 Colin J. McArdle
Colin J. McArdle Alana A. Arnone1,2
Alana A. Arnone1,2 Kimberly F. Raab-Graham
Kimberly F. Raab-Graham- 1Department of Physiology and Pharmacology, Wake Forest University School of Medicine, Winston-Salem, NC, United States
- 2Department of General Surgery, Wake Forest University School of Medicine, Winston-Salem, NC, United States
Gamma-aminobutyric acid (GABA) is the primary inhibitory neurotransmitter in the central nervous system. In the mature brain, inhibitory GABAergic signaling is critical in maintaining neuronal homeostasis and vital human behaviors such as cognition, emotion, and motivation. While classically known to inhibit neuronal function under physiological conditions, previous research indicates a paradoxical switch from inhibitory to excitatory GABAergic signaling that is implicated in several neurological disorders. Various mechanisms have been proposed to contribute to the excitatory switch such as chloride ion dyshomeostasis, alterations in inhibitory receptor expression, and modifications in GABAergic synaptic plasticity. Of note, the hypothesized mechanisms underlying excitatory GABAergic signaling are highlighted in a number of neurodevelopmental, substance use, stress, and neurodegenerative disorders. Herein, we present an updated review discussing the presence of excitatory GABAergic signaling in various neurological disorders, and their potential contributions towards disease pathology.
Introduction
Gamma-aminobutyric acid (GABA), the primary inhibitory neurotransmitter in the adult brain, modulates the inhibitory-excitatory balance necessary for proper brain function predominantly through ionotropic GABAA receptors (GABAARs) and metabotropic GABAB receptors (GABABRs) (1, 2). Both GABA receptor subtypes play a critical role in neurotransmission and proper neuronal functioning. GABAARs are pentameric ligand-gated chloride (Cl−) channels located within the postsynaptic density (PSD) that rapidly hyperpolarize neurons (3, 4). Heterodimeric G-protein coupled receptors (GPCR) GABABRs are located extrasynaptically and generate prolonged and sustained inhibition by activating such channels like G-protein-coupled inwardly rectifying potassium (GIRK) channels and inhibiting voltage-gated calcium channels (VGCC) (1, 5).
The effects of GABAAR activation rely on the concentration gradient of permeable ions and the membrane potential of the cell, preventing an innate drive towards either excitation or inhibition (3). Specifically, the electrophysiological effects of GABAAR signaling highly rely on the intra/extracellular concentrations of chloride (Cl−) ions (Figure 1). During early development, GABAergic transmission is excitatory due to an increased concentration of intracellular Cl− (6). Increased intracellular Cl− results in an efflux of chloride upon GABAAR activation, raising the membrane potential to depolarize the neuron. This developmental profile of Cl− is driven by a high expression of the Na+-K+-Cl− 1 (NKCC1) cotransporter that maintains higher intracellular Cl-concentration during early development. Excitatory GABAergic transmission can additionally increase calcium concentration by activating N-methyl-D-aspartate receptors (NMDARs) (7), resulting in an increased probability for action potential firing (8). These GABA-mediated processes develop prior to the establishment of functional ionotropic glutamate receptors (9) and help to facilitate the maturation of the nervous system by promoting cell proliferation, migration, differentiation, and the expression of brain-derived neurotrophic factor (4, 6, 10, 11). Throughout later courses of development, the expression of the K+/Cl-cotransporter 2 (KCC2) maintains a lower intracellular Cl− concentration. Lowered intracellular Cl− results in an influx of chloride upon GABAAR activation, lowering the membrane potential to hyperpolarize the neuron (12, 13). The switch from excitatory to inhibitory GABAergic signaling results in a maturation of synaptic and network function in the brain, governing normal physiological behaviors into adulthood.
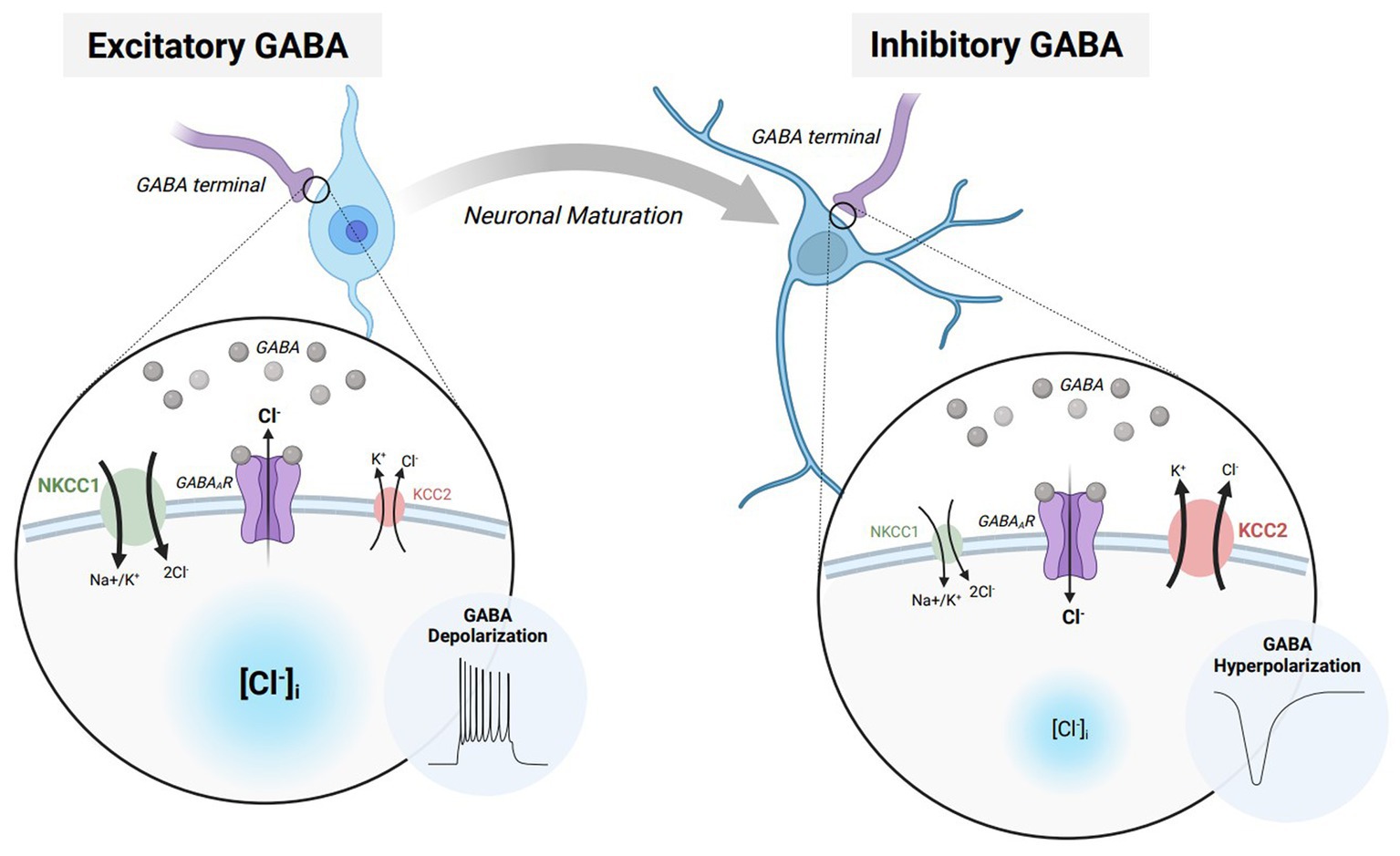
Figure 1. The excitatory/inhibitory transition of GABA during neurodevelopment and maturation. Immature neurons express elevated levels of Na-K-2Cl cotransporter 1 (NCKK1 – green) which allows for a greater intracellular chloride concentration while the neuron is at rest. When GABA binds to GABAA receptors (GABAARs – purple), chloride flows down its respective concentration gradient, resulting in an efflux of chloride ions. The elevation in membrane potential furthermore causes the neuron to depolarize and elicit an action potential. Throughout neuronal maturation, NKCC1 expression is diminished while K-Cl cotransporter 2 (KCC2 – red) is elevated. Heighted KCC2 expression in mature neurons allow for a lower intracellular chloride concentration while the neuron is at rest. When GABA binds to GABAARs, the result is an influx of chloride ions. The decrease in membrane potential moreover causes the neuron to hyperpolarize and prevent action potential propagation. Figure created with BioRender.com.
While excitatory GABAergic transmission is usually only observed during early development, specific populations of adult neurons continue to display excitatory responses to GABAAR signaling (14–16). However, pathological conditions can also influence the continued presence of GABAAR signaling in the mature brain (17). A number of mechanisms have been proposed that could underlie changes in GABA activity, including brain-region specific alterations in GABAARs, changes in Cl− homeostasis, or plasticity of GABAergic synapses. The mechanisms responsible for disrupting the switch from excitatory to inhibitory GABAergic signaling additionally have the potential to exacerbate the various molecular and behavioral pathologies observed in neurological disorders. Herein, this review will highlight the current literature discussing the presence of paradoxical, excitatory GABAergic signaling in the mature nervous system and their contributions towards the hallmarks of several neurological disorders.
Stress related disorders
Stress, either acute or chronic, correlates with an increased prevalence of several behavioral phenotypes including anxiety, depression, learning impairments, seizure susceptibility, and substance use (18). The physiological response to stress is regulated through the Hypothalamus-Pituitary–Adrenal (HPA) Axis where the release of corticotropin-releasing hormone (CRH) from the paraventricular nucleus (PVN) of the hypothalamus stimulates the release of adrenocorticotropic hormone (ACTH) from the pituitary gland to further release cortisol from the adrenal glands (18). Several brain regions, neurotransmitters, and hormones influence the HPA-axis activity, however, GABAergic projecting neurons that synapse onto neurons in the PVN are the primary regulator of the physiological stress response (19).
The addition of one or multiple stressors results in persistent activation of the HPA-axis and subsequent negative behaviors (20–22). The downstream hyperactivation of this axis is furthermore hypothesized to result from alterations in GABAergic signaling. In preclinical models, acute and chronic stress has been shown to alter the shift from excitatory to inhibitory GABAergic signaling in the brain. Previous research utilizing several stress paradigms in rodents including restraint stress, maternal separation, hyperosmotic stress, prenatal stress, early-life stress, or a combination of different stressors all denote a depolarizing shift in GABAAR-mediated signaling in the hypothalamus (23–32). Herein, will focus on stress induced mechanisms that shift GABAAR signaling from hyperpolarizing to depolarizing.
Stress and neurosteroids
The stress-induced switch in GABAAR function is hypothesized to be mediated through several mechanisms: interactions from neurosteroids, altered GABA synthesis/release, as well as chloride (Cl−) dyshomeostasis. Neurosteroids, or steroids synthesized in the brain, adrenals, and gonads, modulate neuronal excitability by allosterically binding to GABAARs (33). Previous work has shown that neurosteroidogenesis, or the synthesis of neurosteroids, exacerbates the excitatory GABAergic drive in response to stress (32). Acute stress can elevate neurosteroids, which can bind and modulate GABAAR function (17). In a model of acute restraint stress, GABA depolarizes CRH-positive neurons in the PVN and further increases corticosterone levels. Acute stress additionally elevates the neurosteroid, tetrahydrodeoxycorticosterone (THDOC), which exacerbates excitability of CRH-positive neurons. Furthermore, inhibiting neuosteroidogenesis with finasteride reduces stress-induced anxiety-like behaviors in mice.
Stress and transsynaptic GABA signaling
Excitatory GABAAR-mediated responses following stress can also be mediated through changes in transsynaptic GABA signaling. This can include alterations in GABA synthesis/transport, as well as changes in GABAAR protein expression. Previous studies denote a reduction in endogenous GABA throughout the nervous system following stress (17, 34, 35). It is also important to note that GABA itself acts as a limiting factor to promote the transition from depolarizing to hyperpolarizing GABAAR-mediated responses by upregulating KCC2 mRNA expression (6). Changes in stress-induced GABA levels can furthermore be attributed to its synthesis, and transport. Glutamic acid decarboxylase 67 and 65 (GAD67 and GAD65), which synthesize GABA from glutamic acid, is downregulated during periods of stress (34, 36, 37). Vesicular GABA transport (vGAT), which imports presynaptic GABA into vehicles, is also reduced following chronic stress (38, 39). Finally, gamma-aminobutyric acid transporter-1 (GAT-1), which is responsible for the reuptake of GABA, is increased after stress (36). Furthermore, these molecular alterations can result in a net decrease in GABA levels, potentiating the presence of stress-induced depolarizing GABAAR-mediated responses (Figure 2).
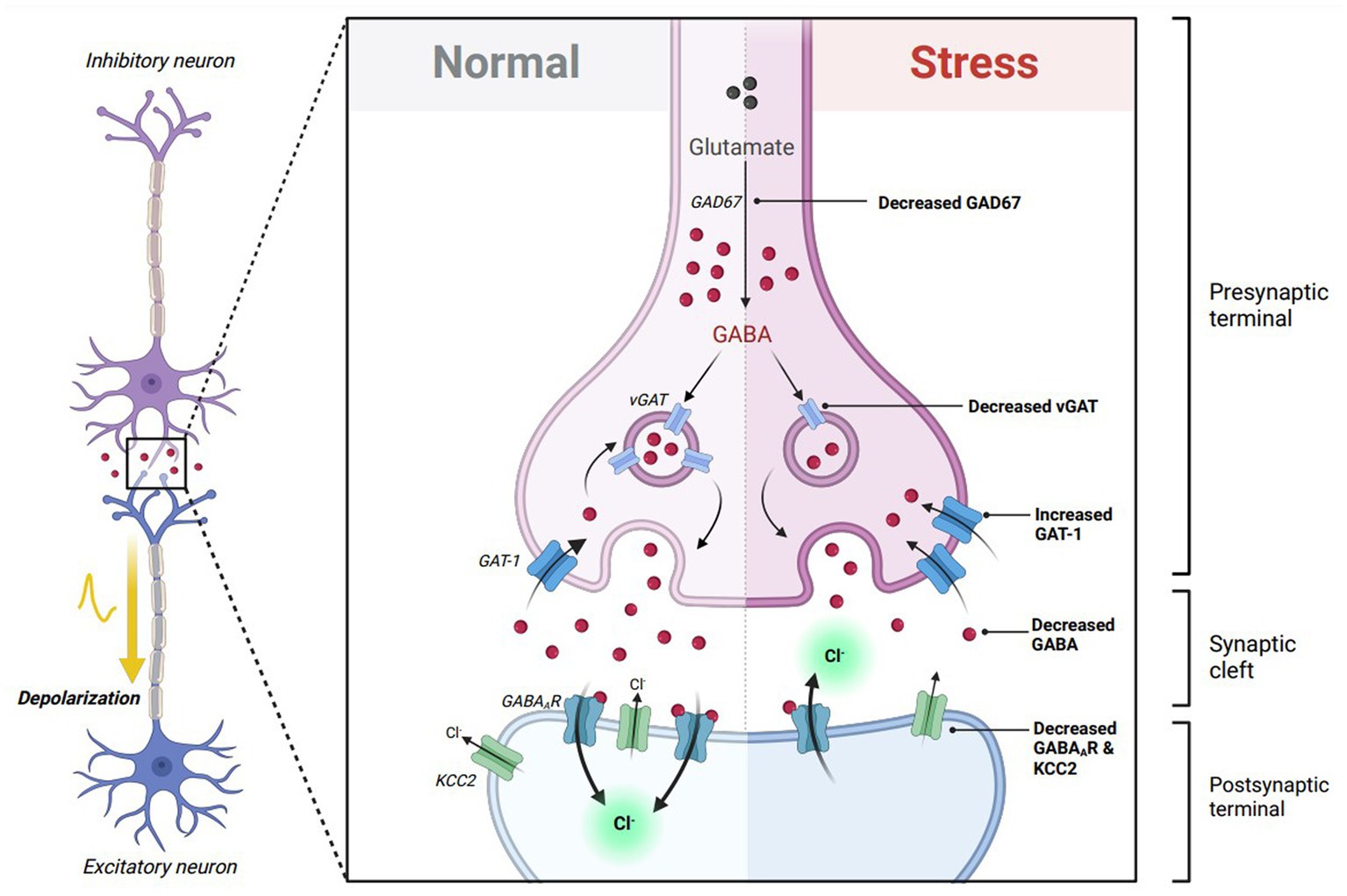
Figure 2. Theoretical effects of GABA transsynaptic signaling on excitatory GABAergic responses. GABA itself is critical in transitioning GABAA receptor (GABAAR) responses from depolarizing to hyperpolarizing throughout neuronal maturation. In immature neurons, GABA binding to excitatory GABAARs promotes the expression of K-Cl cotransporter 2 (KCC2), which decreases intracellular chloride concentration, allowing future GABAAR-mediated responses to become inhibitory in mature neurons. Stress exposure diminishes GABA transsynaptic signaling by reducing glutamic acid decarboxylase 67 (GAD67) and vesicular GABA transporter (vGAT) expression, while elevating GABA transporter type 1 (GAT-1) expression. This results in lowered extracellular GABA to freely bind to its respective receptors. Stress also reduces the expression of GABAARs via downregulation of several GABAAR subunits. The lack of available GABA to bind to GABAARs following stress hypothetically reduces KCC2 expression, which disturbs chloride homeostasis and promotes an excitatory switch in GABAergic signaling. Figure adapted from “Mechanism of action of Selective Serotonin Reuptake Inhibitors,” by BioRender.com (2023). Retrieved from https://app.biorender.com/biorender-templates.
Different stress paradigms alter GABAAR expression in different areas of the brain (17). Various stress paradigms in preclinical models exhibit a reduction in GABAAR subunit expression (39–43), as well as hyposensitivity of these receptors towards GABA binding (34, 35). To note, postsynaptic GABAARs and KCC2 co-transporters are shown to display a bidirectional crosstalk that influences their function. As both colocalize along dendrites (44), GABAAR activity promotes the surface stability of KCC2 via the chloride-sensing kinase, WNK1 (45). Conversely, elevated KCC2 activity potentiates the expression and function of hyperpolarizing GABAARs (46–48). Furthermore, it can be hypothesized that stress-induced reductions in GABAAR expression can lead to reduced KCC2 stability and chloride dyshomeostasis, allowing for the presence of excitatory GABAergic signaling.
Stress and chloride homeostasis
Chloride (Cl−) homeostasis, which is essential for maintaining effective GABAergic inhibition, is also affected by stress. Several reports have indicated that stress-induced Cl- dyshomeostasis is linked to either decreases in KCC2 or increases in NKCC1 within the hypothalamus (24, 25, 27, 28, 49, 50). As previously discussed, downregulation of the KCC2 cotransporter results in elevated intracellular chloride concentrations, allowing GABAAR-mediated responses to switch from hyperpolarizing to depolarizing. Additional studies also note dephosphorylation of the KCC2 residue Ser940 following stress (17, 30–32). The phosphorylation of Ser940 is dependent on Protein kinase C (PKC) activity and enhances KCC2 surface stability and activity by reducing endocytosis (51). Furthermore, reduced phosphorylation and surface expression of KCC2 provides a molecular mechanism underlying Cl- dyshomeostasis and excitatory GABAergic signaling following stress.
One study, however, indicates that stress accelerates the excitatory GABA switch in the infralimbic cortex at postnatal day (PND) 9 (52). Additionally, NKCC1 mRNA expression was downregulated at an earlier developmental timepoint following stress. An explanation for the differences between this study and those previously discussed is that the neurological effects of stress may not be uniform across each brain region. Stress has been shown to reduce PFC function in both humans and animal models that furthermore impairs learning and memory (53). These changes in the PFC are additionally correlated with alterations in network connectivity and dendritic remodeling. Interestingly, the PFC has previously been shown to inhibit HPA-axis activity (54). An acceleration towards hyperpolarizing GABAergic signaling and NKCC1 downregulation moreover could contribute to reductions in PFC activity following stress. These molecular alterations in the PFC could therefore exacerbate HPA-axis activity in response to stress. Additionally, the stress paradigm used in this study did not show elevations in cortisol, which is an output measure for HPA-axis activity. Therefore, the effects from this paradigm may not classically influence HPA-axis activity as compared to the previously discussed research, resulting in a differential physiological and molecular response to stress.
‘Anti-stress’ mechanisms and KCC2 stability
Mechanisms altering GABAergic signaling are involved in both promoting and counteracting the negative behavioral effects of stress. Oxytocin, a hormone released from the pituitary gland, has been previously shown to have therapeutic properties by initiating anxiolytic responses following stress (55, 56). These ‘regenerative’ properties of oxytocin promote learning and memory, cognition, and social behaviors (55, 57, 58). Oxytocin counterbalances the stress response by modulating the GABA switch from excitatory to inhibitory. As previously discussed, stress paradigms promote depolarizing GABAAR-mediated responses by downregulating KCC2 expression. On the contrary, oxytocin initiates the switch from excitatory to inhibitory GABAergic signaling by regulating KCC2 activity. In the hippocampus, oxytocin was previously shown to increase the phosphorylation of KCC2 via the Oxtr/Gq/PKC pathway, increasing KCC2 surface stability to shift GABAergic signaling towards inhibitory during early stages of development (59). Hippocampal neurons lacking oxytocin receptors (Oxtr−/−) exhibit a delay in switching from excitatory to inhibitory GABAergic responses, as well as a net elevation in neuronal excitability (59). During childbirth, a surge in maternal levels of oxytocin allows the fetal brain to transition from depolarizing to hyperpolarizing GABAAR-mediated signaling (60), promoting healthy development and maturation of the brain during early development. In human populations, diminished oxytocin has been shown in patients with anxiety and post-traumatic stress disorder (PTSD) (61–63). The molecular mechanisms underlying oxytocin’s effects on restoring inhibitory GABAergic signaling can furthermore validate oxytocin’s therapeutic potential in alleviating negative behaviors and symptoms in patients with stress related disorders.
Alzheimer’s disease and dementia
The most common form of dementia, Alzheimer’s disease (AD), is marked by memory impairment and cognitive decline (64). These behavioral abnormalities are simultaneously paired with molecular hallmarks including amyloid-beta (Aβ) plaque and neurofibrillary tangles. In addition to these major hallmarks, patients and preclinical models display profound neuronal hyperexcitability and glutamate excitatory that is aversive towards neuronal and synaptic function (65–68). The majority of research hypothesizes overactive NMDAR receptors signaling as the primary contributor towards hyperexcitability in AD (69, 70), however, several studies indicate excitatory GABAergic signaling as an additional contender towards hyperexcitability and the pathology present during the AD disease progression.
Switches in GABAergic signaling have previously been characterized in a preclinical rodent model of AD-related pathology. 2 and 3-month-old rats injected with Aβ1-40 fibrils exhibit a reduction in hippocampal IPSC amplitude compared to wildtypes (71). Of note, Aβ1-40 treatment also results in a positive shift in the equilibrium potential of chloride ions (ECl-), indicating that Aβ1-40 fibrils promote an excitatory GABAergic shift in the hippocampus. This study additionally found similar electrophysiological trends in a 9-month-old APP/PS1 transgenic model of AD. APP/PS1 hippocampal slices also show reduced IPSC amplitude and a positive ECl- shift, emphasizing the conservation of excitatory GABAergic signaling across multiple preclinical AD models.
Previous studies have also uncovered the presence of excitatory GABA switches in additional models of AD-related pathologies. A transgenic mouse line expressing recombinant neutralizing anti-nerve growth factor (NGF) antibodies (AD11 mice) display similar pathological hallmarks to AD such as amyloid plaque and neurofibrillary tangle deposition, neurodegeneration, long-term potentiation (LTP) deficits, and behavioral abnormalities (72–75). 6-month-old AD11 hippocampal slices with isoguvacine, a GABAAR agonist, increased action potential frequency in CA1 hippocampal neurons (76). The increase in firing was further abolished using bicuculine or gabazine, two GABAAR antagonists. To determine the presence of an excitatory switch in GABAergic signaling, hippocampal GABAAR-mediated postsynaptic currents (EGPSC) were recorded via perforated patch clamping following stimulation of local GABAergic interneurons. In wild-type mice, EGPSCs are reduced in comparison to the respective resting membrane potential. In AD11 mice, however, EGPSCs are increased with respect to the resting membrane potential. The positive shift in GABAAR-mediated current in AD11 mice indicates an aberrant shift in hyperpolarizing GABAergic activity to depolarizing.
In conjunction to the electrophysiological findings that GABA acts to depolarize neurons in several models of AD-related pathology, previous research has investigated molecular changes that could underlie the GABA switch. As noted before, a developmental switch from NKCC1 to KCC2 expression underlies the excitatory to inhibitory shift in GABAergic signaling under normal conditions. However, data from both preclinical models and patients with AD show that this transition is disturbed. Imbalances between NKCC1 and KCC2 have recently been shown in patients with AD (77, 78) that are exacerbated with the onset of epileptic activity, indicating that hyperexcitability can act as a feed-forward mechanism to promote excitatory GABAergic signaling. Previous findings in preclinical models of amyloid-beta deposition (APP/PS1 transgenic mice and Aβ1-40 fibril treatment) note reduced KCC2 protein expression in the hippocampus and cortex (71, 76, 79, 80) with increases in NKCC1 protein expression (81). One study, however, indicates increases in KCC2 in a different model of AD-related pathology. Discrepancies between results can be due to contrasting animal models between these studies. KCC2 expression is shown to increase with treatment of AβO oligomers while it decreases with Aβ1–40 fibril treatment. Aβ oligomers and fibrils display different chemical structures and have been shown to have differential effects on neuronal function throughout the progression of Aβ plaque deposition (82). KCC2 expression could alter based on the current changes in neuronal activity induced by either Aβ oligomers or fibrils. Therefore, further exploration is required to definitively confirm the effects of Aβ plaque and different structural forms of Aβ on chloride homeostasis and excitatory GABAergic signaling. Interestingly, treatment with γ-secretase inhibitor, LY411575, restored KCC2 levels in APP/PS1 mice, highlighting the direct impact of Aβ plaque on KCC2 expression (80). Finally, in an APOE4-KI mouse, a model for the largest genetic risk factor of AD, also presents with an increased ratio between NKCC1 and KCC2 expression (83). Moreover, the consensus amongst the current literature argues a predominance towards NKCC1 expression that can underlie the depolarizing actions of GABA in models of AD-related pathology.
It is important to note that while studies have explored the relationship between AD-related pathology and excitatory GABAergic switches, previous research has explored the link between non-pathogenic amyloid precursor protein (APP) and alterations in GABAergic signaling. APP is the precursor protein to Aβ plaque and is metabolized through several steps to produce its subsequent products. The presence of either α-secretase or β-secretase can influence APP’s processing into non-pathogenic or pathogenic products, respectively. While APP itself is not considered toxic in the brain, studies have shown contradicting results in APP’s role in GABAergic transmission. In an APP−/− model, CA1 hippocampal slices display a positive shift of the GABA reversal potential (EGABA), reduced IPSC amplitude, and reduced GABAARα1 expression. The findings additionally show that APP−/− mice exhibit reduced KCC2 surface expression. This is further linked to APP’s ability to physically interact and stabilize KCC2 to the plasma membrane to prevent its ubiquitination and degradation (82). In opposition, in vitro cortical neurons transfected to express full-length neuronal wildtype human APP increase [Ca2+]i and action potential frequency in response to treatment with GABA. Of note, overexpression of hAPP in cortical neurons resulted in decreased KCC2 expression with no differences in NKCC1 in both in vitro and in vivo models (78). Differences in these results may be due to the age in which KCC2 expression was examined in mice. The first study (82) utilized an older APP−/− mouse model while the second study (78) utilized younger hAPP transfected cortical cells and mouse models. Neurons were transduced to overexpress APP prior to when GABA is predicted to shift from depolarizing to hyperpolarizing, while measurements in the APP−/− model were done after the switch. Furthermore, it can be hypothesized that APP has differential effects on KCC2 expression and neuronal hyperexcitability pre- and post-GABA switch.
To further probe the contributions of excitatory GABAergic signaling towards memory and cognitive dysfunction in AD, several studies have investigated the impacts of restoring GABAergic transmission as a potential therapeutic target in preclinical and clinical settings. In rodent models of AD-related pathology, treatment with bumetanide, an NKCC1 inhibitor, significantly enhances contextual fear learning and spatial learning (71, 81). Interestingly, a study (84) identified bumetanide as a top ranking therapeutic for APOE4-related AD according to a Connectivity Map (CMap) database. Bumetanide treatment in an APOE4-KI mouse model mitigated neuronal hyperexcitability, restored LTP deficits, decreased Aβ plaque deposition, and rescued spatial learning deficits (Figure 3). APOE4-KI mice treated with bumetanide also attenuated transcriptomic deficits in genes relating to GABAergic synaptic function and circadian rhythms, two biological processes that are affected by AD-related pathology. This study furthermore highlighted that exposure to bumetanide in humans was correlated with lowered AD prevalence according to electronic health record databases. Of note, a previous study (85) showed that a 7-day regimen of daily injections of 25 mg/kg Memantine, an NMDA receptor antagonist that is a commonly prescribed to patients with AD, reduced cortical and hippocampal KCC2 expression while simultaneously, attenuated the behavioral response to GABAAR activation with diazepam. This data brings forth the use of alternative FDA-approved medications, such as bumetanide, that can restore homeostatic GABAergic signaling as a potential therapeutic avenue to prevent memory and cognitive impairments in AD patients.
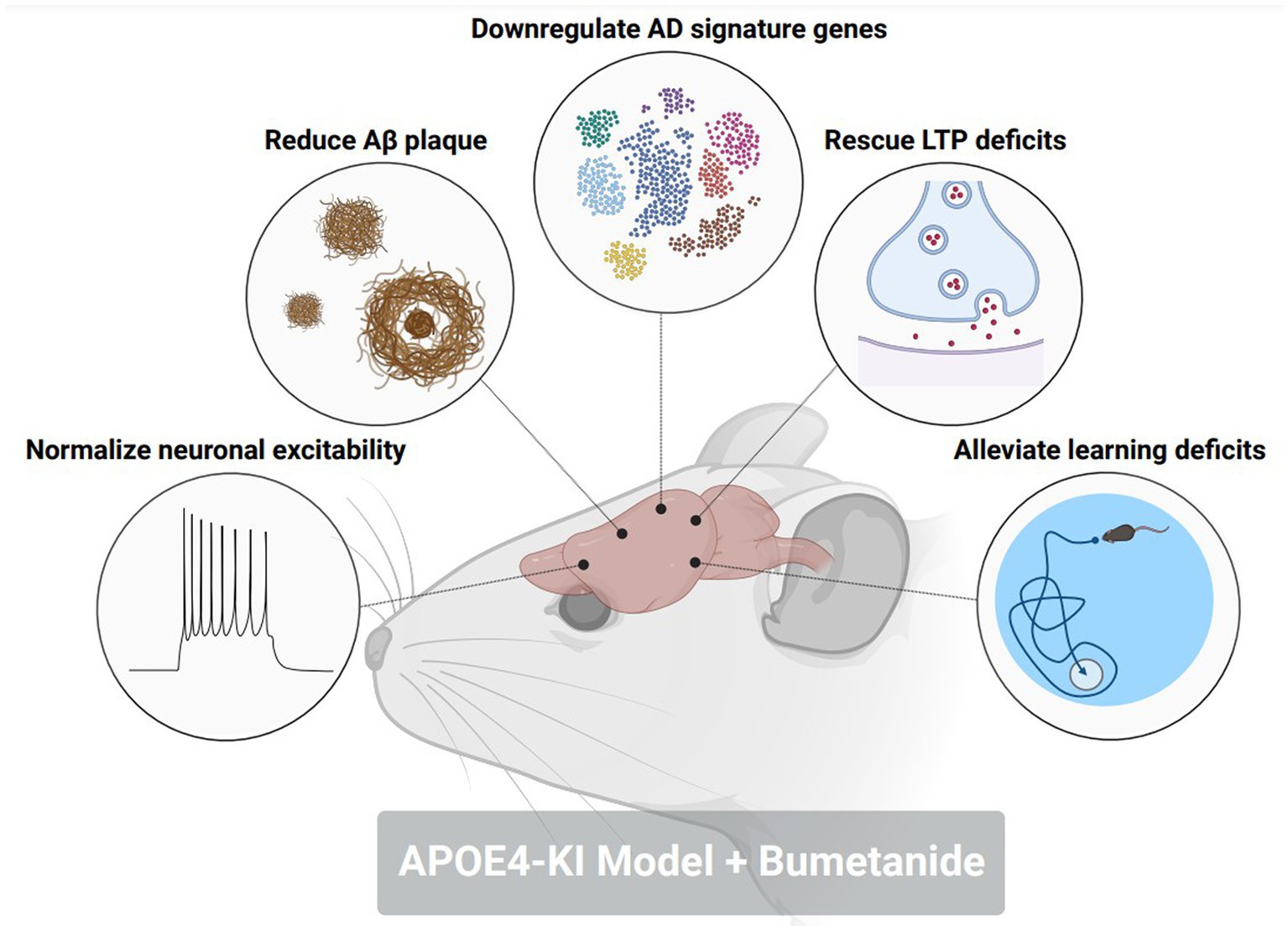
Figure 3. Bumetanide alleviates pathological, electrophysiological, and behavioral hallmarks in an AD-related pathology mouse model. Treatment with NKCC1 inhibitor, bumetanide, in an aged APOE4-KI mouse corrected the following pathological phenotypes in a model of AD-related pathology: (1) normalized neuronal excitability in CA1 pyramidal neurons. (2) reduced amyloid-beta plaque size and number in the hippocampus and cortex. (3) downregulated the expression of AD signature genes that promote disease pathology. (4) rescued deficits in long-term potentiation (LTP) within the CA1 of the hippocampus. (5) Alleviated deficits in spatial memory through a Morris water maze. Figure created with BioRender.com.
Although previous research investigating excitatory GABAergic signaling in AD has predominately focused on models of Aβ deposition, there is a lack of evidence stating whether an excitatory GABA switch is present in models of hyperphosphorylated tau and neurofibrillary tangles. Currently, there are sufficient data to argue for a reduced inhibitory GABAergic tone in preclinical models of tau tangles (86, 87); however, no findings have been presented as to whether a shift to excitatory GABAergic neurotransmission contributes to diminished inhibitory neuronal signaling.
Autism spectrum disorders
Autism spectrum disorder – clinical studies
Autism Spectrum Disorder (ASD) is a neurodevelopmental disorder commonly presented with symptoms such as repetitive behaviors and impairments in social communication (88). Due to the heterogeneity of this disorder, patients display a spectrum of symptoms that is accompanied by a specialized regimen of combinational therapies and pharmaceutical treatments. Previous research has interestingly noted a presence of excitatory GABAergic signaling that can potentially underlie behavioral symptoms (89). Children with ASD were previously treated with diazepam, a GABAAR positive allosteric modulator, that typically exerts an anxiolytic effect. On the contrary, diazepam paradoxically increased aggressive behavior in ASD patients (89), indicating a dysfunction in GABAergic signaling.
KCC2 variants have furthermore been identified in human populations with ASD (90). Genomics and exosome sequencing identified a SLC12A5 variant, the gene encoding for KCC2, that is prevalent in patients with ASD. These variants include R952H, R1O49C, and R1048W within the C-terminal regulatory domain of KCC2, compromising the ability of KCC2 to maintain low concentrations of intracellular chloride and hyperpolarizing GABAergic responses. These KCC2 variants are also predicted to target the CpG sites on the SLC12A5 gene, which can affect the likelihood of DNA methylation and repressed gene expression. Moreover, this data highlights a distinct molecular mechanism in which depolarizing GABAergic responses are preserved in ASD patients. An overabundance of non-functional KCC2 variants can result in disturbed chloride homeostasis, promoting an excitatory GABAergic drive. Of note, several clinical studies have utilized bumetanide as a novel therapeutic for patients with ASD (91–93). Collectively, the evidence shows promising effects of bumetanide to alleviate behavioral and electrophysiological abnormalities. These studies furthermore highlight the significant contributions of excitatory GABAergic towards the pathogenesis of ASD.
Fragile X syndrome
Fragile X Syndrome (FXS) is the most common genetic form of intellectual disability and autism spectrum disorder (ASD). FXS is caused by an overabundance of CGG motif repeats on the Fmr1 gene, silencing its corresponding protein product, Fragile X Ribonuclear Messenger Protein 1 (FMRP) (94). FMRP is an RNA-binding protein that regulates a large domain of proteins critical for transsynaptic signaling (95). The loss of FMRP furthermore results in synaptic dysfunction as well as imbalances in excitatory/inhibitory transmission. This imbalance could furthermore be linked to excitatory GABAergic signaling. Pyramidal neurons in layers IV and V of the somatosensory cortex from WT mice display a shift from depolarizing to hyperpolarizing GABAergic signaling at approximately postnatal day (PND) 9 (96). Neurons from Fmr1 KO mice, however, display a delay in this shift, switching to hyperpolarizing GABAergic responses at PND 14 (96). Additional reports have stated similar delays in hyperpolarizing GABAergic signaling or disrupted responses to GABA (96–99). One study does report a hyperpolarizing GABA switch to occur earlier in Fmr1 KO slices (~P21–25) in auditory cortical neurons as compared to wildtypes (100). However, the region of interest differs from the previous literature investigating the somatosensory cortex. Disturbances to shift towards hyperpolarizing GABA can potentially differ across cortical subregions, explaining the discrepancy of exactly when GABA is no longer excitatory throughout the FXS disease progression.
Depolarizing GABA response in FXS is further hypothesized to be contributed from chloride ion imbalances, via upregulated NKCC1 expression (96), with either decreases or no changes in KCC2 expression (96, 99) in Fmr1 KO mice. FMRP is known to repress the majority of its target synaptic mRNAs (95). Two of its targets include SLC12A5 and SLC12A2 mRNAs, which encode for KCC2 and NKCC1, respectively. However, no conclusive evidence states whether FMRP in fact represses the synthesis of KCC2 and NKCC1. Silencing of FMRP in FXS could furthermore result in an overabundance of either KCC2 or NKCC1, leading to disturbances in chloride homeostasis and alterations in excitatory GABAergic signaling. Previous work also denotes reduced membrane expression of KCC2 in Fmr1 KO cortical tissue. Further analysis of the Fmr1 KO cortices indicate an absence of several phosphorylation sites within the intracellular C-terminus of KCC2 (101). Reduction in KCC2 phosphorylation in FXS could result in compromised membrane trafficking or surface stability.
Studies have additionally targeted NKCC1 in FXS to alleviate symptoms in preclinical models. Treatment with bumetanide, an NKCC1 inhibitor, during a critical period of development (P0-P10) restored hyperpolarizing GABAergic responses in the somatosensory cortex of Fmr1 KO mice, normalized neuronal hyperexcitability, and rescued synaptic function amongst thalamocortical excitatory synapses (102). Bumetanide also altered the proteome in the somatosensory cortex in FMR1 KO mice to overexpress proteins relating to inhibitory interneuron development while downregulating at-risk proteins of several developmental disorders (102). Interestingly, an additional study corroborated these findings by pretreating pregnant FXS female mice with bumetanide and monitoring electrophysiological behavioral phenotypes of their offspring. Maternal bumetanide pretreatment restores hyperpolarizing GABA responses and reduces excitability in the hippocampus of offspring (99). Ultrasonic vocalization behaviors and brain oscillations are also normalized in pretreated offspring (99).
Rett syndrome
Rett Syndrome is a developmental disorder predominately characterized by speech dysfunctions, gait abnormalities, motor impairment, and disturbed breathing patterns (103). The disease is caused by mutations in the Methyl-CpG-binding protein 2 (MeCP2) which under physiological conditions, is critical in regulating gene expression in the brain (103). Previous research denotes an imbalance between excitatory and inhibitory transmission that can underlie the behavioral symptoms in Rett patients. In both in vitro and in vivo models of Rett syndrome where MeCP2 is absent (Mecp2y/−), neurons exhibit a depolarizing response to GABA (104–106). Studies have additionally found a similar pattern compared to other ASD-related diseases such that KCC2 mRNA and protein expression is reduced in preclinical models and Rett patients (104, 106–108). One study identified a potential mechanism underlying reduced KCC2 expression. REST, a transcriptional repressor competes with MeCP2 for the RE-1 binding site on the SLC12A5 gene (109). In Rett neurons, where there is an absence of MeCP2, REST can occupy the RE-1 binding site, preventing the transcription of KCC2 (SLC12A5) mRNA (106).
Several studies have utilized different therapeutic approaches to alleviate excitatory GABAergic signaling and pathological symptoms in preclinical models of Rett Syndrome. Similar to studies done in FXS mice (99), maternal pretreatment with bumetanide 1 day prior to delivery restored the GABA switch, normalized excitability, increased hypersynchronization, and rescued impaired mGluR-induced LTD in CA3 pyramidal neurons of Mecp2−/y mice (105). Another previous study developed a high-throughput drug screening platform to identify pharmaceuticals that could enhance KCC2 expression. A subset of small molecules coined KCC2 expression-enhancing compounds (KEECs) were selected which have a range of molecular targets including fms-like tyrosine kinase 3 (FLT3), glycogen synthase kinase 3 (GSK3), sirtuin 1 (SIRT1), and transient receptor potential cation channel subfamily V member 1 (TRPV1). These molecules were successful in elevating KCC2 expression. Treatment with KEECs not only restored hyperpolarizing GABAergic responses and excitability in MeCP2 KO cultured neurons, but also prevented morphological deficits in these neurons as well. Further treatment of KEECs in MeCP2-mutant mice also prevented the onset of locomotor and breathing abnormalities (110). Interestingly, several studies have utilized Insulin-like Growth Factor 1 (IGF-1) as a novel therapeutic alternative. Previous reports show IGF-1 treatment prevents behavioral, synaptic, and electrophysiological abnormalities in MeCP2-mutant mice (111, 112). Further evidence shows that IGF-1 mitigates its therapeutics effects in MeCP2-mutanat mice by elevated KCC2 and downregulated NKCC1 expression, therefore, lowering intracellular chloride levels needed to induce a hyperpolarizing GABAergic response. IGF-1 has previously been reported to accelerate the maturation of neuronal networks via KCC2 upregulation (113), granting this as a potential therapeutic avenue to alleviate developmental delays in Rett Syndrome.
Tuberous sclerosis complex
Tuberous Sclerosis Complex (TSC) is an autosomal dominant disorder that results in the loss of either the Tsc1 or Tsc2 gene. Patients with TSC present with several autistic-like behaviors, but more importantly, experience at least one epileptic event in their lifetime (114, 115). Studies in clinical and preclinical models point to an imbalance between excitation and inhibition that favors a shift towards cortical hyperexcitability as the hypothesized cause of seizure susceptibility (114, 116, 117). Studies note alterations in GABAergic responses can contribute to aberrant excitability in TSC. In both TSC human cortical neurons and Xenopus oocytes that express membranes from TSC cortical tubers display either depolarizing GABAA receptor-mediated responses or a significant delay in the excitatory GABA switch (118, 119). As a common mechanism between the previously discussed autism spectrum disorders, KCC2 expression is downregulated while NKCC1 expression is elevated in TSC neurons (118, 119). Of note, immunohistochemical evidence shows an elevated abundance of NKCC1 expression amongst dysplastic neurons in TSC corticies, while KCC2 expression is lowered within the soma and proximal dendrites (119). Several studies have utilized bumetanide to target NKCC1 function in TSC patients and noticed an attenuation in both behavioral and electrophysiological abnormalities with treatment (120–122).
TSC is considered an mTORopathy, meaning that mammalian target of rapamycin complex 1 (mTORC1) activity is elevated in preclinical and clinical models of TSC (123, 124). mTORC1 is classically known as a master regulator of mRNA translation to promote protein synthesis (123, 125); however, previous research denotes mTORC1 has equal responsibilities in promoting and repressing mRNA translation (126). Treatment with rapamycin, an mTORC1 inhibitor, caused an upregulation of 166 proteins. Termed mTOR-OFF proteins, this select population of mRNAs are translated when mTORC1 is in its inactive form. One of those mTOR-OFF proteins is KCC2 where the SLC12A5 mRNA is translated when mTORC1 is repressed. Since mTORC1 activity is aberrant in TSC, it can be hypothesized that reduced KCC2 protein synthesis is under direct influence of dysregulated mTORC1 activation (Figure 4). However, previous evidence also indicates that rapamycin treatment represses the protein expression of KCC2 (127). Discrepancies between these two findings can be due to the dosage and frequency of rapamycin treatments. The first study (126) used a one-hour treatment of 10 mg/kg rapamycin prior to tissue collection while the second study (127) used a three-day regimen of 5 mg/kg rapamycin. A single treatment of rapamycin can be considered ‘acute,’ only inhibiting mTORC1 of the mTOR complex, while doses across multiple days can be considered ‘chronic’ inhibiting both mTORC1 and mTORC2 (128). Further experimentation to inhibit only mTORC2 activity will properly address the confounding results.
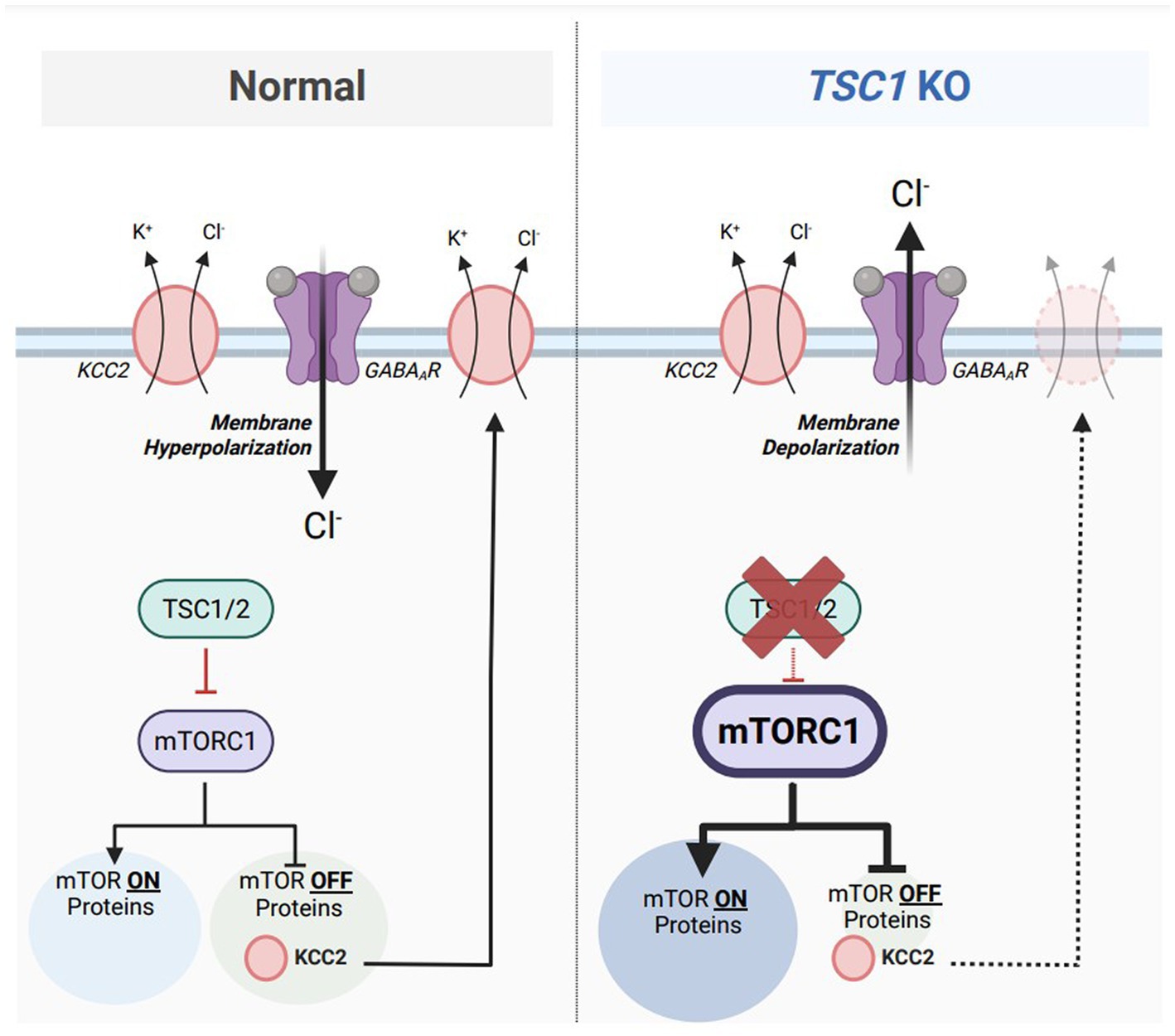
Figure 4. Aberrant mTORC1 activity represses KCC2 expression in a model of Tuberous Sclerosis Complex (TSC). Under normal conditions (left), mammalian target of rapamycin complex 1 (mTORC1) equally activates the translation of 155 target mRNAs (mTOR ON proteins) and represses the translations of 166 target mRNAs (mTOR OFF proteins). Tuberous sclerosis proteins 1 and 2 (TSC1/2) acts upstream of mTORC1 and represses its activity. One particular mTOR OFF protein is KCC2, which is synthesized when mTORC1 is inactive. KCC2 is responsible for maintaining a low intracellular chloride concentration to maintain inhibitory GABAAR-mediated responses. In TSC1 KO neurons, a model of Tuberous Sclerosis Complex (TSC) (right), TSC1/2 are absent which results in aberrant mTORC1 activity. The population of mTOR ON proteins are upregulated while the population of mTOR-OFF proteins are downregulated. Furthermore, KCC2 downregulation from elevated mTORC1 activity results in an accumulation of intracellular chloride, resulting in depolarizing GABAAR-mediated responses. Figure created with Biorender.com.
Interestingly, previous studies highlight a distinct relationship between NKCC1 and mTORC1 activity. NKCC1 activity represses mTORC1 through the Leu transporter LAT1-4F2hc (LAT1), as well as the Akt and Erk signaling pathways (129). In models of TSC where mTORC1 activity is upregulated, NKCC1 upregulation as denoted by previous research (118, 119) could serve as a compensatory mechanism to normalize mTORC1 activity to homeostatic levels. This study also denotes that NKCC1 activity positively correlates with cell size. Cellular hypertrophy is a recognized phenotype in models of TSC (130), however, this study indicates that elevated NKCC1 expression could contribute to increased soma size.
Additional idiopathic forms of autism spectrum disorder
In conjunction with the previously discussed research, other idiopathic forms of ASD have been identified to display disturbances in GABAergic signaling. Preclinical models of Down Syndrome (Ts65Dn) and DiGeorge Syndrome (Lgdel+/−) exhibit a depolarizing GABAA receptor mediated response or a delay to switch to hyperpolarizing GABAergic responses (131, 132). These electrophysiological findings are also correlated with an increased ratio between NKCC1 and KCC2 expression (131). Treatment with bumetanide in Ts65Dn mice furthermore restored inhibitory GABAergic responses while simultaneously ameliorated LTP deficits and cognitive dysfunction (133). While there is a strong argument supporting the presence and impacts of disturbed GABAergic signaling in ASD, further exploration in other idiopathic forms and preclinical models of ASD can corroborate a conserved mechanisms underlying depolarizing GABAergic responses across heterogenous ASD populations.
Substance use disorders
Substance Use Disorders (SUD) are classically characterized by compulsive drug-seeking behaviors, loss of control to limit substances, as well as negative emotional behaviors during withdrawal states (134). The underlying neurocircuitry is executed through the mesolimbic dopaminergic reward pathway which involves projecting dopaminergic neurons from the ventral tegmental area (VTA) to the nucleus accumbens (NAc) (135). While dopamine is responsible for instigating the reinforcing properties of substances, local GABAergic interneurons that synapse onto dopaminergic neurons in the VTA tightly regulate the activity of the mesolimbic pathway (136, 137). Previous reports indicate several substances inhibit GABAergic interneuron activity, disinhibiting dopaminergic neurons to elevate dopamine levels (135, 138–140). However, prolonged exposure to substances results in a ‘hypodopaminergic state,’ promoting drug-seeking behaviors to normalize dopamine levels during withdrawal (141–144). Moreover, collective evidence suggests that substance-induced hypodopaminergia could result from sustained activation of VTA GABAergic interneurons via an excitatory GABA switch.
Alcohol use disorder
Alcohol Use Disorder (AUD) is broadly characterized by repetitive consumption of ethanol with a heighted vulnerability to seek alcohol following the cessation of drinking (145, 146). The reinforcing properties of ethanol on the nervous system are highly controlled by dopaminergic signaling, as described previously (135). However, repetitive drinking behaviors and subsequent alterations in dopaminergic circuity in preclinical models are hypothesized to be driven by excitatory GABAergic signaling.
A previous case study (147) highlighted a 50-year-old male patient undergoing alcohol withdrawal. Treatment with 1 mg lorazepam, a GABAAR positive allosteric modulator, resulted in paradoxical behavioral outcomes that included anxiety, agitation, and eccentric behaviors. In conjunction to this human study, several contributions have been made in preclinical models to understand the molecular mechanisms underlying a paradoxical GABAergic effect following alcohol consumption. Previous research (29, 148) shows that alcohol promotes an excitatory switch amongst GABAergic neurons in the ventral tegmental area (VTA). Following both two-bottle choice paradigms and acute ethanol injections, mice displayed a positive shift in the GABA reversal potential as well as dysfunctions in chloride transport in VTA GABAergic neurons. Additional studies have pointed to reductions in KCC2 and pKCC2 expression in the VTA following ethanol exposure (149). Of note, ethanol exposed rodents treated with CLP290, a KCC2 activator, diminished further ethanol consumption, while consumption was enhanced following treatment with VU0240551, a KCC2 inhibitor. These results furthermore highlight the importance of KCC2 expression in maintaining inhibitory GABAergic responses and regulating the reinforcing properties of ethanol.
Imbalances in chloride homeostasis have also been reported in other brain regions following ethanol exposure. Studies have shown increases in NKCC1 expression in the hippocampus (150, 151). Disturbances in excitability and the presence of epileptiform activity have been reported in the limbic system following ethanol exposure (152), however, the previously discussed data hypothesizes an underlying molecular mechanism relating to chloride dyshomeostasis.
Nicotine
Similar to preclinical studies following ethanol exposure, previous research has reported disturbances in GABAergic signaling following nicotine exposure. Reports indicate that nicotine treatment in rodents is followed by a depolarizing shift in GABAergic signaling in VTA GABAergic neurons (153, 154). The presence of excitatory GABAergic responses is supported with results showing a reduction in KCC2 expression within the VTA following nicotine exposure. Interestingly, cotreatment of nicotine and CLP290 restores hyperpolarizing GABAergic signaling and further diminishes self-administration of reinforcing substances. Chronic nicotinic exposure during neonatal development (P1–P5) was additionally shown to increase hippocampal KCC2 mRNA expression in only males, and not females (155), indicating a potential interaction of sex on the effects of nicotine on chloride homeostasis.
Although nicotine’s effect on promoting excitatory GABAergic signaling is primarily hypothesized to be linked to imbalances in chloride homeostasis, it is important to also consider the contributions of nicotinic acetylcholine receptors (nAChRs). In the immature brain, nAChRs aid in transitioning GABAergic signaling from excitatory to inhibitory. Specifically, previous research indicates α7-containing nAChRs (α7-nAChRs) are key players in terminating the depolarizing effects of GABA. In a α7KO preclinical model, the switch from depolarizing GABA is delayed with subsequent increases in NKCC1 and decreases in KCC2 expression in the mature brain. Of note, a loss of α7-nAChRs further results in defects in neuronal migration and maturation within both the central and peripheral nervous system (156, 157).
The α7-nAChRs are highly susceptible to desensitization following chronic exposure to nicotine (158). As previously noted (154), adolescent exposure to nicotine drastically influences neuronal function and drug-seeking behaviors into later stages of adulthood. Nicotine’s effects on these detrimental processes are mediated by disturbing chloride homeostasis during adolescence. Furthermore, it is important to consider the potential effects of nACR desensitization, following chronic nicotine exposure, as an alternative mechanism promoting excitatory GABAergic signaling in the reward neurocircuitry and the overall reinforcing properties of nicotine.
Opiates
Opiates are classically considered a highly addictive substance and exert their effects through the mesolimbic dopaminergic reward pathway (135, 159). The reinforcing properties of opiates on the reward pathways are hypothesized to be linked to excitatory GABAergic signaling in the VTA. Previous studies report a population of VTA GABAergic neurons that depolarize upon GABAAR activation following opiate withdrawal (160–162). The depolarizing GABAergic effects of opiates are also paired with reduced inhibitory GABAergic tone (163), as well as deficits in chloride exportation (164). Moreover, several molecular mechanisms are hypothesized to promote an opiate-induced excitatory switch. One, opiate-dependent/withdrawn models display reduced KCC2 expression in VTA GABAergic neurons (163, 164), reducing extracellular chloride concentration upon GABAAR activation. Two, increased phosphorylation of cAMP Response Element-Binding Protein (CREB) in the VTA during opiate-dependence/withdrawal (160), can promote excitatory actions of GABAARs (165). Three, opiate exposure can elevate carbonic anhydrase activity (160), leading to an accumulation of intracellular bicarbonate ions. Greater intracellular bicarbonate ions are therefore exported from the neuron through activated GABAAR, further resulting in a depolarizing GABAergic response.
Excitatory GABAB receptor signaling and substance use disorders
Acute exposure to ethanol has been previously shown to result in an anti-depressant behavioral effect (Figure 5) (166–168). Patients with AUD are hypothesized to continue drinking in an attempt to avoid negative affect states as the anti-depressive properties of ethanol shift to depressive properties during withdrawal or chronic use (134, 169). Ethanol has been shown to have very similar effects to rapid antidepressants as both antagonize excitatory NMDARs and affect GABAergic neurotransmission (166). Rapid antidepressants that are NMDAR antagonists stabilize GABAB receptors (GABABRs) via an intracellular adaptor protein, 14-3-3η. Stabilization of 14-3-3η leads to the decoupling of GABABRs from inwardly rectifying potassium channels (Kir/GIRK), causing GABABRs to increase dendritic calcium through L- type voltage-gated calcium channels (170). Increasing dendritic calcium additionally activates the mammalian target of rapamycin (mTOR) kinase activity in dendrites. mTOR, which is a serine/threonine protein kinase, promotes the synthesis of plasticity-related proteins, which is required for the antidepressant efficacy of NMDAR antagonists (Figure 6) (170). Studies have demonstrated that blocking GABABRs prevent the effects of rapid anti-depressants, decrease the activity of mTOR, and reduce the synthesis of plasticity-related proteins such as brain-derived neurotrophic factor (BDNF) (171). Thus, an excitatory switch in GABABRs play a critical role in the efficacy of antidepressants by increasing protein synthesis, as well as increasing dendritic calcium signaling (166, 170).
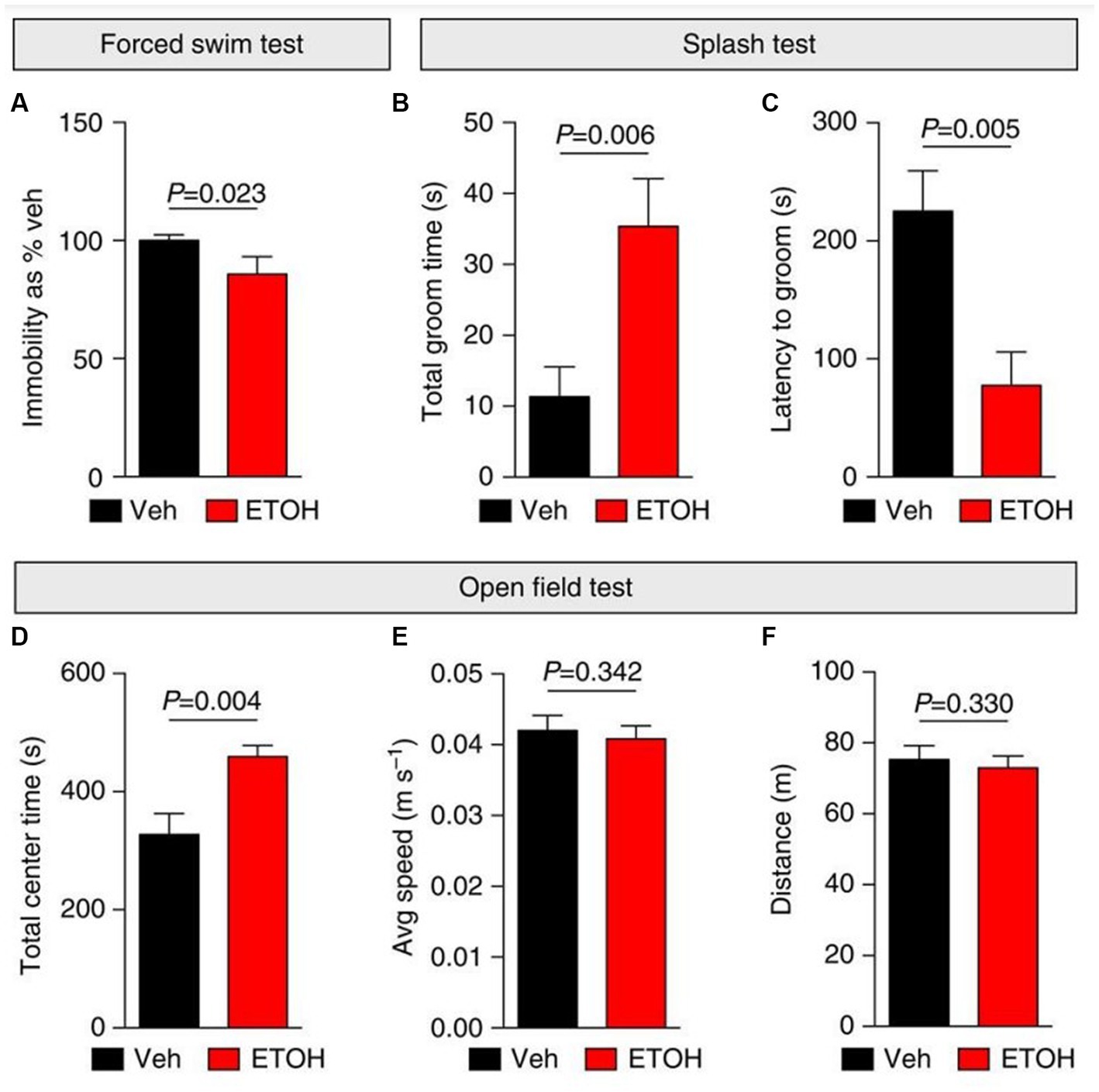
Figure 5. Ethanol elicits anti-depressant behavioral effects in mice. (A–F) Depressive-like (self-care and behavioral despair) and anxiety-like behaviors were measured 24 h following i.p. injections with vehicle (Veh; saline) or ethanol (ETOH; 2.5 g kg−1) in mice. (A) Ethanol elicited anti-depressant behavioral effects by decreasing immobility during a forced swim test (FST) and increasing self-grooming behaviors during a splash test (B,C). (D–F) An open field test was used to measure anxiety-like behaviors following ethanol treatment. (D) Ethanol induced anxiolytic behaviors by increasing the time spent in the center of the open field. Additionally, ethanol did not affect mobility in mice (E,F). Figure reused from (166).
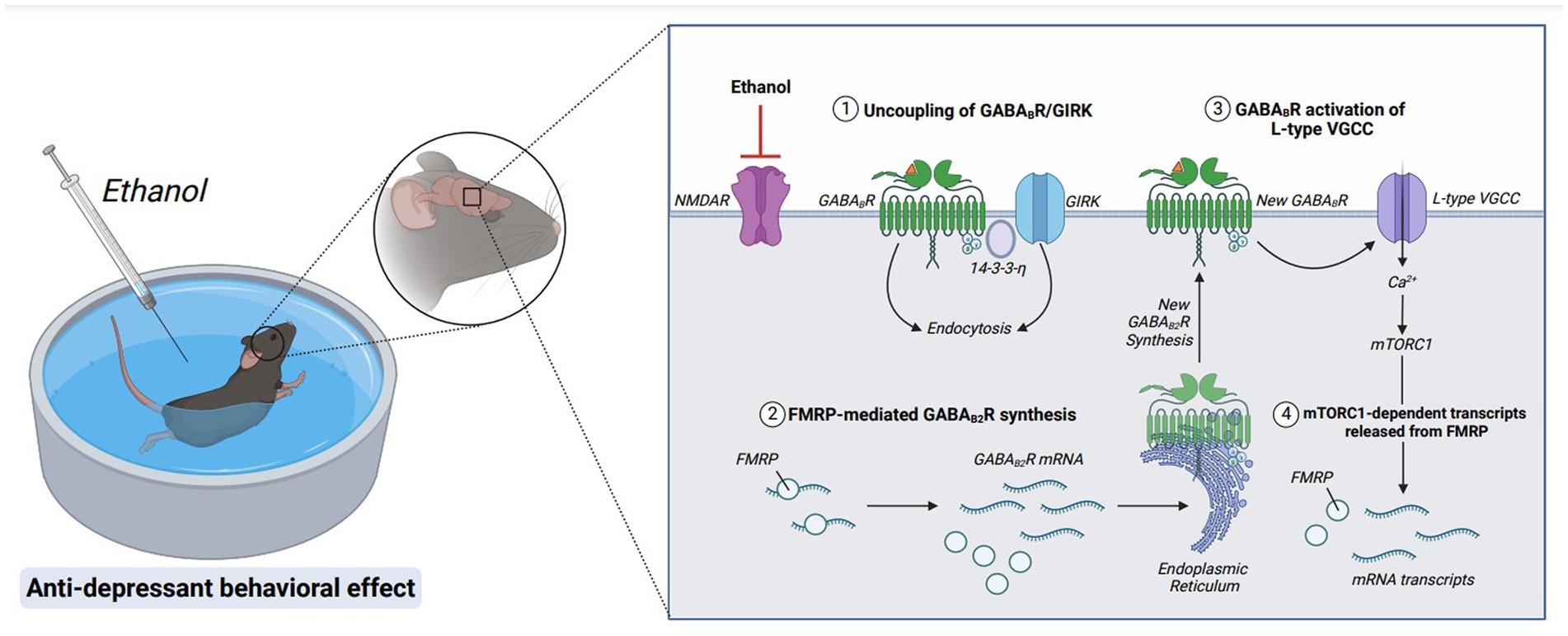
Figure 6. Molecular mechanisms underlying the rapid antidepressant properties of ethanol. Acute ethanol exposure antagonizes NMDAR signaling. GABABRs are then uncoupled from GIRK channels and endocytosed via stabilization of adaptor protein, 14-3-3-η. GABAB2R mRNA, a target of RNA-binding protein FMRP, is released from translational repression, and new GABAB2R protein is synthesized. A new population of surface GABABRs activate L-type voltage-gated calcium channels (L-type VGCC), depolarizing the membrane via calcium influx and activating mTORC1 signaling. Furthermore, mTORC1-dependent transcripts are released from FMRP suppression, upregulating transsynaptic proteins required for anti-depressant behavioral effects. Figure created with BioRender.com.
These data suggest that ethanol exerts its rapid antidepressant effects through similar cellular mechanisms as NMDAR antagonists: promoting a depolarizing response from GABABRs. In addition to altering GABABRs to switch from opening inwardly rectifying potassium channels to increasing dendritic calcium, ethanol also promotes upregulation of GABABRs (166). Of note, the increased surface expression of GABABRs requires FMRP (Figure 6). Without FMRP, the ethanol-induced changes in GABABR expression and excitatory signaling are absent, suggesting that FMRP is important in enhancing the antidepressant properties of ethanol (166).
Moreover, these studies provide a novel mechanism demonstrating an unprecedented role of excitatory GABABR signaling towards the reinforcing properties of ethanol.
Discussion
The data discussed in this review highlights the presence of paradoxical, excitatory GABAergic signaling as a potential contributor toward disease pathology in various neurological disorders. Although excitatory GABAergic responses are critical in the maturation of the nervous system, several disorders can arise due to the prolonged display or inability to terminate depolarizing GABAergic signaling. The predominant mechanism underlying an inability to transition from excitatory to inhibitory GABA across the discussed neurological disorders can be traced to imbalances in chloride ion homeostasis. Moreover, the common denominator contributing to chloride dyshomeostasis is the downregulation of KCC2 and an upregulation of NKCC1. Table 1 outlines potential mechanisms underlying excitatory GABAergic signaling across neurological disorders.
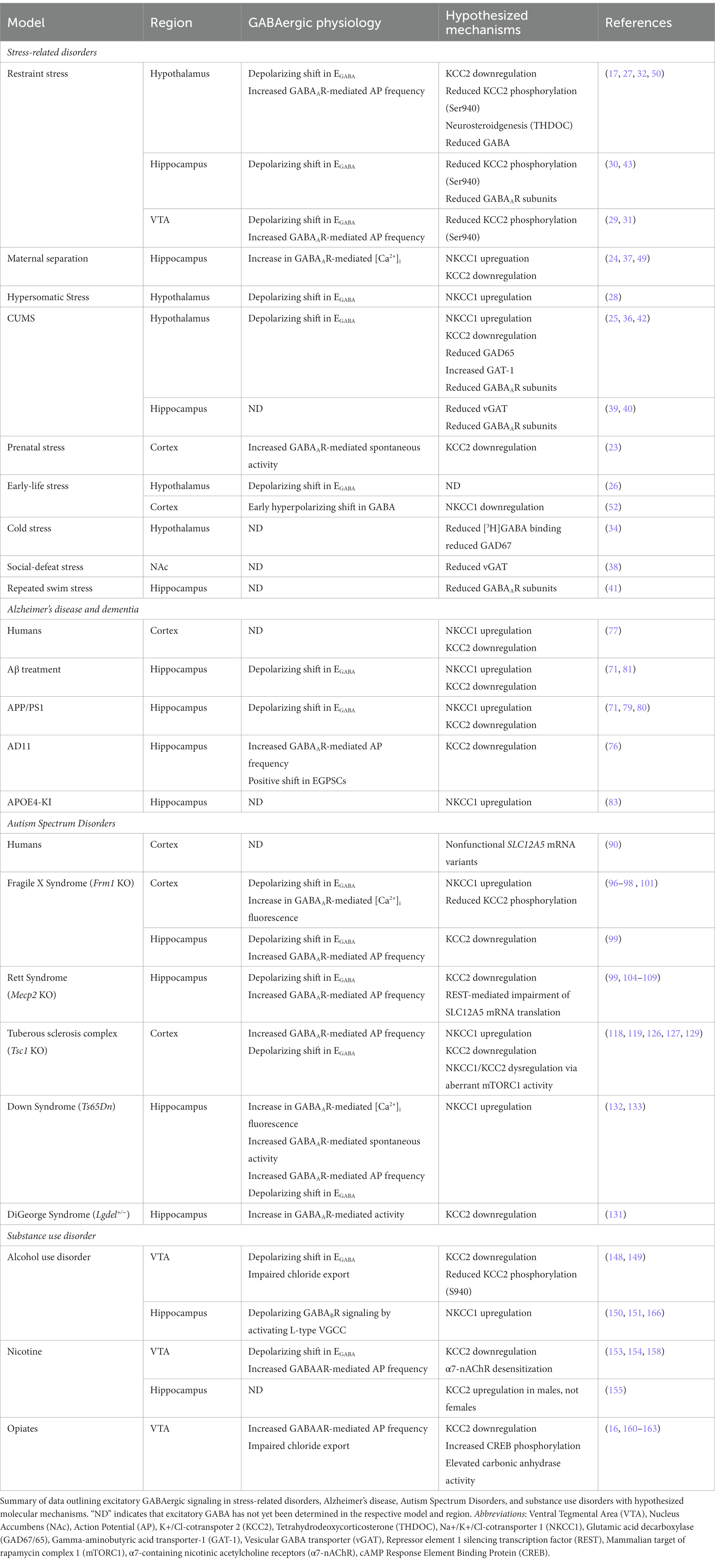
Table 1. Comparing mechanisms underlying excitatory GABAergic signaling in neurological disorders.
Several disorders discussed have comorbid symptoms that could be caused by dysregulated KCC2 and NKCC1 expression. For example, stress is associated with anxiety and substance use disorders (172, 173), cognitive dysfunction is present in both stress-related disorders and AD (174, 175), and epileptogenesis occurs in both autism spectrum disorders (ASD) and Alzheimer’s disease patients (176–178). All these overlapping symptoms could be hypothesized to have common molecular mechanisms linked to KCC2/NKCC1 expression and chloride homeostasis. It is important to note that bumetanide, an NKCC1 inhibitor, has shown positive therapeutic effects in alleviating symptoms in ASD, Alzheimer’s disease, and stress-related disorders (49, 84, 93, 120–122). Furthermore, NKCC1 inhibition could serve as a universal target to prevent pathologies and symptom onset in clinical populations for various neurological disorders.
It is also critical to discuss that while chloride dyshomeostasis could be the predominant cause of excitatory GABAergic signaling in disorders, reductions in transsynaptic GABAergic signaling should also be considered a potential factor. While the data presented in this review outlined the contributions of downregulated GABA signaling towards stress-induced excitatory GABAAR-mediated responses, reductions in GABAergic signaling are also present in Alzheimer’s disease (179), ASD (100, 180), SUD (181, 182). It can be hypothesized that repurposing therapeutics that enhance endogenous levels of GABA could therefore alleviate excitatory GABAAR signaling throughout various disorders.
Although the studies included in this review focused on the excitatory actions of GABA via ionotropic GABAARs, there is a lack of understanding indicating whether metabotropic GABABRs also behave in a similar fashion in neurological disorders. As noted, the antidepressive properties of both ethanol and rapid antidepressants share similar molecular mechanisms in which GABABRs are uncoupled from GIRK channels to L-type voltage-gated calcium channels, resulting in depolarization. Ethanol and rapid antidepressants, moreover, require FMRP to switch GABABRs from inhibitory to excitatory (166, 183). Following exposure to ethanol, FMRP levels are additionally reduced in neuronal dendrites (166). It is important to also include that SLC12A2 mRNA, the transcript encoding NKCC1, is a target of FMRP (95). It could therefore be hypothesized that ethanol-induced downregulation of FMRP could subsequently increase NKCC1 to cause a disturbance in chloride homeostasis, exacerbating the reinforcing properties for ethanol. Previous work (183) presents a novel therapeutic strategy to alleviate synapse loss in a model where FMRP is absent. A combination of both Ro-25-6981 (Ro), a GluN2B-specific antagonist, and CGP35348 (CGP), a GABABR receptor antagonist, increased synapse number and rescued depressive-like symptoms in Fmr1 KO mice (Figure 7). Changes in synapse number and behavior could theoretically be linked to the efficacy of Ro + CGP combinational therapies to rescue chloride ion imbalances via FMRP. Reduced FMRP levels have been shown in the neurological disorders discussed in this review (184–188), but also several other disorders not mentioned such as major depressive disorder (189), Parkinson’s disease (190), epilepsy (185, 191), and schizophrenia (192, 193). Furthermore, a novel combinational therapy antagonizing both NMDARs and GABABRs could universally alleviate pathologies and symptoms across various diseases.
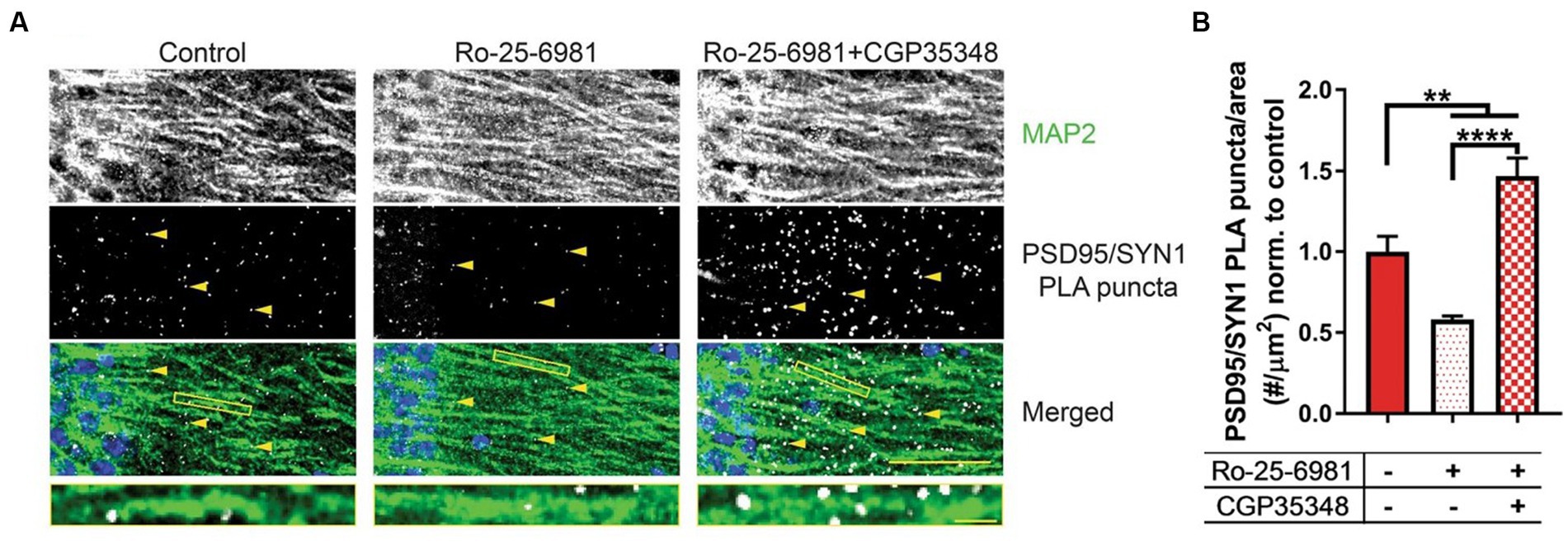
Figure 7. Combinational treatment between Ro-25-6981 and CGP35348 increases hippocampal synapse number in an Fmr1 KO mouse. (A) Representative images of CA1 hippocampal sections from Fmr1 KO mice. MAP 2, a marker of dendrites, is denoted in the first row, while synapses are denoted by the yellow arrows in the second row (PSD95/SYN1 PLA puncta). Synapses are detected in slice using a novel method, DetectSyn, which uses proximity ligation assay (PLA) technology and immunofluorescence to note the presence of synaptic engagement. Bottom row includes a merged image with MAP2 in green, PLA puncta (synapses) in white, and DAPI in blue. (B) Quantification of synapse number following treatments in Fmr1 KO hippocampal slice. Compared to control (red), treatment with Ro-25-6981 (light red), NR2B antagonist, reduced the number of synapses in Fmr1 KO slice. Treatment with Ro-25-6981 and CGP35348, GABABR antagonist, increases synapse number as compared to control (checkered red). Figure reused from (183).
The literature discussed presents an understanding towards the underlying molecular alterations resulting in excitatory GABAergic signaling in neurological disease states. Furthermore, the comorbid mechanisms affecting chloride homeostasis, GABA transsynaptic signaling, and GABAergic plasticity can prompt the repurposing of current FDA-approved therapeutics to alleviate symptom severity and pathogenesis within varieties of neurological disorders.
Author contributions
CM: Writing – original draft, Writing – review & editing. AA: Conceptualization, Writing – original draft. CH: Conceptualization, Writing – original draft. KR-G: Conceptualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. We would like to acknowledge the following grants: NIH NIAAA R01 AA029691 (KR-G), NIH NINDS R01 NS105005 (KR-G), R01 NS105005-03S1 (KR-G); USAMRMC Award W81XWH-14-1-0061 and W81XWH-19-1-0202 (KR-G); Alzheimer’s Association AARG-NTF-21-852843 (KR-G); NIA Wake Forest School of Medicine Alzheimer’s Disease Research Center Pilot Grant P30AG049638 (KR-G), NIAAA T32AA007565 (CH), a grant from FRAXA Research (CH and CM).
Acknowledgments
The authors would like to thank all their colleagues for their contributions and careful review of the manuscript. We additionally would like to thank those colleges for their permission to reuse and republish data from their previous peer-reviewed publications. Sarah Wolfe: FMRP regulates an ethanol-dependent shift in GABABR function and expression with rapid antidepressant properties (2016) – Figure 5. Link to license: https://creativecommons.org/licenses/by/4.0/. Chelcie F. Heaney: Role of FMRP in rapid antidepressant effects and synapse regulation (2021) – Figure 7. Link to license: https://creativecommons.org/licenses/by/4.0/.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Couve, A, Moss, SJ, and Pangalos, MN. GABAB receptors: a new paradigm in G protein signaling. Mol Cell Neurosci. (2000) 16:296–312. doi: 10.1006/mcne.2000.0908
2. Koós, T, and Tepper, JM. Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat Neurosci. (1999) 2:467–72. doi: 10.1038/8138
3. Herbison, AE, and Moenter, SM. Depolarising and hyperpolarising actions of GABA (A) receptor activation on gonadotrophin-releasing hormone neurones: towards an emerging consensus. J Neuroendocrinol. (2011) 23:557–69. doi: 10.1111/j.1365-2826.2011.02145.x
4. Luján, R, Shigemoto, R, and López-Bendito, G. Glutamate and GABA receptor signalling in the developing brain. Neuroscience. (2005) 130:567–80. doi: 10.1016/j.neuroscience.2004.09.042
5. Jones, KA, Borowsky, B, Tamm, JA, Craig, DA, Durkin, MM, Dai, M, et al. GABAB receptors function as a heteromeric assembly of the subunits GABABR1 and GABABR2. Nature. (1998) 396:674–9. doi: 10.1038/25348
6. Ganguly, K, Schinder, AF, and Wong, ST. GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cells. (2001) 105:521–32. doi: 10.1016/S0092-8674(01)00341-5 Poo, M.-m
7. Leinekugel, X, Medina, I, Khalilov, I, Ben-Ari, Y, and Khazipov, R. Ca2+ oscillations mediated by the synergistic excitatory actions of GABAA and NMDA receptors in the neonatal hippocampus. Neuron. (1997) 18:243–55. doi: 10.1016/S0896-6273(00)80265-2
8. Mueller, A, Taube, J, and Schwartzkroin, P. Development of hyperpolarizing inhibitory postsynaptic potentials and hyperpolarizing response to gamma-aminobutyric acid in rabbit hippocampus studied in vitro. J Neurosci. (1984) 4:860–7. doi: 10.1523/JNEUROSCI.04-03-00860.1984
9. Chancey, JH, Adlaf, EW, Sapp, MC, Pugh, PC, Wadiche, JI, and Overstreet-Wadiche, LS. GABA depolarization is required for experience-dependent synapse unsilencing in adult-born neurons. J Neurosci. (2013) 33:6614–22. doi: 10.1523/JNEUROSCI.0781-13.2013
10. Ben-Ari, Y. The GABA excitatory/inhibitory developmental sequence: a personal journey. Neuroscience. (2014) 279:187–219. doi: 10.1016/j.neuroscience.2014.08.001
11. Ben-Ari, Y, Tseeb, V, Raggozzino, D, Khazipov, R, and Gaiarsa, JL. gamma-Aminobutyric acid (GABA): a fast excitatory transmitter which may regulate the development of hippocampal neurones in early postnatal life. Prog Brain Res. (1994) 102:261–73. doi: 10.1016/s0079-6123(08)60545-2
12. Rivera, C, Voipio, J, Payne, JA, Ruusuvuori, E, Lahtinen, H, Lamsa, K, et al. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. (1999) 397:251–5. doi: 10.1038/16697
13. Williams, JR, Sharp, JW, Kumari, VG, Wilson, M, and Payne, JA. The neuron-specific K-Cl cotransporter, KCC2. Antibody development and initial characterization of the protein. J Biol Chem. (1999) 274:12656–64. doi: 10.1074/jbc.274.18.12656
14. Haam, J, Popescu, IR, Morton, LA, Halmos, KC, Teruyama, R, Ueta, Y, et al. GABA is excitatory in adult vasopressinergic neuroendocrine cells. J Neurosci. (2012) 32:572–82. doi: 10.1523/JNEUROSCI.3826-11.2012
15. Sun, D, and Murali, SG. Na+-K+-2Cl− cotransporter in immature cortical neurons: a role in intracellular Cl−regulation. J Neurophysiol. (1999) 81:1939–48. doi: 10.1152/jn.1999.81.4.1939
16. Taylor-Burds, C, Cheng, P, and Wray, S. Chloride Accumulators NKCC1 and AE2 in Mouse GnRH Neurons: Implications for GABAA Mediated Excitation. PLoS One. (2015) 10:e0131076. doi: 10.1371/journal.pone.0131076
17. Maguire, J. Stress-induced plasticity of GABAergic inhibition. Front Cell Neurosci. (2014) 8. doi: 10.3389/fncel.2014.00157
18. Chrousos, GP. Stress and disorders of the stress system. Nat Rev Endocrinol. (2009) 5:374–81. doi: 10.1038/nrendo.2009.106
19. Cullinan, WE, Ziegler, DR, and Herman, JP. Functional role of local GABAergic influences on the HPA axis. Brain Struct Funct. (2008) 213:63–72. doi: 10.1007/s00429-008-0192-2
20. Creutzberg, KC, Sanson, A, Viola, TW, Marchisella, F, Begni, V, Grassi-Oliveira, R, et al. Long-lasting effects of prenatal stress on HPA axis and inflammation: a systematic review and multilevel meta-analysis in rodent studies. Neurosci Biobehav Rev. (2021) 127:270–83. doi: 10.1016/j.neubiorev.2021.04.032
21. Faravelli, C, Lo Sauro, C, Godini, L, Lelli, L, Benni, L, Pietrini, F, et al. Childhood stressful events, HPA axis and anxiety disorders. World J Psychiatry. (2012) 2:13–25. doi: 10.5498/wjp.v2.i1.13
22. Kudielka, BM, Buske-Kirschbaum, A, Hellhammer, DH, and Kirschbaum, C. HPA axis responses to laboratory psychosocial stress in healthy elderly adults, younger adults, and children: impact of age and gender. Psychoneuroendocrinology. (2004) 29:83–98. doi: 10.1016/S0306-4530(02)00146-4
23. Baek, H, Yi, MH, Pandit, S, Park, JB, Kwon, HH, Zhang, E, et al. Altered expression of KCC2 in GABAergic interneuron contributes prenatal stress-induced epileptic spasms in infant rat. Neurochem Int. (2016) 97:57–64. doi: 10.1016/j.neuint.2016.05.006
24. Furukawa, M, Tsukahara, T, Tomita, K, Iwai, H, Sonomura, T, Miyawaki, S, et al. Neonatal maternal separation delays the GABA excitatory-to-inhibitory functional switch by inhibiting KCC2 expression. Biochem Biophys Res Commun. (2017) 493:1243–9. doi: 10.1016/j.bbrc.2017.09.143
25. Gao, Y, Zhou, JJ, Zhu, Y, Kosten, T, and Li, DP. Chronic Unpredictable Mild Stress Induces Loss of GABA Inhibition in Corticotrophin-Releasing Hormone-Expressing Neurons through NKCC1 Upregulation. Neuroendocrinology. (2017) 104:194–208. doi: 10.1159/000446114
26. Gunn, BG, Cunningham, L, Cooper, MA, Corteen, NL, Seifi, M, Swinny, JD, et al. Dysfunctional astrocytic and synaptic regulation of hypothalamic glutamatergic transmission in a mouse model of early-life adversity: relevance to neurosteroids and programming of the stress response. J Neurosci. (2013) 33:19534–54. doi: 10.1523/JNEUROSCI.1337-13.2013
27. Hewitt, SA, Wamsteeker, JI, Kurz, EU, and Bains, JS. Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat Neurosci. (2009) 12:438–43. doi: 10.1038/nn.2274
28. Kim, JS, Kim, WB, Kim, YB, Lee, Y, Kim, YS, Shen, FY, et al. Chronic hyperosmotic stress converts GABAergic inhibition into excitation in vasopressin and oxytocin neurons in the rat. J Neurosci. (2011) 31:13312–22. doi: 10.1523/JNEUROSCI.1440-11.2011
29. Kimmey, BA, Ostroumov, A, and Dani, JA. 5-HT (2A) receptor activation normalizes stress-induced dysregulation of GABAergic signaling in the ventral tegmental area. Proc Natl Acad Sci U S A. (2019) 116:27028–34. doi: 10.1073/pnas.1911446116
30. Mac Kenzie, G, and Maguire, J. Chronic stress shifts the GABA reversal potential in the hippocampus and increases seizure susceptibility. Epilepsy Res. (2015) 109:13–27. doi: 10.1016/j.eplepsyres.2014.10.003
31. Ostroumov, A, Thomas, AM, Kimmey, BA, Karsch, JS, Doyon, WM, and Dani, JA. Stress increases ethanol self-administration via a shift toward excitatory GABA signaling in the ventral tegmental area. Neuron. (2016) 92:493–504. doi: 10.1016/j.neuron.2016.09.029
32. Sarkar, J, Wakefield, S, Mac Kenzie, G, Moss, SJ, and Maguire, J. Neurosteroidogenesis is required for the physiological response to stress: role of neurosteroid-sensitive GABAA receptors. J Neurosci. (2011) 31:18198–210. doi: 10.1523/JNEUROSCI.2560-11.2011
33. Reddy, DS, Kim, H-Y, and Rogawski, MA. Neurosteroid withdrawal model of perimenstrual catamenial epilepsy. Epilepsia. (2001) 42:328–36. doi: 10.1046/j.1528-1157.2001.10100.x
34. Acosta, GB, Otero Losada, ME, and Rubio, MC. Area-dependent changes in GABAergic function after acute and chronic cold stress. Neurosci Lett. (1993) 154:175–8. doi: 10.1016/0304-3940(93)90200-5
35. Losada, MEO. Changes in central GABAergic function following acute and repeated stress. Br J Pharmacol. (1988) 93:483–90. doi: 10.1111/j.1476-5381.1988.tb10302.x
36. Bangsumruaj, J, Kijtawornrat, A, and Kalandakanond-Thongsong, S. Effects of chronic mild stress on GABAergic system in the paraventricular nucleus of hypothalamus associated with cardiac autonomic activity. Behav Brain Res. (2022) 432:113985. doi: 10.1016/j.bbr.2022.113985
37. Dent, G, Choi, DC, Herman, JP, and Levine, S. GABAergic circuits and the stress hyporesponsive period in the rat: Ontogeny of glutamic acid decarboxylase (GAD) 67 mRNA expression in limbic–hypothalamic stress pathways. Brain Res. (2007) 1138:1–9. doi: 10.1016/j.brainres.2006.04.082
38. Heshmati, M, Christoffel, DJ, LeClair, K, Cathomas, F, Golden, SA, Aleyasin, H, et al. Depression and social defeat stress are associated with inhibitory synaptic changes in the nucleus accumbens. J Neurosci. (2020) 40:6228–33. doi: 10.1523/JNEUROSCI.2568-19.2020
39. Tabassum, S, Misrani, A, Huo, Q, Ahmed, A, Long, C, and Yang, L. Minocycline ameliorates chronic unpredictable mild stress-induced neuroinflammation and abnormal mPFC-HIPP oscillations in mice. Mol Neurobiol. (2022) 59:6874–95. doi: 10.1007/s12035-022-03018-8
40. Cullinan, WE, and Wolfe, TJ. Chronic stress regulates levels of mRNA transcripts encoding beta subunits of the GABA (A) receptor in the rat stress axis. Brain Res. (2000) 887:118–24. doi: 10.1016/S0006-8993(00)03000-6
41. Montpied, P, Weizman, A, Weizman, R, Kook, KA, Morrow, AL, and Paul, SM. Repeated swim-stress reduces GABAA receptor alpha subunit mRNAs in the mouse hippocampus. Brain Res Mol Brain Res. (1993) 18:267–72. doi: 10.1016/0169-328X(93)90199-Y
42. Verkuyl, M, Hemby, S, and Joels, M. Chronic stress attenuates GABAergic inhibition and alters gene expression of parvocellular neurons in rat hypothalamus. Eur J Neurosci. (2004) 20:1665–73. doi: 10.1111/j.1460-9568.2004.03568.x
43. Zheng, G, Zhang, X, Chen, Y, Zhang, Y, Luo, W, and Chen, J. Evidence for a role of GABAA receptor in the acute restraint stress-induced enhancement of spatial memory. Brain Res. (2007) 1181:61–73. doi: 10.1016/j.brainres.2007.08.077
44. Wan, L, Chen, L, Yu, J, Wang, G, Wu, Z, Qian, B, et al. Coordinated downregulation of KCC2 and GABAA receptor contributes to inhibitory dysfunction during seizure induction. Biochem Biophys Res Commun. (2020) 532:489–95. doi: 10.1016/j.bbrc.2020.08.082
45. Heubl, M, Zhang, J, Pressey, JC, Al Awabdh, S, Renner, M, Gomez-Castro, F, et al. GABAA receptor dependent synaptic inhibition rapidly tunes KCC2 activity via the Cl−−sensitive WNK1 kinase. Nat Commun. (2017) 8:1776. doi: 10.1038/s41467-017-01749-0
46. Cardarelli, RA, Jones, K, Pisella, LI, Wobst, HJ, McWilliams, LJ, Sharpe, PM, et al. The small molecule CLP257 does not modify activity of the K+–Cl− co-transporter KCC2 but does potentiate GABAA receptor activity. Nat Med. (2017) 23:1394–6. doi: 10.1038/nm.4442
47. Chudotvorova, I, Ivanov, A, Rama, S, Hübner, CA, Pellegrino, C, Ben-Ari, Y, et al. Early expression of KCC2 in rat hippocampal cultures augments expression of functional GABA synapses. J Physiol. (2005) 566:671–9. doi: 10.1113/jphysiol.2005.089821
48. Lee, HH, Deeb, TZ, Walker, JA, Davies, PA, and Moss, SJ. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat Neurosci. (2011) 14:736–43. doi: 10.1038/nn.2806
49. Hu, D, Yu, Z-L, Zhang, Y, Han, Y, Zhang, W, Lu, L, et al. Bumetanide treatment during early development rescues maternal separation-induced susceptibility to stress. Sci Rep. (2017) 7:11878. doi: 10.1038/s41598-017-12183-z
50. Melón, LC, Hooper, A, Yang, X, Moss, SJ, and Maguire, J. Inability to suppress the stress-induced activation of the HPA axis during the peripartum period engenders deficits in postpartum behaviors in mice. Psychoneuroendocrinology. (2018) 90:182–93. doi: 10.1016/j.psyneuen.2017.12.003
51. Lee, HHC, Walker, JA, Williams, JR, Goodier, RJ, Payne, JA, and Moss, SJ. Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2 *. J Biol Chem. (2007) 282:29777–84. doi: 10.1074/jbc.M705053200
52. Karst, H, Droogers, WJ, van der Weerd, N, Damsteegt, R, van Kronenburg, N, Sarabdjitsingh, RA, et al. Acceleration of GABA-switch after early life stress changes mouse prefrontal glutamatergic transmission. Neuropharmacology. (2023) 234:109543. doi: 10.1016/j.neuropharm.2023.109543
53. Vogel, S, and Schwabe, L. Learning and memory under stress: implications for the classroom. NPJ Sci Learn. (2016) 1:16011.
54. Diorio, D, Viau, V, and Meaney, MJ. The role of the medial prefrontal cortex (cingulate gyrus) in the regulation of hypothalamic-pituitary-adrenal responses to stress. J Neurosci. (1993) 13:3839–47. doi: 10.1523/JNEUROSCI.13-09-03839.1993
55. Cochran, DM, Fallon, D, Hill, M, and Frazier, JA. The role of oxytocin in psychiatric disorders: a review of biological and therapeutic research findings. Harv Rev Psychiatry. (2013) 21:219–47. doi: 10.1097/HRP.0b013e3182a75b7d
56. Macdonald, K, and Feifel, D. Helping oxytocin deliver: considerations in the development of oxytocin-based therapeutics for brain disorders. Front Neurosci. (2013) 7. doi: 10.3389/fnins.2013.00035
57. Selles, MC, Fortuna, JTS, de Faria, YPR, Siqueira, LD, Lima-Filho, R, Longo, BM, et al. Oxytocin attenuates microglial activation and restores social and non-social memory in APP/PS1 Alzheimer model mice. iScience. (2023) 26:106545. doi: 10.1016/j.isci.2023.106545
58. Striepens, N, Kendrick, KM, Maier, W, and Hurlemann, R. Prosocial effects of oxytocin and clinical evidence for its therapeutic potential. Front Neuroendocrinol. (2011) 32:426–50. doi: 10.1016/j.yfrne.2011.07.001
59. Leonzino, M, Busnelli, M, Antonucci, F, Verderio, C, Mazzanti, M, and Chini, B. The Timing of the Excitatory-to-Inhibitory GABA Switch Is Regulated by the Oxytocin Receptor via KCC2. Cell Rep. (2016) 15:96–103. doi: 10.1016/j.celrep.2016.03.013
60. Tyzio, R, Cossart, R, Khalilov, I, Minlebaev, M, Hübner, CA, Represa, A, et al. Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science. (2006) 314:1788–92. doi: 10.1126/science.1133212
61. Carson, DS, Berquist, SW, Trujillo, TH, Garner, JP, Hannah, SL, Hyde, SA, et al. Cerebrospinal fluid and plasma oxytocin concentrations are positively correlated and negatively predict anxiety in children. Mol Psychiatry. (2015) 20:1085–90. doi: 10.1038/mp.2014.132
62. Donadon, MF, Martin-Santos, R, and Osório, FL. The associations between oxytocin and trauma in humans: a systematic review. Front Pharmacol. (2018) 9:154. doi: 10.3389/fphar.2018.00154
63. Frijling, JL. Preventing PTSD with oxytocin: effects of oxytocin administration on fear neurocircuitry and PTSD symptom development in recently trauma-exposed individuals. Eur J Psychotraumatol. (2017) 8:1302652. doi: 10.1080/20008198.2017.1302652
64. Goedert, M, and Spillantini, MG. A century of Alzheimer’s disease. Science. (2006) 314:777–81. doi: 10.1126/science.1132814
65. Dickerson, B, Salat, D, Greve, D, Chua, E, Rand-Giovannetti, E, Rentz, D, et al. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology. (2005) 65:404–11. doi: 10.1212/01.wnl.0000171450.97464.49
66. Šišková, Z, Justus, D, Kaneko, H, Friedrichs, D, Henneberg, N, Beutel, T, et al. Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer’s disease. Neuron. (2014) 84:1023–33. doi: 10.1016/j.neuron.2014.10.024
67. Anastacio, TD, Matosin, N, and Ooi, L. Neuronal hyperexcitability in Alzheimer’s disease: what are the drivers behind this aberrant phenotype? Transl Psychiatry. (2022) 12:257. doi: 10.1038/s41398-022-02024-7
68. Yu, T, Liu, X, Wu, J, and Wang, Q. Electrophysiological biomarkers of epileptogenicity in Alzheimer’s disease. Front Hum Neurosci. (2021) 15. doi: 10.3389/fnhum.2021.747077
69. Lerdkrai, C, Asavapanumas, N, Brawek, B, Kovalchuk, Y, Mojtahedi, N, Olmedillas del Moral, M, et al. Intracellular Ca2+ stores control in vivo neuronal hyperactivity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci. (2018) 115:E1279–88. doi: 10.1073/pnas.1714409115
70. Parsons, MP, and Raymond, LA. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron. (2014) 82:279–93. doi: 10.1016/j.neuron.2014.03.030
71. Bie, B, Wu, J, Lin, F, Naguib, M, and Xu, J. Suppression of hippocampal GABAergic transmission impairs memory in rodent models of Alzheimer’s disease. Eur J Pharmacol. (2022) 917:174771. doi: 10.1016/j.ejphar.2022.174771
72. Capsoni, S, and Cattaneo, A. On the molecular basis linking Nerve Growth Factor (NGF) to Alzheimer’s disease. Cell Mol Neurobiol. (2006) 26:619–33. doi: 10.1007/s10571-006-9112-2
73. Capsoni, S, Giannotta, S, and Cattaneo, A. β-Amyloid plaques in a model for sporadic Alzheimer’s disease based on transgenic anti-nerve growth factor antibodies. Mol Cell Neurosci. (2002) 21:15–28. doi: 10.1006/mcne.2002.1163
74. Capsoni, S, Ugolini, G, Comparini, A, Ruberti, F, Berardi, N, and Cattaneo, A. Alzheimer-like neurodegeneration in aged antinerve growth factor transgenic mice. Proc Natl Acad Sci. (2000) 97:6826–31. doi: 10.1073/pnas.97.12.6826
75. Houeland, G, Romani, A, Marchetti, C, Amato, G, Capsoni, S, Cattaneo, A, et al. Transgenic mice with chronic NGF deprivation and Alzheimer’s disease-like pathology display hippocampal region-specific impairments in short-and long-term plasticities. J Neurosci. (2010) 30:13089–94. doi: 10.1523/JNEUROSCI.0457-10.2010
76. Lagostena, L, Rosato-Siri, M, D’Onofrio, M, Brandi, R, Arisi, I, Capsoni, S, et al. In the Adult Hippocampus, Chronic Nerve Growth Factor Deprivation Shifts GABAergic Signaling from the Hyperpolarizing to the Depolarizing Direction. J Neurosci. (2010) 30:885–93. doi: 10.1523/JNEUROSCI.3326-09.2010
77. Barbour, AJ, Gourmaud, S, Lancaster, E, Li, X, Stewart, DA, Irwin, DJ, et al. Excitatory: inhibitory imbalance in Alzheimer’s disease is exacerbated by seizures with attenuation after rapamycin treatment in 5XFAD mice. bio Rxiv, 2023.2003.2002.530499. (2023). doi: 10.1101/2023.03.02.530499
78. Doshina, A, Gourgue, F, Onizuka, M, Opsomer, R, Wang, P, Ando, K, et al. Cortical cells reveal APP as a new player in the regulation of GABAergic neurotransmission. Sci Rep. (2017) 7:370. doi: 10.1038/s41598-017-00325-2
79. Shen, Q, Wu, X, Zhang, Z, Zhang, D, Yang, S, and Xing, D. Gamma frequency light flicker regulates amyloid precursor protein trafficking for reducing β-amyloid load in Alzheimer’s disease model. Aging Cell. (2022) 21:e13573. doi: 10.1111/acel.13573
80. Zhou, Y, Cheng, Y, Li, Y, Ma, J, Wu, Z, Chen, Y, et al. Soluble β-amyloid impaired the GABA inhibition by mediating KCC2 in early APP/PS1 mice. Bio Science Trends. (2021) 15:330–40. doi: 10.5582/bst.2021.01245
81. Lam, P, Vinnakota, C, Guzmán, BC-F, Newland, J, Peppercorn, K, Tate, WP, et al. Beta-amyloid (Aβ1-42) increases the expression of NKCC1 in the mouse hippocampus. Molecules. (2022) 27:2440. doi: 10.3390/molecules27082440
82. Chen, M, Wang, J, Jiang, J, Zheng, X, Justice, NJ, Wang, K, et al. APP modulates KCC2 expression and function in hippocampal GABAergic inhibition. eLife. (2017) 6:e20142. doi: 10.7554/eLife.20142
83. Nova, PW. Enhanced Inhibitory Neurotransmission as a Therapeutic Target in ApoE4-Related Alzheimer’s Disease. San Francisco: University of California (2017).
84. Taubes, A, Nova, P, Zalocusky, KA, Kosti, I, Bicak, M, Zilberter, MY, et al. Experimental and real-world evidence supporting the computational repurposing of bumetanide for APOE4-related Alzheimer’s disease. Nature Aging. (2021) 1:932–47. doi: 10.1038/s43587-021-00122-7
85. Molinaro, G, Battaglia, G, Riozzi, B, Di Menna, L, Rampello, L, Bruno, V, et al. Memantine treatment reduces the expression of the K+/Cl− cotransporter KCC2 in the hippocampus and cerebral cortex, and attenuates behavioural responses mediated by GABAA receptor activation in mice. Brain Res. (2009) 1265:75–9. doi: 10.1016/j.brainres.2009.02.016
86. Levenga, J, Krishnamurthy, P, Rajamohamedsait, H, Wong, H, Franke, TF, Cain, P, et al. Tau pathology induces loss of GABAergic interneurons leading to altered synaptic plasticity and behavioral impairments. Acta Neuropathol Commun. (2013) 1:34. doi: 10.1186/2051-5960-1-34
87. Zheng, J, Li, H-L, Tian, N, Liu, F, Wang, L, Yin, Y, et al. Interneuron accumulation of phosphorylated tau impairs adult hippocampal neurogenesis by suppressing GABAergic transmission. Cell Stem Cell. (2020) 26:331–45. e336. doi: 10.1016/j.stem.2019.12.015
88. Lord, C, Brugha, TS, Charman, T, Cusack, J, Dumas, G, Frazier, T, et al. Autism spectrum disorder. Nat Rev Dis Primers. (2020) 6:5. doi: 10.1038/s41572-019-0138-4
89. Marrosu, F, Marrosu, G, Rachel, MG, and Biggio, G. Paradoxical reactions elicited by diazepam in children with classic autism. Funct Neurol. (1987) 2:355–61.
90. Merner, ND, Chandler, MR, Bourassa, C, Liang, B, Khanna, AR, Dion, P, et al. Regulatory domain or CpG site variation in SLC12A5, encoding the chloride transporter KCC2, in human autism and schizophrenia. Front Cell Neurosci. (2015) 9:386. doi: 10.3389/fncel.2015.00386
91. Ben-Ari, Y, and Cherubini, E. The GABA polarity shift and bumetanide treatment: making sense requires unbiased and undogmatic analysis. Cells. (2022) 11:396. doi: 10.3390/cells11030396
92. Lemonnier, E, Degrez, C, Phelep, M, Tyzio, R, Josse, F, Grandgeorge, M, et al. A randomised controlled trial of bumetanide in the treatment of autism in children. Transl Psychiatry. (2012) 2:e202. doi: 10.1038/tp.2012.124
93. Zhang, L, Huang, C-C, Dai, Y, Luo, Q, Ji, Y, Wang, K, et al. Symptom improvement in children with autism spectrum disorder following bumetanide administration is associated with decreased GABA/glutamate ratios. Transl Psychiatry. (2020) 10:9.
94. Garber, KB, Visootsak, J, and Warren, ST. Fragile X syndrome. Eur J Hum Genet. (2008) 16:666–72. doi: 10.1038/ejhg.2008.61
95. Darnell, JC, Driesche, V, Sarah, J, Zhang, C, and Hung Ka Ying, S. FMRP Stalls Ribosomal Translocation on mRNAs Linked to Synaptic Function and Autism. Cells. (2011) 146:247–61. doi: 10.1016/j.cell.2011.06.013 Mele, A., Fraser, Claire E., … Darnell, Robert B
96. He, Q, Nomura, T, Xu, J, and Contractor, A. The developmental switch in GABA polarity is delayed in fragile X mice. J Neurosci. (2014) 34:446–50. doi: 10.1523/JNEUROSCI.4447-13.2014
97. Brighi, C, Salaris, F, Soloperto, A, Cordella, F, Ghirga, S, de Turris, V, et al. Novel fragile X syndrome 2D and 3D brain models based on human isogenic FMRP-KO iPSCs. Cell Death Dis. (2021) 12:498.
98. Telias, M, Segal, M, and Ben-Yosef, D. Immature responses to GABA in fragile X neurons derived from human embryonic stem cells. Front Cell Neurosci. (2016) 10:121. doi: 10.3389/fncel.2016.00121
99. Tyzio, R, Nardou, R, Ferrari, DC, Tsintsadze, T, Shahrokhi, A, Eftekhari, S, et al. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science. (2014) 343:675–9. doi: 10.1126/science.1247190
100. Song, YJ, Xing, B, Barbour, AJ, Zhou, C, and Jensen, FE. Dysregulation of GABAA receptor-mediated neurotransmission during the auditory cortex critical period in the fragile X syndrome mouse model. Cereb Cortex. (2022) 32:197–215.
101. Smalley, JL, Kontou, G, Choi, C, Ren, Q, Albrecht, D, Abiraman, K, et al. The K-Cl co-transporter 2 is a point of convergence for multiple autism spectrum disorder and epilepsy risk gene products. bio Rxiv. (2020) 2020.2003.2002.973859
102. He, Q, Arroyo, ED, Smukowski, SN, Xu, J, Piochon, C, Savas, JN, et al. Critical period inhibition of NKCC1 rectifies synapse plasticity in the somatosensory cortex and restores adult tactile response maps in fragile X mice. Mol Psychiatry. (2019) 24:1732–47. doi: 10.1038/s41380-018-0048-y
103. Ip, JPK, Mellios, N, and Sur, M. Rett syndrome: insights into genetic, molecular and circuit mechanisms. Nat Rev Neurosci. (2018) 19:368–82. doi: 10.1038/s41583-018-0006-3
104. Banerjee, A, Rikhye, RV, Breton-Provencher, V, Tang, X, Li, C, Li, K, et al. Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome. Proc Natl Acad Sci. (2016) 113:E7287–96. doi: 10.1073/pnas.1615330113
105. Lozovaya, N, Nardou, R, Tyzio, R, Chiesa, M, Pons-Bennaceur, A, Eftekhari, S, et al. Early alterations in a mouse model of Rett syndrome: the GABA developmental shift is abolished at birth. Sci Rep. (2019) 9:9276.
106. Tang, X, Kim, J, Zhou, L, Wengert, E, Zhang, L, Wu, Z, et al. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc Natl Acad Sci. (2016) 113:751–6. doi: 10.1073/pnas.1524013113
107. Duarte, ST, Armstrong, J, Roche, A, Ortez, C, Perez, A, O’Callaghan, M, et al. Abnormal expression of cerebrospinal fluid cation chloride cotransporters in patients with Rett syndrome. PLoS One. (2013) 8:e68851. doi: 10.1371/journal.pone.0068851
108. Hinz, L, Torrella Barrufet, J, and Heine, VM. KCC2 expression levels are reduced in post mortem brain tissue of Rett syndrome patients. Acta Neuropathol Commun. (2019) 7:196. doi: 10.1186/s40478-019-0852-x
109. Ballas, N, et al. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cells. (2005) 121:645–57. doi: 10.1016/j.cell.2005.03.013
110. Tang, X, Drotar, J, Li, K, Clairmont, CD, Brumm, AS, Sullins, AJ, et al. Pharmacological enhancement of KCC2 gene expression exerts therapeutic effects on human Rett syndrome neurons and Mecp2 mutant mice. Sci Transl Med. (2019) 11:eaau 0164.
111. Castro, J, Garcia, RI, Kwok, S, Banerjee, A, Petravicz, J, Woodson, J, et al. Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett Syndrome. Proc Natl Acad Sci. (2014) 111:9941–6. doi: 10.1073/pnas.1311685111
112. Tropea, D, Giacometti, E, Wilson, NR, Beard, C, McCurry, C, Fu, DD, et al. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci. (2009) 106:2029–34. doi: 10.1073/pnas.0812394106
113. Gigliucci, V, Teutsch, J, Woodbury-Smith, M, Luoni, M, Busnelli, M, Chini, B, et al. Region-Specific KCC2 Rescue by rhIGF-1 and Oxytocin in a Mouse Model of Rett Syndrome. Cereb Cortex. (2021) 32:2885–94. doi: 10.1093/cercor/bhab388
114. Bateup, HS, Johnson, CA, Denefrio, CL, Saulnier, JL, Kornacker, K, and Sabatini, BL. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron. (2013) 78:510–22. doi: 10.1016/j.neuron.2013.03.017
115. Henske, EP, Jóźwiak, S, Kingswood, JC, Sampson, JR, and Thiele, EA. Tuberous sclerosis complex. Nat Rev Dis Primers. (2016) 2:16035. doi: 10.1038/nrdp.2016.35
116. Wang, Y, Greenwood, JSF, Calcagnotto, ME, Kirsch, HE, Barbaro, NM, and Baraban, SC. Neocortical hyperexcitability in a human case of tuberous sclerosis complex and mice lacking neuronal expression of TSC1. Ann Neurol. (2007) 61:139–52. doi: 10.1002/ana.21058
117. Wu, X, Sosunov, AA, Lado, W, Teoh, JJ, Ham, A, Li, H, et al. Synaptic hyperexcitability of cytomegalic pyramidal neurons contributes to epileptogenesis in tuberous sclerosis complex. Cell Rep. (2022) 40. doi: 10.1016/j.celrep.2022.111085
118. Ruffolo, G, Iyer, A, Cifelli, P, Roseti, C, Mühlebner, A, van Scheppingen, J, et al. Functional aspects of early brain development are preserved in tuberous sclerosis complex (TSC) epileptogenic lesions. Neurobiol Dis. (2016) 95:93–101. doi: 10.1016/j.nbd.2016.07.014
119. Talos, DM, Sun, H, Kosaras, B, Joseph, A, Folkerth, RD, Poduri, A, et al. Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann Neurol. (2012) 71:539–51. doi: 10.1002/ana.22696
120. Juarez-Martinez, EL, van Andel, DM, Sprengers, JJ, Avramiea, A-E, Oranje, B, Scheepers, FE, et al. Bumetanide effects on resting-state EEG in tuberous sclerosis complex in relation to clinical outcome: an open-label study. Front Neurosci. (2022) 16:879451. doi: 10.3389/fnins.2022.879451
121. Van Andel, D, Sprengers, J, Oranje, B, Scheepers, F, Jansen, F, and Bruining, H. Effects of bumetanide on neurodevelopmental impairments in patients with tuberous sclerosis complex: An open-label pilot study. Mol Autism. (2019, 2020) 11:30.
122. Vlaskamp, C, Poil, S-S, Jansen, F, Linkenkaer-Hansen, K, Durston, S, Oranje, B, et al. Bumetanide as a candidate treatment for behavioral problems in tuberous sclerosis complex. Front Neurol. (2017) 8:469.
123. Inoki, K, Corradetti, MN, and Guan, K-L. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. (2005) 37:19–24. doi: 10.1038/ng1494
124. Magri, L, Cambiaghi, M, Cominelli, M, Alfaro-Cervello, C, Cursi, M, Pala, M, et al. Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell. (2011) 9:447–62. doi: 10.1016/j.stem.2011.09.008
125. Laplante, M, and Sabatini, DM. mTOR Signaling in Growth Control and Disease. Cells. (2012) 149:274–93. doi: 10.1016/j.cell.2012.03.017
126. Niere, F, Namjoshi, S, Song, E, Dilly, GA, Schoenhard, G, Zemelman, BV, et al. Analysis of proteins that rapidly change upon mechanistic/mammalian target of rapamycin complex 1 (mTORC1) repression identifies Parkinson protein 7 (PARK7) as a novel protein aberrantly expressed in tuberous sclerosis complex (TSC). Mol Cell Proteomics. (2016) 15:426–44. doi: 10.1074/mcp.M115.055079
127. Huang, X, McMahon, J, Yang, J, Shin, D, and Huang, Y. Rapamycin down-regulates KCC2 expression and increases seizure susceptibility to convulsants in immature rats. Neuroscience. (2012) 219:33–47. doi: 10.1016/j.neuroscience.2012.05.003
128. Foster, DA, and Toschi, A. Targeting mTOR with rapamycin: One dose does not fit all. Cell Cycle. (2009) 8:1026–9. doi: 10.4161/cc.8.7.8044
129. Demian, WL, Persaud, A, Jiang, C, Coyaud, É, Liu, S, Kapus, A, et al. The Ion Transporter NKCC1 Links Cell Volume to Cell Mass Regulation by Suppressing mTORC1. Cell Rep. (2019) 27:1886–96. doi: 10.1016/j.celrep.2019.04.034
130. Tavazoie, SF, Alvarez, VA, Ridenour, DA, Kwiatkowski, DJ, and Sabatini, BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc 1 and Tsc2. Nat Neurosci. (2005) 8:1727–34. doi: 10.1038/nn1566
131. Amin, H, Marinaro, F, De Pietri Tonelli, D, and Berdondini, L. Developmental excitatory-to-inhibitory GABA-polarity switch is disrupted in 22q11.2 deletion syndrome: a potential target for clinical therapeutics. Sci Rep. (2017) 7:15752. doi: 10.1038/s41598-017-15793-9
132. Lysenko, LV, Kim, J, Madamba, F, Tyrtyshnaia, AA, Ruparelia, A, and Kleschevnikov, AM. Developmental excitatory-to-inhibitory GABA polarity switch is delayed in Ts65Dn mice, a genetic model of Down syndrome. Neurobiol Dis. (2018) 115:1–8. doi: 10.1016/j.nbd.2018.03.005
133. Deidda, G, Parrini, M, Naskar, S, Bozarth, IF, Contestabile, A, and Cancedda, L. Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat Med. (2015) 21:318–26. doi: 10.1038/nm.3827
134. Koob, GF, and Volkow, ND. Neurocircuitry of Addiction. Neuropsychopharmacology. (2010) 35:217–38. doi: 10.1038/npp.2009.110
135. Nestler, EJ. Is there a common molecular pathway for addiction? Nat Neurosci. (2005) 8:1445–9. doi: 10.1038/nn1578
136. Bocklisch, C, Pascoli, V, Wong, JCY, House, DRC, Yvon, C, de Roo, M, et al. Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science. (2013) 341:1521–5. doi: 10.1126/science.1237059
137. Creed, MC, Ntamati, NR, and Tan, KR. VTA GABA neurons modulate specific learning behaviors through the control of dopamine and cholinergic systems. Front Behav Neurosci. (2014) 8:8. doi: 10.3389/fnbeh.2014.00008
138. Guan, Y-Z, and Ye, J-H. Ethanol blocks long-term potentiation of GABAergic synapses in the ventral tegmental area involving μ-opioid receptors. Neuropsychopharmacology. (2010) 35:1841–9. doi: 10.1038/npp.2010.51
139. Johnson, S, and North, R. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. (1992) 12:483–8. doi: 10.1523/JNEUROSCI.12-02-00483.1992
140. Steffensen, SC, Stobbs, SH, Colago, EEO, Lee, R-S, Koob, GF, Gallegos, RA, et al. Contingent and non-contingent effects of heroin on mu-opioid receptor-containing ventral tegmental area GABA neurons. Exp Neurol. (2006) 202:139–51. doi: 10.1016/j.expneurol.2006.05.023
141. Bailey, CP, O’Callaghan, MJ, Croft, AP, Manley, SJ, and Little, HJ. Alterations in mesolimbic dopamine function during the abstinence period following chronic ethanol consumption. Neuropharmacology. (2001) 41:989–99. doi: 10.1016/S0028-3908(01)00146-0
142. George, BE, Dawes, MH, Peck, EG, and Jones, SR. Altered accumbal dopamine terminal dynamics following chronic heroin self-administration. Int J Mol Sci. (2022) 23:8106. doi: 10.3390/ijms23158106
143. Goutaudier, R, Joly, F, Mallet, D, Bartolomucci, M, Guicherd, D, Carcenac, C, et al. Hypodopaminergic state of the nigrostriatal pathway drives compulsive alcohol use. Mol Psychiatry. (2023) 28:463–74. doi: 10.1038/s41380-022-01848-5
144. Siciliano, CA, Ferris, MJ, and Jones, SR. Cocaine self-administration disrupts mesolimbic dopamine circuit function and attenuates dopaminergic responsiveness to cocaine. Eur J Neurosci. (2015) 42:2091–6. doi: 10.1111/ejn.12970
145. Heilig, M, Augier, E, Pfarr, S, and Sommer, WH. Developing neuroscience-based treatments for alcohol addiction: A matter of choice? Transl Psychiatry. (2019) 9:255.
146. Heilig, M, Goldman, D, Berrettini, W, and O’brien, CP. Pharmacogenetic approaches to the treatment of alcohol addiction. Nat Rev Neurosci. (2011) 12:670–84. doi: 10.1038/nrn3110
147. Gonzalez, J, Upadhyaya, VD, Manna, ZT, Sharma, AR, Christopher, J, Douedi, S, et al. Paradoxical excitation following intravenous lorazepam administration for alcohol withdrawal–a case presentation and literature review. J Pharm Pract. (2022), 1244–1248. 08971900221097182
148. Kimmey, BA, Wittenberg, RE, Croicu, A, Shadani, N, Ostroumov, A, and Dani, JA. The serotonin 2A receptor agonist TCB-2 attenuates heavy alcohol drinking and alcohol-induced midbrain inhibitory plasticity. Addict Biol. (2022) 27:e13147. doi: 10.1111/adb.13147
149. Zhang, H, Xu, L, Xiong, J, Li, X, Yang, Y, Liu, Y, et al. Role of KCC2 in the regulation of brain-derived neurotrophic factor on ethanol consumption in rats. Mol Neurobiol. (2023) 60:1040–9. doi: 10.1007/s12035-022-03126-5
150. Santos, LE, Rodrigues, AM, Lopes, MR, Costa, VD, Scorza, CA, Scorza, FA, et al. Long-term alcohol exposure elicits hippocampal nonsynaptic epileptiform activity changes associated with expression and functional changes in NKCC1, KCC2 co-transporters and Na+/K+-ATPase. Neuroscience. (2017) 340:530–41. doi: 10.1016/j.neuroscience.2016.11.015
151. Santos, LEC, da Silveira, GA, Costa, VDC, Batista, AG, Madureira, AP, Rodrigues, AM, et al. Alcohol abuse promotes changes in non-synaptic epileptiform activity with concomitant expression changes in cotransporters and glial cells. PLoS One. (2013) 8:e78854. doi: 10.1371/journal.pone.0078854
152. Costa, VDC, Santos, LEC, Rodrigues, AM, Scorza, FA, Scorza, CA, de Andrade, AG, et al. Alcohol and hippocampal epileptiform activity In: Neuroscience of alcohol : Elsevier (2019). 131–41.
153. Ostroumov, A, Wittenberg, RE, Kimmey, BA, Taormina, MB, Holden, WM, McHugh, AT, et al. Acute nicotine exposure alters ventral tegmental area inhibitory transmission and promotes diazepam consumption. Eneuro. (2020) 7. doi: 10.1523/ENEURO.0348-19.2020
154. Thomas, AM, Ostroumov, A, Kimmey, BA, Taormina, MB, Holden, WM, Kim, K, et al. Adolescent nicotine exposure alters GABAA receptor signaling in the ventral tegmental area and increases adult ethanol self-administration. Cell Rep. (2018) 23:68–77. doi: 10.1016/j.celrep.2018.03.030
155. Damborsky, JC, and Winzer-Serhan, UH. Effects of sex and chronic neonatal nicotine treatment on NKCC1, KCC2, BDNF, NR2A and NR2B mRNA expression in the postnatal rat hippocampus. Neuroscience. (2012) 225:105. doi: 10.1016/j.neuroscience.2012.09.002
156. Campbell, NR, Fernandes, CC, Halff, AW, and Berg, DK. Endogenous signaling through α7-containing nicotinic receptors promotes maturation and integration of adult-born neurons in the hippocampus. J Neurosci. (2010) 30:8734–44. doi: 10.1523/JNEUROSCI.0931-10.2010
157. Liu, Z, Neff, RA, and Berg, DK. Sequential interplay of nicotinic and GABAergic signaling guides neuronal development. Science. (2006) 314:1610–3. doi: 10.1126/science.1134246
158. Giniatullin, R, Nistri, A, and Yakel, JL. Desensitization of nicotinic ACh receptors: shaping cholinergic signaling. Trends Neurosci. (2005) 28:371–8. doi: 10.1016/j.tins.2005.04.009
159. Maldonado, R, Saiardi, A, Valverde, O, Samad, TA, Roques, BP, and Borrelli, E. Absence of opiate rewarding effects in mice lacking dopamine D2 receptors. Nature. (1997) 388:586–9. doi: 10.1038/41567
160. Laviolette, SR, Gallegos, RA, Henriksen, SJ, and van der Kooy, D. Opiate state controls bi-directional reward signaling via GABAA receptors in the ventral tegmental area. Nat Neurosci. (2004) 7:160–9. doi: 10.1038/nn1182
161. Ting-A-Kee, R, and van der Kooy, D. The neurobiology of opiate motivation. Cold Spring Harb Perspect Med. (2012) 2. doi: 10.1101/cshperspect.a012096
162. Vargas-Perez, H, Ting-A-Kee, R, Walton, CH, Hansen, DM, Razavi, R, Clarke, L, et al. Ventral tegmental area BDNF induces an opiate-dependent–like reward state in naïve rats. Science. (2009) 324:1732–4. doi: 10.1126/science.1168501
163. Barbour, AJ, Hauser, KF, McQuiston, AR, and Knapp, PE. HIV and opiates dysregulate K+-Cl− cotransporter 2 (KCC2) to cause GABAergic dysfunction in primary human neurons and Tat-transgenic mice. Neurobiol Dis. (2020) 141:104878. doi: 10.1016/j.nbd.2020.104878
164. Taylor, AM, Castonguay, A, Ghogha, A, Vayssiere, P, Pradhan, AA, Xue, L, et al. Neuroimmune regulation of GABAergic neurons within the ventral tegmental area during withdrawal from chronic morphine. Neuropsychopharmacology. (2016) 41:949–59. doi: 10.1038/npp.2015.221
165. Auger, AP, Perrot-Sinal, TS, and McCarthy, MM. Excitatory versus inhibitory GABA as a divergence point in steroid-mediated sexual differentiation of the brain. Proc Natl Acad Sci. (2001) 98:8059–64. doi: 10.1073/pnas.131016298
166. Wolfe, SA, Workman, ER, Heaney, CF, Niere, F, Namjoshi, S, Cacheaux, LP, et al. FMRP regulates an ethanol-dependent shift in GABABR function and expression with rapid antidepressant properties. Nat Commun. (2016) 7:12867.
167. Jain, NS, Kannamwar, U, and Verma, L. Ethanol induced antidepressant-like effect in the mouse forced swimming test: modulation by serotonergic system. Psychopharmacology. (2017) 234:447–59. doi: 10.1007/s00213-016-4478-4
168. Wolfe, SA, Farris, SP, Mayfield, JE, Heaney, CF, Erickson, EK, Harris, RA, et al. Ethanol and a rapid-acting antidepressant produce overlapping changes in exon expression in the synaptic transcriptome. Neuropharmacology. (2019) 146:289–99. doi: 10.1016/j.neuropharm.2018.11.007
169. Koob, GF, and Moal, ML. Drug abuse: hedonic homeostatic dysregulation. Science. (1997) 278:52–8. doi: 10.1126/science.278.5335.52
170. Workman, ER, Haddick, PC, Bush, K, Dilly, GA, Niere, F, Zemelman, BV, et al. Rapid antidepressants stimulate the decoupling of GABAB receptors from GIRK/Kir 3 channels through increased protein stability of 14-3-3η. Mol Psychiatry. (2015) 20:298–310. doi: 10.1038/mp.2014.165
171. Workman, ER, Niere, F, and Raab-Graham, KF. mTORC1-dependent protein synthesis underlying rapid antidepressant effect requires GABABR signaling. Neuropharmacology. (2013) 73:192–203. doi: 10.1016/j.neuropharm.2013.05.037
172. Butler, TR, Karkhanis, AN, Jones, SR, and Weiner, JL. Adolescent social isolation as a model of heightened vulnerability to comorbid alcoholism and anxiety disorders. Alcohol Clin Exp Res. (2016) 40:1202–14. doi: 10.1111/acer.13075
173. Carlson, HN, and Weiner, JL. Chapter three-the neural, behavioral, and epidemiological underpinnings of comorbid alcohol use disorder and post-traumatic stress disorder In: ES Calipari and NW Gilpin, editors. International review of neurobiology, vol. 157: Academic Press (2021). 69–142.
174. Lazarov, O, and Marr, RA. Of mice and men: neurogenesis, cognition and Alzheimer’s disease. Front Aging Neurosci. (2013) 5:43. doi: 10.3389/fnagi.2013.00043
175. Shansky, RM, and Lipps, J. Stress-induced cognitive dysfunction: hormone-neurotransmitter interactions in the prefrontal cortex. Front Hum Neurosci. (2013) 7:123. doi: 10.3389/fnhum.2013.00123
176. Costa-Mattioli, M, and Monteggia, LM. mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat Neurosci. (2013) 16:1537–43. doi: 10.1038/nn.3546
177. Palop, JJ, Chin, J, Roberson, ED, Wang, J, Thwin, MT, Bien-Ly, N, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. (2007) 55:697–711. doi: 10.1016/j.neuron.2007.07.025
178. Specchio, N, Pietrafusa, N, Trivisano, M, Moavero, R, De Palma, L, Ferretti, A, et al. Autism and epilepsy in patients with tuberous sclerosis complex. Front Neurol. (2020) 11:639.
179. Carello-Collar, G, Bellaver, B, Ferreira, PC, Ferrari-Souza, JP, Ramos, VG, Therriault, J, et al. The GABAergic system in Alzheimer’s disease: a systematic review with meta-analysis. Mol Psychiatry. (2023):1–12.
180. Cellot, G, and Cherubini, E. GABAergic signaling as therapeutic target for autism spectrum disorders. Front Pediatr. (2014) 2:70. doi: 10.3389/fped.2014.00070
181. Alasmari, F, Alotibi, FM, Alqahtani, F, Alshammari, TK, Kadi, AA, Alghamdi, AM, et al. Effects of Chronic Inhalation of Electronic Cigarette Vapor Containing Nicotine on Neurobehaviors and Pre/Postsynaptic Neuron Markers. Toxics. (2022) 10:338. doi: 10.3390/toxics10060338
182. Liang, J, and Olsen, RW. Alcohol use disorders and current pharmacological therapies: the role of GABAA receptors. Acta Pharmacol Sin. (2014) 35:981–93. doi: 10.1038/aps.2014.50
183. Heaney, CF, Namjoshi, SV, Uneri, A, Bach, EC, Weiner, JL, and Raab-Graham, KF. Role of FMRP in rapid antidepressant effects and synapse regulation. Mol Psychiatry. (2021) 26:2350–62. doi: 10.1038/s41380-020-00977-z
184. Bleuzé, L, Triaca, V, and Borreca, A. FMRP-Driven Neuropathology in Autistic Spectrum Disorder and Alzheimer’s disease: A Losing Game. Front Mol Biosci. (2021) 8. doi: 10.3389/fmolb.2021.699613
185. Hagerman, RJ, Berry-Kravis, E, Hazlett, HC, Bailey, DB, Moine, H, Kooy, RF, et al. Fragile X syndrome. Nat Rev Dis Primers. (2017) 3:17065. doi: 10.1038/nrdp.2017.65
186. Krasteva, M, Koycheva, Y, Racheva, R, Taseva, T, Raycheva, T, Simeonova, S, et al. Decreased FMR1 mRNA levels found in men with substance use disorders. Heliyon. (2020) 6. doi: 10.1016/j.heliyon.2020.e05270
187. Sundberg, M, Tochitsky, I, Buchholz, DE, Winden, K, Kujala, V, Kapur, K, et al. Purkinje cells derived from TSC patients display hypoexcitability and synaptic deficits associated with reduced FMRP levels and reversed by rapamycin. Mol Psychiatry. (2018) 23:2167–83. doi: 10.1038/s41380-018-0018-4
188. Uneri, A, Niere, F, Macauley, SL, Ma, T, Keene, CD, Craft, S, et al. mRNA-Binding Protein DJ-1 as a pivotal protein in AD pathology. Alzheimers Dement. (2021) 17:e058602. doi: 10.1002/alz.058602
189. Fatemi, SH, Kneeland, RE, Liesch, SB, and Folsom, TD. Fragile X mental retardation protein levels are decreased in major psychiatric disorders. Schizophr Res. (2010) 124:246–47. doi: 10.1016/j.schres.2010.07.017
190. Tan, Y, Sgobio, C, Arzberger, T, Machleid, F, Tang, Q, Findeis, E, et al. Loss of fragile X mental retardation protein precedes Lewy pathology in Parkinson’s disease. Acta Neuropathol. (2020) 139:319–45. doi: 10.1007/s00401-019-02099-5
191. Hagerman, PJ, and Stafstrom, CE. Origins of epilepsy in fragile X syndrome. Epilepsy Curr. (2009) 9:108–12. doi: 10.1111/j.1535-7511.2009.01309.x
192. Fatemi, SH, Folsom, TD, Rooney, RJ, and Thuras, PD. mRNA and protein expression for novel GABAA receptors θ and ρ2 are altered in schizophrenia and mood disorders; relevance to FMRP-mGluR5 signaling pathway. Transl Psychiatry. (2013) 3:e271. doi: 10.1038/tp.2013.46
Keywords: GABA, depolarizing, excitatory, diseases, GABAARs, neurological
Citation: McArdle CJ, Arnone AA, Heaney CF and Raab-Graham KF (2024) A paradoxical switch: the implications of excitatory GABAergic signaling in neurological disorders. Front. Psychiatry. 14:1296527. doi: 10.3389/fpsyt.2023.1296527
Edited by:
Roberto Ciccocioppo, University of Camerino, ItalyReviewed by:
Holly Hunsberger, Rosalind Franklin University of Medicine and Science, United StatesDaniel Vogt, Michigan State University, United States
Copyright © 2024 McArdle, Arnone, Heaney and Raab-Graham. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kimberly F. Raab-Graham, a3JhYWJncmFAd2FrZWhlYWx0aC5lZHU=