anti-apoptotic elements might enable the increased mitochondrial function in mania and the cyclical nature of BD (132). A recent study found an association between the downregulation of 20 genes related to the apoptosis pathway, TNF-α, TLR, and NFκβ signaling pathways and major depressive disorder (76). Moreover, NFkβ causes an increase in cytoplasmic CREB levels in BD patients, which is of interest as the activity of BDNF against ROS is mediated via CREB transcription, and BDNF levels are lower in mania than in depression and lower in BD patients compared to controls (133). Studies also demonstrated that CREB is involved in neurogenesis and is reduced in depression (134).
Inflammatory Changes
Chronic inflammation has been found to be present in all phases of BD (135), since it promotes a pro-inflammatory environment with an increase in cytokine levels, such as IL-1β, IL-6, and TNF-α, and increased nitric oxide in brain and plasma (2). These changes are higher in bipolar depression than in unipolar depression and highest in mania. A meta-analysis showed that patients with major depressive disorder had higher levels of plasma IL-6, TNF-α, and soluble interleukin-2 receptors (sIL-2R) (2).
The aforementioned evidence on mitochondrial bioenergetics pathophysiology in mood disorders is summarized in Figure 2, which represents the biphasic mitochondrial model in BD in depression (reduced mitochondrial biogenesis) and mania (increased mitochondrial biogenesis), and the derived biological processes in the mitochondria, including oxidative stress, inflammation, genetic damage, increased permeability, cytotoxicity, and apoptosis.
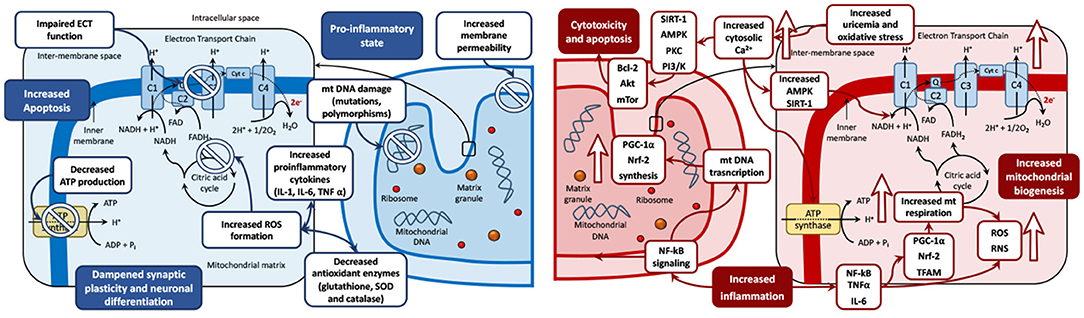
Figure 2. The biphasic mitochondrial model in bipolar disorders. Left: Depression mitochondrial model—Decreased mitochondrial biogenesis. Changes during depressive phases include mitochondrial DNA damage (including mutations and polymorphisms), membrane permeability, and increased formation of ROS. These imbalances lead to a pro-inflammatory state, with increased levels of pro-inflammatory cytokines (IL-1, IL-6 and TNFα) and decreased antioxidant enzymes (glutathione, SOD and catalase). These disturbances can cause cytotoxicity, increased apoptosis, and dampened synaptic plasticity and neuronal differentiation. Antidepressant drugs have shown the capacity to restore mitochondrial disregulation by reestablishing the oxidant/antioxidant balance and counteract the negative effects of depression on the mitochondria. Right: Mania mitochondrial model—Increased mitochondrial biogenesis. Upward arrows symbolize an increase. Changes during manic phases include increased inflammation and elevated production of ROS and RNS, driven by increased activity of the NF-kB signaling pathway. NF-kB signaling stimulates mitochondrial biogenesis via the upregulation of PGC-1α, Nrf-2, and TFAM. PGC-1α and Nrf-2 stimulate mitochondrial respiration, which is a further source of oxidative stress via ROS and RNS production. Increased oxidative stress could induce an increase in the levels of cytosolic Ca2+ ions seen in mania compared to other phases of the disease. Elevated Ca2+ levels can stimulate oxidative phosphorylation and ATP production and may lead to the activation of AMPK and SIRT1, which may increase the activity of NAD+. In an environment of increasing oxidative stress, the activity of SIRT1, AMPK, PKC PI3/K are increased. This can foster mitochondrial survival leading to cytotoxicity and cell death via activation of proapoptotic pathway cascades (Bcl-2, Akt and mTor among others). Increased uric acid levels increase the uptake of Ca2+ ions by mitochondria, increase the mitochondrial membrane potential and therefore enhance ATP production. Antimanic drugs including mood stabilizers and antipsychotics may restore mitochondrial dysregulation by counteracting the mitochondrial imbalance leading to neurogenesis, neuroplasticity, and cell survival. ADP, adenosine diphosphate; ATP, adenosine triphosphate; BDNF, brain-derived neurotrophic factor; C, complex; Cyt c, cytochrome c; e-, electron; ECT, electron transport chain; FAD, flavin adenine dinucleotide; IL, Interleukin; Q, coenzyme Q; mt DNA, mitochondrial DNA; mTOR, mechanistic target of rapamycin; NAD, nicotinamide adenine dinucleotide; NF, nuclear factor; Pi, inorganic phosphate; PI3/K, Inositol 1,4,5 triphosphate; PKC, Protein Kinase C; ROS, reactive oxygen species; RNS, reactive nitrogen species; TFAM, mitochondrial transcription factor A; TNFα, tumor necrosis factor-alpha.
Other specific changes have been observed in mood disorders, which are mentioned in the next sections.
Genetic Changes
Genetic findings also support mitochondrial dysfunction in BD. Some studies have shown that subjects suffering from mitochondrial diseases frequently develop psychiatric symptoms, especially mood symptoms (134).
Increased expression of mitochondrial fission genes and a decreased expression of mitochondrial fusion genes have been associated with depressive behavior in mice (126, 136). Pathological isoforms of DISC1 lead to abnormal neuronal development and mood disorders (137). Genome-wide association studies (GWASs) studies have identified multiple loci, with a small effect, associated with BD risk, including CACNA1C, ANK3, ODZ4, SYNE1, and TRANK1 (34, 122, 138, 139). In addition, Kataoka et al. (140, 141) demonstrated the potential roles of de novo protein-altering mutations and calcium-related genes in BD. Considering the relationship between de novo mutations and clinical phenotypes, the same authors observed significantly earlier disease onset among the BD probands with de novo protein-altering mutations when compared with non-carriers. Although no specific mutations in mtDNA have been associated with BD (142), some mtDNA haplogroups showed significantly lower cerebellar pH, which is also seen in the disorder. Moreover, a rare gene variant of mtDNA, 3644T>C, might be associated with BD, since patients showed a prevalence of 1.43% of the gene variant whereas the prevalence was 0.13% in healthy controls (34, 143). On the other hand, deletions of mtDNA were more commonly found in post-mortem cerebral cortex of patients with BD compared to controls (34, 144), and also in a patient who suffered from depression (145–147). However, other studies did not replicate these findings, which may be due to different methodologies and different brain regions studied (148).
Another study reported higher levels of circulating cell-free mtDNA in patients with MDD compared to healthy controls, while mtDNA content was not significantly different (149–151). Moreover, a recent study found a higher mtDNA copy number and a decreased DNA methylation status in the peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC1α) promoter in patients with MDD, which leads to reduced expression of mitochondrial genes (2, 152). In contrast, Czarny et al. (153) showed that the cellular mtDNA copy number did not differ between healthy and depressed subjects, but it showed a lower capacity for degradation and a higher number of lesions compared to controls (154).
In BD, a meta-analysis for BD-mtDNA copy number studies with a low level of heterogeneity revealed a significant lower mtDNA copy number in patients (154). In contrast, another meta-analysis with a higher level of heterogeneity identified no significant differences between mtDNA copy numbers in BD patients. A recent study showed a decrease in mtDNA copy number and an epigenetic aging acceleration in post-mortem hippocampus from BD patients (155). Focusing on mood-specific states, Wang et al. (156) found that during the depressed and manic states, patients with BD had significantly lower mtDNA copy numbers (157), with the degree of DNA damage associated with the severity of manic and depressive symptoms (157).
Purinergic Dysfunction
The purinergic system appears dysregulated in patients with depression and BD (158). In oxidative stress, the activity of SIRT-1, AMPK, PKA, PKC, GSK, and inositol triphosphate are increased, as well as antiapoptotic proteins, such as Bcl-2, PI3K, mTOR, Akt, and uric acid. Their activation stimulates oxidative phosphorylation via different routes. As the mitochondrial function is increased, oxidative stress is higher and different pathways are activated in order to mitigate the cytotoxic effects of oxidative stress without inducing apoptosis.
Uric acid levels seem to be increased in all phases of BD but are higher in mania than depression or euthymia, which reflects an increase in energy production (159). Increased uric acid levels allow a greater uptake of Ca2+ ions by mitochondria, increased mitochondrial membrane potential, and thus higher ATP production. Lowered levels of uric acid have been described as a risk factor for developing mood disorders. Uric acid acts as a scavenger of peroxynitrite, which has high mitotoxic activity (2). It has other neuroprotective effects, such as increasing AMPK activity, which regulates the function of the CLOCK:BMAL-1 complex and upregulates the activity of SIRT-1, leading also to adaptative responses to oxidative stress for mitochondrial survival and functioning.
Other studies have found that cAMP and PKA are upregulated in BD and regulate the rate of oxidative phosphorylation through the phosphorylation of proteins and enzymes involved in ATP synthesis, such as cytochrome c oxidase, enhancing mitochondrial protection. Cytochrome c oxidase, the terminal respiratory enzyme, key for ATP synthesis, is a metabolic marker for neuronal functional activity (160), with its alterations related to depressive symptoms. The cAMP response element-binding protein (CREB) stimulates cAMP-dependent transcription of ETC enzyme complexes and other proteins from mtDNA, thus stimulating oxidative phosphorylation. The activity of CREB, which enhances the upregulation of CK, key for neuroprotection and energy production, is altered in BD, leading to higher or lower levels of CK in mania and lower levels in mixed states (76).
Genetic variations in the purinergic system and in a number of genes involved in cAMP signaling have been found in BD, which highlights the role of cAMP/CREB on circadian clock genes and to maintain ATP production. Higher activity of antiapoptotic proteins, enzymes and signaling cascades has been observed in mania, which enhances mitochondrial activity (2, 76).
Circadian Clock Genes and Oxidative Phosphorylation
Oxidative stress can enhance changes in circadian clock systems, although chronic oxidative stress provokes pro-survival effects. High levels of ROS resets circadian clocks and induces a range of prosurvival responses and different expression of clock genes secondarily to a pro-inflammatory environment, such as activation of cAMP/CREB signaling. Polymorphisms in clock genes can modify cellular sensitivity to oxidative stress or genotoxic insults. Dysregulation of systems involved in oxidative stress and genetic changes in clock proteins could explain some of the observations in circadian systems in BD.
PKC and inositol triphosphate play a role in the pathogenesis of BD, being associated with and downstream of intracellular Ca2+ levels. In mania, elevated functioning of PKC has been found, which acts by stimulating and protecting mitochondria. Cytosolic Ca2+ activates ATP synthesis enhancing the activity of AMPK through different routes, such as increasing NAD+ and the activity of SIRT-1 (2).
A number of gene variations have shown increased susceptibility for developing more severe forms of BD. These genes control circadian NAD+ concentrations, which increase the activity of SIRT-1 and SIRT-3, and this stimulates oxidative phosphorylation. NAD+ and SIRT-1 directly activate ATP production and upregulate circadian genes, suggesting a pathway of influence in mood disorders (2).
Hypothalamic-Pituitary-Adrenal Axis
Depression is linked to hyperactivity of the hypothalamic-pituitary-adrenal (HPA) axis due to an impairment of the corticosteroid receptor-mediated feedback. This leads to increased secretion of corticotropin-releasing factor (CRF) in the hypothalamus and causes increased levels of glucocorticoids both in the brain and peripherally, being translated into increased mitochondrial activity (2).
In the mitochondria, glucocorticoids form a complex with the antiapoptotic protein Bcl-2 to inhibit the formation of Bax-containing pores on the mitochondrial outer membrane. They also reduce the release of Ca2+ and cytochrome c from the mitochondria, inhibiting apoptosis (134, 161). Nevertheless, a chronic increase in glucocorticoid levels can cause neuronal toxicity and respiratory chain dysfunction, excessive ROS generation, apoptosis, and cell death in skeletal muscle cells and hippocampus (134, 162). Studies in rats treated with lipopolysaccharide have found sex-specific alterations in glucocorticoid receptors, which could be explained by changes in inflammation-induced expression of genes involved in oxidative metabolism (15, 163, 164).
Glutamate and Dopamine in Mitochondrial Dysfunction
Glutamate
Glutamate is implicated in mood disorders. Ketamine, an N-methyl-D-aspartate (NMDA) receptor antagonist, causes a rapid antidepressant effect in patients with MDD (165, 166). This effect might be due to increased BDNF expression (134, 163), modulation by 5-HT receptors, and interactions with inflammatory processes (134, 164). Glutamate levels are higher in brains of patients with mania than other phases of illness, suggestive of dysfunction of the glutamatergic system. In depression, astrocyte density is reduced and, as a consequence, the ratio of glutamine to glutamate is not properly maintained by the glutamate recycling pathway (134, 167). Moreover, mitochondrial energy production is reduced in glutamatergic neurons in patients with MDD (168). High glutamate levels and consequently high intracellular Ca2+ levels promote apoptosis. On the other hand, activation of glutamate receptors also stimulates ATP production, and expression of p53, which can produce an increase of mitochondrial respiration, production of ROS and reduction of GSH (169).
Dopamine
Increased dopamine levels have been noted in mania. Some studies report lower dopamine transporter (DAT) binding in the striatum in unmedicated depressed or euthymic bipolar patients (2). Higher dopamine transmission and impaired DAT function in mania could be explained by elevated oxidative and nitrosative stress, which is higher in mania than in other phases of illness (170). Excessive dopamine levels in mania can also cause damage to nuclear and mtDNA by chronic nitrosative and oxidative stress. However, this is repaired by high dopamine and uric acid levels, which act in a synergistic way to repair free radical-mediated damage (76). In this environment, dopamine can protect neurons against glutamate-induced excitotoxicity, stimulate oxidative phosphorylation, and activate p53, which induces anti-apoptotic activity and inactivates tyrosine hydroxylase, which is necessary for the synthesis of dopamine. Consequently, high dopamine and glutamate levels together with high uric acid levels may not have the expected detrimental effects. Moreover, pro-apoptotic signals may induce the expression of anti-apoptotic genes such as BCL-2, inhibiting the protein Bcl-2 toxicity and apoptosis and stimulating oxidative phosphorylation (76).
All this evidence suggests that changes in mitochondrial function in MDD and BD could be key elements in order to better understand the role of the currently used pharmacotherapy and also to develop novel therapies and new treatment strategies, which will be covered in the next section.
Take-Home Message
Mitochondrial dysfunction may result from different causes, being some of the alterations related with several network processes in which mitochondria are indispensable.
Some of the changes observed in mood disorders include alterations in mitochondrial neurometabolites and metabolic dysfunction, decreased expression and activity of the ETC complexes, increased oxidative damage, altered calcium homeostasis, and changes in mitochondrial morphology, distribution, and degradation. In addition, increased apoptosis, chronic inflammation, Increased expression of mitochondrial fission genes and other genetic changes, including polymorphisms in clock genes, have been observed in mood disorders. Increased mtDNA degradation, purinergic dysfunction, and hyperactivity of the HPA axis, with higher glucocorticoid levels, are other findings reported in mood disorders. Finally, increased glutamate and dopamine levels have been reported in manic episodes. Nevertheless, current evidence is scarce and further studies are needed to assess these changes in mood disorders.
Effects of Pharmacotherapy on Mitochondrial Functions
Since mitochondrial dysfunction has been related to the pathophysiology of mood disorders, including factors such as increased oxidative stress, decreased ATP production, and dysregulation of Ca2+ homeostasis (2), numerous studies have focused on their role as possible drug targets for pharmacological treatments (171, 172). In this regard, conventional psychotropic drugs for mood disorders, including mood stabilizers, antidepressants, and antipsychotics, have demonstrated to have molecular mitochondrial properties, such as neuroprotection, enhancement of mitochondrial function or prevention of cellular apoptosis (173), illustrated in Table 1. In addition, novel interventions are being studied and developed to be used as adjunctive therapies for mood disorders, as noted in Table 2.
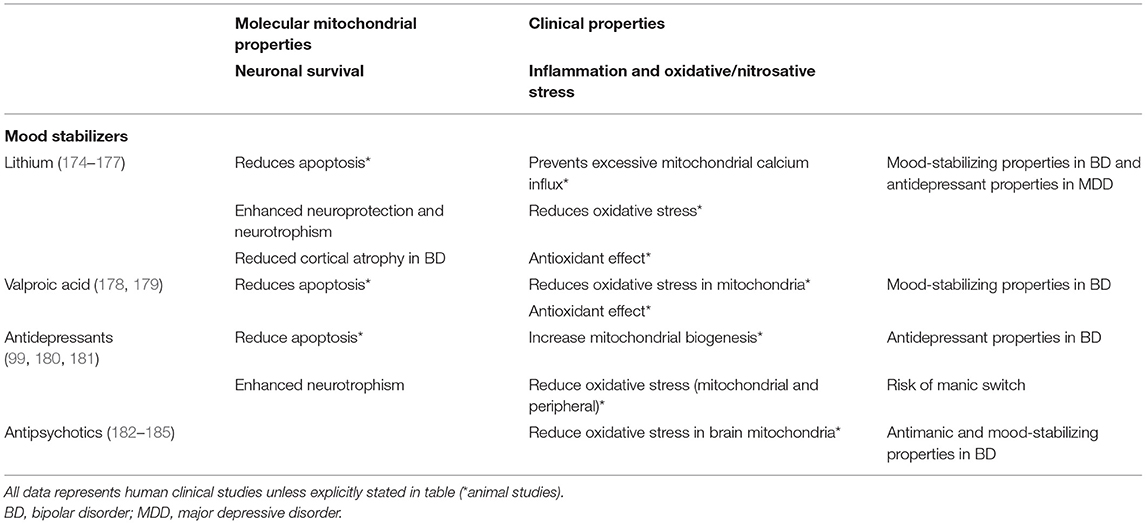
Table 1. Effects of conventional pharmacotherapy on mitochondrial functions.
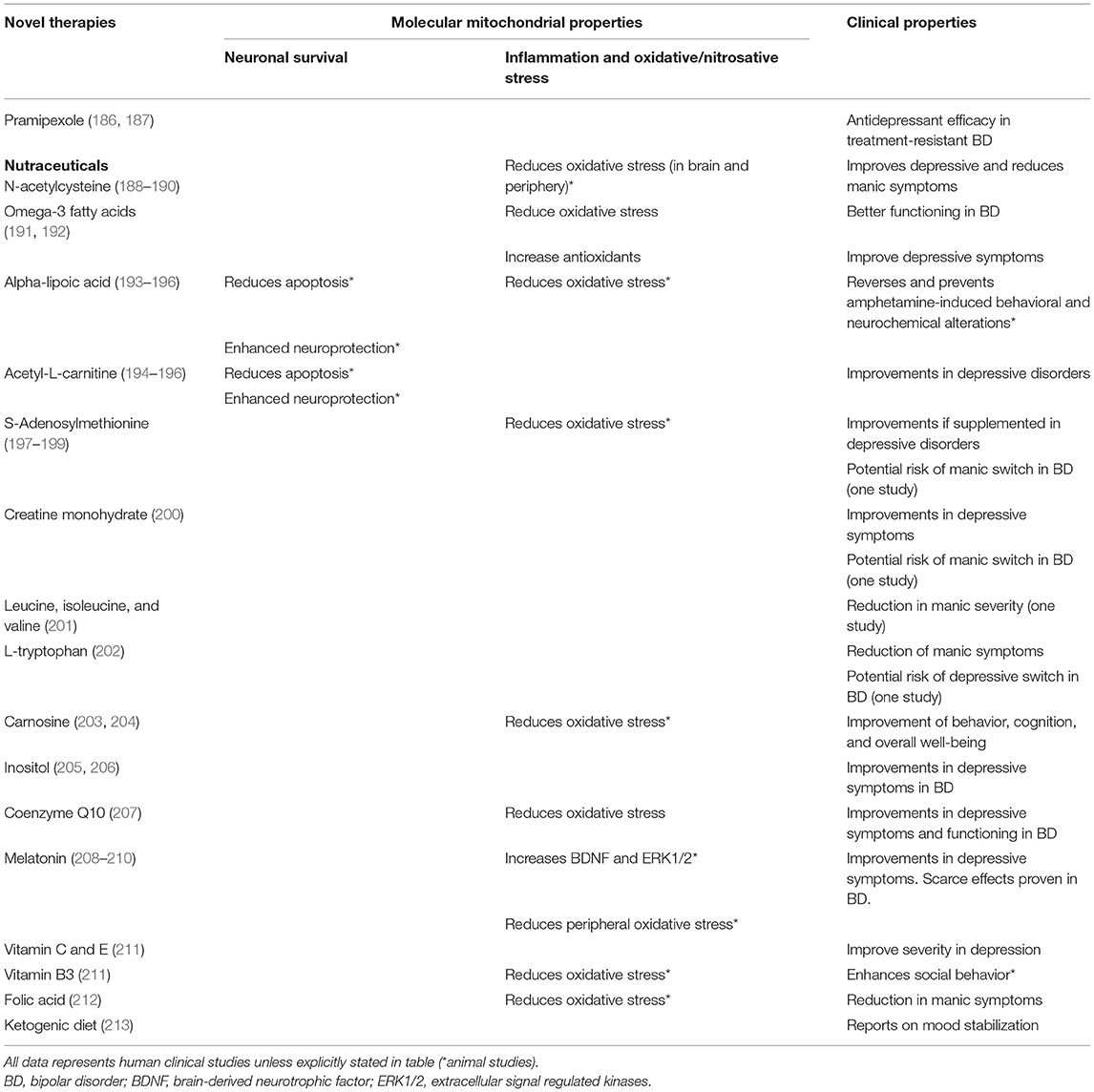
Table 2. Effects of novel therapies on mitochondrial function.
Mood-Stabilizing Drugs
Mood stabilizers are considered first-line drugs in BD to either treat mood episodes or to prevent future recurrences (214). Although the mechanism of action of mood-stabilizing drugs is not clear, some studies suggest that mitochondrial dysfunction and oxidative stress may be therapeutic targets of these drugs.
Some studies have reported that mood stabilizers, apart from altering glutamatergic neurotransmission, decrease intracellular pH, increase expression of the anti-apoptotic gene BCL-2 (by blocking the inhibition produced by histones), regulate expression of other genes, reduce elevated intracellular Ca2+ and increase Ca2+ storage capacity in the ER, and also induce mitochondrial migration to synaptic terminals, modulating neurotransmission (174, 215–217). These findings in aggregate suggest that they may reduce the symptoms of mood disorders at least in part by augmenting mitochondrial activity (63, 214). Lithium and valproate inhibit glutamate-induced apoptosis and oxidative damage to lipid and protein in cerebral cortical cells (218, 219). They also inhibit cytochrome c release from mitochondria. This reduces oxidative stress by stabilizing the inner transmembrane potential of mitochondria and prevents caspase-2 and caspase-3 activation, and cell death (174). Chronic treatment with lithium and valproate has also been shown to inhibit amphetamine-induced hyperactivity (24, 219). Lithium, valproate and carbamazepine are known to reduce inositol levels (220) and augment autophagy in cell cultures (221).
Other animal studies on the antioxidative properties of mood stabilizers have shown that chronic treatment with valproate or lithium is associated with increased ER stress proteins and related proteins, such as calreticulin (222), in cortical and PC12 cells (215, 223). These proteins are involved in antioxidative effects and mitochondrial functioning (216, 224). Moreover, lithium and valproate have other neuroprotective functions, such as regulation of the expression of GST isoenzymes in cerebral cortex, which is a group of detoxification enzymes that inhibit oxidative damage to lipid and protein in cerebral cortical cells, and GSH levels, accelerating conjugation processes (225, 226). However, with low levels of the rate-limiting synthesis enzyme (glutamate-cysteine ligase) and low levels of GSH, lithium and valproate neuroprotective effects are inhibited, which indicates that adequate GSH levels could be important for efficacy (227, 228). Other studies have shown increased oxidative damage to lipids in patients with BD, with higher lipid peroxidation ameliorated after mood-stabilizer treatment, which supports the previous findings (24, 229). Together this evidence indicates that mood stabilizers may reduce the symptoms of BD by enhancing mitochondrial activity (24).
Lithium
Apart from its neuroprotective functions mentioned above, lithium ameliorates BD associated cortical atrophy and maintains cortical thickness (219). Regarding Ca2+ regulation, lithium prevents excessive Ca2+ influx triggered by the N-methyl-D-aspartate receptor. In contrast, animal studies suggest that lithium can allow an increase of Ca2+ concentrations by desensitizing mitochondria against Ca2+, preventing a further response leading to apoptosis. Lithium is also correlated with increased activity of complexes I, II and III, enhances the expression of the scavenger glutathione transferase (184), facilitates mitochondrial respiration, and has other antiapoptotic properties previously mentioned (177, 230).
In rat studies lithium has been shown to inhibit inflammatory signaling pathways related to toll-like receptor 4 (TLR4), which may reduce phosphorylation of NFkβ, reducing inflammatory gene expression and also levels of caspase-3, which might prevent neuronal apoptosis (175, 231). Other studies have shown that the c-Jun N-terminal kinase, which is known to mediate oxidative toxicity, is inhibited by lithium (184, 232). Apart from enhancing this antioxidant defense, lithium has also been reported to increase the activity of SOD (233), of GPx, and total antioxidant reactivity levels in the brain (234).
Lithium also inhibits GSK-3b, a phosphorylating kinase that inhibits the conversion of pyruvate into acetyl-CoA by pyruvate dehydrogenase and also activates BAX (235). Its mutations have been shown to alter lithium response in BD. The inhibition produced by lithium may enhance ATP production and inhibit apoptosis (235, 236). Moreover, in rat models where amphetamines were used, GSK-3b was shown to enhance dopamine activation, leading to the hypothesis that lithium may contribute to maintaining normal levels of dopamine (94).
GSK-3b and phosphoinositide signaling pathways regulate BDNF, which has a complex role in mood disorders. Nevertheless, it is thought to be a potential drug target, since neurotrophic effects of lithium have been related to the increase in hippocampal BDNF in the presence of a neurotoxic insult (235, 237). Phosphoinositol is increased in patients with BD in the central nervous system and is reduced by lithium (184), causing lower levels of myoinositol in this group of patients (238, 239). In contrast, few animal studies have shown contrary effects of lithium treatment, such as the lowering of complex II and IV activity and enhanced ROS formation (240), with reduced antioxidant levels (241). It has also been shown to enhance the activity of caspase-3, leading to apoptosis (242).
Valproic Acid
In rats, valproate has shown to lower amphetamine-induced citrate synthase and to inhibit succinate dehydrogenase, thought to be related to its mood-stabilizing effects (243). Valproate also protects mitochondria from ouabain-induced lipid peroxidation and superoxide formation (176, 244). It could act as a cytoprotective agent in the presence of cytotoxic factors, but alone could inhibit mitochondrial functions (245, 246).
Nevertheless, other studies have shown valproate lowers levels of some cofactors, such as creatine and CoA, involved in the uptake of long-chain fatty acids into mitochondria, which leads to reduced beta-oxidation (214). Valproate has also been shown to enhance ROS generation by inhibition of complex II, and to induce mPTP opening, with a reduction of membrane potential, leading to the release of cytochrome c and apoptosis. Valproate inhibits ATP synthesis when pyruvate is used as a substrate (214). In rat studies, valproate has shown to inhibit glutamate-driven oxidative phosphorylation (247, 248). In cases of impaired ETC structure, valproate inhibits complexes I and IV activity and SOD levels (179).
Other Mood Stabilizers
Although there is scant evidence regarding other mood stabilizers apart from lithium and valproate, changes found in animal studies with carbamazepine treatment include the reduction in mitochondrial respiration, ATP synthesis, and membrane potential, and also the inhibition of Ca2+-induced swelling of liver cells (241, 249, 250). Lamotrigine has been shown to inhibit the effects of rotenone, a cytotoxic agent, and maintain mitochondrial membrane potential, preventing mPTP opening and increasing GSH levels (251). Its neuroprotective effects could be due to complex I inhibition (252).
Antidepressants
In animal models of depression (214, 253), antidepressants seem to increase mitochondrial biogenesis and enhance antioxidative capacity against oxidative stress (139, 184). For instance, venlafaxine increases expression of anti-apoptotic and antioxidant mitochondrial genes (254), and agomelatine may similarly scavenge free radicals (11, 255). As with electroconvulsive therapy, they reduce peripheral inflammatory cytokines (33), which is supported by the reported antidepressant activity of celecoxib, a cyclooxygenase 2 (COX-2) inhibitor (256, 257). Antidepressants also increase autophagy and neural plasticity (134, 258). Since mitochondrial dysfunction has been linked to mental disorders (184), cytochrome c oxidase and apoptosis inhibition have been studied as potential new treatment approaches (259).
In patients treated with antidepressants, there is an increase in the levels of BDNF mRNA (260) and a reversion of the decrease in CREB levels seen in patients with depression (261), which could be mechanisms of action in mood disorders (262). Animal studies have also shown that some antidepressants inhibit complex I in brain mitochondria, reducing its metabolic function (263). A number of studies have shown that some antidepressants, including fluvoxamine, fluoxetine, sertraline, paroxetine, nortriptyline, and venlafaxine, alter ETC activity in mitochondria (241, 264). The reduction of ROS production could explain their beneficial effects (265–267).
Fluoxetine also promotes cytochrome c oxidase and glutamate dehydrogenase activity in presynaptic mitochondria of rat hippocampus (160, 268), and inhibits multiple other enzymes in mitochondria (11, 181). Increased cytochrome c oxidase activity in the female hippocampus by fluoxetine could improve outcomes in women (269–271). Apart from altering mitochondrial energy production, fluoxetine might affect the mitochondrial processes via the glucocorticoid receptor (GR) (33, 272).
Antidepressants are also involved in apoptosis, playing a complex role that depends on cell and brain structure type. One study reported that paroxetine, fluoxetine and clomipramine increased levels of apoptotic markers (cytochrome c and DNA fragments), but imipramine did not have any effect (24, 273). Desipramine induced apoptosis by activating the caspase pathway in glioma cells (274), while fluoxetine and amitriptyline protected PC12 cells from cell death (275). Nortriptyline inhibited neuronal cell death, protecting isolated mitochondria against programmed cell death, inhibiting the release of apoptotic mitochondrial factors and caspases (276). Fluoxetine has been shown to prevent stress-induced apoptosis in the hippocampus, but not in the prefrontal cortex (277, 278). In summary, different mitochondrial functions, such as ATP synthesis, generation of ROS, and cell death, are important targets of antidepressants.
Antipsychotics
Few studies have explored mitochondrial modulation by antipsychotics (279, 280). Olanzapine has shown to increase SOD activity and protect PC12 cells from oxidative damage by H2O2 (20, 281), and also to prevent the decrease in membrane potential and ROS overproduction induced by beta-amyloid peptide (282). In two mice studies, quetiapine increased mitochondrial ETC activity and reduced markers of oxidative stress in the prefrontal cortex, nucleus accumbens, amygdala, and hippocampus (24, 282–284). Scaini et al. [183, 185, 186) showed a significant decrease in all the functional parameters of mitochondrial oxygen consumption after treatment with clozapine and olanzapine in lymphoblastoid cell lines (LCLs) from healthy controls, and these effects were more prominent in cells treated with olanzapine. The same authors also demonstrated that the treatment with clozapine and olanzapine at high doses further decreased mRNA expression of Mfn-2 and Drp-1 in LCLs, supporting the notion that clozapine and olanzapine can potentiate mitochondrial dysfunction.
Novel Therapies
In the last years, a number of agents have been studied as potential therapeutic factors aimed to treat and improve the course of mood disorders, including factors involved in the glutamatergic pathway, insulin transduction pathway, melatoninergic system, purinergic system, endopeptidases, and also mitochondrial modulators (183). A number of the latter agents have been developed or studied with the aim of enhancing antioxidant defenses or mitochondrial functioning as adjuvant therapy to antidepressants (285).
Studies of Ca2+ channel blockers, such as diltiazem and verapamil, have been conducted as potential treatments for BD, but results in the literature are still controversial. It was hypothesized that their therapeutic effect may be due to the protection of neurons against the damage induced by excessive Ca2+ levels (2).
Pramipexole, a D2/D3 agonist approved for the treatment of Parkinson's disease and restless legs syndrome, upregulates Bcl-2 (286). It has also shown antidepressant efficacy in treatment-resistant bipolar patients (287), with a superior response rate compared to placebo and similar to SSRIs.
Some dietary supplements (or nutraceuticals) have been assessed as potential treatments in mood disorders (186, 187), since they may enhance mitochondrial function and brain energy metabolism and prevent ROS-induced damage. These include N-acetylcysteine (NAC), alpha-lipoic acid (ALA), acetyl-L-carnitine (ALCAR), S-adenosylmethionine (SAMe), coenzyme Q10 (CoQ10), creatine monohydrate (CM), and melatonin (201).
The molecular mitochondrial properties shown by novel therapies for mood disorders are summarized in Table 2.
Nutraceuticals
N-Acetylcysteine
N-acetylcysteine (NAC) is a GSH precursor, the major antioxidant agent in the brain (288) for preventing oxidative damage in the mitochondrial ETC (289). By increasing GSH levels, NAC may increase mitochondrial respiratory capacity and have neuroprotective functions by other mechanisms (285, 290). It can prevent oxidative damage to complex I (184), can enhance GST activity, and act directly against oxidant radicals (290, 291).
Some studies in BD have demonstrated that treatment with NAC can improve depressive symptoms, clinical response rates, symptom remission, quality of life and functioning (292, 293). Few clinical trials assessing the efficacy of NAC as adjunctive treatment in patients with BD have shown promising results (189, 281, 289, 294–298), with benefits in depressive symptoms of BD patients (63, 189, 285, 289), but not as maintenance treatment (189, 294). Post-hoc analyses suggested that NAC might be effective in later stages of BD (289) and also to reduce manic symptoms (281). However, there are recent negative trials, albeit smaller and of a shorter duration (298).
Clinical trials in depressive disorders also suggest the potential of NAC as adjunctive treatment in depression (299, 300). Although no differences in depressive symptoms were found in another clinical trial comparing NAC with placebo (189, 301), the NAC group showed a better response at the 16-week post-discontinuation endpoint. A meta-analysis including five studies assessing depressive symptoms with a follow-up of 12–24 weeks revealed significantly greater improvements in depressive symptoms and functionality with NAC compared to placebo (190).
Omega-3 Fatty Acids
Some studies have demonstrated modulatory effects of omega-3 fatty acids on mitochondria. Eicosapentaenoic acid (EPA; 20:5n-3) is a fatty acid that seems to protect against oxidative stress by replenishing oxidized lipids and increasing oxygen and glucose supply to the brain (301). Diets rich in omega-3 fatty acids have shown to upregulate cytochrome c oxidase, cytochrome b, and ATP synthases, leading to increased ATP formation (302, 303).
In a study of rodents with methylphenidate-induced mania, omega-3 fatty acids alone and in combination with lithium and aripiprazole reduced levels of SOD, CAT, and lipid peroxidation products (304). Stanley et al. (63, 305) demonstrated that docosahexaenoic acid (DHA; 22:6n-3) changes mitochondrial membrane phospholipid composition and mitochondrial function, protecting mitochondria against damage (306). A pilot study found significantly higher remission, greater improvements in depressive symptoms, and better global functioning in bipolar patients supplemented with omega-3 fatty acids, while no benefits in mania were found (306). Clinical improvements associated with omega-3 fatty acids intake are produced at least in part by modulation of BDNF levels (192). A study that included 10 different countries found a correlation between lower fish or seafood consumption with a higher prevalence of bipolar spectrum disorders (307, 308). Patients with BD have shown lower levels of erythrocyte DHA, ALA and EPA when compared to healthy controls (63, 309). Another study showed a trend toward lower levels of omega-3 fatty acids in relatives of patients with BD (191).
A systematic review of clinical trials assessing nutraceuticals showed positive and statistically significant results on depression in four out of nine studies (201), but none showed positive findings in mania. However, sample sizes were small, reducing the chance of positive results (192, 310). The previous evidence suggests that supplementation or increased consumption of omega-3 fatty acids may be beneficial in mood disorders, but additional studies are necessary to define their clinical efficacy more accurately.
Alpha-Lipoic Acid
Alpha-lipoic acid (ALA) is an antioxidant found in red meats, spinach, yeast, and other products (201). It facilitates glucose entrance into cells for ATP synthesis and recycling of endogenous antioxidants, such as CoQ10, vitamins C and E, and GSH (193). ALA has been demonstrated to reduce metabolic deficits, oxidative stress and apoptosis (by preventing glutamate-induced Ca2+ cellular influx) (311, 312), stimulate glucose uptake into cells, improve cognitive function and enhance neuroprotection (312, 313) and to stimulate mitochondrial biogenesis (195), but studies in mood disorders are lacking (312).
Acetyl-L-Carnitine
L-carnitine (ALCAR) is a compound obtained through the diet (314) that is biosynthesised from lysine and methionine. It enhances the entrance of fatty acids into the mitochondria for ROS scavenging and beta-oxidation, which leads to ATP and acyl-coenzyme A (acyl-CoA) production (315). Acyl-CoA enters the citric acid cycle (316, 317). Reported functions of ALCAR include neuroprotection, anti-apoptotic properties (315), inhibition of GABA production (316, 318), and enhancing of mitochondrial functioning (319). Animal studies of ALCAR show increased levels of ATP and PCr (195). Results regarding the reduction of oxidative stress are mixed, but coadministration with NAC or ALA has shown benefits (320). Moreover, supplementation with ALA and ALCAR may promote mitochondrial integrity in the hippocampus of aged rats (321). Some early clinical trials suggest the ALCAR has significantly greater efficacy than placebo as an augmentation treatment depressive disorders (63, 194). However, another study found no significant differences in depressive scores of ALCAR/ALA treatment compared to placebo (322–324).
S-Adenosylmethionine
S-Adenosylmethionine (SAMe) is formed from ATP and methionine and is needed for the synthesis of many neurotransmitters and for repairment and degradation of dysfunctional proteins. It is a precursor for GSH production, which plays an important role in reducing oxidative stress. It is also used for homocysteine synthesis, which in turn can regenerate SAMe (196).
Some studies suggest the efficacy of SAMe supplementation for depressive episodes as adjunctive therapy with a number of antidepressants SSRI, venlafaxine, or SNRIs (288). Nevertheless, studies with older antidepressants, including phenelzine, mianserin, and maprotiline, showed inconsistent results (197, 199, 325–327). One study in patients with BD showed SAMe might pose a risk for a manic switch (288), but randomized clinical trials in BD are still lacking (328). A recent large scale trial showed a numerical but not statistically significant benefit of SAMe in depression (198).
Creatine Monohydrate
Creatine is an antioxidant agent synthesized by the liver and kidneys which is also found in meat and fish (288). It can be obtained as a supplement in the form of creatine monohydrate (CM). Creatine is the precursor of PCr, a reservoir of inorganic phosphate, used for ATP synthesis by donating a phosphate to ADP (329). In the context of high-energy demand, PCr is rapidly converted to creatine to donate a high-energy phosphate to ADP to obtain ATP (330).
Creatine also attenuates the decreases in N-acetyl-aspartate (NAA), which acts as a marker of impaired mitochondrial function, and inhibits the activation of the mitochondrial permeability transition, suggesting neuroprotective effects (331, 332). Other neuroprotective effects are intracellular Ca2+ buffering, extracellular glutamate reduction, and antioxidant effects (333–335). PCr and NAA concentrations are reduced in BD patients and this reduction correlates with clinical severity (336). Where PCr levels are diminished, CM supplementation may increase PCr and NAA production to promote neuroprotection (70, 337).
Despite limited evidence in mood disorders, CM has been associated with improvement in depressive symptoms in case studies (63). Benefits were seen in treatment-resistant depression in a small open-label study (338), which also suggested a risk of a manic switch after CM treatment in patients with BD (200). Before CM might be considered as an adjunctive treatment for the management of BD and depressive disorders, RCTs are necessary.
Other Aminoacids
In one study assessing the efficacy of leucine, isoleucine, and valine combination vs. placebo in 25 patients with BD, positive results were seen with significant reductions of the severity of mania within 6 h in the verum group, whose activity may be explained by competitive inhibition of phenylalanine and tyrosine, which are necessary for dopamine synthesis (288). L-tryptophan reduced manic symptoms in a study of 24 patients (339, 340). Moreover, a meta-analysis reported significantly reduced plasma tryptophan levels in patients with MDD (202). This aminoacid was shown to reduce depression scores in people with unipolar depression in methodologically limited studies, so further evidence is required in order to consider it as an adjunctive therapy (341).
Carnosine is a dipeptide made up of the amino acids beta-alanine and histidine that protects brain mitochondria and regulates the immune system (201, 342). It has also been studied for adjuvant treatment of depression (343, 344). So far, it has been demonstrated to reduce the effects of chronic stress in animal studies and to improve behavior, cognition, and overall well-being in human studies (204, 345).
Inositol
A pilot study using inositol, a glucose isomer, in 24 patients with BD found a significant reduction in depression scores after 3 weeks of treatment but not after 6 weeks (2, 203). Another 6-week study in 17 subjects with BD showed no significant reduction in depression or mania scores (206). Notwithstanding, both studies found a greater clinical response with inositol compared to controls, which suggests a potential benefit of this agent in BD (205).
Coenzyme Q10
Coenzyme Q10 is a component of the ETC complex involved in ATP synthesis (201). It acts as an antioxidant in mitochondria and lipid membranes (346). CoQ10 has been suggested to stabilize the mitochondrial membrane in the context of oxidative stress (63, 347). It also inhibits the activity of mPTP and increases complex I activity (348). One open-label placebo-controlled trial reported clinical improvement in depressive symptoms in older adults with bipolar depression using this supplement (63).
A randomized controlled trial comparing nutraceutical treatment (including ALC, CoQ10 and ALA, in addition to co-factors involved in mitochondrial function) with NAC and placebo in patients with depression did not show a significant difference between groups at the primary endpoint. However, the rate of change between baseline and week 20 post-discontinuation was significantly greater in the group previously treated with nutraceuticals compared with the placebo group on depression scores, and also on functioning. This suggests a delayed benefit of the combination or improvement of symptoms on withdrawal, which should be assessed in future studies (285). Thus, current evidence suggests that CoQ10 might be beneficial in mood disorders (288), but further clinical trials in mood disorders are necessary to confirm these early promising but non-definitive signals.
Melatonin
Melatonin is a hormone released in a circadian pattern by the pineal gland and other tissues in the body, including the brain. It has a number of functions and is an important antioxidant free radical scavenger (207, 288). Specifically, it stimulates the production of GSH (349) and increases the expression of genes related to antioxidative functions, such as glutathione peroxidase and SOD (210). Melatonin also seems to directly enhance mitochondrial function since it activates ETC complexes, increases mitochondrial membrane fluidity, and closes the mPTP. It also protects mtDNA against degradation, promotes the expression of mitochondrial genes coding for ETC complex subunits, and has neuroprotective properties (209). The beneficial effects of melatonin seem to be those related to ROS scavenging and actions linked to cytosolic proteins (209, 288).
Clinical studies in mood disorders do not show conclusive results. So far, melatonin has shown benefits improving depressive symptoms in patients with “winter depression” (209, 288) compared to placebo, whereas a controlled study in seasonal affective disorder did not show changes in atypical depressive symptoms (208), and a crossover study on patients with severe depression showed that patients taking melatonin had worsened dysphoria, sleeping patterns and weight gain (350).
Regarding evidence assessing melatonin for BD, one open-label study showed no significant effects on mood or sleep in rapid-cycling patients (351), whereas in another small open-label study it showed sleep-enhancing and antimanic effects in manic patients (352). As mentioned previously, agomelatine, an agonist of melatonin MT1 and MT2 receptors, has demonstrated preliminary evidence of efficacy in bipolar depression (353), but agomelatine has other actions on the serotonin system.
Vitamins and Minerals
Supplementation with vitamins C and E was shown to significantly improve severity in depression. One study where they were combined with monoaminergic antidepressants for 12 weeks showed they improved oxidative stress in subjects with MDD (354), but it was not a placebo-controlled design. Nicotinamide is a form of vitamin B3 found in food, used as an antioxidative substance, and is also a precursor of NAD+. It is hypothesized to be effective for the treatment of mood disorders (211), since it increases oxidative phosphorylation in the brain and enhances social behavior in high-anxiety rats (355). A small clinical trial evaluating magnesium as a potential adjunctive therapy for treating acute mania or rapid cycling BD showed a greater reduction of manic symptoms compared to controls (356), which might be due to the modulation of Ca2+ channel activity and its participation in neurotransmitter release (357). One study using folic acid for 17 BD participants showed no statistically significant differences on symptoms of depression compared to controls (201, 358), but in another where 88 patients with acute mania were initiated on valproate, folic acid at doses of 3 g showed a significant reduction of manic symptoms at week 3 compared to placebo (359). Thus, assessing folate levels and administering supplementation in patients with mood disorders could be beneficial for the clinical course of the acute episodes.
Ketogenic Diet
The effects of specific diets in mood disorders are still not clear despite evidence reporting that they can alter several biological processes. The exception is the Mediterranean diet, which has been associated with antioxidative properties and has shown antidepressant effects in a RCT (201, 360). One study assessing rats on a calorie-restricted diet showed that mitochondrial efficiency and oxidative damage in skeletal muscle were significantly increased in these rats, while antioxidant effects were significantly lowered in food-restricted rats that followed a high-fat diet. Thus, caloric restriction seems to predispose to higher mitochondrial efficiency and also to high-fat induced oxidative damage (361). Other studies have shown that the ketogenic diet (KD) upregulates mitochondrial antioxidant status and protects mtDNA from oxidant-induced damage (362). It has also shown effective anticonvulsant properties and has been suggested as a potential adjunctive therapy as a mood stabilizer.
The ketogenic diet consists of a low-carbohydrate diet that substantially changes the energetic source of the organism (213, 363), which switches from glucose to ketones bodies, obtained by breakdown of fatty acids. This causes alterations in neurotransmitter levels, hormones, and peptides (364), and an increase in oxidative phosphorylation and ATP synthesis (365, 366), increased GSH levels, reduced ROS production (367), reduced inflammatory levels and neuroprotection (368). A ketogenic diet seems to influence epigenetic changes involved in increased mitochondrial function and biogenesis (369), which might also be responsible for the increase of BDNF (370). The ketogenic diet stimulates the endogenous antioxidant system through the activation of nuclear factor erythroid-derived 2 (NF-E2)-related factor 2 (Nrf2), the major inducer of detoxification genes (371, 372), especially in the hippocampus (373). Despite the limited data regarding the ketogenic diet for the treatment of mood disorders, early reports support the hypothesis about its beneficial effects on mood stabilization (374).
Physical Exercise
Physical activity is directly related to increased mitochondrial biogenesis, increased mitochondrial content and oxygen utilization capabilities, and that aerobic exercise in the elderly ameliorates loss of skeletal muscle mitochondrial content (369). One study assessing the efficacy of fluoxetine and exercise in muscle cells of rats reported that physical activity increased cytochrome c oxidase activity compared with the group treated only with fluoxetine. Exercise increased citrate synthase activity in both fluoxetine and non-fluoxetine groups, and fluoxetine increased its activity only in the exercise group. On the other hand, exercise significantly decreased ROS levels in both fluoxetine and non-fluoxetine groups, with this reduction higher in the fluoxetine group (375). Post-hoc analysis of a trial of a mitochondrial combination therapy found the greatest benefits in those with the highest levels of physical activity (376). There is a meta-analytic level of evidence from RCT's that exercise has antidepressant effects. Hence, enhancing mitochondrial function through physical activity may provide a novel way to treat mood disorders (377).
Take-Home Message
Numerous studies have focused on the role of therapeutic agents targeting different mitochondrial functions that are altered in mood disorders. On one hand, mood stabilizers, antidepressants and antipsychotics have shown to promote neuroprotection, reduce oxidative stress and enhance mitochondrial function. On the other hand, novel interventions have been assessed as potential adjunctive therapies for mood disorders.
Some mitochondrial modulators have been developed or studied with the aim of enhancing antioxidant defenses or mitochondrial functioning as adjuvant therapies in mood disorders. Pramipexole has shown antidepressant effects by the upregulation of Bcl-2. Some dietary supplements or nutraceuticals have been found to enhance mitochondrial function and brain energy metabolism mainly by the reduction of oxidative stress. These include N-acetylcysteine (NAC), alpha-lipoic acid (ALA), acetyl-L-carnitine (ALCAR), S-adenosylmethionine (SAMe), coenzyme Q10 (CoQ10), creatine monohydrate (CM), and melatonin. Even though current evidence suggests they might be beneficial in mood disorders, further clinical trials are necessary to confirm these findings.
Melatonin has antioxidative functions and also enhances mitochondrial function. However, clinical studies in mood disorders have not shown positive results. Vitamin supplementation, ketogenic diet and physical exercise have also shown positive effects in mitochondrial function and mood disorders, with scarce evidence.
Conclusion
Mitochondria play a key role in different cellular functions, especially those related to energy production. A number of studies indicate the possible role of mitochondria in the pathophysiology of mood disorders, raising the possibility that the processes of energy generation and oxidative damage could be significant therapeutic targets for the treatment of BD with mood-stabilizing or other kinds of drugs as well as lifestyle approaches. A better knowledge of mitochondrial functioning could help identify impaired processes and specific treatment targets. This would increase the understanding of mechanisms of action of the drugs currently used and aid the development of novel effective therapies to treat specific mitochondrial functions that might be used as the main therapy or as adjunctive treatment, especially for subjects that do not fully respond to conventional therapies. Research on changes in mitochondrial processes in patients with mood disorders might clarify how mitochondrial dysfunction can be considered a biological target. Further studies are needed to confirm that pharmacological treatments reduce or delay neuroprogressive changes in mood disorders, and to demonstrate the potential benefits of putative antioxidant substances.
Author Contributions
All authors contributed in the preparation of the manuscript and gave approval for the final version.
Funding
This study has been supported by the Spanish Ministry of Science and Innovation (PI15/00283, PI18/00805, PI19/00394, PI18/01001, CPII19/00009) integrated into the Plan Nacional de I+D+I and co-financed by the Instituto de Salud Carlos III (ISCIII)-Subdirección General de Evaluación and the Fondo Europeo de Desarrollo Regional (FEDER); the ISCIII; the CIBER of Mental Health (CIBERSAM); the Secretaria d'Universitats i Recerca del Departament d'Economia i Coneixement (2017 SGR 1365), the CERCA Programme, and the Departament de Salut de la Generalitat de Catalunya for the PERIS grant SLT006/17/00357. GA's research was supported by a Pons Bartran 2020 grant (PI046549). MB has been supported by a NHMRC Senior Principal Research Fellowship (1059660 and 1156072). Translational Psychiatry Program (USA) was funded by a grant from the National Institute of Health/National Institute of Mental Health (1R21MH117636-01A1, to JQ) and a research supplement form the Faillace Department of Psychiatry and Behavioral Sciences. Center of Excellence on Mood Disorders (USA) was funded by the Pat Rutherford Jr. Chair in Psychiatry, John S. Dunn Foundation and Anne and Don Fizer Foundation Endowment for Depression Research. Translational Psychiatry Laboratory (Brazil) was funded by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação de Amparo à Pesquisa e Inovação do Estado de Santa Catarina (FAPESC), and Instituto Cérebro e Mente, which support JQ's research. GS was partially supported by Linda Gail Behavioral Health Research Fund.
Conflict of Interest
AG-P has received support from Janssen-Cilag and Otsuka-Lundbeck, and declares no support related to the subject of this article. SD has received grant support from Stanley Medical Research Institute, NHMRC, Beyond Blue, ARHRF, Simons Foundation, Geelong Medical Research Foundation, Fondation FondaMental, Eli Lilly, Glaxo SmithKline, Organon, Mayne Pharma and Servier. He has received speaker's fees from Eli Lilly, advisory board fees from Eli Lilly and Novartis and conference travel support from Servier, with no financial or other relationship relevant to the subject of this article. GA has received CME-related honoraria, or consulting fees from Janssen-Cilag, Lundbeck and Angelini with no financial or other relationship relevant to the subject of this article. JQ received clinical research support from LivaNova; has speaker bureau membership with Myriad Neuroscience, Janssen Pharmaceuticals, and Abbvie; is consultant for Eurofarma; is stockholder at Instituto de Neurociencias Dr. Joao Quevedo; and receives copyrights from Artmed Editora, Artmed Panamericana, and Elsevier/Academic Press. IP has received CME-related honoraria, or consulting fees from ADAMED, Janssen-Cilag and Lundbeck. EV has received research support from or served as consultant, adviser or speaker for AB-Biotics, Abbott, Allergan, Angelini, Dainippon Sumitomo Pharma, Ferrer, Gedeon Richter, Janssen, Lundbeck, Otsuka, Sage pharmaceuticals, Sanofi-Aventis, Shire, Sunovion, Takeda, and reports no financial or other relationship relevant to the subject of this article. MB has received Grant/Research Support from the NIH, Cooperative Research Centre, Simons Autism Foundation, Cancer Council of Victoria, Stanley Medical Research Foundation, Medical Benefits Fund, National Health and Medical Research Council, Medical Research Futures Fund, Beyond Blue, Rotary Health, A2 milk company, Meat and Livestock Board, Woolworths, Avant and the Harry Windsor Foundation, has been a speaker for Abbot, Astra Zeneca, Janssen and Janssen, Lundbeck and Merck and served as a consultant to Allergan, Astra Zeneca, Bio- advantex, Bionomics, Collaborative Medicinal Development, Janssen and Janssen, Lundbeck Merck, Pfizer and Servier, and has licences with Allen and Unwin and Cambridge University Press. MB has received patents for agents that modulate physiological processes and diseases of the central nervous system and related processes, including xanthone-rich plant extracts. All are unrelated to this work.
All other authors report no financial or other relationship relevant to the subject of this article.
References
1. Andreazza AC, Young LT. The neurobiology of bipolar disorder: identifying targets for specific agents and synergies for combination treatment. Int J Neuropsychopharmacol. (2014) 17:1039–52. doi: 10.1017/S1461145713000096
2. Caruso G, Benatti C, Blom JMC, Caraci F, Tascedda F. The many faces of mitochondrial dysfunction in depression: from pathology to treatment. Front Pharmacol. (2019) 10:995. doi: 10.3389/fphar.2019.00995
3. Budd SL, Nicholls DG. Mitochondria in the life and death of neurons. Essays Biochem. (1998) 33:43–52. doi: 10.1042/bse0330043
4. Finkel T. Radical medicine: treating ageing to cure disease. Nat Rev Mol Cell Biol. (2005) 6:971–6. doi: 10.1038/nrm1763
5. Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron. (2012) 75:762–77. doi: 10.1016/j.neuron.2012.08.019
6. Todorova V, Blokland A. Mitochondria and synaptic plasticity in the mature and aging nervous system. Curr Neuropharmacol. (2016) 15:166–73. doi: 10.2174/1570159x14666160414111821
7. Erecinska M, Cherian S, Silver IA. Energy metabolism in mammalian brain during development. Prog Neurobiol. (2004) 73:397–445. doi: 10.1016/j.pneurobio.2004.06.003
8. Pieczenik SR, Neustadt J. Mitochondrial dysfunction and molecular pathways of disease. Exp Mol Pathol. (2007) 83:84–92. doi: 10.1016/j.yexmp.2006.09.008
9. Devine MJ, Kittler JT. Mitochondria at the neuronal presynapse in health and disease. Nat Rev Neurosci. (2018) 19:63–80. doi: 10.1038/nrn.2017.170
10. Kann O, Kovács R. Mitochondria and neuronal activity. Am J Physiol Cell Physiol. (2007) 292:C641–57. doi: 10.1152/ajpcell.00222.2006
11. Allen J, Romay-Tallon R, Brymer KJ, Caruncho HJ, Kalynchuk LE. Mitochondria and mood: mitochondrial dysfunction as a key player in the manifestation of depression. Front Neurosci. (2018) 12:386. doi: 10.3389/fnins.2018.00386
12. Kato T. Neurobiological basis of bipolar disorder: mitochondrial dysfunction hypothesis and beyond. Schizophr Res. (2017) 187:62–6. doi: 10.1016/j.schres.2016.10.037
13. Howarth C, Peppiatt-Wildman CM, Attwell D. The energy use associated with neural computation in the cerebellum. J Cereb Blood Flow Metab. (2010) 30:403–14. doi: 10.1038/jcbfm.2009.231
14. Wen Q, Chklovskii DB. Segregation of the brain into gray and white matter: a design minimizing conduction delays. PLoS Comput Biol. (2005) 1:e78. doi: 10.1371/journal.pcbi.0010078
15. Bansal Y, Kuhad A. Mitochondrial dysfunction in depression. Curr Neuropharmacol. (2016) 14:610–8. doi: 10.2174/1570159x14666160229114755
16. Chakrabarty S, Kabekkodu SP, Singh RP, Thangaraj K, Singh KK, Satyamoorthy K. Mitochondria in health and disease. Mitochondrion. (2018) 43:25–9. doi: 10.1016/j.mito.2018.06.006
17. Kadenbach B, Ramzan R, Wen L, Vogt S. New extension of the mitchell theory for oxidative phosphorylation in mitochondria of living organisms. Biochim Biophys Acta. (2010) 1800:205–12. doi: 10.1016/j.bbagen.2009.04.019
18. Cuperfain AB, Zhang ZL, Kennedy JL, Gonçalves VF. The complex interaction of mitochondrial genetics and mitochondrial pathways in psychiatric disease. Mol Neuropsychiatry. (2018) 4:52–69. doi: 10.1159/000488031
19. Gómez-Serrano M, Camafeita E, Loureiro M, Peral B. Mitoproteomics: tackling mitochondrial dysfunction in human disease. Oxid Med Cell Longev. (2018) 2018:1435934. doi: 10.1155/2018/1435934
20. Vélot C, Srere PA. Reversible transdominant inhibition of a metabolic pathway. In vivo evidence of interaction between the sequential tricarboxylic acid cycle enzymes in yeast. J Biol Chem. (2000) 275:12926–33. doi: 10.1074/jbc.275.17.12926
21. Davies KM, Strauss M, Daum B, Kief JH, Osiewacz HD, Rycovska A, et al. Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc Natl Acad Sci USA. (2011) 108:14121–6. doi: 10.1073/pnas.1103621108
22. Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. (2013) 155:160–71. doi: 10.1016/j.cell.2013.08.032
23. Strauss M, Hofhaus G, Schröder RR, Kühlbrandt W. Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. (2008) 27:1154–60. doi: 10.1038/emboj.2008.35
24. Wang JF. Defects of mitochondrial electron transport chain in bipolar disorder: implications for mood-stabilizing treatment. Can J Psychiatry. (2007) 52:753–62. doi: 10.1177/070674370705201202
25. Lenaz G, Genova ML. Structure and organization of mitochondrial respiratory complexes: a new understanding of an old subject. Antioxid Redox Signal. (2010) 12:961–1008. doi: 10.1089/ars.2009.2704
26. Lenaz G. The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life. (2001) 52:159–64. doi: 10.1080/15216540152845957
27. Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radic Biol Med. (2009) 47:333–43. doi: 10.1016/j.freeradbiomed.2009.05.004
28. Kishida KT, Klann E. Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid Redox Signal. (2007) 9:233–44. doi: 10.1089/ars.2007.9.ft-8
29. Kornmann B. Quality control in mitochondria: use it, break it, fix it, trash it. F1000Prime Rep. (2014) 6:15. doi: 10.12703/P6-15
30. Fischer F, Hamann A, Osiewacz HD. Mitochondrial quality control: an integrated network of pathways. Trends Biochem Sci. (2012) 37:284–92. doi: 10.1016/j.tibs.2012.02.004
31. Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. (2004) 305:626–9. doi: 10.1126/science.1099320
32. Lindsten T, Zong WX, Thompson CB. Defining the role of the Bcl-2 family of proteins in the nervous system. Neuroscientist. (2005) 11:10–5. doi: 10.1177/1073858404269267
33. Adzic M, Brkic Z, Bulajic S, Mitic M, Radojcic MB. Antidepressant action on mitochondrial dysfunction in psychiatric disorders. Drug Dev Res. (2016) 77:400–6. doi: 10.1002/ddr.21332
34. de Sousa RT, Machado-Vieira R, Zarate CA, Manji HK. Targeting mitochondrially mediated plasticity to develop improved therapeutics for bipolar disorder. Expert Opin Ther Targets. (2014) 18:1131–47. doi: 10.1517/14728222.2014.940893
35. Patergnani S, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, et al. Calcium signaling around mitochondria associated membranes (MAMs). Cell Commun Signal. (2011) 9:19. doi: 10.1186/1478-811X-9-19
36. van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta Mol Cell Res. (2014) 1843:2253–62. doi: 10.1016/j.bbamcr.2014.03.009
37. Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. (2006) 86:369–408. doi: 10.1152/physrev.00004.2005
38. Baron KT, Wang GJ, Padua RA, Campbell C, Thayer SA. NMDA-evoked consumption and recovery of mitochondrially targeted aequorin suggests increased Ca2+ uptake by a subset of mitochondria in hippocampal neurons. Brain Res. (2003) 993:124–32. doi: 10.1016/j.brainres.2003.09.022
39. Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J. (1972) 128:617–30. doi: 10.1042/bj1280617
40. Liesa M, Palacín M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev. (2009) 89:799–845. doi: 10.1152/physrev.00030.2008
41. Busch KB, Kowald A, Spelbrink JN. Quality matters: how does mitochondrial network dynamics and quality control impact on mtDNA integrity? Philos Trans R Soc Lond B Biol Sci. (2014) 369:20130442. doi: 10.1098/rstb.2013.0442
42. Courchet J, Lewis TL, Lee S, Courchet V, Liou D-Y, Aizawa S, et al. Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell. (2013) 153:1510–25. doi: 10.1016/j.cell.2013.05.021
43. Sheng ZH. Mitochondrial trafficking and anchoring in neurons: new insight and implications. J Cell Biol. (2014) 204:1087–98. doi: 10.1083/jcb.201312123
44. Chang DTW, Reynolds IJ. Mitochondrial trafficking and morphology in healthy and injured neurons. Prog Neurobiol. (2006) 80:241–68. doi: 10.1016/j.pneurobio.2006.09.003
45. Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. (2004) 430:631–9. doi: 10.1038/nature02621
46. Burkhalter J, Fiumelli H, Allaman I, Chatton J-Y, Martin J-L. Brain-derived neurotrophic factor stimulates energy metabolism in developing cortical neurons. J Neurosci. (2003) 23:8212–20. doi: 10.1523/JNEUROSCI.23-23-08212.2003
47. Markham A, Cameron I, Franklin P, Spedding M. BDNF increases rat brain mitochondrial respiratory coupling at complex I, but not complex II. Eur J Neurosci. (2004) 20:1189–96. doi: 10.1111/j.1460-9568.2004.03578.x
48. Kuromi H, Kidokoro Y. Exocytosis and endocytosis of synaptic vesicles and functional roles of vesicle pools: lessons from the Drosophila neuromuscular junction. Neuroscientist. (2005) 11:138–47. doi: 10.1177/1073858404271679
49. Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, et al. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. (2005) 47:379–93. doi: 10.1016/j.neuron.2005.06.027
50. Verstreken P, Ly CV, Venken KJT, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. (2005) 47:365–78. doi: 10.1016/j.neuron.2005.06.018
51. Kathleen Baxter K, Uittenbogaard M, Yoon J, Chiaramello A. The neurogenic basic helix-loop-helix transcription factor NeuroD6 concomitantly increases mitochondrial mass and regulates cytoskeletal organization in the early stages of neuronal differentiation. ASN Neuro. (2009) 1:195–211. doi: 10.1042/AN20090036
52. Calingasan NY, Ho DJ, Wille EJ, Campagna MV, Ruan J, Dumont M, et al. Influence of mitochondrial enzyme deficiency on adult neurogenesis in mouse models of neurodegenerative diseases. Neuroscience. (2008) 153:986–96. doi: 10.1016/j.neuroscience.2008.02.071
53. Kirby DM, Rennie KJ, Smulders-Srinivasan TK, Acin-Perez R, Whittington M, Enriquez J-A, et al. Transmitochondrial embryonic stem cells containing pathogenic mtDNA mutations are compromised in neuronal differentiation. Cell Prolif. (2009) 42:413–24. doi: 10.1111/j.1365-2184.2009.00612.x
54. Bahat A, Gross A. Mitochondrial plasticity in cell fate regulation. J Biol Chem. (2019) 294:13852–63. doi: 10.1074/jbc.REV118.000828
55. Lees JG, Gardner DK, Harvey AJ. Pluripotent stem cell metabolism and mitochondria: beyond ATP. Stem Cells Int. (2017) 2017:2874283. doi: 10.1155/2017/2874283
56. Lorenz C, Prigione A. Mitochondrial metabolism in early neural fate and its relevance for neuronal disease modeling. Curr Opin Cell Biol. (2017) 49:71–6. doi: 10.1016/j.ceb.2017.12.004
57. Toker L, Agam G. Mitochondrial dysfunction in psychiatric morbidity: current evidence and therapeutic prospects. Neuropsychiatr Dis Treat. (2015) 11:2441–7. doi: 10.2147/NDT.S70346
58. Blommaart EFC, Luiken JJFP, Blommaart PJE, Van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem. (1995) 270:2320–6. doi: 10.1074/jbc.270.5.2320
59. Palikaras K, Daskalaki I, Markaki M, Tavernarakis N. Mitophagy and age-related pathologies: development of new therapeutics by targeting mitochondrial turnover. Pharmacol Ther. (2017) 178:157–74. doi: 10.1016/j.pharmthera.2017.04.005
60. Maria Michel T, Pulschen D, Thome J. The role of oxidative stress in depressive disorders. Curr Pharm Des. (2012) 18:5890–9. doi: 10.2174/138161212803523554
61. Scaglia F. The role of mitochondrial dysfunction in psychiatric disease. Dev Disabil Res Rev. (2010) 16:136–43. doi: 10.1002/ddrr.115
62. Fattal O, Budur K, Vaughan AJ, Franco K. Review of the literature on major mental disorders in adult patients with mitochondrial diseases. Psychosomatics. (2006) 47:1–7. doi: 10.1176/appi.psy.47.1.1
63. Scaini G, Rezin GT, Carvalho AF, Streck EL, Berk M, Quevedo J. Mitochondrial dysfunction in bipolar disorder: evidence, pathophysiology and translational implications. Neurosci Biobehav Rev. (2016) 68:694–713. doi: 10.1016/j.neubiorev.2016.06.040
64. Kato T, Takahashi S, Shioiri T, Inubushi T. Brain phosphorous metabolism in depressive disorders detected by phosphorus-31 magnetic resonance spectroscopy. J Affect Disord. (1992) 26:223–30. doi: 10.1016/0165-0327(92)90099-R
65. Mayberg HS, Brannan SK, Mahurin RK, Jerabek PA, Brickman JS, Tekell JL, et al. Cingulate function in depression: a potential predictor of treatment response. Neuroreport. (1997) 8:1057–61. doi: 10.1097/00001756-199703030-00048
66. Baxter LR, Schwartz JM, Phelps ME, Mazziotta JC, Guze BH, Selin CE, et al. Reduction of prefrontal cortex glucose metabolism common to three types of depression. Arch Gen Psychiatry. (1989) 46:243–50. doi: 10.1001/archpsyc.1989.01810030049007
67. Brody AL, Saxena S, Silverman DHS, Alborzian S, Fairbanks LA, Phelps ME, et al. Brain metabolic changes in major depressive disorder from pre- to post-treatment with paroxetine. Psychiatry Res Neuroimaging. (1999) 91:127–39. doi: 10.1016/S0925-4927(99)00034-7
68. Little JT, Ketter TA, Kimbrell TA, Dunn RT, Benson BE, Willis MW, et al. Bupropion and venlafaxine responders differ in pretreatment regional cerebral metabolism in unipolar depression. Biol Psychiatry. (2005) 57:220–8. doi: 10.1016/j.biopsych.2004.10.033
69. Milak MS, Parsey RV, Lee L, Oquendo MA, Olvet DM, Eipper F, et al. Pretreatment regional brain glucose uptake in the midbrain on PET may predict remission from a major depressive episode after three months of treatment. Psychiatry Res Neuroimaging. (2009) 173:63–70. doi: 10.1016/j.pscychresns.2008.09.004
70. Chang K, Adleman N, Dienes K, Barnea-Goraly N, Reiss A, Ketter T. Decreased N-acetylaspartate in children with familial bipolar disorder. Biol Psychiatry. (2003) 53:1059–65. doi: 10.1016/s0006-3223(02)01744-4
71. Deicken RF, Pegues MP, Anzalone S, Feiwell R, Soher B. Lower concentration of hippocampal N-acetylaspartate in familial bipolar I disorder. Am J Psychiatry. (2003) 160:873–82. doi: 10.1176/appi.ajp.160.5.873
72. Du F, Yuksel C, Chouinard VA, Huynh P, Ryan K, Cohen BM, et al. Abnormalities in high-energy phosphate metabolism in first-episode bipolar disorder measured using 31P-magnetic resonance spectroscopy. Biol Psychiatry. (2018) 84:797–802. doi: 10.1016/j.biopsych.2017.03.025
73. MacDonald ML, Naydenov A, Chu M, Matzilevich D, Konradi C. Decrease in creatine kinase messenger RNA expression in the hippocampus and dorsolateral prefrontal cortex in bipolar disorder. Bipolar Disord. (2006) 8:255–64. doi: 10.1111/j.1399-5618.2006.00302.x
74. Dogan AE, Yuksel C, Du F, Chouinard VA, Öngür D. Brain lactate and pH in schizophrenia and bipolar disorder: a systematic review of findings from magnetic resonance studies. Neuropsychopharmacology. (2018) 43:1681–90. doi: 10.1038/s41386-018-0041-9
75. Kuang H, Duong A, Jeong H, Zachos K, Andreazza AC. Lactate in bipolar disorder: a systematic review and meta-analysis. Psychiatry Clin Neurosci. (2018) 72:546–55. doi: 10.1111/pcn.12671
76. Morris G, Walder K, McGee SL, Dean OM, Tye SJ, Maes M, et al. A model of the mitochondrial basis of bipolar disorder. Neurosci Biobehav Rev. (2017) 74:1–20. doi: 10.1016/j.neubiorev.2017.01.014
77. Iwamoto K, Bundo M, Kato T. Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum Mol Genet. (2005) 14:241–53. doi: 10.1093/hmg/ddi022
78. Dror N, Klein E, Karry R, Sheinkman A, Kirsh Z, Mazor M, et al. State-dependent alterations in mitochondrial complex I activity in platelets: a potential peripheral marker for schizophrenia. Mol Psychiatry. (2002) 7:995–1001. doi: 10.1038/sj.mp.4001116
79. Taurines R, Thome J, Duvigneau JC, Forbes-Robertson S, Yang L, Klampfl K, et al. Expression analyses of the mitochondrial complex I 75-kDa subunit in early onset schizophrenia and autism spectrum disorder: increased levels as a potential biomarker for early onset schizophrenia. Eur Child Adolesc Psychiatry. (2010) 19:441–8. doi: 10.1007/s00787-009-0074-z
80. Mehler-Wex C, Duvigneau JC, Hartl RT, Ben-Shachar D, Warnke A, Gerlach M. Increased mRNA levels of the mitochondrial complex I 75-kDa subunit: a potential peripheral marker of early onset schizophrenia? Eur Child Adolesc Psychiatry. (2006) 15:504–7. doi: 10.1007/s00787-006-0560-5
81. Akarsu S, Torun D, Bolu A, Erdem M, Kozan S, Ak M, et al. Mitochondrial complex I and III gene mRNA levels in schizophrenia, and their relationship with clinical features. J Mol psychiatry. (2014) 2:6. doi: 10.1186/s40303-014-0006-9
82. Valvassori SS, Bavaresco DV, Feier G, Cechinel-Recco K, Steckert AV, Varela RB, et al. Increased oxidative stress in the mitochondria isolated from lymphocytes of bipolar disorder patients during depressive episodes. Psychiatry Res. (2018) 264:192–201. doi: 10.1016/j.psychres.2018.03.089
83. Zvěrová M, Hroudová J, Fišar Z, Hansíková H, Kališová L, Kitzlerová E, et al. Disturbances of mitochondrial parameters to distinguish patients with depressive episode of bipolar disorder and major depressive disorder. Neuropsychiatr Dis Treat. (2019) 15:233–40. doi: 10.2147/NDT.S188964
84. de Sousa RT, Streck EL, Forlenza OV, Brunoni AR, Zanetti MV, Ferreira GK, et al. Regulation of leukocyte tricarboxylic acid cycle in drug-naïve bipolar disorder. Neurosci Lett. (2015) 605:65–8. doi: 10.1016/j.neulet.2015.08.022
85. Gubert C, Stertz L, Pfaffenseller B, Panizzutti BS, Rezin GT, Massuda R, et al. Mitochondrial activity and oxidative stress markers in peripheral blood mononuclear cells of patients with bipolar disorder, schizophrenia, and healthy subjects. J Psychiatr Res. (2013) 47:1396–402. doi: 10.1016/j.jpsychires.2013.06.018
86. Fišar Z, Hansíková H, KríŽová J, Jirák R, Kitzlerová E, Zvěrová M, et al. Activities of mitochondrial respiratory chain complexes in platelets of patients with Alzheimer's disease and depressive disorder. Mitochondrion. (2019) 48:67–77. doi: 10.1016/j.mito.2019.07.013
87. Ben-Shachar D, Zuk R, Gazawi H, Reshef A, Sheinkman A, Klein E. Increased mitochondrial complex I activity in platelets of schizophrenic patients. Int J Neuropsychopharmacol. (1999) 2:245–53. doi: 10.1017/S1461145799001649
88. Kapczinski F, Dal-Pizzol F, Teixeira AL, Magalhaes PVS, Kauer-Sant'Anna M, Klamt F, et al. Peripheral biomarkers and illness activity in bipolar disorder. J Psychiatr Res. (2011) 45:156–61. doi: 10.1016/j.jpsychires.2010.05.015
89. Banerjee U, Dasgupta A, Rout JK, Singh OP. Effects of lithium therapy on Na +-K +-ATPase activity and lipid peroxidation in bipolar disorder. Prog Neuro-Psychopharmacology Biol Psychiatry. (2012) 37:56–61. doi: 10.1016/j.pnpbp.2011.12.006
90. Andreazza AC, Kauer-Sant'Anna M, Frey BN, Bond DJ, Kapczinski F, Young LT, et al. Oxidative stress markers in bipolar disorder: a meta-analysis. J Affect Disord. (2008) 111:135–44. doi: 10.1016/j.jad.2008.04.013
91. Bajpai A, Verma AK, Srivastava M, Srivastava R. Oxidative stress and major depression. J Clin Diagnostic Res. (2014) 8:CC04. doi: 10.7860/JCDR/2014/10258.5292
92. Moretti A, Gorini A, Villa RF. Affective disorders, antidepressant drugs and brain metabolism. Mol Psychiatry. (2003) 8:773–85. doi: 10.1038/sj.mp.4001353
93. Wang JF, Shao L, Sun X, Young LT. Increased oxidative stress in the anterior cingulate cortex of subjects with bipolar disorder and schizophrenia. Bipolar Disord. (2009) 11:523–9. doi: 10.1111/j.1399-5618.2009.00717.x
94. Andreazza AC, Shoo L, Wang JF, Trevor Young L. Mitochondrial complex i activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch Gen Psychiatry. (2010) 67:360–8. doi: 10.1001/archgenpsychiatry.2010.22
95. Andreazza AC, Kapczinski F, Kauer-Sant'Anna M, Walz JC, Bond DJ, Gonçalves CA, et al. 3-Nitrotyrosine and glutathione antioxidant system in patients in the early and late stages of bipolar disorder. J Psychiatry Neurosci. (2009) 34:263–71.
96. Brown NC, Andreazza AC, Young LT. An updated meta-analysis of oxidative stress markers in bipolar disorder. Psychiatry Res. (2014) 218:61–8. doi: 10.1016/j.psychres.2014.04.005
97. Reddy R, Sahebarao MP, Mukherjee S, Murthy JN. Enzymes of the antioxidant defense system in chronic schizophrenic patients. Biol Psychiatry. (1991) 30:409–12. doi: 10.1016/0006-3223(91)90298-z
98. Eren I, Naziroglu M, Demirdaş A. Protective effects of lamotrigine, aripiprazole and escitalopram on depression-induced oxidative stress in rat brain. Neurochem Res. (2007) 32:1188–95. doi: 10.1007/s11064-007-9289-x
99. Eren I, Naziroglu M, Demirdaş A, Celik O, Uguz AC, Altunbaşak A, et al. Venlafaxine modulates depression-induced oxidative stress in brain and medulla of rat. Neurochem Res. (2007) 32:497–505. doi: 10.1007/s11064-006-9258-9
100. Zhang D, Wen X, Wang X, Shi M, Zhao Y. Antidepressant effect of Shudihuang on mice exposed to unpredictable chronic mild stress. J Ethnopharmacol. (2009) 123:55–60. doi: 10.1016/j.jep.2009.02.029
101. Sun X, Wang JF, Tseng M, Young LT. Downregulation in components of the mitochondrial electron transport chain in the postmortem frontal cortex of subjects with bipolar disorder. J Psychiatry Neurosci. (2006) 31:189–196.
102. Bezchlibnyk YB, Wang JF, McQueen GM, Trevor Young L. Gene expression differences in bipolar disorder revealed by cDNA array analysis of post-mortem frontal cortex. J Neurochem. (2001) 79:826–34. doi: 10.1046/j.1471-4159.2001.00628.x
103. Benes FM, Matzilevich D, Burke RE, Walsh J. The expression of proapoptosis genes is increased in bipolar disorder, but not in schizophrenia. Mol Psychiatry. (2006) 11:241–51. doi: 10.1038/sj.mp.4001758
104. Kuloglu M, Ustundag B, Atmaca M, Canatan H, Ertan Tezcan A, Cinkilinc N. Lipid peroxidation and antioxidant enzyme levels in patients with schizophrenia and bipolar disorder. Cell Biochem Funct. (2002) 20:171–5. doi: 10.1002/cbf.940
105. Ranjekar PK, Hinge A, Hegde MV, Ghate M, Kale A, Sitasawad S, et al. Decreased antioxidant enzymes and membrane essential polyunsaturated fatty acids in schizophrenic and bipolar mood disorder patients. Psychiatry Res. (2003) 121:109–22. doi: 10.1016/S0165-1781(03)00220-8
106. Savas HA, Gergerlioglu HS, Armutcu F, Herken H, Yilmaz HR, Kocoglu E, et al. Elevated serum nitric oxide and superoxide dismutase in euthymic bipolar patients: impact of past episodes. World J Biol Psychiatry. (2006) 7:51–5. doi: 10.1080/15622970510029993
107. Andreazza AC, Cassini C, Rosa AR, Leite MC, de Almeida LMV, Nardin P, Cunha ABN, et al. Serum S100B and antioxidant enzymes in bipolar patients. J Psychiatr Res. (2007) 41:523–9. doi: 10.1016/j.jpsychires.2006.07.013
108. Machado-Vieira R, Andreazza AC, Viale CI, Zanatto V, Cereser V, da Silva Vargas R, et al. Oxidative stress parameters in unmedicated and treated bipolar subjects during initial manic episode: a possible role for lithium antioxidant effects. Neurosci Lett. (2007) 421:33–6. doi: 10.1016/j.neulet.2007.05.016
109. Gergerlioglu HS, Savas HA, Bulbul F, Selek S, Uz E, Yumru M. Changes in nitric oxide level and superoxide dismutase activity during antimanic treatment. Prog Neuro-Psychopharmacology Biol Psychiatry. (2007) 31:697–702. doi: 10.1016/j.pnpbp.2006.12.020
110. de Sousa RT, Zarate CA, Zanetti MV, Costa AC, Talib LL, Gattaz WF, et al. Oxidative stress in early stage bipolar disorder and the association with response to lithium. J Psychiatr Res. (2014) 50:36–41. doi: 10.1016/j.jpsychires.2013.11.011
111. Hahn C-G, Gomez G, Restrepo D, Friedman E, Josiassen R, Pribitkin EA, et al. Aberrant intracellular calcium signaling in olfactory neurons from patients with bipolar disorder. Am J Psychiatry. (2005) 162:616–8. doi: 10.1176/appi.ajp.162.3.616
112. Yoon IS, Li PP, Siu KP, Kennedy JL, Cooke RG, Parikh SV. Altered IMPA2 gene expression and calcium homeostasis in bipolar disorder. Mol Psychiatry. (2001) 6:678–83. doi: 10.1038/sj.mp.4000901
113. Kato T. Role of mitochondrial DNA in calcium signaling abnormality in bipolar disorder. Cell Calcium. (2008) 44:92–102. doi: 10.1016/j.ceca.2007.11.005
114. Uemura T, Green M, Corson TW, Perova T, Li PP, Warsh JJ. Bcl-2 SNP rs956572 associates with disrupted intracellular calcium homeostasis in bipolar I disorder. Bipolar Disord. (2011) 13:41–51. doi: 10.1111/j.1399-5618.2011.00897.x
115. Mertens J, Wang QW, Kim Y, Yu DX, Pham S, Yang B, et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature. (2015) 527:95–9. doi: 10.1038/nature15526
116. Greer PL, Greenberg ME. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron. (2008) 59:846–60. doi: 10.1016/j.neuron.2008.09.002
117. Marcelo KL, Means AR, York B. The Ca2+/calmodulin/CaMKK2 axis: nature's metabolic camshaft. Trends Endocrinol Metab. (2016) 27:706–18. doi: 10.1016/j.tem.2016.06.001
118. Grimaldi B, Sassone-Corsi P. Metabolic clockwork. Nature. (2007) 447:386–7. doi: 10.1038/447386a
119. Atakhorrami M, Rahimi-Aliabadi S, Jamshidi J, Moslemi E, Movafagh A, Ohadi M, et al. A genetic variant in CAMKK2 gene is possibly associated with increased risk of bipolar disorder. J Neural Transm. (2016) 123:323–8. doi: 10.1007/s00702-015-1456-7
120. O'Byrne SN, Scott JW, Pilotte JR, da Santiago AS, Langendorf CG, Oakhill JS, et al. In depth analysis of kinase cross screening data to identify CaMKK2 inhibitory scaffolds. Molecules. (2020) 25:325. doi: 10.3390/molecules25020325
121. Tokumitsu H, Iwabu M, Ishikawa Y, Kobayashi R. Differential regulatory mechanism of ca2+/calmodulin-dependent protein kinase kinase isoforms. Biochemistry. (2001) 40:13925–32. doi: 10.1021/bi010863k
122. Kato T. Molecular genetics of bipolar disorder and depression. Psychiatry Clin Neurosci. (2007) 61:3–19. doi: 10.1111/j.1440-1819.2007.01604.x
123. Park SJ, Lee SB, Suh Y, Kim S-J, Lee N, Hong J-H, et al. DISC1 Modulates neuronal stress responses by gate-keeping ER-mitochondria Ca2+ transfer through the MAM. Cell Rep. (2017) 21:2748–59. doi: 10.1016/j.celrep.2017.11.043
124. Dwivedi Y, Janicak PG, Pandey GN. Elevated [3H]inositol 1,4,5-trisphosphate binding sites and expressed inositol 1,4,5-trisphosphate receptor protein level in platelets of depressed patients. Psychopharmacology (Berl). (1998) 138:47–54. doi: 10.1007/s002130050644
125. Scaini G, Barichello T, Fries GR, Kennon EA, Andrews T, Nix BR, et al. TSPO upregulation in bipolar disorder and concomitant downregulation of mitophagic proteins and NLRP3 inflammasome activation. Neuropsychopharmacology. (2019) 44:1291–9. doi: 10.1038/s41386-018-0293-4
126. Cataldo AM, McPhie DL, Lange NT, Punzell S, Elmiligy S, Ye NZ, et al. Abnormalities in mitochondrial structure in cells from patients with bipolar disorder. Am J Pathol. (2010) 177:575–85. doi: 10.2353/ajpath.2010.081068
127. Safiulina D, Kaasik A. Energetic and dynamic: how mitochondria meet neuronal energy demands. PLoS Biol. (2013) 11:e1001755. doi: 10.1371/journal.pbio.1001755
128. Scaini G, Fries GR, Valvassori SS, Zeni CP, Zunta-Soares G, Berk M, et al. Perturbations in the apoptotic pathway and mitochondrial network dynamics in peripheral blood mononuclear cells from bipolar disorder patients. Transl Psychiatry. (2017) 7:e1111. doi: 10.1038/tp.2017.83
129. Kim H-W, Rapoport SI, Rao JS. Altered expression of apoptotic factors and synaptic markers in postmortem brain from bipolar disorder patients. Neurobiol Dis. (2010) 37:596–603. doi: 10.1016/j.nbd.2009.11.010
130. Moutsatsou P, Tsoporis JN, Salpeas V, Bei E, Alevizos B, Anagnostara C, et al. Peripheral blood lymphocytes from patients with bipolar disorder demonstrate apoptosis and differential regulation of advanced glycation end products and S100B. Clin Chem Lab Med. (2014) 52:999–1007. doi: 10.1515/cclm-2013-0978
131. Roberts DJ, Tan-Sah VP, Smith JM, Miyamoto S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J Biol Chem. (2013) 288:23798–806. doi: 10.1074/jbc.M113.482026
132. Mansur RB, Rizzo LB, Santos CM, Asevedo E, Cunha GR, Noto MN, et al. Impaired glucose metabolism moderates the course of illness in bipolar disorder. J Affect. (2016) 195:57–62. doi: 10.1016/j.jad.2016.02.002
133. Liu Y, Ho RCM, Mak A. Interleukin (IL)-6, tumour necrosis factor alpha (TNF-α) and soluble interleukin-2 receptors (sIL-2R) are elevated in patients with major depressive disorder: a meta-analysis and meta-regression. J Affect Disord. (2012) 139:230–9. doi: 10.1016/j.jad.2011.08.003
134. Sharma S, Akundi RS. Mitochondria: a connecting link in the major depressive disorder jigsaw. Curr Neuropharmacol. (2018) 17:550–62. doi: 10.2174/1570159x16666180302120322
135. Wang H, Zhang M, Xie Q, Yu J, Qi Y, Yue Q. Identification of diagnostic markers for major depressive disorder by cross-validation of data from whole blood samples. PeerJ. (2019) 7:e7171. doi: 10.7717/peerj.7171
136. Petschner P, Gonda X, Baksa D, Eszlari N, Trivaks M, Juhasz G, et al. Genes linking mitochondrial function, cognitive impairment and depression are associated with endophenotypes serving precision medicine. Neuroscience. (2018) 370:207–17. doi: 10.1016/j.neuroscience.2017.09.049
137. Chen C, Wang Y, Zhang J, Ma L, Gu J, Ho G. Contribution of neural cell death to depressive phenotypes of streptozotocin-induced diabetic mice. Dis Model Mech. (2014) 7:723–30. doi: 10.1242/dmm.016162
138. Norkett R, Modi S, Birsa N, Atkin TA, Ivankovic D, Pathania M, et al. DISC1-dependent regulation of mitochondrial dynamics controls the morphogenesis of complex neuronal dendrites. J Biol Chem. (2016) 291:613–29. doi: 10.1074/jbc.M115.699447
139. Culmsee C, Michels S, Scheu S, Arolt V, Dannlowski U, Alferink J. Mitochondria, microglia, and the immune system-how are they linked in affective disorders? Front psychiatry. (2018) 9:739. doi: 10.3389/fpsyt.2018.00739
140. Shinozaki G, Potash JB. New developments in the genetics of bipolar disorder. Curr Psychiatry Rep. (2014) 16:493. doi: 10.1007/s11920-014-0493-5
141. Ou X, Crane DE, MacIntosh BJ, Young LT, Arnold P, Ameis S, et al. CACNA1C rs1006737 genotype and bipolar disorder: focus on intermediate phenotypes and cardiovascular comorbidity. Neurosci Biobehav Rev. (2015) 55:198–210. doi: 10.1016/j.neubiorev.2015.04.022
142. Kataoka M, Matoba N, Sawada T, Kazuno A-A, Ishiwata M, Fujii K, et al. Exome sequencing for bipolar disorder points to roles of de novo loss-of-function and protein-altering mutations. Mol Psychiatry. (2016) 21:885–93. doi: 10.1038/mp.2016.69
143. Rollins B, Martin MV, Sequeira PA, Moon EA, Morgan LZ, Watson SJ, et al. Mitochondrial variants in schizophrenia, bipolar disorder, and major depressive disorder. PLoS ONE. (2009) 4:e4913. doi: 10.1371/journal.pone.0004913
144. Munakata K, Tanaka M, Mori K, Washizuka S, Yoneda M, Tajima O, et al. Mitochondrial DNA 3644T–>C mutation associated with bipolar disorder. Genomics. (2004) 84:1041–50. doi: 10.1016/j.ygeno.2004.08.015
145. Kato T, Stine OC, McMahon FJ, Crowe RR. Increased levels of a mitochondrial DNA deletion in the brain of patients with bipolar disorder. Biol Psychiatry. (1997) 42:871–5. doi: 10.1016/S0006-3223(97)00012-7
146. Sequeira A, Martin MV, Rollins B, Moon EA, Bunney WE, Macciardi F, et al. Mitochondrial mutations and polymorphisms in psychiatric disorders. Front Genet. (2012) 3:103. doi: 10.3389/fgene.2012.00103
147. Shao L, Martin MV, Watson SJ, Schatzberg A, Akil H, Myers RM, et al. Mitochondrial involvement in psychiatric disorders. Ann Med. (2008) 40:281–95. doi: 10.1080/07853890801923753
148. Suomalainen A, Peltonen L, Paetau A, Leinonen H, Majander A, Somer H. Inherited idiopathic dilated cardiomyopathy with multiple deletions of mitochondrial DNA. Lancet. (1992) 340:1319–20. doi: 10.1016/0140-6736(92)92496-3
149. Kakiuchi C, Ishiwata M, Kametani M, Nelson C, Iwamoto K, Kato T. Quantitative analysis of mitochondrial DNA deletions in the brains of patients with bipolar disorder and schizophrenia. Int J Neuropsychopharmacol. (2005) 8:515–22. doi: 10.1017/S1461145705005213
150. Sabunciyan S, Kirches E, Krause G, Bogerts B, Mawrin C, Llenos IC, et al. Quantification of total mitochondrial DNA and mitochondrial common deletion in the frontal cortex of patients with schizophrenia and bipolar disorder. J Neural Transm. (2007) 114:665–74. doi: 10.1007/s00702-006-0581-8
151. Fuke S, Kametani M, Kato T. Quantitative analysis of the 4977-bp common deletion of mitochondrial DNA in postmortem frontal cortex from patients with bipolar disorder and schizophrenia. Neurosci Lett. (2008) 439:173–7. doi: 10.1016/j.neulet.2008.05.009
152. Lindqvist D, Dhabhar FS, James SJ, Hough CM, Jain FA, Bersani FS, et al. Oxidative stress, inflammation and treatment response in major depression. Psychoneuroendocrinology. (2017) 76:197–205. doi: 10.1016/j.psyneuen.2016.11.031
153. Chung JK, Lee SY, Park M, Joo E-J, Kim SA. Investigation of mitochondrial DNA copy number in patients with major depressive disorder. Psychiatry Res. (2019) 282:112616. doi: 10.1016/j.psychres.2019.112616
154. Czarny P, Wigner P, Strycharz J, Swiderska E, Synowiec E, Szatkowska M, et al. Mitochondrial DNA copy number, damage, repair and degradation in depressive disorder. World J Biol Psychiatry. (2019) 21:1–11. doi: 10.1080/15622975.2019.1588993
155. Yamaki N, Otsuka I, Numata S, Yanagi M, Mouri K, Okazaki S, et al. Mitochondrial DNA copy number of peripheral blood in bipolar disorder: the present study and a meta-analysis. Psychiatry Res. (2018) 269:115–7. doi: 10.1016/j.psychres.2018.08.014
156. Fries GR, Zamzow MJ, Andrews T, Pink O, Scaini G, Quevedo J. Accelerated aging in bipolar disorder: A comprehensive review of molecular findings and their clinical implications. Neurosci. Biobehav. Rev. (2020) 112:107–16. doi: 10.1016/j.neubiorev.2020.01.035
157. Wang Y-C, Tai P-A, Poly TN, Islam MM, Yang H-C, Wu C-C, et al. Increased risk of dementia in patients with antidepressants: a meta-analysis of observational studies. Behav Neurol. (2018) 2018:5315098. doi: 10.1155/2018/5315098
158. Chang C-C, Jou S-H, Lin T-T, Liu C-S. Mitochondrial DNA variation and increased oxidative damage in euthymic patients with bipolar disorder. Psychiatry Clin Neurosci. (2014) 68:551–7. doi: 10.1111/pcn.12163
159. Berk M, Plein H, Ferreira D, Jersky B. Blunted adenosine A2a receptor function in platelets in patients with major depression. Eur Neuropsychopharmacol. (2001) 11:183–6. doi: 10.1016/S0924-977X(01)00074-8
160. Adzic M, Lukic I, Mitic M, Djordjevic J, Elaković I, Djordjevic A, et al. Brain region- and sex-specific modulation of mitochondrial glucocorticoid receptor phosphorylation in fluoxetine treated stressed rats: effects on energy metabolism. Psychoneuroendocrinology. (2013) 38:2914–24. doi: 10.1016/j.psyneuen.2013.07.019
161. Anderson G, Maes M. Oxidative/nitrosative stress and immuno-inflammatory pathways in depression: treatment implications. Curr Pharm Des. (2014) 20:3812–47. doi: 10.2174/13816128113196660738
162. Du J, Wang Y, Hunter R, Wei Y, Blumenthal R, Falke C, et al. Dynamic regulation of mitochondrial function by glucocorticoids. Proc Natl Acad Sci USA. (2009) 106:3543–8. doi: 10.1073/pnas.0812671106
163. Zarate CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. (2006) 63:856–64. doi: 10.1001/archpsyc.63.8.856
164. Hare BD, Ghosal S, Duman RS. Rapid acting antidepressants in chronic stress models: molecular and cellular mechanisms. Chronic Stress. (2017) 1:247054701769731. doi: 10.1177/2470547017697317
165. Brkic Z, Milosavljevic M, Glavonic E, Adzic M. Mitochondrial signaling in inflammation-induced depressive behavior in female and male rats: the role of glucocorticoid receptor. Brain Res Bull. (2019) 150:317–27. doi: 10.1016/j.brainresbull.2019.06.016
166. Brkic Z, Francija E, Petrovic Z, Franic D, Lukic I, Mitic M, et al. Distinct modifications of hippocampal glucocorticoid receptor phosphorylation and FKBPs by lipopolysaccharide in depressive female and male rats. J Psychopharmacol. (2017) 31:1234–49. doi: 10.1177/0269881117725914
167. Ghasemi M, Phillips C, Fahimi A, McNerney MW, Salehi A. Mechanisms of action and clinical efficacy of NMDA receptor modulators in mood disorders. Neurosci Biobehav Rev. (2017) 80:555–72. doi: 10.1016/j.neubiorev.2017.07.002
168. Rajkowska G, Stockmeier C. Astrocyte pathology in major depressive disorder: insights from human postmortem brain tissue. Curr Drug Targets. (2013) 14:1225–36. doi: 10.2174/13894501113149990156
169. Abdallah CG, Jiang L, De Feyter HM, Fasula M, Krystal JH, Rothman DL, et al. Glutamate metabolism in major depressive disorder. Am J Psychiatry. (2014) 171:1320–7. doi: 10.1176/appi.ajp.2014.14010067
170. Anand A, Barkay G, Dzemidzic M, Albrecht D, Karne H, Zheng Q-H, et al. Striatal dopamine transporter availability in unmedicated bipolar disorder. Bipolar Disord. (2011) 13:406–13. doi: 10.1111/j.1399-5618.2011.00936.x
171. Quiroz JA, Gray NA, Kato T, Manji HK. Mitochondrially mediated plasticity in the pathophysiology and treatment of bipolar disorder. Neuropsychopharmacology. (2008) 33:2551–65. doi: 10.1038/sj.npp.1301671
172. Srivastava R, Faust T, Ramos A, Ishizuka K, Sawa A. Dynamic changes of the mitochondria in psychiatric illnesses: new mechanistic insights from human neuronal models. Biol Psychiatry. (2018) 83:751–60. doi: 10.1016/j.biopsych.2018.01.007
173. Clay HB, Sillivan S, Konradi C. Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int J Dev Neurosci. (2011) 29:311–24. doi: 10.1016/j.ijdevneu.2010.08.007
174. Shalbuyeva N, Brustovetsky T, Brustovetsky N. Lithium desensitizes brain mitochondria to calcium, antagonizes permeability transition, and diminishes cytochrome C release. J Biol Chem. (2007) 282:18057–68. doi: 10.1074/jbc.M702134200
175. Ghribi O, Herman MM, Spaulding NK, Savory J. Lithium inhibits aluminum-induced apoptosis in rabbit hippocampus, by preventing cytochrome c translocation, Bcl-2 decrease, Bax elevation and caspase-3 activation. J Neurochem. (2002) 82:137–45. doi: 10.1046/j.1471-4159.2002.00957.x
176. Feier G, Valvassori SS, Varela RB, Resende WR, Bavaresco DV, Morais MO, et al. Lithium and valproate modulate energy metabolism in an animal model of mania induced by methamphetamine. Pharmacol Biochem Behav. (2013) 103:589–96. doi: 10.1016/j.pbb.2012.09.010
177. Maurer IC, Schippel P, Volz H-P. Lithium-induced enhancement of mitochondrial oxidative phosphorylation in human brain tissue. Bipolar Disord. (2009) 11:515–22. doi: 10.1111/j.1399-5618.2009.00729.x
178. Michaelis M, Suhan T, Michaelis UR, Beek K, Rothweiler F, Tausch L, et al. Valproic acid induces extracellular signal-regulated kinase 1/2 activation and inhibits apoptosis in endothelial cells. Cell Death Differ. (2006) 13:446–53. doi: 10.1038/sj.cdd.4401759
179. Luís PBM, Ruiter JPN, Aires CCP, Soveral G, de Almeida IT, Duran M. Valproic acid metabolites inhibit dihydrolipoyl dehydrogenase activity leading to impaired 2-oxoglutarate-driven oxidative phosphorylation. Biochim Biophys Acta. (2007) 1767:1126–33. doi: 10.1016/j.bbabio.2007.06.007
180. Cumurcu BE, Ozyurt H, Etikan I, Demir S, Karlidag R. Total antioxidant capacity and total oxidant status in patients with major depression: impact of antidepressant treatment. Psychiatry Clin Neurosci. (2009) 63:639–45. doi: 10.1111/j.1440-1819.2009.02004.x
181. Villa RF, Ferrari F, Bagini L, Gorini A, Brunello N, Tascedda F. Mitochondrial energy metabolism of rat hippocampus after treatment with the antidepressants desipramine and fluoxetine. Neuropharmacology. (2017) 121:30–8. doi: 10.1016/j.neuropharm.2017.04.025
182. Ignácio ZM, Réus GZ, Abelaira HM, Titus SE, Carlessi AS, da Luz JR, et al. Acute and chronic treatments with quetiapine increase mitochondrial respiratory chain complex activity in the rat brain. Curr Neurovasc Res. (2015) 12:283–92. doi: 10.2174/1567202612666150603140912
183. Scaini G, Quevedo J, Velligan D, Roberts DL, Raventos H, Walss-Bass C. Second generation antipsychotic-induced mitochondrial alterations: implications for increased risk of metabolic syndrome in patients with schizophrenia. Eur Neuropsychopharmacol. (2018) 28:369–80. doi: 10.1016/j.euroneuro.2018.01.004
184. Robertson OD, Coronado NG, Sethi R, Berk M, Dodd S. Putative neuroprotective pharmacotherapies to target the staged progression of mental illness. Early Interv Psychiatry. (2019) 13:1032–49. doi: 10.1111/eip.12775
185. Ignácio ZM, Réus GZ, Abelaira HM, de Moura AB, de Souza TG, Matos D, et al. Acute and chronic treatment with quetiapine induces antidepressant-like behavior and exerts antioxidant effects in the rat brain. Metab Brain Dis. (2017) 32:1195–208. doi: 10.1007/s11011-017-0028-y
186. Zarate CA, Payne JL, Singh J, Quiroz JA, Luckenbaugh DA, Denicoff KD, et al. Pramipexole for bipolar II depression: a placebo-controlled proof of concept study. Biol Psychiatry. (2004) 56:54–60. doi: 10.1016/j.biopsych.2004.03.013
187. Burdick KE, Braga RJ, Nnadi CU, Shaya Y, Stearns WH, Malhotra AK. Placebo-controlled adjunctive trial of pramipexole in patients with bipolar disorder: targeting cognitive dysfunction. J Clin Psychiatry. (2012) 73:103–12. doi: 10.4088/JCP.11m07299
188. Bush AI, Cotton SM, Berk M, Malhi GS, Schapkaitz I, Moss K, et al. The efficacy of adjunctive N-acetylcysteine in major depressive disorder. J Clin Psychiatry. (2014) 75:628–36. doi: 10.4088/jcp.13m08454
189. Berk M, Copolov DL, Dean O, Lu K, Jeavons S, Schapkaitz I, et al. N-acetyl cysteine for depressive symptoms in bipolar disorder-a double-blind randomized placebo-controlled trial. Biol Psychiatry. (2008) 64:468–75. doi: 10.1016/j.biopsych.2008.04.022
190. Berk M, Dean OM, Cotton SM, Jeavons S, Tanious M, Kohlmann K, et al. The efficacy of adjunctive N-acetylcysteine in major depressive disorder: a double-blind, randomized, placebo-controlled trial. J Clin Psychiatry. (2014) 75:628–36. doi: 10.4088/JCP.13m08454
191. Balanzá-Martínez V, Fries GR, Colpo GD, Silveira PP, Portella AK, Tabarés-Seisdedos R, et al. Therapeutic use of omega-3 fatty acids in bipolar disorder. Expert Rev Neurother. (2011) 11:1029–47. doi: 10.1586/ern.11.42
192. Stoll AL, Severus WE, Freeman MP, Rueter S, Zboyan HA, Diamond E, et al. Omega 3 fatty acids in bipolar disorder: a preliminary double-blind, placebo-controlled trial. Arch Gen Psychiatry. (1999) 56:407–12. doi: 10.1001/archpsyc.56.5.407
193. Packer L, Witt EH, Tritschler HJ. Alpha-lipoic acid as a biological antioxidant. Free Radic Biol Med. (1995) 19:227–50. doi: 10.1016/0891-5849(95)00017-R
194. Aliev G, Liu J, Shenk JC, Fischbach K, Pacheco GJ, Chen SG, et al. Neuronal mitochondrial amelioration by feeding acetyl-L-carnitine and lipoic acid to aged rats. J Cell Mol Med. (2009) 13:320–33. doi: 10.1111/j.1582-4934.2008.00324.x
195. Hagen TM, Liu J, Lykkesfeldt J, Wehr CM, Ingersoll RT, Vinarsky V, et al. Feeding acetyl-L-carnitine and lipoic acid to old rats significantly improves metabolic function while decreasing oxidative stress. Proc Natl Acad Sci USA. (2002) 99:1870–5. doi: 10.1073/pnas.261708898
196. Brennan BP, Jensen JE, Hudson JI, Coit CE, Beaulieu A, Pope HG, et al. A placebo-controlled trial of acetyl-L-carnitine and α-lipoic acid in the treatment of bipolar depression. J Clin Psychopharmacol. (2013) 33:627–35. doi: 10.1097/JCP.0b013e31829a83f5
197. Alpert JE, Papakostas G, Mischoulon D, Worthington JJ, Petersen T, Mahal Y, et al. S-adenosyl-L-methionine (SAMe) as an adjunct for resistant major depressive disorder: an open trial following partial or nonresponse to selective serotonin reuptake inhibitors or venlafaxine. J Clin Psychopharmacol. (2004) 24:661–4. doi: 10.1097/01.jcp.0000145339.45794.cd
198. Sarris J, Murphy J, Stough C, Mischoulon D, Bousman C, MacDonald P, et al. S-Adenosylmethionine (SAMe) monotherapy for depression: an 8-week double-blind, randomised, controlled trial. Psychopharmacology (Berl). (2020) 237:209–18. doi: 10.1007/s00213-019-05358-1
199. Lipinski JF, Cohen BM, Frankenburg F, Tohen M, Waternaux C, Altesman R, et al. Open trial of S-adenosylmethionine for treatment of depression. Am J Psychiatry. (1984) 141:448–50. doi: 10.1176/ajp.141.3.448
200. Levine J, Green T, Osher Y, Karni N, Levine J. Creatine monohydrate in resistant depression: a preliminary study. Bipolar Disord. (2007) 9:754–8. doi: 10.1111/j.1399-5618.2007.00532.x
201. Sarris J, Mischoulon D, Schweitzer I. Adjunctive nutraceuticals with standard pharmacotherapies in bipolar disorder: a systematic review of clinical trials. Bipolar Disord. (2011) 13:454–65. doi: 10.1111/j.1399-5618.2011.00945.x
202. Chouinard G, Young SN, Annable L. A controlled clinical trial of l-tryptophan in acute mania. Biol Psychiatry. (1985) 20:546–57. doi: 10.1016/0006-3223(85)90026-5
203. Hipkiss AR. Possible benefit of dietary carnosine towards depressive disorders. Aging Dis. (2015) 6:300–3. doi: 10.14336/AD.2014.1211
204. Caruso G, Fresta CG, Musso N, Giambirtone M, Grasso M, Spampinato SF, et al. Carnosine prevents Aβ-induced oxidative stress and inflammation in microglial cells: a key role of TGF-β1. Cells. (2019) 8:64. doi: 10.3390/cells8010064
205. Evins AE, Demopulos C, Yovel I, Culhane M, Ogutha J, Grandin LD, et al. Inositol augmentation of lithium or valproate for bipolar depression. Bipolar Disord. (2006) 8:168–74. doi: 10.1111/j.1399-5618.2006.00303.x
206. Chengappa KNR, Levine J, Gershon S, Mallinger AG, Hardan A, Vagnucci A, et al. Inositol as an add-on treatment for bipolar depression. Bipolar Disord. (2000) 2:47–55. doi: 10.1034/j.1399-5618.2000.020107.x
207. Forester BP, Zuo CS, Ravichandran C, Harper DG, Du F, Kim S, et al. Coenzyme Q10 effects on creatine kinase activity and mood in geriatric bipolar depression. J Geriatr Psychiatry Neurol. (2012) 25:43–50. doi: 10.1177/0891988712436688
208. Lewy AJ, Bauer VK, Cutler NL, Sack RL. Melatonin treatment of winter depression: a pilot study. Psychiatry Res. (1998) 77:57–61. doi: 10.1016/S0165-1781(97)00128-5
209. Acuña-Castroviejo D, Escames G, León J, Carazo A, Khaldy H. Mitochondrial regulation by melatonin and its metabolites. Adv Exp Med Biol. (2003) 527:549–57. doi: 10.1007/978-1-4615-0135-0_63
210. Winiarska K, Fraczyk T, Malinska D, Drozak J, Bryla J. Melatonin attenuates diabetes-induced oxidative stress in rabbits. J Pineal Res. (2006) 40:168–76. doi: 10.1111/j.1600-079X.2005.00295.x
211. Ghodake SR, Suryakar AN, Kulhalli PM, Padalkar RK, Shaikh AK. A study of oxidative stress and influence of antioxidant vitamins supplementation in patients with major depression. Curr Neurobiol. (2012) 3:107–11.
212. Behzadi AH, Omrani Z, Chalian M, Asadi S, Ghadiri M. Folic acid efficacy as an alternative drug added to sodium valproate in the treatment of acute phase of mania in bipolar disorder: a double-blind randomized controlled trial. Acta Psychiatr Scand. (2009) 120:441–5. doi: 10.1111/j.1600-0447.2009.01368.x
213. Jarrett SG, Milder JB, Liang L-P, Patel M. The ketogenic diet increases mitochondrial glutathione levels. J Neurochem. (2008) 106:1044–51. doi: 10.1111/j.1471-4159.2008.05460.x
214. L'upták M, Hroudová J. Important role of Mitochondria and the effect of mood stabilizers on mitochondrial function. Physiol Res. (2019) 68:s3–15. doi: 10.33549/physiolres.934324
215. Wang JF, Bown C, Young LT. Differential display PCR reveals novel targets for the mood-stabilizing drug valproate including the molecular chaperone GRP78. Mol Pharmacol. (1999) 55:521–7.
216. Shao L, Sun X, Xu L, Young LT, Wang J-F. Mood stabilizing drug lithium increases expression of endoplasmic reticulum stress proteins in primary cultured rat cerebral cortical cells. Life Sci. (2006) 78:1317–23. doi: 10.1016/j.lfs.2005.07.007
217. Quiroz JA, Machado-Vieira R, Zarate CA, Manji HK. Novel insights into lithium's mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology. (2010) 62:50–60. doi: 10.1159/000314310
218. Hough CJ, Irwin RP, Gao XM, Rogawski MA, Chuang DM. Carbamazepine inhibition of N-methyl-D-aspartate-evoked calcium influx in rat cerebellar granule cells. J Pharmacol Exp Ther. (1996) 276:143–9.
219. Cihangir Uguz A, Demirci K, Espino J. The importance of melatonin and mitochondria interaction in mood disorders and schizophrenia: a current assessment. Curr Med Chem. (2016) 23:2146–58. doi: 10.2174/0929867323666160428105849
220. Bachmann RF, Wang Y, Yuan P, Zhou R, Li X, Alesci S, et al. Common effects of lithium and valproate on mitochondrial functions: protection against methamphetamine-induced mitochondrial damage. Int J Neuropsychopharmacol. (2009) 12:805–22. doi: 10.1017/S1461145708009802
221. Williams RSB, Cheng L, Mudge AW, Harwood AJ. A common mechanism of action for three mood-stabilizing drugs. Nature. (2002) 417:292–5. doi: 10.1038/417292a
222. Zschocke J, Zimmermann N, Berning B, Ganal V, Holsboer F, Rein T. Antidepressant drugs diversely affect autophagy pathways in astrocytes and neurons–dissociation from cholesterol homeostasis. Neuropsychopharmacology. (2011) 36:1754–68. doi: 10.1038/npp.2011.57
223. Chen B, Wang JF, Young LT. Chronic valproate treatment increases expression of endoplasmic reticulum stress proteins in the rat cerebral cortex and hippocampus. Biol Psychiatry. (2000) 48:658–64. doi: 10.1016/s0006-3223(00)00878-7
224. Hiroi T, Wei H, Hough C, Leeds P, Chuang D-M. Protracted lithium treatment protects against the ER stress elicited by thapsigargin in rat PC12 cells: roles of intracellular calcium, GRP78 and Bcl-2. Pharmacogenomics J. (2005) 5:102–11. doi: 10.1038/sj.tpj.6500296
225. Yu Z, Luo H, Fu W, Mattson MP. The endoplasmic reticulum stress-responsive protein GRP78 protects neurons against excitotoxicity and apoptosis: suppression of oxidative stress and stabilization of calcium homeostasis. Exp Neurol. (1999) 155:302–14. doi: 10.1006/exnr.1998.7002
226. Nigam SK, Goldberg AL, Ho S, Rohde MF, Bush KT, Sherman MYu. A set of endoplasmic reticulum proteins possessing properties of molecular chaperones includes Ca(2+)-binding proteins and members of the thioredoxin superfamily. J Biol Chem. (1994) 269:1744–9.
227. Wang JF, Shao L, Sun X, Young LT. Glutathione S-transferase is a novel target for mood stabilizing drugs in primary cultured neurons. J Neurochem. (2004) 88:1477–84. doi: 10.1046/j.1471-4159.2003.02276.x
228. Sharma R, Yang Y, Sharma A, Awasthi S, Awasthi YC. Antioxidant role of glutathione S-transferases: protection against oxidant toxicity and regulation of stress-mediated apoptosis. Antioxidants Redox Signal. (2004) 6:289–300. doi: 10.1089/152308604322899350
229. Cui J, Shao L, Young LT, Wang J-F. Role of glutathione in neuroprotective effects of mood stabilizing drugs lithium and valproate. Neuroscience. (2007) 144:1447–53. doi: 10.1016/j.neuroscience.2006.11.010
230. Valvassori SS, Rezin GT, Ferreira CL, Moretti M, Gonçalves CL, Cardoso MR, et al. Effects of mood stabilizers on mitochondrial respiratory chain activity in brain of rats treated with d-amphetamine. J Psychiatr Res. (2010) 44:903–9. doi: 10.1016/j.jpsychires.2010.02.009
231. Machado-Vieira R, Manji HK, Zarate CA. The role of lithium in the treatment of bipolar disorder: convergent evidence for neurotrophic effects as a unifying hypothesis. Bipolar Disord. (2009) 11(Suppl. 2):92–109. doi: 10.1111/j.1399-5618.2009.00714.x
232. Khan MS, Ali T, Abid MN, Jo MH, Khan A, Kim MW, et al. Lithium ameliorates lipopolysaccharide-induced neurotoxicity in the cortex and hippocampus of the adult rat brain. Neurochem Int. (2017) 108:343–54. doi: 10.1016/j.neuint.2017.05.008
233. Chen R-W, Qin Z-H, Ren M, Kanai H, Chalecka-Franaszek E, Leeds P, et al. Regulation of c-Jun N-terminal kinase, p38 kinase and AP-1 DNA binding in cultured brain neurons: roles in glutamate excitotoxicity and lithium neuroprotection. J Neurochem. (2003) 84:566–75. doi: 10.1046/j.1471-4159.2003.01548.x
234. De Vasconcellos APS, Nieto FB, Crema LM, Diehl LA, De Almeida LM, Prediger ME, et al. Chronic lithium treatment has antioxidant properties but does not prevent oxidative damage induced by chronic variate stress. Neurochem Res. (2006) 31:1141–51. doi: 10.1007/s11064-006-9139-2
235. Alsaif M, Haenisch F, Guest PC, Rahmoune H, Bahn S. Challenges in drug target discovery in bipolar disorder. Expert Opin Ther Targets. (2013) 17:565–77. doi: 10.1517/14728222.2013.771169
236. Bijur GN, Jope RS. Glycogen synthase kinase-3 beta is highly activated in nuclei and mitochondria. Neuroreport. (2003) 14:2415–9. doi: 10.1097/00001756-200312190-00025
237. Bachmann RF, Schloesser RJ, Gould TD, Manji HK. Mood stabilizers target cellular plasticity and resilience cascades: implications for the development of novel therapeutics. Mol Neurobiol. (2005) 32:173–202. doi: 10.1385/MN:32:2:173
238. Gould TD, Einat H, Bhat R, Manji HK. AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int J Neuropsychopharmacol. (2004) 7:387–90. doi: 10.1017/S1461145704004535
239. McNamara RK, Ostrander M, Abplanalp W, Richtand NM, Benoit SC, Clegg DJ. Modulation of phosphoinositide-protein kinase C signal transduction by omega-3 fatty acids: implications for the pathophysiology and treatment of recurrent neuropsychiatric illness. Prostaglandins Leukot Essent Fatty Acids. (2006) 75:237–57. doi: 10.1016/j.plefa.2006.07.009
240. Manji HK, Bersudsky Y, Chen G, Belmaker RH, Potter WZ. Modulation of protein kinase C isozymes and substrates by lithium: the role of myo-inositol. Neuropsychopharmacology. (1996) 15:370–81. doi: 10.1016/0893-133X(95)00243-7
241. Hroudova J, Fisar Z. Activities of respiratory chain complexes and citrate synthase influenced by pharmacologically different antidepressants and mood stabilizers. Neuroendocrinol Lett. (2010) 31:336–42.
242. Eskandari MR, Fard JK, Hosseini M-J, Pourahmad J. Glutathione mediated reductive activation and mitochondrial dysfunction play key roles in lithium induced oxidative stress and cytotoxicity in liver. Biometals. (2012) 25:863–73. doi: 10.1007/s10534-012-9552-8
243. Salimi A, Gholamifar E, Naserzadeh P, Hosseini M-J, Pourahmad J. Toxicity of lithium on isolated heart mitochondria and cardiomyocyte: a justification for its cardiotoxic adverse effect. J Biochem Mol Toxicol. (2017) 31:e21836. doi: 10.1002/jbt.21836
244. Corrêa C, Amboni G, Assis LC, Martins MR, Kapczinski F, Streck EL, et al. Effects of lithium and valproate on hippocampus citrate synthase activity in an animal model of mania. Prog Neuropsychopharmacol Biol Psychiatry. (2007) 31:887–91. doi: 10.1016/j.pnpbp.2007.02.005
245. Lopes-Borges J, Valvassori SS, Varela RB, Tonin PT, Vieira JS, Gonçalves CL, et al. Histone deacetylase inhibitors reverse manic-like behaviors and protect the rat brain from energetic metabolic alterations induced by ouabain. Pharmacol Biochem Behav. (2015) 128:89–95. doi: 10.1016/j.pbb.2014.11.014
246. Valvassori SS, Resende WR, Lopes-Borges J, Mariot E, Dal-Pont GC, Vitto MF, et al. Effects of mood stabilizers on oxidative stress-induced cell death signaling pathways in the brains of rats subjected to the ouabain-induced animal model of mania: mood stabilizers exert protective effects against ouabain-induced activation of the cell death pathway. J Psychiatr Res. (2015) 65:63–70. doi: 10.1016/j.jpsychires.2015.04.009
247. Silva MFB, Ruiter JPN, Ijlst L, Jakobs C, Duran M, Tavares De Almeida I, et al. Valproate inhibits the mitochondrial pyruvate-driven oxidative phosphorylation in vitro. J Inherited Metabolic Dis. (1997) 20:397–400. doi: 10.1023/A:1005398516208
248. Aires CCP, Soveral G, Luís PBM, ten Brink HJ, de Almeida IT, Duran M. Pyruvate uptake is inhibited by valproic acid and metabolites in mitochondrial membranes. FEBS Lett. (2008) 582:3359–66. doi: 10.1016/j.febslet.2008.08.028
249. Finsterer J, Scorza FA. Effects of antiepileptic drugs on mitochondrial functions, morphology, kinetics, biogenesis, and survival. Epilepsy Res. (2017) 136:5–11. doi: 10.1016/j.eplepsyres.2017.07.003
250. Finsterer J, Zarrouk Mahjoub S. Mitochondrial toxicity of antiepileptic drugs and their tolerability in mitochondrial disorders. Expert Opin Drug Metab Toxicol. (2012) 8:71–9. doi: 10.1517/17425255.2012.644535
251. Cikankova T, Sigitova E, Zverova M, Fisar Z, Raboch J, Hroudova J. Mitochondrial dysfunctions in bipolar disorder: effect of the disease and pharmacotherapy. CNS Neurol Disord Drug Targets. (2017) 16:176–86. doi: 10.2174/1871527315666161213110518
252. Kim YJ, Ko HH, Han ES, Lee CS. Lamotrigine inhibition of rotenone- or 1-methyl-4-phenylpyridinium-induced mitochondrial damage and cell death. Brain Res Bull. (2007) 71:633–40. doi: 10.1016/j.brainresbull.2006.12.006
253. Hroudová J, Fišar Z. Connectivity between mitochondrial functions and psychiatric disorders. Psychiatry Clin Neurosci. (2011) 65:130–41. doi: 10.1111/j.1440-1819.2010.02178.x
254. Yang S-J, Kim S-Y, Stewart R, Kim J-M, Shin I-S, Jung S-W, et al. Gender differences in 12-week antidepressant treatment outcomes for a naturalistic secondary care cohort: the CRESCEND study. Psychiatry Res. (2011) 189:82–90. doi: 10.1016/j.psychres.2010.12.027
255. Tamási V, Petschner P, Adori C, Kirilly E, Ando RD, Tothfalusi L, et al. Transcriptional evidence for the role of chronic venlafaxine treatment in neurotrophic signaling and neuroplasticity including also Glutamatergic [corrected]—and insulin-mediated neuronal processes. PLoS ONE. (2014) 9:e113662. doi: 10.1371/journal.pone.0113662
256. Hestad KA, Tønseth S, Støen CD, Ueland T, Aukrust P. Raised plasma levels of tumor necrosis factor α in patients with depression: normalization during electroconvulsive therapy. J ECT. (2003) 19:183–8. doi: 10.1097/00124509-200312000-00002
257. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. (2009) 65:732–41. doi: 10.1016/j.biopsych.2008.11.029
258. Müller N, Schwarz MJ, Dehning S, Douhe A, Cerovecki A, Goldstein-Müller B, et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry. (2006) 11:680–4. doi: 10.1038/sj.mp.4001805
259. Gottschalk WK, Lutz MW, He YT, Saunders AM, Burns DK, Roses AD, et al. The broad impact of TOM40 on neurodegenerative diseases in aging. J Park Dis Alzheimers Dis. (2014) 1:1–25. doi: 10.13188/2376-922X.1000003
260. Malkesman O, Austin DR, Tragon T, Henter ID, Reed JC, Pellecchia M, et al. Targeting the BH3-interacting domain death agonist to develop mechanistically unique antidepressants. Mol Psychiatry. (2012) 17:770–80. doi: 10.1038/mp.2011.77
261. Cattaneo A, Gennarelli M, Uher R, Breen G, Farmer A, Aitchison KJ, et al. Candidate genes expression profile associated with antidepressants response in the GENDEP study: differentiating between baseline “predictors” and longitudinal “targets.” Neuropsychopharmacology. (2013) 38:377–85. doi: 10.1038/npp.2012.191
262. Aguiar AS, Tuon T, Pinho CA, Silva LA, Andreazza AC, Kapczinski F, et al. Mitochondrial IV complex and brain neurothrophic derived factor responses of mice brain cortex after downhill training. Neurosci Lett. (2007) 426:171–4. doi: 10.1016/j.neulet.2007.08.058
263. Kara NZ, Toker L, Agam G, Anderson GW, Belmaker RH, Einat H. Trehalose induced antidepressant-like effects and autophagy enhancement in mice. Psychopharmacology (Berl). (2013) 229:367–75. doi: 10.1007/s00213-013-3119-4
264. Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. (2010) 329:959–64. doi: 10.1126/science.1190287
265. Ferreira GK, Cardoso MR, Jeremias IC, Gonçalves CL, Freitas KV, Antonini R, et al. Fluvoxamine alters the activity of energy metabolism enzymes in the brain. Rev Bras Psiquiatr. (2014) 36:220–6. doi: 10.1590/1516-4446-2013-1202
266. Scaini G, Maggi DD, De-Nês BT, Gonçalves CL, Ferreira GK, Teodorak BP, et al. Activity of mitochondrial respiratory chain is increased by chronic administration of antidepressants. Acta Neuropsychiatr. (2011) 23:112–8. doi: 10.1111/j.1601-5215.2011.00548.x
267. Jeong J, Park M, Yoon JS, Kim H, Lee SK, Lee E, et al. Requirement of AMPK activation for neuronal metabolic-enhancing effects of antidepressant paroxetine. Neuroreport. (2015) 26:424–8. doi: 10.1097/WNR.0000000000000365
268. Agostinho FR, Réus GZ, Stringari RB, Ribeiro KF, Ferraro AK, Benedet J, et al. Treatment with olanzapine, fluoxetine and olanzapine/fluoxetine alters citrate synthase activity in rat brain. Neurosci Lett. (2011) 487:278–81. doi: 10.1016/j.neulet.2010.10.037
269. Vaynman S, Ying Z, Wu A, Gomez-Pinilla F. Coupling energy metabolism with a mechanism to support brain-derived neurotrophic factor-mediated synaptic plasticity. Neuroscience. (2006) 139:1221–34. doi: 10.1016/j.neuroscience.2006.01.062
270. El Idrissi AE, Trenkner E. Growth factors and taurine protect against excitotoxicity by stabilizing calcium homeostasis and energy metabolism. J Neurosci. (1999) 19:9459–68. doi: 10.1523/jneurosci.19-21-09459.1999
271. Mitic M, Simic I, Djordjevic J, Radojcic MB, Adzic M. Gender-specific effects of fluoxetine on hippocampal glucocorticoid receptor phosphorylation and behavior in chronically stressed rats. Neuropharmacology. (2013) 70:100–11. doi: 10.1016/j.neuropharm.2012.12.012
272. Villa RF, Ferrari F, Gorini A, Brunello N, Tascedda F. Effect of desipramine and fluoxetine on energy metabolism of cerebral mitochondria. Neuroscience. (2016) 330:326–34. doi: 10.1016/j.neuroscience.2016.05.051
273. Pandi-Perumal SR, Srinivasan V, Maestroni GJM, Cardinali DP, Poeggeler B, Hardeland R. Melatonin: nature's most versatile biological signal? FEBS J. (2006) 273:2813–38. doi: 10.1111/j.1742-4658.2006.05322.x
274. Levkovitz Y, Gil-Ad I, Zeldich E, Dayag M, Weizman A. Differential induction of apoptosis by antidepressants in glioma and neuroblastoma cell lines: evidence for p-c-Jun, cytochrome c, and caspase-3 involvement. J Mol Neurosci. (2005) 27:29–42. doi: 10.1385/JMN:27:1:029
275. Ma YG, Dong L, Ye XL, Deng CL, Cheng JH, Liu WC, et al. Activation of cloned BKCa channels in nitric oxide-induced apoptosis of HEK293 cells. Apoptosis. (2010) 15:426–38. doi: 10.1007/s10495-009-0423-x
276. Kolla N, Wei Z, Richardson JS, Li XM. Amitriptyline and fluoxetine protect PC12 cells from cell death induced by hydrogen peroxide. J Psychiatry Neurosci. (2005) 30:196–201.
277. Souza MEJ, Polizello ACM, Uyemura SA, Castro-Silva O, Curti C. Effect of fluoxetine on rat liver mitochondria. Biochem Pharmacol. (1994) 48:535–41. doi: 10.1016/0006-2952(94)90283-6
278. Curti C, Mingatto FE, Polizello ACM, Galastri LO, Uyemura SA, Santos AC. Fluoxetine interacts with the lipid bilayer of the inner membrane in isolated rat brain mitochondria, inhibiting electron transport and F1F0-ATPase activity. Mol Cell Biochem. (1999) 199:103–9. doi: 10.1023/A:1006912010550
279. Djordjevic A, Djordjevic J, Elaković I, Adzic M, Matić G, Radojcic MB. Effects of fluoxetine on plasticity and apoptosis evoked by chronic stress in rat prefrontal cortex. Eur J Pharmacol. (2012) 693:37–44. doi: 10.1016/j.ejphar.2012.07.042
280. Djordjevic A, Djordjevic J, Elaković I, Adzic M, Matić G, Radojcic MB. Fluoxetine affects hippocampal plasticity, apoptosis and depressive-like behavior of chronically isolated rats. Prog Neuro-Psychopharmacology Biol Psychiatry. (2012) 36:92–100. doi: 10.1016/j.pnpbp.2011.10.006
281. Magalhães PV, Dean OM, Bush AI, Copolov DL, Malhi GS, Kohlmann K, et al. N-acetyl cysteine add-on treatment for bipolar II disorder: a subgroup analysis of a randomized placebo-controlled trial. J Affect Disord. (2011) 129:317–20. doi: 10.1016/j.jad.2010.08.001
282. Wei Z, Bai O, Richardson JS, Mousseau DD, Li X-M. Olanzapine protects PC12 cells from oxidative stress induced by hydrogen peroxide. J Neurosci Res. (2003) 73:364–8. doi: 10.1002/jnr.10668
283. Wang H, Xu H, Dyck LE, Li XM. Olanzapine and quetiapine protect PC12 cells from β-amyloid peptide25-35-induced oxidative stress and the ensuing apoptosis. J Neurosci Res. (2005) 81:572–80. doi: 10.1002/jnr.20570
284. Li XM, Chlan-Fourney J, Juorio AV, Bennett VL, Shrikhande S, Keegan DL, et al. Differential effects of olanzapine on the gene expression of superoxide dismutase and the low affinity nerve growth factor receptor. J Neurosci Res. (1999) 56:72–5. doi: 10.1002/(SICI)1097-4547(19990401)56:1<72::AID-JNR9>3.0.CO;2-0
285. Hoertel N, de Maricourt P, Gorwood P. Novel routes to bipolar disorder drug discovery. Expert Opin Drug Discov. (2013) 8:907–18. doi: 10.1517/17460441.2013.804057
286. Bown CD, Wang J-F, Chen B, Young LT. Regulation of ER stress proteins by valproate: therapeutic implications. Bipolar Disord. (2002) 4:145–51. doi: 10.1034/j.1399-5618.2002.t01-1-40201.x
287. Mathew SJ, Manji HK, Charney DS. Novel drugs and therapeutic targets for severe mood disorders. Neuropsychopharmacology. (2008) 33:2080–92. doi: 10.1038/sj.npp.1301652
288. Nierenberg AA, Kansky C, Brennan BP, Shelton RC, Perlis R, Iosifescu DV. Mitochondrial modulators for bipolar disorder: a pathophysiologically informed paradigm for new drug development. Aust N Z J Psychiatry. (2013) 47:26–42. doi: 10.1177/0004867412449303
289. Berk M, Dean OM, Cotton SM, Gama CS, Kapczinski F, Fernandes B, et al. Maintenance N-acetyl cysteine treatment for bipolar disorder: a double-blind randomized placebo controlled trial. BMC Med. (2012) 10:91. doi: 10.1186/1741-7015-10-91
290. Nicoletti VG, Marino VM, Cuppari C, Licciardello D, Patti D, Spina Purrello V, et al. Effect of antioxidant diets on mitochondrial gene expression in rat brain during aging. Neurochem Res. (2005) 30:737–52. doi: 10.1007/s11064-005-6867-7
291. Mayer M, Noble M. N-acetyl-L-cysteine is a pluripotent protector against cell death and enhancer of trophic factor-mediated cell survival in vitro. Proc Natl Acad Sci USA. (1994) 91:7496–500. doi: 10.1073/pnas.91.16.7496
292. Aruoma OI, Halliwell B, Hoey BM, Butler J. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic Biol Med. (1989) 6:593–7. doi: 10.1016/0891-5849(89)90066-X
293. Benrahmoune M, Thérond P, Abedinzadeh Z. The reaction of superoxide radical with N-acetylcysteine. Free Radic Biol Med. (2000) 29:775–82. doi: 10.1016/S0891-5849(00)00380-4
294. Berk M, Dean O, Cotton SM, Gama CS, Kapczinski F, Fernandes BS, et al. The efficacy of N-acetylcysteine as an adjunctive treatment in bipolar depression: an open label trial. J Affect Disord. (2011) 135:389–94. doi: 10.1016/j.jad.2011.06.005
295. Dean OM, Bush AI, Copolov DL, Kohlmann K, Jeavons S, Schapkaitz I, et al. Effects of N-acetyl cysteine on cognitive function in bipolar disorder. Psychiatry Clin Neurosci. (2012) 66:514–7. doi: 10.1111/j.1440-1819.2012.02392.x
296. Magalhães PV, Dean OM, Bush AI, Copolov DL, Malhi GS, Kohlmann K, et al. N-acetylcysteine for major depressive episodes in bipolar disorder. Rev Bras Psiquiatr. (2011) 33:374–8. doi: 10.1590/s1516-44462011000400011
297. Magalhães PV, Dean OM, Bush AI, Copolov DL, Weisinger D, Malhi GS, et al. Systemic illness moderates the impact of N-acetyl cysteine in bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. (2012) 37:132–5. doi: 10.1016/j.pnpbp.2011.11.011
298. Da Silva Magalhães PV, Dean OM, Bush AI, Copolov DL, Malhi GS, Kohlmann K, et al. A preliminary investigation on the efficacy of N-acetyl cysteine for mania or hypomania. Aust N Z J Psychiatry. (2013) 47:564–8. doi: 10.1177/0004867413481631
299. Nasca C, Bigio B, Zelli D, Nicoletti F, McEwen BS. Mind the gap: glucocorticoids modulate hippocampal glutamate tone underlying individual differences in stress susceptibility. Mol Psychiatry. (2015) 20:755–63. doi: 10.1038/mp.2014.96
300. Nasca C, Bigio B, Zelli D, de Angelis P, Lau T, Okamoto M, et al. Role of the astroglial glutamate exchanger xCT in ventral hippocampus in resilience to stress. Neuron. (2017) 96:402–13.e5. doi: 10.1016/j.neuron.2017.09.020
301. Fernandes BS, Dean OM, Dodd S, Malhi GS, Berk M. N-Acetylcysteine in depressive symptoms and functionality: a systematic review and meta-analysis. J Clin Psychiatry. (2016) 77:e457–66. doi: 10.4088/JCP.15r09984
302. Katsumata T, Katayama Y, Obo R, Muramatsu H, Ohtori T, Terashi A. Delayed administration of ethyl eicosapentate improves local cerebral blood flow and metabolism without affecting infarct volumes in the rat focal ischemic model. Eur J Pharmacol. (1999) 372:167–74. doi: 10.1016/S0014-2999(99)00171-5
303. Young G, Conquer J. Omega-3 fatty acids and neuropsychiatric disorders. Reprod Nutr Dev. (2005) 45:1–28. doi: 10.1051/rnd:2005001
304. Sobczak S, Honig A, Christophe M, Maes M, Helsdingen RWC, De Vriese S, et al. Lower high-density lipoprotein cholesterol and increased omega-6 polyunsaturated fatty acids in first-degree relatives of bipolar patients. Psychol Med. (2004) 34:103–12. doi: 10.1017/S0033291703001090
305. Kitajka K, Puskás LG, Zvara A, Hackler L, Barceló-Coblijn G, Yeo YK, et al. The role of n-3 polyunsaturated fatty acids in brain: modulation of rat brain gene expression by dietary n-3 fatty acids. Proc Natl Acad Sci USA. (2002) 99:2619–24. doi: 10.1073/pnas.042698699
306. Stanley WC, Khairallah RJ, Dabkowski ER. Update on lipids and mitochondrial function: impact of dietary n-3 polyunsaturated fatty acids. Curr Opin Clin Nutr Metab Care. (2012) 15:122–6. doi: 10.1097/MCO.0b013e32834fdaf7
307. Maes M, Delange J, Ranjan R, Meltzer HY, Desnyder R, Cooremans W, et al. Acute phase proteins in schizophrenia, mania and major depression: modulation by psychotropic drugs. Psychiatry Res. (1997) 66:1–11. doi: 10.1016/S0165-1781(96)02915-0
308. Shaltiel G, Chen G, Manji HK. Neurotrophic signaling cascades in the pathophysiology and treatment of bipolar disorder. Curr Opin Pharmacol. (2007) 7:22–6. doi: 10.1016/j.coph.2006.07.005
309. Noaghiul S, Hibbeln JR. Cross-national comparisons of seafood consumption and rates of bipolar disorders. Am J Psychiatry. (2003) 160:2222–7. doi: 10.1176/appi.ajp.160.12.2222
310. Frangou S, Lewis M, McCrone P. Efficacy of ethyl-eicosapentaenoic acid in bipolar depression: randomised double-blind placebo-controlled study. Br J Psychiatry. (2006) 188:46–50. doi: 10.1192/bjp.188.1.46
311. Estrada DE, Ewart HS, Tsakiridis T, Volchuk A, Ramlal T, Tritschler H, et al. Stimulation of glucose uptake by the natural coenzyme-lipoic acid/thioctic acid: participation of elements of the insulin signaling pathway. Diabetes. (1996) 45:1798–804. doi: 10.2337/diab.45.12.1798
312. Liu J. The effects and mechanisms of mitochondrial nutrient alpha-lipoic acid on improving age-associated mitochondrial and cognitive dysfunction: an overview. Neurochem Res. (2008) 33:194–203. doi: 10.1007/s11064-007-9403-0
313. Liu J, Atamna H, Kuratsune H, Ames BN. Delaying brain mitochondrial decay and aging with mitochondrial antioxidants and metabolites. Ann N Y Acad Sci. (2002) 959:133–66. doi: 10.1111/j.1749-6632.2002.tb02090.x
314. Macêdo DS, Medeiros CD, Cordeiro RC, Sousa FC, Santos JV, Morais TA, et al. Effects of alpha-lipoic acid in an animal model of mania induced by D-amphetamine. Bipolar Disord. (2012) 14:707–18. doi: 10.1111/j.1399-5618.2012.01046.x
315. Hoppel C. The role of carnitine in normal and altered fatty acid metabolism. Am J Kidney Dis. (2003) 41:S4–12. doi: 10.1016/s0272-6386(03)00112-4
316. Al-Majed AA, Sayed-Ahmed MM, Al-Omar FA, Al-Yahya AA, Aleisa AM, Al-Shabanah OA. Carnitine esters prevent oxidative stress damage and energy depletion following transient forebrain ischaemia in the rat hippocampus. Clin Exp Pharmacol Physiol. (2006) 33:725–33. doi: 10.1111/j.1440-1681.2006.04425.x
317. Rebouche CJ. Kinetics, pharmacokinetics, and regulation of L-Carnitine and acetyl-L-carnitine metabolism. Ann N Y Acad Sci. (2004) 1033:30–41. doi: 10.1196/annals.1320.003
318. Virmani A, Gaetani F, Binienda Z. Effects of metabolic modifiers such as carnitines, coenzyme Q10, and PUFAs against different forms of neurotoxic insults: metabolic inhibitors, MPTP, and methamphetamine. Ann N Y Acad Sci. (2005) 1053:183–91. doi: 10.1196/annals.1344.016
319. Hao Y, Basile AS, Chen G, Zhang L. Glutamate-induced over-expression of GAD is down-regulated by acetyl-L-carnitine in rat islet cells. Endocr Res. (2004) 30:107–16. doi: 10.1081/ERC-120029890
320. Aureli T, Miccheli A, Ricciolini R, Di Cocco ME, Ramacci MT, Angelucci L, et al. Aging brain: effect of acetyl-l-carnitine treatment on rat brain energy and phospholipid metabolism. A study by 31P and 1H NMR spectroscopy. Brain Res. (1990) 526:108–12. doi: 10.1016/0006-8993(90)90255-A
321. Ames BN, Liu J. Delaying the mitochondrial decay of aging with acetylcarnitine. Ann N Y Acad Sci. (2004) 1033:108–16. doi: 10.1196/annals.1320.010
322. Pettegrew JW, Levine J, Gershon S, Stanley JA, Servan-Schreiber D, Panchalingam K, et al. 31P-MRS study of acetyl-L-carnitine treatment in geriatric depression: preliminary results. Bipolar Disord. (2002) 4:61–6. doi: 10.1034/j.1399-5618.2002.01180.x
323. Tempesta E, Casella L, Pirrongelli C, Janiri L, Calvani M, Ancona L. L-acetylcarnitine in depressed elderly subjects. A cross-over study vs. placebo. Drugs Exp Clin Res. (1987) 13:417–23.
324. Zanardi R, Smeraldi E. A double-blind, randomised, controlled clinical trial of acetyl-L-carnitine vs. amisulpride in the treatment of dysthymia. Eur Neuropsychopharmacol. (2006) 16:281–7. doi: 10.1016/j.euroneuro.2005.10.005
325. Bottiglieri T, Laundy M, Crellin R, Toone BK, Carney MWP, Reynolds EH. Homocysteine, folate, methylation, and monoamine metabolism in depression. J Neurol Neurosurg Psychiatry. (2000) 69:228–32. doi: 10.1136/jnnp.69.2.228
326. Papakostas GI, Mischoulon D, Shyu I, Alpert JE, Fava M. S-adenosyl methionine (SAMe) augmentation of serotonin reuptake inhibitors for antidepressant nonresponders with major depressive disorder: a double-blind, randomized clinical trial. Am J Psychiatry. (2010) 167:942–8. doi: 10.1176/appi.ajp.2009.09081198
327. Berlanga C, Ortega-Soto HA, Ontiveros M, Senties H. Efficacy of S-adenosyl-L-methionine in speeding the onset of action of imipramine. Psychiatry Res. (1992) 44:257–62. doi: 10.1016/0165-1781(92)90029-3
328. Carney MWP, Chary TKN, Bottiglieri T, Reynolds EH. The switch mechanism and the bipolar/unipolar dichotomy. Br J Psychiatry. (1989) 154:48–51. doi: 10.1192/bjp.154.1.48
329. Juhn MS, Tarnopolsky M. Oral creatine supplementation and athletic performance: a critical review. Clin J Sport Med. (1998) 8:286–97. doi: 10.1097/00042752-199810000-00006
330. Ames A. CNS energy metabolism as related to function. Brain Res Rev. (2000) 34:42–68. doi: 10.1016/S0165-0173(00)00038-2
331. Andres RH, Ducray AD, Schlattner U, Wallimann T, Widmer HR. Functions and effects of creatine in the central nervous system. Brain Res Bull. (2008) 76:329–43. doi: 10.1016/j.brainresbull.2008.02.035
332. Sahlin K, Harris RC. The creatine kinase reaction: a simple reaction with functional complexity. Amino Acids. (2011) 40:1363–7. doi: 10.1007/s00726-011-0856-8
333. Ferrante RJ, Andreassen OA, Jenkins BG, Dedeoglu A, Kuemmerle S, Kubilus JK, et al. Neuroprotective effects of creatine in a transgenic mouse model of Huntington's disease. J Neurosci. (2000) 20:4389–97. doi: 10.1523/JNEUROSCI.20-12-04389.2000
334. Hemmer W, Wallimann T. Functional aspects of creatine kinase in brain. Dev Neurosci. (1993) 15:249–60. doi: 10.1159/000111342
335. O'Gorman E, Beutner G, Wallimann T, Brdiczka D. Differential effects of creatine depletion on the regulation of enzyme activities and on creatine-stimulated mitochondrial respiration in skeletal muscle, heart, and brain. Biochim Biophys Acta. (1996) 1276:161–70. doi: 10.1016/0005-2728(96)00074-6
336. Cunha MP, Lieberknecht V, Ramos-Hryb AB, Olescowicz G, Ludka FK, Tasca CI, et al. Creatine affords protection against glutamate-induced nitrosative and oxidative stress. Neurochem Int. (2016) 95:4–14. doi: 10.1016/j.neuint.2016.01.002
337. Kato T, Takahashi S, Shioiri T, Murashita J, Hamakawa H, Inubushi T. Reduction of brain phosphocreatine in bipolar II disorder detected by phosphorus-31 magnetic resonance spectroscopy. J Affect Disord. (1994) 31:125–33. doi: 10.1016/0165-0327(94)90116-3
338. Amital D, Vishne T, Rubinow A, Levine J. Observed effects of creatine monohydrate in a patient with depression and fibromyalgia. Am J Psychiatry. (2006) 163:1840–1. doi: 10.1176/ajp.2006.163.10.1840b
339. Scarnà A, Gijsman HJ, McTavish SFB, Harmer CJ, Cowen PJ, Goodwin GM. Effects of a branched-chain amino acid drink in mania. Br J Psychiatry. (2003) 182:210–3. doi: 10.1192/bjp.182.3.210
340. Roiser JP, McLean A, Ogilvie AD, Blackwell AD, Bamber DJ, Goodyer I, et al. The subjective and cognitive effects of acute phenylalanine and tyrosine depletion in patients recovered from depression. Neuropsychopharmacology. (2005) 30:775–85. doi: 10.1038/sj.npp.1300659
341. Ogawa S, Fujii T, Koga N, Hori H, Teraishi T, Hattori K, et al. Plasma l-tryptophan concentration in major depressive disorder: new data and meta-analysis. J Clin Psychiatry. (2014) 75:e906–15. doi: 10.4088/JCP.13r08908
342. Sarris J, Schoendorfer N, Kavanagh DJ. Major depressive disorder and nutritional medicine: a review of monotherapies and adjuvant treatments. Nutr Rev. (2009) 67:125–31. doi: 10.1111/j.1753-4887.2009.00180.x
343. Caruso G, Caraci F, Jolivet RB. Pivotal role of carnosine in the modulation of brain cells activity: multimodal mechanism of action and therapeutic potential in neurodegenerative disorders. Prog Neurobiol. (2019) 175:35–53. doi: 10.1016/j.pneurobio.2018.12.004
344. Fresta CG, Hogard ML, Caruso G, Melo Costa EE, Lazzarino G, Lunte SM. Monitoring carnosine uptake by RAW 264.7 macrophage cells using microchip electrophoresis with fluorescence detection. Anal Methods. (2017) 9:402–8. doi: 10.1039/C6AY03009B
345. Caruso G, Fresta CG, Grasso M, Santangelo R, Lazzarino G, Lunte SM, et al. Inflammation as the common biological link between depression and cardiovascular diseases: can carnosine exert a protective role? Curr Med Chem. (2019) 26:1783–1900. doi: 10.2174/0929867326666190712091515
346. Marriage BJ, Clandinin MT, Macdonald IM, Glerum DM. Cofactor treatment improves ATP synthetic capacity in patients with oxidative phosphorylation disorders. Mol Genet Metab. (2004) 81:263–72. doi: 10.1016/j.ymgme.2003.12.008
347. Noack H, Kube U, Augustin W. Relations between tocopherol depletion and coenzyme Q during lipid peroxidation in rat liver mitochondria. Free Radic Res. (1994) 20:375–86. doi: 10.3109/10715769409145637
348. Somayajulu M, McCarthy S, Hung M, Sikorska M, Borowy-Borowski H, Pandey S. Role of mitochondria in neuronal cell death induced by oxidative stress; neuroprotection by Coenzyme Q10. Neurobiol Dis. (2005) 18:618–27. doi: 10.1016/j.nbd.2004.10.021
349. Korkmaz A, Reiter RJ, Topal T, Manchester LC, Oter S, Tan D-X. Melatonin: an established antioxidant worthy of use in clinical trials. Mol Med. (2009) 15:43–50. doi: 10.2119/molmed.2008.00117
350. Leppämäki S, Partonen T, Vakkuri O, Lönnqvist J, Partinen M, Laudon M. Effect of controlled-release melatonin on sleep quality, mood, and quality of life in subjects with seasonal or weather-associated changes in mood and behaviour. Eur Neuropsychopharmacol. (2003) 13:137–45. doi: 10.1016/S0924-977X(02)00175-X
351. Carman JS, Post RM, Buswell R, Goodwin FK. Negative effects of melatonin on depression. Am J Psychiatry. (1976) 133:1181–6. doi: 10.1176/ajp.133.10.1181
352. Leibenluft E, Feldman-Naim S, Turner EH, Wehr TA, Rosenthal NE. Effects of exogenous melatonin administration and withdrawal in five patients with rapid-cycling bipolar disorder. J Clin Psychiatry. (1997) 58:383–8. doi: 10.4088/JCP.v58n0902
353. Bersani G, Garavini A. Melatonin add-on in manic patients with treatment resistant insomnia. Prog Neuro-Psychopharmacology Biol Psychiatry. (2000) 24:185–91. doi: 10.1016/S0278-5846(99)00097-4
354. Calabrese JR, Guelfi JD, Perdrizet-Chevallier C, Abbar M, Gay C, Jouan A, et al. Agomelatine adjunctive therapy for acute bipolar depression: preliminary open data. Bipolar Disord. (2007) 9:628–35. doi: 10.1111/j.1399-5618.2007.00507.x
355. Schiavone S, Trabace L. Pharmacological targeting of redox regulation systems as new therapeutic approach for psychiatric disorders: a literature overview. Pharmacol Res. (2016) 107:195–204. doi: 10.1016/j.phrs.2016.03.019
356. Hollis F, van der Kooij MA, Zanoletti O, Lozano L, Cantó C, Sandi C. et al. Mitochondrial function in the brain links anxiety with social subordination. Proc Natl Acad Sci. (2015) 112:15486–91. doi: 10.1073/pnas.1512653112
357. Giannini AJ, Nakoneczie AM, Melemis SM, Ventresco J, Condon M. Magnesium oxide augmentation of verapamil maintenance therapy in mania. Psychiatry Res. (2000) 93:83–7. doi: 10.1016/S0165-1781(99)00116-X
358. Eby GA, Eby KL. Rapid recovery from major depression using magnesium treatment. Med Hypotheses. (2006) 67:362–70. doi: 10.1016/j.mehy.2006.01.047
359. Coppen A, Chaudhry S, Swade C. Folic acid enhances lithium prophylaxis. J Affect Disord. (1986) 10:9–13. doi: 10.1016/0165-0327(86)90043-1
360. Jacka FN, O'Neil A, Opie R, Itsiopoulos C, Cotton S, Mohebbi M, et al. A randomised controlled trial of dietary improvement for adults with major depression (the “SMILES” trial). BMC Med. (2017) 15:1–13. doi: 10.1186/s12916-017-0791-y
361. Billingsley HE, Carbone S. The antioxidant potential of the Mediterranean diet in patients at high cardiovascular risk: an in-depth review of the PREDIMED. Nutr Diabetes. (2018) 8:13. doi: 10.1038/s41387-018-0025-1
362. Crescenzo R, Bianco F, Coppola P, Mazzoli A, Liverini G, Iossa S. Caloric restriction followed by high fat feeding predisposes to oxidative stress in skeletal muscle mitochondria. Horm Metab Res. (2013) 45:874–9. doi: 10.1055/s-0033-1351280
363. Lopresti AL, Jacka FN. Diet and bipolar disorder: a review of its relationship and potential therapeutic mechanisms of action. J Altern Complement Med. (2015) 21:733–9. doi: 10.1089/acm.2015.0125
364. Brietzke E, Mansur RB, Subramaniapillai M, Balanzá-Martínez V, Vinberg M, González-Pinto A, et al. Ketogenic diet as a metabolic therapy for mood disorders: evidence and developments. Neurosci Biobehav Rev. (2018) 94:11–6. doi: 10.1016/j.neubiorev.2018.07.020
365. Schwartzkroin PA. Mechanisms underlying the anti-epileptic efficacy of the ketogenic diet. Epilepsy Res. (1999) 37:171–80. doi: 10.1016/S0920-1211(99)00069-8
366. Giordano C, Marchiò M, Timofeeva E, Biagini G. Neuroactive peptides as putative mediators of antiepileptic ketogenic diets. Front Neurol. (2014) 5:63. doi: 10.3389/fneur.2014.00063
367. Devivo DC, Leckie MP, Ferrendelli JS, McDougal DB. Chronic ketosis and cerebral metabolism. Ann Neurol. (1978) 3:331–7. doi: 10.1002/ana.410030410
368. Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience. (2007) 145:256–64. doi: 10.1016/j.neuroscience.2006.11.065
369. Campbell IH, Campbell H. Ketosis and bipolar disorder: controlled analytic study of online reports. BJP Sych Open. (2019) 5:e58. doi: 10.1192/bjo.2019.49
370. Hughes SD, Kanabus M, Anderson G, Hargreaves IP, Rutherford T, O'Donnell M, et al. The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J Neurochem. (2014) 129:426–33. doi: 10.1111/jnc.12646
371. Koppel SJ, Swerdlow RH. Neuroketotherapeutics: a modern review of a century-old therapy. Neurochem Int. (2018) 117:114–25. doi: 10.1016/j.neuint.2017.05.019
372. Genzer Y, Dadon M, Burg C, Chapnik N, Froy O. Effect of dietary fat and the circadian clock on the expression of brain-derived neurotrophic factor (BDNF). Mol Cell Endocrinol. (2016) 430:49–55. doi: 10.1016/j.mce.2016.04.015
373. Pinto A, Bonucci A, Maggi E, Corsi M, Businaro R. Anti-oxidant and anti-inflammatory activity of ketogenic diet: new perspectives for neuroprotection in Alzheimer's disease. Antioxidants. (2018) 7:63. doi: 10.3390/antiox7050063
374. Milder JB, Liang LP, Patel M. Acute oxidative stress and systemic Nrf2 activation by the ketogenic diet. Neurobiol Dis. (2010) 40:238–44. doi: 10.1016/j.nbd.2010.05.030
375. Mikkelsen K, Stojanovska L, Polenakovic M, Bosevski M, Apostolopoulos V. Exercise and mental health. Maturitas. (2017) 106:48–56. doi: 10.1016/j.maturitas.2017.09.003
376. Tutakhail A, Nazari QA, Khabil S, Gardier A, Coudore F. Muscular and mitochondrial effects of long-term fluoxetine treatment in mice, combined with physical endurance exercise on treadmill. Life Sci. (2019) 232:116508. doi: 10.1016/j.lfs.2019.05.064
377. Ashton MM, Mohebbi M, Turner A, Marx W, Berk M, Malhi GS, et al. Physical activity as a predictor of clinical trial outcomes in bipolar depression: a subanalysis of a mitochondrial-enhancing nutraceutical randomized controlled trial. Can J Psychiatry. (2020) 65:306–18. doi: 10.1177/0706743719889547
Keywords: mitochondrial dysfunction, mood disorders, bipolar disorder, major depressive disorder, novel therapies
Citation: Giménez-Palomo A, Dodd S, Anmella G, Carvalho AF, Scaini G, Quevedo J, Pacchiarotti I, Vieta E and Berk M (2021) The Role of Mitochondria in Mood Disorders: From Physiology to Pathophysiology and to Treatment. Front. Psychiatry 12:546801. doi: 10.3389/fpsyt.2021.546801
Received: 29 March 2020; Accepted: 24 May 2021;
Published: 06 July 2021.
Edited by:
Xiancang Ma, First Affiliated Hospital of Xi'an Jiaotong University, ChinaReviewed by:
Aislinn Joanmarie Williams, The University of Iowa, United StatesMara Zilocchi, University of Regina, Canada
Copyright © 2021 Giménez-Palomo, Dodd, Anmella, Carvalho, Scaini, Quevedo, Pacchiarotti, Vieta and Berk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Berk, bWljaGFlbC5iZXJrQGJhcndvbmhlYWx0aC5vcmcuYXU=