 George R. Uhl1,2*
George R. Uhl1,2*- 1Departments of Neurology and Pharmacology, University of Maryland School of Medicine, Neurology Service, VA Maryland Healthcare System, Baltimore, MD, United States
- 2Department of Mental Health, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, United States
Substance use disorders provide challenges for development of effective medications. Use of abused substances is likely initiated, sustained and “quit” by complex brain and pharmacological mechanisms that have both genetic and environmental determinants. Medical utilities of prescribed stimulants and opioids provide complex challenges for prevention: how can we minimize their contribution to substance use disorders while retaining medical benefits for pain, restless leg syndrome, attention deficit hyperactivity disorder, narcolepsy and other indications. Data required to support assessments of reduced abuse liability and resulting regulatory scheduling differs from information required to support licensing of novel prophylactic or therapeutic anti-addiction medications, adding further complexity and challenges. I describe some of these challenges in the context of our current efforts to develop pentilludin as a novel anti-addiction therapeutic for a target that is strongly supported by human and mouse genetic and pharmacologic studies, the receptor type protein tyrosine phosphatase D (PTPRD).
Introduction
Urgent public health needs
Development of safe and effective medications to aid prevention and treatment of stimulant, opioid and stimulant + opioid use disorders are urgent public health needs. I can describe some of the statistics using published materials and some via links to websites for material that is not published.
Stimulants
Almost 20 metric tons of amphetamines and almost 9 metric tons of lisdexamphetamine are prescribed via >30 million annual prescriptions in the US (1). Many were prescribed chronically for indications including ADHD and narcolepsy.1 1.8% of the US population reported missuse of a prescribed stimulant.2 0.7% of the US population used an amphetamine from a licit or illicit source, almost 7.5 million Americans reported cocaine or methamphetamine use and almost 1.8 million reported a cocaine or amphetamine use disorder (see text footnote 2). Despite the lack of any FDA-approved medication for cocaine or methamphetamine use disorders, there are about 200,000 annual admissions to a stimulant use disorder treatment program (see text footnote 2).
Opioids
More than 142 million opioid prescriptions are written annually in the US,3 many prescribed chronically for chronic pain. More than 10 million Americans misuse opioids/year (745,000 using heroin and 9.7M prescription pain relievers) and almost 50,000/year die from this use.4
Stimulants + opioids
There is increasing co use of opioids along with stimulants (though not necessarily from “speedball” preparations that are intentionally co-injected). Past-year methamphetamine use among people using heroin rose from 23 to 37% between 2015 and 2018, while past year of methamphetamine use among people using prescription opioids increased from 5 to 8% over this period (2). Among people reporting past-month heroin use, past-month methamphetamine use increased from 9 to 30% between 2015 and 2017 (3). Fifty Percent of a 2015 sample of people who inject drugs reported injecting both heroin and methamphetamine (4).
Drug overdoses, both fatal and non-fatal, now include increasing numbers of individuals who use both opioids and stimulants (5, 6). Opioid users experience adverse features that are even more prevalent in opioid + stimulant co-users. Co-users have higher prevalence of injection drug use, serious mental illness, hepatitis B or C (7) emergency department visits, days hospitalized, utilization of social services, involvement with the criminal justice system (8) as well as overdose (4, 6).
Treatment
Such consequences are among the reasons that many individuals with opioid or opioid + stimulant use disorders attempt to quit. More than 750,000, 560,000, 490,000 and 118,000 Americans seek treatment for disorders of use of heroin, amphetamines, cocaine and “stimulants” annually, respectively.5 Agonist-like, antagonist and other therapeutics for opioid use disorders provide benefits but remain suboptimal for many for reasons that include variable adherence to these regimens (9, 10). More than 300,000 and 175,000 Americans receive methadone or buprenorphine annually, respectively;6 about 20,000 receive naltrexone.
Despite available treatments, relapses (especially early in treatment) are frequent. Relapse occurred within a week in 59% of a group of individuals seeking to quit opioid use (11) and within a month in 37% of a group of individuals seeking to quit methamphetamine use (12). Rates of relapse decline during subsequent periods.
Current FDA approved agonist and antagonist therapies for opioid use disorder remain suboptimal for many. There are no FDA-approved medications for preventing disorders of use of opioids or stimulants, none for treating stimulant abuse disorders and none approved for disorders of combined opioid + stimulant use.
Prevention and treatment
Prevention
Pharmacologic strategies to prevent development of substance use disorders in those who are prescribed opioids or stimulants could reasonably focus on goals that include reducing the abuse liability of these substances while maintaining their therapeutic benefits for indications that include reductions in pain, symptoms of restless leg syndrome (RLS), symptoms of attention deficit-hyperactivity disorder (ADHD) and the daytime sleepiness of narcolepsy.
Animal model data
Tests in experimental animals and humans can evaluate abuse liability with sufficient reliability that they are used to place new substances in the appropriate regulatory schedule (13, 14). There are good animal models for several types of pain that are often validated in humans (15). Although animal models for ADHD or RLS are perhaps not as well validated (16, 17), there is still reasonably good ability to identify compounds that reduce abuse liability of opioid analgesics and stimulants while preserving many likely therapeutic benefits. Such identification in animal models can lead to assessments of such selectivity in human laboratory studies that test aspects of human abuse liability (14). Taken together, human and animal data that indicate lower abuse liability might support less restrictive scheduling of combined stimulant + agent that reduces abuse liability and/or opioid + agent that reduces abuse liability. Below, I consider some of the hurdles that a candidate antiaddiction therapeutic, pentilludin, might need to clear to be used in these contexts. Our discussion overlaps with recent work considering different “endpoints” for antiaddiction therapeutics (18–23) but approaches this topic in a different way (e.g., from the perspective of a novel agent's development).
Treatment
Pharmacologic strategies for treatment include facilitating initial abstinence, often in the face of withdrawal symptoms. Sustaining abstinence often requires success in the face of pharmacologic and behavioral effects of “lapse” doses of an abused substance as well as behavioral effects of exposures to stimuli that have been strongly associated with prior drug experiences (24). Current animal models cover some of these features (25). Studies of behavioral components of reinstatement triggered by experimenter-administered drug doses, pain or stress provide significant data (26–28). Below, I consider the hurdles that pentilludin would need to clear for use in reducing the reinstatement-promoting effects of lapse doses of stimulants or opioids and contrast this with hurdles required for less-targeted use in other aspects required to achieve and sustain abstinence.
A number of recent publications note interest in using reductions in use and/or quantity frequency as an indication for treatments for stimulant use disorders (18–22). I thus also consider the hurdles that pentilludin would need to clear should similar consensus develop re endpoints of reduced use in individuals with disorders of use of stimulants, opioids or stimulants + opioids.
PTPRD and pentilludin
PTPRD's phosphatase as a target for novel antiaddiction therapeutics
PTPRD, the receptor type protein tyrosine phosphatase D, is now a strongly-supported target for antiaddiction medication based on human, mouse model and in vitro data. Human genetic results (29) associate common variation in PTPRD with vulnerability to develop a substance use disorder [e.g., polysubstance (30–32), opioid (33), alcohol (34)]. PTPRD variation is also associated with the abilities to quit (smoking (35, 36), use of opioids (37) and alcohol when aided by naltrexone [though not acamprosate (38)]. There are PTPRD associations with individual differences in a specific constellation of rewarding responses to amphetamine administration (39).
PTPRD is a highly-expressed, largely-neuronal, substantially synaptic, single transmembrane protein (Figure 1) that (likely) transduces signals from binding to extracellular ligands (40) to alter activity of its intracellular phosphatase (41). Synaptosomal proteomic, in situ hybridization, single cell RNAseq and electron and light microscopic immuohistochemical data support these conclusions re localization (42–44).7 Reported PTPRD extracellular binding partners include slit/trk, interleukin-1 receptor like and accessory proteins, synaptic adhesion-like molecules (SALMs) and the peptide asprosin (45–50). Substrates for PTPRD's phosphatase include proteins that regulate synaptic strength and maturation (51). Processes of PTPRD-expressing neurons grow when their PTPRD makes homomeric bonds with PTPRD expressed by adjacent cells (52). Cerebral cortical, ventral midbrain, striatal/ accumbens, reticular thalamic and other circuits that express PTPRD mRNA in likely glutamatergic, GABAergic, cholinergic and dopaminergic neurons are likely to develop and adapt differently when they express PTPRD at differing levels (53).
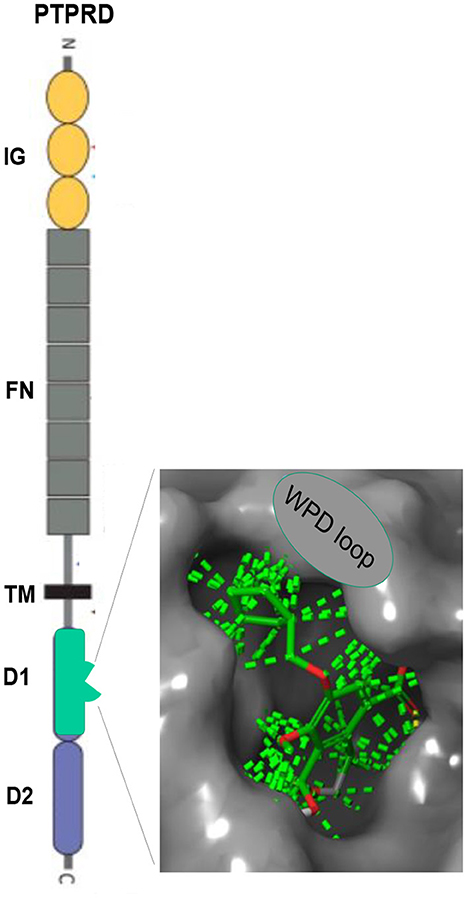
Figure 1. PTPRD. Extracellular immunoglobulin (IG) and fibronectin (FN) domains, transmembrane (TM) domain, D1 intracellular phosphatase domain. Enlargement: Pentilludin (NHB1109) bound to PTPRD phosphatase catalytic site and WPD loop. Dashed lines: van der Walls interactions.
Results from mouse models of common human allelic PTPRD variation
We have identified robust, 60- 70% individual differences in brain levels of expression of PTPRD mRNA (54) from human subjects with major vs. minor PTPRD SNP alleles. By contrast, PTPRD lacks common missense variants (53).
PTPRD knockout mice with only one wildtype gene copy and 50% constitutive alterations in levels of PTPRD expression (e.g., heterozygotes) thus model effects of common human PTPRD allelic variation. These mice are similar to wildtype littermates in tests of nociception (hotplate, tailflick), memory (Morris water maze), fear/anxiety (dark box emergence, thigmotaxis) and motor abilities (screen hang time, locomotion, rotarod) (54).
Mice with reduced PTPRD expression display sizable reductions in stimulant reward as assessed by conditioned place preference (CPP, 10 mg/kg cocaine) or self-administration (55).
Results from synthesis and testing of 7-BIA and 70 novel analogs identification of pentilludin (NHB1109)
Despite concerns that phosphatases were “undruggable” (56), we reported a lead compound PTPRD phosphatase inhibitor, 7-BIA, in 2018 (55). We followed this discovery with further structure-activity work testing more than 70 analogs. We identified 10 congeners with greater potency than 7-BIA and several that are more selective (57).
NHB1109, which I now name pentilludin, is a 7-position substituted cyclopentyl analog that, like 7-BIA, appears to provide pseudoirreversible inhibition of PTPRD's phosphatase (Figure 2) (57). It displays more potency and more selectivity vs. 7-BIA with respect to both close family members (PTPRS and PTPRF) and other phosphatases at which 7-BIA displays some potency, including PTPRJ and PTPN1/PTP1B. IC50 values are ≥10−4 M at 12 other receptor- and non-receptor type protein tyrosine phosphatases tested and ≥10−5 M in EUROFINS screen for targets of current drugs.
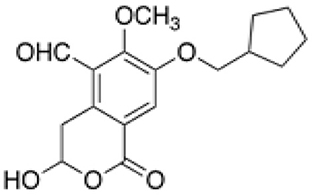
Figure 2. Pentilludin (NHB1109).
Initial hurdles for pentilludin (NHB1109)
Pentilludin has cleared many initial hurdles in addition to the in vitro specificity noted above. Dose limiting toxicity comes from doses >10 x those that reduce cocaine reward. Mice treated daily with 200- 2,000 mg/kg gavage doses reduce food/water intake over serval days and lose weight (57). These results fit with recent observations that PTPRD's phosphatase serves as a receptor for the orexigenic actions of a “positive feedback” signal from fat cells, asprosin (50).
Initial effects of pentilludin (NHB1109) pretreatments on stimulant and opioid reward
Pentilludin has replicated and extended the reward-reducing effects of acute PTPRD phosphatase inhibition displayed by our initial lead compound PTPRD phosphatase inhibitor 7-BIA (55).
The lack of deal-breaking toxicities and the presence of evidence for reduced stimulant reward suggest that pentilludin has cleared significant hurdles and encouraged us to move forward with its development as an antiaddiction compound.
Evidence bearing on hurdles for development as an antiaddiction therapeutic
Selecting the appropriate next hurdles for pentilludin
Development of novel small molecules for use in clinical addiction-related contexts requires substantial good laboratory practice (GLP) studies to seek evidence for toxicities investigational new drug application (IND), first doses in human research volunteers, repeated doses in humans, and dosing in humans along with addictive substance doses.
For purposes of this article, I focus on the ways in pentilludin or any other reward-reducing pharmacotherapeutic might be developed and deployed in light of the regulatory and other hurdles that these pathways place before its development. Another frame for this discussion: what useful endpoints might be most appropriately targeted by the actions that I believe PTPRD phosphatase inhibitors will provide? Prior to this discussion, I return to details of genetic and other evidence that might provide clues to which subsequent hurdles pentilludin might face with the most confidence, a priori.
Some details from human evidence
Reevaluating details of selected human genetic studies supports testable hypotheses concerning the likely clinical influences of inhibitors that reduce the effects mediated by PTPRD. I evaluate these ideas in the context of the associations of common PTPRD haplotypes with substantial individual differences in levels of brain PTPRD expression (54).
Stimulant reward
Hart et al. provided a clinical composite “factor 1” derived from responses of genotyped research volunteers as they experienced the effects of 10 mg oral amphetamine doses in a laboratory setting (39). Factor 1 came from sparse factor analyses of responses to items on the Profile of Mood States, Drug Effects Questionnaire and Addiction Research Center Inventory questionnaires. These authors generously confirmed to us that their 9p genomic association with factor 1 identified PTPRD. Pharmacologic reductions in PTPRD activity are thus likely to reduce “factor 1” effects of amphetamine in a) increasing friendliness, elation, vigor, feel high, want more, like, amphetamine-like, benzedrine-like, marijuana-like and morphine/benzedrine group-like responses and b) decreasing ratings of depression, fatigue, confusion and pentobarbitol-chlorpromazine-alcohol group sedation.
Ability to quit use of addictive substances from different classes
The settings in which ability to reduce or quit substance use has been associated with variation in PTPRD may also provide clues. We identified polygenic association of PTPRD variation with biochemically-confirmed success in smoking cessation in participants in three clinical trials aided by nicotine replacement or bupropion (36), in a trial aided by denicotinized cigarettes (58), in a trial aided by precessation nicotine replacement (59) and in a nicotine replacement community trial (60). We also identified PTPRD associations in comparisons of former vs. current smokers (58). Cox et al. identified PTPRD associations in comparisons of individuals who displayed lifetime opioid dependence diagnoses and who (a) self-reported abstinence >1 year vs. (b) continued to use (e.g., any abstinence <6 mos) (37). Biernacka et al. evaluated data from studies of pharmacologic effects on alcohol abstinence and identified robust PTPRD associations with time to relapse and time to relapse to heavy drinking in analyses of data from naltrexone treated subjects, though not in analyses of subjects treated with acamprosate (38). Interestingly, naltrexone responsiveness has been associated with drinking associated with reward seeking, while acamprosate responsiveness has been associated with drinking to seek relief from negative affect (61). These alcohol results, combined with data from smoking cessation and ability to quit opioid use, are consistent with the idea that antiaddiction agents that reduce PTPRD activity should aid reduction in use and/or abstinence from addictive substances of several classes.
Vulnerability to develop a substance use disorder of several classes
Vulnerability to develop a substance use disorder has been repeatedly associated with genomic variants at the PTPRD locus, beginning with our initial identification of a SNP in this chromosomal region using 10,000 SNP microarrays (30). We found PTPRD associations in comparisons of amphetamine dependent subjects to matched controls (62), research volunteers dependent on at least one illicit substance vs. corecruited or convenience controls (32, 63) and in more population- representative samples (64, 65). There are PTPRD copy number variant associations with vulnerability to develop opioid dependence (33). Agents that reduce PTPRD activity might thus be able to reduce development of dependence on addictive substances with which they were coadministered.
Evidence against toxicities
There is also human evidence that speaks to the likelihood that pharmacologically-modified effects mediated by PTPRD would be oncogenic or provide irreversible toxicities. Several papers term PTPRD a “tumor suppressor” gene based on e.g., its abilities to alter cancer-related phosphorylation pathways (66). However, PTPRD variation has not been reproducibly associated with any common cancer in genetic association studies [bladder (67), breast (68), colon (69), endometrial (70), kidney (71), leukemia (72), liver (73), lung (74), melanoma (75), non-Hodgkin lymphomas [e.g., (76)], pancreatic (77), prostate (78) or thyroid (79)]. Mice with reduced PTPRD expression fail to develop tumors at ages up to 24 mos. Carcinogenicity risks of agents that alter PTPRD seem unlikely to be greater than those of agents that influence any novel target.
There is also evidence from accidental human ingestions of Jack o'lantern mushrooms (Omphalotus illudens) (80, 81). These mushrooms contain the illudalic acid compounds that have activities at PTPRD-related phosphatases and provide the core of pentilludin's structure (82). They also contain compounds with muscarinic cholinergic activities (83). There has been no lethality or persisting sequelae noted after > 60 reported cases of accidental ingestion of these mushrooms. The ingestions do produce nausea/emesis. These symptoms have been attributed to the mushrooms' muscarinic effects by authors including a physician who ingested them (81). It is also possible that pentilludin, especially at high doses, will act at PTPRD to produce anorexia/nausea in humans. PTPRD is expressed in the arcuate hypothalamic neurons that express the orexigenic agouti related peptide AgRP (50) as well as in human enteric neurons (84) and in brainstem sites of cholinergic inputs to the cervical ganglia that innervate the stomach and small intestine. We have identified reduced food intake in mice treated with single doses of pentilludin that are >10 times higher than those that reduce cocaine and opioid reward (57). Data from accidental human Ingestions of Omphalotus illudens do provide evidence for the lack of other idiosyncratic human responses to illudalic acid doses that is likely to be pertinent for the illudalic acid analog pentilludin.
Human evidence limitations
The cumulative likelihood that all of the nominally-significant genetic associations cited above is due to chance is exceedingly small. When combined with the compelling mouse model and pharmacologic data, the a posteriori probabilities that PTPRD associations are due to chance are even lower. Nevertheless, none of these individual associations reproducibly meet the ultraconservative p < 10−8 Bonferroni corrected p-value required to declare “genome wide significance”. The PTPRD SNPs or copy number variants that provide these associations are not the same across all studies. All studies of addiction genetics do not identify PTPRD. The size of these human associations also provides a caution: the effects of common PTPRD genomic variation are likely to be modest when compared to the cumulative effects of other genetic and environmental variation on individual differences in vulnerability to develop a substance use disorder, reward from administration of addictive substances or ability to reduce use/abstain from use.
All in all, with the human genetic, mouse genetic and human mushroom experience, cited above we thus have substantial confidence that “on target” pentilludin actions at PTPRD are likely to be well tolerated at the proposed therapeutic doses, with an acceptable therapeutic index.
Synthesis of human and animal evidence
The large effects on stimulant reward observed in our animal model studies (55), when we control other genetic and environmental features, suggest that both genetic and pharmacologic modulation of PTPRD activities can display robust effects. We thus seek a developmental pathway for the PTPRD phosphatase inhibitor pentilludin on which we can demonstrate clinical benefits in a setting in which these benefits will be less likely to be obscured by the effects of variation in other genetic and environmental influences.
Pragmatically, we wish to select the hurdles that pentilludin will be most likely to clear during further development with the support that is available to us. Another way to frame this: we need to select the most appropriate endpoint for pentilludin's initial use. We provide several possible examples below.
Hurdles for development of antiaddiction therapeutic in several contexts
Potentially-modest hurdle: Reducing reward from “lapse” doses of stimulants during the initial period of abstinence
Reward from “lapse” doses of stimulants is likely to contribute to the reasons why relapse is so frequent when individuals with stimulant use disorders attempt to quit. A relapse occurred within a month in 37% of a group of individuals seeking to quit methamphetamine use (12). Rates of relapse decline during subsequent periods. Interventions that reduce the reward from “lapse” doses of stimulants taken during the key 1st weeks of a quit attempt could thus reduce relapse rates and aid longer term abstinence. Quantitative tests can biochemically confirm abstinence, exposure to modest “lapse” doses or relapse in appropriately-collected urine samples (85, 86). In this context, endpoints could be both (1) number of positive urine tests and (2) number of strikingly positive tests indicative of relapse in individuals who have previously displayed a modest positive indicative of lapse dosing.
“Lapse” is perhaps best defined in the smoking field, where smoking a single experimenter-administered cigarette can more than double the risk of continued smoking within the next 24 h (87). Such human evidence is complemented by animal studies that document how robustly experimenter-administered “priming” doses increase subsequent relapse-like efforts to self-administer drugs during periods of “abstinence” (28). Reducing the reward from “lapse” stimulant doses taken during the 1st week of attempted stimulant abstinence should thus provide a significant benefit for individuals seeking to quit use of stimulants.
IND-enabling studies for development of pentilludin to reduce reward from “lapse” doses of stimulants during the initial period of abstinence could be relatively tractable. Several weeks' pentilludin dosing in two species could be coupled with simulated “lapse” doses of stimulants. Since there are likely fewer than 200,000 individuals treated with any pharmacological adjunct to aid abstinence from a stimulant use disorder8 in the US, use of pentilludin to aid abstinence by reducing the reward from lapse doses of stimulants sampled during the 1st week of abstinence might even provide an “orphan” indication. Achieving orphan designation could encourage commercial partnerships to continue development by providing tax benefits and a period of exclusivity.9
The benefits of tractable IND-enabling work, possible orphan indication, ability to biochemically confirm both lapse dosing and abstinence and a plausible link to the reward conferred by stimulants appears to raise only moderately-high hurdles to development of pentilludin for this indication. However, this indication would target a market of only modest size. We do not have a clear indication of the fraction of the total variance in relapse to stimulant use that comes from PTPRD-sensitive stimulant reward during lapse doses vs. the fraction that comes from other genetic and environmental variables. The hurdle provided by this indication for pentilludin may thus be higher than we anticipate, a priori.
Potentially-higher hurdle: Reducing abuse liability from prescribed stimulants and/or opioids
Prevention of the substance use disorders that arise from use of prescribed drugs would provide a large clinical impact. This impact could come from strategies that reduced the reward that these prescribed drugs provide while maintaining their therapeutic benefits. A practical manifestation of this impact could be lower scheduling of combination products (e.g., pentilludin + stimulant or pentilludin + opioid).
Human laboratory and experimental animal assessments of reward have been key to Drug Enforcement Administration (DEA) assignment of appropriate regulatory schedules to new drugs (Box 110), with consultation by FDA (13). Postmarketing data identifying frequencies of missuse and abuse in the community add valuable information (13). Possible endpoints in this developmental pathway would be demonstration of lower signs of abuse liability in standard testing paradigms and thus reduced scheduling for combination products.
Box 1. Schedules for controlled substances.
Schedule I: Drugs with no currently accepted medical use and a high potential for abuse. (ex: heroin, LSD, marijuana (cannabis), 3,4-methylenedioxy-methamphetamine (ecstasy), methaqualone, peyote.
Schedule II: Drugs with a high potential for abuse, potentially leading to severe psychological or physical dependence, that are considered dangerous. (ex: combination products with <15 mg hydrocodone per dosage, cocaine, methamphetamine, amphetamine, methadone, hydromorphone, meperidine, oxycodone, fentanyl, methylphenidate.
Schedule III: Drugs with a moderate to low potential for physical and psychological dependence, less than Schedule I/ II drugs but more than Schedule IV. (ex: products containing < 90 mg codeine per dosage, ketamine, anabolic steroids, testosterone.
Schedule IV: Drugs with a low potential for abuse and low risk of dependence. (ex: diazepam, tramadol, alprazolam, carisoprodol, propoxyphene, lorazepam, pentazocine, zolpidem).
Schedule V: Drugs with lower potential for abuse than Schedule IV, many containing limited quantities of certain narcotics and often used for antidiarrheal, antitussive, and analgesic purposes. (ex: cough preparations with <200 mg codeine/100 ml, diphenoxylate/atropine, difenoxin/atropine, pregabalin, attapulgite).
One set of hurdles for developing pentilludin for use in this context relates to the magnitude of its effects: could co-administration of pentilludin with a Schedule II stimulant or opioid reduce abuse liability sufficiently to allow the combination product to be marketed as Schedule III? It is fortunate that we do have genetic association of PTPRD variation with individual differences in rewarding responses to laboratory-administered amphetamine doses (noted above). Assessments using many of the same instruments has provided data that has been accepted by regulatory agencies to schedule new drugs in the past.11
Another set of hurdles results from the chronicity of treatment: can IND-enabling and other studies adequately reflect the years-long patterns of use of prescribed stimulants and, for some, opioids?
An additional hurdle come from the differences in pharmacodynamic properties of pentilludin (a pseudoirreversible agent with an apparent days-long physiological half-life) vs. those of e.g., amphetamine or oxycodone, with shorter physiological half-lives.
Producing a robust abuse-resistant combination formulation that deters extraction of the opioid or stimulant provides another hurdle.
And, finally, assuring that combination formulations retain human benefits in reducing pain, combatting ADHD or RLS symptoms or reducing daytime sleepiness of narcoleptics provides a significant hurdle as well.
Despite these hurdles, the potential for preventing development of substance use disorders by marketing pentilludin-containing stimulant and opioid combination products with reduced abuse liability and correspondingly-less restrictive scheduling remains a powerfully attractive idea. Such marketing would then have to clear a final hurdle: Real world post marketing surveillance data that demonstrated less abuse (13).
Potentially-higher hurdle: Aiding initiation and maintenance of abstinence in individuals with ongoing stimulant or opioid use disorders
Established disorders of stimulant and opioid use are likely to be maintained by complex polygenic genetic and environmental factors.12 A pharmacologic “magic bullet” that could arrest such ongoing disorders without unacceptable side effects in all abusers would provide huge societal benefits and has therefore been sought. However, the complex interplay of habitual and learned behaviors with the pulls exerted by both pharmacological reward and reduction in withdrawal's aversive features provides a daunting hurdle for any antiaddiction pharmaceutic to clear.
One conceptual approach to this “magic bullet” thinking considers the different ways in which individuals with substance use disorders might sustain their use and thus come to treatment via different pathways. Studies of alcohol use disorders have sought to separate drinkers who largely drink for the reward that alcohol provides and those who largely drink to mitigate negative features of their lives and affects (88). As noted above, these studies have identified more prominent benefits of naltrexone for those who drink to experience reward and more benefits of acamprosate for those who drink to reduce negative affect and feelings. PTPRD variants provide strong association with ability to reduce alcohol use with naltrexone, without any evidence for a strong or even moderate-strength association with ability to reduce alcohol use with acamprosate (38). PTPRD effects could thus plausibly be more prominent in aiding reductions in or abstinence from stimulants or opioids in the subset of individuals whose use was the most strongly maintained by the reward that they obtain from these substances. Possible endpoints for this approach could include reduced or absent urinary levels of abused substances in unselected treatment-seeking substance users. Another endpoint could focus on the individuals whose substance use disorders had been most maintained by the rewarding effects of the substance.
Hurdles to development of pentilludin to aid initiation of and/or maintenance of reductions in or abstinence from use of addictive substances are thus daunting, but perhaps not impossible. In an ideal scenario, drug addiction investigators would identify the stimulant or opioid users whose use was most dependent on the reward that these drugs provide vs. habitual use or use to relieve negative features. In an even more ideal scenario, drug addiction investigators would also provide solid data (re benefits) that would lower barriers to licensing novel agents based on reductions in use of addictive drugs rather that complete abstinence. In such a setting, one of the highest hurdles to testing pentilludin to aid initiation of reduced use/abstinence in the most reward-dependent users might be studies identifying little pentilludin toxicity with co-use of the multiple addictive substance that are characteristic of many with substance use disorders. Aid in maintaining abstinence, once achieved, could be analogous to reducing reward from “lapse” doses, as noted above. Hurdles to aiding initiation and maintenance of abstinence in individuals with the most reward-dependent ongoing stimulant or opioid use disorders thus might not be as high as they appeared a priori.
Conclusions
Congressional recognition of the need for pharmacotherapeutics for prevention and treatment of substance use disorders and the difficulties of such development led to establishment of the medications development program at the National Institute on Drug Abuse in 1990, and to its increasing funding and sophistication during subsequent years.13 Bases for regulation of substances with abuse liability dates to the early 1900's and currently centers on the Controlled Substance Act and related legislation.14 Despite this regulatory sophistication and support for medication development, there is still no licensed pharmacotherapeutic that can prevent development of substance use disorders in those who are prescribed stimulants or opioids and none that can effectively treat established stimulant use disorders.
In these contexts, development of pentilludin, a novel pharmacotherapeutic that acts at a novel addiction-associated site to reduce reward from stimulants and opioids, provides an example of the promises and hurdles that face antiaddiction medication development. Categorizing pentilludin's strengths and limitations in several potential settings and with several different sets of endpoints outlines some of the ways in which choosing the correct set of hurdles and endpoints could increase the likelihood that this compound will be able to reach general clinical use.
Author contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was funded by NIH grants U01DA047713, UG3DA056039 and NIA supplements to these grants.
Conflict of interest
VA intellectual property covering pentilludin with GU as a coinventor.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
1. ^https://clincalc.com/drugstats/drugs/amphetamine
2. ^https://www.samhas.gov/data/data-we-collect/teds-treatment-episode-data-set
3. ^https://www.cdc.gov/drugoverdose/rxrate-maps/index.html
4. ^https://www.hhs.gov/opioids/statistics/index.html; https://nida.nih.gov/research-topics/opioids
5. ^https://samhsa.gov/data/release/2020-national-survey-drug-use-health-nsduh-release
6. ^https://samhsa.gov/data/data-we-collect/n-ssats-national-survey-substance-abuse-treatment-services
7. ^https://mouse.brain-map.org
8. ^https://www.samhas.gov/data/data-we-collect/teds-treatment-episode-data-set
9. ^https://www.fda.gov/industry/developing-products-rare-diseases-conditions/designating-orphan-product-drugs-and-biological-products
10. ^https://www.dea.gov/drug-information/drug-scheduling
11. ^https://www.fda.gov/media/116739/download
12. ^https://nida.nih.gov/publications/drugs-brains-behavior-science-addiction/drug-misuse-addiction
13. ^https://nida.nih.gov/about-nida/organization/divisions/division-therapetics-medical-consesquences-dtmc/research-programs
14. ^https://uscode.house.gov/view.xhtml?path=/prelim@title21/chapter13&edition=prelim
References
1. Piper BJ, Ogden CL, Simoyan OM, Chung DY, Caggiano JF, Nichols SD, et al. Trends in use of prescription stimulants in the United States and Territories, 2006 to 2016. PLoS ONE. (2018) 13:e0206100. doi: 10.1371/journal.pone.0206100
2. Palamar JJ, Han BH, Keyes KM. Trends in characteristics of individuals who use methamphetamine in the United States, 2015–2018. Drug Alcohol Depend. (2020) 213:108089. doi: 10.1016/j.drugalcdep.2020.108089
3. Strickland JC, Havens JR, Stoops WW. A nationally representative analysis of “twin epidemics”: rising rates of methamphetamine use among persons who use opioids. Drug Alcohol Depend. (2019) 204:107592. doi: 10.1016/j.drugalcdep.2019.107592
4. Al-Tayyib A, Koester S, Langegger S, Raville L. Heroin and methamphetamine injection: an emerging drug use pattern. Subst Use Misuse. (2017) 52:1051–8. doi: 10.1080/10826084.2016.1271432
5. Ciccarone D. The rise of illicit fentanyls, stimulants and the fourth wave of the opioid overdose crisis. Curr Opin Psychiatry. (2021) 34:344–50. doi: 10.1097/YCO.0000000000000717
6. Korthuis PT, Cook RR, Foot CA, Leichtling G, Tsui JI, Stopka TJ, et al. Association of methamphetamine and opioid use with nonfatal overdose in rural communities. JAMA Netw Open. (2022) 5:e2226544. doi: 10.1001/jamanetworkopen.2022.26544
7. Shearer RD, Howell BA, Bart G, Winkelman TNA. Substance use patterns and health profiles among US adults who use opioids, methamphetamine, or both, 2015–2018. Drug Alcohol Depend. (2020) 214:108162. doi: 10.1016/j.drugalcdep.2020.108162
8. Howell BA, Bart G, Wang EA, Winkelman TNA. Service involvement across multiple sectors among people who use opioids, methamphetamine, or both, United States-2015–2018. Med Care. (2021) 59:238–44. doi: 10.1097/MLR.0000000000001460
9. T. P. s. C. o. C. D. A. a. t. O. Crisis. Report of the President's Comission on Combatting Drug Addiction and the Opioid Crisis [2017].
10. Hasan MM, Noor EAM, Shi J, Young LD, Young GJ. Long-term patient outcomes following buprenorphine/naloxone treatment for opioid use disorder: a retrospective analysis in a commercially insured population. Am J Drug Alcohol Abus. (2022) 48:481–491. doi: 10.1080/00952990.2022.2065638
11. Smyth BP, Barry J, Keenan E, Ducray K. Lapse and relapse following inpatient treatment of opiate dependence. Ir Med J. (2010) 103:176–9.
12. Brecht ML, Herbeck D. Time to relapse following treatment for methamphetamine use: a long-term perspective on patterns and predictors. Drug Alcohol Depend. (2014) 139:18–25. doi: 10.1016/j.drugalcdep.2014.02.702
13. Johanson C-E, Balster RL, Henningfield JE, Schuster CR, Anthony JC, Barthwell AG, et al. Risk management and post-marketing surveillance for the abuse of medications acting on the central nervous system: expert panel report. Drug Alcohol Depend. (2009) 105 Suppl 1:S65–71. doi: 10.1016/j.drugalcdep.2009.08.006
14. Carter LP, Griffiths RR. Principles of laboratory assessment of drug abuse liability and implications for clinical development. Drug Alcohol Depend. (2009) 105 Suppl 1:S14–25. doi: 10.1016/j.drugalcdep.2009.04.003
15. Abboud C, Duveau A, Bouali-Benazzouz R, Massé K, Mattar J, Brochoire L, et al. Animal models of pain: diversity and benefits. J Neurosci Methods. (2021) 348:108997. doi: 10.1016/j.jneumeth.2020.108997
16. Kantak KM. Rodent models of attention-deficit hyperactivity disorder: an updated framework for model validation and therapeutic drug discovery. Pharmacol Biochem Behav. (2022) 216:173378. doi: 10.1016/j.pbb.2022.173378
17. Salminen AV, Silvani A, Allen RP, Clemens S, Garcia-Borreguero D, Ghorayeb I, et al. Consensus guidelines on rodent models of restless legs syndrome. Mov Disord. (2021) 36:558–569. doi: 10.1002/mds.28401
18. Loflin MJE, Kiluk BD, Huestis MA, Aklin WM, Budney AJ, Carroll KM, et al. The state of clinical outcome assessments for cannabis use disorder clinical trials: a review and research agenda. Drug Alcohol Depend. (2020) 212:107993. doi: 10.1016/j.drugalcdep.2020.107993
19. Kiluk BD, Carroll KM, Duhig A, Falk DE, Kampman K, Lai S, et al. Measures of outcome for stimulant trials: ACTTION recommendations and research agenda. Drug Alcohol Depend. (2016) 158:1–7. doi: 10.1016/j.drugalcdep.2015.11.004
20. Roos CR, Nich C, Mun CJ, Babuscio TA, Mendonca J, Miguel AQC, et al. Clinical validation of reduction in cocaine frequency level as an endpoint in clinical trials for cocaine use disorder. Drug Alcohol Depend. (2019) 205:107648. doi: 10.1016/j.drugalcdep.2019.107648
21. Kiluk BD, Fitzmaurice GM, Strain EC, Weiss RD. What defines a clinically meaningful outcome in the treatment of substance use disorders: reductions in direct consequences of drug use or improvement in overall functioning? Addiction. (2019) 114:9–15. doi: 10.1111/add.14289
22. Kiluk BD, Babuscio TA, Nich C, Carroll KM. Initial validation of a proxy indicator of functioning as a potential tool for establishing a clinically meaningful cocaine use outcome. Drug Alcohol Depend. (2017) 179:400–7. doi: 10.1016/j.drugalcdep.2017.07.020
23. Sandberg B. In search of a stimulant use disorder treatment: “We've been doing it all wrong” Pink Sheet. Available online at: https://pink.pharmaintelligence.informa.com/PS145216/IN-search-of-a-stimulant-use-disorder-treatment-weve-been-doing-it-all-wrong (accessed November 11, 2021).
24. Garavan H, Brennan KL, Hester R, Whelan R. The neurobiology of successful abstinence. Curr Opin Neurobiol. (2013) 23:668–74. doi: 10.1016/j.conb.2013.01.029
25. Fredriksson I, Venniro M, Reiner DJ, Chow JJ, Bossert JM, Shaham Y. Animal models of drug relapse and craving after voluntary abstinence: a review. Pharmacol Rev. [2021] 73:1050–83. doi: 10.1124/pharmrev.120.000191
26. Vanderschuren L, Ahmed SH. Animal models of the behavioral symptoms of substance use disorders. Cold Spring Harb Perspect Med. (2021) 1:a040287. doi: 10.1101/cshperspect.a040287
27. Reiner DJ, Fredriksson I, Lofaro OM, Bossert JM, Shaham Y. Relapse to opioid seeking in rat models: behavior, pharmacology and circuits. Neuropsychopharmacology. (2019) 44:465–77. doi: 10.1038/s41386-018-0234-2
28. Farrell MR, Schoch H, Mahler SV. Modeling cocaine relapse in rodents: behavioral considerations and circuit mechanisms. Prog Neuropsychopharmacol Biol Psychiatry. (2018) 87:33–47. doi: 10.1016/j.pnpbp.2018.01.002
29. Uhl GR, Drgonova J. Cell adhesion molecules: druggable targets for modulating the connectome and brain disorders? Neuropsychopharmacology. (2014) 39:235. doi: 10.1038/npp.2013.240
30. Lima YO, Costa EA. Pooled association genome scanning: validation and use to identify addiction vulnerability loci in two samples. Proc Natl Acad Sci USA. (2005) 102:11864–9. doi: 10.1073/pnas.0500329102
31. Liu Q-R, Drgon T, Johnson C, Walther D, Hess J, Uhl GR. Addiction molecular genetics: 639, 401 SNP whole genome association identifies many “cell adhesion” genes. Am J Med Genet B Neuropsychiatr Genet. (2006) 14:918–25. doi: 10.1002/ajmg.b.30436
32. Drgon T, Johnson CA, Nino M, Drgonova J, Walther DM, Uhl GR. “Replicated” genome wide association for dependence on illegal substances: genomic regions identified by overlapping clusters of nominally positive SNPs. Am J Med Genet B Neuropsychiatr Genet. (2011) 156:125–38. doi: 10.1002/ajmg.b.31143
33. Li D, Zhao H, Kranzler HR, Li MD, Jensen KP, Zayats T, et al. Genome-wide association study of copy number variations (CNVs) with opioid dependence. Neuropsychopharmacology. (2015) 40:1016–26. doi: 10.1038/npp.2014.290
34. Jung ZH. J, Grant BF, Chou P. Identification of novel genetic variants of DMS5 alcohol use disorder: genome wide association study in National Epidemiological Survey on Alcohol Related Conditions-III Abstracts, World Congress of Psychaitroc Genetics. [2017].
35. Uhl GR, Liu Q-R, Drgon T, Johnson C, Walther D, Rose JE. Molecular genetics of nicotine dependence and abstinence: whole genome association using 520,000 SNPs. BMC Genet. (2007) 8:10. doi: 10.1186/1471-2156-8-10
36. Uhl GR, Liu Q-R, Drgon T, Johnson C, Walther D, Rose JE, et al. Molecular genetics of successful smoking cessation: convergent genome-wide association study results. Arch Gen Psychiatry. (2008) 65:683–93. doi: 10.1001/archpsyc.65.6.683
37. Cox JW, Sherva RM, Lunetta KL, Johnson EC, Martin NG, Degenhardt L, et al. Genome-wide association study of opioid cessation. J Clin Med. (2020) 9:180. doi: 10.3390/jcm9010180
38. Biernacka JM, Coombes BJ, Batzler A, Ho AM-C, Geske JR, Frank J, et al. Genetic contributions to alcohol use disorder treatment outcomes: a genome-wide pharmacogenomics study. Neuropsychopharmacology. (2021) 46:2132–9. doi: 10.1038/s41386-021-01097-0
39. Hart AB, Engelhardt BE, Wardle MC, Sokoloff G, Stephens M, de Wit H, et al. Genome-wide association study of d-amphetamine response in healthy volunteers identifies putative associations, including cadherin 13 (CDH13). PLoS ONE. (2012) 7:e42646. doi: 10.1371/journal.pone.0042646
40. Um JW, Ko J. LAR-RPTPs: synaptic adhesion molecules that shape synapse development. Trends Cell Biol. (2013) 23:465–75. doi: 10.1016/j.tcb.2013.07.004
41. Takahashi H, Craig AM. Protein tyrosine phosphatases PTPdelta, PTPsigma, and LAR: presynaptic hubs for synapse organization. Trends Neurosci. (2013) 36:522–34. doi: 10.1016/j.tins.2013.06.002
42. Kohansal-Nodehi M, Chua JJ, Urlaub H, Jahn R, Czernik D. Analysis of protein phosphorylation in nerve terminal reveals extensive changes in active zone proteins upon exocytosis. Elife. (2016) 5:e14530. doi: 10.7554/eLife.14530
43. Mizuno K, Hasegawa K, Ogimoto M, Katagiri T, Yakura H. Developmental regulation of gene expression for the MPTP delta isoforms in the central nervous system and the immune system. FEBS Lett. (1994) 355:223–8. doi: 10.1016/0014-5793(94)01188-5
44. Park H, Choi Y, Jung H, Kim S, Lee S, Han H, et al. Splice-dependent trans-synaptic PTPdelta-IL1RAPL1 interaction regulates synapse formation and non-REM sleep. EMBO J. (2020) 39:e104150. doi: 10.15252/embj.2019104150
45. Yamagata A, Sato Y, Goto-Ito S, Uemura T, Maeda A, Shiroshima T, et al. Structure of Slitrk2-PTPdelta complex reveals mechanisms for splicing-dependent trans-synaptic adhesion. Sci Rep. (2015) 5:9686. doi: 10.1038/srep09686
46. Yoshida T, Shiroshima T, Lee S-J, Yasumura M, Uemura T, Chen X, et al. Interleukin-1 receptor accessory protein organizes neuronal synaptogenesis as a cell adhesion molecule. J Neurosci. (2012) 32:2588–600. doi: 10.1523/JNEUROSCI.4637-11.2012
47. Kwon SK, Woo J, Kim SY, Kim H, Kim E. Trans-synaptic adhesions between netrin-G ligand-3 (NGL-3) and receptor tyrosine phosphatases LAR, protein-tyrosine phosphatase delta (PTPdelta), and PTPsigma via specific domains regulate excitatory synapse formation. J Biol Chem. (2010) 285:13966–78. doi: 10.1074/jbc.M109.061127
48. Lin Z, Liu J, Ding H, Xu F, Liu H. Structural basis of SALM5-induced PTPdelta dimerization for synaptic differentiation. Nat Commun. (2018) 9:268. doi: 10.1038/s41467-017-02414-2
49. Choi Y, Nam J, Whitcomb DJ, Song YS, Kim D, Jeon S, et al. SALM5 trans-synaptically interacts with LAR-RPTPs in a splicing-dependent manner to regulate synapse development. Sci Rep. (2016) 6:26676. doi: 10.1038/srep26676
50. Mishra I, Xie WR, Bournat JC, He Y, Wang C, Silva ES, et al. Protein tyrosine phosphatase receptor delta serves as the orexigenic asprosin receptor. Cell Metab. (2022) 34:549–563e548. doi: 10.1016/j.cmet.2022.02.012
51. Henderson IM, Marez C, Dokladny K, Smoake J, Martinez M, Johnson D, et al. Substrate-selective positive allosteric modulation of PTPRD's phosphatase by flavonols. Biochem Pharmacol. (2022) 202:115109. doi: 10.1016/j.bcp.2022.115109
52. Wang J, Bixby JL. Receptor tyrosine phosphatase-delta is a homophilic, neurite-promoting cell adhesion molecular for CNS neurons. Mol Cell Neurosci. (1999) 14:370–84. doi: 10.1006/mcne.1999.0789
53. Uhl GR, Martinez MJ. PTPRD neurobiology, genetics, and initial pharmacology of a pleiotropic contributor to brain phenotypes. Ann N Y Acad Sci. (2019) 1451:112–29. doi: 10.1111/nyas.14002
54. Drgonova J, Walther D, Wang KJ, Hartstein GL, Lochte B, Troncoso J, et al. Mouse model for PTPRD associations with WED/RLS and addiction: reduced expression alters locomotion, sleep behaviors and cocaine-conditioned place preference. Mol Med. (2015) 21:717–25. doi: 10.2119/molmed.2015.00017
55. Uhl GR, Martinez MJ, Paik P, Sulima A, Bi G-H, Iyer MR, et al. Cocaine reward is reduced by decreased expression of receptor-type protein tyrosine phosphatase D (PTPRD) and by a novel PTPRD antagonist. Proc Natl Acad Sci USA. (2018) 115:11597–602. doi: 10.1073/pnas.1720446115
56. Stanford SM, Bottini N. Targeting tyrosine phosphatases: time to end the stigma. Trends Pharmacol Sci. (2017) 38:524–40. doi: 10.1016/j.tips.2017.03.004
57. Henderson IM, Zeng F, Bhuiyan NH, Luo D, Martinez M, Smoake J, et al. Structure-activity studies of PTPRD phosphatase inhibitors identify a 7-cyclopentymethoxy illudalic acid analog candidate for development. Biochem Pharmacol. (2022) 195:114868. doi: 10.1016/j.bcp.2021.114868
58. Drgon T, Montoya I, Johnson C, Liu Q-R, Walther D, Hamer D, et al. Genome-wide association for nicotine dependence and smoking cessation success in NIH research volunteers. Mol Med. (2009) 15:21–7. doi: 10.2119/molmed.2008.00096
59. Uhl GR, Drgon T, Johnson C, Ramoni MF, Behm FM, Rose JE. Genome-wide association for smoking cessation success in a trial of precessation nicotine replacement. Mol Med. (2010) 16:513–26. doi: 10.2119/molmed.2010.00052
60. Uhl GR, Drgon T, Johnson C, Walther D, David SP, Aveyard P, et al. Genome-wide association for smoking cessation success: participants in the Patch in Practice trial of nicotine replacement. Pharmacogenomics. (2010) 11:357–67. doi: 10.2217/pgs.09.156
61. Roos CR, Bold KW, Witkiewitz K, Leeman RF, DeMartini KS, Fucito LM, et al. Reward drinking and naltrexone treatment response among young adult heavy drinkers. Addiction. (2021) 116:2360–71. doi: 10.1111/add.15453
62. Uhl GR, Drgon T, Liu Q-R, Johnson C, Walther D, Komiyama T, et al. Genome-wide association for methamphetamine dependence: convergent results from 2 samples. Arch Gen Psychiatry. (2008) 65:345–55. doi: 10.1001/archpsyc.65.3.345
63. Johnson C, Drgon T, Walther D, Uhl GR. Genomic regions identified by overlapping clusters of nominally-positive SNPs from genome-wide studies of alcohol and illegal substance dependence. PLoS One. (2011) 6:e19210. doi: 10.1371/journal.pone.0019210
64. Uhl GR, Walther D, Musci R, Fisher C, Anthony JC, Storr CL, et al. Smoking quit success genotype score predicts quit success and distinct patterns of developmental involvement with common addictive substances. Mol Psychiatry. (2014) 19:50–4. doi: 10.1038/mp.2012.155
65. Johnson C, Drgon T, Liu Q-R, Zhang P-W, Walther D, Li C-Y, et al. Genome wide association for substance dependence: convergent results from epidemiologic and research volunteer samples. BMC Med Genet. (2008) 9:113. doi: 10.1186/1471-2350-9-113
66. Lin Y, Zhou X, Yang K, Chen Y, Wang L, Luo W, et al. Protein tyrosine phosphatase receptor type D gene promotes radiosensitivity via STAT3 dephosphorylation in nasopharyngeal carcinoma. Oncogene. (2021) 40:3101–17. doi: 10.1038/s41388-021-01768-8
67. Figueroa JD, Ye Y, Siddiq A, Garcia-Closas M, Chatterjee N, Prokunina-Olsson L, et al. Genome-wide association study identifies multiple loci associated with bladder cancer risk. Hum Mol Genet. (2014) 23:1387–98.
68. Adedokun B, Du Z, Gao G, Ahearn TU, Lunetta KL, Zirpoli G, et al. Cross-ancestry GWAS meta-analysis identifies six breast cancer loci in African and European ancestry women. Nat Commun. (2021) 12:4198.
69. Tanskanen T, van den Berg L, Välimäki N, Aavikko M, Ness-Jensen E, Hveem K, et al. Genome-wide association study and meta-analysis in Northern European populations replicate multiple colorectal cancer risk loci. Int J Cancer. (2018) 142:540–6. doi: 10.1002/ijc.31076
70. O'Mara TA, Glubb DM, Amant F, Annibali D, Ashton K, Attia J, et al. Identification of nine new susceptibility loci for endometrial cancer. Nat Commun. (2018) 9:3166.
71. Scelo G, Purdue MP, Brown KM, Johansson M, Wang Z, Eckel-Passow JE, et al. Genome-wide association study identifies multiple risk loci for renal cell carcinoma. Nat Commun. (2017) 8:15724.
72. Lin W-Y, Fordham SE, Hungate E, Sunter NJ, Elstob C, Xu Y, et al. Genome-wide association study identifies susceptibility loci for acute myeloid leukemia. Nat Commun. (2021) 12:6233. doi: 10.1038/s41467-020-20822-9
73. Wang Z, Budhu AS, Shen Y, Wong LL, Hernandez BY, Tiirikainen M, et al. Genetic susceptibility to hepatocellular carcinoma in chromosome 22q13.31. findings of a genome-wide association study. JGH Open. (2021) 5:1363–72. doi: 10.1002/jgh3.12682
74. Lesseur C, Ferreiro-Iglesias A, McKay JD, Bossé Y, Johansson M, Gaborieau V, et al. Genome-wide association meta-analysis identifies pleiotropic risk loci for aerodigestive squamous cell cancers. PLoS Genet. (2021) 17:e1009254. doi: 10.1371/journal.pgen.1009254
75. Liyanage UE, MacGregor S, Bishop DT, Shi J, An J, Ong JS, et al. Multi-trait genetic analysis identifies autoimmune loci associated with cutaneous melanoma. J Invest Dermatol. (2021) 142:1607–16. doi: 10.1016/j.jid.2021.08.449
76. Cerhan JR, Berndt SI, Vijai J, Ghesquières H, McKay J, Wang SS, et al. Genome-wide association study identifies multiple susceptibility loci for diffuse large B cell lymphoma. Nat Genet. (2014) 46:1233–8.
77. Lin Y, Nakatochi M, Hosono Y, Ito H, Kamatani Y, Inoko A, et al. Genome-wide association meta-analysis identifies GP2 gene risk variants for pancreatic cancer. Nat Commun. (2020) 11:3175.
78. Conti DV, Darst BF, Moss LC, Saunders EJ, Sheng X, Chou A, et al. Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nat Genet. (2021) 53:65–75.
79. Figlioli G, Elisei R, Romei C, Melaiu O, Cipollini M, Bambi F, et al. A comprehensive meta-analysis of case-control association studies to evaluate polymorphisms associated with the risk of differentiated thyroid carcinoma. Cancer Epidemiol Biomarkers Prev. (2016) 25:700–13. doi: 10.1158/1055-9965.EPI-15-0652
80. Vanden Hoek TL, Erickson T, Hryhorczuk D, Narasimhan K. Jack o'lantern mushroom poisoning. Ann Emerg Med. (1991) 20:559–61. doi: 10.1016/S0196-0644(05)81617-8
81. Masters EJ. Personal experience with jack o'lantern mushroom toxicity. Wilderness Environ Med. (2002) 13:182–3. doi: 10.1580/1080-6032(2002)013[0183:LTTE]2.0.CO;2
82. Nair MS, Takeshita H, McMorris TC, Anchel M. Metabolites of clitocybe illudens. IV Illudalic acid, a sesquiterpenoid, and illudinine, a sesquiterpenoid alkaloid. J Org Chem. (1969) 34:240–3. doi: 10.1021/jo00838a058
83. Clark CD, Smith CS. Toxicologic studies of the mushrooms clitocybe illudens and inocybe infida. Mycologia. (1913) 5:224–32. doi: 10.1080/00275514.1913.12018521
84. May-Zhang AA, Tycksen E, Southard-Smith AN, Deal KK, Benthal JT, Buehler DP, et al. Combinatorial transcriptional profiling of mouse and human enteric neurons identifies shared and disparate subtypes in situ. Gastroenterology. (2021) 160:755–70e726. doi: 10.1053/j.gastro.2020.09.032
85. Cody JT, Valtier S, Nelson SL. Amphetamine excretion profile following multidose administration of mixed salt amphetamine preparation. J Anal Toxicol. (2004) 28:563–74. doi: 10.1093/jat/28.7.563
86. Elkashef AM, Rawson RA, Anderson AL, Li SH, Holmes T, Smith EV, et al. Bupropion for the treatment of methamphetamine dependence. Neuropsychopharmacology. (2008) 33:1162–70. doi: 10.1038/sj.npp.1301481
87. Muench C, Malloy EJ, Juliano LM. Lower self-efficacy and greater depressive symptoms predict greater failure to recover from a single lapse cigarette. J Consult Clin Psychol. (2020) 88:965–70. doi: 10.1037/ccp0000605
Keywords: receptor type protein tyrosine phosphatase, lapse doses, relapse, antiaddiction drug development, inhibitors of protein tyrosine phosphatase
Citation: Uhl GR (2023) Selecting the appropriate hurdles and endpoints for pentilludin, a novel antiaddiction pharmacotherapeutic targeting the receptor type protein tyrosine phosphatase D. Front. Psychiatry 14:1031283. doi: 10.3389/fpsyt.2023.1031283
Received: 29 August 2022; Accepted: 30 January 2023;
Published: 17 April 2023.
Edited by:
Cristina Núñez, University of Murcia, SpainReviewed by:
M. Foster Olive, Arizona State University, United StatesKabirullah Lutfy, Western University of Health Sciences, United States
Copyright © 2023 Uhl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George R. Uhl, R2VvcmdlLlVobEB2YS5nb3Y=