 Susanne Edelmann
Susanne Edelmann Christine Kummer
Christine Kummer Sarah Pasche
Sarah Pasche Milan Zimmermann1
Milan Zimmermann1 Vanessa Nieratschker
Vanessa Nieratschker- 1Department of Psychiatry and Psychotherapy, University Hospital of Tuebingen, Eberhard Karls University of Tuebingen, Tuebingen, Germany
- 2German Center for Mental Health (DZPG), Tuebingen, Germany
Epigenetic regulation is significantly altered in individuals with alcohol use disorder (AUD), representing a promising avenue for understanding its pathomechanisms and developing new therapies. In an earlier epigenome-wide study of CD3+ T cells, we identified SYNGAP1–a critical regulator of synaptic plasticity that influences neuronal communication and network remodeling–as epigenetically dysregulated, with significantly lower DNA methylation (DNAm) in patients than controls. After three weeks of inpatient withdrawal, SYNGAP1 DNAm increased to control levels. In the present study, we aimed to validate these differential SYNGAP1 DNAm levels in an independent cohort of 64 AUD patients and 83 healthy controls in peripheral blood and saliva, to assess its potential as a biomarker. Using a linear mixed-effects model including AUD status and covariates, no significant differences were observed. Post hoc analyses revealed an unexpected pattern: In blood, SYNGAP1 DNAm was higher in patients before treatment than controls, with no difference after withdrawal; in saliva, no differences or therapy effects were detected. Overall, these results did not confirm our previous findings, suggesting limited value of SYNGAP1 DNAm as a biomarker for AUD. While blood methylation showed some association, the effect direction contradicted earlier results, and saliva showed no signal. Further research is needed to clarify SYNGAP1 epigenetic regulation in AUD and its potential relevance for biomarkers or therapy.
1 Introduction
Alcohol Use Disorder (AUD) is a severe chronic disorder contributing substantially to the global burden of disease (1). The development of AUD underlies both genetic and environmental factors (2, 3), and gene-environment interactions, such as epigenetic mechanisms, play a pivotal role (4). Epigenetics describes the – reversible – modulation of genomic activity and gene function without changing the DNA sequence itself. One of the most studied epigenetic mechanisms is DNA methylation (DNAm) (5). AUD has been widely described as being associated with altered DNAm (4, 6, 7). Several investigations conducting epigenome-wide association studies (EWAS) in blood and brain provided evidence for altered DNAm patterns, e.g. in genes involved in glutamate signaling (8), immune-related pathways (9, 10), and glucocorticoid and inflammation-related signaling (11). Recently, White et al. (2024) identified 105 AUD-associated CpGs annotated to 120 genes within and across brain regions that were enriched in histone marks tagging active promoters (12). In a previous epigenome-wide study in our group, we identified decreased DNAm levels of the CpG site cg02652579 present in the promotor region of Synaptic Ras-GTPase-activating protein gene (SYNGAP1) in CD3+ T-cells of male AUD patients compared to matched control individuals. Interestingly, following three weeks of inpatient withdrawal treatment, SYNGAP1 DNAm levels increased and reached levels observed in healthy control individuals (13). SYNGAP1 encodes for the SynGAP protein (14, 15) which is part of complex networks located on the postsynaptic density (PSD), mainly in the cortex and hippocampus. SynGAP fulfills several functions in neurotransmitter signaling, morphology of synapses and scaffolding of protein networks (15, 16). Furthermore, SynGAP promotes, via various intracellular signal cascades, AMPAR (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor) insertion and long-term potentiation (LTP) induction in activated neurons while providing for a stable number of AMPARs during baseline activity (15, 17–23). An association between alcohol consumption and SYNGAP1 has been described recently in mice, where SynGAP protein was significantly downregulated in animals undergoing alcohol withdrawal (24).
The aim of the current study was to validate our earlier finding of significantly altered DNAm patterns of SYNGAP1 (i.e. cg02652579) in more easily accessible somatic tissue – peripheral venous whole blood and saliva – as well as female AUD patients. SYNGAP1 was prioritized for validation as it was among the top hits exhibiting this therapy-associated reversal pattern, suggesting its potential involvement in AUD pathophysiology and response to treatment. Therefore, we investigated the potential of SYNGAP1 DNAm to serve as a novel epigenetic biomarker for AUD diagnosis as well as withdrawal therapy outcome. Our study may support the understanding of underlying molecular processes, which could open new perspectives on SynGAP as a possible therapeutic target, enabling personalized therapy options and a more effective health care.
2 Methods
2.1 Study subjects
In total, 147 participants were included in the study between 2020 and 2023. The patient group consisted of 64 individuals diagnosed with a severe form of AUD (Alcohol dependence) according to the International Statistical Classification of Diseases and Related Health Problems, 10th Revision (ICD-10 (25),). Patients underwent a three-weeks inpatient qualified withdrawal treatment according to the German S3 guideline on alcohol related disorders (26) at the Department of Psychiatry and Psychotherapy of the University Hospital Tübingen. Samples and data have been collected at hospital admission (T1), as well as after three-weeks of therapy (T2). Samples and data of 83 control individuals have also been collected. At T2, 134 participants (N = 53 AUD patients, N = 81 healthy control individuals, Supplementary Table S1) remained in the study. Of both groups, individuals with comorbid substance use disorder other than nicotine or alcohol and with comorbid psychiatric disorders other than Major Depressive Disorder were excluded. At both time points, the following self-administered questionnaires were assessed: alcohol consumption using the Alcohol Use Disorder Identification Test (AUDIT (27),) for alcohol consumption and Obsessive-Compulsive Drinking Scale (OCDS (28),) for alcohol craving (Supplementary Table S1).
All participants were of European descent and aged between 20 and 71, sampling numbers and details are shown in Supplementary Table S1. They provided informed written consent. The study was approved by the ethics committee of the University of Tübingen (Reference number 264/2018 BO2) and was conducted in accordance with the Declaration of Helsinki.
2.2 DNA methylation analysis
Ethylenediaminetetraacetic (EDTA) blood and saliva samples (in Oragene® DNA Collection Kits, DNA Genotek, Ottawa, Ontario, Canada) were collected at both time points (T1 and T2). The DNA was extracted from blood samples using the QIAamp® DNA Blood-Maxi Kit (Qiagen, Hilden, Germany) and with Oragene® prepIT•L2P (DNA Genotek, Ottawa, Ontario, Canada) for saliva samples, respectively, according to the manufacturer’s instructions. The DNA was stored at -20 °C until proceeding and bisulfite converted with EpiTect® Fast DNA Bisulfit Kit (Qiagen, Hilden, Germany). The region of interest within the promotor region of SYNGAP1 (hg19, chr6:33386818-33387117) was amplified using the PyroMark PCR Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. PCR primers (Metabion, Planegg, Germany) were as follows: PCR forward primer: 5 ́-GAG GGG TTA ATG AGA GGT AGA GAG GTG-3 ́; PCR reverse primer: Biotin-5’- - CCC CAC TTC CCT ACC CTA AAA CC -’3. The PCR products were quality-controlled on an agarose gel and subsequently pyrosequenced with the PyroMark® Q24 using the Pyromark Gold Q24 reagents (Qiagen, Hilden, Germany) and the following sequencing primer: 5’-TGG TTT GGT GGT GGG GAT GTT-3’. The analyzed CpG site (cg02652579) is located at chr6:33386967 (hg 19). The DNAm level was analyzed using the PyroMark® software (Version Q24 2.0.7). At least two replicates of the PCR and sequencing reaction were performed for each sample. Only replicates with a deviation of ≤ 3% between runs were further analyzed. In all steps of the protocol, samples were arranged in a balanced order to avoid batch effects.
2.3 Statistical analysis and visualization
All analyses were performed using the software environment R and Python. Statistical tests, that are available within the R package ggpubr (version 0.6.0) (29) or the Python package stat.test (30) were used depending on the analysis specified in the following sections.
Distribution of the values per group, variable (such as age and questionnaire scores) and time point of sampling was analyzed applying the Shapiro-Wilk-test (Supplementary Table S2). To investigate the effects of AUD and its therapy on SYNGAP1 DNA methylation levels, a linear mixed-effects model (using the R package lme4 (31)) was fitted including age, sex and smoking as covariates using the following formula: DNAm ~ group*time + group*smoking + age + sex + (1|ID).
For the post-hoc tests, normally distributed values (i.e., DNAm data of blood samples) were analyzed with parametric student´s t-test. Non-parametric tests (Mann-Whitney U test for independent samples and Wilcoxon signed rank test for paired data) were applied for not-normally distributed data. Benjamini-Hochberg correction (32) was performed to correct for multiple testing and therefore, protect against false positive or Type 1 errors. An adjusted p-value was calculated for the respective number of tests for time-wise demographic/clinical variables as well as DNAm data of blood and saliva independently. An adjusted p-value (p.adj.) <.050 was considered as significant. Effect sizes were calculated using Cohen’s d (33).
3 Results
The study sample included 64 AUD patients and 83 healthy control individuals (Table 1, Supplementary Table S1). Although age, sex and smoking behavior of both groups were aimed to be matched throughout the recruitment process, the two groups still revealed significant differences: Healthy control individuals (HC) were significantly younger (40.64 ± 13.73, W = 3708, p.adj. < 0.001, Table 1) and included more females (66% females, X-squared = 9.71, df = 1, p = 0.002, Table 1) than patients (age: 49.8 ± 11.47 years, 39% females, Table 1). Although assessed, it was not possible to match the groups for smoking status resulting in a large overlap of the variables AUD status and smoking status (77% of the AUD patients were smokers and 95% of the healthy control group were non-smokers, X-squared = 74.38, df = 1, p < 0.001).
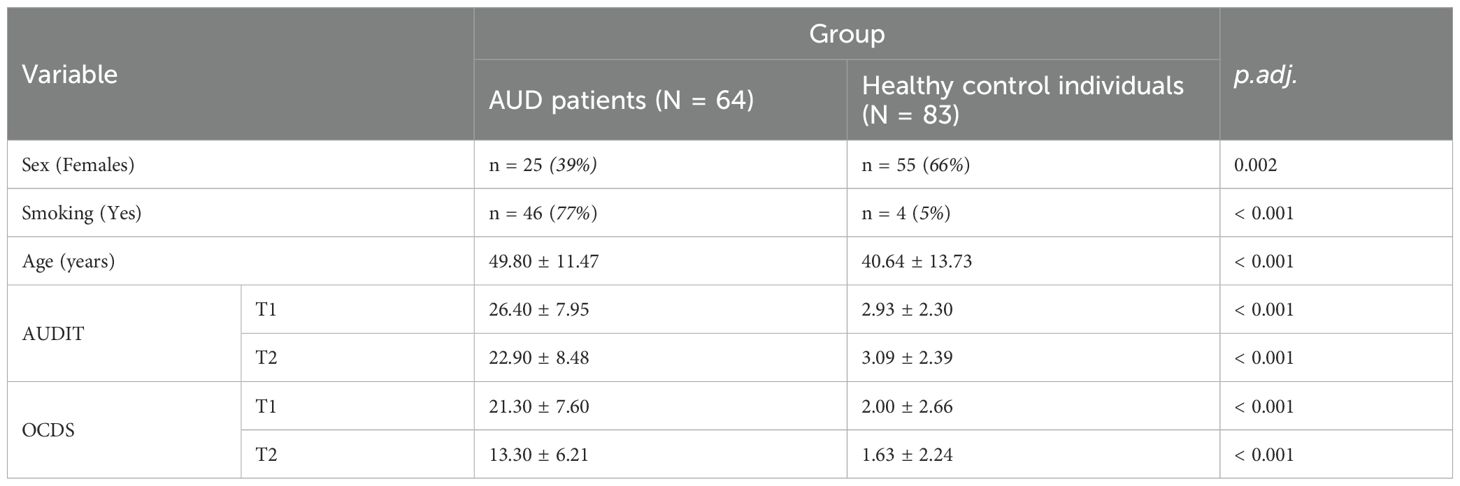
Table 1. Demographic and clinical information of the study cohort.
AUDIT scores were significantly higher in patients at both time points (T1: AUD: 26.40 ± 7.95, HC: 2.93 ± 2.30, p < 0.001; T2: AUD: 22.90 ± 8.48, HC: 3.09 ± 2.39, WT1 = 4573, p.adj.T1 < 0.001; WT2 = 1064, p.adj.T2 < 0.001; Table 1). OCDS scores were also significantly higher at both time points (T1: AUD: 21.30 ± 7.60, HC: 2.00 ± 2.66, WT1 = 4946, p.adj.T1 < 0.001; T2: AUD: 13.30 ± 6.21, HC: 1.63 ± 2.24; WT2 = 4059, p.adj.T2 < 0.001; Table 1), which shows elevated craving and obsessive tendencies towards alcohol in AUD patients. All questionnaire scores significantly improved post therapy in patients (OCDS: V = 1154, p.adj. < 0.001 (nT1 = 60, nT2 = 52); AUDIT: V = 198, p.adj. = 0.027 (nT1 = 56, nT2 = 24)), showing a tendency of positive effects of the detoxification treatment on drinking behavior and withdrawal of AUD patients.
To investigate the effects of AUD and its therapy on SYNGAP1 DNAm in blood while accounting for potential effects of demographic variables, a linear mixed-effects model with the factors group (AUD patients vs. healthy control individuals), time (pre and post withdrawal treatment) as well as smoking status and their interaction together with age and sex was fitted. A significant effect of time was revealed (Std. Error = 0.366, p = 0.012, Supplementary Table S3). However, neither a significant effect of AUD status (Std. Error = 1.202, p = 0.758, Supplementary Table S3) nor of the interaction of AUD status and time (reflecting withdrawal treatment, Std. Error = 1.108, p = 0.488) was observed.
As previously noted, unfortunately, smoking status was strongly overlapping with AUD status in the cohort (Table 1). To address this, we included both smoking status and the interaction between AUD and smoking status in the model. However, neither smoking status (Std. Error = 1.918, p = 0.163) nor the interaction term reached significance (Std. Error = 1.098, p = 0.644, Supplementary Table S3). Furthermore, neither age (Std. Error = 0.026, p = 0.611) nor sex (Std. Error = 0.736, p = 0.556) had a significant effect on SYNGAP1 DNAm in blood.
However, post-hoc tests comparing AUD patients and healthy control individuals revealed that prior to the three-weeks inpatient withdrawal treatment, SYNGAP1 DNAm of patients was significantly higher with an average of 77.8 ± 4.78% compared to that of healthy control individuals with an average of 76.1 ± 3.77% at T1 (t = 2.30, p.adj. = 0.047; Cohen’s d = 0.41, AUD patients: n = 64, Healthy controls: n =83, Figure 1A).
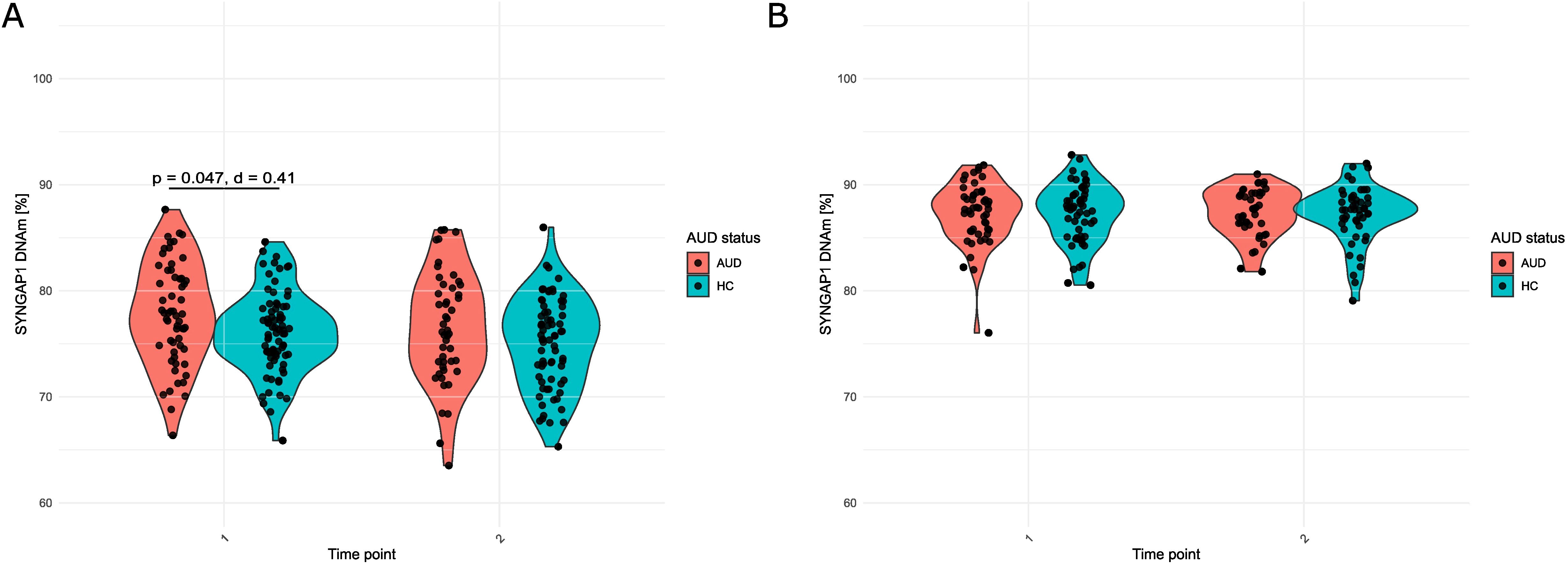
Figure 1. SYNGAP1 DNAm (%) in (A) Blood and (B) Saliva of patients (AUD) and healthy control individuals (HC) at T1 and T2. For A, student’s t-test with Benjamini-Hochberg correction was used. For B, Mann-Whitney U test with Benjamini-Hochberg correction was used. For A, Cohen’s d is additionally reported.
After the three-weeks inpatient withdrawal treatment (T2), SYNGAP1 DNAm of AUD patients remained without significant changes compared to T1 (t = 1.20, p.adj. = 0.237; Cohen’s d = 0.16, n = 46). Also, in healthy control individuals, SYNGAP1 DNAm levels in blood did not significantly change compared to T1 (t = 1.71, p.adj. = .184; Cohen’s d = 0.18, n = 72). However, the difference in DNAm between the groups at T1 was no longer observed at T2 (t = 1.68, p.adj. = 0.097, Cohen’s d = 0.32).
The same way, we analyzed SYNGAP1 DNAm in saliva of AUD patients in comparison to healthy controls before and after withdrawal treatment. SYNGAP1 DNAm of saliva was in average higher compared to blood SYNGAP1 DNAm (DNAmsaliva(AUD): 87.3 ± 2.91% and DNAmsaliva(HC): 87.3 ± 2.78% at T1). The linear mixed effects modelling did not reveal any significant effects of AUD status or any other tested variable (smoking, age, sex and time as well as the interaction of AUD status and time or smoking status, respectively) on saliva DNAm (Supplementary Table S3). Replacing AUD status with smoking status in the model revealed similar results (Supplementary Table S4).
Posthoc tests furthermore confirmed stable saliva DNAm levels throughout treatment (DNAmsaliva(AUD): 87.4 ± 2.26% and DNAmsaliva(HC): 87.2 ± 2.72% at T2 compared to T1 mentioned before; WAUD = 458, p.adj.AUD = 0.700, Cohen’s dAUD = 0.14, WHC = 585, p.adj.HC = 0.830, Cohen’s dHC = 0.00) without an influence of AUD (WT1 = 1388, p.adj.T1 = 0.921, Cohen’s dT1 = 0.00, WT2 = 1064, p.adj.T2 = 0.921, Cohen’s dT2 = 0.07, Figure 1B).
4 Discussion
In the present study, we investigated DNA methylation of a CpG site (cg02652579) in the promotor region of SYNGAP1 in whole blood and saliva of AUD patients in a longitudinal study design – before (T1) and after (T2) a three-week inpatient withdrawal treatment – compared to healthy control individuals. Analyzing SYNGAP1 DNAm in whole blood of 64 AUD patients and 83 controls, we could not confirm our previous results of lower DNA methylation levels in CD3+ T cells in patients than in control individuals. While a linear mixed-effects model including AUD status and relevant covariates, revealed no significant differences in SYNGAP1 DNAm, post-hoc analyses showed higher SYNGAP1 DNAm in patients prior to treatment compared to controls. After withdrawal therapy, this difference was no longer evident. In saliva, no significant differences in SYNGAP1 DNAm were detected between groups, and therapy showed no effect. Altered DNAm in association with AUD has been shown before by several studies on an epigenome-wide ( (8, 9, 34) as well as candidate gene level (35, 36). In a previous epigenome-wide study, we showed reduced methylation of the same CpG site (cg02652579) associated with SYNGAP1 in CD3+ cells of AUD patients (13). Moreover, SYNGAP1 expression has been identified to be correlated with alcohol withdrawal in mice brains (24). Interestingly, Witt et al. (2022) observed a significantly hypomethylated CpG site (cg07573985), which is 500 bp upstream of cg02652579, in blood of AUD patients (37). Although they measured hypomethylation rather than the hypermethylation we identified for CpG site cg02652579, their data support the notion that SYNGAP1 DNAm is influenced by AUD.
Statistically significant effects of the three-week inpatient withdrawal therapy on the blood DNAm levels were not observed. Therefore, a potential dysregulation of SYNGAP1 on the DNA methylation levels as revealed by the groupwise post hoc test could be either consistent or – as the differential methylation of SYNGAP1 observed at T1 was no longer present at T2 – the small size of our sample does not allow definitive conclusions, but leaves the trend of reversing towards healthy levels after therapy. Brückmann et al. identified such a reversal of cg02652579 methylation in their epigenome-wide approach in CD3+ cells of AUD patients undergoing withdrawal therapy, although in this case, the initially lower methylation increased with therapy approximating the healthy control levels (13). In our study, the initial higher cg02652579 methylation showed a tendency of decreasing towards control levels. Moreover, the general tendency of SYNGAP1 DNAm reversal after withdrawal therapy is supported by the findings of Witt et al., who identified two other CpG sites within the SYNGAP1 gene body, whose methylation levels changed with therapy: cg01069468 (first intron) and cg26257411 (third intron), both of which were higher methylated post treatment compared to prior (37).
Taken together, we were not able to validate the findings of Brückmann et al. (2017) in our study. The opposite direction of alteration observed in our recent data could be attributed to differences in the study materials analyzed, as DNAm varies widely across tissues (38). This is further supported by our data from whole blood and saliva that show different methylation levels of the same CpG site within the same individuals. It is plausible that SYNGAP1 DNA methylation does not exhibit a uniform pattern of dysregulation across tissues in AUD, but instead reflects heterogeneous or context-specific changes. Furthermore, Brückmann et al. studied DNAm in a cohort only consisting of males. Therefore, even if the AUD diagnosis is the same in male and female patients in our cohort, different drinking patterns may induce differential DNAm of SYNGAP1. For example, women with AUD may demonstrate a telescoping pattern—initiating drinking later than men but advancing more rapidly to dependence and treatment in clinical samples (39). Furthermore, due to sex-specific biological differences in alcohol metabolism (e.g., lower total body water, reduced dehydrogenase activity), women tend to reach higher blood alcohol levels than men from equivalent intake and are more prone to harm, even at lower drinking levels (40, 41). However, males and females revealed no differences in SYNGAP1 DNAm in our cohort.
SynGAP, encoded by SYNGAP1, plays a central role in excitatory synaptic networks, including the postsynaptic density and NMDAR complexes, where it regulates excitability and plasticity (13–16, 38, 39). Because the analyzed CpG site is located in the promoter region, higher DNA methylation could suppress SYNGAP1 expression (42, 43), possibly leading to reduced SynGAP protein and downstream signaling changes involving Ras/Rab/Rap, ERK, and AMPAR insertion (15, 17, 18, 20–22, 44). This may hypothetically resemble chronic ethanol effects, which have been linked to altered NMDAR activity, AMPAR expression, and increased hippocampal excitability in rodents (45–52). These interpretations remain highly speculative and require direct experimental validation. While direct evidence linking SYNGAP1 DNAm to AUD symptoms remains limited, dysregulation of synaptic gene methylation is increasingly recognized in AUD pathophysiology (53, 54). Studies have shown that alcohol exposure alters DNA methylation in genes related to synaptic function and neuronal communication, which may influence AUD-related behaviors (55). Although SYNGAP1 methylation itself has not been extensively studied in the context of AUD, its role in synaptic plasticity suggests potential involvement in molecular mechanisms underlying addiction and symptom severity. Further targeted studies are warranted to explore SYNGAP1 methylation changes in AUD and their clinical implications.
Epigenetic marks vary fundamentally between individuals and different somatic tissues (56, 57). The choice of tissue and cell type to analyze in order to provide robust information about epigenetic mechanisms concerning the respective research object is substantial and challenging (38). In an online tool created by Hannon et al., a trend of correlation between SYNGAP1 DNAm in blood and the prefrontal cortex was displayed (r = 0.219, p = 0.061 (58)). The prefrontal cortex is especially intertwined in the neurocircuitry of addiction and it is ascribed a central position in the controlling of craving (59). Simultaneously, its activation decreases and impedes decision making and self-regulation (59). Therefore, a neuronal activation during craving would be correlated with an increase in SYNGAP1 DNAm, which would enable glutamatergic activity. This is coherent with our finding of a significantly higher SYNGAP1 DNAm in blood of patients compared to control individuals and substantiates the potential as a possible diagnostic biomarker. However, as we, as well as Brückmann et al. (2017), examined peripheral tissues, we are not able to draw final conclusions on the regulation of SYNGAP1 in the brain of AUD patients through differential DNAm. A potentially tissue dependent epigenetic regulation of SYNGAP1 is supported by our findings in saliva, where we did not identify any effects of AUD on SYNGAP1 DNAm. Although in an earlier study, an impact of hazardous drinking behavior on DNA methylation was observed in saliva (60), SYNGAP1 sites were not among the differentially methylated CpG sites. We therefore conclude that SYNGAP1 DNAm in saliva cannot be used as a biomarker for AUD diagnosis or therapy outcome. However, saliva DNA methylation analysis faces unique technical challenges, including contamination with bacterial DNA, DNA fragmentation, and variability in cell types, which can affect data quality and sensitivity. Therefore, technical limitations may contribute to the null findings for SYNGAP1 methylation in saliva, warranting cautious interpretation and further methodological refinement.
Interestingly, Brückmann et al. restricted their analyses to smokers and observed different SYNGAP1 methylation patterns. Smoking is known to exert widespread epigenetic effects, including changes in DNA methylation across multiple loci (Zillich 2022), which could interact with or mask alcohol-related methylation signals. Thus, differences between studies may partly reflect the inclusion of non-smokers in our sample, highlighting a potential modulatory role of smoking on SYNGAP1 DNA methylation.
Taken together, this study has several limitations: Given the small sample, the study was likely underpowered to detect effects of small magnitude. Additionally, sample size (especially of patients) decreased from T1 to T2, leading to reduced sample sizes over time. While the longitudinal mixed models applied can accommodate missing data, the smaller numbers remain a limitation for post hoc comparisons of change between time points. Moreover, the AUD patient group and the healthy control group were not properly matched concerning age and sex. Although we have examined these variables for their potential to confound our results in a mixed-effects model, hidden effects cannot be excluded. In addition, smoking was assessed as a binary yes/no variable, which may have obscured differences in intensity, duration, or recency of use. This simplification could reduce statistical power, mask dose–response relationships, and introduce residual confounding. Furthermore, smoking status largely overlapped with AUD status. Therefore, it is not possible to distinguish between AUD and smoking and the effects of these variables on SYNGAP1 DNAm. In order to tackle this problem within our data, we included not only smoking status, but also the interaction of smoking and AUD status (to analyze potential additive or interactive effects) in our linear mixed-effects models, which did not reveal any notable effect. Moreover, in this study, cell-type composition measures were not available for the blood or saliva samples analyzed. As methylation levels can vary substantially across cell types, this represents a potential confounding factor that may influence interpretation of DNA methylation results. While computational deconvolution methods exist for genome-wide methylation data, they are not applicable for targeted, single-gene methylation assays due to limited coverage. Therefore, the effects of cell-type heterogeneity could not be directly assessed or corrected in our analyses. The SYNGAP1 DNA methylation differences observed in our study (~1%) are substantially smaller than the 6% reported by Brückmann et al., which may limit their potential functional impact; however, a 1% difference in DNAm is small but not necessarily negligible, as its significance depends on CpG location, tissue/cell type, and the biological context of the gene, and for dosage-sensitive neural genes even minor changes could theoretically influence protein levels and downstream signaling. Moreover, gene expression underlies a complex network of regulatory factors (61) of which DNAm represents only one (62). Unfortunately, literature has been limited to gene expression or DNAm of SYNGAP1. Investigations into additional mechanisms related to SYNGAP1 expression represent a necessary topic of research to provide a more complete picture of its regulation in general and specifically in association with alcohol consumption and AUD.
In conclusion, differential DNAm of SYNGAP1 could not be reliably validated in comparison to the previous study of Brückmann et al. (13) in whole blood, although differential methylation levels were observed when not including potential confounding factors. When extending the analysis to saliva, we observed no differences in SYNGAP1 DNAm comparing AUD patients and healthy control individuals. We neither observed an effect of withdrawal therapy on SYNGAP1 DNAm in whole blood, nor in saliva. As the effects in blood were small and there were no effects in saliva, we conclude that SYNGAP1 DNAm provides restricted potential as a biomarker for AUD diagnosis – perhaps as part of a panel – but not therapy. An important challenge for future studies is the identification of biomarkers with stronger effects in sample materials that meet the requirement for both informative value and convenient access and analysis.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by Ethics committee of the University of Tuebingen Gartenstraße 47 72074 Tübingen 264/2018BO2. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
SE: Conceptualization, Formal analysis, Investigation, Supervision, Visualization, Writing – original draft, Writing – review & editing. CK: Investigation, Methodology, Writing – review & editing. SP: Methodology, Writing – review & editing. MZ: Methodology, Writing – review & editing. VN: Conceptualization, Funding acquisition, Investigation, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was funded by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) under DFG NI 1332/13-1. We acknowledge support by Open Access Publishing Fund of University of Tuebingen.
Acknowledgments
We would like to express our appreciation to all participants for their contributions. Furthermore, we thank all residents present during the time of recruitment for contributing to the recruitment process. We furthermore thank the German Center for Mental Health (DZPG) for infrastructural support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2025.1661760/full#supplementary-material
References
1. World Health Organization. Global status report on alcohol and health 2018 (2018). Available online at: https://iris.who.int/handle/10665/274603 (Accessed June 1, 2025).
2. Nieratschker V, Batra A, and Fallgatter AJ. Genetics and epigenetics of alcohol dependence. J Mol Psychiatry. (2013) 1:11. doi: 10.1186/2049-9256-1-11
3. Reilly MT, Noronha A, Goldman D, and Koob GF. Genetic studies of alcohol dependence in the context of the addiction cycle. Neuropharmacology. (2017) 122:3–21. doi: 10.1016/j.neuropharm.2017.01.017
4. Longley MJ, Lee J, Jung J, and Lohoff FW. Epigenetics of alcohol use disorder-A review of recent advances in DNA methylation profiling. Addict Biol. (2021) 26:e13006. doi: 10.1111/adb.13006
5. Campagna MP, Xavier A, Lechner-Scott J, Maltby V, Scott RJ, Butzkueven H, et al. Epigenome-wide association studies: current knowledge, strategies and recommendations. Clin Epigenet. (2021) 13:214. doi: 10.1186/s13148-021-01200-8
6. Berkel TD and Pandey SC. Emerging role of epigenetic mechanisms in alcohol addiction. Alcoholism: Clin Exp Res. (2017) 41:666–80. doi: 10.1111/acer.13338
7. Zhou H and Gelernter J. Human genetics and epigenetics of alcohol use disorder. J Clin Invest. (2024) 134:1–13. doi: 10.1172/JCI172885
8. Lohoff FW, Clarke TK, Kaminsky ZA, Walker RM, Bermingham ML, Jung J, et al. Epigenome-wide association study of alcohol consumption in N = 8161 individuals and relevance to alcohol use disorder pathophysiology: identification of the cystine/glutamate transporter SLC7A11 as a top target. Mol Psychiatry. (2022) 27:1754–64. doi: 10.1038/s41380-021-01378-6
9. Zillich L, Frank J, Streit F, Friske MM, Foo JC, Sirignano L, et al. Epigenome-wide association study of alcohol use disorder in five brain regions. Neuropsychopharmacology. (2022) 47:832–9. doi: 10.1038/s41386-021-01228-7
10. Zillich L, Poisel E, Frank J, Foo JC, Friske MM, Streit F, et al. Multi-omics signatures of alcohol use disorder in the dorsal and ventral striatum. Transl Psychiatry. (2022) 12:190. doi: 10.1038/s41398-022-01959-1
11. Lohoff FW, Roy A, Jung J, Longley M, Rosoff DB, Luo A, et al. Epigenome-wide association study and multi-tissue replication of individuals with alcohol use disorder: evidence for abnormal glucocorticoid signaling pathway gene regulation. Mol Psychiatry. (2020) 26:2224–37. doi: 10.1038/s41380-020-0734-4
12. White JD, Rojas-Soto M, Oguni A, and Kennedy MB. Alcohol use disorder-associated DNA methylation in the nucleus accumbens and dorsolateral prefrontal cortex. medRxiv. (2024) 17:2024.01.17.23300238. doi: 10.1101/2024.01.17.23300238
13. Bruckmann C, Islam SA, MacIsaac JL, Morin AM, Karle KN, Di Santo A, et al. DNA methylation signatures of chronic alcohol dependence in purified CD3(+) T-cells of patients undergoing alcohol treatment. Sci Rep. (2017) 7:6605.
14. Chen HJ, et al. A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron. (1998) 20:895–904. doi: 10.1016/S0896-6273(00)80471-7
15. Kim JH, Liao D, Lau LF, and Huganir RL. SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron. (1998) 20:683–91. doi: 10.1016/S0896-6273(00)81008-9
16. Araki Y, Hong I, Gamache TR, Ju S, Collado-Torres L, Shin JH, et al. SynGAP isoforms differentially regulate synaptic plasticity and dendritic development. Elife. (2020) 9:e56273. doi: 10.7554/eLife.56273
17. Araki Y, Zeng M, Zhang M, and Huganir RL. Rapid dispersion of SynGAP from synaptic spines triggers AMPA receptor insertion and spine enlargement during LTP. Neuron. (2015) 85:173–89. doi: 10.1016/j.neuron.2014.12.023
18. Kilinc M, Creson T, Rojas C, Aceti M, Ellegood J, Vaissiere T, et al. Species-conserved SYNGAP1 phenotypes associated with neurodevelopmental disorders. Mol Cell Neurosci. (2018) 91:140–50. doi: 10.1016/j.mcn.2018.03.008
19. Cornelia Koeberle S, Tanaka S, Kuriu T, Iwasaki H, Koeberle A, Schulz A, et al. Developmental stage-dependent regulation of spine formation by calcium-calmodulin-dependent protein kinase IIalpha and Rap1. Sci Rep. (2017) 7:13409. doi: 10.1038/s41598-017-13728-y
20. Krapivinsky G, Medina I, Krapivinsky L, Gapon S, and Clapham DE. SynGAP-MUPP1-CaMKII synaptic complexes regulate p38 MAP kinase activity and NMDA receptor-dependent synaptic AMPA receptor potentiation. Neuron. (2004) 43:563–74. doi: 10.1016/j.neuron.2004.08.003
21. Rumbaugh G, Adams JP, Kim JH, and Huganir RL. SynGAP regulates synaptic strength and mitogen-activated protein kinases in cultured neurons. Proc Natl Acad Sci U S A. (2006) 103:4344–51. doi: 10.1073/pnas.0600084103
22. Wang CC, Held RG, and Hall BJ. SynGAP regulates protein synthesis and homeostatic synaptic plasticity in developing cortical networks. PLoS One. (2013) 8:e83941. doi: 10.1371/journal.pone.0083941
23. Zhu JJ, Qin Y, Zhao M, Van Aelst L, and Malinow R. Ras and rap control AMPA receptor trafficking during synaptic plasticity. Cell. (2002) 110:443–55. doi: 10.1016/S0092-8674(02)00897-8
24. Kong W, Huang S, Chen Z, Li X, Liu S, Zhang Z, et al. Proteomics and weighted gene correlated network analysis reveal glutamatergic synapse signaling in diazepam treatment of alcohol withdrawal. Front Pharmacol. (2022) 13:1111758. doi: 10.3389/fphar.2022.1111758
25. World Health Organization. International Statistical Classification of Diseases and Related Health Problems. 10th ed. Geneva: World Health Organization (2016).
26. Guenthner A, Weissinger V, Fleischmann H, Veltrup C, Jäpel B, Längle G, et al. Health care organization-the new German S3-guideline on alcohol-related disorders and its relevance for health care. Die Rehabil. (2018) 57:314–20. doi: 10.1055/s-0043-118955
27. Babor TF, Higgins-Biddle JC, Saunders JB, and Monteiro MG. AUDIT - The Alcohol Use Disorders Identification Test: Guidelines for Use in Primary Care. 2nd ed. Geneva (2001).
28. Mann K and Ackermann K. Die OCDS-G: Psychometrische Kennwerte der deutschen Version der Obsessive Compulsive Drinking Scale. Sucht. (2000) 46:90–100. doi: 10.1024/suc.2000.46.2.90
29. Kassambara A. ggpubr: ‘ggplot2’ Based Publication Ready Plots. R package version 0.6.0. Vienna, Austria: Comprehensive R Archive Network (CRAN) (2023).
30. Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods. (2020) 17:261–72. doi: 10.1038/s41592-019-0686-2
31. Bates D, Maechler M, Bolker B, and Walker S. Fitting linear mixed-effects models usinglme4. J Stat Software. (2015) 67:1–48. doi: 10.18637/jss.v067.i01
32. Benjamini Y and Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Society: Ser B (Methodological). (2018) 57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
33. Cohen J. Statistical power analysis for the behavioral sciences. New York, NY: Routledge (2013).
34. Philibert RA, Penaluna B, White T, Shires S, Gunter T, Liesveld J, et al. A pilot examination of the genome-wide DNA methylation signatures of subjects entering and exiting short-term alcohol dependence treatment programs. Epigenetics. (2014) 9:1212–9. doi: 10.4161/epi.32252
35. Hillemacher T, Frieling H, Luber K, Yazici A, Muschler MAN, Lenz B, et al. Epigenetic regulation and gene expression of vasopressin and atrial natriuretic peptide in alcohol withdrawal. Psychoneuroendocrinology. (2009) 34:555–60. doi: 10.1016/j.psyneuen.2008.10.019
36. Biermann T, Reulbach U, Lenz B, Frieling H, Muschler MAN, Hillemacher T, et al. N-methyl-D-aspartate 2b receptor subtype (NR2B) promoter methylation in patients during alcohol withdrawal. J Neural Transm (Vienna). (2009) 116:615–22. doi: 10.1007/s00702-009-0212-2
37. Witt SH, Frank J, Frischknecht U, Treutlein J, Streit F, Foo JC, et al. Acute alcohol withdrawal and recovery in men lead to profound changes in DNA methylation profiles: a longitudinal clinical study. Addiction. (2020) 115:2034–44. doi: 10.1111/add.15020
38. Harlaar N and Hutchison KE. Alcohol and the methylome: Design and analysis considerations for research using human samples. Drug Alcohol Depend. (2013) 133:305–16. doi: 10.1016/j.drugalcdep.2013.07.026
39. Randall CL, Roberts JS, Del Broca FK, Carroll KM, Connors GJ, and Mattson ME. Telescoping of landmark events associated with drinking: a gender comparison. J Stud Alcohol. (1999) 60:252–60. doi: 10.15288/jsa.1999.60.252
40. Frezza M, di Padova C, Pozzato G, Terpin M, Baraona E, and Lieber CS. High blood alcohol levels in women: the role of decreased gastric alcohol dehydrogenase activity and first-pass metabolism. New Engl J Med. (1990) 322:95–9. doi: 10.1056/NEJM199001113220205
41. Ely M, Hardy R, Longford NT, and Wadsworth ME. Gender differences in the relationship between alcohol consumption and drink problems are largely accounted for by body water. Alcohol Alcoholism. (1999) 34:894–902. doi: 10.1093/alcalc/34.6.894
42. Reik W and Dean W. DNA methylation and mammalian epigenetics. Electrophoresis. (2001) 22:2838–43. doi: 10.1002/1522-2683(200108)22:14<2838::AID-ELPS2838>3.0.CO;2-M
43. Brenet F, Moh M, Funk P, Feierstein E, Viale AJ, Socci ND, et al. DNA methylation of the first exon is tightly linked to transcriptional silencing. PloS One. (2011) 6:e14524. doi: 10.1371/journal.pone.0014524
44. Oh JS, Manzerra P, and Kennedy MB. Regulation of the neuron-specific ras GTPase-activating protein, synGAP, by Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. (2004) 279:17980–8. doi: 10.1074/jbc.M314109200
45. Lovinger DM, White G, and Weight FF. NMDA receptor-mediated synaptic excitation selectively inhibited by ethanol in hippocampal slice from adult rat. J Neurosci. (1990) 10:1372–9. doi: 10.1523/JNEUROSCI.10-04-01372.1990
46. Blitzer RD, Gil O, and Landau EM. Long-term potentiation in rat hippocampus is inhibited by low concentrations of ethanol. Brain Res. (1990) 537:203–8. doi: 10.1016/0006-8993(90)90359-J
47. Fujii S, Yamazaki Y, Sugihara T, and Wakabayashi I. Acute and chronic ethanol exposure differentially affect induction of hippocampal LTP. Brain Res. (2008) 1211:13–21. doi: 10.1016/j.brainres.2008.02.052
48. Nelson TE, Ur CL, and Gruol DL. Chronic intermittent ethanol exposure enhances NMDA-receptor-mediated synaptic responses and NMDA receptor expression in hippocampal CA1 region. Brain Res. (2005) 1048:69–79. doi: 10.1016/j.brainres.2005.04.041
49. Bruckner MK, Rossner S, and Arendt T. Differential changes in the expression of AMPA receptor genes in rat brain after chronic exposure to ethanol. an in situ hybridization study. J Hirnforsch. (1997) 38:369–76.
50. Chandler LJ, Norwood D, and Sutton G. Chronic ethanol upregulates NMDA and AMPA, but not kainate receptor subunit proteins in rat primary cortical cultures. Alcoholism: Clin Exp Res. (2006) 23:363–70. doi: 10.1111/j.1530-0277.1999.tb04123.x
51. Haugbol SR, Ebert B, and Ulrichsen J. Upregulation of glutamate receptor subtypes during alcohol withdrawal in rats. Alcohol Alcohol. (2005) 40:89–95. doi: 10.1093/alcalc/agh117
52. Ewin SE, Morgan JW, Niere F, McMullen NP, Barth SH, Almonte AG, et al. Chronic intermittent ethanol exposure selectively increases synaptic excitability in the ventral domain of the rat hippocampus. Neuroscience. (2019) 398:144–57. doi: 10.1016/j.neuroscience.2018.11.028
53. Jarczak J, Miszczak M, and Radwanska K. Is DNA methylation in the brain a mechanism of alcohol use disorder? Front Behav Neurosci. (2023) 17:957203. doi: 10.3389/fnbeh.2023.957203
54. Schuch JB, Bandeira CE, Junior JLS, Mueller D, Charao MF, da Silva BS, et al. Global DNA methylation patterns in Alcohol Use Disorder. Genet Mol Biol. (2024) 46:e20230139. doi: 10.1590/1678-4685-GMB-2023-0139
55. Meng W, Sjoeholm LK, Kononenko O, Tay N, Zhang D, Sarkisyan D, et al. Genotype-dependent epigenetic regulation of DLGAP2 in alcohol use and dependence. Mol Psychiatry. (2021) 26:4367–82. doi: 10.1038/s41380-019-0588-9
56. Hannon E, Lunnon K, Schalkwyk L, and Mill J. Interindividual methylomic variation across blood, cortex, and cerebellum: implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics. (2015) 10:1024–32. doi: 10.1080/15592294.2015.1100786
57. Cabrera-Mendoza B, Stertz L, Najera K, Selvaraj S, Teixeira AL, Meyer TD, et al. Within subject cross-tissue analyzes of epigenetic clocks in substance use disorder postmortem brain and blood. Am J Med Genet Part B: Neuropsychiatr Genet. (2023) 192:13–27. doi: 10.1002/ajmg.b.32920
58. Hannon E, et al. Blood Brain DNA Methylation Comparison Tool (2015). Available online at: https://epigenetics.essex.ac.uk/bloodbrain/ (Accessed January 16, 2024).
59. Koob GF and Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry. (2016) 3:760–73. doi: 10.1016/S2215-0366(16)00104-8
60. Harlaar N, Bryan AD, Thayer RE, Karoly HC, Oien N, and Hutchison KE. Methylation of a CpG site near the ALDH1A2 gene is associated with loss of control over drinking and related phenotypes. Alcohol Clin Exp Res. (2014) 38:713–21. doi: 10.1111/acer.12312
61. Pope SD and Medzhitov R. Emerging principles of gene expression programs and their regulation. Mol Cell. (2018) 71:389–97. doi: 10.1016/j.molcel.2018.07.017
Keywords: alcohol use disorder (AUD), SYNGAP1 gene, DNA methylation, epigenetics, validation
Citation: Edelmann S, Kummer C, Pasche S, Zimmermann M and Nieratschker V (2025) Epigenetic regulation of SYNGAP1 in alcohol use disorder in whole blood and saliva. Front. Psychiatry 16:1661760. doi: 10.3389/fpsyt.2025.1661760
Received: 08 July 2025; Accepted: 06 October 2025;
Published: 06 November 2025.
Edited by:
Mauro Ceccanti, Sapienza University of Rome, ItalyReviewed by:
Bhagyalakshmi Shankarappa, National Institute of Mental Health and Neurosciences (NIMHANS), IndiaRudong Li, Purdue University Indianapolis, United States
Copyright © 2025 Edelmann, Kummer, Pasche, Zimmermann and Nieratschker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susanne Edelmann, c3VzYW5uZS5lZGVsbWFubkBtZWQudW5pLXR1ZWJpbmdlbi5kZQ==