 Maximiliano Elgueta-Reyes1,2*
Maximiliano Elgueta-Reyes1,2* Sergio Hidalgo
Sergio Hidalgo Jorge M. Campusano
Jorge M. Campusano- 1Centro Interdisciplinario de Neurociencia UC, Facultad de Ciencias Biológicas, Pontificia Universidad Católica de Chile, Santiago, Chile
- 2Facultad de Ciencias Biológicas, Pontificia Universidad Católica de Chile, Santiago, Chile
- 3Department of Integrative Physiology and Neuroscience, Washington State University, Pullman, WA, United States
Schizophrenia is a complex neuropsychiatric disorder characterized by positive, negative, and cognitive symptoms. While positive symptoms have been extensively studied, negative symptoms—such as anhedonia, social withdrawal, and apathy—remain challenging to model and treat. Vertebrate animal models for schizophrenia have provided insights into some of the underlying mechanisms associated with this disorder. Recently, Drosophila melanogaster has emerged as a valuable model due to its genetic tractability, conserved neurochemical pathways as compared to vertebrates, and suitability for high-throughput behavioral analyses. Mutations in genes such as dysb1, Rim, and Neuroligins have been linked to behaviors in flies resembling negative symptoms of schizophrenia, supporting the relevance of this animal model in psychiatric research. Moreover, behavioral paradigms aimed at assessing social interaction, motivation, and anhedonia in Drosophila are being refined to better capture schizophrenia-related deficits. The use of Drosophila enables precise investigation of neural circuits and molecular pathways underlying negative symptoms of schizophrenia, research that has the potential to lead to novel therapeutic targets.
Introduction
Schizophrenia is a complex and multidimensional neuropsychiatric disorder, affecting approximately 1% of the global population, which exhibits a higher prevalence in males (1–3). Globally, costs associated to schizophrenia are estimated between US$94 and US$102 billion annually. This represents an economic burden equivalent to 0.02% to 1.65% of a country’s gross domestic product (GDP), with indirect costs—such as lost productivity and social security expenses—accounting for 50% to 85% of that total (4, 5). This is relevant as the health, social, and economic burden associated to this disorder is substantial, impacting patients but also families, caregivers and society at large.
By 1908, Eugen Bleuler first introduced the term “schizophrenia”, describing personality, perception, and cognitive symptoms in a group of patients (6). Schizophrenia was later categorized into positive (hallucinations, delusions) and negative symptoms (blunted affect, avolition, anhedonia, asociality, and alogia) (6–8). It is currently known that schizophrenia also involves cognitive impairment, including alterations in language, executive function, verbal memory, spatial memory, among other features (9, 10) (Figure 1).
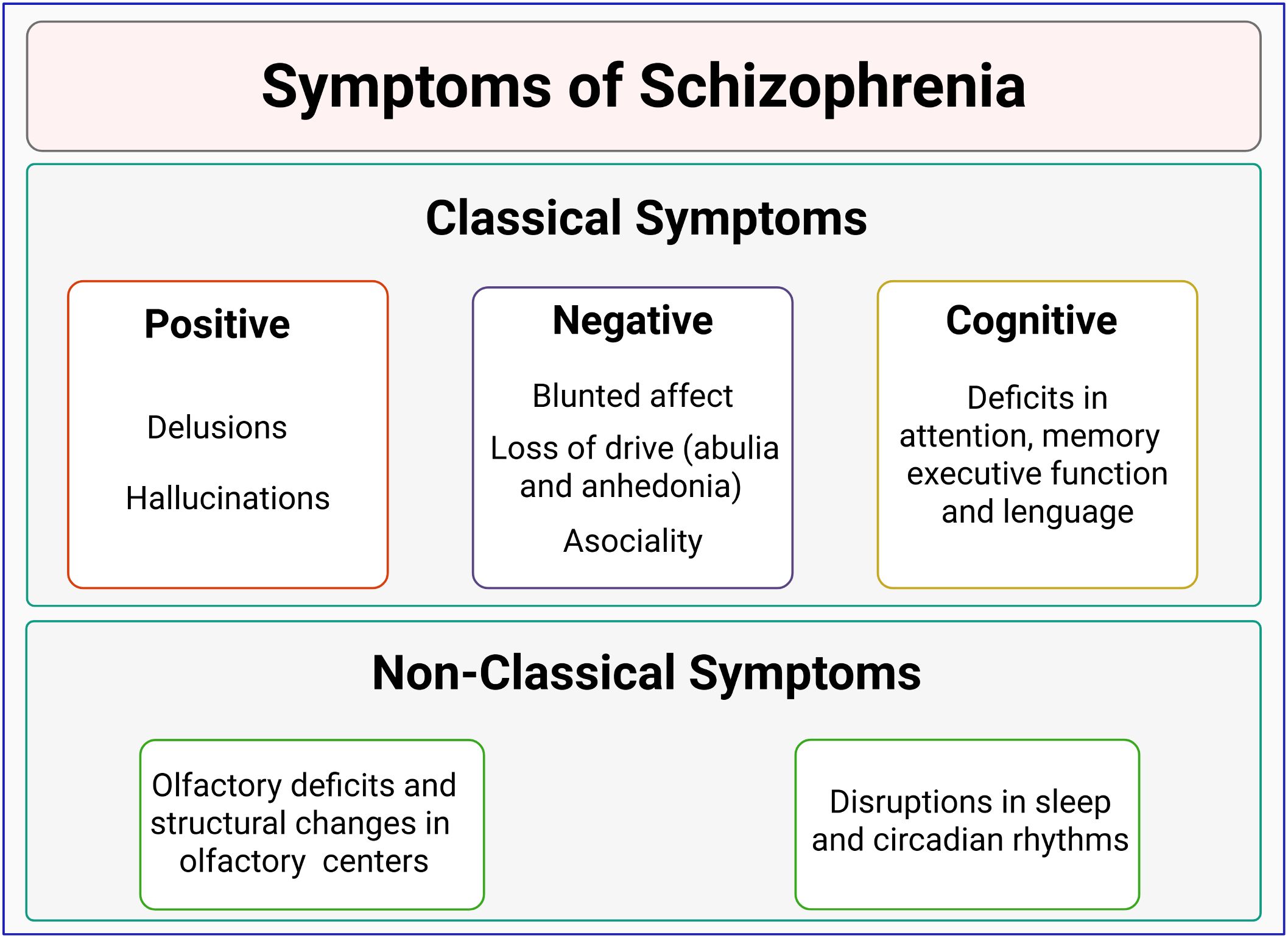
Figure 1. Classification of schizophrenia symptoms. Schizophrenia is characterized by positive symptoms (such as delusions and hallucinations) and negative symptoms (including blunted affect, poverty of speech, and anhedonia). Cognitive impairments, such as deficits in language, memory, and executive function, are also common. Non-classical symptoms, involving olfactory discrimination deficits and sleep/circadian disruptions, are emerging as potential prodromal markers of schizophrenia.
On the other hand, non-classical symptoms, such as olfactory impairments (11) and circadian disruptions (12), have been observed in 80% of schizophrenia cases (12–14), and have gained attention as prodromal symptoms or markers of this disorder (Figure 1).
The study of schizophrenia has largely focused on positive symptoms due to the effectiveness of antipsychotics on them (15, 16). However, although negative symptoms seem critical in determining the loss in the quality of life of people with a diagnosis of schizophrenia, they remain a major therapeutic challenge.
Negative symptoms of schizophrenia
Negative symptoms involve behavioral features that are absent or undermined in patients. They are classified into two primary domains: abulia/apathy and diminished emotional expression (17, 18). The first domain is understood as deficits in motivation and pleasure. It involves reduced motivation and goal-directed behavior and decreased pleasure when facing positive experiences (17). Thus, this domain includes individual symptoms (or subdomains) of abulia, asociality, and anhedonia (19).
The second domain involves a decrease in the external expression of emotions (blunted affect) and speech (alogia) (18). Blunted affect or affective flattening is linked with diminished quality of life, depressive symptoms, poor social functioning, emotional withdrawal, negative self-evaluation, and suicide ideation (20), while alogia has been associated with cognitive deficits, such as alterations in semantic memory (21).
Importantly, negative symptoms of schizophrenia are little responsive to dopaminergic agents, which are more effective towards positive symptoms of this disorder (22). Thus, there is a need for a better comprehension of the mechanisms underlying the negative symptoms of schizophrenia, in the search for new treatments and therapeutical approaches.
Schizophrenia etiology and negative symptoms
The etiology of this disorder involves multifactorial elements ranging from genetic features to risk factors in brain development to environmental influences, which accumulate and interact to produce a wide range of symptoms, mainly in adolescence and youth (23, 24).
Several genetic linkage and GWAS studies have tried to identify genes that could play a role in the disorder, and some of these reports have pointed out a genetic contribution to negative symptoms. Thus, for instance, a strong link has been found between negative symptoms of schizophrenia and chromosome 22q11 microdeletions, as well as with alterations in the NKAIN2 gene, which encodes a protein that interacts with subunits of the sodium/potassium ATPase (25). Additionally, these studies have identified an association between haplotypes of the DTNBP1 gene (Dystrobrevin Binding Protein 1, also known as Dysbindin-1), and cognitive and negative symptoms of the disorder (26–28). Notably, dysbindin-1 deficiency affects glutamatergic, GABAergic, and dopaminergic neurotransmission (29, 30), some of the neurochemical systems mostly associated with schizophrenia etiology (31). Similarly, haplotypes and polymorphisms in the gene that encodes COMT, an enzyme involved in dopamine metabolism, have been linked to the severity of schizoaffective negative symptoms (32–35).
The serotonergic system plays a well-established role in regulating mood and affect, some of the features associated with negative symptoms of schizophrenia. Considering this, it was proposed that the serotonergic neurochemical system could play a role in these symptoms and early studies supported this idea (36). Accordingly, pharmacological treatments targeting serotonin receptors have been shown to prevent the loss of gray matter typically observed in schizophrenia patients and to improve cognitive and negative symptoms of this disorder (37, 38).
Importantly, most of these studies support that the interaction of genetic and environmental factors during early neurodevelopment contributes to brain vulnerability and predisposition to develop schizophrenia (31, 39). However, what is the contribution of genes and environment, or what are the exact mechanisms responsible for this effect, is an open question that is difficult to study in humans. In this regard, animal models seem better suited to advance on this issue (40).
Animal models in the study of negative symptoms of schizophrenia
Despite the inherent limitations of studying a complex human disorder like schizophrenia in vertebrate animal models, research in non-human primates, rodents, and zebrafish has provided valuable insights into the cellular, molecular, and circuit-level underpinnings of some behavioral features of schizophrenia (41–43). Positive symptoms, for example, are often modeled through non-verbal indicators such as hyperlocomotion or stereotypy, while negative symptoms are inferred from behaviors like impaired thigmotaxis, reduced exploration, or diminished social interaction (44).
Rather than replicating the full disorder, animal studies focus on isolating specific symptoms or symptom clusters to explore their underlying causes (45, 46). This strategy has been instrumental in identifying or understanding environmental, genetic, and pharmacological factors contributing to schizophrenia, helping to dissect the complex interplay of elements involved in its pathophysiology (Figure 2).

Figure 2. Animal models in the study of negative symptoms of schizophrenia. Experimental models are categorized into three main approaches: neurodevelopmental, pharmacological, and genetic models. Neurodevelopmental models involve prenatal and postnatal stress paradigms or maternal malnutrition during gestation. Pharmacological models utilize NMDA receptor and serotonin receptor antagonists to induce schizophrenia-like phenotypes. Genetic models include mutations in schizophrenia-associated genes such as DISC1, DTNBP1 (dysbindin), COMT, and deletions in the 22q11.2 region. These models contribute to understanding the neurobiological basis of schizophrenia and its negative symptoms.
Genetic models in mice to generate schizophrenia-like symptoms include mutants for the DISC1, DTNBP1, and COMT genes (47–50), as well as deletions in the equivalent to 22q11.2 region, among others (51, 52). Most of these tools have been successful in modeling some of the positive symptoms of the disorder.
Environmental models primarily focus on neurodevelopmental disruptions, such as prenatal and postnatal stress paradigms, including maternal exposure to adverse conditions that elevate corticosterone levels, maternal malnutrition during gestation, and maternal separation, resulting in behavioral alterations and schizophrenia-related symptoms in the offspring (53–55).
Pharmacological models offer another widely used approach, employing acute or chronic exposure of animals to specific compounds. For instance, administration of methamphetamine or amphetamine induces hyperlocomotion and stereotypy in rodents, mimicking positive symptoms of schizophrenia and providing support for the dopaminergic hypothesis of this disorder (56, 57). However, these models poorly replicate negative symptoms of the disorder (58, 59). To overcome this limitation, researchers have used NMDA receptor antagonists, such as phencyclidine (PCP) and MK-801, which can induce negative-like symptoms, including social withdrawal, reduced social interaction, and increased immobility in the forced swim test (60–62).
Another pharmacological approach to model negative symptoms of schizophrenia is based on the serotonergic hypothesis for this disorder. This is based, as stated above, on the fact that serotonin dysregulation induces behavioral features that resemble negative symptoms of this disorder and that serotonergic agents show some efficacy against schizophrenia’s negative symptoms (37, 38). Thus, serotonin receptor antagonists, alone or in combination with glutamatergic antagonists or dopaminergic agonists, have been used to generate rodent models for negative symptoms of schizophrenia (63–65) (Figure 2).
Although vertebrate models have provided valuable insights into the neurobiological basis of schizophrenia, many of these approaches, particularly pharmacological and neurodevelopmental models, carry the risk of inducing widespread, non-specific alterations in several body organs, multiple neural circuits, and signaling pathways (66–68). Thus, if the goal of these animal models is to understand the contribution of specific circuits or neurochemical systems to the disorder, these global approaches might hinder the precise dissection of the mechanisms contributing to the onset and progression of schizophrenia. Moreover, they might lead to the misidentification of contributing factors. In this regard, the use of models that allow for a more precise spatial and temporal dissection of neural activity and dysfunction is necessary.
Drosophila models for schizophrenia and the study of negative symptoms
In the study of complex human disorders, the use of invertebrate models including Drosophila melanogaster, has gained increasing attention. Drosophila offers several advantages as an animal model for the study of disorders, including its fully sequenced genome (69), its short life cycle, the possibility to obtain and study a large number of animals, and a high proportion of conserved genes when compared to the human genome (70, 71). Notably, approximately 75% of human disease-related genes have functional orthologs in Drosophila (70, 71), including many implicated in schizophrenia. Although exist evident anatomical and structural differences between the brains of flies and vertebrates, the basic principles that govern their development and operation are conserved. Moreover, Drosophila connectomics, which has been well established and refined (72, 73), further support using this animal in modeling anatomical and functional features of complex psychiatric and neurodevelopmental disorders like schizophrenia. Furthermore, several binary expression systems have enabled the study of human gene homologs linked to this disorder in Drosophila (70, 71). This animal model is particularly valuable because it encompasses the same major neurochemical systems associated with schizophrenia in humans, including dopaminergic, serotonergic, GABAergic, and glutamatergic systems, although some differences in their respective enzymes, receptor subtypes, transporters, and metabolizing proteins need to be considered (74, 75).
Several Drosophila models for schizophrenia have been developed and characterized, exhibiting key features observed in other animal models of the disorder and patients. These include altered circadian rhythms, hyperlocomotion, and some cognitive deficits, such as impaired learning and memory (76–80). These models have become valuable tools for exploring the cellular and molecular processes underlying the pathophysiological aspects of schizophrenia, including its negative symptoms, and have provided important information on the human disorder.
Thus, for instance, one of the first studies that explored the molecular underpinnings underlying schizophrenia pathophysiology in Drosophila was that of Sawamura et al. (77). In this work, authors generated transgenic flies expressing the human gene Disrupted in schizophrenia 1, DISC1, which resulted in alterations in sleep homeostasis. Importantly, it was demonstrated that DISC1 modulates CRE-mediated gene transcription by interacting with ATF4/CREB2 (77), an important factor in a broad range of brain conditions (81–83).
Other studies used Drosophila to provide further support to the dopamine ontogenic hypothesis for schizophrenia (76, 84). In these works, activation of the dopaminergic system in specific early developmental windows resulted in behavioral alterations in adult animals, including changes in sleep patterns, and behavioral responses to mechanic and visual stimuli, which could reflect an effect on salience allocation, a characteristic of the positive symptoms of schizophrenia (85).
These and other studies demonstrate the validity of Drosophila models to assess the mechanisms underlying complex human disorders including schizophrenia. Nevertheless, one of the challenges in schizophrenia research is the difficulty in replicating negative symptoms in animals -including Drosophila- to study the molecular, cellular, and circuital underpinnings underlying their onset. Importantly, new tests and social and cognitive paradigms have been developed over recent years to assess complex behavioral, social, and cognitive functions in Drosophila relevant to neurological and psychiatric conditions. For instance, the flies’ clustering behavior, which consists of flies aggregating in groups, has been linked to social coordination and has provided insights into collective behavior dynamics (86). In addition, Drosophila exhibits attention-like processes, allowing them to prioritize certain stimuli above others, an aspect of cognition observed in more complex organisms (87). Research has further revealed that Drosophila engages in goal-driven behavioral adaptations, modifying their actions based on environmental conditions or experiences, a process akin to motivation, learning, and behavior modifications seen in vertebrates (88). Furthermore, Drosophila has been tested in their ability to make choices, which somehow resembles basic decision-making processes (89). These findings highlight the potential of Drosophila as a model for studying multifaceted brain processes underlying complex behaviors and foster support that it is possible to study the mechanisms underpinning schizophrenia negative symptoms in this animal.
In this regard, our lab advanced the previous characterization of the hypomorphic mutant dysb1, which represents a loss-of-function mutation in the fly orthologue of DTNBP1/Dysbindin-1 (90, 91). Our findings revealed several behavioral phenotypes reminiscent of schizophrenia’s negative symptoms in humans. In particular, dysb1 flies exhibit increased social spacing compared to controls (92). This is in agreement with previous studies in the “sandy” mouse (mutant for dysbindin-1) (93) and in schizophrenia patients, which demonstrate alterations in social distance (94, 95), supporting the notion that social space is a good marker or probe for negative symptoms of schizophrenia.
We further showed neurochemical alterations in the dysb1 mutant flies, including reduced serotonin levels and a two-fold increase in dSERT expression (92). Interestingly, the administration of 4-MTA, a serotonin-releasing agent, effectively increased social behaviors in control flies but failed to elicit the same effect in dysb1 mutants, providing further support for the idea that the serotonergic system plays a role in the expression of negative symptoms of schizophrenia (92).
In a different work (80) we investigated the role of the orthologue for the Rab-3 interacting molecule-1 (RIM1) gene, called Rim in Drosophila, to some of the behavioral anatomical and functional phenotypes observed in schizophrenia patients. In this work, Rim mutants displayed impaired social behavior, which is similar to the social impairment described in RIM1α−/− mutant mice (96, 97). Moreover, the Rim mutant flies showed impaired olfactory acuity and circadian defects, including a loss of circadian rhythmicity and decreased period length phenotypes, that mapped to the pacemaker ventral lateral clock neurons. Importantly, haloperidol, a typical antipsychotic, efficiently rescued Rim mutant deficits to normal levels further validating the Drosophila model for investigating the mechanisms underlying schizophrenia-related behaviors (80). Other studies have used Drosophila to assess the role that could play alterations in Neuroligins (NLGs) to schizophrenia symptoms. NLGs are a family of proteins that form protein-protein complexes essential for the proper formation, maturation, and functional adjustment of chemical synaptic connections between neurons (98, 99). Several alterations in genes coding for NLGs have been associated with changes in social behavior in disorders such as autism and schizophrenia (100). Specifically, mutations in orthologs for these genes in Drosophila (dlng2 and dlng4) have shown alterations in the sleep rhythms, altered acoustic communication signals, as well as a reduced tendency to form groups and social interactions (101–103), phenotypes that parallel negative symptoms observed in schizophrenia.
Recent studies have expanded the scope of Drosophila schizophrenia models to study endophenotypes, heritable and quantifiable traits that serve as intermediate markers linking genetic risks to clinical symptoms of a disorder. For instance, Foka et al. (104) demonstrated that Drosophila furin1 mutants exhibit defective habituation to repeated stimuli, a phenotype that mirrors impaired habituation observed in schizophrenia patients (105). In that work, it was also demonstrated that the deficit observed in flies can be reversed by antipsychotic treatment, validating the translational relevance of this model (104). Likewise, Schiöth et al. (106) provided the first evidence of prepulse inhibition (PPI) for visual stimuli in adult Drosophila, an endophenotype sensitive to NMDA receptor antagonists in flies that has been reported in people with this disorder (107).
Conclusion
Drosophila melanogaster has proven to be a valuable model for investigating some of the neurobiological underpinnings of schizophrenia, particularly its negative symptoms, which remain one of the most challenging aspects to study in this disorder. These findings not only affirm the relevance of these genes to the disorder but also underscore Drosophila as a model system for investigating the mechanisms involved in psychiatric conditions. Moreover, the development of new behavioral paradigms, such as sucrose preference to assess anhedonia (108), and the forced swim test to measure despair-related behavior (109), further expands the utility of this model. As research continues to refine these approaches, Drosophila holds significant potential for deepening our understanding on the cellular and molecular mechanisms driving schizophrenia and for identifying new therapeutic targets to alleviate its debilitating negative symptoms.
Author contributions
ME-R: Writing – review & editing, Writing – original draft. SH: Writing – review & editing. JC: Funding acquisition, Writing – review & editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. ME-R received a doctoral fellowship from ANID (No. 21231956). JC is supported by Fondecyt No. 1231556, ANID (Chile) and SH is supported by NIH K99NS133470 (US).
Acknowledgments
We thank the Campusano Lab for comments on the idea of this mini review.
Conflict of interest
The authors declare that this work was conducted in the absence of any commercial or financial relationships that could be considered a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ripke S, Neale BM, Corvin A, Walters JTR, Farh K-H, Holmans PA, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. (2014) 511:421–7. doi: 10.1038/nature13595
2. Rossler W, Salize HJ, van Os J, and Riecher-Rossler A. Size of burden of schizophrenia and psychotic disorders. Eur Neuropsychopharmacol. (2005) 15:399–409. doi: 10.1016/j.euroneuro.2005.04.009
3. Tamminga CA and Holcomb HH. Phenotype of schizophrenia: a review and formulation. Mol Psychiatry. (2005) 10:27–39. doi: 10.1038/sj.mp.4001563
4. Evensen S, Wisloff T, Lystad JU, Bull H, Ueland T, and Falkum E. Prevalence, Employment Rate, and Cost of Schizophrenia in a High-Income Welfare Society: A Population-Based Study Using Comprehensive Health and Welfare Registers. Schizophr Bull. (2016) 42:476–83. doi: 10.1093/schbul/sbv141
5. Chong HY, Teoh SL, Wu DB, Kotirum S, Chiou CF, and Chaiyakunapruk N. Global economic burden of schizophrenia: a systematic review. Neuropsychiatr Dis Treat. (2016) 12:357–73. doi: 10.2147/NDT.S96649
6. Dollfus S and Lyne J. Negative symptoms: History of the concept and their position in diagnosis of schizophrenia. Schizophr Res. (2017) 186:3–7. doi: 10.1016/j.schres.2016.06.024
7. Andreasen NC. Negative Symptoms in Schizophrenia: Definition and Reliability. Arch Gen Psychiatry. (1982) 39:784–8. doi: 10.1001/archpsyc.1982.04290070020005
8. Crow TJ. The Two-syndrome Concept: Origins and Current Status. Schizophr Bull. (1985) 11:471–88. doi: 10.1093/schbul/11.3.471
9. Takano H. Cognitive Function and Monoamine Neurotransmission in Schizophrenia: Evidence From Positron Emission Tomography Studies. Front Psychiatry. (2018) 9:228. doi: 10.3389/fpsyt.2018.00228
10. Millan MJ, Andrieux A, Bartzokis G, Cadenhead K, Dazzan P, Fusar-Poli P, et al. Altering the course of schizophrenia: progress and perspectives. Nat Rev Drug Discovery. (2016) 15:485–515. doi: 10.1038/nrd.2016.28
11. Moberg PJ and Turetsky BI. Scent of a disorder: olfactory functioning in schizophrenia. Curr Psychiatry Rep. (2003) 5:311–9. doi: 10.1007/s11920-003-0061-x
12. Ferrarelli F. Sleep Abnormalities in Schizophrenia: State of the Art and Next Steps. Am J Psychiatry. (2021) 178:903–13. doi: 10.1176/appi.ajp.2020.20070968
13. Sakurai T, Gamo NJ, Hikida T, Kim S-H, Murai T, Tomoda T, et al. Converging models of schizophrenia – Network alterations of prefrontal cortex underlying cognitive impairments. Prog Neurobiol. (2015) 134:178–201. doi: 10.1016/j.pneurobio.2015.09.010
14. Zurlo L, Dal Bò E, Gentili C, and Cecchetto C. Olfactory dysfunction in schizophrenia and other psychotic disorders: A comprehensive and updated meta-analysis. Schizophr Res. (2025) 275:62–75. doi: 10.1016/j.schres.2024.12.001
15. Lee J, Takeuchi H, Fervaha G, Sin GL, Foussias G, Agid O, et al. Subtyping Schizophrenia by Treatment Response: Antipsychotic Development and the Central Role of Positive Symptoms. Can J Psychiatry. (2015) 60:515–22. doi: 10.1177/070674371506001107
16. Leucht S, Leucht C, Huhn M, Chaimani A, Mavridis D, Helfer B, et al. Sixty Years of Placebo-Controlled Antipsychotic Drug Trials in Acute Schizophrenia: Systematic Review, Bayesian Meta-Analysis, and Meta-Regression of Efficacy Predictors. Am J Psychiatry. (2017) 174:927–42. doi: 10.1176/appi.ajp.2017.16121358
17. Messinger JW, Trémeau F, Antonius D, Mendelsohn E, Prudent V, Stanford AD, et al. Avolition and expressive deficits capture negative symptom phenomenology: Implications for DSM-5 and schizophrenia research. Clin Psychol Rev. (2011) 31:161–8. doi: 10.1016/j.cpr.2010.09.002
18. Kirkpatrick B. Developing concepts in negative symptoms: primary vs secondary and apathy vs expression. J Clin Psychiatry. (2014) 75 Suppl 1:3–7. doi: 10.4088/JCP.13049su1c.01
19. Ang MS, Rekhi G, and Lee J. Validation of the Brief Negative Symptom Scale and its association with functioning. Schizophr Res. (2019) 208:97–104. doi: 10.1016/j.schres.2019.04.005
20. Grigoriou M and Upthegrove R. Blunted affect and suicide in schizophrenia: A systematic review. Psychiatry Res. (2020) 293:113355. doi: 10.1016/j.psychres.2020.113355
21. Chang X, Zhao W, Kang J, Xiang S, Xie C, Corona-Hernández H, et al. Language abnormalities in schizophrenia: binding core symptoms through contemporary empirical evidence. Schizophrenia. (2022) 8:95. doi: 10.1038/s41537-022-00308-x
22. Sabe M, Kirschner M, and Kaiser S. Prodopaminergic Drugs for Treating the Negative Symptoms of Schizophrenia: Systematic Review and Meta-analysis of Randomized Controlled Trials. J Clin Psychopharmacol. (2019) 39:658–64. doi: 10.1097/JCP.0000000000001124
23. Liang SG and Greenwood TA. The impact of clinical heterogeneity in schizophrenia on genomic analyses. Schizophr Res. (2015) 161:490–5. doi: 10.1016/j.schres.2014.11.019
24. Sawa A and Snyder SH. Schizophrenia: diverse approaches to a complex disease. Science. (2002) 296:692–5. doi: 10.1126/science.1070532
25. Edwards AC, Bigdeli TB, Docherty AR, Bacanu S, Lee D, de Candia TR, et al. Meta-analysis of Positive and Negative Symptoms Reveals Schizophrenia Modifier Genes. Schizophr Bull. (2015) 42:279–87. doi: 10.1093/schbul/sbv119
26. DeRosse P, Funke B, Burdick KE, Lencz T, Ekholm JM, Kane JM, et al. Dysbindin Genotype and Negative Symptoms in Schizophrenia. Am J Psychiatry. (2006) 163:532–4. doi: 10.1176/appi.ajp.163.3.532
27. Fanous AH, E.J.v.d. Oord BP, Aggen SH, Neale MC, O’Neill FA, et al. Relationship Between a High-Risk Haplotype in the DTNBP1 (Dysbindin) Gene and Clinical Features of Schizophrenia. Am J Psychiatry. (2005) 162:1824–32. doi: 10.1176/appi.ajp.162.10.1824
28. Wessman J, Paunio T, Tuulio-Henriksson A, Koivisto M, Partonen T, Suvisaari J, et al. Mixture model clustering of phenotype features reveals evidence for association of DTNBP1 to a specific subtype of schizophrenia. Biol Psychiatry. (2009) 66:990–6. doi: 10.1016/j.biopsych.2009.05.034
29. Papaleo F, Yang F, Garcia S, Chen J, Lu B, Crawley JN, et al. Dysbindin-1 modulates prefrontal cortical activity and schizophrenia-like behaviors via dopamine/D2 pathways. Mol Psychiatry. (2012) 17:85–98. doi: 10.1038/mp.2010.106
30. Trantham-Davidson H and Lavin A. Loss of dysbindin-1 affects GABAergic transmission in the PFC. Psychopharmacology. (2019) 236:3291–300. doi: 10.1007/s00213-019-05285-1
31. Howes OD, Bukala BR, and Beck K. Schizophrenia: from neurochemistry to circuits, symptoms and treatments. Nat Rev Neurol. (2024) 20:22–35. doi: 10.1038/s41582-023-00904-0
32. Molero P, Ortuño F, Zalacain M, and Patiño-García A. Clinical involvement of catechol-O-methyltransferase polymorphisms in schizophrenia spectrum disorders: influence on the severity of psychotic symptoms and on the response to neuroleptic treatment. Pharmacogenomics J. (2007) 7:418–26. doi: 10.1038/sj.tpj.6500441
33. Wang Y, Fang Y, Shen Y, and Xu Q. Analysis of association between the catechol-O-methyltransferase (COMT) gene and negative symptoms in chronic schizophrenia. Psychiatry Res. (2010) 179:147–50. doi: 10.1016/j.psychres.2009.03.029
34. Chen C-Y, Lu R-B, Yeh Y-W, Shih M-C, and Huang S-Y. Association study of catechol-O-methyltransferase gene polymorphisms with schizophrenia and psychopathological symptoms in Han Chinese. Genes Brain Behav. (2011) 10:316–24. doi: 10.1111/j.1601-183X.2010.00670.x
35. Pelayo-Terán JM, Pérez-Iglesias R, Vázquez-Bourgon J, Mata I, Carrasco-Marín E, Vázquez-Barquero JL, et al. Catechol-O-methyltransferase Val158Met polymorphism and negative symptoms after acute antipsychotic treatment in first-episode non-affective psychosis. Psychiatry Res. (2011) 185:286–9. doi: 10.1016/j.psychres.2010.06.006
36. Spina E, De Domenico P, Ruello C, Longobardo N, Gitto C, Ancione M, et al. Adjunctive fluoxetine in the treatment of negative symptoms in chronic schizophrenic patients. Int Clin Psychopharmacol. (1994) 9:281–5. doi: 10.1097/00004850-199400940-00007
37. Meltzer HY. New Trends in the Treatment of Schizophrenia. CNS Neurol Disord Drug Targets. (2017) 16:900–6. doi: 10.2174/1871527316666170728165355
38. Štrac DŠ, Pivac N, and Mück-Šeler D. The serotonergic system and cognitive function. Trans Neurosci. (2016) 7:35–49. doi: 10.1515/tnsci-2016-0007
39. Birnbaum R and Weinberger DR. Genetic insights into the neurodevelopmental origins of schizophrenia. Nat Rev Neurosci. (2017) 18:727–40. doi: 10.1038/nrn.2017.125
40. Damianidou E, Mouratidou L, and Kyrousi C. Research models of neurodevelopmental disorders: The right model in the right place. Front Neurosci. (2022), 16. doi: 10.3389/fnins.2022.1031075
41. Blackman RK, MacDonald AW, and Chafee MV. Effects of Ketamine on Context-Processing Performance in Monkeys: A New Animal Model of Cognitive Deficits in Schizophrenia. Neuropsychopharmacology. (2013) 38:2090–100. doi: 10.1038/npp.2013.118
42. Demin KA, Meshalkina DA, Volgin AD, Yakovlev OV, de Abreu MS, Alekseeva PA, et al. Developing zebrafish experimental animal models relevant to schizophrenia. Neurosci Biobehav Rev. (2019) 105:126–33. doi: 10.1016/j.neubiorev.2019.07.017
43. Langova V, Vales K, Horka P, and Horacek J. The Role of Zebrafish and Laboratory Rodents in Schizophrenia Research. Front Psychiatry. (2020), 11. doi: 10.3389/fpsyt.2020.00703
44. Tordjman S, Drapier D, Bonnot O, Graignic R, Fortes S, Cohen D, et al. Animal Models Relevant to Schizophrenia and Autism: Validity and Limitations. Behav Genet. (2007) 37:61–78. doi: 10.1007/s10519-006-9120-5
45. Yee BK and Singer P. A conceptual and practical guide to the behavioural evaluation of animal models of the symptomatology and therapy of schizophrenia. Cell Tissue Res. (2013) 354:221–46. doi: 10.1007/s00441-013-1611-0
46. Nani JV, Muotri AR, and Hayashi MAF. Peering into the mind: unraveling schizophrenia’s secrets using models. Mol Psychiatry. (2025) 30:659–78. doi: 10.1038/s41380-024-02728-w
47. Clapcote SJ, Lipina TV, Millar JK, Mackie S, Christie S, Ogawa F, et al. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron. (2007) 54:387–402. doi: 10.1016/j.neuron.2007.04.015
48. Cox MM, Tucker AM, Tang J, Talbot K, Richer DC, Yeh L, et al. Neurobehavioral abnormalities in the dysbindin-1 mutant, sandy, on a C57BL/6J genetic background. Genes Brain Behav. (2009) 8:390–7. doi: 10.1111/j.1601-183X.2009.00477.x
49. Hikida T, Jaaro-Peled H, Seshadri S, Oishi K, Hookway C, Kong S, et al. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci. (2007) 104:14501–6. doi: 10.1073/pnas.0704774104
50. Pletnikov MV, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov MV, Huang H, et al. Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry. (2008) 13:173–86. doi: 10.1038/sj.mp.4002079
51. Managò F, Mereu M, Mastwal S, Mastrogiacomo R, Scheggia D, Emanuele M, et al. Genetic Disruption of Arc/Arg3.1 in Mice Causes Alterations in Dopamine and Neurobehavioral Phenotypes Related to Schizophrenia. Cell Rep. (2016) 16:2116–28. doi: 10.1016/j.celrep.2016.07.044
52. Sumitomo A, Horike K, Hirai K, Butcher N, Boot E, Sakurai T, et al. A mouse model of 22q11.2 deletions: Molecular and behavioral signatures of Parkinson’s disease and schizophrenia. Sci Adv. (2018) 4:eaar6637. doi: 10.1126/sciadv.aar6637
53. Marsden CA, King MV, and Fone KC. Influence of social isolation in the rat on serotonergic function and memory–relevance to models of schizophrenia and the role of 5-HT(6) receptors. Neuropharmacology. (2011) 61:400–7. doi: 10.1016/j.neuropharm.2011.03.003
54. Meyer U and Feldon J. Epidemiology-driven neurodevelopmental animal models of schizophrenia. Prog Neurobiol. (2010) 90:285–326. doi: 10.1016/j.pneurobio.2009.10.018
55. Weiss IC and Feldon J. Environmental animal models for sensorimotor gating deficiencies in schizophrenia: a review. Psychopharmacol (Berl). (2001) 156:305–26. doi: 10.1007/s002130100800
56. Ceretta APC, Schaffer LF, de Freitas CM, Reinheimer JB, Dotto MM, and Fachinetto R. Gabapentin prevents behavioral changes on the amphetamine-induced animal model of schizophrenia. Schizophr Res. (2016) 175:230–1. doi: 10.1016/j.schres.2016.04.044
57. Tenn CC, Fletcher PJ, and Kapur S. Amphetamine-sensitized animals show a sensorimotor gating and neurochemical abnormality similar to that of schizophrenia. Schizophr Res. (2003) 64:103–14. doi: 10.1016/S0920-9964(03)00009-4
58. Cohen-Laroque J, Grangier I, Perez N, Kirschner M, Kaiser S, and Sabé M. Positive and negative symptoms in methamphetamine-induced psychosis compared to schizophrenia: A systematic review and meta-analysis. Schizophr Res. (2024) 267:182–90. doi: 10.1016/j.schres.2024.03.037
59. Voce A, Burns R, Castle D, Calabria B, and McKetin R. Is there a discrete negative symptom syndrome in people who use methamphetamine? Compr Psychiatry. (2019) 93:27–32. doi: 10.1016/j.comppsych.2019.06.002
60. Sams-Dodd F. Phencyclidine in the social interaction test: an animal model of schizophrenia with face and predictive validity. Rev Neurosci. (1999) 10:59–90. doi: 10.1515/REVNEURO.1999.10.1.59
61. Neill JC, Harte MK, Haddad PM, Lydall ES, and Dwyer DM. Acute and chronic effects of NMDA receptor antagonists in rodents, relevance to negative symptoms of schizophrenia: a translational link to humans. Eur Neuropsychopharmacol. (2014) 24:822–35. doi: 10.1016/j.euroneuro.2013.09.011
62. Lim AL, Taylor DA, and Malone DT. Consequences of early life MK-801 administration: long-term behavioural effects and relevance to schizophrenia research. Behav Brain Res. (2012) 227:276–86. doi: 10.1016/j.bbr.2011.10.052
63. Aghajanian GK and Marek GJ. Serotonin model of schizophrenia: emerging role of glutamate mechanisms. Brain Res Rev. (2000) 31:302–12. doi: 10.1016/S0165-0173(99)00046-6
64. Galici R, Boggs JD, Miller KL, Bonaventure P, and Atack JR. Effects of SB-269970, a 5-HT7 receptor antagonist, in mouse models predictive of antipsychotic-like activity. Behav Pharmacol. (2008) 19:153–9. doi: 10.1097/FBP.0b013e3282f62d8c
65. Maxwell J, Gleason SD, Falcone J, Svensson K, Balcer OM, Li X, et al. Effects of 5-HT7 receptor antagonists on behaviors of mice that detect drugs used in the treatment of anxiety, depression, or schizophrenia. Behav Brain Res. (2019) 359:467–73. doi: 10.1016/j.bbr.2018.11.019
66. Deutsch SI, Mastropaolo J, and Rosse RB. Neurodevelopmental consequences of early exposure to phencyclidine and related drugs. Clin Neuropharmacol. (1998) 21:320–32.
67. Feifel D and Shilling PD. Promise and pitfalls of animal models of schizophrenia. Curr Psychiatry Rep. (2010) 12:327–34. doi: 10.1007/s11920-010-0122-x
68. Winship IR, Dursun SM, Baker GB, Balista PA, Kandratavicius L, Maia-de-Oliveira JP, et al. An overview of animal models related to schizophrenia. Can J Psychiatry. (2019) 64:5–17. doi: 10.1177/0706743718773728
69. Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, et al. The genome sequence of Drosophila melanogaster. Science. (2000) 287:2185–95. doi: 10.1126/science.287.5461.2185
70. Jennings BH. Drosophila – a versatile model in biology & medicine. Materials Today. (2011) 14:190–5. doi: 10.1016/S1369-7021(11)70113-4
71. Victor Atoki A, Maduabuchi AP, Salihu ST, Nyakundi OE, Ismahil AA, Victor FI, et al. Exploring the versatility of Drosophila melanogaster as a model organism in biomedical research: a comprehensive review. Fly. (2025) 19:2420453. doi: 10.1080/19336934.2024.2420453
72. Dorkenwald S, Matsliah A, Sterling AR, Schlegel P, Yu S-c, McKellar CE, et al. Neuronal wiring diagram of an adult brain. Nature. (2024) 8032):124–38:634. doi: 10.1038/s41586-024-07558-y
73. Schlegel P, Yin Y, Bates AS, Dorkenwald S, Eichler K, Brooks P, et al. Whole-brain annotation and multi-connectome cell typing of Drosophila. Nature. (2024) 634:139–52. doi: 10.1038/s41586-024-07686-5
74. Carvajal-Oliveros A and Campusano JM. Studying the contribution of serotonin to neurodevelopmental disorders. Can This Fly? Front Behav Neurosci. (2020) 14:601449. doi: 10.3389/fnbeh.2020.601449
75. Yoshihara M, Ensminger AW, and Littleton JT. Neurobiology and the Drosophila genome. Funct Integr Genomics. (2001) 1:235–40. doi: 10.1007/s101420000029
76. Calcagno B, Eyles D, van Alphen B, and van Swinderen B. Transient activation of dopaminergic neurons during development modulates visual responsiveness, locomotion and brain activity in a dopamine ontogeny model of schizophrenia. Transl Psychiatry. (2013) 3:e206. doi: 10.1038/tp.2012.139
77. Sawamura N, Ando T, Maruyama Y, Fujimuro M, Mochizuki H, Honjo K, et al. Nuclear DISC1 regulates CRE-mediated gene transcription and sleep homeostasis in the fruit fly. Mol Psychiatry. (2008) 13:1138–48, 1069. doi: 10.1038/mp.2008.101
78. Shao L, Shuai Y, Wang J, Feng S, Lu B, Li Z, et al. Schizophrenia susceptibility gene dysbindin regulates glutamatergic and dopaminergic functions via distinctive mechanisms in Drosophila. Proc Natl Acad Sci USA. (2011) 108:18831–6. doi: 10.1073/pnas.1114569108
79. Hidalgo S, Campusano JM, and Hodge JJL. The Drosophila ortholog of the schizophrenia-associated CACNA1A and CACNA1B voltage-gated calcium channels regulate memory, sleep and circadian rhythms. Neurobiol Dis. (2021) 155:105394. doi: 10.1016/j.nbd.2021.105394
80. Hidalgo S, Campusano JM, and Hodge JJL. Assessing olfactory, memory, social and circadian phenotypes associated with schizophrenia in a genetic model based on Rim. Transl Psychiatry. (2021) 11:292. doi: 10.1038/s41398-021-01418-3
81. Chen L, Tang J, Liu XQ, Li QQ, Li JY, Li YY, et al. TIGAR Suppresses ER Stress-Induced Neuronal Injury through Targeting ATF4 Signaling in Cerebral Ischemia/Reperfusion. J Neurosci. (2025) 45. doi: 10.1523/JNEUROSCI.1406-24.2025
82. Goswami P, Akhter J, Mangla A, Suramya S, Jindal G, Ahmad S, et al. Downregulation of ATF-4 attenuates the endoplasmic reticulum stress-mediated neuroinflammation and cognitive impairment in experimentally induced alzheimer's disease model. Mol Neurobiol. (2024) 61:5071–82. doi: 10.1007/s12035-023-03861-3
83. Mamdani F, Alda M, Grof P, Young LT, Rouleau G, and Turecki G. Lithium response and genetic variation in the CREB family of genes. Am J Med Genet B Neuropsychiatr Genet. (2008) 147b. doi: 10.1002/ajmg.b.v147b:4
84. Ferguson L, Petty A, Rohrscheib C, Troup M, Kirszenblat L, Eyles DW, et al. Transient dysregulation of dopamine signaling in a developing drosophila arousal circuit permanently impairs behavioral responsiveness in adults. Front Psychiatry. (2017) 8:22. doi: 10.3389/fpsyt.2017.00022
85. Kapur S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry. (2003) 160:13–23. doi: 10.1176/appi.ajp.160.1.13
86. Simon AF, Chou MT, Salazar ED, Nicholson T, Saini N, Metchev S, et al. A simple assay to study social behavior in Drosophila: measurement of social space within a group. Genes Brain Behav. (2012) 11:243–52. doi: 10.1111/j.1601-183X.2011.00740.x
87. van Swinderen B and Flores KA. Attention-like processes underlying optomotor performance in a Drosophila choice maze. Dev Neurobiol. (2007) 67:129–45. doi: 10.1002/dneu.20334
88. Pick S and Strauss R. Goal-driven behavioral adaptations in gap-climbing Drosophila. Curr Biol. (2005) 15:1473–8. doi: 10.1016/j.cub.2005.07.022
89. Zhang K, Guo JZ, Peng Y, Xi W, and Guo A. Dopamine-mushroom body circuit regulates saliency-based decision-making in Drosophila. Science. (2007) 316:1901–4. doi: 10.1126/science.1137357
90. Dickman DK and Davis GW. The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science. (2009) 326:1127–30. doi: 10.1126/science.1179685
91. Mullin AP, Sadanandappa MK, Ma W, Dickman DK, VijayRaghavan K, Ramaswami M, et al. Gene dosage in the dysbindin schizophrenia susceptibility network differentially affect synaptic function and plasticity. J Neurosci. (2015) 35:325–38. doi: 10.1523/JNEUROSCI.3542-14.2015
92. Hidalgo S, Castro C, Zarate RV, Valderrama BP, Hodge JJL, and Campusano JM. The behavioral and neurochemical characterization of a Drosophila dysbindin mutant supports the contribution of serotonin to schizophrenia negative symptoms. Neurochem Int. (2020) 138:104753. doi: 10.1016/j.neuint.2020.104753
93. Hattori S, Murotani T, Matsuzaki S, Ishizuka T, Kumamoto N, Takeda M, et al. Behavioral abnormalities and dopamine reductions in sdy mutant mice with a deletion in Dtnbp1, a susceptibility gene for schizophrenia. Biochem Biophys Res Commun. (2008) 373:298–302. doi: 10.1016/j.bbrc.2008.06.016
94. Deus V and Jokic-Begic N. Personal space in schizophrenic patients. Psychiatr Danub. (2006) 18:150–8.
95. Di Cosmo G, Costantini M, Salone A, Martinotti G, Di Iorio G, Di Giannantonio M, et al. Peripersonal space boundary in schizotypy and schizophrenia. Schizophr Res. (2018) 197:589–90. doi: 10.1016/j.schres.2017.12.003
96. Blundell J, Kaeser PS, Südhof TC, and Powell CM. RIM1α and interacting proteins involved in presynaptic plasticity mediate prepulse inhibition and additional behaviors linked to schizophrenia. J Neurosci. (2010) 30:5326–33. doi: 10.1523/JNEUROSCI.0328-10.2010
97. Haws ME, Kaeser PS, Jarvis DL, Südhof TC, and Powell CM. Region-specific deletions of RIM1 reproduce a subset of global RIM1α–/– phenotypes. Genes Brain Behav. (2012) 11:201–13. doi: 10.1111/j.1601-183X.2011.00755.x
98. Chubykin AA, Atasoy D, Etherton MR, Brose N, Kavalali ET, Gibson JR, et al. Activity-dependent validation of excitatory versus inhibitory synapses by Neuroligin-1 versus Neuroligin-2. Neuron. (2007) 54:919–31. doi: 10.1016/j.neuron.2007.05.029
99. Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, et al. Neuroligins determine synapse maturation and function. Neuron. (2006) 51:741–54. doi: 10.1016/j.neuron.2006.09.003
100. Kenny EM, Cormican P, Furlong S, Heron E, Kenny G, Fahey C, et al. Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol Psychiatry. (2014) 19:872–9. doi: 10.1038/mp.2013.127
101. Corthals K, Heukamp AS, Kossen R, Großhennig I, Hahn N, Gras H, et al. Neuroligins Nlg2 and Nlg4 Affect Social Behavior in Drosophila melanogaster. Front Psychiatry. (2017) 8. doi: 10.3389/fpsyt.2017.00113
102. Hahn N, Geurten B, Gurvich A, Piepenbrock D, Kästner A, Zanini D, et al. Monogenic heritable autism gene neuroligin impacts Drosophila social behaviour. Behav Brain Res. (2013) 252:450–7. doi: 10.1016/j.bbr.2013.06.020
103. Li Y, Zhou Z, Zhang X, Tong H, Li P, Zhang ZC, et al. Drosophila neuroligin 4 regulates sleep through modulating GABA transmission. J Neurosci. (2013) 33:15545–54. doi: 10.1523/JNEUROSCI.0819-13.2013
104. Foka K, Georganta EM, Semelidou O, and Skoulakis EMC. Loss of the schizophrenia-linked furin protein from drosophila mushroom body neurons results in antipsychotic-reversible habituation deficits. J Neurosci. (2022) 42:7496–511. doi: 10.1523/JNEUROSCI.1055-22.2022
105. Williams LE, Blackford JU, Luksik A, Gauthier I, and Heckers S. Reduced habituation in patients with schizophrenia. Schizophr Res. (2013) 151:124–32. doi: 10.1016/j.schres.2013.10.017
106. Schioth HB, Donzelli L, Arvidsson N, Williams MJ, and Moulin TC. Evidence for prepulse inhibition of visually evoked motor response in drosophila melanogaster. Biol (Basel). (2023) 12:635. doi: 10.3390/biology12040635
107. Swerdlow NR, Talledo J, Sutherland AN, Nagy D, and Shoemaker JM. antipsychotic effects on prepulse inhibition in normal ‘low gating’ humans and rats. Neuropsychopharmacology. (2006) 31:2011–21. doi: 10.1038/sj.npp.1301043
108. Ries A-S, Hermanns T, Poeck B, and Strauss R. Serotonin modulates a depression-like state in Drosophila responsive to lithium treatment. Nat Commun. (2017) 8:15738. doi: 10.1038/ncomms15738
Keywords: Drosophila, schizophrenia, negative symptom, dysbindin-1 (DTNBP1), Rim1, neuroligin
Citation: Elgueta-Reyes M, Hidalgo S and Campusano JM (2025) Beyond vertebrates: Drosophila melanogaster as a model to study negative symptoms of schizophrenia. Front. Psychiatry 16:1622281. doi: 10.3389/fpsyt.2025.1622281
Received: 03 May 2025; Accepted: 16 June 2025;
Published: 23 July 2025.
Edited by:
Diana Rodrigues, Ludwig Maximilian University of Munich, GermanyReviewed by:
Zoltan Asztalos, Aktogen Limited, United KingdomCopyright © 2025 Elgueta-Reyes, Hidalgo and Campusano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maximiliano Elgueta-Reyes, bWF4LmVsZ3VldGExN0BnbWFpbC5jb20=; Jorge M. Campusano, am1jYW1wdXNAdWMuY2w=