 Aruna Jain
Aruna Jain Elizabeth VanSickle
Elizabeth VanSickle Lia Zitano
Lia Zitano Timothy Moss2
Timothy Moss2- 1College of Human Medicine, Michigan State University College of Human Medicine, Grand Rapids, MI, United States
- 2Medical Genetics, Corewell Health Helen DeVos Children’s Hospital, Grand Rapids, MI, United States
Introduction: We describe a 43-year-old man with neurodevelopmental disorder (NDD) with features of autism spectrum disorder (ASD) due to a rare pathogenic variant in the NCKAP1 gene. There are only 5 young adults described in the literature with NCKAP1-related NDD; there are currently no reports of middle-aged or elderly adults with the condition. The most common clinical characteristics include ASD, intellectual disability (ID), speech-language problems, repetitive behaviors, and seizures.
Conclusion: This case report highlights an adult phenotype of NCKAP1-related NDD with goals to 1.) contribute insight into a rare genetic variant leading to NDD with ASD features and 2.) highlight adult manifestations of NCKAP1-related NDD as a patient in middle adulthood with the condition has not yet been reported.
Introduction
Neurodevelopmental disorders (NDD) are a group of complex conditions from atypical brain development that produce impairments or variations in behavior, cognition and communication (1). Autism spectrum disorder (ASD) is characterized by impaired social communication, restricted and repetitive behaviors, and sensory sensitivities. ASD affects individuals from early childhood and persists throughout their lives, and is an incredibly heterogeneous disorder with a wide range of severity and symptom presentation amongst patients. Thus, ASD is not a single clinical disorder, but rather a broad spectrum of phenotypes, with various genetic and environmental factors contributing to its etiology (2). The genetics of ASD are complex; there has thus far been over 1100 genes identified that are associated with ASD (3). Recent technological advances in genetics have solidified the notion that ASD is a product of a single gene variant, copy number variants, or chromosomal syndromes, rather than of Mendelian inheritance (4, 5). Due to the advent of exome sequencing, the contribution of single nucleotide variants has become clear as a large contributor to the etiology of ASD (6).
In this case report, we present an analysis of an adult with NDD associated with ASD who has a pathogenic single nucleotide variant in the NCKAP1 gene. NCKAP1 is a gene known to play a crucial role in neuronal development and migration as well as in neuronal cytoskeletal dynamics and neuronal differentiation (7). NCKAP1 also is largely involved in formation of protein complexes essential for effective synaptic inhibition as well as actin dynamics (8). NCKAP1 also interacts directly with CYFIP2, which has been linked to intellectual disability and ASD (9, 13). While the exact role of NCKAP1 in the pathogenesis of NDDs and ASD is still not fully understood, research and documentation of NCKAP1 variants as well as literature on intact NCKAP1 supports that it is a gene that is likely to impact NDD and ASD risk. However, compared to other well-known genetic variants that lead to ASD, the literature on NCKAP1 variants is limited, thus the neuromolecular findings on the role of intact NCKAP1 is important to consider as it can help us to better understand why variants may lead to NDDs and ASD.
There are 36 reported individuals who have NCKAP1 gene variants that explain their ASD (3). However, the majority of these cases are in children and adolescents; only 5 are adults, and they are from ages 20-23. Guo et al. (10) reported 21 of these 36 individuals, and they reported the core features of NCKAP1-related NDD to be ASD features, speech-language problems, childhood motor delay, intellectual disability (ID), and learning disabilities. Neuropsychiatric behaviors such as repetitive behavior, aggressive behavior, and attention deficit hyperactivity disorder (ADHD) were present in the majority of their participants as well. The summary and analysis of phenotypes by Guo et al. (10) has been useful in providing insight into the specific manifestations of an NCKAP1 gene variant. This case report highlights an adult patient with a previous diagnosis of ASD, who was diagnosed with NCKAP1-related NDD in adulthood (due to recent inquiry of genetic testing). We aim to provide further insight into the possible clinical manifestations of NCKAP1-related NDD, specifically in an adult patient.
Case presentation
The patient is a 43-year-old male who was adopted from first-degree consanguineous parents and was diagnosed with ASD at 17 years of age. Chart documentation indicates a diagnosis of Asperger’s syndrome at that time; however, the specific diagnostic assessments were not recorded. The delayed diagnosis may be attributed to barriers in accessing healthcare services during childhood. The patient also had been diagnosed with mild cognitive impairment, ID, ADHD, anxiety, and depression. These diagnoses were made through a community mental health agency external to our healthcare system, and detailed records regarding diagnostic criteria, assessment measures, severity, and longitudinal course were not available for review. An IQ score was likewise not recorded in the accessible medical record. Although the onset and severity of anxiety and depression could not be determined, medication history documents initiation of clonazepam and bupropion at 33 years of age. The only available quantitative measure of current symptom burden was a PHQ-4 administered at 41 years of age, which yielded a score of 4, suggestive of mild symptoms at that time.
At his medical genetics appointment, vital measurements were as follows: height 1.676 m (5’6”); weight 108 kg (238 lb); head circumference 55 cm (21.65”); BMI 38.41 kg/m². On physical examination, no craniofacial dysmorphic features were noted. His left upper extremity was larger than his right arm due to boggy subcutaneous tissue leading to increased girth. Neurological examination revealed a facial motor tic, no motor or focal deficits, and normal speech and comprehension.
The facial motor tic was first documented at 36 years of age. He is not receiving pharmacological treatment for the facial motor tic. The patient also had class 2 obesity, essential hypertension, obstructive sleep apnea, left upper extremity lymphedema, hyperlipidemia, and left ventricular hypertrophy (LVH). Obesity, hypertension, and obstructive sleep apnea were first noted at 33 years of age; hyperlipidemia and LVH at 34 years; and lymphedema at 36 years.
LVH was identified on both echocardiogram and cardiac magnetic resonance imaging (MRI).
The patient was diagnosed with non-congenital bilateral sensorineural hearing loss at 39 years of age following a hearing screen. He reported significant improvement with bilateral hearing aids. Audiometry was performed at 41 years of age, though no results were documented; however, his otolaryngologist described the hearing loss as bilateral sensorineural. Computed tomography (CT) of the internal auditory canals and posterior fossa at 40 years of age showed no structural abnormalities, supporting the diagnosis of sensorineural hearing loss. A summary of our patient's phenotypes is described in Table 1.
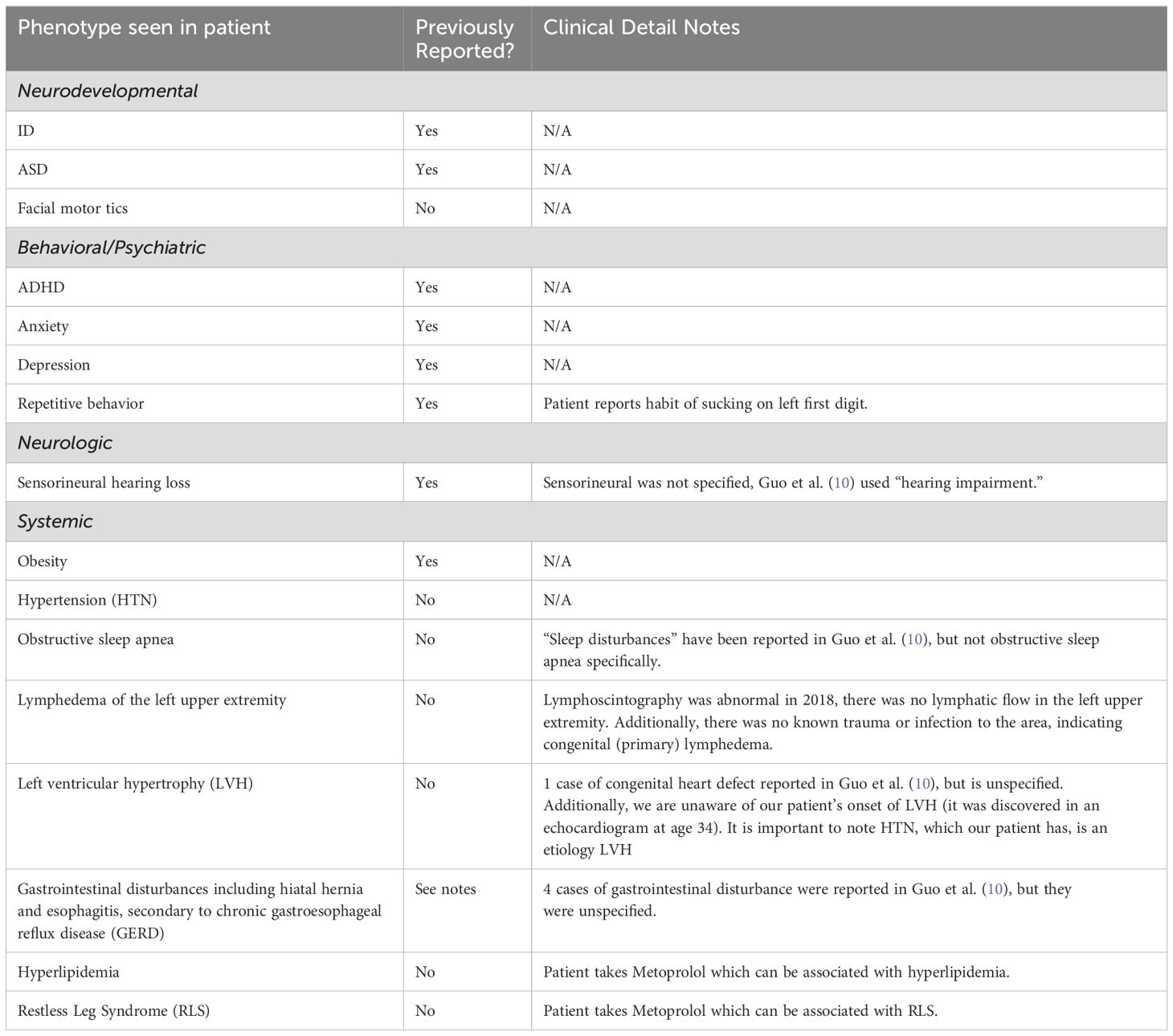
Table 1. Summary of our patient’s phenotypes in comparison to previous reports and unreported clinical manifestations of NCKAP1-Related Neurodevelopmental Disorder.
The patient initially desired genetic testing for a better understanding of his sensorineural hearing loss. Chromosome microarray was performed, and it showed no clinically significant copy number variants. However, the absence of heterozygosity was 18.69%, indicating first-degree relative consanguinity. The patient’s adoptive mother confirmed that his biological parents were siblings, and reported his biological father had a learning disability. Further details about the health status of biological parents is unknown. Due to the fact that the chromosome microarray did not indicate a cause for the patient’s hearing loss, ASD, or other medical conditions combined with the knowledge of his biological parents’ first-degree relationship, the patient and the medical genetics team opted to have exome sequencing performed.
Once the variant in NCKAP1 was identified, the medical genetics team was prompted to further investigate manifestations of NCKAP1 variants in order to best inform him on approaches to management and treatment. Because there is limited research on NCKAP1 variants, interventions taken were focused on preventative care and included patient education and establishing care with specialists. The patient was counseled on the phenotypes reported in the literature, as well as the possibility of future children inheriting this genetic change. The risk of seizures stood out as a potentially urgent clinical feature for our patient to be aware of, and thus our team informed him of this potential risk and signs to monitor. Additionally, he is seeing a cardiologist routinely for his LVH. He also began seeing lifestyle medicine with goals to lose weight and modify his diet. Since the diagnosis, our patient is scheduled to follow up with medical genetics in two years, as no additional interventions beyond coordinating care with other specialties was necessary.
Discussion
Individuals who have variants in NCKAP1 may display variable expressivity, including NDD with ASD, ADHD, ID, neuropsychiatric behaviors, and seizures (10). Our patient’s clinical presentation aligns with some of the common phenotypes reported in literature on NCKAP1 variants. Guo et al. (10) described two brothers, aged 21 and 22 years, with the same variant as our patient, c.2410C>T [p.Arg804*]. The variants were classified as likely pathogenic for the brothers, whereas the clinical significance of our patient’s variant is deemed pathogenic, likely due to the evidence from Guo et al. (10). The phenotypes that overlap between our patient and these brothers include ID and anxiety. Additionally, the proband was noted to be overweight, which aligns with our patient’s clinical profile. However, there are notable differences in the phenotypic expressions. The brothers exhibited features not present in our patient, including speech and language difficulties, aggressive behavior, and tall stature. The proband had seizures and skeletal abnormalities, while his brother had self-injurious behavior and microcephaly—none of which our patient demonstrated. Furthermore, ASD and ADHD, were either absent or unreported in the proband and his brother, respectively, highlighting the variability in expression even among individuals with the same genetic variant.
Many of the non-neurodevelopmental phenotypes observed in our patient have not yet been reported in the existing literature on NCKAP1 variants. We describe them here to provide additional insights into the potential phenotypic spectrum of NCKAP1 variants, particularly as individuals progress into adulthood. Of note, our patient’s presentation of localized lymphedema is significant, as primary lymphedema is a rare congenital condition most commonly associated with Turner syndrome. At 37 years of age, our patient underwent a lymphoscintigraphy study, which demonstrated absent lymphatic flow in the left upper extremity, consistent with congenital lymphedema. The patient reported no history of trauma, surgery, or infection involving the affected limb that might otherwise account for secondary lymphedema. He has experienced recurrent flares of lymphedema throughout his life, further supporting a diagnosis of primary lymphedema. Lymphedema associated with NCKAP1 variants has not been previously reported. Based on the patient’s clinical presentation, absence of alternative etiologies, and lack of other pathogenic genetic variants, we propose localized lymphedema as a potential associated phenotype of NCKAP1 variation.
Our patient’s diagnoses of hyperlipidemia, hiatal hernia, LVH, hypertension, and obstructive sleep apnea could potentially be secondary to obesity as well as advancing in age. Thus, we cannot say with certainty that he developed these diagnoses solely due to a NCKAP1 variation. This highlights a limitation in the internal validity of our observations– namely, that multiple plausible etiologies exist for his findings, making it difficult to isolate the effect of the NCKAP1 variant. However, obesity is supported as an association of NCKAP1 variation in past reports, and thus, these conditions are essential to highlight, as screening for obesity-related comorbidities may be considered advisable. The external validity of our findings is limited by the fact that adult phenotypes associated with NCKAP1 variation remain poorly characterized. Longitudinal follow-up studies, such as tracking the participants from Guo et al. (10) into adulthood, would strengthen the generalizability of observations like ours. More comprehensive data could help determine whether obesity and its related health outcomes are consistently part of the NCKAP1-related NDD spectrum or more coincidental in isolated cases.
It is also important to consider that our patient’s significant consanguinity, identified through microarray analysis and confirmed by his adoptive mother, may contribute to his phenotypic features through mechanisms that are not fully understood. This represents a potential confounding factor, as his clinical presentation may result from genetic influences independent of, or interacting with, the NCKAP1 variant.
In addition to providing more insight into the adult clinical manifestations of NCKAP1 variants, we wanted to note how this finding of the variant can be highlighted during genetic counseling of patients. Guo et al. (10) and Anazi et al. (11) show support for the variant arising de novo or inherited in the autosomal dominant pattern, so this would be a beneficial point to raise with patients as they consider having children of their own. Another point for genetic counselors to convey in the interest of informed decision-making and education is the variable expressivity of NCKAP1-related NDD seen in the literature, as limited information on the condition is available to the public.
Finally, this case highlights the fact that many adult patients with NDDs, even those diagnosed earlier in life, often have not undergone genetic testing. As a result, they may not have received condition-specific education, treatment, or preventative measures. As genetic testing becomes more accessible, the development of targeted intervention strategies for associated phenotypes, including preventative measures and specialty care not typically included in general health maintenance guidelines, becomes possible.
Exome sequencing, which identifies specific underlying gene variants responsible for NDDs, has proven beneficial for our patient’s treatment and for advancing the clinical understanding of the NDD spectrum (12). During discussions of the previously reported features of NCKAP1-related NDD, our patient shared that this aligns with his own experiences and feels that this explains the challenges he has faced. He understood the importance of following up with specialists and to seek care if new concerns, such as seizures, arise. He expressed enthusiasm and gratitude for having a biological explanation for his conditions and has a desire to connect with others affected by NCKAP1-related NDD.
Methods
Exome sequencing was performed with GeneDx, and a single nucleotide variant was identified in the NCKAP1 NM_205842 gene (c.2410 C>T in exon 23 p. [Arg804*]. This variant causes truncation in exon 24 in NCKAP1. This was interpreted by GeneDx as a pathogenic variant. The differential diagnosis supported by the GeneDx report as well as our patients phenotypes is NCKAP1-related neurodevelopmental disorder. Single nucleotide variants in NCKAP1 have been reported in individuals with NCKAP1-related neurodevelopmental and neuropsychiatric disorders. Given this knowledge and the absence of other variants in exome sequencing, this patient’s NCKAP1 variant was thought to be causative for his clinical symptoms and his diagnosis was felt to be consistent with a NCKAP1-related NDD. Our patient’s biological parents were not available for testing, so we were unable to determine if this patient’s NCKAP1 variant was inherited or de novo.
Conclusion
We describe the phenotype of an adult man who has NKCAP1-related NDD. By conducting exome sequencing and comparing his phenotype with the current literature, we were able to pinpoint the involvement of the NCKAP1 gene in our patient’s NDD. The identification of NCKAP1 as a risk gene for NDD highlights the need for continued research and reporting of its clinical presentation. Our patient’s case provides continued evidence for the association of NCKAP1 gene variants leading to NDD associated with ASD, as well as unreported adult phenotypes. This finding not only has the potential to contribute to the growing body of knowledge surrounding the pathogenicity of NCKAP1 variants, but it also helped to provide better self-understanding for our patient.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
Ethical approval was not required for the studies involving humans because this case only involved one patient, thus it was exempt from the requirement for IRB approval. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
AJ: Writing – original draft, Writing – review & editing. EV: Writing – review & editing. LZ: Writing – review & editing. TM: Writing – review & editing. ES: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Funding for publication was provided by Corewell Health Research Institute.
Acknowledgments
We would like to thank our patient and his family for participating in this research and the Corewell Health Medical Genetics Team.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Thapar A, Cooper M, and Rutter M. Neurodevelopmental disorders. Lancet Psychiatry. (2017) 4:339–46. doi: 10.1016/S2215-0366(16)30376-5
2. Tordjman S, Somogyi E, Coulon N, Kermarrec S, Cohen D, Bronsard G, et al. Gene×environment interactions in autism spectrum disorders: Role of epigenetic mechanisms. Front Psychiatry. (2014) 5:53. doi: 10.3389/fpsyt.2014.00053
3. SFARI human Gene Module (2024). Available online at: https://gene.sfari.org/database/human-gene%20-/ (Accessed November 22, 2024).
4. Codina-Solà M, Rodríguez-Santiago B, Homs A, Santoyo J, Rigau M, Aznar-Laín G, et al. Integrated analysis of whole-exome sequencing and transcriptome profiling in males with autism spectrum disorders. Mol Autism. (2015) 6:21. doi: 10.1186/s13229-015-0017-0
5. Rosti RO, Sadek AA, Vaux KK, and Gleeson JG. The genetic landscape of autism spectrum disorders. Dev Med Child Neurol. (2014) 56:12–8. doi: 10.1111/dmcn.12278
6. Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. (2012) 485:237–41. doi: 10.1038/nature10945
7. Yokota Y, Ring C, Cheung R, Pevny L, and Anton ES. Nap1-regulated neuronal cytoskeletal dynamics is essential for the final differentiation of neurons in cerebral cortex. Neuron. (2007) 54:429–45. doi: 10.1016/j.neuron.2007.04.016
8. Smalley JL, Kontou G, Choi C, Ren Q, Albrecht D, and Abiraman K. The K-Cl co-transporter 2 is a point of convergence for multiple autism spectrum disorder and epilepsy risk gene products. bioRxiv. (2020). doi: 10.1101/2020.03.02.973859
9. Zweier M, Begemann A, McWalter K, Cho MT, Abela L, Banka S, et al. Spatially clustering de novo variants in CYFIP2, encoding the cytoplasmic FMRP interacting protein 2, cause intellectual disability and seizures. Eur J Hum Genet. (2019) 27:747–59. doi: 10.1038/s41431-018-0331-z
10. Guo H, Zhang Q, Dai R, Yu B, Hoekzema K, and Tan J. NCKAP1 disruptive variants lead to a neurodevelopmental disorder with core features of autism. Am J Hum Genet. (2020) 107:963–76. doi: 10.1016/j.ajhg.2020.10.002
11. Anazi S, Maddirevula S, Asi YT, Alsahli S, Alhashem A, and Shamseldin HE. Expanding the genetic heterogeneity of intellectual disability. Hum Genet. (2017) 136:1419–29. doi: 10.1007/s00439-017-1843-2
12. Srivastava S, Love-Nichols JA, Dies KA, Ledbetter DH, Martin CL, and Chung WK. Meta-analysis and multidisciplinary consensus statement: Exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. (2019) 21:2413–21. doi: 10.1038/s41436-019-0554-6
Keywords: NCKAP1, neurodevelopmental disorder, autism spectrum disorder, nonsense variant, case report
Citation: Jain A, VanSickle E, Zitano L, Moss T and Schrader E (2025) Case Report: An adult with NCKAP1-related neurodevelopmental disorder and autism spectrum disorder. Front. Psychiatry 16:1532982. doi: 10.3389/fpsyt.2025.1532982
Received: 22 November 2024; Accepted: 16 May 2025;
Published: 09 June 2025.
Edited by:
Joana M. Gaspar, Federal University of Santa Catarina, BrazilReviewed by:
Magdalena Budisteanu, Prof. Dr. Alexandru Obregia Psychiatry Hospital, RomaniaAndrew Sobering, Augusta University/University of Georgia Medical Partnership, United States
Copyright © 2025 Jain, VanSickle, Zitano, Moss and Schrader. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aruna Jain, amFpbmFydTFAbXN1LmVkdQ==